

Abstract
Mutant p53 and p73 have been shown to form a complex, which renders p73 inactive, but it has been previously reported that wild-type p53 does not interact with p73. Mutations in the DNA binding domain of p53 result in structural changes that are permissive for interaction with p73. In a mouse model, p73 and p53 are important to effectively induce apoptosis, and there are several specific domains of p53 that are important for its apoptotic activity including the Proline Rich Domain (PRD). Within this domain, phosphorylation of threonine 81 (Thr81) mediated by c-Jun N-terminal kinase (JNK) is important for apoptosis. We examined if phosphorylation of Thr81 was important for mutant or wild-type p53 to bind to p73. Our data shows that phosphorylation of Thr81 results in both mutant and wild-type p53 complex formation with p73. Structurally, the phosphorylation of Thr81 exposes the DNA binding domain of which is important for binding to p73. The dimerization of p53 and p73 is important for the induction of apoptotic targets such as p53 upregulated modulator of apoptosis (PUMA) and Bcl-2-associated X protein (Bax), and the induction of apoptosis in response to JNK activation. We describe how JNK phosphorylation of mutant and wild-type p53 promotes a p53/p73 complex that dictates cell fate. These findings refine the current understanding of the role of p53 and p73 and reveal a new functional role for Thr81 phosphorylation.
One Sentence Summary:
JNK phosphorylation of mutant or wild type p53 increases the interaction of p73 with mutant p53, as well as p73 and wild-type p53, leading to the induction of apoptosis (WT p53).
Introduction
Mutant p53 was first described to have a dominant-negative effect on wild-type p73 by altering the ability of p73 to bind to DNA(1). It was found that most of the gain of function p53 mutants, commonly found in human tumors, bound to p73(1). Additional studies have shown that this complex prevented the induction of apoptosis mediated by p73 (2). Later work examined the requirement of having intact p53, p73, and p63 genes in mice (3). Intriguingly, knockout of one or both alleles of individual genes of p53, p73, or p63 resulted in a loss of cell death in response to apoptotic stimuli (3). This data suggested that p53, p73, and p63 are necessary to invoke a robust apoptotic response to DNA damaging stimuli. However, further work showed that wild-type p73 and p63 would form heterodimeric (or higher order) complexes while wild-type p53 did not bind to these family members (4, 5).
Wild-type p53 has two transactivation domains, TAD1 and TAD2. TAD2 (amino acids 43–54) is located proximal to the proline rich domain (PRD) (amino acids 63–97) (6, 7). The PRD has been shown to be important for the induction of apoptotic genes(8, 9). Serine 46 (Ser46) in the second transactivation domain, and Threonine 81 (Thr81) within the PRD are two key phosphorylation sites in these domains (10–14). Several kinases have been reported to phosphorylate Ser46 and cause the transactivation of genes responsible for apoptosis (12, 15, 16). JNK has been reported to phosphorylate Thr81 and is responsible for the induction of apoptosis (17), yet the understanding of how modification of Thr81 regulates p53 activity is limited. While TAD1 of p53 has been shown to be dispensable for apoptosis (18), Thr81 and Ser46 are required.
The PRD is an important structural domain as prolyl isomerization in this domain regulates the structure of other domains of p53 (19). We rationalized that phosphorylation within this domain may also be sufficient to alter the structure of other domains of p53. We found that when wild-type p53 was bound to p73, Thr81 in the PRD was phosphorylated. Blockage of JNK signaling diminished the endogenous mtp53-p73 interaction, while activation of JNK promoted wtp53-p73 binding. Further analysis showed that p53-p73 complex formation requires phosphorylation of Thr81 of p53, and this complex resulted in the induction of apoptosis through PUMA and Bax. Thus, our findings support previously published work on genetic knockout mice (3), the proline rich domain (20, 21), and mutant p53/p73 complex formation (1, 2), but most importantly¸ improve the current understanding of the interaction between p53 and p73.
Results
JNK phosphorylation of p53 results in induction of PUMA
Since JNK is a key mediator of p53 induced apoptosis, we were curious if Thr81 was phosphorylated in mutant p53 cells. In mutant p53 cells, MDA-MB-468 (Arg273), TMD231 (Lys280), and MDA-MB-157 (truncated p53), JNK was active, as determined by phospho-Tyr183-Tyr185 JNK antibody (Fig. 1A). In addition, we found that mutant p53 was phosphorylated at Thr81, the known JNK phosphorylation site, under growth conditions. Treatment with camptothecin and anisomycin increased phosphorylation of JNK and phos-Thr81. Next we examined wild-type p53 3T3 and HME1 (human mammary epithelial 1) cells to determine if untransformed cell lines showed similar induction of JNK and p53 Thr81 phosphorylation. We found that there was limited activation of JNK or Thr81 under growth conditions, but upon camptothecin and anisomycin exposure there was a robust activation of JNK and p53 phos-Thr81 (Fig. 1B). Moreover, we examined additional wild-type p53 human foreskin fibroblast cells (BJ) and glioblastoma cell lines (U87 and SF767). In Figure 1B, we show that JNK was indeed activated and p53 was phosphorylated at Thr81 in response to treatment in all of the cell lines tested. Thus in normal and tumor cells, in response to camptothecin or anisomycin, JNK is activated and p53 (wild-type or mutant) is phosphorylated at Thr81.
Fig. 1. Phosphorylation of Thr81 of p53 in response to JNK activation.

(A) Western blot of phosphorylated JNK, phosphorylated Thr81 of p53, and actin in cellular extracts from TMD231, MDA-MB-468 and MDA-MB-157 cells treated with either camptothecin or anisomycin for the indicated times. Blots are representative of three independent experiments. (B) Western Blot of phosphorylated JNK or Thr81 of p53 and actin from cellular extract of 3T3, SF767, HME-1, BJ and U87 treated with anisomycin or camptothecin for the indicated times. Blots are representative of three independent experiments. (C) U87 or SF767 cellular extracts from DMSO, SP600125, anisomycin or pretreatment with SP600125 then anisomycin, and western blotted for PUMA and actin. Blots are representative of two independent experiments.
We examined the dependence of JNK activation on the induction of the apoptotic downstream target of p53, PUMA. PUMA was increased in cells that maintained wild-type p53 by anisomycin treatment (Fig. 1C). To show this was dependent on JNK signaling, a small molecule inhibitor to JNK (SP600125) was used. Cells were pretreated with SP600125 and then anisomycin was added. In U87 and SF767 cells (wild-type p53), PUMA was induced in response to anisomycin and this induction was blocked with pretreatment of SP600125, thereby confirming that activation of JNK was important for induction of the p53 target PUMA. To ensure that this increase in PUMA resulted in an increase in cell death, we performed a cell death assay on U87 cells using similar conditions and saw a significant increase in cell death in response to JNK activation (Supplementary Figure 1).
The predicted structure of a Thr81 phosphomimic shows a potential mode of binding to p73
To gain insight into the structural implications of phosphorylation at Thr81 of p53, we took advantage of a protein structure prediction algorithm, I-TASSER (Iterative Threading ASSEmbly Refinement) (22–24). Using I-TASSER, we generated predicted protein structure models for wild-type p53, R248W mutant p53 (a common DNA binding domain mutant), and a Thr81 phosphomimic p53 (T81E) (Fig. 2). Analysis of wild-type and mutant p53 shows distinct structural differences in the amino and carboxy termini. Thr81 phosphorylation (T81E) resulted in a substantial structural change in the tetramerization/regulatory (TET/REG) domain compared to unphosphorylated wild-type p53. The TET/REG domain of the T81E model more closely resembled the R248W mutant p53 model than the wild-type p53 model. The R248W mutation of p53 is one of the mutants that was shown to bind to p73 (1). It is probable that it is this structural change in the TET/REG domain that allows p53 to interact with p73, which explains the original finding that wild-type p53 was unable to bind to p73. The results of our structural analysis implies that phosphorylation at Thr81 of p53 is permissive of binding to p73. We also produced in silico structural models of the S46D phosphomimic p53 and a R248W/T81E mutant p53 phosphomimic (Fig. 2) to examine if there were any structural changes. The TAD2 of the S46D phosphomimic is structurally similar to the T81E phosphomimic, but distinct from either of the R248W mutant p53 models. Phosphorylation at Ser46 of p53 is needed for a robust apoptotic response, and phosphorylation at Thr81 of p53 causes a structural change similar to that of phosphorylation at Ser46. These data suggest that TAD2 and PRD are integrated into important structural changes that are associated with induction of apoptotic genes. Compared to mutant p53 alone (R248W), the Thr81 phosphomimic of R248W mutant p53 (R248W/T81E) causes an additional structural change in almost every single functional domain. This finding would suggest that the use of a JNK inhibitor may reduce the binding of mutant p53 to p73 without completely ablating the interaction.
Fig. 2. Predicted structure for a Thr81 phosphomimic reveals a possible mode of binding to p73.

(A) Whole protein represents amino acids 1–393 of p53. Transactivation domains 1 and 2 (TAD1/TAD2) represent amino acids 1–40 and 43–54. The proline rich domain (PRD) represents amino acids 63–97. The tetramerization and regulatory domains (TET/REG) represent amino acids 323–393. Thr81 is circled in the WT and R248W images of the PRD, and the phosphomimic glutamic acid is circles in the T81E image of the PRD. Arrows represent the region of a conformational change permissive of binding. (B) Protein map of p53 highlighting relevant domains and amino acids.
Phosphorylation of Thr81 of p53 promotes a p53-p73 complex
Our structural data in Fig. 2, combined with previous reports that mutant p53 bound to wild-type p73, and the observation of high basal Thr81 phosphorylation of mtp53 (Fig.1) led us to examine if blocking JNK using SP600125 could alter the mutant p53-p73 interaction endogenously. Mutant p53 was immunoprecipitated from DMSO or SP600125 treated TMD231 cellular extracts to examine the association with p73. We found that p73 co-purified with mutant p53, and this binding was decreased by 40% when JNK activity was blocked with SP600125 (Fig. 3A). This data suggests that JNK only has a modest effect on the complex formation between mutant p53 and p73 which was predicted in the structural data (Fig.2). We next tested if wild-type p53 and p73 would be affected by JNK phosphorylation. H1299 cells were transiently transfected with wild-type p53 and HA-tagged p73, and were then treated with DMSO or anisomycin. Immunoprecipitation of wild-type p53 from cellular extracts shows that p73 co-purified only when JNK was activated with anisomycin (Fig. 3B). To provide evidence that phosphorylation of Thr81 was necessary for wild-type p53 to co-purify p73, we transiently transfected wild-type p53 or a phospho-mimic where Thr81 was changed to glutamic acid (T81E) in H1299 cells. Immunoprecipitation of exogenous p53 shows that T81E, and not wild-type p53, was able to co-purify endogenous p73 (Fig. 3C) Next we immunoprecipitated endogenous p73 from U87 cell extracts treated with DMSO, anisomycin, SP600125, or pretreatment with SP600125 and then anisomycin. The data shows that endogenous wild-type p53 co-purifed with p73, only when Thr81 was phosphorylated (Fig. 3D). These data provide strong evidence that Thr81 phosphorylation is necessary for a wild-type p53-p73 complex. To definitively show the requirement of phosphorylation of Thr81, we used p53 phospho-mimics (T81E, S46D, T81E-S46D) and p73 produced in bacteria and performed a far western to examine the requirement of phosphorylation of Thr81. We fixed recombinant wild-type p53 and phospho-mimics (lane 1), as well as two positive controls (Mdm2 and MdmX, lane 2) to nitrocellulose, then incubated the membrane with recombinant p73 and blotted for p73 (Fig. 3E). In support of our cellular data shown in the previous panels, p73 only bound to T81E and not wild-type p53 or S46D p53 (Fig. 3E). We also probed for p53 in a separate lane to show the relative amounts of p53 loaded per well (Fig. 3E). Collectively, through overexpression, endogenous, and recombinant approaches we show that p73 will bind to wild-type p53 when p53 is phosphorylated at Thr81.
Fig. 3. p53-p73 complex formation and activity.

(A) Immunoprecipitiation of p53 or control IgG from TMD231 cells treated with DMSO or SP600125 and western blotted for p73 and p53. Blots are representative of four independent experiments. (B) Transient transfection of H1299 cells with p53, HA-p73 and treated with either DMSO or Anisomycin. Western blot of p53 and HA from p53 immunoprecipitated from cellular extracts. Blots are representative of two independent experiments. (C) Immunoprecipitiation of p53 or control IgG and western blot of p53 and p73 from H1299 cells transfected with wild type p53 or T81Ep53. WCL is whole cell lysate from H1299 cells. Blots are representative of two independent experiments. (D) Immunoprecipitation of endogenous p73 from U87 cells treated with SP600125, anisomycin or both and western blot of phospho-Thr81, p53, and p73. Blots are representative of two independent experiments. (E) Far western analysis of p73 binding to p53. Lane 1 corresponds to p53 and the phosphomimics of p53, and lane 2 is Mdm2 and Mdmx. Blotting for p73 is detects binding to p53 and Mdm2 and Mdmx (right panel). The relative amount of p53 on the membrane is the left panel. Blots are representative of two independent experiments.
To gain insight into where p53 and p73 were interacting, MDA157, HME, or TMD231 cells were grown on coverslips and treated with anisomycin, SP600125, or DMSO and fixed for staining with antibodies specific for p53 and p73 for confocal immunofluorescence microscopy. Anisomycin treatment caused a significant increase in colocolization between p53 and p73 in the nucleus of and HME cells (wild-type p53), but no significant increase in the nuclei of MDA157 (mutant p53) (Fig. 4A and B and Supplementary Table 1). Since TMD231 cells have mutant p53 and high basal JNK activation, we treated with the JNK inhibitor SP600125. Blockage of JNK activation caused a significant decrease in colocolization of mutant p53 and p73 in the nucleus (Fig. 4C and Supplementary Table 1), which is in accordance with the decrease in binding of mutant p53 and p73 (Fig. 3). To ensure that anisomycin was indeed activating JNK and increasing phosphorylation at Thr81, and that SP600125 was inhibiting JNK and decreasing phosphorylation at Thr81 of p53, the same cells were treated as described in the confocal experiments and lysates were western blotted for active JNK and phosphorylated Thr81 p53 (Supplementary Figure 2).
Fig. 4. p53 activation and colocalization with p73.
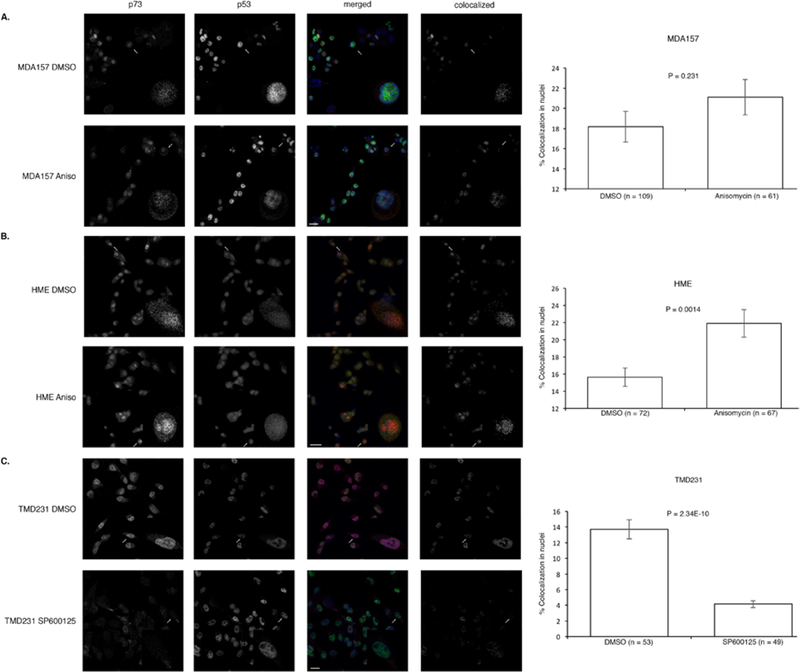
(A-C) Immunofluorescence of p53-p73 colocalization in MDA157 and HME cells treated with anisomycin or DMSO and TMD231 cells treated with SP600125 or DMSO. Error bars represent standard error of the mean. Significance was determined using a two-tailed unpaired t test. Data is representative of three independent experiments.
The JNK initiated p53-p73 pathway is important for apoptosis
We showed that phosphorylation of Thr81 of p53 resulted in an increase in PUMA (Fig. 1C) and complex formation with p73 (Fig. 3). We next examined if p53, p73, and activation of JNK was necessary for the induction of apoptosis. We showed that the removal of p53 in BJ (wild-type p53) cells resulted in resistance to JNK activated cell death by anisomycin treatment (Fig. 5A). Treatment with anisomycin for 9 hours initiated a steady increase of Bax and PUMA protein in wild-type p53 BJ cells. However, shRNA knockdown of p53 resulted in no induction in PUMA over 9 hours of anisomycin treatment. Bax protein began to increase after 6 hours of anisomycin treatment. This may be due to p53 overcoming the shRNA knockdown at 6 hours in response to prolonged anisomycin induced stress.
Fig. 5. Downstream effects of p53-p73 pathway.

(A) Western blot of Bax, PUMA, p53 and actin in cellular extracts from Empty Vector control (E.V) and knockdown p53 (shp53) BJ cells treated with anisomycin for the indicated time. Both E.V. and shp53 lanes where developed from the same western blot. Blots are representative of three independent experiments. (B) Cell viability assay of Empty Vector control (E.V.) and knockdown p53 (shp53) BJ cells stained with Methylene Blue. Cells were treated with DMSO or anisomycin for 9 hours. Data is representative of three independent experiments. (C) Relative GFP fluorescence was measured in GFP shp53 and LVTHM empty vector BJ cells. Cells were treated with DMSO or anisomycin for 48 hours. Relative fluorescence was derived from DMSO controls. Data is representative of four independent experiments. Significance was determined by a two tailed unpaired t test (***, P<0.005) (D) GFP fluorescence was measured in H1299 cells transfected with GFP alone or GFP and either wild-type or T81A p53, then treated with DMSO or anisomycin. Fold change survival was calculated as the change in survival from DMSO to anisomycin treatment and normalized to the GFP alone control. Error bars represent standard error of the mean. Data is representative of three independent experiments.
The apoptotic resistance inferred to shp53 BJ cells was further investigated by a cell death assay (Fig. 5B). shp53 BJ cells showed an improvement in cell viability compared to wild-type p53 BJ cells over 9 hours of anisomycin treatment. These BJ cells are GFP positive (shRNA vector) and fluorescence of shp53 and empty vector BJ cells showed a significant difference in response to anisomycin treatment (Fig. 5C). shp53 cell lines remained viable with anisomycin treatment, whereas vector controls showed a drastic drop in viability after treatment for 48 hours. This data suggests that the lack of the p53-p73 complex results in the resistance to apoptosis. In order to test the impact of Thr81 phosphorylation on cell death, we used a T81A point mutant and examined survival in a cell death assay (Fig. 5D). H1299 cells were transfected with GFP and either wild-type p53 or the T81A p53 point mutant and survival was determined by GFP fluorescence after anisomycin treatment. Our results show an increased survival with the T81A point mutation compared to wild-type p53, and this trend is supported by previously published data showing increased survival with p53 T81A (17).
Discussion
The tumor suppressor p53 is a highly studied protein, which has a defined role in regulating many cellular responses: cell cycle arrest, DNA repair, cell death, and metabolism. Its family member p73 has some described overlapping functions yet, unlike p53, p73 is rarely mutated in human cancer. While p73 is rarely mutated, mutation of p53 may function as a dominant negative to ablate p73 activity (1). The regulation of p53 is extremely important as it can dictate binding partners and consequently transcription of different targets, such as induction of the pten gene in response to apoptotic stimuli versus the induction of mdm2 gene expression for re-entry into the cell cycle and survival (25, 26). Increased PTEN can protect p53 from Mdm2 through the Akt pathway (10, 27–30). Thus, p53 can selectively induce PTEN expression and programmed cell death over the autoregulatory feedback loop with Mdm2 to promote cell survival (10, 31). Moreover, PTEN can form a complex with p73 and bind to the PUMA promoter to increase expression leading to the induction of apoptosis (32). These molecular and biochemical studies are supported by genetic studies in mice where the loss of p53, p73, or PTEN confers resistance to apoptotic stimuli and an increase in neoplastic development (3, 33–36). Thus, an effective response to apoptotic stimuli requires the integration of numerous intact tumor suppressors, yet how these tumor suppressors are signaled to form a complex has not previously been clearly defined.
There are well-characterized kinase signaling pathways that phosphorylate p53 and p73 in response to DNA damage, but some phosphorylation sites appear to be dispensable for the induction of apoptosis. It has been reported by several groups that Ser46 and Thr81 are important for the induction of apoptosis (10, 12, 14, 16). HIPK2 and p38 have been shown to phosphorylate Ser46 of p53 and JNK has been shown to phosphorylate Thr81 (10, 12, 14, 16). JNK is known to play a dual role in cellular proliferation as well as being activated to mediate cell death (37). p73 is a substrate for JNK as well, which leads to apoptosis (38), but the mechanism remains unclear. Analysis of apoptosis by the JNK-p73 axis in that study was performed in cells that maintained wild-type p53 (38). Notably, p53 abundance is not directly correlated with activity. In fact, p53 can be neddylated in response to growth factors, which renders p53 stable but inactive (39). Primary cells have the apoptotic pathway intact and there are deneddylating enzymes, tumor suppressors, active kinases, and post-translational modification enzymes that are activated in response to genotoxic stress that provide a robust induction of apoptosis.
There has been a renewed interest in p53 family member interactions, and several studies have highlighted the importance of the interactions between different family members (40–42). Our studies have linked the JNK-p53-p73 proteins showing that phosphorylation of p53 at Thr81 by JNK is permissive of a p53-p73 interaction (Fig. 3). Remarkably, our structural modeling analysis shows that the structure of phosphorylated Thr81 is nearly identical to that of R248W (Fig. 2). Therefore, activated JNK can convert wild-type p53 to mimic mutant p53 with regards to binding to p73 by exposing the DNA binding domain. Ser46 is proximal to the proline rich domain in the second transactivation domain, which may be important for additional structural changes to recruit other factors for gene expression. However, we also found that phosphorylation of Ser46 has no effect on binding to p73 (Fig. 3), supporting that this phosphorylation may instead be required for recruiting additional coactivators. The interaction between wild-type p53 and p73 mediated by JNK provides biochemical evidence in support of the existing animal models and gene dose sensitivity of p53 and p73 to apoptosis (33). We also show that in mutant p53 cells, JNK activity is increased resulting in a mutant p53/wild-type p73 complex with mutant p53 acting as a dominant negative on p73. These data are consistent with previous reports showing that mutant p53/p73 form a complex (2). This pathway is important biologically for apoptosis in cells that maintain wild type p53, however when p53 is mutant this pathway is engaged for survival (Fig. 6). Thus, this pathway helps to refine the understanding of how cells promote or circumvent the p53 apoptotic-signaling axis.
Fig. 6. Cell fate is dictated by p53/p73 complex formation.
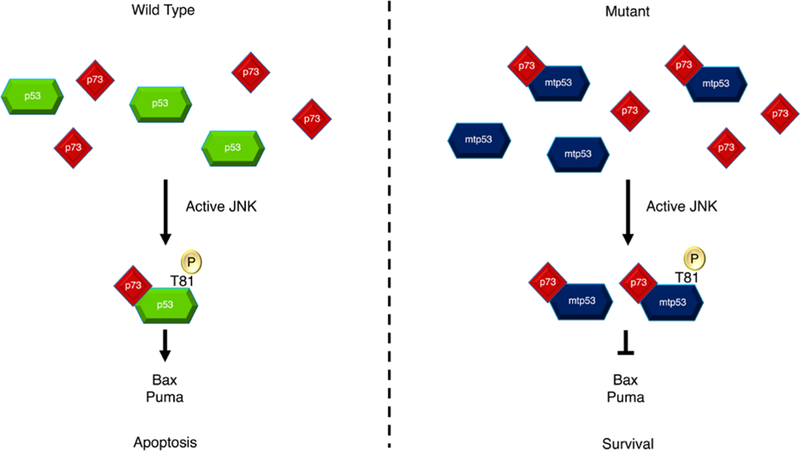
Apoptotic stimuli, for example DNA damage, results in the phosphorylation and activation of JNK. JNK phosphorylates wild-type p53 at Thr81 causing a structural change in p53 and the subsequent binding of p73. This complex drives the transcription of PUMA and Bax genes leading to apoptosis. Mutant p53 does not require phosphorylation by JNK to form a complex with p73. Phosphorylation of mutant p53 instead serves to improve the ability of p53 to bind with p73, hence increasing total complex formation. However, PUMA and Bax are not transcribed due to mutant p53’s dysfunctional DNA binding domain as well as the sequestering of p73 by complex formation. This decreases the apoptotic signal and promotes tumor survival.
Materials and Methods
Cell Culture and transfection.
H1299, TMD231, U87, HME1, SF767, 3T3, hTert BJ, MDA-MB-468 and MDA-MB-157 were cultured in Dulbecco’s Modified Eagle Media (DMEM) with 10% fetal bovine serum (FBS) supplemented with penicillin and streptomycin at 37° C humidified incubator in 5% CO2. Anisomycin, SP600125, and Camptothecin were purchased and suspended as described by the manufacturer (Sigma-Aldrich). Anisomycin was used at 30 μM, Camptothecin was used at 30 μM, and SP600125 was used at 25 μM unless otherwise noted. Transient transfections were performed using lipofectamine per manufacturer’s instructions (Life Technologies). Annexin V cell dead kit from Millipore was used to stain cells which were analyzed on a MUSE cytometer.
Plasmids, Protein purification, and Western blot.
We generated mutant p53 at Thr81 with a site directed mutagenesis kit (Life technologies). Recombinant p73 and p53 proteins were produced in BL21DE3 cells and purified as previously described (30, 32). Far Western was completed by 96 well dot blot system and recombinant proteins were placed wells and fixed to nitrocellulose by drying. Blot was blocked with 5% nonfat dry milk and incubated with recombinant p73. p73 was then detected by p73 antibody followed by secondary and an enhanced chemiluminescence (ECL) reagent. Western blot analysis used whole cell lysates lysed under denaturing conditions (6 M urea buffer), or immunoprecipitated proteins extracted with lysis buffer (32). Antibodies used for this analysis were p73 (OP1008 Millipore), phos-T81, phos-Y183-Y185-JNK, PUMA (Cell Signaling), and p53 (D0–1), Actin and GAPDH (Santa Cruz Biotech).
I-TASSER Protein Structure Prediction.
Amino acid sequences for wild-type p53, R248W p53, T81E p53, S46D, and R248W/T81E p53 were input to the I-TASSER web server as FASTA sequences. Predicted protein structures were generated using a previously described method (22–24) and analyzed using the CCP4 Molecular Graphics Program (CCP4MG) v. 2.10.6 (43). Structures were superposed using a built in function within the software.
Immunofluorescence microscopy.
MDA157, HME, or TMD231 cells were grown on glass coverslips and fixed in 4% paraformaldehyde in phospho-buffered saline (PBS) for 15 minutes, washed with PBS, and permeabilized in 1% Triton X-100 in PBS for 15 minutes. Coverslips were blocked in 5% bovine serum albumin for 1 hour in PBS/Tween before being stained with p73 (D3G10, rabbit monoclonal, Cell Signaling Technologies) and p53 (DO-1, mouse monoclonal, Santa Cruz) antibodies. Coverslips were then incubated with anti-rabbit AlexaFluor 647 and anti-mouse AlexaFluor 488 secondary antibodies before being fixed to slides with ProLong Diamond Antifade Mountant with DAPI (Life Technologies). Slides were visualized on a Leica SP8 MP microscope. Colocolization analysis was done using Imaris v9.0.2 and colocalized images were created using the “RG2B_Colocolization” plugin for ImageJ. Scale bar, 25 μm (MDA157, TMD231) or 50 μm (HME).
Colony forming assay.
Empty Vector and shp53 hTert BJ cells where plated in triplicate and treated with Anisomycin for 9 hrs. Dimethyl sulfoxide (DMSO) was used as a vehicle control. Cells were stained with equal parts methanol and methylene blue for 15 minutes. The plate was then washed 3 times with deionized water to remove any excess stain and left to dry at room temperature.
GFP florescence assay.
Green fluorescent protein (GFP) shp53 and LVTHM Empty Vector control BJ cells were plated in quadruplet and treated with DMSO or 10 μM of Anisomycin for 48 hours. Fluorescence emitted by GFP in each well was measured at 509 nm using a SpectraMax M5 spectrophotometer (Molecular Devices).
Supplementary Material
Acknowledgments:
We would like to thank members of the Mayo laboratory for critical reading of the manuscript. Phil Wubbolding and Paula M. Hauck for production and purification of recombinant proteins.
Funding: This work was supported in part through funds provided by: the Jeff Gordon Children’s Foundation, the Riley Children’s Foundation, and NIH CA172256 to LDM.
Footnotes
Competing interests: The authors declare that they have no competing interests.
References and Notes:
- 1.Di Como CJ, Gaiddon C, Prives C, p73 function is inhibited by tumor-derived p53 mutants in mammalian cells. Mol Cell Biol 19, 1438–1449 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gaiddon C, Lokshin M, Ahn J, Zhang T, Prives C, A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol Cell Biol 21, 1874–1887 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Flores ER et al. , Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell 7, 363–373 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Davison TS et al. , p73 and p63 are homotetramers capable of weak heterotypic interactions with each other but not with p53. J Biol Chem 274, 18709–18714 (1999). [DOI] [PubMed] [Google Scholar]
- 5.Joerger AC et al. , Structural evolution of p53, p63, and p73: implication for heterotetramer formation. Proc Natl Acad Sci U S A 106, 17705–17710 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu J, Jiang J, Zhou W, Zhu K, Chen X, Differential regulation of cellular target genes by p53 devoid of the PXXP motifs with impaired apoptotic activity. Oncogene 18, 2149–2155 (1999). [DOI] [PubMed] [Google Scholar]
- 7.Zhu J, Zhang S, Jiang J, Chen X, Definition of the p53 functional domains necessary for inducing apoptosis. J Biol Chem 275, 39927–39934 (2000). [DOI] [PubMed] [Google Scholar]
- 8.Ruaro EM et al. , A proline-rich motif in p53 is required for transactivation-independent growth arrest as induced by Gas1. Proc Natl Acad Sci U S A 94, 4675–4680 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Venot C et al. , The requirement for the p53 proline-rich functional domain for mediation of apoptosis is correlated with specific PIG3 gene transactivation and with transcriptional repression. EMBO J 17, 4668–4679 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mayo LD et al. , Phosphorylation of human p53 at serine 46 determines promoter selection and whether apoptosis is attenuated or amplified. J Biol Chem 280, 25953–25959 (2005). [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, Debatin KM, Hug H, HIPK2 overexpression leads to stabilization of p53 protein and increased p53 transcriptional activity by decreasing Mdm2 protein levels. BMC Mol Biol 2, 8 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.D’Orazi G et al. , Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nat Cell Biol 4, 11–19 (2002). [DOI] [PubMed] [Google Scholar]
- 13.Hofmann TG et al. , Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nat Cell Biol 4, 1–10 (2002). [DOI] [PubMed] [Google Scholar]
- 14.Fuchs SY, Adler V, Pincus MR, Ronai Z, MEKK1/JNK signaling stabilizes and activates p53. Proc Natl Acad Sci U S A 95, 10541–10546 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saito S et al. , ATM mediates phosphorylation at multiple p53 sites, including Ser(46), in response to ionizing radiation. J Biol Chem 277, 12491–12494 (2002). [DOI] [PubMed] [Google Scholar]
- 16.Bulavin DV et al. , Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. EMBO J 18, 6845–6854 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buschmann T et al. , Jun NH2-terminal kinase phosphorylation of p53 on Thr-81 is important for p53 stabilization and transcriptional activities in response to stress. Mol Cell Biol 21, 2743–2754 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu J, Zhou W, Jiang J, Chen X, Identification of a novel p53 functional domain that is necessary for mediating apoptosis. J Biol Chem 273, 13030–13036 (1998). [DOI] [PubMed] [Google Scholar]
- 19.Berger M, Stahl N, Del Sal G, Haupt Y, Mutations in proline 82 of p53 impair its activation by Pin1 and Chk2 in response to DNA damage. Mol Cell Biol 25, 5380–5388 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walker KK, Levine AJ, Identification of a novel p53 functional domain that is necessary for efficient growth suppression. Proc Natl Acad Sci U S A 93, 15335–15340 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baptiste N, Friedlander P, Chen X, Prives C, The proline-rich domain of p53 is required for cooperation with anti-neoplastic agents to promote apoptosis of tumor cells. Oncogene 21, 9–21 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Roy A, Kucukural A, Zhang Y, I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5, 725–738 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang J et al. , The I-TASSER Suite: protein structure and function prediction. Nat Methods 12, 7–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9, 40 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mayo LD, Dixon JE, Durden DL, Tonks NK, Donner DB, PTEN protects p53 from Mdm2 and sensitizes cancer cells to chemotherapy. J Biol Chem 277, 5484–5489 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Stambolic V et al. , Regulation of PTEN transcription by p53. Mol Cell 8, 317–325 (2001). [DOI] [PubMed] [Google Scholar]
- 27.Eitel JA et al. , PTEN and p53 are required for hypoxia induced expression of maspin in glioblastoma cells. Cell Cycle 8, 896–901 (2009). [DOI] [PubMed] [Google Scholar]
- 28.Mayo LD, Donner DB, A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci U S A 98, 11598–11603 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li AG et al. , Mechanistic insights into maintenance of high p53 acetylation by PTEN. Mol Cell 23, 575–587 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Maehama T, Dixon JE, PTEN: a tumour suppressor that functions as a phospholipid phosphatase. Trends Cell Biol 9, 125–128 (1999). [DOI] [PubMed] [Google Scholar]
- 31.Wu X, Bayle JH, Olson D, Levine AJ, The p53-mdm-2 autoregulatory feedback loop. Genes Dev 7, 1126–1132 (1993). [DOI] [PubMed] [Google Scholar]
- 32.Lehman JA et al. , Induction of apoptotic genes by a p73-phosphatase and tensin homolog (p73-PTEN) protein complex in response to genotoxic stress. J Biol Chem 286, 36631–36640 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flores ER et al. , p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature 416, 560–564 (2002). [DOI] [PubMed] [Google Scholar]
- 34.Talos F, Nemajerova A, Flores ER, Petrenko O, Moll UM, p73 suppresses polyploidy and aneuploidy in the absence of functional p53. Mol Cell 27, 647–659 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP, Pten is essential for embryonic development and tumour suppression. Nat Genet 19, 348–355 (1998). [DOI] [PubMed] [Google Scholar]
- 36.Suzuki A et al. , High cancer susceptibility and embryonic lethality associated with mutation of the PTEN tumor suppressor gene in mice. Curr Biol 8, 1169–1178 (1998). [DOI] [PubMed] [Google Scholar]
- 37.Davis RJ, Signal transduction by the c-Jun N-terminal kinase. Biochem Soc Symp 64, 1–12 (1999). [DOI] [PubMed] [Google Scholar]
- 38.Jones EV, Dickman MJ, Whitmarsh AJ, Regulation of p73-mediated apoptosis by c-Jun N-terminal kinase. Biochem J 405, 617–623 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Batuello CN, Hauck PM, Gendron JM, Lehman JA, Mayo LD, Src phosphorylation converts Mdm2 from a ubiquitinating to a neddylating E3 ligase. Proc Natl Acad Sci U S A 112, 1749–1754 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kehrloesser S et al. , Intrinsic aggregation propensity of the p63 and p73 TI domains correlates with p53R175H interaction and suggests further significance of aggregation events in the p53 family. Cell Death Differ 23, 1952–1960 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gebel J et al. , Mechanism of TAp73 inhibition by DeltaNp63 and structural basis of p63/p73 hetero-tetramerization. Cell Death Differ 23, 1930–1940 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chillemi G et al. , Structural Evolution and Dynamics of the p53 Proteins. Cold Spring Harb Perspect Med 7, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McNicholas S, Potterton E, Wilson KS, Noble ME, Presenting your structures: the CCP4mg molecular-graphics software. Acta Crystallogr D Biol Crystallogr 67, 386–394 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.