

Abstract
In countries infected with HIV clade B, some patients develop a rapidly progressive dementia that if untreated results in death. In regions of the world infected with HIV clade C, only milder forms of cognitive impairment have been recognized. HIV-infected macrophages are the principal mediators of dementia. HIV clade C, however, efficiently infects macrophages and HIV-infected macrophages are found in the brains of clade C-infected patients. HIV-infected macrophages release Tat protein, which may act directly on neurons to cause toxicity. We found that Tat released from Tat-expressing cells was at least 1000-fold more toxic than recombinant Tat protein. We determined whether Tat could interact with NMDA receptors and whether these interactions are clade dependent. It is demonstrated that Tat binds directly to the NMDA receptor leading to excitotoxicity. The Cys 30-Cys 31 motif in Tat is critical for exciting the NMDA receptor and the Cys31Ser mutation found in clade C Tat has a significantly attenuated neurotoxic response. Through molecular modeling and site-directed mutagenesis, we predict that Cys 31 disrupts the disulfide bond between Cys 744 and Cys 798 on the NR1 subunit of the NMDA receptor by directly interacting with Cys 744 leading to a free thiol group on Cys 798 and subsequent persistent activation of the NMDA receptor.
Keywords: injury, NMDA receptor, neuron, neuronal death, nitric oxide, neurotoxicity
Introduction
It has been recognized since the beginning of the pandemic that some patients with HIV infection develop a dementing illness. If left untreated, the dementia may rapidly progress and result in death (Navia et al., 1986). This pattern of dementia has been recognized in North America, Western Europe, and Australia, which are also the regions infected with HIV clade B, and continues to have vast socioeconomic consequences in these regions. As the virus has evolved and spread to different regions of the world, it has become apparent that there may be geographical differences in the neurological manifestations of HIV infection. In regions of the world infected with HIV clade C, only milder forms of cognitive impairment have been recognized, even in patients with advanced immune suppression in the absence of antiretroviral therapy (Riedel et al., 2006; Gupta et al., 2007). Although it is possible that this may be a result of selection bias in these studies, it is also important to determine whether these clades may vary in their neurovirulent properties and to identify the molecular determinants for such differences.
HIV-infected macrophages are considered to be the principal mediators of neuronal injury and subsequent development of dementia (González-Scarano and Martín-García, 2005). HIV clade C however, efficiently infects macrophages. Further, HIV-infected macrophages are found in the brains of clade C-infected patients (Mahadevan et al., 2007). One of the mechanisms by which HIV-infected macrophages cause neuronal injury involves the extracellular release of Tat protein, which may then act directly on neurons to cause toxicity (Mattson et al., 2005). Previous studies have shown that HIV clade B-derived Tat protein can cause excitotoxicity, which involves the activation of the NMDA receptors (Song et al., 2003), however, it remains unknown whether Tat can directly bind to the NMDA receptor. In this study, we compared the neurotoxic potential of clade B and C Tat protein. We also determined whether Tat could interact with NMDA receptors and whether these interactions were clade dependent. We also identified the molecular determinants for Tat-NMDA receptor binding.
Materials and Methods
Construction of plasmids.
Plasmid pTat72 that encodes Tat1–72 protein of HIV-1 strain HXBII was obtained from the NIH AIDS repository and pTat101 encodes Tat1–101 protein from the same virus was gifted by Dr. Ashok Chauhan (University of South Carolina, Columbia, SC). Plasmid pGST-Tat72 was constructed by cutting Tat72 gene from pTat72 and inserting it into pGEX-4T2 vector (GE Healthcare). Plasmids pNR1, pNR2A, pNR2B, pNR2C, and pNR2D encode rat NMDAR subunits NR1, NR2A, NR2B, NR2C, and NR2D, respectively, which were gifted by Dr. Richard O'Brien at Johns Hopkins University (Baltimore, MD). Glu-R1 plasmid (pRK5-gluR1-GFP) was gifted by Dr. Richard Huganir at Johns Hopkins University. Plasmid pVenus encodes a brighter variant of green fluorescent protein and was gifted by Dr. Atsushi Miyawaki at RIKEN Brain Science Institute (Wako, Japan). Plasmid pLTR-Venus was constructed by replacing CAT gene in pLTR-CAT (NIH AIDS Reagent Repository) with the Venus gene.
Cloning of clade C HIV-1 Tat cDNA.
DNA was extracted from the frontal lobe of the brain obtained at autopsy from three HIV-infected patients (A68-01, A89-02, and A69-03) from the National Institute of Mental Health and Neurosciences (Bangalore, India). A68-01 and A89-02 died of cerebral toxoplasmosis and A69-03 from cerebral malaria. HIV was detected in infiltrating macrophages by immunostaining for p24 in each of these patients. The neuropathological findings of the first two patients have been previously reported (Mahadevan et al., 2007). A small piece of the tissue was placed in 700 μl of Tris buffer (50 mm Tris pH 8.0, 100 mm EDTA, 100 mm NaCl, 1% SDS) and 20 μl of proteinase K (20 mg/ml). This was allowed to incubate overnight at 55°C. The next day, 350 μl of 6 m NaCl was added to each tube and gently vortexed. Samples were place on ice for 5 min, then centrifuged at 4° for 10 min at 13,200 rpm. The supernatant was transferred to a 2 ml tube, mixed with 900 μl of isopropanol, and then centrifuged at 13,200 rpm for 15 min to pellet the DNA. The DNA pellets were washed with 70% ethanol, allowed to air dry, and redissolved in 8 mm NaOH. Five hundred nanograms of DNA were used for the initial step of the nested PCR. The primers used were 5′-ACC ATG GCT TCA TGG CTT AG-3′ (forward) and 5′-TCA TTG CCA CTG TCT TCT GC-3′ (backward). The second step of the nested PCR used 5 μl from the previous step as starting material with the following primers: 5′-ATG GGG ATA CTT GGA CAG GA-3′ (forward) and 5′-TGC TAC TAT CAA TGC CCC T-3′ (backward). The region amplified represents the first exon of Tat that encodes for the first 72 aa. The PCR products were subcloned into pcDNA3 vector and sequenced. They were designated pTatA68-01, pTatA89-02, and pTatA69-03.
Cell culture and DNA transfection.
HEK 293 and SVGA cells were maintained in DMEM supplemented with 10% fetal bovine serum (FBS) and antibiotics (100 U/ml penicillin and 100 μg/ml streptomycin). For transfection, cells were seeded into six-well plates at a density of 1 × 106 cells/cm2. After 24 h, SVGA cells were transfected with 5 μg of pTat72, pTat101, pTatA68-01, pTatA89-02, or pTatA69-03 using Lipofectamine 2000 (Invitrogen). The culture media from these SVGA-Tat cells were collected 24 h after transfection and used as a source of secreted Tat. The amount of Tat released was estimated by measuring the optical density of the bands on an autoradiograph after Western blot analysis using TR-1 antibody. A standard curve was generated by using recombinant Tat protein. The amount of Tat released was 1.1 ng/106 cells over 24 h after transfection. For NMDA receptor expression, HEK 293 cells were cotransfected with 2.5 μg of pNR1 and 2.5 μg of pNR2A/well. These cells were designated HEK-NMDAR cells. For cell viability assay, additional 0.5 μg of pVenus was used together with pNR1 and pNR2A. Three hours after transfection, NMDA receptor antagonist, APV (0.05 mm) was added to the HEK-NMDAR cells to inhibit excitotoxicity through NMDA receptors.
Toxicity assay.
HEK 293 cells were transfected with 2.5 μg of pNR1, 2.5 μg of pNR2A, and 0.5 μg of pVenus. After 24 h, the cells were treated with either NMDA for another 6 h or culture media from SVGA-Tat cells for 24 h. The numbers of cells expressing Venus were counted using a florescent microscope, which represented the numbers of viable cells. Culture media from empty vector-transfected cells was used as a control.
Primary rat hippocampal neuronal cultures were prepared from E18 embryos as previously described (Kruman et al., 1998) and were seeded in 96-well plate at 1 × 106 cells/cm2. After 7 d in culture, the neurons were treated with recombinant Tat protein in Opti-MEM media for 18 h. Neuronal cell viability was assessed with an MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay. This assay is based on the ability of a mitochondrial dehydrogenase enzyme from viable cells to cleave the tetrazolium rings of the pale yellow MTT and form dark blue formazan crystals. These crystals are largely impermeable to cell membranes, and thus accumulate within healthy cells. The resultant formazan precipitates are solubilized with dimethyl sulfoxide and read on a multiwell scanning spectrophotometer. The number of surviving cells is directly proportional to the level of the formazan product created (Mosmann, 1983). All recombinant Tat proteins used in this manuscript were produced in our laboratory as previously described (Hollman et al., 2005).
S-nitrosylation of Tat with nitric oxide donor.
Recombinant Tat72 was dissolved in HEN buffer (250 mm HEPES, 1 mm EDTA, 0.1 mm neocuproine, pH 7.7) to 1 μg/μl. S-nitrosyl glutathione (GSNO) was then added to the clade B Tat72 solution to a final concentration of 100 μm. The reaction was protected from light and allowed to take place at room temperature for 30 min. All the following steps involving GSNO and reaction product were performed in the dark. After the reaction finished, the remaining GSNO was removed by Micro Bio-Spin P6 column preequilibrated with PBS. S-nitrosylation of Tat was then detected by a biotin-switch assay (Jaffrey et al., 2001). For assess the effect of secreted clade B Tat72, culture medium from SVGA-Tat cells was treated with 100 μm GSNO for 30 min at room temperature. The remaining GSNO was removed by Bio-Spin column preequilibrated with fresh culture media. For negative control, glutathione (GSH) was used in the place of GSNO. Cellular proteins modified by S-nitrosylation were then subjected to biotin-switching and immunoprecipitated with anti-biotin agarose beads (Sigma-Aldrich). The precipitated proteins were separated by SDS-PAGE and probed with an anti-Tat monoclonal antibody (NIH AIDS Reagent Repository).
Binding of NMDA receptor to GST-Tat.
Recombinant fusion protein GST-Tat72 was bound to glutathione-agarose beads. Cell lysate from HEK293 cells transfected with pNR1 and pNR2A were incubated with GST-Tat72/glutathione agarose beads for 1 h at room temperature. The agarose beads were extensively washed with lysis buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Triton X-100) and then boiled with 1× SDS loading buffer (50 mm Tris-HCl, pH 6.8, 100 mm DTT, 2% SDS, 0.1% bromophenol blue, 12.5% glycerol). The eluted proteins were subjected to immunoblotting with anti-NR1 (Invitrogen) or anti-NR2A antibodies (BD Biosciences).
Coimmunoprecipitation of NMDA receptor or LRP and Tat.
After 24 h of transfection with NMDA receptor subunits, NR1 and NR2A, the HEK 293 cells were washed with fresh medium and then incubated with secreted clade B Tat72 at room temperature. After 30 min of incubation, the cells were washed with PBS, pH 7.4, and lysed with 1% Triton X-100 in PBS. The lysate was precleared by protein-G-coupled agarose beads and then incubated with TR-1 antibody (1 μg/ml) for 60 min. Protein-G-coupled agarose beads were added into the lysate and incubated for another 60 min. The precipitated complex was then washed and lysed with protein sample buffer. The coprecipitated NMDA receptor was then detected by Western blot analysis. A similar protocol was used for detection of LRP using HEK293 cells. A monoclonal antisera to LPR (Calbiochem) was used for Western blot analysis.
DNA mutagenesis.
DNA mutagenesis was performed using GeneEditor in vitro site-directed mutagenesis system from Promega. To generate Cys31Ser mutation in clade B Tat72, primer GTAAAAAGTGTAGCTTTCATTGC was used. To generate Ser31Cys mutation in clade C Tat72, primer GTAAAAGATGTTGCTACCATTG was used. The respective mutation products were named Tat72B/C31S and Tat72C/S31C. A deletion mutant of Tat72B missing amino acids 48–56 (Chauhan et al., 2003) was subcloned into pcDNA3.1(−) (Invitrogen) and was named Tat72BΔ.
Long terminal repeat transactivation assay.
The transactivation of long terminal repeat (LTR)-CAT was measured using a CAT ELISA kit (Roche Applied Science). SVGA LTR-CAT cells were incubated with secreted Tat for 48 h. Cells were then harvested, and CAT protein in the cell lysate was determined following the manufacturer's instructions. The amount of CAT was expressed as picograms per microgram of total protein.
Modeling of Tat-NMDA receptor interactions.
The modeling was done with Swiss-PdbViewer DeepView available at http://spdbv.vital-it.ch/. HIV-1 clade B-Tat used was an NMR model from protein data bank ID: 1TBC (model 1 of ensemble). The model of NR 1 subunit of the NMDA receptor was protein data bank ID: 1PB7. Tat was rigidly docked on to NR1 and the side chains of Cys 31 of Tat and Cys 744 and Cys 798 of NR1 adjusted to model the change in disulfides.
Statistical analysis.
All data are represented as mean + SEM and analyzed by one-way ANOVA and Newman–Keul's pairwise comparison.
Results
Clade B and C Tat are released equally extracellularly
We used a monoclonal antibody to Tat that showed equal affinity to clade B and C Tat (Fig. 1 A) to detect the amount of Tat released extracellularly. SVGA cells were transfected with plasmids from clade B Tat72, clade C Tat72 (A68-01), and their respective mutants Tat72B/C31S and Tat72C/S31C. In each instance equal amounts of Tat were detected by Western blot analysis. By comparison to a known amount of recombinant clade B Tat72 protein, we estimated that the amount of Tat released extracellularly was ∼0.5 ng of Tat/ml (Fig. 1 B). Similarly, equal amount of Tat was released from each of the brain-derived clade C Tat proteins (Fig. 1 C). Thus the release of Tat extracellularly was independent of the clade from which it was derived.
Figure 1.

Quantitation and comparison of different types of Tat secreted from transfected cells. A, Ten nanograms of recombinant clade B or clade C (A68-01) Tat proteins were analyzed in triplicate by Western blot using TR-1 antibody. TR-1 antibody shows similar affinity for both clade B and clade C Tat. B, Semiquantitative measurement of secreted Tat. Different Tat proteins as shown were immunoprecipitated from 10 ml of culture medium and analyzed by comparison with serial dilution of recombinant Tat by Western blot. Secreted Tat over 24 h is ∼1.1 ng/106 cells in conditioned medium. C, Similar amount of Tat was secreted from SVGA cells, which was transfected with different types of Tat constructs. Conditioned culture medium (2.5 ml) was immunoprecipitated with Tat antibody and analyzed by Western blot.
Clade differences in Tat-induced HIV-LTR activation and neurotoxicity
Tat protein is encoded by two exons. The first exon forms 72 aa and most functional properties of Tat are attributed to this region. The second exon forms the remaining 29 aa. We determined whether there were any differences in the release of first exon derived Tat or full-length Tat. When a glial cell line was transfected with the respective tat plasmids both, first exon Tat and full-length Tat were equally expressed in the cells and equally released extracellularly. (Fig. 2 A). No differences were found in the ability of these Tat proteins to transactivate HIV-LTR (Fig. 2 B,C). Similarly, there was no significant difference in the ability of clade B or C Tat to transactivate the HIV-LTR (Fig. 2 D). To determine whether there were any clade-dependent differences in Tat-induced neurotoxicity, we produced recombinant clade B and C Tat protein and exposed neuronal cultures to these proteins. Significantly, lower toxicity was seen with clade C Tat (Fig. 2 E).
Figure 2.
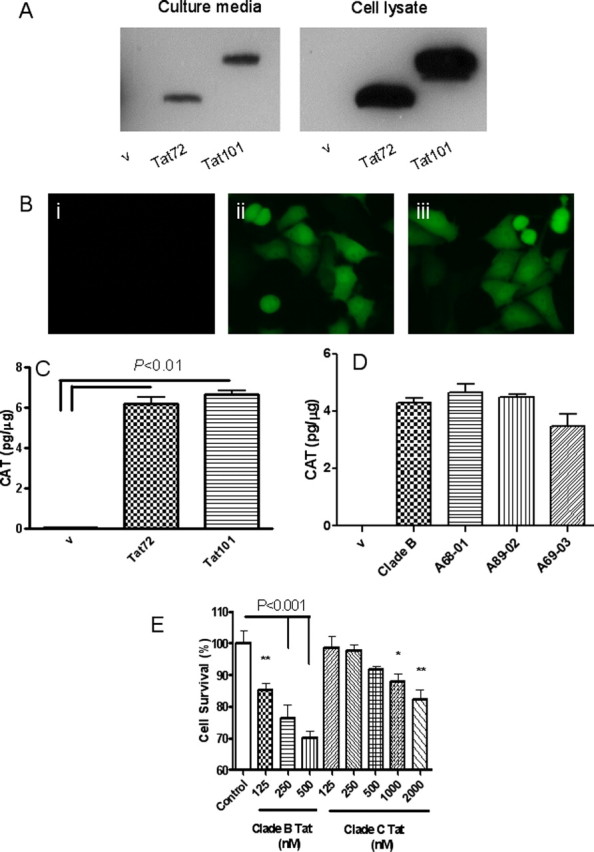
Extracellularly released Tat causes LTR activation and neurotoxicity. A, SVGA cells were transfected with empty vector (v), pTat72, or pTat101 for 24 h. Culture media (i) and cell lysate from transfected cells (ii) were immunoblotted with anti-Tat monoclonal antibody (TR-1). B, Stable SVGA (LTR-Venus) cells were cultured in conditioned media from SVGA cells transfected with v (i), pTat72 (ii), and pTat101 (iii) for 48 h, and the expression of Venus was examined under the fluorescent microscope. C, D, Stable SVGA (LTR-CAT) cells were cultured in conditioned media from SVGA cells transfected with v, Tat72, and Tat101 (C) or v, clade B-Tat72, and clade C-Tat from A68-01, A89-02, or A69-03 (D) for 48 h, and the expression of CAT was determined by ELISA and presented as picograms of CAT per milligram of total protein. E, Primary rat hippocampal neurons were exposed to recombinant clade B-Tat72 or recombinant clade C-Tat from A68-01. Cell viability was assessed by MTT assay. Clade B was at least 15 times more potent than clade C Tat. *p < 0.05; **p < 0.01.
Tat neurotoxicity is mediated via NMDA receptors
To determine whether the differences between the clades were attributable to differences in their interactions with the NMDA receptors, we first transfected HEK293 cells with NMDA receptor subunits NR1 and NR2A and confirmed their expression in cellular extracts by Western blot analysis (Fig. 3 A) and by immunostaining (Fig. 3 B). Treatment of these cells with NMDA resulted in nearly 50% cell death, which could be blocked by NMDA antagonists MK-801 and APV, confirming the functional expression of the NMDA receptors (Fig. 3 C). We used these cells (HEK-NMDAR) for all subsequent experiments. When these cells were treated with clade B-Tat, cell death was noted at levels comparable to that caused by NMDA. We compared the effect of Tat on other NMDA receptor subtypes by expressing them in HEK293 cells and treating them with clade B-Tat. The combination of NR1A with either NR2A or NR2B produced maximal toxicity (Fig. 3 D). This toxicity could be blocked by NMDA receptor antagonists (Fig. 4 A). The toxicity was specific for Tat, because it could be attenuated by antibody to Tat but not by an isotype control antibody (Fig. 4 A). Similar findings were noted when primary hippocampal neurons were treated with clade B-Tat (Fig. 4 B). To determine whether Tat could bind to the NMDA receptor, we immunoprecipitated the NMDA receptor subunits using either recombinant GST-Tat or Tat conjugated to a monoclonal antibody. Both subunits, NR1 and NR2A, could be immunoprecipitated by Tat (Fig. 4 C). We also expressed Glu-R1 in HEK293 cells and repeated the immunoprecipitation. No binding of Tat to Glu-R1 was noted (data not shown). All subsequent experiments were conducted with HEK cells expressing the NR1–NR2A complex.
Figure 3.
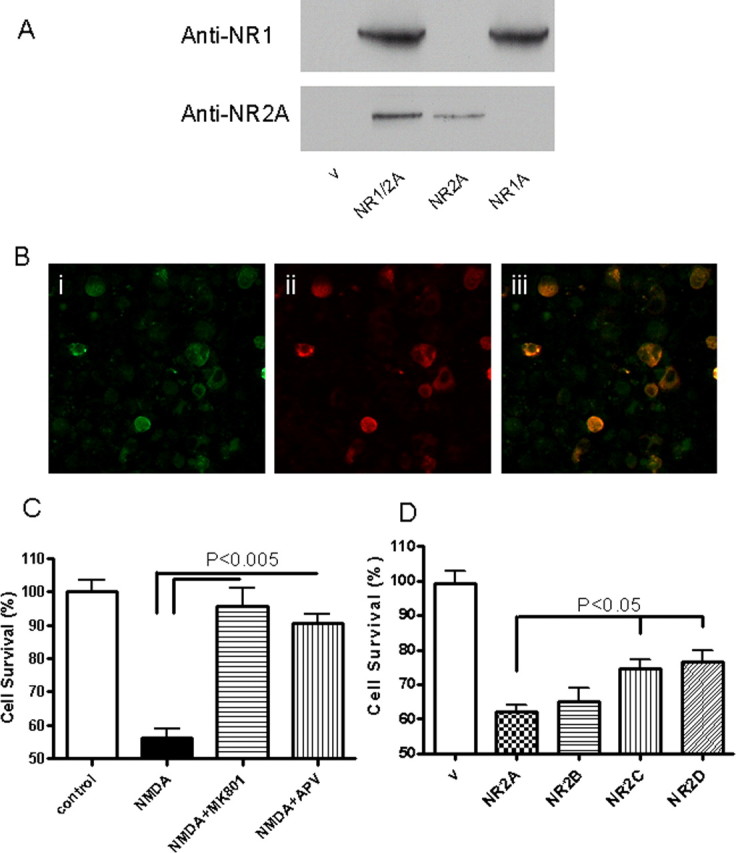
Expression of functional NR1A and NR2A complex in HEK293 cells. A, HEK293 cells were transfected with pNR1A and pNR2A, and 24 h later the proteins were extracted and analyzed by Western blots. Vehicle (V) control shows no immunoreactivity. Both NR1 and NR2A are expressed in these cells. B, HEK293 cells transfected with pNR1 and pNR2A were immunostained with antisera to the respective subunits. Bi shows immunostaining for NR1, Bii shows immunostaining for NR2A, and Biii shows overlap of both images. C, HEK293 cells transfected with pNR1 and pNR2A show extreme sensitivity to NMDA (300 μm NMDA with 50 mm glycine for 6 h) toxicity, which could be blocked by NMDA receptor antagonists MK-801 (10 μm) and APV (500 μm). D, HEK293 cells were transfected with the combination of NR1 and NR2A, NR2B, NR2C, or NR2D. The cells were treated with clade B Tat72 for 18 h. Equal amount of toxicity was seen with NR2A and NR2B; however, there was significantly less toxicity with NR2C and NR2D.
Figure 4.
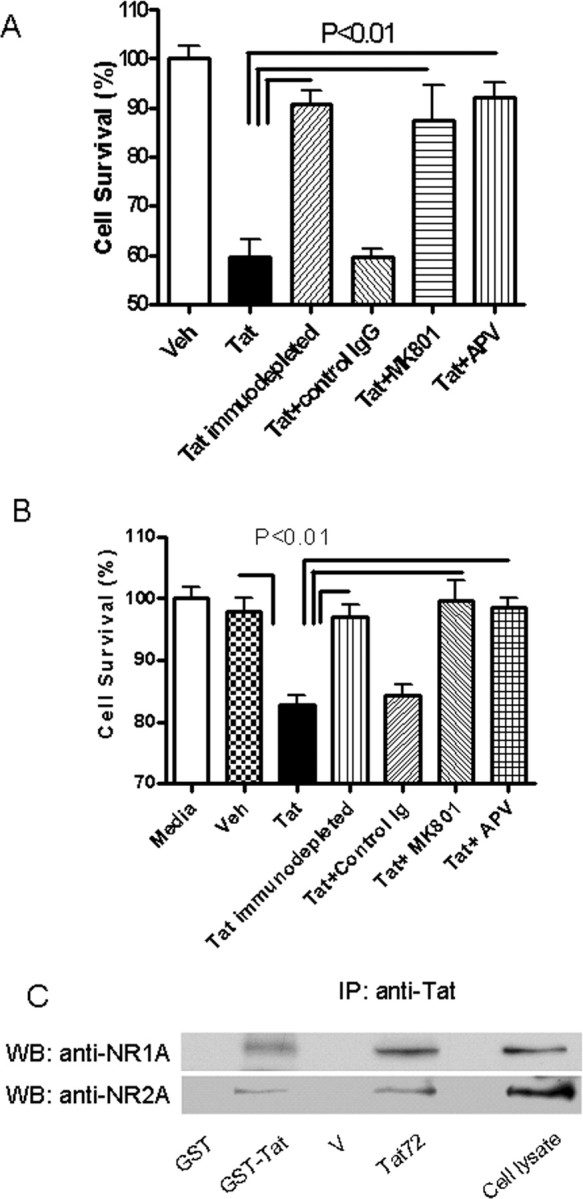
Tat toxicity is mediated via NMDA receptors. A, HEK-NMDAR cells were cultured in conditioned media with Tat72 protein (0.5 ng/ml) for 24 h. Cell survival was determined by counting cells expressing an indicator protein (Venus). B, Primary rat hippocampal neurons were incubated with secreted Tat (0.5 ng/ml) for 24 h, and cell viability was assessed by MTT assay. Tat caused significant neuronal death in both cell types. After immunodepletion of Tat from the media, the toxicity is significantly diminished. Coincubation with 10 μm MK-801 or 500 μm APV also significantly blocked cell death induced by Tat protein. C, Interaction between Tat and NMDA receptor. Recombinant fusion protein GST-Tat bound to Glutathione Sepharose 4B resin could pull down NR1 and NR2A subunits from HEK-NMDAR cells but GST alone could not. Similarly, when HEK-NMDAR cells were incubated in conditioned media with Tat protein for 30 min, NR1 and NR2A could be coimmunoprecipitated by a monoclonal Tat antibody (TR-1).
Interactions of Tat with NMDA receptor are independent of LRP
Tat has been previously shown to bind to low-density lipoprotein receptor-related protein (LRP), which forms a complex with postsynaptic density protein-95 (PSD-95) and NMDA receptors (Eugenin et al., 2007), hence we compared LRP expression in the HEK293 cells and neurons and found only small amounts of LRP expression in the HEK293 cells (Fig. 5 A). Tat was unable to immunoprecipitate LRP from the HEK-NMDAR cells (Fig. 5 B). Further, LRP ligands RAP, α2 macroglobulin, and lactoferrin failed to block Tat-induced cell death in the HEK-NMDAR cells (Fig. 5 C). Further, HEK293 cells do not express PSD-95. These observations suggest that clade B-Tat binds directly to the NMDA receptor.
Figure 5.

Tat interacts with NMDAR independent of LRP. A, Expression of LRP in HEK293 was compared with that in primary rat hippocampal neurons by Western blot analysis. GAPDH was used as a control. B, Lane 1 shows expression of LRP and NR1 in the HEK293 cells. The top panel was overexposed to maximize LRP detection. Lane 2 is a vehicle (v) control in which Tat was omitted from the immunoprecipitation experiment. In lane 3, recombinant clade B-Tat bound to Tat antibody (TR-1) was able to immunoprecipitate NR1 subunit but not LRP from HEK-NMDAR cells. C, Clade B Tat-induced toxicity in HEK-NMDAR cells could not be blocked by LRP ligands RAP (500 nm), lactoferrin (50 units/ml), or α2-macroglobulin (500 nm), but could be blocked by NMDA receptor antagonists MK-801 (10 μm) or APV (500 μm).
Nitrosylation of Tat blocks its neurotoxic potential but not its binding to NMDA receptor
Tat has a Cys-rich region with 7 Cys in clade B-Tat, whereas there is a Cys31Ser mutation in clade C-Tat. To determine the role of the Cys in the interactions with the NMDA receptor, the Cys in clade B-Tat were nitrosylated using S-nitrosyl-glutathione (GSNO) and detected by a biotin-switch assay. Nitrosylation of recombinant Tat was confirmed by Western blot analysis (Fig. 6 A). Nitrosylated Tat failed to cause neurotoxicity in hippocampal neurons (Fig. 6 B). This observation was also verified using secreted Tat from glial cells (Fig. 6 C) and the HEK-NMDAR cells (Fig. 6 D). Immunoprecipitation experiments showed that Tat was unable to bind to the NMDA receptor subunits after nitrosylation (Fig. 6 E). These experiments suggest that the Cys in clade B-Tat are critical in mediating interactions with the NMDA receptor.
Figure 6.

Nitrosylation of recombinant Tat attenuates its toxic properties. A, Recombinant Tat was treated with 100 μm S-nitrosyl-glutathione (GSNO) and S-nitrosylation on Tat was detected by biotin-switch labeling and examined by anti-biotin antibody. Glutathione (GSH; 100 μm) was used as a negative control. B, Rat primary hippocampal neurons were treated with 500 nm Tat (untreated rTat, GSH-treated rTat, or GSNO-treated rTat) for 24 h. Cell viability was determined by MTT assay. Treatment with GSH had no effect on toxicity of Tat. However, treatment with GSNO completely abolished toxicity of Tat protein. C, SVGA cells transfected with pTat72 were treated with 100 μm GSNO for 30 min. Cell lysate was subjected to biotin-switch labeling, and the labeled proteins were immunoprecipitated with anti-biotin antisera conjugated to agarose beads. The precipitated proteins were examined with anti-Tat antibody. Cells treated with GSNO showed S-nitrosylation of Tat. D, The effect of S-nitrosylation of secreted Tat was examined in HEK-NMDAR cells. Treatment with GSH had no effect on Tat-induced toxicity, but treatment with GSNO showed a significant reduction in toxicity of Tat. E, After treatment with GSNO, secreted Tat was incubated with HEK-NMDAR cells for 30 min, and cell lysate was immunoprecipitated with anti-Tat antibody. The precipitated proteins were analyzed by Western blot using both anti-NR1 and anti-NR2A antibodies. Treatment of Tat with GSNO completely abolished its binding with NMDA receptor.
Cys31Ser mutation in clade C Tat causes attenuation of neurotoxicity without affecting NMDA receptor binding
To determine the biological relevance of clade differences in Tat sequences, we sequenced the tat gene from the brain of several individuals with HIV clade-C infection. We chose three sequences to further study their neurotoxic properties. Two of these patients had a Cys31Ser mutation, whereas patient A89-02 had several of the mutations similar to the other two patients except that the Cys31 was preserved (Fig. 7 A). The neurotoxic properties of each of these proteins were tested in the HEK-NMDAR cells. Tat protein from patients A68-01 and A69-03 showed significant attenuation of toxicity compared with clade B-Tat, whereas Tat protein from A89-02 caused toxicity similar to that seen with clade B-Tat (Fig. 7 B). To further confirm the role of the Cys31, a Cys31Ser mutation was created in clade B-Tat and a Ser31Cys mutation in clade C-Tat from A68-01 (Fig. 7 C). It was found that the Cys31Ser mutation attenuated the toxicity of clade B-Tat and the Ser31Cys mutation resulted in gain of toxic properties of clade C-Tat (Fig. 7 D). Interestingly, however, the point mutation at Cys31Ser had no effect on its binding to the NMDA receptor subunits (Fig. 7 E). We considered the possibility that other regions of the Tat may be important in binding to the cell membrane, whereas the Cys may be critical for NMDA receptor activation.
Figure 7.

Role of clade C-Tat with Cys31Ser mutation in neurotoxicity and binding to NMDAR. A, Alignment of brain-derived clade C tat sequences from patients A68-01, A89-01, and A69-03 with HIV clade B Tat from HIV-HXB2 strain. An important difference is that A68-01 and A69-03 have Cys31Ser point mutation in the Cys-rich domain. B, Expression plasmids were constructed from these Tat sequences using pcDNA3 vector. The resulting plasmids were used to transfect SVGA cells under the same conditions. The culture supernatant containing secreted Tat was harvested, and their toxicity was determined in HEK-NMDAR cells. A68-01 and A69-03 had significantly less toxicity than clade B Tat and A89-02. C, Cys31Ser mutation was made in clade B Tat, and a Ser31Cys mutation was made in clade C Tat from patient A68-01. D, Using HEK-NMDAR cells, the toxicity of each of the secreted Tat proteins were compared. Proteins with Cys31 were more toxic than proteins with Ser31, indicating the importance of Cys31 in Tat-induced toxicity. E, The secreted Tat protein derived from clade B virus, each of the clade C-infected patients (A68-01, A89-02, and A69-03), the clade B Tat with Cys31Ser mutation (mClade B), and the clade C with Ser31Cys mutation (mA68-01) were individually bound to anti-Tat antisera and used for immunoprecipitating the NMDA receptor from HEK-NMDAR cells. The immunoprecipitates were analyzed by Western blots. The Cys31Ser mutation had no significant effect on the Tat-NMDA receptor binding.
Deletion of the Arg-rich domain of Tat eliminates binding and neurotoxicity
Previous studies have suggested that the Arg-rich domain is necessary for membrane binding because of its highly basic charge (Sabatier et al., 1991; Schwarze et al., 1999). We created a deletion mutant from which the Arg region (amino acids 48–56) that was homologous in clade B- and C-Tat, and its neurotoxic properties and binding potential to NMDA receptor were determined. The deletion mutant failed to bind to the NMDAR subunits (Fig. 8 A) or cause toxicity in HEK-NMDAR cells (Fig. 8 B).
Figure 8.

Arg-rich domain is critical for Tat-NMDAR interaction and Tat-induced toxicity through NMDAR. A, Culture media from SVGA cells transfected with vector, pTatΔ, or pTat72 were incubated with HEK-NMDAR cells for 30 min. The HEK-NMDAR cells were then lysed and subjected to immunoprecipitation using anti-Tat antibody. The precipitated proteins were examined by Western blot analysis using anti-NR1A and anti-Tat antibody. Although the Tat antibody can recognize and precipitate TatΔ from the culture media, it failed to immunoprecipitate NR1A. B, The toxicity of TatΔ was measured and compared with clade B-Tat using HEK-NMDAR cells. No significant cell death was induced by TatΔ.
Modeling of Tat-NR1 subunit interactions
We modeled the interactions of Tat with the NR1 subunit because Tat could cause toxicity when NR1 was expressed in combination with each of the other subunits. In this model, Cys31 of Tat forms a disulfide with Cys744 of NR1 of NMDA (Fig. 9). The disulfide 744–798 in NR1 has been proposed to be important for persistent activation of the NMDA receptor (Choi et al., 2001). To test this model, we mutated the Cys744 of NR1 to Ala and expressed it in HEK cells with NR2A. Tat neurotoxicity was attenuated in these cells compared with the HEK-NMDAR cells. Although the expression of NR1 and NR1-Cys744Ala was similar, significantly less Tat was bound to NR1-Cys744-Ala (Fig. 10). These findings confirm the interactions of Tat with Cys 744 on NR1.
Figure 9.
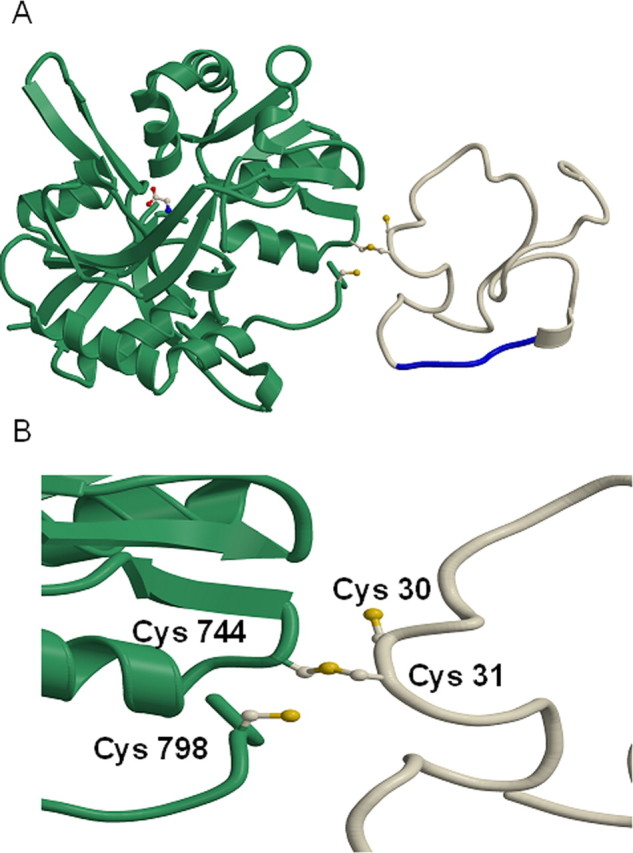
Modeling of NR1 and Tat interactions. A, Model of the HIV-1 Tat protein interacting with the NR1 subunit of the NMDA receptor through a disulfide bond between Cys 31 of Tat and Cys 744 of NR1, which disrupts an existing disulfide bond between Cys 744 and Cys 798 of NMDA. HIV-1 clade B-Tat (PDB entry 1TBC) was rigidly docked on to NR1 (PDB entry 1PB7) and the side chains of Cys 31 of Tat and Cys 744 and Cys 798 of NR1 adjusted to model the change in disulfides. NR1 is shown in green and Tat in white, with the Arg-rich region (residues 48–56) of Tat shown in blue that binds to the cell membrane. A glycine bound as an agonist to NR1 is shown in a ball and stick representation. B, Close up of the modeled disulfide between Tat and NR1 with the Cys 30 and 31 of Clade B-Tat and 744 and 798 of NR1 labeled.
Figure 10.

C744A mutation in NR1 attenuates Tat neurotoxicity and binding. A, HEK 293 cells were transfected with either NR1-wt/NR2A or NR1-C744A/NR2A. The transfected cells were then incubated with secreted Tat for 24 h. Tat caused significantly less cell death in HEK 293 cells with NR1-C744A than in cells with NR1-wt. B, The transfected HEK 293 cells were incubated with secreted Tat for 30 min, and cell lysate was immunoprecipitated with anti-Tat antibody. The precipitated proteins were analyzed by Western blot using anti-NR1 antibody. Although the expression level of NR1-wt and NR1-C744A was equal (Input), Tat precipitated less NR1-C744A (70.5% relative to NR1-wt).
Discussion
Although recent epidemiological data suggest that the regional differences in HIV clade infection may parallel the differences in severity and frequency of cognitive impairment, the host and viral genetic factors that account for these differences remain unknown. Numerous studies have shown that the presence of HIV-infected macrophages in the brain is critical for mediating the pathophysiological cascade of events that lead to neuronal injury and cognitive impairment. Interestingly, however, HIV clade C infects and replicates in macrophages. We further show that there are no differences in the ability of the Tat protein of clade B and C viruses in transactivating the HIV-LTR, which is critical for viral replication. Because viral evolution may occur in the brain, we used brain-derived clade C Tat proteins. Although we identified several mutations between the two clades, this lack of differences for LTR transactivation is likely because the Arg-rich basic domain of Tat and the Cys in positions 22, 25, 27, and 37 are conserved in the two clades (Jeang et al., 1999). Further, there were no differences in the extracellular release of clade B and C Tat proteins.
Most previous studies of Tat neurotoxicity have been performed using recombinant Tat protein. In this study, we used Tat released from cells transfected with the Tat plasmid. We found this form of Tat to be at least 1000-fold more toxic than recombinant protein (Nath et al., 2000). This is likely because Tat may get oxidized or denatured during the process of purification. We have previously shown that extracellular Tat is excitotoxic to neurons, hence we compared the ability of both clades to cause neurotoxicity. We found that clade C-derived Tat was significantly less toxic to neurons compared with clade B-derived Tat. Electrophysiological studies have shown that clade B Tat causes a train of action potentials in hippocampal neurons in slice cultures, it depolarizes the cell membrane in whole cell patch and outside out patch preparations. These effects of Tat can be blocked by NMDA receptor blockers (Cheng et al., 1998; Song et al., 2003). These effects of Tat occur within milliseconds suggesting a direct interaction between Tat and the NMDA receptor, however, binding studies require prolonged incubation of Tat to the cells which causes interactions of Tat with other cell surface molecules (Eugenin et al., 2007). Hence to study direct interactions between Tat and the NMDA receptor, we used a cell line in which the NMDA receptor subunits could be expressed without the other binding molecules. To determine whether the differences in Tat effects could be caused by differences in interaction with the NMDA receptor, we initially characterized the clade B Tat binding properties to the NMDA receptor. Tat-induced toxicity was most significant when NR1 subunit of the NMDA receptor was expressed with the NR2A or NR2B subunit. The NMDA receptor is a heteromeric receptor channel that is formed of two subunits of NR1 and two subunits of either NR2A or NR2B. Immunoprecipitation experiments showed that Tat could bind to the NR1–NR2A complex. Tat protein has been shown to bind to other cell surface proteins in other cell types this includes interactions with LRP on neurons (Liu et al., 2000). We show that when NMDA receptor complex was expressed in HEK cells, Tat induced toxicity could not be blocked by specific blockers of LRP and Tat did not immunoprecipitate LPR from these cells. But instead Tat-induced toxicity could be blocked by pharmacological blockers of NMDA receptor, which also suggests that the interactions of Tat with NMDA receptor are specific. These findings are consistent with studies showing that Tat can be colocalized to the NMDA receptor by immunostaining on the cell surface of neurons (Chandra et al., 2005) and suggest that the binding properties of Tat are specific for the NMDA receptor.
To determine the regions of Tat that mediate the interactions with the NMDA receptor, we initially nitrosylated the cysteines and found that this blocked both the toxicity and the binding to NMDA receptors. Although we did not further pursue the role of nitrosative stress in the context of this study, it has been shown that there is significantly increased nitrosative stress during HIV infection in the brain and several host proteins may be nitrosylated (Adamson et al., 1996). Because nitrosylation of Tat obliterates its neurotoxicity, it is possible that nitrosative stress may also be a host defense mechanism. Further, these experiments suggest an important role for the Cys residues in interaction with the NMDA receptor. Clade B Tat has seven Cys residues and comparison with the brain-derived clade C Tat shows that only one of the Cys residues at position 31 was mutated to a Ser. Interestingly, in one of the patients, the Cys residue was preserved in position 31 but had all the other mutations similar to those seen in the other clade C Tat sequences. When we compared the neurotoxic potential of each of these Tat proteins, we found that the presence of Cys in position 31 caused increased toxicity. This was further confirmed by site specific mutagenesis in which we mutated the Cys31 in clade B Tat to a Ser and mutated Ser31 to Cys in clade C Tat and then compared their neurotoxic potential. This is consistent with a recent study that implicated the role of Cys31 in Tat-mediated neurotoxicity in primary neurons (Mishra et al., 2008). Interestingly, however, both clade B and C Tat could similarly immunoprecipitate the NMDA receptor. This suggested that although Cys31 may be critical in mediating excitation of the NMDA receptor, it may not be important in mediating binding to the receptor or the cell membrane.
The Tat protein has a Zn2+ binding site and the possibility that Tat may chelate Zn2+ and thus cause disinhibition of the NMDA receptor has been proposed. However, previous studies have shown that Tat can cause persistent activation of the NMDA receptor in the presence of a Zn2+ specific chelator and also after mutation of the Zn2+ specific site on NR1 (Cys61Ala), which suggests that Tat may act at another site on the NMDA receptor (Chandra et al., 2005). In modeling the interactions of Tat with the NMDA receptor we considered the possibility that a large ligand such as Tat protein might anchor itself to the cell membrane and then Cys31 might interact with the receptor to activate it. The Arg-rich region of Tat has been previously shown to be important for interaction with cell membranes (Schwarze et al., 1999). We thus deleted this region from Tat and found that Tat failed to immunoprecipitate the NMDA receptor or cause toxicity. Our model thus proposes that once Tat is anchored to the cell membrane by the Arg-rich domain, Cys 31 interacts with Cys 744 on NR1 forming a disulfide bond. We tested this model by mutating Cys 744 on NR1 to Ala and found that as a result Tat neurotoxicity was significantly attenuated. Under physiological conditions, Cys 744 forms a disulfide bond with Cys 798 on the NR1 subunit. Reducing agents and hypoxic conditions lead to a free thiol state at these sites leading to potentiation of NMDA currents. Conversely, oxidizing agents favor disulfide formation and inhibit NMDA activity (Takahashi et al., 2007). In our model, the interaction of the Cys 31 on Tat with Cys 744 on NR1 would lead to a free thiol group on Cys 798 which may cause a persistent or abnormal activation of the receptor resulting in neuronal degeneration. These findings have important implications. Known ligands of NMDA receptors are all small molecules. Proteins have not been previously shown to bind or activate this class of receptors. Viruses, however, have evolved to interact with host receptors. Here, we show that the Tat protein of HIV can act as an agonist of the NMDA receptor by direct interactions with the receptor. Accordingly, interactions of clade B-Tat with NR1 would lead to persistent NMDA receptor activation and a single amino acid mutation at Cys 31 would alter its excitatory properties. Interestingly, such a mutation occurs naturally in regions of the world infected with HIV clade C virus. In these regions, severe forms of HIV dementia do not occur. Because NMDA receptors play a critical role in learning and memory and excitotoxicity at this receptor can lead to cognitive dysfunction, it is possible that these clade differences in the Tat protein may in part contribute to the regional differences in HIV dementia. Our observations do not exclude the role of other host or viral genetic factors that may also contribute to the regional differences in neurological manifestations of HIV infection.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants P01MH070306, R01NS039253, and R01NS056884 and by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences. We are grateful to Dr. Joan Berman for critically reading this manuscript, Dr. Richard Huganir for helpful comments, and Dr. Solomon Snyder's laboratory for assistance with the biotin switch assay.
References
- Adamson DC, Wildemann B, Sasaki M, Glass JD, McArthur JC, Christov VI, Dawson TM, Dawson VL. Immunologic NO synthase: Elevation in severe AIDS dementia and induction by HIV-1 gp41. Science. 1996;274:1917–1921. doi: 10.1126/science.274.5294.1917. [DOI] [PubMed] [Google Scholar]
- Chandra T, Maier W, König HG, Hirzel K, Kögel D, Schüler T, Chandra A, Demirhan I, Laube B. Molecular interactions of the type 1 human immunodeficiency virus transregulatory protein Tat with N-methyl-d-aspartate receptor subunits. Neuroscience. 2005;134:145–153. doi: 10.1016/j.neuroscience.2005.02.049. [DOI] [PubMed] [Google Scholar]
- Chauhan A, Turchan J, Pocernich C, Bruce-Keller A, Roth S, Butterfield DA, Major EO, Nath A. Intracellular human immunodeficiency virus Tat expression in astrocytes promotes astrocyte survival but induces potent neurotoxicity at distant sites via axonal transport. J Biol Chem. 2003;278:13512–13519. doi: 10.1074/jbc.M209381200. [DOI] [PubMed] [Google Scholar]
- Cheng J, Nath A, Knudsen B, Hochman S, Geiger JD, Ma M, Magnuson DSK. Neuronal excitatory properties of human immunodeficiency virus type 1 tat protein. Neuroscience. 1998;82:97–106. doi: 10.1016/s0306-4522(97)00174-7. [DOI] [PubMed] [Google Scholar]
- Choi Y, Chen HV, Lipton SA. Three pairs of cysteine residues mediate both redox and Zn2+ modulation of the NMDA receptor. J Neurosci. 2001;21:392–400. doi: 10.1523/JNEUROSCI.21-02-00392.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eugenin EA, King JE, Nath A, Calderon TM, Zukin RS, Bennett MV, Berman JW. HIV-tat induces formation of an LRP-PSD-95-NMDAR-nNOS complex that promotes apoptosis in neurons and astrocytes. Proc Natl Acad Sci U S A. 2007;104:3438–3443. doi: 10.1073/pnas.0611699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Scarano F, Martín-García J. The neuropathogenesis of AIDS. Nat Rev Immunol. 2005;5:69–81. doi: 10.1038/nri1527. [DOI] [PubMed] [Google Scholar]
- Gupta JD, Satishchandra P, Gopukumar K, Wilkie F, Waldrop-Valverde D, Ellis R, Ownby R, Subbakrishna DK, Desai A, Kamat A, Ravi V, Rao BS, Satish KS, Kumar M. Neuropsychological deficits in human immunodeficiency virus type 1 clade C-seropositive adults from South India. J Neurovirol. 2007;13:195–202. doi: 10.1080/13550280701258407. [DOI] [PubMed] [Google Scholar]
- Hollman AM, Christian DA, Ray PD, Galey D, Turchan J, Nath A, Bhattacharyya D. Selective isolation and purification of tat protein via affinity membrane separation. Biotechnol Prog. 2005;21:451–459. doi: 10.1021/bp049804z. [DOI] [PubMed] [Google Scholar]
- Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- Jeang KT, Xiao H, Rich EA. Multifaceted activities of the HIV-1 transactivator of transcription, Tat. J Biol Chem. 1999;274:28837–28840. doi: 10.1074/jbc.274.41.28837. [DOI] [PubMed] [Google Scholar]
- Kruman II, Nath A, Mattson MP. HIV protein Tat induces apoptosis by a mechanism involving mitochondrial calcium overload and caspase activation. Exp Neurol. 1998;154:276–288. doi: 10.1006/exnr.1998.6958. [DOI] [PubMed] [Google Scholar]
- Liu Y, Jones M, Hingtgen CM, Bu G, Laribee N, Tanzi RE, Moir RD, Nath A, He JJ. LRP-mediated uptake of HIV-1 Tat protein disrupts neuronal metabolic balance of LRP ligands. Nat Med. 2000;6:1380–1387. doi: 10.1038/82199. [DOI] [PubMed] [Google Scholar]
- Mahadevan A, Shankar SK, Satishchandra P, Ranga U, Chickabasaviah YT, Santosh V, Vasanthapuram R, Pardo CA, Nath A, Zink MC. Characterization of human immunodeficiency virus (HIV)-infected cells in infiltrates associated with CNS opportunistic infections in patients with HIV clade C infection. J Neuropathol Exp Neurol. 2007;66:799–808. doi: 10.1097/NEN.0b013e3181461d3e. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Haughey NJ, Nath A. Cell death in HIV dementia. Cell Death Differ. 2005;12(Suppl 1):893–904. doi: 10.1038/sj.cdd.4401577. [DOI] [PubMed] [Google Scholar]
- Mishra M, Vetrivel S, Siddappa NB, Ranga U, Seth P. Clade-specific differences in neurotoxicity of human immunodeficiency virus-1 B and C Tat of human neurons: significance of dicysteine C30C31 motif. Ann Neurol. 2008;63:366–376. doi: 10.1002/ana.21292. [DOI] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Nath A, Haughey NJ, Jones M, Anderson C, Bell JE, Geiger JD. Synergistic neurotoxicity by human immunodeficiency virus proteins Tat and gp120: protection by memantine. Ann Neurol. 2000;47:186–194. [PubMed] [Google Scholar]
- Navia BA, Jordan BD, Price RW. The AIDS dementia complex: I. Clinical features. Ann Neurol. 1986;19:517–524. doi: 10.1002/ana.410190602. [DOI] [PubMed] [Google Scholar]
- Riedel D, Ghate M, Nene M, Paranjape R, Mehendale S, Bollinger R, Sacktor N, McArthur J, Nath A. Screening for human immunodeficiency virus (HIV) dementia in an HIV clade C-infected population in India. J Neurovirol. 2006;12:34–38. doi: 10.1080/13550280500516500. [DOI] [PubMed] [Google Scholar]
- Sabatier JM, Vives E, Mabrouk K, Benjouad A, Rochat H, Duval A, Hue B, Bahraoui E. Evidence for neurotoxicity of tat from human immunodeficiency virus type 1. J Virol. 1991;65:961–967. doi: 10.1128/jvi.65.2.961-967.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- Song L, Nath A, Geiger JD, Moore A, Hochman S. Human immunodeficiency virus type 1 Tat protein directly activates neuronal N-methyl-d-aspartate receptors at an allosteric zinc-sensitive site. J Neurovirol. 2003;9:399–403. doi: 10.1080/13550280390201704. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Shin Y, Cho SJ, Zago WM, Nakamura T, Gu Z, Ma Y, Furukawa H, Liddington R, Zhang D, Tong G, Chen HS, Lipton SA. Hypoxia enhances S-nitrosylation-mediated NMDA receptor inhibition via a thiol oxygen sensor motif. Neuron. 2007;53:53–64. doi: 10.1016/j.neuron.2006.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]