

Abstract
ATRX, a chromatin remodeling protein of the Snf2 family, participates in diverse cellular functions including regulation of gene expression and chromosome alignment during mitosis and meiosis. Mutations in the human gene cause alpha thalassemia mental retardation, X-linked (ATR-X) syndrome, a rare disorder characterized by severe cognitive deficits, microcephaly and epileptic seizures. Conditional inactivation of the Atrx gene in the mouse forebrain leads to neonatal lethality and defective neurogenesis manifested by increased cell death and reduced cellularity in the developing neocortex and hippocampus. Here, we show that Atrx-null forebrains do not generate dentate granule cells due to a reduction in precursor cell number and abnormal migration of differentiating granule cells. In addition, fewer GABA-producing interneurons are generated that migrate from the ventral telencephalon to the cortex and hippocampus. Staining for cleaved caspase 3 demonstrated increased apoptosis in both the hippocampal hem and basal telencephalon concurrent with p53 pathway activation. Elimination of the tumor suppressor protein p53 in double knock-out mice rescued cell death in the embryonic telencephalon but only partially ameliorated the Atrx-null phenotypes at birth. Together, these findings show that ATRX deficiency leads to p53-dependent neuronal apoptosis which is responsible for some but not all of the phenotypic consequences of ATRX deficiency in the forebrain.
Keywords: dentate gyrus, GABAergic neurons, migration, apoptosis, p53, ATRX, brain development, X-linked mental retardation
Introduction
The normal formation of the hippocampus is critical for learning and memory. It is positioned at the caudal-medial border of the neocortex and consists of the dentate gyrus, Cornus Ammonis fields CA1, CA3 and the subicular cortex. The dentate gyrus is the major input pathway into the hippocampus and dentate precursors continue dividing in the adult brain (Altman and Bayer, 1990a,b; Kempermann et al., 1997a,b, 1998; Gage et al., 1998). Limited information is available on the molecular aspects of dentate granule cell development but the study of mouse mutants has yielded important clues and helped to identify specific regulators. Mice lacking NeuroD1 exhibit proliferation and differentiation defects and excessive apoptosis of dentate granule cells (Liu et al., 2000a; Schwab et al., 2000). Empty spiracles homolog 2 (Emx2) knock-out mice lack migrating precursors and display abnormal glial scaffolding (Pellegrini et al., 1996; Oldekamp et al., 2004). Wnt pathway molecules including the lymphoid enhancer-binding factor (LEF1) and the Wnt coreceptor LRP6 also play a role in forming a normal hippocampus and dentate gyrus (Galceran et al., 2000; Zhou et al., 2004). Last, the CXCR4 receptor and its ligand CXCL12 (Sdf-1) form a chemokine signaling pathway required for dentate progenitor differentiation and migration (Bagri et al., 2002; Lu et al., 2002).
The hippocampus also contains a population of GABAergic interneurons that are derived from subpallial progenitors in the ganglionic eminences of the ventral telencephalon. These inhibitory interneurons follow a tangential migration route before reaching the cortex and hippocampus (Pleasure et al., 2000b). They are thought to modulate the excitability of principal neurons, and defective GABAergic transmission is believed to contribute to cognitive disabilities and epilepsy. Dlx1, Dlx2, Lhx6, and Nkx2.1 deficiency can inhibit the migration of GABAergic interneurons from the medial ganglionic eminence (MGE) to the neocortex and hippocampus (Pleasure et al., 2000b; Cobos et al., 2005; Liodis et al., 2007).
ATRX gene mutations in humans cause the alpha thalassemia mental retardation, X-linked (ATR-X) syndrome and other X-linked mental retardation syndromes (Gibbons et al., 1995; Villard et al., 1996, 2000; Abidi et al., 1999; Lossi et al., 1999; Liu et al., 2002). Decreased ATRX activity gives rise to severe learning disabilities, indicating a vital role for the development of brain regions involved in learning and memory processing. We previously reported that mice with ATRX deficiency in the forebrain are born with a hypocellular cerebral cortex and hippocampus and fail to form a normal dentate gyrus (Bérubé et al., 2005). More recently, we identified ATRX as an important regulator of mitosis and found evidence of chromosome mis-segregation in ATRX-null mitotic neuroprogenitors in the developing telencephalon (Bérubé et al., 2000; Ritchie et al., 2008). In the present study, we show that Atrx deficiency results in loss of dentate granule and GABAergic neuron precursors combined with incomplete migration to their appropriate target sites. Neuronal cell death was accompanied by activation of the p53 pathway and removal of the p53 tumor suppressor protein rescued apoptotic cell death and but was not sufficient to allow complete recovery of the defects at birth.
Materials and Methods
Mouse husbandry, genotyping and tissue preparation.
The AtrxFl/Fl mice have been described previously (Bérubé et al., 2005; Garrick et al., 2006) AtrxFl/Fl mice, when mated to mice expressing Cre recombinase under the control of the Foxg1 promoter (Foxg1KiCre/+) (Hébert and McConnell, 2000), result in male progeny (AtrxFl/YFoxg1KiCre/+) with Atrx-deficiency in the forebrain (AtrxFoxg1Cre). Trp53tm1Tyj/J mice were obtained from Jackson Laboratories and the mutant allele introduced into the AtrxFl and the Foxg1KiCre/+ mice. Subsequent matings of this progeny yielded mice that are p53-null in all tissues and Atrx/p53 dKO in the forebrain. Atrx and Cre genotyping was performed by PCR as previously described (Bérubé et al., 2005). Yolk sac or tail DNA was used for p53 genotyping. One set of primers (5′-GATCGGCCATTGAACAAGAT-3′ and 5′-ATA GGT CGG CGG TTC AT-3′) amplified a 1.5 kb fragment of neo gene, whereas the other set (5′-CCC GAG TAT CTG GAA GAC AG-3′ and 5′-ATA GGT CGG CGG TTC AT-3′) amplified a 600 bp fragment of wild type. PCR conditions were as follows: 95°C for 3 min (95°C for 30 s, 55°C for 30 s, and 72°C for 1 min) × 35, 72°C for 5 min.
BrdU labeling.
Pregnant homozygous AtrxFl/Fl female mice were injected intraperitoneally with 50 μg BrdU per gram body weight (Sigma-Aldrich). Animals were killed after 1 h by cervical dislocation and the embryos were recovered in ice-cold PBS, pH 7.4, and were fixed in 4% paraformaldehyde. Tissue was equilibrated in 30% sucrose-PBS, and frozen in 1:1 30% sucrose-PBS:O.C.T. (Tissue Tek). Sections (10 μm) were treated with 2N HCl to denature the DNA, neutralized with 0.1 m Na2B4O7, pH 8.5, incubated with the primary antibodies overnight at 4°C, washed in PBS, and incubated with the secondary antibodies for 1 h. Sections were counterstained with DAPI (Invitrogen) and mounted in Vectashield (Vector Laboratories). Four adjacent areas covering the hem were examined in two different control/AtrxFoxg1Cre littermate pairs and the percentage of cells that incorporated BrdU was assessed.
Histology and immunostaining.
For histological studies, slides were stained with hematoxylin and eosin (H&E). For immunofluorescence staining, sections were incubated with the primary antibody overnight at 4°C, washed for 1 h in PBS, and incubated with the secondary antibody for 1 h. Sections were counterstained with DAPI (Sigma; D9542) and mounted in Vectashield (Vector Laboratories). Primary antibodies used were as follows: anti-ATRX (1:100; Santa Cruz Biotechnology) anti-BrdU (1:100; Santa Cruz Biotechnology), RSC2 (1:20; Hybridoma bank), cleaved caspase 3 - Asp175 (1:100; Cell Signaling Technology; #9661), anti-Prox1 (1:100; Covance; PRB-238C), anti-neuropeptide Y (1:2000; Millipore; AB1583). Secondary antibodies used were as follows: goat-anti rabbit Alexa 594 (1:1500; Invitrogen) and goat anti-mouse Alexa 488 (1:1500; Invitrogen). Volocity software (PerkinElmer) was used to quantify distance and area measurements.
In situ hybridization.
For developmental studies, midday of the day of vaginal plug discovery was considered E0.5. At scheduled times, pregnant females were anesthetized by CO2 and killed by cervical dislocation. Embryos were fixed overnight in 4% paraformaldehyde, equilibrated in 30% sucrose-PBS, and frozen in 1:1 30% sucrose-PBS: O.C.T. (O.C.T., Tissue Tek). Sections (10 μm) were incubated with a DIG-labeled RNA antisense probe (1:1000 in hybridization buffer) overnight at 65°C, washed in 1× SSC/50% formamide/0.1% Tween 20 3 × 30 min at 65°C then MABT for 30 min at room temperature. Sections were blocked with 20% heat-inactivated sheep serum/2% blocking reagent/1× MABT for 1 h then incubated with anti-DIG antibody (Roche; 1:1500) overnight. Sections were washed with MABT 4 × 20 min, prestained with 100 mm NaCl/50 mm MgCl2/100 mm Tris pH 9.4/0.1% Tween 20 for 2 × 10 min, then stained with NBT and BCIP (Roche; 4.5 μl/ml and 3.5 μl/ml, respectively, in prestaining buffer) overnight. Sections were washed with PBS 3 × 15 min, and mounted with VectaMount (Vector Laboratories). Source of plasmids used for making gene-specific antisense probes are listed in supplemental Table 1, available at www.jneurosci.org as supplemental material.
Real-time PCR.
Total RNA was isolated using the RNeasy Mini kit (QIAGEN). First-strand cDNA was synthesized from 3 μg of total RNA using the SuperScriptTM II Reverse Transcriptase kit (Invitrogen) with 25 mm dNTPs (GE Healthcare), porcine RNAguard (GE Healthcare) and random primers (GE Healthcare). PCRs were performed on a Chromo4 Continuous Fluorescence Detector (Bio-Rad) in the presence of iQ SYBR Green supermix (Bio-Rad) and recorded using the Opticon Monitor 3 software (Bio-Rad). Results were normalized to β-actin expression and relative gene expression levels were calculated using GeneX software (Bio-Rad). Samples were amplified as follows: 95°C for 10 s, annealed for 20 s, 72°C for 30 s (see supplemental Table 2 for primer sequences, available at www.jneurosci.org as supplemental material). After amplification, a melting curve was generated, and samples were run on a 1.5% agarose gel (75V for 1 h) to visualize amplicon purity. Standard curves were generated for each primer pair using fivefold serial dilutions of control cDNA. Primer efficiency was calculated as E = [10(−1/slope) − 1]*100, where a desirable slope is −3.32 and r2>0.99.
p53-mediated signaling pathways in cortical progenitor cultures.
Cortical progenitor cells were cultured as described previously (Callaghan et al., 1999). Briefly, cortices were dissected from AtrxFoxg1Cre mutant and littermate control embryos at E13.5. Cortices were mechanically dissociated by trituration, and cell aggregates were plated on polyornithine-coated 96-well dishes and cultured in media containing Neurobasal medium (Invitrogen), 0.5 mm glutamine, 0.5% penicillin-streptomycin, 1% N2 supplement (Invitrogen) and 10 ng/ml FGF-2 (Sigma). After 48 h, cells were transfected with an inducible p53-responsive firefly luciferase reporter and constitutively expressing Renilla construct with appropriate controls of the p53 Cignal Reporter Assay kit, as described by the manufacturer (SABiociences). One day after transfection, the activation of p53-dependent pathways was measured using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's instructions.
Results
Atrx expression in the developing telencephalon and hippocampus
To gain insight into the role of ATRX in the developing telencephalon, we first determined its pattern of expression in the developing forebrain by immunofluorescence. ATRX is present in many areas of the neuroepithelium at E13.5 (Fig. 1A–D). The protein is enriched on condensed mitotic chromosomes in precursors that line the lateral ventricle (LV) (Fig. 1E). It is also expressed in cycling cells of the ventricular zone (VZ), at densely stained nuclear DAPI+ bundles characteristic of pericentromeric heterochromatin (Fig. 1E). Atrx becomes highly expressed in the first differentiating cortical neurons at E13.5 (Fig. 1A, arrows), as we had previously described (Bérubé et al., 2005). At later stages (E16.5 and P0.5) it is widespread throughout the hippocampus, with staining in the CA1 and CA3 fields, the dentate gyrus and the subiculum, the transition zone between neocortex and hippocampus (Fig. 1F,G). The ubiquitous expression of ATRX and characteristic patterns of expression in mitotic precursors, cycling or differentiated cells suggests that ATRX could function in numerous aspects of hippocampal development.
Figure 1.
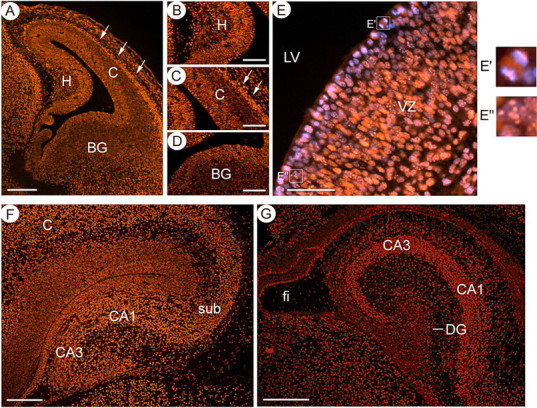
Expression profile of ATRX in the developing hippocampus. A, Immunofluorescence detection of ATRX in the telencephalon at E13.5 reveals that ATRX is expressed in the mitotic and ventricular layers that line the lateral ventricle and is also highly expressed in the differentiated neurons that form the cortical plate (white arrows). Scale bar, 200 μm. Expression is detected in the ventricular zone of the hippocampal primordium (B), the cortex (C) and the basal ganglia (D). Scale bar, 100 μm. E, ATRX immunofluorescence at E13.5 shows that the protein colocalizes with condensed chromosomes in the mitotic layer (E′) and to DAPI-rich heterochromatin bundles in cycling cells of the ventricular zone (E″). Scale bar, 50 μm. F, ATRX is expressed in most cells of the hippocampus at E15.5. Scale bar, 200 μm. G, In the newborn brain, ATRX expression is maintained in the hippocampus in all cell types. Scale bar, 200 μm. H, Hippocampal primordium; C, cortex; BG, basal ganglia; sub, subiculum; fi, fibrium.
Targeted inactivation of Atrx does not affect proliferation in the hippocampal neuroepithelium
To study the role of ATRX in hippocampal development, mice that contain floxed alleles of Atrx were intercrossed to mice that express Cre recombinase under the control of the Forkhead box G1 promoter (Foxg1) (Hébert and McConnell, 2000). Foxg1Cre expression starting at E8.5 induces nearly complete recombination of the Atrx floxed allele in the forebrain (Bérubé et al., 2005). There is a significant reduction in the size of the hippocampus including the subiculum, in the AtrxFoxg1Cre mice. A remarkable outcome of ATRX-deficiency in the hippocampus is the loss of granule neurons of the dentate gyrus that are replaced by a few disorganized cells (Bérubé et al., 2005). To establish whether the reduction of the precursor pool in the hippocampal primordium was due to a problem in cell proliferation early in hippocampal development, we performed acute BrdU labeling at E13.5 and assessed the proportion of BrdU+ cells in the hippocampal hem by immunostaining (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The percentage of cells in S phase was not changed in mutant embryos (28.2%) compared with controls (27.2%) in the cortical hem (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). These results confirmed that precursor proliferation is maintained in the Atrx-null hippocampal neuroepithelium, similar to what we had previously observed in the cortical neuroepithelium (Bérubé et al., 2005).
Incomplete migration of Atrx-null NeuroD1+ dentate precursors
To further investigate the hippocampal phenotypes, we evaluated the ability of Atrx-null granule neurons to differentiate and migrate normally. To determine whether granule cells could commit to the granule cell fate and attain a mature differentiated phenotype, we examined the expression of Prox1, a marker of committed dentate granule cells (Galceran et al., 2000; Liu et al., 2000a; Elliott et al., 2001; Lu et al., 2002; Oldekamp et al., 2004) and the NeuroD-related factor (NDRF), a marker of terminal differentiation (Ohtsuka et al., 1998; Liao et al., 1999; Oda et al., 2000). In the Atrx-null hippocampus, Prox1 RNA and protein was not detectable at E16.5 and at birth (Fig. 2) but remained high in the thalamus, a region that is still expressing Atrx in the AtrxFoxg1Cre mice (data not shown). NDRF expression was decreased at E16.5 and in the newborn hippocampus (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), suggesting that precursors can only achieve early stages of differentiation and never attain a full granule cell phenotype. We also examined the expression of NeuroD, an early differentiation marker expressed in postmitotic immature neurons (Liu et al., 2000a,b). At E13.5, we observed reduced expression of NeuroD in the hippocampal primordium of AtrxFoxg1Cre mutant mice (Fig. 3A–D). Conversely, NeuroD was highly expressed in the Atrx-null dentate precursors at E16.5, but with evidence of defective tangential migration toward the dentate anlage compared with controls (Fig. 3E,F). In newborn pups, we detected very high levels of NeuroD expression in the AtrxFoxg1Cre hippocampus. However, there were fewer NeuroD positive cells and they were abnormally localized within the dentate subventricular zone and the migratory stream rather than the dentate granule cell layer (Fig. 3G,H). Therefore, fewer NeuroD-expressing granule cells are produced and lack the capacity to migrate normally to the site of the dentate gyrus, indicating that the dentate granule cell differentiation program is delayed. Together, these results show that ATRX deficiency reduces the pool of granule cell precursors and prevents migration and full commitment to the dentate granule cell fate.
Figure 2.
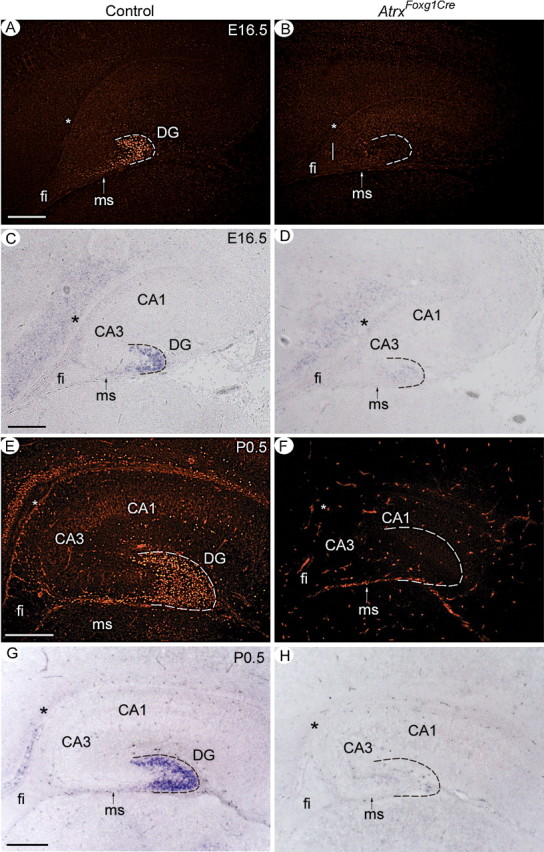
The granule cell marker Prox1 is not expressed in the ATRX-null hippocampus. Prox1 protein and transcript levels were analyzed by immunofluorescence (A, B, E, F) and by in situ hybridization (C, D, G, H), respectively. Prox1 expression was observed as early as E15.5 in the control hippocampus and can be detected in a subset of migrating precursors in the migratory stream and in differentiated granule cells that form the dentate gyrus at E16.5 and P0.5. Prox1 transcripts and protein were not detected in the ATRX-null hippocampus (AtrxFoxg1Cre). ms, Migratory stream; fi, fibrium. Asterisks indicate the site of the dentate notch. Hatched lines outline the shape of the dentate gyrus in control sections and its expected location in the AtrxFoxg1Cre sections. Scale bars, 200 μm.
Figure 3.
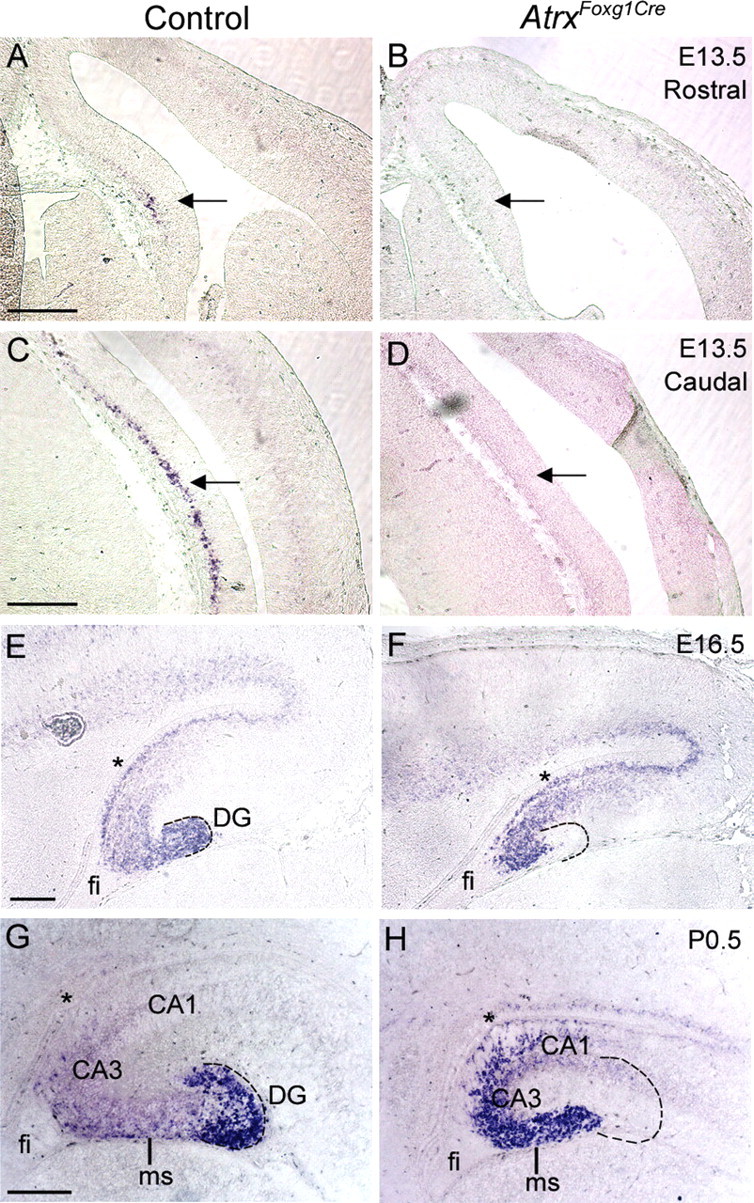
Delayed differentiation and incomplete migration of ATRX-null dentate precursors. In situ hybridization of control and AtrxFoxg1Cre sections using a DIG-labeled antisense probe specific for NeuroD1, a marker of dentate granule cell differentiation. NeuroD1 expression was detectable in the caudal and rostral hippocampal hem in control, but not AtrxFoxg1Cre sections at E13.5 (A–D, arrows). Precursor cells in both the control and ATRX-null hippocampus express NeuroD1 at E16.5 (E, F) and at P0.5 (G, H) but have not migrated to the site of the DG in the ATRX-deficient hippocampus. fi, Fibrium; ms, migratory stream. Asterisks indicate the site of the dentate notch. Hatched lines outline the shape of the dentate gyrus in control sections and its expected location in the AtrxFoxg1Cre sections. Scale bars, 200 μm.
BrdU birthdating experiments previously showed that only a subset of mitotic cells born at E15.5 in the dentate ventricular zone eventually reach the dentate gyrus (Bérubé et al., 2005). These results, combined with our present observation of NeuroD+ precursors arrested in the migratory stream, suggest that the majority of granule cell progenitors born at E15.5 are unable to reach their destination in the dentate gyrus. We examined whether dentate precursors displayed problems in migration at this developmental time. We observed extensive Cre recombinase expression of AtrxFoxg1cre mice but not control mice at this time point, as expected (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). A reduction in hippocampal size was already apparent from the pattern of Tbr1 gene expression at this stage (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Migrating precursors of dentate granule cells are characterized by the expression of basic helix-loop-helix (bHLH) transcription factors. We observed that at E15.5, Hes5, Ngn2 and Id2-positive precursors have not migrated tangentially into the migratory stream in AtrxFoxg1Cre mutants to the same extent as what is observed in controls, and the precursor pool was smaller within the secondary proliferative zone at this stage (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). We further examined Ngn2 expression 1 d later at E16.5 when more precursors should have entered the migratory path. A striking difference in the number of Ngn2+ cells was observed in the Atrx-null hippocampus, with fewer positive cells having migrated toward the future dentate gyrus (Fig. 4). Lef1 also marks migrating precursors at E16.5 and its expression in the migratory stream was decreased in AtrxFoxg1Cre mutants by in situ hybridization (Fig. 4). Math3 has been implicated in the molecular cascade of events that characterize dentate precursors as they exit the cell cycle and start migrating toward the future dentate gyrus (Pleasure et al., 2000a). We found that Math3 staining was mostly restricted to dentate precursors near the dentate notch at E16.5 and that fewer Math3+ progenitor cells were present in this region in the Atrx-null hippocampus (Fig. 4). These results indicate that the precursor pool is reduced and that fewer precursors populate the migratory stream. Cajal-Retzius cells direct the migration of hippocampal and dentate granule cells (Del Río et al., 1997; Nakajima et al., 1997). Reelin is an extracellular protein expressed by Cajal-Retzius cells that is involved in the radial migration in the cortex and hippocampus (Ogawa et al., 1995). Analysis of Reelin expression in the AtrxFoxg1Cre and control forebrain showed a normal localization in the marginal zone of the cortex and hippocampus at E15.5 (supplemental Fig. 4, available at www.jneurosci.org as supplemental material), ruling it out as a possible cause defective migration.
Figure 4.
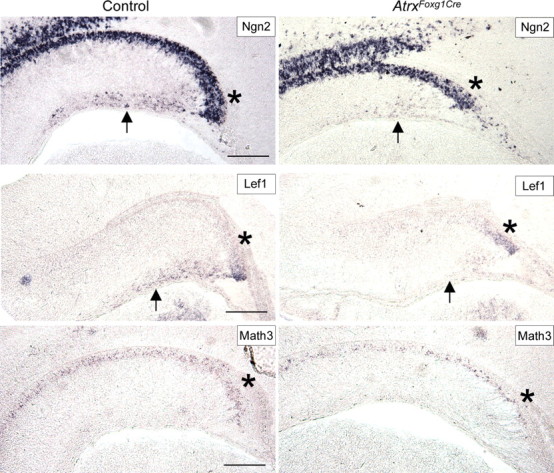
Fewer precursor cells are present in the dentate neuroepithelium and the migratory stream at E16.5. Expression of Ngn2, Lef1 and Math3 that mark granule precursors was analyzed in control and AtrxFoxg1Cre hippocampus at E16.5 by in situ hybridization using specific DIG-labeled antisense probes. Fewer precursor cells are localized at the dentate notch (indicated by asterisks) and fewer Ngn2+ and Lef1+ precursors are present in the migratory stream at E16.5 (indicated by arrows). Scale bar, 200 μm.
ATRX deficiency in the forebrain results in reduction of GABAergic markers
The majority of GABA-producing neurons in the hippocampus originate in the MGE and lateral ganglionic eminence. They represent another group of cells that migrate in a tangential manner through the developing cortical neuroepithelium to eventually settle within the hippocampus(Pleasure et al., 2000b). We detected decreased expression levels of neuropeptides produced by GABAergic neurons in the AtrxFoxg1Cre embryos when compared with controls. Using quantitative real-time PCR, lower levels of somatostatin and neuropeptide Y transcripts at E13.5 and of neuropeptide Y, somatostatin and cholecystokinin at P0.5 were detected in the AtrxFoxg1Cre forebrain compared with control tissue (Fig. 5A). In addition, we tested the expression levels of other GABAergic markers and showed that Dlx5, Gbx2 and Lhx6 all exhibited decreased expression levels in the developing AtrxFoxg1Cre telencephalon, suggesting the loss of GABAergic neurons. Decreased levels of neuropeptide Y were also confirmed by immunofluorescence staining in the ventral telencephalon at E13.5 and at E16.5 (Fig. 5B). Quantification of Npy staining per area (μm2) in the basal ganglia at E16.5 confirmed a decrease in Npy expression in the AtrxFoxg1Cre compared with wild type embryos (60.2 ± SE 45.5 vs 537.7 ± SE 93.9). These results suggest that loss of ATRX in the developing forebrain leads to a loss of a subset of GABAergic neurons in the developing forebrain.
Figure 5.

Reduced expression of GABAergic markers in the ATRX-null forebrain. A, Real-time reverse transcriptase PCR of GABAergic markers in forebrain tissue isolated from E13.5 and P0.5 control and AtrxFoxg1Cre littermate embryos (n = 3). B, Immunofluorescence detection of Npy in the ventral telencephalon at E13.5 and E16.5 shows reduced staining levels in the ATRX-null embryos. Npy, Neuropeptide Y; Sst, somatostatin; Dlx5, distal-less homeobox 5; Gbx2, gastrulation brain homeobox 2; Lhx6, LIM homeobox protein 6; Cck, cholecystokinin. E13.5: Scale bar, 200 μm; E16.5: Scale bar, 150 μm.
p53-dependent cell death in the Atrx-null telencephalon
The hippocampal hem of AtrxFoxg1Cre embryos is characterized by the presence of pyknotic cells often grouped in clusters (Bérubé et al., 2005) (Fig. 6A). We had previously shown increased terminal dUTP nick end labeling staining in the hippocampal hem and cortical neuroepithelium in the AtrxFoxg1cre embryos (Bérubé et al., 2005). The p53 protein is a well known mediator of apoptotic cell death, and we therefore assessed if progenitor cell apoptosis in the Atrx-null hippocampus and ventral telencephalon was mediated via p53. We tested this hypothesis in primary cortical neuroprogenitor cultures by assessing the activation of the p53 signal transduction pathways, using and inducible p53 Cignal Reporter Assay system (SABiosciences). Cortical neuroprogenitor cultures were established from AtrxFoxg1Cre and Cre+ control E13.5 telencephalon, and transfected with the reporter and control plasmids. P53 signaling activity was measured using the dual luciferase assay. The results demonstrated increased activation of p53 pathways in the ATRX-null cortical progenitors (supplemental Fig. 5A, available at www.jneurosci.org as supplemental material). Increased progenitor cell death was accompanied by higher expression levels of cyclin G1 and p21, known targets of the p53 tumor suppressor gene (supplemental Fig. 5B, available at www.jneurosci.org as supplemental material).
Figure 6.
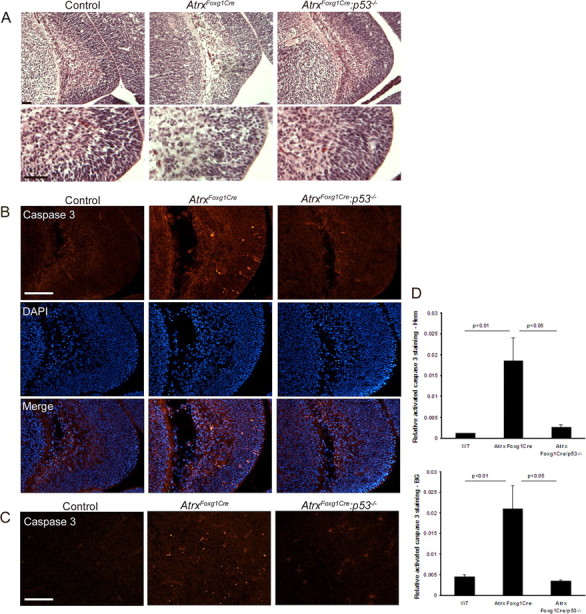
Rescue of neuronal apoptosis in the ATRX-null telencephalon by loss of p53. A, H&E staining of the hippocampal hem at E13.5 reveals pyknotic clusters in the AtrxFoxg1Cre embryos but not in control and AtrxFoxg1Cre:p53−/− embryos. Bottom micrographs show higher magnification of the regions boxed in the top micrographs. Scale bar, 50 μm. B, Immunofluorescence detection of activated caspase 3, a marker of apoptotic cell death, in the hippocampal hem at E13.5. DAPI counterstaining of the sections outlines the nuclei. Apoptotic cells are more frequently observed in AtrxFoxg1Cre compared with control and AtrxFoxg1Cre:p53−/− sections. Scale bar, 100 μm. C, Activated caspase 3 immunofluorescence detection in the basal telencephalon at E13.5 shows increased levels of apoptosis in the AtrxFoxg1Cre compared with control and AtrxFoxg1Cre:p53−/− embryos. Scale bar, 100 μm. D, Quantification of activated caspase 3 staining in the hippocampal hem and in the basal ganglia confirms a statistically significant difference in cell death in the AtrxFoxg1Cre:p53−/− embryos compared with AtrxFoxg1Cre embryos.
To further investigate the involvement of p53 in the apoptotic process, we introduced a mutant p53 allele (Trp53tm1Tyj) in the AtrxloxP and Foxg1Cre mice to generate AtrxFoxg1Cre/p53−/− mice that lack Atrx and p53 expression in the forebrain. Histological (H&E) staining and cleaved caspase 3 staining of the hippocampal hem at E13.5 showed higher levels of pyknotic clusters and increased apoptotic cell death in the AtrxFoxg1cre telencephalon, respectively, that was rescued in the AtrxFoxg1Cre/p53−/− embryos (Fig. 6A–D). These findings show that induction of programmed cell death by deletion of Atrx is dependent on the presence of the p53 protein.
The elevated levels of progenitor apoptosis that characterize the AtrxFoxg1Cre brain at E13.5 results in distinct forebrain size reduction at E16.5 (Fig. 7A,B). To determine whether the increased neuronal survival observed in the AtrxFoxg1Cre/p53−/− at E13.5 resulted in an increase in hippocampal and cortical size, we compared sections from control and AtrxFoxg1Cre/p53−/− or AtrxFoxg1Cre E16.5 embryos (Fig. 7A,B). Most of the measured parameters revealed no significant recovery of cortical or hippocampal size. However, partial recovery of medial cortical size was suggested by the decreased distance between the left and right cingulate cortex in the AtrxFoxg1Cre:p53−/− compound mutants compared with the AtrxFoxg1Cre embryos (Fig. 7A,C, asterisk).
Figure 7.

Minimal size recovery of the E16.5 Atrx-null forebrain in the absence of p53. A, H&E sections of E16.5 forebrain shows that loss of p53 does not ameliorate forebrain size, except in the caudal-medial area (indicated by *). Scale bar, 500 μm. B, Measurements of cortical plate thickness, brain height and hippocampal area confirm size reductions in the AtrxFoxg1Cre embryos but show no significant amelioration in the AtrxFoxg1Cre:p53−/− embryos. C, Partial recovery of medial cortical size is suggested by the decreased distance between the left and right cingulate cortex in the AtrxFoxg1Cre:p53−/− compound mutants compared with the AtrxFoxg1Cre embryos (* in A). H, Hippocampus; Cx, cortex. n = 3 for all genotypes except for AtrxFoxg1Cre:p53+/+ where n = 2. All distances are measured in micrometers unless otherwise indicated.
We next examined embryos at E18.5 and found that loss of p53 partially rescued the defects seen in the AtrxFoxg1Cre embryos (Fig. 8A,B). Quantification of size differences shows gradual amelioration from the Atrx-null, to the p53 heterozygous and p53 homozygous embryos, demonstrating that even a partial increase in p53 levels can rescue ATRX-null defects. While the size of the cortex was largely recovered in the compound mutants, the hippocampal area and the dentate gyrus were only minimally rescued in E18.5 and P0.5 AtrxFoxg1Cre/p53−/− mice (Fig. 8A–C). Closer investigation of AtrxFoxg1Cre/p53−/− sections revealed that more cells accumulate in the dentate neuroepithelium compared with controls (Fig. 8C, bottom), indicating that some of the granule precursor cells rescued by p53 deletion are still unable to migrate normally toward the site of the dentate gyrus. Overall, our findings suggest that while p53 loss of function can prevent neuroprogenitor cell death, it cannot rescue all aspects of ATRX deficiency in the developing forebrain.
Figure 8.
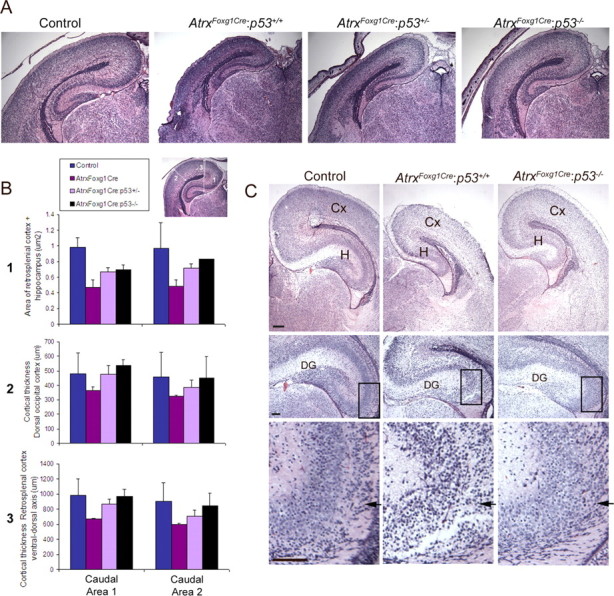
Partial rescue of ATRX-null forebrain size upon loss of p53 at E18.5 and P0.5. A, H&E sections of E18.5 forebrain showing partial size recovery of the hippocampus and cortex in the AtrxFoxg1Cre:p53+/− and AtrxFoxg1Cre:p53−/− compared with AtrxFoxg1Cre embryos. B, Size differences were measured as indicated in the inset (parameters 1–3) in two different caudal areas of the forebrain (C) Partial forebrain size recovery was also observed at P0.5 (top). Higher magnification micrographs show minimal rescue of cellularity in the dentate gyrus in the AtrxFoxg1Cre:p53−/− compound mutants (middle), and accumulation of dentate precursors in the dentate neuroepithelium (bottom, arrows). Scale bar, 100 μm. H, Hippocampus; Cx, cortex.
Discussion
The molecular events that guide the development of the hippocampus are not well understood. We had previously reported that the loss of Atrx expression during early mouse brain development resulted in reduced hippocampal and cortical size and the loss of the dentate gyrus (Bérubé et al., 2005). Our understanding of the expression and function of this chromatin regulator during hippocampal development is limited and we therefore investigated some of the defects that characterize the Atrx-null hippocampus. We found that the ATRX protein is present at all stages of hippocampal development. ATRX protein was restricted to the nuclear compartment with marked enrichment at pericentromeric heterochromatin in both mitotic and interphasic cells. We determined that the progenitor pool in the dentate neuroepithelium is reduced but that this reduction is likely not a result of decreased proliferation but rather increased programmed cell death in the hippocampal primordium and the ventral telencephalon, which is the source of hippocampal GABAergic interneurons. Surviving dentate precursors still express many of the bHLH gene markers that typically characterize migrating precursors as well as the early differentiation marker NeuroD, but incomplete tangential migration was observed as early as E15.5. This lack of migration to the dentate hylus was most apparent at birth, where the precursors expressed high levels of NeuroD but remained completely constrained within the migratory stream whereas the majority of NeuroD+ cells had already reached the dentate gyrus in control embryos. Although these results implicate ATRX in the migration of precursor cells, the lack of Prox1 RNA and protein expression further implies an inability to commit to the dentate granule cell fate.
The majority of neuronal apoptosis observed in the hippocampal primordium and basal ganglia at E13.5 was rescued by loss of p53, demonstrating that Atrx-null neuroprogenitors undergo apoptosis that is mediated by the p53 pathway. An interesting outcome is that despite general rescue of neuronal apoptosis at E13.5, brain size was only partially regained at E18.5 and at birth, suggesting that defects observed in the Atrx-null hippocampus and cortex do not result solely from neuronal cell death. The accumulated pool of dentate precursors in the AtrxFoxg1Cre:p53−/− hippocampus in the newborn forebrain was more suggestive of a migration defect. We speculate that p53 deficiency reduces the amount of apoptosis, but that granule cell precursors are abnormal and fail to migrate properly and completely toward the dentate gyrus.
Normal hippocampal function is crucial for learning and memory processing and, not surprisingly, abnormalities are associated with epilepsy (Sloviter, 1994; Nadler, 2003), learning disorders (Reeves et al., 1995) and mental retardation (Galdzicki et al., 2001; Castrén et al., 2002; Huber et al., 2002; Weeber et al., 2003). The abnormalities that characterize the Atrx conditional-null forebrain demonstrate that ATRX function is required for the normal development of the hippocampus and suggests that hippocampal abnormalities could contribute to learning deficiencies and increased incidence of seizures experienced by a subset of ATR-X patients. Defects in GABAergic function have also been associated with epilepsy and mental retardation in humans. We provide the first evidence that ATRX is expressed in GABAergic neurons in the basal forebrain (Fig. 1A,D) and show data that supports a role in GABAergic neuron production or function, which might contribute to the seizures in ATR-X and related syndromes. Interestingly, ATRX transgenic mice were observed to have spontaneous seizures (Bérubé et al., 2002), and we have observed the same phenomenon in a subset of surviving AtrxFog1Cre heterozygous females (our unpublished data). However, the abnormalities in AtrxFoxg1Cre mice are expected to be more severe than in the human syndrome, since human ATRX human mutations identified thus far are never null. Further experimentation will be required to understand the pathogenesis of cognitive delay and epilepsy in ATR-X syndrome.
A likely molecular explanation for the loss of dentate granule or GABAergic neurons is that ATRX uses ATP-dependent chromatin remodeling activity or alters DNA methylation to regulate gene expression. However, there is mounting evidence that ATRX also controls chromosome dynamics in dividing cells. We recently reported that ATRX is an important regulator of mitotic events in human somatic cells (Ritchie et al., 2008). It is therefore possible that M phase abnormalities in granule or GABAergic progenitors cause death or alter the timing of differentiation resulting in subsequent problems in fate commitment and migration.
Collectively, our findings support the idea that depletion of dentate granule cells within the dentate neuroepithelium and of GABAergic neurons from the basal ganglia is the outcome of increased p53-dependent apoptosis but perhaps also results from incomplete migration of precursor cells. Therefore, ATRX deficiency in the developing telencephalon causes cumulative defects that impinge on precursor cell function as they become committed to the granule or GABAergic fate.
Mutations in several other genes that regulate chromatin structure and organization are associated with cognitive delay. They include the methyl-CpG-binding protein MeCP2 in Rett syndrome (Chen et al., 2001), the histone acetyl transferase Creb-binding protein in Rubinstein-Taybi syndrome (Petrij et al., 1995; Kalkhoven et al., 2003), the histone H3 serine/threonine kinase RSK2 in Coffin-Lowry syndrome (Jacquot et al., 1998; Hanauer and Young, 2002), the SWI/SNF-related ATPase ERCC6 in Cockayne syndrome (Troelstra et al., 1992; Licht et al., 2003) and the DNA methyltransferase DNMT3B in ICF (Immunodeficiency, Centromere instability, Facial anomaly) syndrome. These examples demonstrate that learning disabilities are a common and prominent feature of mutations affecting chromatin dynamics and that chromatin structure plays a key role in neuronal gene regulation and normal brain development and function.
Footnotes
This work was supported by a Canadian Institutes for Health Research (CIHR) operating grant to N.G.B. N.G.B. is a CIHR New Investigator. M.A.L. was supported in part by the Curtis Cadman Foundation.
References
- Abidi F, Schwartz CE, Carpenter NJ, Villard L, Fontés M, Curtis M. Carpenter-Waziri syndrome results from a mutation in XNP. Am J Med Genet. 1999;85:249–251. doi: 10.1002/(sici)1096-8628(19990730)85:3<249::aid-ajmg12>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Altman J, Bayer SA. Migration and distribution of two populations of hippocampal granule cell precursors during the perinatal and postnatal periods. J Comp Neurol. 1990a;301:365–381. doi: 10.1002/cne.903010304. [DOI] [PubMed] [Google Scholar]
- Altman J, Bayer SA. Mosaic organization of the hippocampal neuroepithelium and the multiple germinal sources of dentate granule cells. J Comp Neurol. 1990b;301:325–342. doi: 10.1002/cne.903010302. [DOI] [PubMed] [Google Scholar]
- Bagri A, Gurney T, He X, Zou YR, Littman DR, Tessier-Lavigne M, Pleasure SJ. The chemokine SDF1 regulates migration of dentate granule cells. Development. 2002;129:4249–4260. doi: 10.1242/dev.129.18.4249. [DOI] [PubMed] [Google Scholar]
- Bérubé NG, Smeenk CA, Picketts DJ. Cell cycle-dependant phosphorylation of the ATRX protein correlates with changes in nuclear matrix and chromatin association. Human Molecular Genetics. 2000;9:539–547. doi: 10.1093/hmg/9.4.539. [DOI] [PubMed] [Google Scholar]
- Bérubé NG, Jagla M, Smeenk C, De Repentigny Y, Kothary R, Picketts DJ. Neurodevelopmental defects resulting from ATRX overexpression in transgenic mice. Human Molecular Genetics. 2002;11:253–261. doi: 10.1093/hmg/11.3.253. [DOI] [PubMed] [Google Scholar]
- Bérubé NG, Mangelsdorf M, Jagla M, Vanderluit J, Garrick D, Gibbons RJ, Higgs DR, Slack RS, Picketts DJ. The chromatin-remodelling protein ATRX is critical for neuronal survival during corticogenesis. J Clin Invest. 2005;115:258–267. doi: 10.1172/JCI22329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaghan DA, Dong L, Callaghan SM, Hou YX, Dagnino L, Slack RS. Neural precursor cells differentiating in the absence of Rb exhibit delayed terminal mitosis and deregulated E2F 1 and 3 activity. Dev Biol. 1999;207:257–270. doi: 10.1006/dbio.1998.9162. [DOI] [PubMed] [Google Scholar]
- Castrén M, Lampinen KE, Miettinen R, Koponen E, Sipola I, Bakker CE, Oostra BA, Castrén E. BDNF regulates the expression of fragile X mental retardation protein mRNA in the hippocampus. Neurobiol Dis. 2002;11:221–229. doi: 10.1006/nbdi.2002.0544. [DOI] [PubMed] [Google Scholar]
- Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- Cobos I, Calcagnotto ME, Vilaythong AJ, Thwin MT, Noebels JL, Baraban SC, Rubenstein JL. Mice lacking Dlx1 show subtype-specific loss of interneurons, reduced inhibition and epilepsy. Nat Neurosci. 2005;8:1059–1068. doi: 10.1038/nn1499. [DOI] [PubMed] [Google Scholar]
- Del Río JA, Heimrich B, Borrell V, Förster E, Drakew A, Alcántara S, Nakajima K, Miyata T, Ogawa M, Mikoshiba K, Derer P, Frotscher M, Soriano E. A role for Cajal-Retzius cells and reelin in the development of hippocampal connections. Nature. 1997;385:70–74. doi: 10.1038/385070a0. [DOI] [PubMed] [Google Scholar]
- Elliott RC, Khademi S, Pleasure SJ, Parent JM, Lowenstein DH. Differential regulation of basic helix-loop-helix mRNAs in the dentate gyrus following status epilepticus. Neuroscience. 2001;106:79–88. doi: 10.1016/s0306-4522(01)00198-1. [DOI] [PubMed] [Google Scholar]
- Gage FH, Kempermann G, Palmer TD, Peterson DA, Ray J. Multipotent progenitor cells in the adult dentate gyrus. J Neurobiol. 1998;36:249–266. doi: 10.1002/(sici)1097-4695(199808)36:2<249::aid-neu11>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Galceran J, Miyashita-Lin EM, Devaney E, Rubenstein JL, Grosschedl R. Hippocampus development and generation of dentate gyrus granule cells is regulated by LEF1. Development. 2000;127:469–482. doi: 10.1242/dev.127.3.469. [DOI] [PubMed] [Google Scholar]
- Galdzicki Z, Siarey R, Pearce R, Stoll J, Rapoport SI. On the cause of mental retardation in Down syndrome: extrapolation from full and segmental trisomy 16 mouse models. Brain Res Brain Res Rev. 2001;35:115–145. doi: 10.1016/s0926-6410(00)00074-4. [DOI] [PubMed] [Google Scholar]
- Garrick D, Sharpe JA, Arkell R, Dobbie L, Smith AJH, Wood WG, Higgs DR, Gibbons RJ. Loss of Atrx affects trophoblast development and the pattern of X-inactivation in extraembryonic tissues. PLoS Genet. 2006;2:e58. doi: 10.1371/journal.pgen.0020058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons RJ, Picketts DJ, Villard L, Higgs DR. Mutations in a putative global transcriptional regulator cause X-linked mental retardation with alpha-thalassaemia (ATR-X syndrome) Cell. 1995;80:837–845. doi: 10.1016/0092-8674(95)90287-2. [DOI] [PubMed] [Google Scholar]
- Hanauer A, Young ID. Coffin-Lowry syndrome: clinical and molecular features. J Med Genet. 2002;39:705–713. doi: 10.1136/jmg.39.10.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hébert JM, McConnell SK. Targeting of cre to the Foxg1 (BF-1) locus mediates loxP recombination in the telencephalon and other developing head structures. Dev Biol. 2000;222:296–306. doi: 10.1006/dbio.2000.9732. [DOI] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A. 2002;99:7746–7750. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquot S, Merienne K, De Cesare D, Pannetier S, Mandel JL, Sassone-Corsi P, Hanauer A. Mutation analysis of the RSK2 gene in Coffin-Lowry patients: extensive allelic heterogeneity and a high rate of de novo mutations. Am J Hum Genet. 1998;63:1631–1640. doi: 10.1086/302153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalkhoven E, Roelfsema JH, Teunissen H, den Boer A, Ariyurek Y, Zantema A, Breuning MH, Hennekam RC, Peters DJ. Loss of CBP acetyltransferase activity by PHD finger mutations in Rubinstein-Taybi syndrome. Hum Mol Genet. 2003;12:441–450. doi: 10.1093/hmg/ddg039. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Kuhn HG, Gage FH. Genetic influence on neurogenesis in the dentate gyrus of adult mice. Proc Natl Acad Sci U S A. 1997a;94:10409–10414. doi: 10.1073/pnas.94.19.10409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempermann G, Kuhn HG, Gage FH. More hippocampal neurons in adult mice living in an enriched environment. Nature. 1997b;386:493–495. doi: 10.1038/386493a0. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Brandon EP, Gage FH. Environmental stimulation of 129/SvJ mice causes increased cell proliferation and neurogenesis in the adult dentate gyrus. Curr Biol. 1998;8:939–942. doi: 10.1016/s0960-9822(07)00377-6. [DOI] [PubMed] [Google Scholar]
- Liao J, He J, Yan T, Korzh V, Gong Z. A class of neuroD-related basic helix-loop-helix transcription factors expressed in developing central nervous system in zebrafish. DNA Cell Biol. 1999;18:333–344. doi: 10.1089/104454999315394. [DOI] [PubMed] [Google Scholar]
- Licht CL, Stevnsner T, Bohr VA. Cockayne syndrome group B cellular and biochemical functions. Am J Hum Genet. 2003;73:1217–1239. doi: 10.1086/380399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liodis P, Denaxa M, Grigoriou M, Akufo-Addo C, Yanagawa Y, Pachnis V. Lhx6 activity is required for the normal migration and specification of cortical interneuron subtypes. J Neurosci. 2007;27:3078–3089. doi: 10.1523/JNEUROSCI.3055-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Pleasure SJ, Collins AE, Noebels JL, Naya FJ, Tsai MJ, Lowenstein DH. Loss of BETA2/NeuroD leads to malformation of the dentate gyrus and epilepsy. Proc Natl Acad Sci U S A. 2000a;97:865–870. doi: 10.1073/pnas.97.2.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Pereira FA, Price SD, Chu MJ, Shope C, Himes D, Eatock RA, Brownell WE, Lysakowski A, Tsai MJ. Essential role of BETA2/NeuroD1 in development of the vestibular and auditory systems. Genes Dev. 2000b;14:2839–2854. doi: 10.1101/gad.840500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Gong Y, Chen B, Guo C, Li J, Guo Y. [Linkage analysis of X-linked nuclear protein gene in Smith-Fineman-Myers syndrome] Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2002;19:22–25. [PubMed] [Google Scholar]
- Lossi AM, Millán JM, Villard L, Orellana C, Cardoso C, Prieto F, Fontés M, Martínez F. Mutation of the XNP/ATR-X gene in a family with severe mental retardation, spastic paraplegia and skewed pattern of X inactivation: demonstration that the mutation is involved in the inactivation bias. Am J Hum Genet. 1999;65:558–562. doi: 10.1086/302499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Grove EA, Miller RJ. Abnormal development of the hippocampal dentate gyrus in mice lacking the CXCR4 chemokine receptor. Proc Natl Acad Sci U S A. 2002;99:7090–7095. doi: 10.1073/pnas.092013799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadler JV. The recurrent mossy fiber pathway of the epileptic brain. Neurochem Res. 2003;28:1649–1658. doi: 10.1023/a:1026004904199. [DOI] [PubMed] [Google Scholar]
- Nakajima K, Mikoshiba K, Miyata T, Kudo C, Ogawa M. Disruption of hippocampal development in vivo by CR-50 mAb against reelin. Proc Natl Acad Sci U S A. 1997;94:8196–8201. doi: 10.1073/pnas.94.15.8196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda H, Iwata I, Yasunami M, Ohkubo H. Structure of the mouse NDRF gene and its regulation during neuronal differentiation of P19 cells. Brain Res Mol Brain Res. 2000;77:37–46. doi: 10.1016/s0169-328x(00)00038-3. [DOI] [PubMed] [Google Scholar]
- Ogawa M, Miyata T, Nakajima K, Yagyu K, Seike M, Ikenaka K, Yamamoto H, Mikoshiba K. The reeler gene-associated antigen on Cajal-Retzius neurons is a crucial molecule for laminar organization of cortical neurons. Neuron. 1995;14:899–912. doi: 10.1016/0896-6273(95)90329-1. [DOI] [PubMed] [Google Scholar]
- Ohtsuka T, Asahi M, Matsuura N, Kikuchi H, Hojo M, Kageyama R, Ohkubo H, Hoshimaru M. Regulated expression of neurogenic basic helix-loop-helix transcription factors during differentiation of the immortalized neuronal progenitor cell line HC2S2 into neurons. Cell Tissue Res. 1998;293:23–29. doi: 10.1007/s004410051094. [DOI] [PubMed] [Google Scholar]
- Oldekamp J, Kraemer N, Alvarez-Bolado G, Skutella T. bHLH gene expression in the Emx2-deficient dentate gyrus reveals defective granule cells and absence of migrating precursors. Cereb Cortex. 2004;14:1045–1058. doi: 10.1093/cercor/bhh064. [DOI] [PubMed] [Google Scholar]
- Pellegrini M, Mansouri A, Simeone A, Boncinelli E, Gruss P. Dentate gyrus formation requires Emx2. Development. 1996;122:3893–3898. doi: 10.1242/dev.122.12.3893. [DOI] [PubMed] [Google Scholar]
- Petrij F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RC, Masuno M, Tommerup N, van Ommen GJ, Goodman RH, Peters DJ, et al. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376:348–351. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]
- Pleasure SJ, Collins AE, Lowenstein DH. Unique expression patterns of cell fate molecules delineate sequential stages of dentate gyrus development. J Neurosci. 2000a;20:6095–6105. doi: 10.1523/JNEUROSCI.20-16-06095.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleasure SJ, Anderson S, Hevner R, Bagri A, Marin O, Lowenstein DH, Rubenstein JL. Cell migration from the ganglionic eminences is required for the development of hippocampal GABAergic interneurons. Neuron. 2000b;28:727–740. doi: 10.1016/s0896-6273(00)00149-5. [DOI] [PubMed] [Google Scholar]
- Reeves RH, Irving NG, Moran TH, Wohn A, Kitt C, Sisodia SS, Schmidt C, Bronson RT, Davisson MT. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat Genet. 1995;11:177–184. doi: 10.1038/ng1095-177. [DOI] [PubMed] [Google Scholar]
- Ritchie K, Seah C, Moulin J, Isaac C, Dick F, Bérubé NG. Loss of ATRX leads to chromosome cohesion and congression defects. J Cell Biol. 2008;180:315–324. doi: 10.1083/jcb.200706083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab MH, Bartholomae A, Heimrich B, Feldmeyer D, Druffel-Augustin S, Goebbels S, Naya FJ, Zhao S, Frotscher M, Tsai MJ, Nave KA. Neuronal basic helix-loop-helix proteins (NEX and BETA2/Neuro D) regulate terminal granule cell differentiation in the hippocampus. J Neurosci. 2000;20:3714–3724. doi: 10.1523/JNEUROSCI.20-10-03714.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloviter RS. The functional organization of the hippocampal dentate gyrus and its relevance to the pathogenesis of temporal lobe epilepsy. Ann Neurol. 1994;35:640–654. doi: 10.1002/ana.410350604. [DOI] [PubMed] [Google Scholar]
- Troelstra C, van Gool A, de Wit J, Vermeulen W, Bootsma D, Hoeijmakers JH. ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne's syndrome and preferential repair of active genes. Cell. 1992;71:939–953. doi: 10.1016/0092-8674(92)90390-x. [DOI] [PubMed] [Google Scholar]
- Villard L, Fontès M, Adès LC, Gecz J. Identification of a mutation in the XNP/ATR-X gene in a family reported as Smith-Fineman-Myers syndrome. Am J Med Genet. 2000;91:83–85. [PubMed] [Google Scholar]
- Villard L, Gecz J, Mattéi JF, Fontés M, Saugier-Veber P, Munnich A, Lyonnet S. XNP mutation in a large family with Juberg-Marsidi syndrome. Nat Genet. 1996;12:359–360. doi: 10.1038/ng0496-359. [DOI] [PubMed] [Google Scholar]
- Weeber EJ, Jiang YH, Elgersma Y, Varga AW, Carrasquillo Y, Brown SE, Christian JM, Mirnikjoo B, Silva A, Beaudet AL, Sweatt JD. Derangements of hippocampal calcium/calmodulin-dependent protein kinase II in a mouse model for Angelman mental retardation syndrome. J Neurosci. 2003;23:2634–2644. doi: 10.1523/JNEUROSCI.23-07-02634.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou CJ, Zhao C, Pleasure SJ. Wnt signaling mutants have decreased dentate granule cell production and radial glial scaffolding abnormalities. J Neurosci. 2004;24:121–126. doi: 10.1523/JNEUROSCI.4071-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]