

Abstract
Inflammation influences several steps of adult neurogenesis, but whether it regulates the functional integration of the new neurons is unknown. Here, we explored, using confocal microscopy and whole-cell patch-clamp recordings, whether a chronic inflammatory environment affects the morphological and electrophysiological properties of new dentate gyrus granule cells, labeled with a retroviral vector encoding green fluorescent protein. Rats were exposed to intrahippocampal injection of lipopolysaccharide, which gave rise to long-lasting microglia activation. Inflammation caused no changes in intrinsic membrane properties, location, dendritic arborization, or spine density and morphology of the new cells. Excitatory synaptic drive increased to the same extent in new and mature cells in the inflammatory environment, suggesting increased network activity in hippocampal neural circuitries of lipopolysaccharide-treated animals. In contrast, inhibitory synaptic drive was more enhanced by inflammation in the new cells. Also, larger clusters of the postsynaptic GABAA receptor scaffolding protein gephyrin were found on dendrites of new cells born in the inflammatory environment. We demonstrate for the first time that inflammation influences the functional integration of adult-born hippocampal neurons. Our data indicate a high degree of synaptic plasticity of the new neurons in the inflammatory environment, which enables them to respond to the increase in excitatory input with a compensatory upregulation of activity and efficacy at their afferent inhibitory synapses.
Keywords: adult neurogenesis, inflammation, synaptic plasticity, gephyrin, electrophysiology, hippocampus
Introduction
In the adult brain, neural stem cells in the subgranular zone (SGZ) and the subventricular zone (SVZ) form new dentate granule cells and olfactory bulb interneurons, respectively. Available evidence points to a role for hippocampal neurogenesis in mood regulation, learning, and memory, and for olfactory bulb neurogenesis in olfactory discrimination and memory (Zhao et al., 2008). Neurogenesis is influenced by pathological conditions [e.g., status epilepticus (SE) and stroke], which stimulate the formation of new neurons in SVZ and SGZ (Bengzon et al., 1997; Parent et al., 1997, 2002; Arvidsson et al., 2001, 2002).
Brain inflammation is involved in the pathogenesis of neurological disorders such as stroke (Danton and Dietrich, 2003) and epilepsy (Gorter et al., 2006) and can be both detrimental and beneficial for neurogenesis. Autoimmune T-cells promote SGZ and SVZ progenitor proliferation by interacting with microglia (Ziv et al., 2006). Tumor necrosis factor-α, released by activated microglia, suppresses SGZ and SVZ progenitor proliferation through tumor necrosis factor receptor 1 after SE and stroke (Iosif et al., 2006, 2008). Microglia activated early after stroke or SE, or by administration of the bacterial endotoxin lipopolysaccharide (LPS), compromises survival and differentiation of newly formed hippocampal and striatal neurons (Ekdahl et al., 2003; Monje et al., 2003; Hoehn et al., 2005). However, the newly formed granule cells, which do not die during the first month after SE, survive for 5 months thereafter despite chronic inflammation (Bonde et al., 2006).
Whether brain inflammation alters the functional integration of new neurons into existing neural circuitries is unknown. In the intact brain, the maturation of new granule cells follows distinct morphological stages (Zhao et al., 2006), and they develop synaptic inputs closely resembling those of mature granule cells (van Praag et al., 2002). The functional synaptic connectivity of adult-born granule cells is remarkably similar to that of cells generated during development (Laplagne et al., 2006, 2007). Recent experimental evidence suggests that developing in an inflammatory environment may influence the synaptic connectivity of the new neurons. Activated microglia secrete cytokines and growth factors, which can modulate synaptic transmission (Henneberger et al., 2005; Pickering et al., 2005) and alter dendritic spine morphology (Schratt et al., 2006; von Bohlen und Halbach et al., 2006). Moreover, after SE, new hippocampal neurons exhibit increased inhibitory and decreased excitatory drive at their afferent synapses (Jakubs et al., 2006), which may mitigate abnormal excitability. The epileptic insult causes seizures, neuronal death, and inflammation, and each of these pathologies could underlie the altered synaptic properties (Jakubs et al., 2006).
Here, we created a chronic inflammatory environment by LPS injection into rat hippocampal formation and labeled new granule cells through injection of a retroviral (RV) vector encoding green fluorescent protein (GFP) 1 week thereafter. After 6–8 weeks, we performed detailed microscopical analysis of the new neurons and whole-cell patch-clamp recordings to assess their intrinsic membrane properties and excitatory and inhibitory synaptic inputs. We demonstrate for the first time that inflammation regulates the functional synaptic connectivity of new neurons generated in the adult brain.
Materials and Methods
Animal groups and lipopolysaccharide administration.
One hundred fourteen male Sprague Dawley rats were used, weighing 200–250 g at the beginning of experiments. Experimental procedures were approved by the Malmö-Lund Ethical Committee. Animals were anesthetized with isoflurane (2%) or ketamine/xylazine (90/16.9 mg/kg), and LPS from Salmonella enterica, serotype abortus equi [Sigma-Aldrich; 15 μg in 1.5 μl of artificial CSF (aCSF)] or vehicle (1.5 μl of aCSF) was stereotaxically injected into the left dorsal dentate gyrus (DG) (coordinates: 4.0 mm caudal and 2.5 mm lateral to bregma, 2.9 mm ventral from dura, toothbar at −3.3 mm) (Paxinos and Watson, 1997) using a glass microcapillary. Fifty-three rats were used for electrophysiological recordings (LPS, n = 28; vehicle, n = 25), and 61 animals for morphological analysis (LPS, n = 31; vehicle, n = 30). To explore whether the LPS-induced inflammation had caused epileptic activity, a recording electrode was implanted into the right ventral hippocampus (coordinates: 4.8 mm caudal and 5.2 mm lateral to bregma, 6.3 mm ventral from dura, toothbar at −3.3 mm) in five rats before LPS (n = 3) or vehicle (n = 2) injection for subsequent electroencephalogram (EEG) recording. EEG activity was monitored for 5 min at 1 and 8 weeks after LPS injection.
Labeling of new neurons.
One week after vehicle or LPS injection, rats were anesthetized with isoflurane or ketamine/xylazine and injected with 1.5 μl of retrovirus, containing the GFP gene under the CAG promoter (1.2–1.3 × 108 transducing U/ml) [for details, see Zhao et al. (2006)], ipsilaterally at two dorsal hippocampal sites (coordinates: 3.6 mm caudal and 2.0 mm lateral to bregma, and 2.8 ventral to dura; 4.4 mm caudal and 3.0 mm lateral to bregma, and 3.0 mm ventral to dura; toothbar at −3.3 mm).
Electrophysiological analysis and tissue preparation.
Seven to 9 weeks after LPS or vehicle injection, animals were deeply anesthetized with isoflurane and decapitated. Brains were immediately removed, and ipsilateral hippocampi were dissected and placed in ice-cold modified aCSF containing the following (in mm): 225 sucrose, 2.5 KCl, 0.5 CaCl2, 7.0 MgCl2, 28 NaHCO3, 1.25 NaH2PO4, 7.0 glucose, 1.0 ascorbate, and 3.0 pyruvate, pH 7.2–7.4, 295–300 mOsm. Transverse 225-μm-thick hippocampal slices were cut using a vibratome, transferred to gassed aCSF (95% O2, 5% CO2) containing the following (in mm): 119 NaCl, 2.5 KCl, 1.3 MgSO4, 2.5 CaCl2, 26.2 NaHCO3, 1 NaH2PO4, and 11 glucose, and then allowed to rest for at least 1 h.
Whole-cell patch-clamp recordings were performed at room temperature (RT) in all cases except for spontaneous EPSCs (sEPSCs), which were recorded at 32–34°C. Individual slices were placed in a submerged recording chamber constantly perfused with gassed aCSF. Cells for recording were visualized with Olympus upright microscope and a digital camera using infrared light with differential interference contrast and UV light for identifying GFP-expressing cells in the granule cell layer (GCL). Recording pipettes with a tip resistance of 3–6 MΩ were filled with pipette solution, pH 7.2–7.4 and 295–300 mOsm, containing the following (in mm): 135.0 CsCl, 10.0 CsOH, 0.2 CsOH-EGTA, 2.0 Mg-ATP, 0.3 Na3-GTP, 8.0 NaCl, and 5.0 lidocaine N-ethyl bromide (QX-314) for voltage-clamp recordings of IPSCs, or 117.5 Cs-gluconate, 17.5 CsCl, 8.0 NaCl, 10.0 CsOH-HEPES, 0.2 CsOH-EGTA, 2.0 Mg-ATP, 0.3 Na3GTP, and 5.0 QX-314, for voltage-clamp recordings of EPSCs. For current-clamp recordings, the pipette solution contained the following (in mm): 122.5 K-gluconate, 12.5 KCl, 10.0 KOH-HEPES, 0.2 KOH-EGTA, 2.0 MgATP, 0.3 Na3GTP, and 8.0 NaCl. Biocytin (0.5%; Sigma-Aldrich) was freshly dissolved in the pipette solution before recordings. Spontaneous and miniature IPSCs (sIPSCs and mIPSCs, respectively) were recorded continuously for 3 min while holding the cell membrane potential at −80 mV and blocking NMDA and non-NMDA glutamate receptors with d-AP5 (50 μm) and 1,2,3,4-tetrahydro-6-nitro-2,3-dioxo-benzo[f]quinoxaline-7-sulfonamide (NBQX) (10 μm), respectively, in the aCSF perfusion solution. Tetrodotoxin (TTX) (1 μm)-containing perfusion solution was applied to the slices during 8 min and mIPSCs were then recorded for a 3 min period. Spontaneous EPSCs were recorded for 3 min while blocking GABAA receptors with 100 μm picrotoxin (PTX) in the perfusion solution. Whole-cell access resistance was continuously monitored by a test pulse applied through the patch pipette, and recordings were discarded if access resistance changed >20%. For paired-pulse experiments, a bipolar stimulation electrode was placed in the middle part of the molecular layer (ML) for monosynaptic IPSC induction, or lateral perforant path (outer one-third of ML) for EPSCs induction [for more details, see Jakubs et al. (2006)], and a series of 20 paired stimulations was delivered with 25, 50, 100, or 200 ms interstimulus intervals. For inhibitory experiments, the GABAB receptor agonist baclofen (10 μm) (Wu and Leung, 1997; Gloveli et al., 2003) was added to the aCSF, and the slices were perfused with this solution for 8 min after which another series of 20 paired stimulations was applied. IPSCs were then blocked with 100 μm PTX, confirming that they were mediated by GABAA receptors. For recordings of intrinsic membrane properties, resting membrane potential was estimated in current-clamp mode immediately after breaking the membrane and establishing whole-cell configuration. For measuring current–voltage relationship, 500 ms hyperpolarizing and depolarizing current pulses were delivered in 15 pA increments through the whole-cell pipette. The GFP expression of all recorded new cells was confirmed either by visualization of GFP that had diffused into the pipette during the recording, or with post hoc immunocytochemical detection of GFP colocalization with biocytin. All drugs were from Tocris unless otherwise stated.
Spontaneous and miniature postsynaptic currents were detected and analyzed using MiniAnalysis software (Synaptosoft). The last 100 sIPSCs and first 50 mIPSCs or 50 sEPSCs were analyzed from each cell. Minimum amplitude for detection was set at 5 times root-mean-square noise level as determined by the software for s/mIPSCs, or 5 pA for sEPSCs. Recordings with root-mean-square level of >5 pA were discarded. All detected events were visually controlled. Group interevent intervals (IEIs) and amplitudes were compared using Kolmogorov–Smirnov's statistical test. Approximately 20 stimulation-evoked IPSCs or EPSCs were averaged for each cell (both before and after baclofen application for IPSCs), and peak amplitudes were measured to calculate paired-pulse depression (PPD) or facilitation (PPF) in percentage. Average PPD or PPF values were compared between groups using Student's unpaired t test or ANOVA. Current–voltage relationship was plotted at steady-state potential 100 ms after onset of the test current pulses. Input resistance was calculated from the steady-state membrane potential response to a 15 pA hyperpolarizing current injection. Membrane time constant was calculated by fitting a single exponential curve to the membrane voltage response (5–90 ms from onset) to a 15 pA hyperpolarizing current injection. Action potential threshold was determined at the beginning of the steepest rising phase of the membrane potential. Amplitude of the action potential was estimated as absolute difference between threshold and peak, and duration (half-width) was measured as the time between rising and falling phase at half-amplitude. Level of significance for the statistical tests was set at p < 0.05.
Sections from electrophysiology experiments were fixed for 2–24 h in 4% paraformaldehyde (PFA) immediately after recordings and stored in antifreeze medium at −20°C. For double staining of biocytin and GFP, free-floating sections were preincubated for 1 h in 5% serum in 0.25% Triton X-100 in potassium PBS, and exposed to rabbit anti-GFP primary antibody (1:10,000; Abcam) overnight at RT. FITC-conjugated donkey anti-rabbit secondary antibody and Cy3-streptavidin (1:200; Jackson ImmunoResearch) were then added for 2 h in the dark at RT. Sections were mounted on glass slides and coverslipped.
Tissue preparation and morphological analysis.
At 10, 17, 23 d, and 8 weeks after LPS (n = 6, 6, 6, and 6 animals) or vehicle injection (n = 6, 6, 6, and 4 animals), rats received an overdose of pentobarbital (250 mg/kg, i.p.) and were transcardially perfused with 100 ml of saline followed by 250 ml of 4% ice-cold PFA. Brains were postfixed overnight, dehydrated in 20% sucrose in 0.1 m PBS overnight, cut in 30 μm sections, and stored in cryoprotective solution. For analysis of gephyrin distribution (Schneider Gasser et al., 2006), 10 rats (LPS, n = 6; vehicle, n = 4) were anesthetized and decapitated. Brains were dissected and placed in ice-cold aCSF, cut in transverse 300-μm-thick sections, placed in gassed aCSF for 20 min and in PFA for 10 min, rinsed in PBS, dehydrated in 30% sucrose in 0.1 m PBS, cut in 14 μm sections, and stored at −20°C for at least 1 h.
For immunohistochemistry, incubation with appropriate primary antibodies was performed for 1 h at RT. Primary antibodies were: rabbit anti-Iba1 (1:1000; Wako Chemicals) for active and quiescent microglia (Imai and Kohsaka, 2002), mouse-anti-ED1 (1:200; Serotec) for phagocytic microglia (Damoiseaux et al., 1994), rabbit anti-GFP, mouse anti-NeuN (1:100; Millipore Bioscience Research Reagents), and mouse anti-gephyrin (1:10,000; Synaptic Systems). Free-floating sections were incubated overnight at 4°C. Secondary antibodies were as follows: Cy3-conjugated donkey anti-rabbit, biotinylated horse anti-mouse (both 1:200; Vector Laboratories), Cy3-conjugated donkey anti-mouse (1:300; Jackson ImmunoResearch), and FITC-conjugated donkey anti-rabbit with incubation for 2 h at RT (1 h for gephyrin). Rinsing in potassium PBS with or without 0.25% Triton X-100 was performed between each incubation. For double stainings, biotinylated antibodies were detected with streptavidin-conjugated Alexa Fluor 488 (1:200; Invitrogen) for 2 h at RT, whereas in single stainings, avidin–biotin–peroxidase complex (Elite ABC kit; Vector Laboratories), 3,3′-diaminobenzidine, and hydrogen peroxide were used. Chromogenic visualization included pretreatment with blocking of endogenous peroxidase activity with 3% H2O2 and 10% methanol. Sections were mounted on microscope slides and immersed in xylene and coverslipped with Pertex mounting medium (HistoLab). For terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL)/Hoechst staining of DNA fragmentation in cells with apoptotic morphology, sections were immersed in 99% ethanol for 30 min, proteinase K (10 μg/ml) for 6 min, PFA for 5 min, and in 0.1% Triton X-100 in 0.1% sodium citrate for 2 min on ice, before being incubated for 1 h with terminal deoxynucleotidyl transferase buffer (TdT) containing TdT enzyme (5 μl/section) and TUNEL label solution with fluorescein-conjugated dUTP (45 μl/section; all Roche Diagnostics) and finally labeled with the nuclear marker Hoechst 33342 (1:1000; Invitrogen) for 10 min in the dark followed by mounting and coverslipping with glycerol-based mounting medium. For Fluoro-Jade staining of dying cells (Schmued et al., 1997), mounted sections were pretreated with 0.06% potassium permanganate before being agitated for 30 min in 0.001% Fluoro-Jade (Histochem) in 0.1% acetic acid, immersed in xylene, and coverslipped with Pertex mounting medium.
Cell counts and morphological analyses were performed ipsilaterally to LPS injections in six hippocampal sections per animal located 3.3–5.1 mm posterior to bregma (Paxinos and Watson, 1997) by an observer blind to treatment conditions. Numbers of Iba1+/ED1+, Fluoro-Jade+, and TUNEL+ cells with apoptotic morphology were counted with an Olympus BX61 epifluorescence and light microscope in SGZ/GCL, which for GFP+ cell counts was further subdivided into inner, middle, and outer GCL. GFP+ cell counts also included dentate hilus and ML. The volume of SGZ/GCL was measured in NeuN-stained sections using stereological equipment [Olympus BH-2; 40× objective; CC-IRIS color video camera; CAST-GRID software (Olympus)]. The polarization axis was classified for all GFP+ cells (LPS, n = 130 cells; vehicle, n = 332 cells) into either of three categories: 0–22, 22.5–67, and 67.5–90°, where 90° is perpendicular to the GCL. Numbers of apical dendrites and percentage of GFP+ cells with apical, basal, and recurrent basal dendrites, and basal, medial, and apical axons were analyzed for all GFP+ cells. Dendritic branching of GFP+ cells was analyzed by assessing the cumulative number of branching points when following each dendrite (LPS, n = 60; vehicle, n = 173) from the cell soma in 12.5 μm increments until reaching 75 μm.
Spine density (numbers per micrometer) and morphology (divided into four categories: thin, stubby, filopodia, and mushroom) (Zhao et al., 2006) and gephyrin cluster density (numbers per micrometer) and size (area in square micrometers) were analyzed in a confocal laser scanning microscope (Bio-Rad MRC1021UV) with Kr–Ar 488 and 568 nm excitation filters, 100× objective and 16× digital zoom in six squares (9.38 × 9.38 μm) per animal, on proximal and distal dendrites in the inner and outer ML, respectively. The area of each gephyrin cluster was measured in the z-focal plane.
Data were compared using Student's unpaired nonparametric t test. Results are presented as means ± SEM. Differences are considered significant at p < 0.05.
Results
Lipopolysaccharide induces chronic inflammation in dentate gyrus without neuronal death or seizure activity
Lipopolysaccharide has previously been used to study the effect of inflammation on hippocampal neurogenesis (Ekdahl et al., 2003; Monje et al., 2003). We found here that the single LPS injection into the dorsal DG induced a long-lasting inflammatory response (Fig. 1A–C). At 1 week after LPS injection, the time point at which the newborn neurons were labeled with RV-GFP, both the total number of microglia (Iba1+ cells; LPS, 831.5 ± 8.0; vehicle, 555.7 ± 5.8) and the number of microglia with a phagocytic phenotype (Iba1+/ED1+ cells) were elevated in the SGZ/GCL (Fig. 1A–C) to similar magnitude as we have previously observed 1 week after SE (Ekdahl et al., 2003; Jakubs et al., 2006). The numbers of Iba1+ (LPS, 683.8 ± 7.8; vehicle, 456.3 ± 9.5) and Iba1+/ED1+ cells were still elevated 7 weeks later (approximate time point of electrophysiological recordings) (Fig. 1C). Despite the marked increase in numbers of microglia, NeuN staining revealed that the general cytoarchitecture remained intact (Fig. 1D) and the volume of the SGZ/GCL did not differ between LPS- and vehicle-treated animals at 1 week (LPS, 7.6 ± 0.3 mm3; vehicle, 8.2 ± 0.5 mm3) or 8 weeks (LPS, 8.3 ± 0.4 mm3; vehicle, 7.9 ± 0.4 mm3) after injections. We did not detect any neurodegeneration using Fluoro-Jade staining (data not shown). Also, we found only occasional apoptotic TUNEL+ cells in the SGZ/GCL, with no differences between LPS and vehicle groups at either 1 week (2.5 ± 0.7 and 2.3 ± 0.8 cells, respectively) or 8 weeks (0.5 ± 0.3 and 0.7 ± 0.3 cells, respectively). Abnormal EEG activity (seizure activity or presence of interictal spikes) was not observed at any time point after vehicle or LPS treatment (Fig. 1E).
Figure 1.
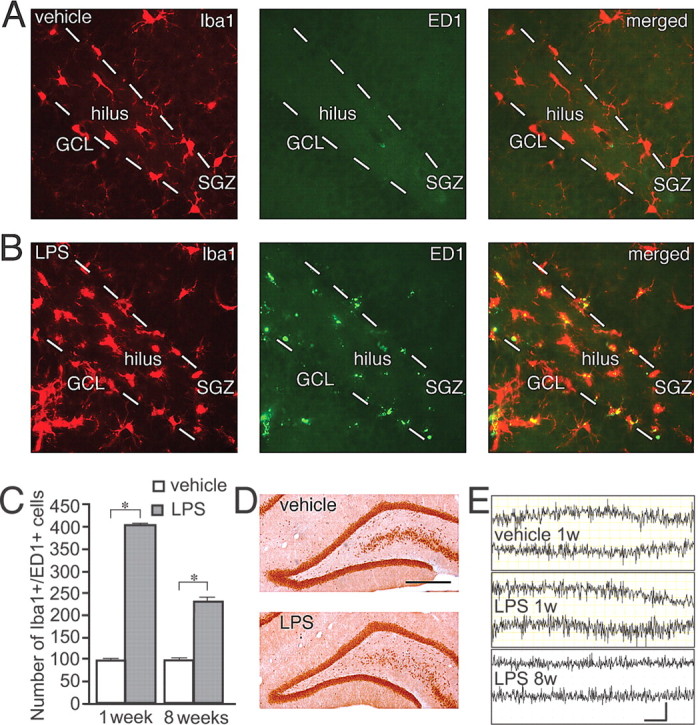
Lipopolysaccharide injection induces a long-lasting inflammatory response in dentate gyrus without neuronal death or seizure activity. Distribution of Iba1+ (red), ED1+ (green), and Iba1+/ED1+ (yellow) microglia in the dentate gyrus 1 week after intrahippocampal vehicle (A) or LPS (B) injection. C, Increased numbers of Iba1+/ED1+, activated microglia in the SGZ/GCL in LPS-treated animals at 1 and 8 weeks after injection. Student's unpaired t test, *p < 0.05 (LPS and vehicle at 1 week, n = 4 and 3, and 8 weeks, n = 6 and 6, respectively). Error bars indicate SEM. D, NeuN expression showing intact cytoarchitecture in the dentate gyrus at 1 week after LPS or vehicle injection. E, EEG recordings after vehicle injection, or at 1 and 8 weeks (1w and 8w) after LPS injection without pathological manifestations. Scale bar (in D): A, B, 50 μm; D, 400 μm. Calibration: E, 4 s, 0.2 mV.
Inflammatory environment does not alter location, dendritic tree, or intrinsic membrane properties of new granule cells
The RV-GFP vector induced stable GFP expression in new neurons in the DG when injected into both vehicle- and LPS-treated animals (Fig. 2A–E). The spatiotemporal maturation of the newborn GFP+ cells in the inflammatory and control environment closely resembled that previously reported for dentate granule cells in intact brain (Zhao et al., 2006; Shapiro et al., 2007). The apical dendrites of GFP+ cells reached into the molecular layer at 3 d (Fig. 2A), and axonal projections toward CA3 were clearly visible in the hilus at 10 d after GFP labeling (Fig. 2B), with no differences between groups. Large dendritic trees (Fig. 2C) with spines (inset) had developed at 16 d. Despite the detrimental effect of inflammation on hippocampal neurogenesis, causing death of many newly formed neurons (Ekdahl et al., 2003), we observed that a substantial number of GFP+ cells had differentiated into mature granule cells 7 weeks later (LPS, 32.5 ± 13.0 cells; vehicle, 55.3 ± 15.4 cells; p > 0.05), with highly arborized dendritic tree throughout the ML, and axons reaching CA3 at 7 weeks after GFP labeling in vehicle-treated (Fig. 2D) and LPS-treated animals (Fig. 2E).
Figure 2.
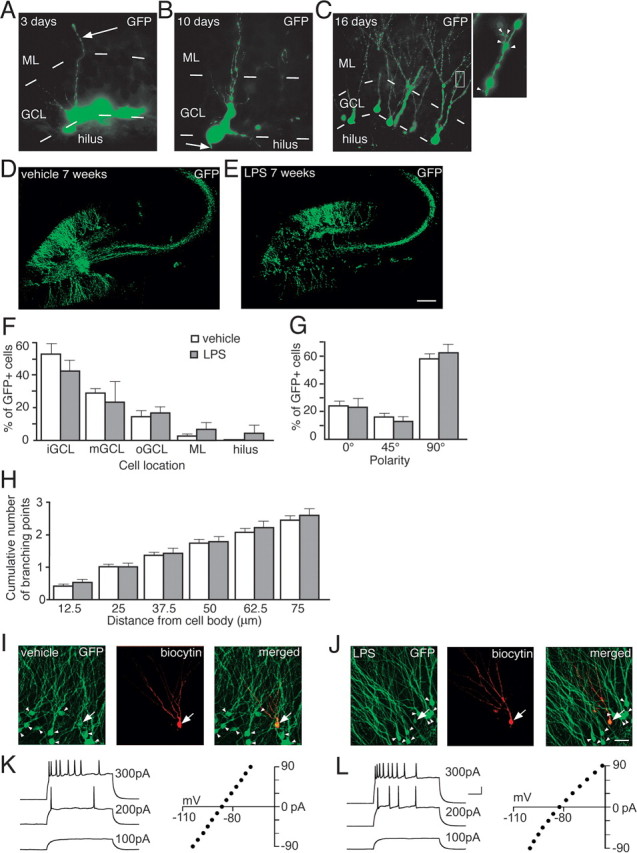
New neurons born into a chronic inflammatory environment differentiate into mature dentate granule cells. A, GFP+ cell located in the GCL with dendrite (arrow) reaching well into the ML 3 d after RV-GFP labeling. B, GFP+ cell with branched dendrites and an axon (arrow) in the hilus 10 d after RV-GFP labeling. Highly branched dendritic trees (C) with spines (inset, arrowheads) on GFP+ cells 16 d after RV-GFP labeling. Distribution of new GFP+ neurons in the GCL of vehicle- (D) or LPS-injected (E) animals 7 weeks after RV-GFP labeling. F, Relative location of GFP+ cells in inner, middle, or outer GCL (iGCL, mGCL, and oGCL, respectively), ML, or hilus. G, Polarity of the GFP+ cell soma, where 90° is perpendicular to GCL direction. 0, 45, and 90° comprise 0–22, 22.5–67, and 67.5–90°, respectively. H, Cumulative number of dendritic branching points at increasing distances from the GFP+ cell body. Microscopical analyses in F–H were performed 8 weeks after LPS or vehicle injection. No differences between LPS- and vehicle-treated animals were observed. Error bars indicate SEM. Student's unpaired t test, p > 0.05. GFP+ neurons visualized for patch-clamp analysis after vehicle (I) and LPS (J) injection. Coexpression of GFP (green) and biocytin (red) that had diffused from the patch pipette into the recorded cell (yellow; arrow) with neighboring, nonrecorded GFP+/biocytin− cells (arrowheads). Cells in I and J are from same animals as in D and E. Action potentials and current–voltage relationship plots of new and mature neurons from vehicle-treated (K) and LPS-treated (L) animals (recordings at 7–8 weeks after RV-GFP injection). Current injections of 500 ms were delivered in 15 pA increments. Traces are shown with 100, 200, and 300 pA injection. LPS, n = 130 cells; vehicle, n = 332 cells (F–H). Scale bars: (in E) A, B, 10 μm; (in E) C, 25 μm; (in E) C, inset, 5 μm; (in E) D, E, 215 μm; (in J) I, J, 30 μm. Calibration: K, L, 100 ms, 20 mV.
The distributional pattern of GFP+ cells in the DG was similar in LPS- and vehicle-treated animals (Fig. 2F). The absolute majority of the new cells were located in the GCL, preferentially in its inner part, but a few GFP+ cells were found in the hilus and the ML. We observed no differences between LPS- and vehicle-treated animals in the polarity of the GFP+ cell soma (Fig. 2G). For most of the cells, the somata were aligned vertically, at a 90° angle to the direction of the GCL. The numbers of apical dendrites per GFP+ cell (LPS, 1.6 ± 0.2; vehicle, 1.4 ± 0.1), dendritic branching points at increasing distances from the soma (Fig. 2H), recurrent basal dendrites (which were very rare), as well as exit points for the axons on the soma did not differ between GFP+ cells in LPS- and vehicle-treated animals. Most axons originated from the basal side, some from the medial side, and very few from the apical side of the GFP+ cell soma (LPS, 68.7 ± 12.4, 29.8 ± 11.3, and 1.5 ± 1.5%; vehicle, 79.8 ± 6.4, 20.0 ± 6.3, and 0.4 ± 0.4%, respectively).
We performed whole-cell patch-clamp recordings from both GFP+ cells (new cells, born at the time of RV-GFP injection) and GFP− cells (mature cells, most likely born before LPS and vehicle injection) (Jakubs et al., 2006) in the GCL at 6–8 weeks after RV-GFP injection. Whole-cell current-clamp recordings revealed that the intrinsic membrane properties (resting membrane potential, membrane time constant, input resistance, and action potential threshold, amplitude, and duration) of the new and mature cells in LPS-treated animals were similar to those of new and mature cells in vehicle-treated animals, and closely resembled those characteristic of dentate granule cells (Fig. 2I–L; supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Inflammatory environment causes similar increase in excitatory synaptic drive on new and mature granule cells
We first analyzed whether the LPS-induced chronic inflammation influenced the excitatory synaptic input to the new and mature neurons in the GCL. Whole-cell voltage-clamp recordings of EPSCs were performed while blocking GABAA receptors with PTX. Both the new and mature cells in the LPS-treated animals exhibited increased frequency but no change in amplitude of sEPSCs compared with the new and mature cells in the vehicle-treated animals (Fig. 3A–E). Cumulative fraction analysis showed that this effect of LPS-induced inflammation was of the same magnitude in new and mature cells (Fig. 3F,G). We then explored whether short-term plasticity at excitatory synapses on new and mature cells was affected by the chronic inflammatory environment. When we delivered paired stimulations to the lateral perforant path, we observed the same level of PPF of EPSCs in the new and mature granule cells in vehicle- and LPS-treated animals at all interstimulus intervals (Fig. 4). In summary, our findings revealed increased overall excitatory synaptic activity because of the LPS-induced chronic inflammation but no specific effect at synapses on the new compared with the mature cells.
Figure 3.
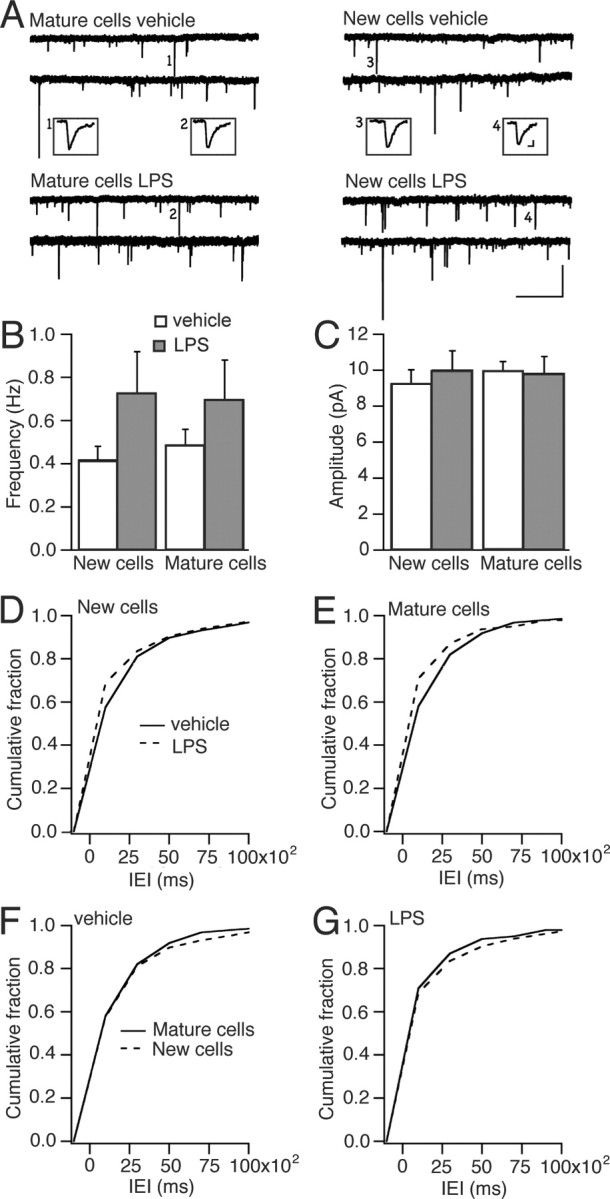
Chronic inflammatory environment causes increased frequency of spontaneous EPSCs in new and mature neurons. A, Continuous whole-cell voltage-clamp recordings of sEPSCs in mature or new cells in hippocampal slices, perfused with aCSF containing PTX, from either vehicle- or LPS-injected rats at 7.5–8 weeks after RV-GFP injection. Mean frequency (B) and amplitude (C) of sEPSCs in new and mature granule cells from LPS- and vehicle-treated animals. Error bars indicate SEM. Cumulative fraction curves of IEIs showing shorter intervals (i.e., higher frequency) in both new (D) (p < 0.001) and mature (E) (p < 0.001) cells in LPS-treated compared with vehicle-treated animals but no differences between new and mature cells in vehicle-treated (F) (p > 0.05) or LPS-treated (G) (p > 0.05) animals (Kolmogorov–Smirnov test). Numbers of cells recorded were as follows: in vehicle, six GFP+, seven GFP−; in LPS, seven GFP+, five GFP−.
Figure 4.

Chronic inflammatory environment does not influence short-term plasticity at excitatory synapses on new and mature neurons. Whole-cell voltage-clamp recordings of paired stimulation-induced EPSCs in new and mature granule cells in hippocampal slices, perfused with aCSF containing PTX, from vehicle- or LPS-treated animals 6.5–7.5 weeks after RV-GFP labeling. Paired stimulations were delivered to lateral perforant path in the outer one-third of the molecular layer with interstimulus intervals (ISIs) of 25, 50, 100, and 200 ms. Average paired-pulse facilitation plotted at each ISI for new (A) and mature cells (B) in slices from vehicle- or LPS-treated animals. Representative EPSCs elicited with 100 ms ISI are shown in insets from vehicle-treated (A1, B1) or LPS-treated (A2, B2) animals. Twenty responses were averaged for each cell at each ISI. Comparisons using one-way ANOVA, or Student's unpaired t test at each ISI, revealed no differences between new cells or between mature cells in vehicle- and LPS-treated animals, or between new and mature cells within the same treatment group. Numbers of cells recorded were as follows: in vehicle, four GFP+, five GFP−; in LPS, five GFP+, five GFP−. Error bars indicate SEM. Calibration: A1, A2, B1, B2, 50 ms, 50 pA.
Inflammatory environment leads to more inhibitory synaptic drive on new compared with mature granule cells
We next determined whether the new neurons exhibited altered inhibitory synaptic input if they had been born into a chronic inflammatory environment. Whole-cell voltage-clamp recordings of sIPSCs were performed while blocking glutamate receptors with NBQX and d-AP5. In animals treated with LPS, the IEIs of sIPSCs recorded in new cells were reduced compared with those in new cells from vehicle-treated animals (Fig. 5A,C). Also, the sIPSCs recorded in the mature cells after LPS treatment occurred with shorter IEIs compared with mature cells in vehicle-injected animals (Fig. 5B,D). The amplitude of sIPSCs in new cells was higher in LPS- than in vehicle-injected animals (Fig. 5E), whereas no such difference was observed in mature cells (data not shown).
Figure 5.

Chronic inflammatory environment causes increased frequency of spontaneous IPSCs in new and mature neurons. Whole-cell voltage-clamp recordings from new (A) and mature (B) cells in slices from vehicle- and LPS-treated animals at 6–7.4 weeks after RV-GFP injection in the presence of the glutamate receptor blockers NBQX and d-AP5. The top two traces in each group represent sIPSCs (“NBQX + d-AP5”), the third trace depicts mIPSCs (“+TTX”), and the bottom trace demonstrates blockade of IPSCs by picrotoxin (“+PTX”). C, Cumulative fraction analysis showing shorter IEIs of sIPSCs recorded in new cells from LPS-treated animals compared with those in new cells from vehicle-treated rats (p < 0.001; 8 cells in each group). D, Spontaneous IPSCs at shorter IEIs in mature cells from LPS- than from vehicle-injected animals (p < 0.05; 8 and 10 cells in LPS and vehicle groups, respectively). E, Higher amplitudes of sIPSCs in new cells from LPS-treated animals than in those from vehicle-treated animals (p < 0.001) (Kolmogorov–Smirnov test). Calibration: A, B, 50 ms, 50 pA.
When comparing the cells within each treatment group, cumulative fraction analysis revealed that the sIPSCs occurred with similar IEIs (Fig. 6A) and amplitude (data not shown) in new and mature cells in vehicle-treated animals. In contrast, the IEIs of sIPSCs were shorter and their amplitude was larger in new cells compared with mature cells from LPS-treated animals (Fig. 6B,C). When action potential-dependent IPSCs were blocked with TTX (thereby isolating mIPSCs) (Fig. 6D–F), IEIs of mIPSCs did not differ between new and old cells in vehicle-injected animals (Fig. 6D). Moreover, the decreased IEIs and increased amplitude of sIPSCs observed in LPS-treated animals were no longer observed in new cells after TTX application (Fig. 6E,F). Together, these data indicate that LPS-induced inflammation causes a more marked action potential-dependent inhibitory drive at afferent synapses on the new compared with the mature dentate granule cells.
Figure 6.

Chronic inflammatory environment leads to more action potential-dependent inhibitory drive in new than in mature neurons. Whole-cell voltage-clamp recordings of sIPSCs (in the presence of NBQX plus d-AP5) and mIPSCs (after addition of TTX) in cells from vehicle-treated (A, D) and LPS-treated (B, C, E, F) animals. Cumulative fraction curves showing sIPSCs at similar IEIs in new and mature cells in vehicle-treated animals (A) (p > 0.05) but with shorter IEIs (B) (p < 0.001) in new cells compared with mature cells in LPS-treated animals. C, Higher amplitude of sIPSCs in new cells compared with old cells in LPS animals (p < 0.001). Similar IEIs of mIPSCs in new and mature cells in vehicle-treated animals (D) (p > 0.05), and similar IEIs (E) (p > 0.05) and amplitudes (F) (p > 0.05) of mIPSCs in new cells compared with mature cells in LPS-treated rats after action potential blockade with TTX (Kolmogorov–Smirnov test). The cells are the same as in Figure 5.
We then analyzed the effect of LPS-induced chronic inflammation on short-term plasticity at inhibitory synapses on the new cells by delivering paired stimulations to the ML of the DG with an interstimulus interval of 100 ms while blocking glutamatergic neurotransmission with NBQX and d-AP5. The first stimulation-induced release of GABA during paired stimulation-induced, monosynaptic IPSCs activates, apart from postsynaptic GABAA receptors, presynaptic GABAB receptors, causing an autoinhibition of the subsequent release of GABA, which results in depression of the second IPSC (Davies et al., 1990; Otis et al., 1993). In our experiment, the PPD of the stimulation-evoked IPSCs recorded in the new cells born into an inflammatory environment closely resembled that of the new cells in vehicle-treated animals (LPS, −48.0 ± 4.8%; vehicle, −50.7 ± 7.2%). We then maximally activated GABAB receptors by exogenous application of the selective agonist baclofen. This treatment strongly suppressed IPSCs and abolished PPD in new cells from vehicle- and LPS-treated animals to the same extent, resulting in similar paired-pulse ratio (Fig. 7A–C). These data indicate that short-term plasticity and presynaptic GABAB receptor sensitivity or expression at afferent inhibitory synapses on new dentate granule cells are not altered in the chronic inflammatory environment.
Figure 7.

Chronic inflammatory environment does not influence short-term plasticity at inhibitory synapses in new neurons. A, Representative recordings (each trace is average of 20 responses from the same cell) from new cells in vehicle- and LPS-treated animals before (“−BAC”; B1) and after (“+BAC”; B2) addition of the GABAB receptor agonist baclofen. Baclofen eliminated PPD similarly in new cells from vehicle (B) and LPS (C) animals. B, C, Depression/facilitation of IPSCs in individual cells; each cell is plotted before and after baclofen addition (connected with dashed lines), and group averages are depicted by the solid line (n = 5 and 7 cells in LPS- and vehicle-treated animals, respectively). *p < 0.05, Student's paired t test. Recordings were performed 6.1–7.7 weeks after RV-GFP injection. Calibration: A1, 50 ms, 100 pA; A2, 50 ms, 50 pA.
Inflammatory environment causes no change of dendritic spines but increased area of gephyrin clusters at inhibitory synapses on new granule cells
We finally wanted to explore whether the chronic inflammation, induced by the LPS treatment, gave rise to structural alterations at excitatory and inhibitory synapses on the new neurons. We first analyzed the dendritic spines, which are the major postsynaptic sites for the excitatory, glutamatergic inputs on dentate granule cells. Using confocal microscopy, we estimated the density and morphology of spines on the GFP+ new granule cells in the ML of the DG. No difference in spine density was observed between LPS- and vehicle-treated animals in inner and outer ML (Fig. 8A,B). We then compared the density of dendritic spines with specific morphology (mushroom, thin, stubby, and filopodia) between new cells born in an inflammatory or control environment. Mushroom spines are likely to be associated with stronger excitatory synapses, and accelerated mushroom spine formation on new granule cells has been reported in running animals (Zhao et al., 2006). No differences were found in the density of mushroom spines (Fig. 8C) or any of the other morphological subtypes of dendritic spines between new cells in LPS- and vehicle-treated animals (data not shown). Together, our data do not reveal any morphological alterations caused by the inflammatory environment at presumed excitatory synapses on the dendrites of the new cells.
Figure 8.
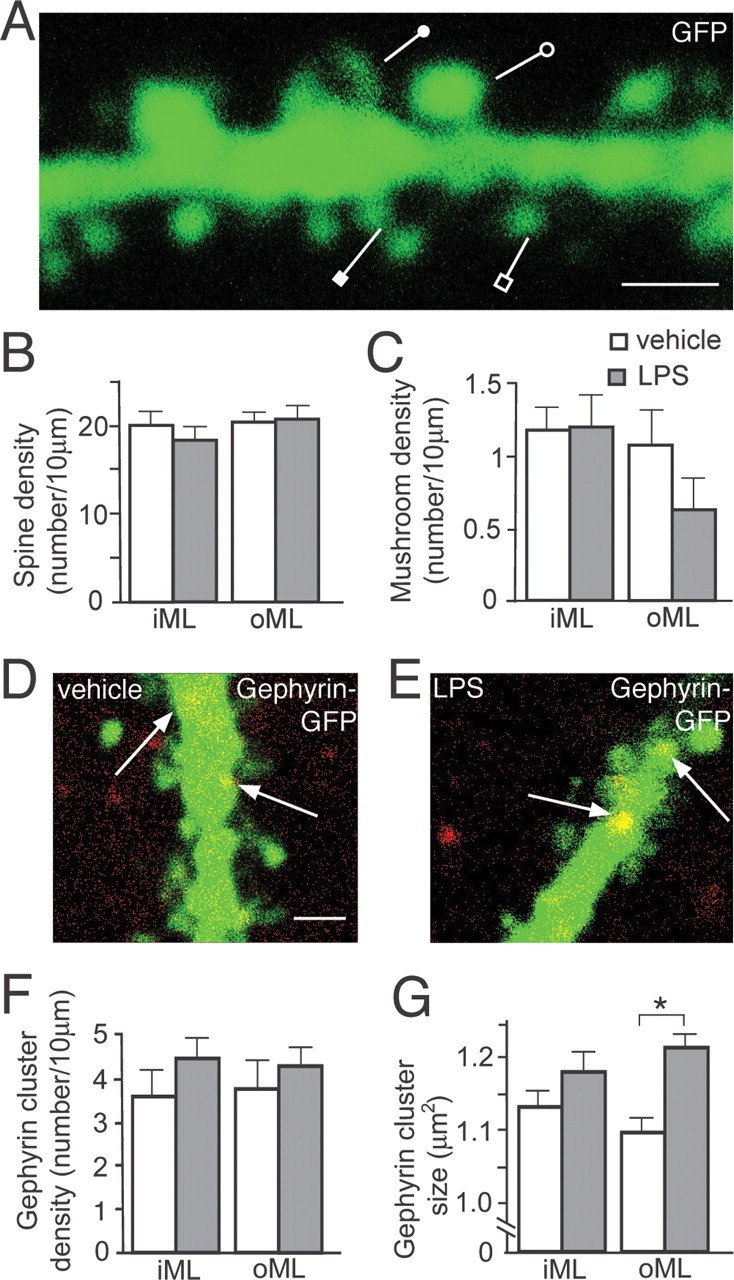
New neurons born into a chronic inflammatory environment develop normal spine density and morphology but increased size of the postsynaptic scaffolding protein gephyrin. New neurons develop dendritic tree with thin (open square), stubby (closed square), filopodia (closed circle), and mushroom spines (open circle) (A), and with similar density of dendritic spines (B) and mushroom spines (C) in vehicle- and LPS-treated animals in inner and outer ML (iML and oML, respectively). New neurons born in vehicle-treated (D) or LPS-treated (E) animals develop gephyrin clusters on dendrites (arrows), with similar density (F) but increased size (G) on distal dendrites in outer ML of LPS-treated compared with vehicle-treated animals (*p < 0.001) (Student's unpaired t test) (LPS, n = 23–34 dendrites; vehicle, n = 21–33 dendrites). Error bars indicate SEM. Scale bars: A, D (for D, E), 1 μm.
To detect inflammation-induced alterations at inhibitory synapses on the new neurons, we analyzed with confocal microscopy the immunohistochemical staining of the postsynaptic scaffolding protein gephyrin in the dendrites of GFP+ cells. Gephyrin is a multifunctional protein involved in the clustering of glycine and GABAA receptors at inhibitory synapses (Fritschy et al., 2008). We found no differences in the density of gephyrin clusters in the inner and outer ML on new cells in vehicle- and LPS-treated animals (Fig. 8D–F). However, the area of the gephyrin clusters was larger on the distal dendrites (located in the outer ML) of the new cells that had matured in the inflammatory environment (Fig. 8D,E,G). This finding indicates that chronic inflammation leads to structural alterations at inhibitory synapses on new dentate granule cells.
Discussion
Here, we demonstrate that new hippocampal neurons, which have developed in a chronic inflammatory environment, respond with more pronounced enhancement of the afferent inhibitory synaptic drive than mature neurons that were born already before the onset of inflammation. The new neurons show normal location, dendritic morphology, and intrinsic membrane properties, and exhibit increased excitatory synaptic drive of the same magnitude as the mature neurons.
Chronic inflammation, without neuronal damage and abnormal EEG activity, was induced by a single intrahippocampal injection of LPS. This treatment caused increased numbers of microglia with phagocytotic activity (Iba1+/ED1+) in the SGZ/GCL, both at the time when the new cells were born (1 week after LPS injection) and when they had reached the mature state (at the time of recording and morphological analysis, 7 weeks later). Consistent with our findings, Herber et al. (2006) reported that microglia remain activated 4 weeks after a single intrahippocampal injection of LPS in mice. Previous studies have shown that LPS-induced inflammation can alter synaptic transmission between mature neurons as well as influence behaviors regulated in the hippocampal formation. Administration of LPS by systemic injection causes impairment of both hippocampal-dependent learning (Shaw et al., 2001; Hennigan et al., 2007) and long-term potentiation (Vereker et al., 2000; Hennigan et al., 2007). Furthermore, injection of LPS in the hippocampal CA1 area of rats gives rise to learning and memory deficits and decreased glutamatergic neurotransmission but no cell death (Tanaka et al., 2006). LPS-evoked inflammation causes increased levels of cyclooxygenase-2, which, via oxidative metabolism of endocannabinoids, induces higher frequency of mEPSCs in hippocampal cultures (Sang et al., 2005, 2007). We find here that the frequency of sEPSCs was increased to the same extent in mature and new cells in the inflammatory environment compared with the control environment. The lack of inflammation-induced changes of spine density and morphology in the new cells argue against the possibility that such alterations would underlie the increased excitatory drive at their afferent excitatory synapses. Moreover, we found no changes of PPF at the excitatory synapses in the new and mature neurons, indicating unaltered overall glutamate release probability (Zucker and Stockbridge, 1983; Debanne et al., 1996; Murthy et al., 1997). Therefore, the increased excitatory drive on new and mature granule cells is most likely attributable to increased network activity in hippocampal neural circuitries caused by the inflammation.
Both the new and the mature granule cells exhibited increased inhibitory synaptic drive in response to LPS-induced inflammation. Exposure to LPS causes increased expression of interleukin-1β in microglia (Lund et al., 2006) and elevated levels of this cytokine in hippocampal organotypic cultures (Hellstrom et al., 2005). Similarly, intrahippocampal LPS administration gives rise to a rapid, severalfold increase of interleukin-1β production from microglia (Tanaka et al., 2006). In line with our observations, LPS-treated organotypic cultures showed increased inhibitory transmission in CA1 pyramidal neurons (larger amplitude of monosynaptic IPSPs during electrical stimulation of GABAergic afferent fibers), which was prevented by coincubation with an IL-1 receptor antagonist (Hellstrom et al., 2005). Thus, the increased inhibitory drive on the new and mature cells in our experiment could have been induced by microglial release of proinflammatory cytokines such as interleukin-1β.
The enhancement of the inhibitory synaptic drive (i.e., the frequency and amplitude of sIPSCs) in the chronic inflammatory environment was more pronounced on the new compared with the mature cells. Inhibitory synaptic inputs are mostly located close to the soma and can be synchronized (Cobb et al., 1995; Miles et al., 1996). Therefore, even small changes in the mean rate of inhibitory synaptic input are likely to significantly modulate the gain of action potential generation by the cell (Mitchell and Silver, 2003). We obtained no evidence in the PPD paradigm for any changes in short-term synaptic plasticity at inhibitory synapses on the new cells. Because no differences in frequency or amplitude of IPSCs in new compared with mature granule cells remained after TTX administration, the higher inhibitory synaptic drive on the new cells in the inflammatory environment is most likely caused by action potential-dependent, presynaptic changes. Hypothetically, the new cells might interact with the afferent inhibitory neurons through putative retrograde messengers, and the presynaptic alterations could occur according to one of several scenarios. First, the new cells in the inflammatory environment may receive inhibitory input from a population of interneurons with higher frequency of action potential generation compared with the mature cells. Second, the inhibitory synapses could be more numerous or have higher overall release probability of GABA on the new cells born in a chronic inflammatory environment. Arguing against this possibility, the differences in sIPSC frequency between new and mature cells were no longer observed after TTX application. Third, the increase in sIPSC frequency could be attributed to enhanced expression or activation of voltage-dependent calcium channels on the inhibitory afferents at the new cells in LPS-treated animals. Increased release of GABA would then primarily be attributable to more activation of voltage-dependent calcium channels and higher calcium influx triggered by action potential-induced depolarization at the presynapses (Mintz et al., 1995). This interpretation is also consistent with the elevated amplitude of sIPSCs but not of mIPSCs in the new cells in LPS-treated animals.
The increased amplitude of sIPSCs could, alternatively, reflect postsynaptic alterations, which is supported by our finding of larger gephyrin clusters on the dendrites of the new cells, which had developed in the chronic inflammatory environment. Gephyrin is an important postsynaptic scaffolding protein that is required for clustering of glycine and GABAA receptors at inhibitory synapses (Fritschy et al., 2008), and is essential for the stability of GABAergic synapses. Disruption of gephyrin clusters causes reduced presynaptic GABAergic innervation and decreased amplitude and frequency of sIPSCs in cultured hippocampal neurons (Yu et al., 2007). The size of gephyrin clusters in hippocampal slice cultures was found to be inversely regulated by neuronal activity (Marty et al., 2004). It is conceivable that the larger gephyrin clusters on the new cells born in the chronic inflammatory environment indicate a more efficacious inhibitory input. In support, Lim et al. (1999) reported a positive correlation between synaptic strength and the size of postsynaptic gephyrin clusters. Inconsistent with a major role of the postsynaptic changes, the difference in amplitude of sIPSCs between new cells in LPS- and vehicle-treated animals did not remain after blockade of action potentials. It should be pointed out, however, that given the lower overall amplitude of mIPSCs compared with sIPSCs, and that the gephyrin clusters were increased in the axodendritic inhibitory synapses on the distal dendrites (i.e., distal to the patch pipette in the cell soma), subtle changes in mIPSC amplitudes could have escaped detection.
Inflammation did not alter the migration or polarity of the new cells, the timing when apical dendrites had reached the ML and the axons had reached CA3, the arborizations of the dendrites, or the development, density, or shape of the spines. Thus, the morphological maturation of the new cells, which has previously been described in adult mouse hippocampus using the same RV-GFP vector (Zhao et al., 2006), was strictly controlled also in the chronic inflammatory environment. Spines on dendrites of new granule cells are highly motile, transforming from one subtype/form to another in a very dynamic manner, as demonstrated by time-lapse imaging (Zhao et al., 2006). We analyzed spine density and morphology only at one time point (∼49 d after RV-GFP injection) and found no differences between LPS-treated and control animals. In agreement, running-induced increased hippocampal activity in mice caused no alterations of spine density on new cells at any time point (Zhao et al., 2006). Mushroom spine density was increased in runners at 56 d but not at earlier and later time points. This transient change of spine morphology occurred in the mice 1 week after our analysis in the rats, which may explain why it was not detected in the present study.
Our findings provide evidence of a high degree of plasticity of the new neurons at their afferent synaptic inputs when exposed to the pathological environment. Recordings were made 6–8 weeks after the new cells had been born (i.e., after the critical period of enhanced synaptic plasticity between 1 and 1.5 months of cell age) (Ge et al., 2007). We find that the new neurons had responded to the increased excitatory synaptic drive, caused by the chronic inflammation, with an additional upregulation of inhibitory activity at their afferent synapses and the development of larger gephyrin clusters. The previously unknown regulation of gephyrin in adult-born neurons reveals another mechanism that can contribute to the plasticity in their functional synaptic connectivity. It remains to be explored whether the observed plastic changes in inhibitory inputs on the new neurons, and to a lesser extent on the mature neurons, are directly induced by the inflammation, or if they are indirect and compensatory, aiming to preserve cellular and/or network homeostasis in a pathological condition with increased excitatory drive on both new and mature granule cells.
The synaptic properties of the new cells in the LPS-induced, chronic inflammatory environment differed from those of cells that had developed after SE (Jakubs et al., 2006). Compared with cells born into a physiological environment, the new cells generated after SE exhibited decreased excitatory drive because of reduced release probability of glutamate, and increased inhibitory drive, most likely because of decreased sensitivity/expression of presynaptic GABAB receptors and increased postsynaptic GABAA receptor expression. It is conceivable that these changes in the afferent synapses on the cells born after SE were compensatory and occurred in response to network hyperexcitability. In the LPS-induced chronic inflammatory environment, both the new and the mature cells exhibited increased excitatory and inhibitory afferent drive, primarily because of changes at the network level and not at their afferent synapses. In addition, the new cells that had developed in a chronic inflammatory environment showed synapse-specific enhancement of the afferent inhibitory drive, but this change was not attributable to alterations in presynaptic GABAB receptors as after SE. Together, our findings provide evidence that the pathological environments induced by SE and LPS lead to different and specific compensatory alterations of the properties of the afferent synapses on the new neurons.
In the present study, inflammation was induced by LPS already before the formation of the new neurons, and there was chronic inflammation in the environment throughout their development/maturation and still at the time of recordings and morphological analysis. Our findings raise several important issues that need to be explored in future experiments: first, to determine when during the development of the new neurons they are particularly sensitive to inflammation and respond with alterations of their functional synaptic connectivity. Ideally, inflammation could be induced by LPS at different time points after the formation of the new neurons. However, a major confounding factor, which would make the findings difficult to compare with the present data, is that the acute effects of LPS, which are unlikely to play a role when cells are born at 1 week after LPS injection, most probably will affect the properties of the new neurons if LPS is delivered when the neurons have already been formed. For example, LPS injection in vivo induces acute, transient increases (returning to baseline within 24 h) of several inflammatory markers such as IL-6, MCP-1, and TNF-α (tumor necrosis factor-α) (Lund et al., 2006). Second, to analyze whether the alterations of excitatory and, in particular, inhibitory synaptic drive onto the new neurons are permanent. This will require assessment of their properties both if the chronic inflammation has declined and if it is maintained. Our hypothesis postulates that the increased excitatory drive onto both new and mature neurons is induced by inflammation, which leads to the enhanced increase of inhibitory drive on new neurons. Because there was a substantial reduction of microglia from 1 to 8 weeks, continued inflammation at later time points would probably require additional LPS injections, which could result in cyclic exacerbations of the inflammatory response. Third is to investigate how the characteristics of the inflammatory environment will affect the development of the functional synaptic connectivity. Microglia can have both detrimental and beneficial effects on adult neurogenesis depending on their morphological and molecular phenotype and whether they are acutely or chronically activated [for references, see, for example, Ekdahl et al. (2008)]. Moreover, autoimmune, CNS-specific T-lymphocytes, by interacting with resident microglia, can promote neurogenesis in the SGZ and possibly also influence neuronal differentiation (Ziv et al., 2006). Here, we only assessed the consequences of one type of chronic inflammatory environment. Hypothetically, if the features of the different cellular and molecular players in the inflammation are altered, this could lead to a different development of the functional synaptic connectivity of the new neurons.
The continuous generation of new dentate granule cells probably plays a role in learning and memory, and deterioration of hippocampal neurogenesis may be linked to the cognitive decline in aging and Alzheimer's disease. Brain inflammation and microglia activation are involved in many disorders associated with cognitive impairment. Our data provide the first experimental evidence that inflammation affects the functional integration of the new adult-born neurons in existing neural circuitries. Thus, clarifying the extent of involvement of adult neurogenesis in pathological conditions associated with inflammation (e.g., in mouse Alzheimer models) mandates functional analysis at the cellular level to complement quantitative data on progenitor proliferation and neuron numbers. Whether the changes of functional synaptic connectivity of new neurons, which have developed in an inflammatory environment, act to mitigate or worsen brain dysfunction remains to be investigated.
Footnotes
This work was supported by the Swedish Research Council, Juvenile Diabetes Research Foundation, European Union Project LSHB-2006-037526 (StemStroke), and The Söderberg, Crafoord, Segerfalk, and Kock Foundations. The Lund Stem Cell Center is supported by a Center of Excellence Grant in Life Sciences from the Swedish Foundation for Strategic Research. We thank Dr. Fred H. Gage and Dr. H. van Praag for RV-GFP, and Bengt Mattsson for technical assistance.
References
- Arvidsson A, Kokaia Z, Lindvall O. N-Methyl-d-aspartate receptor-mediated increase of neurogenesis in adult rat dentate gyrus following stroke. Eur J Neurosci. 2001;14:10–18. doi: 10.1046/j.0953-816x.2001.01611.x. [DOI] [PubMed] [Google Scholar]
- Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat Med. 2002;8:963–970. doi: 10.1038/nm747. [DOI] [PubMed] [Google Scholar]
- Bengzon J, Kokaia Z, Elmér E, Nanobashvili A, Kokaia M, Lindvall O. Apoptosis and proliferation of dentate gyrus neurons after single and intermittent limbic seizures. Proc Natl Acad Sci U S A. 1997;94:10432–10437. doi: 10.1073/pnas.94.19.10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonde S, Ekdahl CT, Lindvall O. Long-term neuronal replacement in adult rat hippocampus after status epilepticus despite chronic inflammation. Eur J Neurosci. 2006;23:965–974. doi: 10.1111/j.1460-9568.2006.04635.x. [DOI] [PubMed] [Google Scholar]
- Cobb SR, Buhl EH, Halasy K, Paulsen O, Somogyi P. Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature. 1995;378:75–78. doi: 10.1038/378075a0. [DOI] [PubMed] [Google Scholar]
- Damoiseaux JG, Döpp EA, Calame W, Chao D, MacPherson GG, Dijkstra CD. Rat macrophage lysosomal membrane antigen recognized by monoclonal antibody ED1. Immunology. 1994;83:140–147. [PMC free article] [PubMed] [Google Scholar]
- Danton GH, Dietrich WD. Inflammatory mechanisms after ischemia and stroke. J Neuropathol Exp Neurol. 2003;62:127–136. doi: 10.1093/jnen/62.2.127. [DOI] [PubMed] [Google Scholar]
- Davies CH, Davies SN, Collingridge GL. Paired-pulse depression of monosynaptic GABA-mediated inhibitory postsynaptic responses in rat hippocampus. J Physiol. 1990;424:513–531. doi: 10.1113/jphysiol.1990.sp018080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debanne D, Guerineau NC, Gahwiler BH, Thompson SM. Paired-pulse facilitation and depression at unitary synapses in rat hippocampus: quantal fluctuation affects subsequent release. J Physiol. 1996;491:163–176. doi: 10.1113/jphysiol.1996.sp021204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekdahl CT, Claasen JH, Bonde S, Kokaia Z, Lindvall O. Inflammation is detrimental for neurogenesis in adult brain. Proc Natl Acad Sci U S A. 2003;100:13632–13637. doi: 10.1073/pnas.2234031100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekdahl CT, Kokaia Z, Lindvall O. Brain inflammation and adult neurogenesis: the dual role of microglia. Neuroscience. 2008 doi: 10.1016/j.neuroscience.2008.06.052. Advance online publication. Retrieved November 3, 2008. [DOI] [PubMed] [Google Scholar]
- Fritschy JM, Harvey RJ, Schwarz G. Gephyrin: where do we stand, where do we go? Trends Neurosci. 2008;31:257–264. doi: 10.1016/j.tins.2008.02.006. [DOI] [PubMed] [Google Scholar]
- Ge S, Yang CH, Hsu KS, Ming GL, Song H. A critical period for enhanced synaptic plasticity in newly generated neurons of the adult brain. Neuron. 2007;54:559–566. doi: 10.1016/j.neuron.2007.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloveli T, Behr J, Dugladze T, Kokaia Z, Kokaia M, Heinemann U. Kindling alters entorhinal cortex-hippocampal interaction by increased efficacy of presynaptic GABA(B) autoreceptors in layer III of the entorhinal cortex. Neurobiol Dis. 2003;13:203–212. doi: 10.1016/s0969-9961(03)00039-1. [DOI] [PubMed] [Google Scholar]
- Gorter JA, van Vliet EA, Aronica E, Breit T, Rauwerda H, Lopes da Silva FH, Wadman WJ. Potential new antiepileptogenic targets indicated by microarray analysis in a rat model for temporal lobe epilepsy. J Neurosci. 2006;26:11083–11110. doi: 10.1523/JNEUROSCI.2766-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellstrom IC, Danik M, Luheshi GN, Williams S. Chronic LPS exposure produces changes in intrinsic membrane properties and a sustained IL-beta-dependent increase in GABAergic inhibition in hippocampal CA1 pyramidal neurons. Hippocampus. 2005;15:656–664. doi: 10.1002/hipo.20086. [DOI] [PubMed] [Google Scholar]
- Henneberger C, Kirischuk S, Grantyn R. Brain-derived neurotrophic factor modulates GABAergic synaptic transmission by enhancing presynaptic glutamic acid decarboxylase 65 levels, promoting asynchronous release and reducing the number of activated postsynaptic receptors. Neuroscience. 2005;135:749–763. doi: 10.1016/j.neuroscience.2005.06.044. [DOI] [PubMed] [Google Scholar]
- Hennigan A, Trotter C, Kelly AM. Lipopolysaccharide impairs long-term potentiation and recognition memory and increases p75NTR expression in the rat dentate gyrus. Brain Res. 2007;1130:158–166. doi: 10.1016/j.brainres.2006.10.066. [DOI] [PubMed] [Google Scholar]
- Herber DL, Maloney JL, Roth LM, Freeman MJ, Morgan D, Gordon MN. Diverse microglial responses after intrahippocampal administration of lipopolysaccharide. Glia. 2006;53:382–391. doi: 10.1002/glia.20272. [DOI] [PubMed] [Google Scholar]
- Hoehn BD, Palmer TD, Steinberg GK. Neurogenesis in rats after focal cerebral ischemia is enhanced by indomethacin. Stroke. 2005;36:2718–2724. doi: 10.1161/01.STR.0000190020.30282.cc. [DOI] [PubMed] [Google Scholar]
- Imai Y, Kohsaka S. Intracellular signaling in M-CSF-induced microglia activation: role of Iba1. Glia. 2002;40:164–174. doi: 10.1002/glia.10149. [DOI] [PubMed] [Google Scholar]
- Iosif RE, Ekdahl CT, Ahlenius H, Pronk CJ, Bonde S, Kokaia Z, Jacobsen SE, Lindvall O. Tumor necrosis factor receptor 1 is a negative regulator of progenitor proliferation in adult hippocampal neurogenesis. J Neurosci. 2006;26:9703–9712. doi: 10.1523/JNEUROSCI.2723-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iosif RE, Ahlenius H, Ekdahl CT, Darsalia V, Thored P, Jovinge S, Kokaia Z, Lindvall O. Suppression of stroke-induced progenitor proliferation in adult subventricular zone by tumor necrosis factor receptor 1. J Cereb Blood Flow Metab. 2008;28:1574–1587. doi: 10.1038/jcbfm.2008.47. [DOI] [PubMed] [Google Scholar]
- Jakubs K, Nanobashvili A, Bonde S, Ekdahl CT, Kokaia Z, Kokaia M, Lindvall O. Environment matters: synaptic properties of neurons born in the epileptic adult brain develop to reduce excitability. Neuron. 2006;52:1047–1059. doi: 10.1016/j.neuron.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Laplagne DA, Espósito MS, Piatti VC, Morgenstern NA, Zhao C, van Praag H, Gage FH, Schinder AF. Functional convergence of neurons generated in the developing and adult hippocampus. PLoS Biol. 2006;4:e409. doi: 10.1371/journal.pbio.0040409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplagne DA, Kamienkowski JE, Espósito MS, Piatti VC, Zhao C, Gage FH, Schinder AF. Similar GABAergic inputs in dentate granule cells born during embryonic and adult neurogenesis. Eur J Neurosci. 2007;25:2973–2981. doi: 10.1111/j.1460-9568.2007.05549.x. [DOI] [PubMed] [Google Scholar]
- Lim R, Alvarez FJ, Walmsley B. Quantal size is correlated with receptor cluster area at glycinergic synapses in the rat brainstem. J Physiol. 1999;516:505–512. doi: 10.1111/j.1469-7793.1999.0505v.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund S, Christensen KV, Hedtjärn M, Mortensen AL, Hagberg H, Falsig J, Hasseldam H, Schrattenholz A, Pörzgen P, Leist M. The dynamics of the LPS triggered inflammatory response of murine microglia under different culture and in vivo conditions. J Neuroimmunol. 2006;180:71–87. doi: 10.1016/j.jneuroim.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Marty S, Wehrlé R, Fritschy JM, Sotelo C. Quantitative effects produced by modifications of neuronal activity on the size of GABAA receptor clusters in hippocampal slice cultures. Eur J Neurosci. 2004;20:427–440. doi: 10.1111/j.1460-9568.2004.03491.x. [DOI] [PubMed] [Google Scholar]
- Miles R, Tóth K, Gulyás AI, Hájos N, Freund TF. Differences between somatic and dendritic inhibition in the hippocampus. Neuron. 1996;16:815–823. doi: 10.1016/s0896-6273(00)80101-4. [DOI] [PubMed] [Google Scholar]
- Mintz IM, Sabatini BL, Regehr WG. Calcium control of transmitter release at a cerebellar synapse. Neuron. 1995;15:675–688. doi: 10.1016/0896-6273(95)90155-8. [DOI] [PubMed] [Google Scholar]
- Mitchell SJ, Silver RA. Shunting inhibition modulates neuronal gain during synaptic excitation. Neuron. 2003;38:433–445. doi: 10.1016/s0896-6273(03)00200-9. [DOI] [PubMed] [Google Scholar]
- Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302:1760–1765. doi: 10.1126/science.1088417. [DOI] [PubMed] [Google Scholar]
- Murthy VN, Sejnowski TJ, Stevens CF. Heterogeneous release properties of visualized individual hippocampal synapses. Neuron. 1997;18:599–612. doi: 10.1016/s0896-6273(00)80301-3. [DOI] [PubMed] [Google Scholar]
- Otis TS, De Koninck Y, Mody I. Characterization of synaptically elicited GABAB responses using patch-clamp recordings in rat hippocampal slices. J Physiol. 1993;463:391–407. doi: 10.1113/jphysiol.1993.sp019600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH. Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci. 1997;17:3727–3738. doi: 10.1523/JNEUROSCI.17-10-03727.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JM, Vexler ZS, Gong C, Derugin N, Ferriero DM. Rat forebrain neurogenesis and striatal neuron replacement after focal stroke. Ann Neurol. 2002;52:802–813. doi: 10.1002/ana.10393. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. San Diego: Academic; 1997. [DOI] [PubMed] [Google Scholar]
- Pickering M, Cumiskey D, O'Connor JJ. Actions of TNF-alpha on glutamatergic synaptic transmission in the central nervous system. Exp Physiol. 2005;90:663–670. doi: 10.1113/expphysiol.2005.030734. [DOI] [PubMed] [Google Scholar]
- Sang N, Zhang J, Marcheselli V, Bazan NG, Chen C. Postsynaptically synthesized prostaglandin E2 (PGE2) modulates hippocampal synaptic transmission via a presynaptic PGE2 EP2 receptor. J Neurosci. 2005;25:9858–9870. doi: 10.1523/JNEUROSCI.2392-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang N, Zhang J, Chen C. COX-2 oxidative metabolite of endocannabinoid 2-AG enhances excitatory glutamatergic synaptic transmission and induces neurotoxicity. J Neurochem. 2007;102:1966–1977. doi: 10.1111/j.1471-4159.2007.04668.x. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Albertson C, Slikker W., Jr Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 1997;751:37–46. doi: 10.1016/s0006-8993(96)01387-x. [DOI] [PubMed] [Google Scholar]
- Schneider Gasser EM, Straub CJ, Panzanelli P, Weinmann O, Sassoè-Pognetto M, Fritschy JM. Immunofluorescence in brain sections: simultaneous detection of presynaptic and postsynaptic proteins in identified neurons. Nat Protoc. 2006;1:1887–1897. doi: 10.1038/nprot.2006.265. [DOI] [PubMed] [Google Scholar]
- Schratt GM, Tuebing F, Nigh EA, Kane CG, Sabatini ME, Kiebler M, Greenberg ME. A brain-specific microRNA regulates dendritic spine development. Nature. 2006;439:283–289. doi: 10.1038/nature04367. [DOI] [PubMed] [Google Scholar]
- Shapiro LA, Upadhyaya P, Ribak CE. Spatiotemporal profile of dendritic outgrowth from newly born granule cells in the adult rat dentate gyrus. Brain Res. 2007;1149:30–37. doi: 10.1016/j.brainres.2006.07.032. [DOI] [PubMed] [Google Scholar]
- Shaw KN, Commins S, O'Mara SM. Lipopolysaccharide causes deficits in spatial learning in the watermaze but not in BDNF expression in the rat dentate gyrus. Behav Brain Res. 2001;124:47–54. doi: 10.1016/s0166-4328(01)00232-7. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Ide M, Shibutani T, Ohtaki H, Numazawa S, Shioda S, Yoshida T. Lipopolysaccharide-induced microglial activation induces learning and memory deficits without neuronal cell death in rats. J Neurosci Res. 2006;83:557–566. doi: 10.1002/jnr.20752. [DOI] [PubMed] [Google Scholar]
- van Praag H, Schinder AF, Christie BR, Toni N, Palmer TD, Gage FH. Functional neurogenesis in the adult hippocampus. Nature. 2002;415:1030–1034. doi: 10.1038/4151030a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vereker E, Campbell V, Roche E, McEntee E, Lynch MA. Lipopolysaccharide inhibits long term potentiation in the rat dentate gyrus by activating caspase-1. J Biol Chem. 2000;275:26252–26258. doi: 10.1074/jbc.M002226200. [DOI] [PubMed] [Google Scholar]
- von Bohlen und Halbach O, Krause S, Medina D, Sciarretta C, Minichiello L, Unsicker K. Regional- and age-dependent reduction in trkB receptor expression in the hippocampus is associated with altered spine morphologies. Biol Psychiatry. 2006;59:793–800. doi: 10.1016/j.biopsych.2005.08.025. [DOI] [PubMed] [Google Scholar]
- Wu C, Leung LS. Partial hippocampal kindling decreases efficacy of presynaptic GABAB autoreceptors in CA1. J Neurosci. 1997;17:9261–9269. doi: 10.1523/JNEUROSCI.17-23-09261.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Jiang M, Miralles CP, Li RW, Chen G, de Blas AL. Gephyrin clustering is required for the stability of GABAergic synapses. Mol Cell Neurosci. 2007;36:484–500. doi: 10.1016/j.mcn.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Teng EM, Summers RG, Jr, Ming GL, Gage FH. Distinct morphological stages of dentate granule neuron maturation in the adult mouse hippocampus. J Neurosci. 2006;26:3–11. doi: 10.1523/JNEUROSCI.3648-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Deng W, Gage FH. Mechanisms and functional implications of adult neurogenesis. Cell. 2008;132:645–660. doi: 10.1016/j.cell.2008.01.033. [DOI] [PubMed] [Google Scholar]
- Ziv Y, Ron N, Butovsky O, Landa G, Sudai E, Greenberg N, Cohen H, Kipnis J, Schwartz M. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat Neurosci. 2006;9:268–275. doi: 10.1038/nn1629. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Stockbridge N. Presynaptic calcium diffusion and the time courses of transmitter release and synaptic facilitation at the squid giant synapse. J Neurosci. 1983;3:1263–1269. doi: 10.1523/JNEUROSCI.03-06-01263.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]