

Abstract
The molecular alterations that induce tau pathology in Alzheimer disease (AD) are not known, particularly whether this is an amyloid-β (Aβ)-dependent or -independent event. We addressed this issue in the 3xTg-AD mice using both genetic and immunological approaches and show that a selective decrease in Aβ42 markedly delays the progression of tau pathology. The mechanism underlying this effect involves alterations in the levels of C terminus of heat shock protein70-interacting protein (CHIP) as we show that Aβ accumulation decreases CHIP expression and increases tau levels. We show that the Aβ-induced effects on tau were rescued by restoring CHIP levels. Our findings have profound clinical implications as they indicate that preventing Aβ accumulation will significantly alter AD progression. These data highlight the critical role CHIP plays as a link between Aβ and tau and identify CHIP as a new potential target not only for AD but for other neurodegenerative disorders characterized by tau accumulation.
Keywords: Aβ, plaques, transgenic, aging, Alzheimer's disease, tangles
Introduction
Alzheimer disease (AD) is the most common age-dependent neurodegenerative disorder. Postmortem diagnosis of AD requires the presence of two major lesions: amyloid plaques, mainly comprised of a small peptide called amyloid-β (Aβ) (Glenner and Wong, 1984; Masters et al., 1985) and neurofibrillary tangles, mainly comprised of hyperphosphorylated tau (Grundke-Iqbal et al., 1986; Ihara et al., 1986; Kosik et al., 1986; Goedert et al., 1988). Aβ is liberated via the proteolytic processing of the amyloid precursor protein (APP), predominantly generating two major isoforms, Aβ40 and Aβ42, that are produced in a ratio of ∼9:1. The Aβ42 species is more hydrophobic and more prone to form plaques than Aβ40 (Jarrett et al., 1993).
Although most forms of AD are sporadic and of unknown etiology, a small proportion of cases are linked to mutations in three different genes: APP, presenilin 1, and presenilin 2 (Chartier-Harlin et al., 1991; Goate et al., 1991; Sherrington et al., 1995). Mutations in these three genes affect Aβ metabolism or its properties, leading to an increase in total Aβ levels, alteration of the Aβ42/40 ratio, or its propensity to aggregate (Hardy, 1997; Selkoe, 2001). This strong genetic evidence led to the formulation of the amyloid cascade hypothesis, which stipulates that Aβ accumulation is the initiating step in a cascade of pathological changes that lead to AD (Hardy and Selkoe, 2002). A key implication of the amyloid cascade hypothesis is that Aβ alters cellular metabolism and triggers other downstream pathological features of AD such as tau hyperphosphorylation/tangle formation.
Experimental support for the amyloid cascade hypothesis is also derived from studies involving AD transgenic models (Götz et al., 2001; Lewis et al., 2001; Oddo et al., 2003b; Bolmont et al., 2007). For example, we developed a transgenic model of AD (3xTg-AD) that harbors three mutant transgenes (PS1, APPswe, TauP301L) that develops both amyloid plaques and neurofibrillary tangles. We previously showed that Aβ accumulation plays a major role in the onset of cognitive decline and tau pathology in the 3xTg-AD mice (Oddo et al., 2003b, 2004, 2006a,b; Billings et al., 2005). A major unresolved question is whether all forms of Aβ can induce these changes or whether a specific form of Aβ, namely Aβ40 or Aβ42, may play a pivotal role. In addition, it remains to be determined whether preventing Aβ accumulation will suffice to stop the development of tau pathology.
Genetic and biochemical evidence suggest a link between the ubiquitin proteasome system (UPS) and AD (Keller et al., 2000; Bertram et al., 2005). This is particularly intriguing, because tau, which is normally a member of the natively unfolded protein family, seems to be degraded by the proteasome. In particular, it has been shown that the C terminus of heat shock protein70-interacting protein (CHIP) plays a crucial role in the removal of tau species that are abnormally hyperphosphorylated (Petrucelli et al., 2004; Shimura et al., 2004; Dickey et al., 2006, 2007). On the other end, there is strong evidence from different laboratories suggesting that Aβ adversely alters UPS function (Gregori et al., 1995; Oddo et al., 2004; Oh et al., 2005; Almeida et al., 2006; Tseng et al., 2008). The molecular mechanisms underlying such effects, however, remain to be determined.
Using a genetic and immunological approach, we show that preventing Aβ accumulation markedly reduces the onset of tau pathology, even though expression of the human tau transgene is unchanged. This finding provides strong support for a pivotal role for Aβ in facilitating the accumulation of tau pathology in the mammalian brain. We further show that Aβ42, and not Aβ40, plays a major role in the induction of tau pathology. One of the molecular mechanisms underlying the effects of Aβ on tau is mediated by CHIP, as we show that Aβ accumulation reduced CHIP levels and, more importantly, we show that the Aβ-induced tau pathology can be rescued by restoring CHIP levels. These data suggest CHIP as a valid therapeutic target and increasing its activity may reduce the Aβ-induced tau pathology.
Materials and Methods
Mice and behavioral tests.
The derivation and characterization of the 3xTg-AD and the PS1/tau mice has been described previously (Oddo et al., 2003b). Briefly, two independent transgenes encoding human APPSwe and the human tauP301L (both under control of the mouse Thy1.2 regulatory element) were comicroinjected into single-cell embryos harvested from homozygous mutant PS1M146V knock-in (PS1-KI) mice. All mice were given ad libitum access to food and water. Morris water maze tests were conducted in a circular tank of 1.5 m in diameter located in a room with several cues on the walls. The water filling the tank was maintained at 25°C. The platform (14 cm in diameter) location was kept constant for each mouse during training, and it was 1.5 cm beneath the surface of the water. During training the mice received 4 trials/d alternated among 4 pseudorandom starting points. If a mouse failed to find the platform within 60 s, it was guided to the platform by the researcher and kept there for 10 s. The intertrial interval was 25 s, during which each mouse was returned in its home cage. This was repeated each day for as many days it took for the mice to reach criterion (arbitrarily defined as <20 s mean escape latency). Probe trials were conducted 1.5 and 24 h after the last training trial. During the probe trials, the platform was removed and mice were free to swim in the tank for 60 s. All the probe trials were recorded by a video camera mounted on the ceiling. The number of platform location crosses and the time spent in the target and the opposite quadrant were recorded.
Surgical procedures.
Male and female 3xTg-AD mice weighing 25–49 g at the time of surgery were group housed and kept on a 12 h light:12 h dark schedule. Surgeries were performed during the light cycle. Twelve- and eighteen-month-old 3xTg-AD mice were anesthetized with avertin (1.3% tribromoethanol, 0.8% amyl alcohol, given 0.6 ml/25 g body weight) and placed in a stereotactic apparatus (MyNeuroLab) with a mouse adaptor. Each mouse received two injections, the CHIP expressing lentivirus (3 μl of a CHIP-expressing lentivirus, 1.2 × 106 transducing particles/ml) was injected into the left hippocampus through a 33-gauge injector attached to a 5 μl Hamilton syringe. The coordinates, with respect to bregma, were −2.7 mm posterior, +2.5 mm lateral, and −3.0 ventral to the skull. A control virus was injected into the right hippocampus, the coordinates, with respect to bregma, were −2.7 mm posterior, −2.5 mm lateral, and −3.0 ventral to the skull. Both viruses were purchased from BioGenova. Injections occurred over the span of 5 min, after which the cannula was left in place for an additional 5 min to allow for diffusion. Animals were kept on a warming pad until they had fully recovered from anesthesia, and were kept in individual cages until they were killed for tissue processing to prevent damage to the scalp sutures. All animal procedures were in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals, and all appropriate measures were taken to minimize pain and discomfort in experimental animals.
Protein extraction, Western blot, and ELISA.
Mice were killed by CO2 asphyxiation, and their brains were extracted and cut in half sagittally. For immunohistochemical analysis, one half was dropped–fixed in 4% paraformaldehyde in PBS for 48 h and then transferred in 0.02% sodium azide in PBS until slicing. The other half was frozen in dry ice for biochemical analysis. Frozen brains were homogenized in a solution of tissue protein extraction reagent (T-PER; Pierce) containing 0.7 mg/ml Pepstatin A supplemented with a complete Mini protease inhibitor tablet (Roche) and phosphatase inhibitors (Invitrogen). The homogenized mixes were briefly sonicated to sheer the DNA and centrifuged at 4°C for 1 h at 100,000 × g. The supernatant was stored as the soluble fraction. The pellet was rehomogenized in 70% formic acid (FA) and centrifuged as above. The supernatant was stored as the insoluble fraction. Western blot analyses were conducted as described by Oddo et al. (2007). Briefly, proteins from the soluble fraction were resolved by SDS/PAGE (10% Bis-Tris from Invitrogen) under reducing conditions and transferred to a nitrocellulose membrane. The membrane was incubated in a 5% solution of nonfat milk for 1 h at 20°C. After overnight incubation at 4°C with primary antibody, the blots were washed in Tween 20-TBS (T-TBS) (0.02% Tween 20, 100 mm Tris pH 7.5; 150 nm NaCl) for 20 min and incubated at 20°C with secondary antibody. The blots were washed in T-TBS for 20 min and incubated for 5 min with Super Signal (Pierce), washed again, and exposed. Aβ40 and Aβ42 levels were measured from the soluble and insoluble fractions using a sandwich ELISA protocol as described by Oddo et al. (2005).
Immunohistochemistry.
For immunohistochemical analysis, 50-μm-thick sections were obtained using a vibratome slicing system, and sections were stored at 4°C in 0.02% sodium azide in PBS. To quench the endogenous peroxidase activity, free-floating sections were incubated for 30 min in H2O2. For the Aβ staining, sections were subsequently incubated in 90% formic acid for 7 min to expose the epitope. The appropriate primary antibody was applied, and sections were incubated overnight at 4°C. After removing the primary antibody in excess, sections were incubated in the appropriate secondary antibody for 1 h at 20°C. After a final wash of 20 min, sections were developed with diaminobenzidine (DAB) substrate using the avidin-biotin horseradish peroxidase system (Vector Laboratories). The primary antibodies were applied at the following dilutions: 1:1000 for 6E10, 8C11, and 16B6; 1:3000 for 1560; 1:1000 for HT7; 1:200 for AT8 and anti-Aβ42; and 1:500 for AT100.
Immunization.
For active immunization, Aβ42 peptide was synthesized at the University of California Core Facility, and fibrillar Aβ42 (fAβ42) was prepared as previously described (Cribbs et al., 2003) and delivered subcutaneously. Blood was collected before the first immunization and 10 d after each boost from the retro-orbital sinus into the EDTA-coated tubes. Tubes were centrifuged for 10 min at 4°C, and the sera were collected as a supernatant and stored at −80°C. Intracerebral antibody injections were performed as previously described (Oddo et al., 2004).
Cell culture experiments.
Cell culture experiments were conducted as described previously (Kitazawa et al., 2003). Briefly, human embryonic kidney (HEK293) cells were maintained in DMEM supplemented with 10% fetal bovine serum (FBS), 50 U of penicillin, and 50 mg/ml streptomycin. HEK cells stably overexpressing human wild-type APP695 (HEK263) were maintained in DMEM supplemented with 10% fetal bovine serum (FBS), 0.2 mg/ml G418, 50 U of penicillin, and 50 mg/ml streptomycin. All cells were incubated at 37°C in humidified atmosphere with 5% CO2.
Statistical analysis.
The data were subsequently analyzed by ANOVA or t test comparison, using Graphpad Prism software.
Results
We previously reported the generation of a transgenic model of AD (3xTg-AD) that progressively manifests accumulation of plaques and tangles, which are associated with age-dependent cognitive decline. To generate the 3xTg-AD mice, two independent transgenes encoding human APPSwe and human tauP301L were injected into single-cell embryos harvested from homozygous mutant PS1M146V knock-in (PS1-KI) mice (Oddo et al., 2003b). The M146V mutation is known to significantly increase Aβ42 levels (Guo et al., 1999). For these studies, we were interested in determining the impact of decreasing Aβ42 levels on the onset and progression of the tau pathology and the subsequent cognitive decline. We first used a genetic approach to replace the mutant PS1 allele with its wild-type counterpart. Because of the approach used to generate the 3xTg-AD mice, the APP and tau transgenes cointegrated at the same locus, thus replacing the mutant PS1 alleles with the wild-type version was readily facile and was achieved by crossing homozygous 3xTg-AD mice (PS1M146V/PS1M146V; APPSwe/APPSwe; TauP301L/TauP301L) with nontransgenic (NonTg) mice. The F1 progeny (PS1M146V/PS1WT; APPSwe/0; TauP301L/0) were intercrossed and 25% of the mice were double transgenic homozygous for the mutant APP and tau transgenes (PS1WT/PS1WT; APPSwe/APPSwe; TauP301L/TauP301L) but now contain two wild-type PS1 alleles. These derivative double transgenic will be referred to as APP/tau mice and are basically the 3xTg-AD without the mutant PS1 allele (Table 1). Notably, the genetic background of the mice (C57BL6/129sv) was not altered.
Table 1.
Genotypes and transgene expression levels among the various transgenic lines
| Name | PS1M146VKI | hAPP | htau | Expression of hAPP over endogenous | Expression of htau over endogenous |
|---|---|---|---|---|---|
| 3xTg-AD | +/+ | +/+ | +/+ | ∼6× | ∼6× |
| APP/tau | −/− | +/+ | +/+ | ∼6× | ∼6× |
| PS1/tau | +/+ | −/− | +/− | N/A | ∼6× |
| NonTg | −/− | −/− | −/− | N/A | N/A |
The table shows the genotype and the relative expression levels of all the mice used in this study. Notably, because of the way we derived the mice, all the groups are on the same genetic background (C57BL6/129sv). The characterization of the 3xTg-AD, the PS1/tau, and the NonTg mice was previously described (Oddo et al., 2003b).
Aβ pathology is reduced in APP/tau mice
The 3xTg-AD mice accumulate ∼10 times more Aβ42 than Aβ40 (Oddo et al., 2003a,b), which is attributable to the presence of the M146V mutation in the PS1 gene (Guo et al., 1999). To define the impact of replacing the mutant PS1 allele with its wild-type counterpart on APP and Aβ levels, we compared their levels between the 3xTg-AD and the APP/tau mice at 6, 12, and 18 months of age (n = 10/genotype/time point). We found that the steady-state levels of APP were similar between the two groups of mice at each time point analyzed as determined by Western blot analysis (Fig. 1A–C). Using sandwich ELISA, however, we found that soluble Aβ42 levels were significantly and selectively decreased, whereas Aβ40 levels remained unaltered in the brains of the APP/tau mice versus age- and gender-matched 3xTg-AD mice (Fig. 1D–F). In particular, we found that Aβ42 levels were 38.92% lower in the brains of 6-month-old APP/tau mice compared with 3xTg-AD mice (Fig. 1D). Notably, the reduction in Aβ42 levels in the APP/tau mice became greater as the mice aged, and at 12 and 18 months of age, levels were decreased by ∼75% and 136%, respectively (Fig. 1E,F). In addition, in the 3xTg-AD mice there is an age-dependent accumulation of insoluble Aβ levels, however, insoluble Aβ levels were below the limit of detection in the APP/tau mice (data not shown).
Figure 1.

Selective reduction of Aβ42 levels in the APP/tau mice. A–C, To define the impact of replacing the mutant PS1 with the wild-type allele on the APP levels, we measured the steady-state levels of APP by semiquantitative Western blot in the brains of 3xTg-AD and APP/tau mice (n = 10 mice/group/time point). Panels A–C show representative Western blots for APP. β-Actin was used as a loading control. Quantification analysis shows that the steady-state levels of APP were similar between 3xTg-AD and APP/tau mice at all ages analyzed (data not shown). D–F, We next used sandwich ELISA to measure the Aβ levels in the brains of the 3xTg-AD and the APP/tau mice. Aβ40 levels were similar between the two groups at all ages analyzed. In contrast, we found that the levels of Aβ42 were selectively decreased in the brains of the APP/tau mice compared with age- and gender-matched 3xTg-AD mice. Notably, this difference is accentuated as the mice age.
We next evaluated the effect this selective decrease in Aβ42 levels on the onset and progression of Aβ deposition in the brains of 6-, 12-, and 18-month-old APP/tau and 3xTg-AD mice. As we previously reported, the Aβ deposition in 6-month-old 3xTg-AD mice is predominantly intraneuronal (Fig. 2A,B). Notably, selective reduction in the Aβ42 levels caused a marked reduction in intraneuronal Aβ immunoreactivity in the APP/tau mice (Fig. 2C,D). As the 3xTg-AD mice age, extracellular Aβ deposit starts to accumulate and are readily apparent in the subiculum of 12-month-old mice (Fig. 2E,F) and throughout the brain in 18-month-old mice (Fig. 2I,J). In contrast, no extracellular Aβ deposits were evident in age- and gender-matched APP/tau mice (Fig. 2G,H,K,L). Notably, the levels of intraneuronal Aβ deposits in 6-month-old 3xTg-AD mice were higher than 18-month-old APP/tau mice (Fig. 2, compare B,L). These results, together with the ELISA data, indicate that in addition to delaying the onset of soluble Aβ accumulation, selectively decreasing Aβ42 prevents the development of insoluble Aβ aggregates. In addition, these results indicate that Aβ40 is insufficient to trigger plaques formation, which is consistent with the data showing that Aβ42 is necessary for extracellular Aβ formation (McGowan et al., 2005; Kim et al., 2007).
Figure 2.

Reduction in Aβ deposition in the APP/tau mice. To determine whether the onset and progression of Aβ deposition were altered by the selective reduction in Aβ42 levels, brain sections from 6-, 12-, and 18-month-old APP/tau and 3xTg-AD mice (n = 10 mice/group/time point) were stained using different anti-Aβ antibodies. A–D, As expected, robust intraneuronal immunoreactivity was detected in the brains of 6-month-old 3xTg-AD mice, whereas we found that the selective reduction of Aβ42 levels led to a significant reduction of the intraneuronal Aβ staining. E–H, Immunohistochemical analysis of sections from 12-month-old mice revealed several extracellular Aβ plaques in the CA1/subiculum region. In contrast, no plaques were detected in the brains of age- and gender-matched APP/tau mice. I–L, We next evaluated the brains of 18-month-old mice. In the 3xTg-AD mice, Aβ plaques were evident throughout the brain, whereas no plaques were detected in the APP/tau mice, and only a weak intraneuronal immunoreactivity was apparent.
Selective reduction in Aβ42 levels markedly delays the onset of tau pathology
A growing body of evidence suggests that Aβ and tau may be mechanistically linked (for review, see Blurton-Jones and LaFerla, 2006). To better understand this relationship, we next assessed whether the onset and progression of tau pathology was affected by genetically reducing Aβ42 levels. If these two pathologies are unrelated, we would expect that the tau pathology would develop along the same time course in the APP/tau mice as in the 3xTg-AD mice.
We first determined whether expression of tau steady-state levels was affected by performing Western blot analysis on brain homogenates from the 3xTg-AD and APP/tau mice, and found that the levels were similar at all ages analyzed (Fig. 3). To determine whether selective reduction of Aβ42 levels altered the onset and progression of the tau pathology, sections from 6-, 12-, and 18-month-old APP/tau mice were immunostained for tau and compared with age- and gender-matched 3xTg-AD mice and PS1/tau mice. The PS1/tau mice are on the same genetic background as the 3xTg-AD mice and expression levels of human tau are similar to those in the 3xTg-AD mice (Oddo et al., 2003b). The use of the PS1/tau mice is an important control as it has been shown that mutant PS1 can directly alter tau pathology independently of Aβ (Gómez-Isla et al., 1999; Boutajangout et al., 2002; Kang et al., 2005; Tanemura et al., 2006). Using a human-specific anti-tau antibody, robust accumulation of tau in the somatodendritic compartment of pyramidal neurons of the 3xTg-AD mice was evident, which progressively increased as a function of age (Fig. 4A–C). In contrast, tau accumulation in the somatodendritic compartment of APP/tau mice was almost absent at 6 and 12 months of age (Fig. 4D,E), although it started to become apparent at 18 months of age (Fig. 4F). To determine whether the robust reduction in tau accumulation was attributable to the reduction of soluble Aβ42 levels or to a direct effect of replacing the mutant PS1 with the wild-type allele, we stained sections from age- and gender-matched PS1/tau mice (Table 1). Only baseline levels of tau immunoreactivity were detected in the brains of these mice at all ages analyzed (Fig. 4G–I), supporting the conclusion that the delay in the buildup of somatodendritic tau accumulation in the APP/tau mice is most likely caused by reduced levels of soluble Aβ42 and not to a direct effect of mutant PS1 on tau. In addition, the paucity of tau staining in the somatodendritic compartment of neurons in the APP/tau and PS1/tau mice with antibody HT7 was further substantiated by using additional tau antibodies (Fig. 4J–O).
Figure 3.

Selective decrease in Aβ42 levels did not alter the steady-state levels of human tau. To assess the effect of reducing Aβ42 levels on the levels of tau, proteins were extracted from the brains of 6-, 12-, and 18-month-old APP/tau and 3xTg-AD mice (n = 10 mice/group/time point). A–C, Western blot analysis indicated that the steady-state levels of human tau were similar between the APP/tau and the 3xTg-AD mice at all the time points analyzed.
Figure 4.

Selective reduction of Aβ42 levels markedly delays the onset of tau pathology. To assess the effects of genetically reducing Aβ42 levels on the onset and progression of tau pathology, sections from 6-, 12-, and 18-month-old APP/tau mice were immunostained using different anti-tau antibodies and compared with age- and gender-matched 3xTg-AD and PS1/tau mice (n = 10 mice/group/time point). A–C, Robust somatodendritic tau accumulation was detected in the brains of 3xTg-AD mice at 6, 12, and 18 months of age using the human specific tau antibody HT7. D–F, In contrast, only a background staining was detected in the brains of 6- and 12-month-old APP/tau mice and some somatodendritic tau deposits start to be apparent in the CA1 regions of 18-month-old mice. G–I, No somatodendritic tau accumulation was detected in the PS1/tau mice with HT7. J–O, The reduction in somatodendritic tau accumulation in the APP/tau and PS1/tau mice was further confirmed using additional anti-tau antibodies. To determine whether the HT7-positive deposits detected in the brains of 18-month-old APP/tau mice (F) were phosphorylated, sections from these mice were stained with AT8 and AT100 and compared with age- and gender-matched 3xTg-AD mice. P–S, A strong immunoreactivity with both AT8 and AT100 was detected in the 3xTg-AD mice whereas such immunoreactivity was absent in the APP/tau mice. Together, these data clearly indicate that reduction of Aβ42 levels delays the onset and progression of tau pathology despite the fact that tau pathology in the APP/tau mice is driven by its own transgene.
Tau immunoreactivity begins to become apparent in the somatodendritic compartment of pyramidal CA1 neurons in 18-month-old APP/tau mice (Fig. 4F). To determine whether these tau deposits are phosphorylated, we used two different phospho-tau-specific antibodies, AT8 (which recognizes tau phosphorylated at Ser202 and Thr205) and AT100 (which recognizes tau phosphorylated at Thr212 and Ser214), and found that tau was not hyperphosphorylated at these sites in the APP/tau mice, whereas pronounced immunoreactivity was apparent in age-matched 3xTg-AD mice (Fig. 4P–S). Although we cannot exclude the possibility that tau may be phosphorylated at other sites, these data indicate that selective reduction of Aβ42 drastically alter the onset and progression of tau pathology, therefore further supporting an upstream role of Aβ and the pathogenesis of tau pathology.
Age-dependent cognitive impairments are delayed by replacing mutant with wild-type PS1
We next evaluated the onset and progression of the cognitive decline in the APP/tau mice at 2, 6, 12, and 18 months of age (n = 10/genotype/time point), using a spatial reference version of the Morris water maze (MWM). The changes in cognition of the APP/tau mice were compared with age- and gender-matched 3xTg-AD, NonTg, and PS1/tau (Table 1). Because the 3xTg-AD mice harbor a disease-associated mutation in the tau gene, it is possible that this by itself can further exacerbate the cognitive decline in the 3xTg-AD mice. The use of the PS1/tau mice in these experiments served as an important control to determine the role of mutant tau on the cognitive impairments in the 3xTg-AD mice and APP/tau mice.
At 2 months of age, Aβ and tau pathology are not apparent in the 3xTg-AD mice, and indeed behavioral testing at this time point indicates that the mice are unimpaired. Likewise, we found that all the groups performed similarly in the spatial learning task at this age (Fig. 5A), and their short- and long-term memory was unaffected, as determined by the probe trials conducted at 1.5 and 24 h after the last training trial (Fig. 5A) (p = 0.3315 for overall learning for all the genotypes and p < 0.05 for all probe trials measured). These data indicate that the 3xTg-AD, APP/tau, and PS1/tau mice have no learning and memory deficits at 2 months as they performed as well as age- and gender-matched NonTg mice.
Figure 5.

Replacing the mutant allele with its wild-type counterpart delays cognitive impairments. Mice of different genotypes were evaluated in the spatial reference versions of the MWM. A, At 2 months of age, all four groups of mice learned the task equally well, and no impairments were found during the probe trials as determined by time spent in the opposite and target quadrant and number of platform location crosses, indicating that short- and long-term memory is similar among the different genotypes. B, At 6 months of age, the 3xTg-AD mice required more trials to reach criterion, as determined by an escape latency of <20 s. Notably, both PS1/tau and APP/tau mice were indistinguishable from the NonTg and learned the task faster than the 3xTg-AD mice. To measure short- and long-term memory, probe trials were conducted 1.5 and 24 h after the last training trial. The 3xTg-AD mice performed significantly worse than the NonTg and PS1/tau mice. Notably, replacing the PS1 mutant with the wild-type allele rescued both short- and long-term memory as the APP/tau mice performed similarly to the NonTg and PS1/tau mice. C, Twelve-month-old 3xTg-AD required 6 d of training to reach criterion, whereas age- and gender-matched APP/tau mice learn the task faster and reach criterion after 5 d of training. Although the APP/tau mice performed better than the 3xTg-AD mice, it took them an extra day to reach criterion compared with NonTg and PS1/tau mice. Notably, short- and long-term memory in the APP/tau mice was similar to the NonTg and PS1/tau and significantly better than the 3xTg-AD mice in all the measurements taken. D, Although 18-month-old 3xTg-AD and APP/tau did not reach criterion after 6 d of training (escape latency 33.7 and 21.7 s, respectively), the escape latency at days 5 and 6 was significantly lower for the APP/tau mice compared with the 3xTg-AD mice, indicating that the APP/tau mice still learn better and faster than the 3xTg-AD mice. The probe trials indicated that short- and long-term memory was significantly better in the APP/tau mice compared with the 3xTg-AD mice in all the measurements taken. *Significant difference between the 3xTg-AD and all the other 3 genotypes; #significant difference between the 3xTg-AD and the APP/tau mice. Error bars indicate SEM.
At 6 months of age, the 3xTg-AD mice are characterized by intraneuronal accumulation of Aβ in the cortex, hippocampus, and amygdala, whereas no overt Aβ or tau pathology is detected in the PS1/tau mice (Oddo et al., 2003a,b). At this age, all groups were able to learn the task (as determined by an escape latency <20 s), although the 3xTg-AD mice required 5 d of training compared with the 3 d for the NonTg, PS1/tau, and APP/tau mice (Fig. 5B). Post hoc analysis indicated that at days 3 and 4, the escape latency of the 3xTg-AD was significantly higher than all the other groups, nevertheless after 5 d of training the escape latency for all the groups was <20 s (Fig. 5B). To measure short- and long-term memory, probe trials were conducted at 1.5 and 24 h after the last training trial. We found that replacing the PS1 mutant allele with its wild-type counterpart rescued the memory deficit present in the 3xTg-AD mice as indicated by the significantly reduced time the APP/tau mice spent in the opposite quadrant and by the significant increase in the number of platform location crosses and time spent in the target quadrant (Fig. 5B). Notably the APP/tau mice performed similarly to the NonTg and the PS1/tau mice. Hence, despite equivalent levels of tau expression between the APP/tau and the PS1/tau mice (Table 1), replacing the mutant PS1 gene with its wild-type counterpart, is sufficient to delay the onset of the cognitive decline.
As the mice age, the cognitive deficits in the 3xTg-AD mice worsen, and at 12 months of age, 6 d of training are required for them to reach criterion (Fig. 5C). The APP/tau mice performed better, reaching criterion after 5 d of training (Fig. 5C). Nevertheless, it took 1 d extra for the 12-month-old compared with 6-month-old APP/tau mice, indicating that there is an age-dependent cognitive decline in these mice. Statistical analysis indicated that the 3xTg-AD performed worse than the NonTg and the PS1/tau mice at days 3, 4, and 5 (p = 0.0051 and p = 0.0041, respectively for day 3, p = 0.004 and p = 0.002, respectively for day 4, p = 0.0013 and p = 0.0079, respectively for day 5). The escape latency for the APP/tau mice was significantly lower compared with the 3xTg-AD mice at days 4 and 5 (p = 0.0236 and p = 0.0221, respectively). Notably, during the probe trials, the APP/tau mice performed similarly to the NonTg and PS1/tau mice but significantly better than the 3xTg-AD mice in all the measurements taken (Fig. 5C).
At 18 months of age, after 6 d of training the 3xTg-AD and the APP/tau did not reach criterion (escape latency 33.7 and 21.7 s, respectively), nevertheless, the escape latency at days 5 and 6 was significantly lower for the APP/tau versus the 3xTg-AD mice (p = 0.432 and p = 0.173, respectively), indicating that the APP/tau mice still learn faster and better than the 3xTg-AD mice (Fig. 5D). During the probe trials, we found that the APP/tau mice performed significantly better than the 3xTg-AD mice in all measurements (Fig. 5D).
Not surprisingly, these data indicate that replacing the mutant PS1 allele with its wild-type counterpart and thus changing the Aβ and tau pathology, delays the onset and progression of cognitive decline. Notably, the APP/tau mice performed similarly to the PS1/tau mice, suggesting that the early cognitive decline occurring in the 3xTg-AD mice is attributable to Aβ buildup and is consistent with our previous work indicating that intraneuronal Aβ accumulation is responsible for the onset of cognitive decline in the 3xTg-AD mice (Oddo et al., 2004, 2006a,b).
Aβ42 immunization delays the development of tau pathology
The data presented here show that the onset and progression of the tau pathology, and the subsequent cognitive decline in the APP/tau mice is markedly different compared with age- and gender-matched 3xTg-AD mice. These changes appear to be mediated by the selective reduction of Aβ42 and not by a direct interaction between mutant PS1 and tau. To more directly access whether this effect is mediated by Aβ, we used a different approach and actively immunized prepathological, 2-month-old 3xTg-AD mice against fibrillar Aβ42 for 12 months (Fig. 6A). Eighteen 2-month-old 3xTg-AD mice were randomly assigned to one of three groups: (1) untreated; (2) adjuvant only; (3) fibrillar Aβ42 (fAβ42). This latter group was injected with 100 μg of fAβ42 formulated with 50 μg (initial injection) or 20 μg (subsequent injections) of QuailA adjuvant in a total volume of 100 μl adjusted with PBS. Antibody response was measured before and at 14 h and 7 d after each immunization, as well as at the end of the experiment. Fortuitously, not all mice produced a good antibody response (Fig. 6B), allowing us to correlate the degree of pathology with the antibody titer. All the mice were 13.5 months old at the end of the immunization paradigm.
Figure 6.
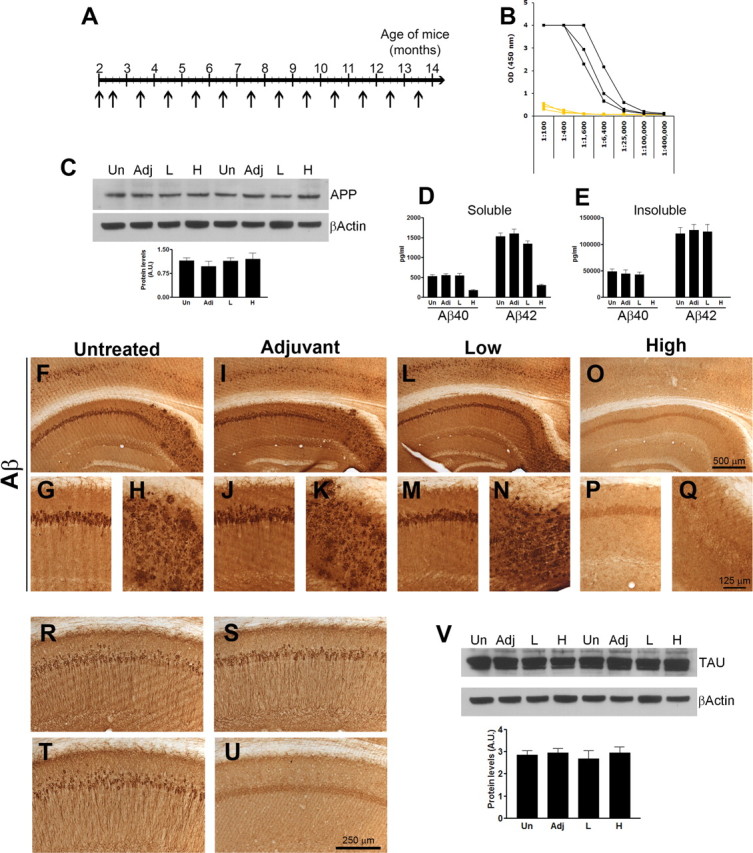
Aβ42 immunization of prepathological 3xTg-AD mice delays the development of tau pathology. To further support the role of Aβ42 in the onset and progression of the tau pathology, we actively immunized 2-month-old 3xTg-AD mice with fibrillar Aβ42 with the goal of preventing Aβ accumulation and determining its effect on the onset and progression of tau pathology. A, Six 2-month-old mice were injected with 100 μg of fAβ42 formulated with 50 μg (initial injection) or 20 μg (subsequent injections) of QuailA adjuvant in a total volume of 100 μl adjusted with PBS. As a control, six mice were injected with adjuvant only and six mice remained untreated. All the mice received a second injection 10 d after the first and monthly thereafter until they were 13.5-month-old. B, Antibody response was only detected in some mice immunized with fAβ42, therefore allowing us to use the low responders as an extra control group. C–E, The steady-state levels of full-length APP were similar among all groups (C), whereas we found a reduction in soluble and insoluble Aβ40 and Aβ42 levels in the brains of the mice with a robust antibody response (D–E). Notably, the insoluble Aβ levels, both 40 and 42, were below detection levels in the immunized mice. F–Q, Whereas the control groups and the low responders show a robust intraneuronal and extracellular Aβ staining, no Aβ deposits were detected in the brains of the mice with a robust antibody response. R–U, Representative photographs indicating that somatodendritic tau levels were drastically reduced in the immunized mice with a robust antibody response (U) compared with untreated (R), adjuvant-treated (S), or low-responder mice (T). V, Western blot analysis indicated that the steady-state levels of tau were similar between all the groups.
We first measured the steady-state levels of APP and found that Aβ immunization did not alter the expression levels of the transgene (Fig. 6C). To determine the effect of the immunization on the Aβ pathology, we then measured the Aβ levels by sandwich ELISA. Our data indicate that the immunized mice with a high antibody titer had a significant decrease in soluble Aβ40 and Aβ42 compared with the control groups and the immunized mice with low antibody response (Fig. 6D). Notably, insoluble Aβ levels, both 40 and 42, were not detected in the brains of high responders (Fig. 6E). To determine how Aβ immunization effected the Aβ deposition, we stained sections from untreated, adjuvant, low-responders and immunized mice with an anti-Aβ antibody. At this age, neurons in the CA1/subiculum region contain robust intracellular Aβ immunoreactivity and also several extracellular Aβ plaques are readily apparent (Fig. 6F–H). Comparable degrees of Aβ pathology was detected in the brains of adjuvant-treated and nonresponder mice (Fig. 6I–N). In contrast, we found that immunization with fAβ42 prevented the accumulation of intraneuronal and extracellular Aβ deposits (Fig. 6O–Q). These data are consistent with other reports showing that immunization of prepathological mice can prevent the buildup of Aβ.
We previously showed that a single intrahippocampal injection of an anti-Aβ antibody reduced not only Aβ deposits but more importantly led to the clearance of early tau pathology, but not late, hyperphosphorylated tau aggregates (Oddo et al., 2004). To determine whether these hyperphosphorylated tau aggregates can be prevented by blocking Aβ accumulation, we stained sections from untreated, adjuvant, low-responders and immunized mice with an anti-tau antibody and found that tau accumulation in the somatodendritic compartment was evident in the brains of untreated, adjuvant-only and nonresponder mice (Fig. 6R–T, respectively), whereas only background staining was detected in the brains of immunized mice (Fig. 6U). These studies clearly show that Aβ immunization in these mice can prevent the development of tau pathology despite the fact that the expression levels of the tau transgene were unaffected by this immunization paradigm (Fig. 6V). These data, together with the genetic data presented earlier, clearly indicate that the development of the tau pathology in the 3xTg-AD mice is highly dependent on Aβ accumulation and suggest that preventing it to occur may have strong therapeutic values as it will have an effect on both Aβ and tau deposits.
CHIP is a key molecular link between Aβ and tau pathologies
Using a genetic and an immunological approach, we showed that reducing Aβ42 levels markedly delays the onset of the tau pathology. The molecular mechanisms underlying the link between Aβ and tau, however, remain to be determined. Evidence from different groups points to ubiquitin/proteasome pathway as playing a critical role in the pathogenesis of AD. In particular, it has been shown that CHIP is a tau ubiquitin ligase and its deletion leads to the accumulation of tau (Dickey et al., 2006). Moreover, we and others have previously shown that Aβ can interfere with proteasomal function (Gregori et al., 1995; Oddo et al., 2004; Oh et al., 2005; Almeida et al., 2006; Tseng et al., 2008). To determine whether CHIP levels are altered by Aβ, we measured CHIP levels in HEK cells stably transfected with APP695 and found that there was a 35.1 ± 7.7% reduction in CHIP levels in cells overexpressing APP compared with control cells (Fig. 7A). Notably, CHIP levels were restored when we reduced APP levels using siRNA (Fig. 7B,C). To determine whether this change was attributable to Aβ or to APP, HEK cells overexpressing APP were treated with a γ-secretase inhibitor. We found that CHIP levels were restored to normal levels by blocking Aβ production (Fig. 7D). Hence, we conclude that Aβ, and not full-length APP, is responsible for the reduction in CHIP levels.
Figure 7.

Inverse relationship between Aβ and CHIP levels. To determine the mechanism by which Aβ and tau interact, we measured CHIP levels (which has previously been involved in tau turnover) in HEK cells stably transfected with APPswe. A, Representative Western blot from proteins obtained from lysates from cells expressing APP or control cells. Densitometric analysis indicates that in the presence of APP there is a 35.1 ± 7.7% reduction in CHIP levels. B, C, CHIP levels were rescued by reducing APP expression using siRNA. To determine whether the effect on CHIP were mediated by full-length APP or Aβ, cells overexpressing APP were treated with a γ-secretase inhibitor to block Aβ formation. D, Western blot analyses showed that blocking Aβ production, CHIP levels were restored to control levels, indicating that Aβ, and not APP, is responsible for the decrease in CHIP levels. *p < 0.05.
To determine how CHIP levels change in vivo, we measured CHIP levels in the brains of the 3xTg-AD mice at different ages, corresponding with different degrees of pathology. We found that in 9- and 12-month-old 3xTg-AD mice the levels of CHIP were significantly lower compared with age-matched NonTg mice (Fig. 8A). Hence, we were next interested in determining whether reducing Aβ levels in vivo could restore CHIP levels to physiological levels. To address this issue, we analyzed CHIP levels from 12- and 18-month-old APP/tau mice and compared their levels to those found in NonTg, PS1/tau, and 3xTg-AD mice. Notably, we found that CHIP levels in the PS1/tau and APP/tau mice were similar to the CHIP levels measured in NonTg mice, which correlates with the low Aβ levels present in the brains of these mice (Fig. 8B). To further understand the relationship between Aβ and CHIP levels, we next use an immunological approach to decrease Aβ levels. Toward this end, 6-month-old 3xTg-AD mice were injected the anti-Aβ antibody 1560 (control mice were injected with PBS) into the cerebral ventricle (4 mice/group). This single antibody injection was sufficient to remove Aβ deposits from the brains of these mice (data not shown), consistent with our previous reports (Oddo et al., 2004, 2006b). We found a 26.7 ± 4.3% increase in CHIP levels in the immunized mice compared with PBS-injected 3xTg-AD mice (Fig. 8C). To further support these data, we next measured CHIP levels in the brains of actively immunized mice and found that preventing Aβ accumulation also restored the CHIP levels to NonTg levels (Fig. 8D). Based on the data presented here, it appears that Aβ42 (either directly or indirectly) reduces CHIP levels, which is sufficient to interfere with tau turnover and degradation, thereby facilitating the buildup of tau aggregates.
Figure 8.

Aβ accumulation decreases CHIP levels in vivo. To further understand the relationship between Aβ and CHIP, we measured CHIP levels in the brains of the 3xTg-AD mice as a function of age. A, Representative Western blots from proteins extracted from brains of 3-, 6-, 9-, and 12-month-old 3xTg-AD mice. Densitometric analysis showed a significant decrease in CHIP levels in the brains of 9- and 12-month-old mice. B, The decrease in CHIP levels are rescued in the APP/tau mice after selectively reducing Aβ42 levels, indicating that even in vivo, Aβ can alter CHIP levels. We next determine whether CHIP levels could be restored by reducing Aβ levels via Aβ immunization. C, Representative Western blot from proteins extracted from the brains of 6-month-old mice injected with the anti-Aβ antibody 1560 or PBS. Densitometric analysis showed a 26.7 ± 4.3% increase in CHIP levels in the immunized mice. D, Representative Western blots from proteins extracted from actively immunized mice with fAβ42, which prevented Aβ accumulation (Fig. 6). Densitometric analysis indicated that CHIP levels were restored to NonTg levels in the brains of immunized mice showing a high antibody-response. *p < 0.05.
To more conclusively demonstrate the critical role that CHIP plays as a molecular mediator of Aβ-induced tau pathology, we sought to determine whether the Aβ-induced effects on tau could be reestablished by elevating CHIP levels even in the presence of Aβ. To address this issue, we injected 3 μl of a CHIP-expressing lentivirus (1.2 × 106 transducing particles/ml) into the left hippocampi of 12- and 18-month-old 3xTg-AD mice. As a control, the right hippocampi received injection of a control lentivirus. Seven days after injection, mice were killed and their hippocampi extracted and processed for analysis (n = 8/time point). In protein extracts from the left hippocampus, which received the CHIP-expressing lentivirus, Western blot analysis showed a significant increase in CHIP steady-state levels compared with protein extract harvested from the right, control-virus injected hippocampi; the effect was apparent in both 12- and 18-month-old mice (Fig. 9A,B). The increase in CHIP levels led to a decrease in phosphorylated tau as detected by the AT180 and AT8 phospho-specific tau antibodies, whereas the steady-state levels of the tau transgene remained unaltered (Fig. 9A,B). We next determined whether the increase in CHIP levels had any effect on the somatodendritic tau accumulation. We first analyzed the brains of 12-month-old mice and found that tau deposits were significantly decreased in sections close to the injection site (left hippocampi), whereas its levels were not altered on the right hippocampi, receiving the control virus (Fig. 9C–E,J). Notably, in sections distal to the site of injection, somatodendritic tau immunoreactivity was comparable between the left and right hippocampi, consistent with the relatively small zone of infectivity exhibited by lentiviral vectors (Fig. 9F–G,K). In 18-month-old mice, where tau phosphorylation and aggregation is more advanced compared with 12-month-old mice (Oddo et al., 2003b), we also observed a significant reduction in somatodendritic tau levels in the left versus right hippocampi (Fig. 9H–I,K), although the effect was not as robust as those saw in 12-month-old mice. Based on the cumulative evidence presented here from our in vitro and in vivo studies, using both genetic and immunological approaches, we conclude that CHIP plays a major role in mediating the Aβ-induced tau pathology.
Figure 9.

Increasing CHIP levels ameliorates tau pathology in vivo. CHIP-expressing lentivirus was injected into the left hippocampi of 12- and 18-month-old 3xTg-AD mice, whereas the right hippocampi were injected with a control lentivirus. A, Representative Western blot indicating that CHIP levels are increased in the hippocampi receiving the CHIP-expressing virus (ipsilateral) compared with the hippocampi injected with the control virus (contralateral), whereas the expression levels of tau, as detected by HT7 remained unchanged. The increase in CHIP levels led to a reduction of tau phosphorylation as detected by AT180 and AT8. β-Actin was used as a loading control. B, Quantification analysis shows that the steady-state levels of CHIP were significantly higher in the hippocampi of the mice receiving the CHIP-expressing lentivirus compared with mice receiving a control virus (n = 4/time point), which led to a significant reduction in AT180 and AT8 level both in 12- and 18-month-old mice. To determine whether the somatodendritic tau deposits were altered by increasing CHIP levels, sections from injected mice were stained for tau. C, Representative microphotograph from 12-month-old mice showing a decrease in somatodendritic tau deposits in the left hippocampus, receiving the CHIP-expressing lentivirus (ipsilateral) compared with the contralateral hippocampus, receiving the control virus. D, E, High-magnification view of C. When sections distal to the injection sites were stained for tau we found no differences in the somatodendritic tau deposits (F, G). In 18-month-old mice, a significant reduction in somatodendritic tau levels was also observed in the left hippocampi (ipsilateral) receiving the CHIP expressing virus compared with the right hippocampi (contralateral) receiving the control virus (H, I). J, Quantification analysis of the immunohistochemical data from sections close to the injection site. Notably, the reduction in tau pathology was more robust in 12-month-old mice compared with 18-month-old mice. K, Quantification analysis of the immunohistochemical data from sections distal to the injection site. *p < 0.05; **p < 0.01.
Discussion
The pivotal role Aβ plays in the pathogenesis of AD is widely accepted and evidence from different laboratories shows that Aβ accumulation underlies the memory loss in transgenic mice (Billings et al., 2005; Cleary et al., 2005; Lesné et al., 2006). Recent evidence points to tau as a necessary component of the Aβ-induced cognitive dysfunction (Roberson et al., 2007), suggesting that either a direct or indirect interaction between Aβ and tau may be central to the development of AD dementia. Hence, exploration of the possible mechanistic links between Aβ and tau pathology may greatly aid our understanding of the disease.
Despite the fact that the 3xTg-AD mice harbor an independent transgene that drives the expression of mutant human tau, here we present experimental evidence showing that the development of tau pathology is strongly dependent on the Aβ42 levels. We first used a genetic approach to replace the mutant PS1 gene with its wild-type counterpart. As expected, compared with the 3xTg-AD mice, the APP/tau mice show a selective decrease in Aβ42 levels, whereas Aβ40 levels remained unaltered. Notably, reducing Aβ42 levels markedly delayed the onset of Aβ deposition, and no plaque deposits were apparent in the brains of 18-month-old APP/tau mice. The most significant finding emerging from these studies is that this selective reduction in Aβ42 levels markedly delayed the onset and progression of tau pathology. Indeed, somatodendritic tau accumulation, which is a sign of early tau pathology, is not detected until 18-month of age in the APP/tau mice and is significantly less in extent compared with 6-month-old 3xTg-AD mice. To our knowledge, this is the first report showing that preventing Aβ42 accumulation alters the onset and progression of tau pathology.
Aβ immunization has been successfully used to decrease Aβ pathology from the brains of transgenic mice (Schenk et al., 1999; Janus et al., 2000; Morgan et al., 2000). We previously showed that Aβ immunotherapy suffices to remove early but not late hyperphosphorylated tau lesions (Oddo et al., 2004, 2006a). In this study, to further understand the relationship between Aβ and tau, we also used an immunological strategy and found that immunizing young prepathological mice with fAβ42 was sufficient to prevent Aβ accumulation. Most importantly, mimicking the results obtained by genetically reducing Aβ42 levels, the prevention of Aβ accumulation markedly delayed the onset of tau pathology despite no changes in the steady-state levels of the tau transgene. This finding is remarkable as the 3xTg-AD mice contain an independent transgene that drives expression of mutant human tau and despite that, the development of the tau pathology is highly dependent on Aβ42 levels. These findings provide clear evidence that Aβ plays a critical role in inducing tau pathology.
Several neurodegenerative disorders (including AD) are characterized by the abnormal accumulation of proteins, implying that alterations of the UPS may play a role in their pathogenesis and indeed, previous studies have implicated UPS dysfunction in AD (Keller et al., 2000; Bertram et al., 2005). In particular, we previously showed that Aβ accumulation may impair proteasomal function and lead to tau accumulation (Oddo et al., 2004; Tseng et al., 2008). The molecular mechanisms underlying the effects of Aβ accumulation on the UPS are not well understood. In this study, we provide the first experimental evidence that Aβ accumulation decreases CHIP levels, which is a link between chaperones and the UPS (McDonough and Patterson, 2003). Moreover, we showed that the Aβ-induced effects on the tau pathology are rescued by overexpressing CHIP, providing evidence that CHIP plays a major role in the Aβ-induced tau pathology. Future studies are needed to determine whether Aβ alters CHIP levels directly or indirectly through its influence on other components of the UPS. Nevertheless, the finding that Aβ alters CHIP levels are highly significant as CHIP, in addition of regulating tau turnover, plays a critical role in cellular protein control (McDonough and Patterson, 2003). Particularly, CHIP positively regulates the stress response by activating heat shock factor 1 (Dai et al., 2003), a key regulator of the cellular stress response that decreases with aging (Hsu et al., 2003). By diminishing CHIP levels, Aβ accumulation may accelerate the normal age-dependent decrease in the heat shock protein response, thus facilitating cellular aging.
One of the main therapeutic targets in AD is Aβ, and a major effort is being conducted to the design β and γ secretase inhibitors and a clinical trial is currently ongoing using passive Aβ immunization. The data presented here have profound clinical implications, supporting the utility of this approach, as compounds designed to lower Aβ production and accumulation are expected to have beneficial effects on the tau pathology as well. This is highly relevant as tau accumulation alone, in the absence of Aβ, can cause cognitive decline (Ballatore et al., 2007). Furthermore, we previously showed that Aβ immunotherapy leads to the clearance of early but not late hyperphosphorylated tau aggregates (Oddo et al., 2004). Here we showed that reestablishing CHIP levels has a beneficial effect even in old mice (18-month-old mice) with advanced pathology, suggesting that a combined therapeutic approach, targeting Aβ and increasing proteasomal function may further the pathological sequelae of AD. Our results suggest that increasing CHIP levels may have beneficial effects as it decreases the tau pathology. This is not only relevant for AD but also for other neurodegenerative disorders such as, frontotemporal dementia characterized by the accumulation of tau.
Footnotes
This work was supported by funding from the National Institute on Aging (AG0212982) to F.M.L. and (AG029729A) to S.O.
References
- Almeida CG, Takahashi RH, Gouras GK. β-Amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J Neurosci. 2006;26:4277–4288. doi: 10.1523/JNEUROSCI.5078-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- Bertram L, Hiltunen M, Parkinson M, Ingelsson M, Lange C, Ramasamy K, Mullin K, Menon R, Sampson AJ, Hsiao MY, Elliott KJ, Velicelebi G, Moscarillo T, Hyman BT, Wagner SL, Becker KD, Blacker D, Tanzi RE. Family-based association between Alzheimer's disease and variants in UBQLN1. N Engl J Med. 2005;352:884–894. doi: 10.1056/NEJMoa042765. [DOI] [PubMed] [Google Scholar]
- Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- Blurton-Jones M, LaFerla FM. Pathways by which Abeta facilitates tau pathology. Curr Alzheimer Res. 2006;3:437–448. doi: 10.2174/156720506779025242. [DOI] [PubMed] [Google Scholar]
- Bolmont T, Clavaguera F, Meyer-Luehmann M, Herzig MC, Radde R, Staufenbiel M, Lewis J, Hutton M, Tolnay M, Jucker M. Induction of tau pathology by intracerebral infusion of amyloid-beta-containing brain extract and by amyloid-beta deposition in APP x Tau transgenic mice. Am J Pathol. 2007;171:2012–2020. doi: 10.2353/ajpath.2007.070403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutajangout A, Leroy K, Touchet N, Authelet M, Blanchard V, Tremp G, Pradier L, Brion JP. Increased tau phosphorylation but absence of formation of neurofibrillary tangles in mice double transgenic for human tau and Alzheimer mutant (M146L) presenilin-1. Neurosci Lett. 2002;318:29–33. doi: 10.1016/s0304-3940(01)02461-2. [DOI] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Crawford F, Houlden H, Warren A, Hughes D, Fidani L, Goate A, Rossor M, Roques P, Hardy J, et al. Early-onset Alzheimer's disease caused by mutations at codon 717 of the beta-amyloid precursor protein gene. Nature. 1991;353:844–846. doi: 10.1038/353844a0. [DOI] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Cribbs DH, Ghochikyan A, Vasilevko V, Tran M, Petrushina I, Sadzikava N, Babikyan D, Kesslak P, Kieber-Emmons T, Cotman CW, Agadjanyan MG. Adjuvant-dependent modulation of Th1 and Th2 responses to immunization with beta-amyloid. Int Immunol. 2003;15:505–514. doi: 10.1093/intimm/dxg049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Q, Zhang C, Wu Y, McDonough H, Whaley RA, Godfrey V, Li HH, Madamanchi N, Xu W, Neckers L, Cyr D, Patterson C. CHIP activates HSF1 and confers protection against apoptosis and cellular stress. EMBO J. 2003;22:5446–5458. doi: 10.1093/emboj/cdg529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey CA, Yue M, Lin WL, Dickson DW, Dunmore JH, Lee WC, Zehr C, West G, Cao S, Clark AM, Caldwell GA, Caldwell KA, Eckman C, Patterson C, Hutton M, Petrucelli L. Deletion of the ubiquitin ligase CHIP leads to the accumulation, but not the aggregation, of both endogenous phospho- and caspase-3-cleaved tau species. J Neurosci. 2006;26:6985–6996. doi: 10.1523/JNEUROSCI.0746-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey CA, Kamal A, Lundgren K, Klosak N, Bailey RM, Dunmore J, Ash P, Shoraka S, Zlatkovic J, Eckman CB, Patterson C, Dickson DW, Nahman NS, Jr, Hutton M, Burrows F, Petrucelli L. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J Clin Invest. 2007;117:648–658. doi: 10.1172/JCI29715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, Mant R, Newton P, Rooke K, Roques P, Talbot C, Pericak-Vance M, Roses A, Williamson R, Rossor M, Owen M, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- Goedert M, Wischik CM, Crowther RA, Walker JE, Klug A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc Natl Acad Sci U S A. 1988;85:4051–4055. doi: 10.1073/pnas.85.11.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Isla T, Growdon WB, McNamara MJ, Nochlin D, Bird TD, Arango JC, Lopera F, Kosik KS, Lantos PL, Cairns NJ, Hyman BT. The impact of different presenilin 1 andpresenilin 2 mutations on amyloid deposition, neurofibrillary changes and neuronal loss in the familial Alzheimer's disease brain: evidence for other phenotype-modifying factors. Brain. 1999;122:1709–1719. doi: 10.1093/brain/122.9.1709. [DOI] [PubMed] [Google Scholar]
- Götz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293:1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- Gregori L, Fuchs C, Figueiredo-Pereira ME, Van Nostrand WE, Goldgaber D. Amyloid beta-protein inhibits ubiquitin-dependent protein degradation in vitro. J Biol Chem. 1995;270:19702–19708. doi: 10.1074/jbc.270.34.19702. [DOI] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q, Fu W, Sopher BL, Miller MW, Ware CB, Martin GM, Mattson MP. Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nat Med. 1999;5:101–106. doi: 10.1038/4789. [DOI] [PubMed] [Google Scholar]
- Hardy J. Amyloid, the presenilins and Alzheimer's disease. Trends Neurosci. 1997;20:154–159. doi: 10.1016/s0166-2236(96)01030-2. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hsu AL, Murphy CT, Kenyon C. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science. 2003;300:1142–1145. doi: 10.1126/science.1083701. [DOI] [PubMed] [Google Scholar]
- Ihara Y, Nukina N, Miura R, Ogawara M. Phosphorylated tau protein is integrated into paired helical filaments in Alzheimer's disease. J Biochem. 1986;99:1807–1810. doi: 10.1093/oxfordjournals.jbchem.a135662. [DOI] [PubMed] [Google Scholar]
- Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Horne P, Heslin D, French J, Mount HT, Nixon RA, Mercken M, Bergeron C, Fraser PE, St George-Hyslop P, Westaway D. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease. Nature. 2000;408:979–982. doi: 10.1038/35050110. [DOI] [PubMed] [Google Scholar]
- Jarrett JT, Berger EP, Lansbury PT., Jr The C-terminus of the beta protein is critical in amyloidogenesis. Ann N Y Acad Sci. 1993;695:144–148. doi: 10.1111/j.1749-6632.1993.tb23043.x. [DOI] [PubMed] [Google Scholar]
- Kang DE, Yoon IS, Repetto E, Busse T, Yermian N, Ie L, Koo EH. Presenilins mediate phosphatidylinositol 3-kinase/AKT and ERK activation via select signaling receptors. Selectivity of PS2 in platelet-derived growth factor signaling. J Biol Chem. 2005;280:31537–31547. doi: 10.1074/jbc.M500833200. [DOI] [PubMed] [Google Scholar]
- Keller JN, Hanni KB, Markesbery WR. Impaired proteasome function in Alzheimer's disease. J Neurochem. 2000;75:436–439. doi: 10.1046/j.1471-4159.2000.0750436.x. [DOI] [PubMed] [Google Scholar]
- Kim J, Onstead L, Randle S, Price R, Smithson L, Zwizinski C, Dickson DW, Golde T, McGowan E. Aβ40 inhibits amyloid deposition in vivo. J Neurosci. 2007;27:627–633. doi: 10.1523/JNEUROSCI.4849-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazawa M, Anantharam V, Kanthasamy AG. Dieldrin induces apoptosis by promoting caspase-3-dependent proteolytic cleavage of protein kinase Cdelta in dopaminergic cells: relevance to oxidative stress and dopaminergic degeneration. Neuroscience. 2003;119:945–964. doi: 10.1016/s0306-4522(03)00226-4. [DOI] [PubMed] [Google Scholar]
- Kosik KS, Joachim CL, Selkoe DJ. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci U S A. 1986;83:4044–4048. doi: 10.1073/pnas.83.11.4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonough H, Patterson C. CHIP: a link between the chaperone and proteasome systems. Cell Stress Chaperones. 2003;8:303–308. doi: 10.1379/1466-1268(2003)008<0303:calbtc>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C, Skipper L, Murphy MP, Beard J, Das P, Jansen K, Delucia M, Lin WL, Dolios G, Wang R, Eckman CB, Dickson DW, Hutton M, Hardy J, Golde T. Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. 2005;47:191–199. doi: 10.1016/j.neuron.2005.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature. 2000;408:982–985. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer's disease. Neurobiol Aging. 2003a;24:1063–1070. doi: 10.1016/j.neurobiolaging.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003b;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43:321–332. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Green KN, Liang K, Tran L, Chen Y, Leslie FM, LaFerla FM. Chronic nicotine administration exacerbates tau pathology in a transgenic model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2005;102:3046–3051. doi: 10.1073/pnas.0408500102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Vasilevko V, Caccamo A, Kitazawa M, Cribbs DH, LaFerla FM. Reduction of soluble Abeta and tau, but not soluble Abeta alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J Biol Chem. 2006a;281:39413–39423. doi: 10.1074/jbc.M608485200. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Tran L, Lambert MP, Glabe CG, Klein WL, LaFerla FM. Temporal profile of amyloid-beta (Abeta) oligomerization in an in vivo model of Alzheimer disease. A link between Abeta and tau pathology. J Biol Chem. 2006b;281:1599–1604. doi: 10.1074/jbc.M507892200. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Cheng D, Jouleh B, Torp R, LaFerla FM. Genetically augmenting tau levels does not modulate the onset or progression of Abeta pathology in transgenic mice. J Neurochem. 2007;102:1053–1063. doi: 10.1111/j.1471-4159.2007.04607.x. [DOI] [PubMed] [Google Scholar]
- Oh S, Hong HS, Hwang E, Sim HJ, Lee W, Shin SJ, Mook-Jung I. Amyloid peptide attenuates the proteasome activity in neuronal cells. Mech Ageing Dev. 2005;126:1292–1299. doi: 10.1016/j.mad.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Petrucelli L, Dickson D, Kehoe K, Taylor J, Snyder H, Grover A, De Lucia M, McGowan E, Lewis J, Prihar G, Kim J, Dillmann WH, Browne SE, Hall A, Voellmy R, Tsuboi Y, Dawson TM, Wolozin B, Hardy J, Hutton M. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet. 2004;13:703–714. doi: 10.1093/hmg/ddh083. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- Shimura H, Schwartz D, Gygi SP, Kosik KS. CHIP-Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J Biol Chem. 2004;279:4869–4876. doi: 10.1074/jbc.M305838200. [DOI] [PubMed] [Google Scholar]
- Tanemura K, Chui DH, Fukuda T, Murayama M, Park JM, Akagi T, Tatebayashi Y, Miyasaka T, Kimura T, Hashikawa T, Nakano Y, Kudo T, Takeda M, Takashima A. Formation of tau inclusions in knock-in mice with familial Alzheimer disease (FAD) mutation of presenilin 1 (PS1) J Biol Chem. 2006;281:5037–5041. doi: 10.1074/jbc.M509145200. [DOI] [PubMed] [Google Scholar]
- Tseng BP, Green KN, Chan JL, Blurton-Jones M, LaFerla FM. Abeta inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol Aging. 2008;29:1607–1618. doi: 10.1016/j.neurobiolaging.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]