

Abstract
Parkinson's disease (PD) is a progressive neurodegenerative disease characterized by the degeneration of the dopaminergic neurons in the substantia nigra pars compacta (SNpc). Activation of the mixed lineage kinase and c-Jun N-terminal kinase (JNK) has been reported in models of PD. Our focus was to discern whether distinct pathways were activated in cell-specific manner within the SNpc. We now demonstrate the selective phosphorylation of p38 MAP kinase within the dopaminergic neurons, whereas JNK activation occurs predominantly in the microglia. p38 activation results in downstream phosphorylation of p53 and increased p53 mediated transcription of Bax and Puma in the ventral midbrain. Treatment with p38 inhibitor, SB239063 protected primary dopaminergic neurons derived from human progenitor cells from MPP+ mediated cell death and prevented the downstream phosphorylation of p53 and its translocation to the nucleus in vivo, in the ventral midbrain. The increased staining of phosphorylated p38 in the surviving neurons of SNpc in human brain sections from patients with PD and in MPTP treated mice but not in the ventral tegmental area provides further evidence suggesting a role for p38 in the degeneration of dopaminergic neurons of SNpc. We thus demonstrate the cell specific activation of MAP kinase pathways within the SNpc after MPTP treatment emphasizing the role of multiple signaling cascades in the pathogenesis and progression of the disease. Selective inhibitors of p38 may therefore, help preserve the surviving neurons in PD and slow down the disease progression.
Keywords: Parkinson's disease, cell death, Bax, oxidative stress, MAP kinase, neurodegeneration
Introduction
Parkinson's disease (PD) is a movement disorder characterized by the selective loss of dopaminergic neurons of substantia nigra pars compacta (SNpc) and their terminals in the striatum. The mechanism underlying the selective vulnerability of SNpc neurons is yet unclear although mitochondrial dysfunction seen as complex I loss is recognized as a key player in the pathogenesis of PD (Mizuno et al., 1995; Olanow, 2007). Dysfunction of complex I seen in brain, muscle and platelets of Parkinson's disease patients (Parker et al., 1989) can generate reactive oxygen species (ROS) leading to substantial oxidative stress (Mancuso et al., 2007).
ROS can activate several signaling pathways in cells including those that regulate cell death. An important signaling pathway that is activated by ROS is the mitogen-activated protein kinase (MAPK) pathway. The family of MAP kinases comprises of extracellular signal-regulated kinases (ERKs) and stress-activated protein kinases (SAPKs), c-Jun N-terminal kinases (JNKs) and p38 kinases. JNK is phosphorylated in models of PD (Klintworth et al., 2007; Saporito et al., 2000) and in the SNpc of PD patients obtained at autopsy (Ferrer et al., 2001) and JNK inhibitors offer neuroprotection in PD models. The downstream events after JNK activation leading to apoptosis could potentially involve both transcriptional and mitochondrial mechanisms. Together, these studies show that JNK directly phosphorylates Bim leading to activation of Bax, which translocates to the mitochondria facilitating cytochrome c release and apoptosis in animal model of PD (Perier et al., 2007). Both transcriptional and post-translational activation of Bax have been observed in SNpc of MPTP treated mice (Duan et al., 2002) and PD patients (Hartmann et al., 2001), and Bax knock-out mice are protected from MPTP toxicity (Vila et al., 2001; Perier et al., 2005). MPTP also activates p53 which induces transcriptional upregulation of Bax and p53 knock-out mice are also protected from MPTP toxicity (Duan et al., 2002; Perier et al., 2007).
In light of these compelling observations, it is indeed surprising that CEP1347, an upstream inhibitor of JNK (which prevents JNK activation through inhibition of mixed lineage kinases, MLK) did not afford any neuroprotection in clinical trials performed in early PD patients (Parkinson Study Group PRECEPT Investigators, 2007) although previous treatment with JNK inhibitor(s) offered neuroprotection in cultured cells (Wang et al., 2004) and animal models of PD (Saporito et al., 1999).
Our focus was to discern whether distinct MAP kinases were activated in a cell-specific manner within the SNpc and identify their contribution to the demise of dopaminergic neurons. We now demonstrate that there is selective phosphorylation of p38 MAP kinase in the SNpc neurons, whereas the activation of JNK occurs predominantly in the microglia. Identification of cell-selective activation of death signaling pathways in distinct cell population could help in the development of specific inhibitors for treatment as disease modifying agents in PD.
Materials and Methods
Materials.
Antibodies to Bax (B-9, monoclonal), p38 (C-20, polyclonal, α p38), pJNK (G-7, monoclonal), and p53 (DO-1, monoclonal) were purchased from Santa Cruz Biotechnology. Antibody to β-tubulin was obtained from Sigma-Aldrich. Antibody to pp53 (Ser15) was obtained from Abcam. Antibodies to pp38 MAPK (Thr180/Tyr182), JNK and lamin A/C were purchased from Cell Signaling Technology. Antibody to tyrosine hydroxylase was obtained from Chemicon. Anti-Histone H3 was obtained from Upstate. Antibodies to neuronal class III β-tubulin (Tuj1) and Iba1 were obtained from Covance and Wako Pure Chemical Industries, respectively. Vectastain-ABC Elite kit was purchased from Vector Labs. p38 inhibitor, SB239063 was obtained from Calbiochem. In situ Cell Death Detection Kit, TMR red kit was purchased from Roche. All other chemicals and reagents were of analytical grade and were obtained from Sigma-Aldrich or Qualigens.
Animals.
All animal experiments were performed as per the institutional guidelines for the use and care of animals. All efforts were made to minimize animal suffering, to reduce the number of animals used and to use alternatives to in vivo techniques if available. Male C57BL6J (2–3 months, 25–30 g) were obtained from Central Animal Research Facility of National Brain Research Centre (NBRC). Male C57BL6J mice were administered MPTP (30 mg/kg body weight/d, s.c.). Control animals received saline alone. Animals were treated with a single dose of MPTP and killed 1, 4, 12, and 24 h later. Some animals also received the above dose of MPTP daily for 8 d and were killed 24 h after the last dose. Animals had access to pelleted diet and water ad libitum. At the end of the experimental period, mice were anesthetized with ether and perfused transcardially with ice-cold normal saline before decapitation. Ventral midbrain and striatum were dissected as described earlier (Karunakaran et al., 2007) and frozen in liquid nitrogen for immunoblotting. In some experiments, animals were perfused transcardially with buffered paraformaldehyde (4% w/v) and the brain was dissected out and processed for immunohistochemistry. For examining the effect of p38 kinase inhibitor in vivo, mice were divided into four groups. Two groups received the vehicle DMSO (3% in normal saline; 100 μl) intrathecally and this was followed by normal saline or MPTP (30 mg/kg body weight; s.c.) 1 h later. The other two groups received SB239063 (92 μg dissolved in 100 μl of 3% DMSO) intrathecally before MPTP or saline treatment. Mice were killed 12 h after MPTP treatment and the ventral midbrain was dissected out.
Processing of tissue.
Tissue was homogenized in 0.25 m sucrose and centrifuged at 1000 × g for 10 min to obtain postnuclear supernatant. The postnuclear supernatant was used for immunoblotting. In some experiments the cytosol was isolated from the postnuclear supernatant by centrifugation at 100,000 × g for 1 h. Protein concentration was estimated by a dye-binding method (Bradford, 1976). Nuclear extracts were prepared as described (Korner et al., 1989).
Human brain samples.
Brain tissue from male traffic accident victims (control) and PD patients were obtained from the Human Brain Tissue Repository of the National Institute of Mental Health and Neurosciences, Bangalore, India, in compliance with the ethical guidelines of the Government of India. Autopsy was performed after obtaining informed consent from the next of kin and the protocol for use of autopsy tissue was cleared by the institutional human ethics committee of National Institute of Mental Health and Neurosciences, Bangalore and National Brain Research Centre, Manesar. After autopsy, brains were washed in ice-cold saline and substantia nigra was dissected out. The substantia nigra was fixed in buffered formalin and processed for paraffin embedding.
Immunohistochemistry.
Male C57BL6J mice were administered MPTP (30 mg/kg body weight/d, s.c.) once daily for 1 or 8 d. Control animals received saline. Animals were anesthetized with ether 24 h after the last injection and perfused transcardially with phosphate buffered saline (PBS) followed by paraformaldehyde (4%, w/v) in PBS. Coronal sections (30 μm thick) were cut throughout the entire midbrain using a cryostat. Immunostaining was visualized using FITC labeled secondary antibody and counterstained with DAPI or HRP labeled secondary antibody followed by staining with Nova Red.
Serial sections (5–7 μm thick) were cut from paraffin embedded autopsy tissue from human brain. Sections were dewaxed, hydrated in graded alcohol and PBS (10 mm, pH 7.4) and processed for immunohistochemical staining in a similar manner as described above. Additional treatment with hydrogen peroxide followed by potassium permanganate/oxalic acid was performed to avoid interference caused by lipofuchsin (Barden, 1984; Schnell et al., 1999). Negative controls, wherein the primary antibody was omitted were run along with the samples and processed and photographed under the same conditions.
Immunoblotting.
The postnuclear supernatant prepared from ventral midbrain or striatum (20 μg of protein) of vehicle and MPTP treated mice were resolved on 10% SDS polyacrylamide gel. Proteins were transferred to nitrocellulose membranes (Towbin et al., 1979), incubated with primary antibody (1:1000) followed by secondary antibody (1:2000) labeled with alkaline phosphatase or secondary antibody (1:5000) labeled with horseradish peroxidase. Immunostained bands were detected using nitroblue tetrazolium and 5-bromo 4-chloro 3-indolyl phosphate as chromogens (Roche) or using chemiluminescence kit (ECL, Amersham Pharmacia Biotech). Blots were normalized with β-tubulin, glyceraldehyde 3-phosphate dehydrogenase or lamin/histone as appropriate.
Quantitative real-time PCR.
Total RNA was isolated from mouse ventral midbrain and cDNA was synthesized from 1 μg of total RNA by using random hexamers. Real-time PCR was performed using Power SYBR Green PCR Master Mix from Applied Biosystems according to the manufacturer's instructions. The sequences of the primers used were: forward 5′-CCACCTGAGTTCGCAGCTCAA-3′, reverse 5′-GTTGAGCACACTCGTCCTTCAA-3′ for Noxa and forward 5′-ACGACCTCAACGCACAGTACG-3′, reverse 5′-TGGGTAAGGGCAGGAGTCC-3′ for Puma. All reactions were performed in triplicate and negative controls contained no template DNA. The PCR conditions were: 95°C for 10 min, followed by 40 cycles of 95°C for 20 s, 67°C for 30 s and 72°C for 40 s. We used 18S rRNA as an endogenous control for normalization. To verify that the primer pair produced only a single product, a dissociation protocol was added after thermocycling, determining dissociation of the PCR products from 60°C to 95°C. Data were analyzed using the comparative threshold cycle (ΔΔCt) method.
Studies with primary human CNS progenitor cells.
Human CNS progenitor cell cultures were prepared from 8- to 12-week-old embryos obtained from elective medical termination of first trimester pregnancies performed at the local hospital after informed consent as per the approved protocol of the institutional human ethics committee in compliance with the recommendations of the Indian Council of Medical Research, Government of India. Primary cultures of human CNS progenitor cells were prepared and cultured as monolayers on poly-d-lysine coated chamber slides in serum-free neurobasal medium and neurons were derived from them as described previously (Mishra et al., 2008). Neuronal differentiation involved changing the growth factors in the progenitor media to brain-derived neurotrophic factor (BDNF, 10 ng/ml) and platelet-derived growth factor (PDGF)-A/B (10 ng/ml) for 3 weeks. Almost 90% cells were positive for neuronal marker, Tuj-1. Cells were seeded in 8 well chamber slides to a total count of 10,000 cells per well and were immunostained for Tuj-1, glial fibrillary acidic protein (GFAP) and tyrosine hydroxylase (TH) after fixation with 4% PFA. Cells were treated with p38/JNK MAP kinase inhibitor (SB239063, 1 μm; SP600125, 5 μm) or p53 inhibitor, pifithrin-α (250 nm) 60 min before treatment with MPP+ (10 μm) or vehicle for 24 h. Cells were then washed with PBS and fixed with 4% paraformaldehyde and immunostained for p53. Finally the cells were mounted in medium containing propidium iodide (PI) and visualized under fluorescence microscope. In some experiments the cell death was assessed using terminal deoxynucleotidyl transferase mediated dUTP-biotin nick end-labeling (TUNEL) method following manufacturer's protocol. The cells were observed using fluorescence microscopy and both the TUNEL positive cells and DAPI stained nuclei were counted in four independent experiments in each case. On average 300 cells were scored in four different fields for each sample.
Statistical analysis.
Statistical analysis of the data were performed using ANOVA or repeated measures of ANOVA followed by post hoc tests (Student–Newman–Keuls or Dunnet's test). Student's t test or paired t test were used when 2 groups were compared. Values of p < 0.05 were taken as being statistically significant.
Results
MPTP stimulates p38 MAPK phosphorylation in substantia nigra neurons
The phospho-MAPK signal was normalized to the total MAPK signal in each sample to assess MAPK activation, as described earlier (Guan et al., 2003; Sharma et al., 2006). In the ventral midbrain sustained phosphorylation of p38 MAP kinase was observed 12 and 24 h (3- to 3.8-fold) after a single dose of MPTP (Fig. 1A). In the striatum, although increased phosphorylation of p38 was seen after 12 h of MPTP treatment, total p38 levels (α p38) increased significantly such that the pp38/p38 ratio was not significantly altered in the striatum (Fig. 1A).
Figure 1.

A–D, MPTP-induced activation of p38 and JNK in the ventral midbrain and striatum. Animals were treated with a single dose of vehicle or MPTP and killed 1, 4, 12, and 24 h later or treated with a daily dose of MPTP for 8 d and killed on the ninth day. Representative immunoblots from ventral midbrain (MB) and striatum (ST) of animals treated with saline (C) and MPTP (lanes 1, 4, 12, 24 h, 8 d) depicting (A) the levels of phospho-p38 (pp38) and p38 and (B) JNK54, JNK46, phospho-JNK54 (pJNK54), phospho-JNK46 (pJNK46) in the MB and ST after MPTP treatment. C and D depict the level of JNK54, JNK46, phospho-JNK54 (pJNK54), phospho-JNK46 (pJNK46) in the MB and ST after 8 d of MPTP treatment. β-Tubulin levels were measured as loading controls. Densitometric analysis of the immunoblots representing the relative intensity of the immunoreactive bands from MB (n = 6) and ST (n = 3) are shown below the respective blots. They are represented as solid line for pJNK54 and JNK54, whereas pJNK46 and JNK46 are depicted as dotted lines in A and B. Activation of p38 MAPK and JNK is indicated by their respective pMAPK/MAPK ratio. The increase in ratio is expressed as fold increase with respect to the control ratio (1.0). Values are mean ± SD. Asterisks indicate values significantly different from corresponding control (p < 0.05). Repeated measures of ANOVA followed by Dunnet's test was performed for A and B, whereas paired t test was performed for C and D.
We colocalized pp38 and tyrosine hydroxylase in the midbrain after acute and subchronic exposure to MPTP (Fig. 2A). Increased phosphorylation of p38 was seen selectively in the SNpc (Fig. 2B) as diffuse granular staining but not in the ventral tegmental area (VTA) (Fig. 2C) after a single exposure or subchronic exposure to MPTP for 8 d. Vehicle treated controls showed sparse staining for pp38 (Fig. 2A–C). pp38 did not show cell specific staining in the striatum after acute (Fig. 2D) or subchronic MPTP treatment (Fig. 2D). However, the fiber terminals projecting toward the caudate and putamen showed increased pp38 staining after MPTP treatment (Fig. 2D).
Figure 2.

A–D, Activation of p38 in substantia nigra pars compacta (SNpc) and their terminals in the striatum after administration of MPTP to mice. Animals were treated with a single dose of MPTP and killed 24 h later or treated with a daily dose of MPTP for 8 d and killed on the ninth day. A, Immunohistochemical colocalization revealed the presence of pp38 (green) in the soma of tyrosine hydroxylase (TH) positive (red) SNpc neurons of the ventral midbrain after MPTP exposure. pp38 is present at low levels in the SNpc neurons of control animals while not detectable in ventral tegmental area (VTA) neurons. After single dose for 24 h and 8 d of MPTP treatment, there was an increase in pp38 in the SNpc but not VTA (arrowhead). Negative control for colocalization of pp38 and TH in the ventral midbrain is depicted. Corresponding magnified images of the neurons in SNpc (B) and VTA (C) are depicted. D, Images from the striatum show pp38 in the nerve terminals projecting toward the striatum in control, after a single dose and sub chronic exposure to MPTP. Scale bars: 25 μm; magnified images, 10 μm.
Phosphorylation of JNK is seen predominantly in microglia in the substantia nigra
MPTP administration increased the levels of p46 and p54 phospho-JNK proteins by 1.6-fold in the ventral midbrain at 12–24 h (Fig. 1B). At early time periods, namely 1–4 h after a single dose of MPTP, the ratio of phosphorylated to total JNK54 increased significantly. In the striatum, the activation of JNK 46 and 54 occurred soon after MPTP exposure (1 h) and was sustained up to 24 h. Concomitant increase in the levels of the JNK protein occurred and the ratio of pJNK/JNK was increased to 1.9-fold at 4–24 h for JNK46 and 1.6-fold at 4–12 h for JNK54 (Fig. 1B). The activation of JNK54 was attenuated by 24 h, whereas JNK46 levels remained sustained. After exposure of MPTP for 8 d the ratio of pJNK/JNK was increased for JNK 54 and 46 in the striatum (Fig. 1D) but not in midbrain (Fig. 1C). In the midbrain although the pJNK immunostaining persisted (Fig. 3A) and pJNK54 levels were increased as seen by immunoblotting, we did not see increased ratio of pJNK54/JNK54 because of increase in the level of total JNK 54. JNK46 was not activated in midbrain (Fig. 1B,C).
Figure 3.
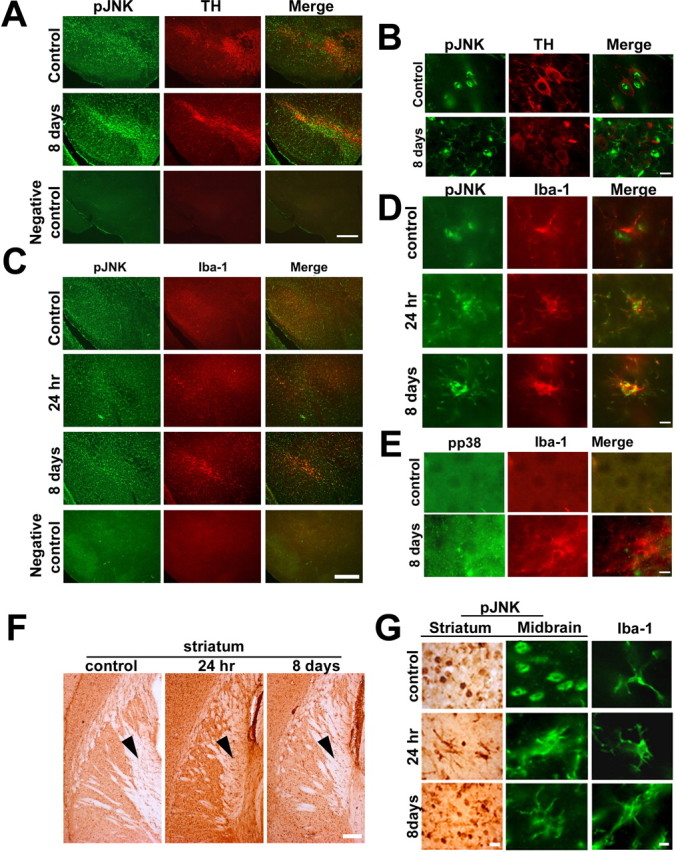
Activation of JNK in microglial cells of the ventral midbrain and striatum after administration of MPTP to mice. Animals were treated with a single dose of MPTP and killed 24 h later or a daily dose of MPTP for 8 d and killed on the ninth day. A, Immunohistochemical colocalization revealed the presence of pJNK (green) throughout the brain including tyrosine hydroxylase positive neurons of the SNpc (red). Negative control for colocalization of pJNK and TH in the ventral midbrain is depicted. B, JNK activation was observed predominantly in the microglia in SNpc after 8 d of MPTP treatment. C, pJNK (green) colocalized with Iba1(red) after single and 8 d of MPTP treatment. Negative control for colocalization of pJNK and Iba1in the ventral midbrain is depicted. D, Magnified images depicting the colocalization of Iba1(red) with pJNK (green) after single dose and 8 d of MPTP treatment. E, pp38 does not colocalize with Iba1 after MPTP treatment. F, JNK activation was also observed specifically in microglia in the striatum after single dose and 8 d of MPTP treatment. The magnified representative images of cells indicated by arrowheads in F is depicted in G (first column). G, Change in morphology of cells positive for pJNK to microglial morphology in the ventral midbrain after MPTP treatment is also depicted. Neuron specific staining of pJNK is seen in the control section. Scale bars: 25 μm; magnified images, 10 μm.
Immunohistochemical localization of pJNK revealed that in vehicle treated control animals pJNK was discernible in the neurons of substantia nigra (Fig. 3A,B). After MPTP exposure (single dose or subchronic for 8 d) pJNK immunostaining increased in the substantia nigra and the VTA (Fig. 3A,B) and the activation seemed to occur predominantly in microglial cells (Fig. 3C,D), which was in contrast to p38 activation that occurred in neurons (Fig. 2B) but not in the microglia (Fig. 3E). pJNK colocalized with Iba 1 immunopositive cells after acute or subchronic MPTP (Fig. 3C). In the striatum, pJNK immunostaining was substantially increased after a single dose or subchronic exposure to MPTP (Fig. 3F,G). pJNK immunostaining in the neurons seen in the vehicle-treated controls persisted after MPTP exposure; however, in addition, extensive staining was seen in microglial cells in the striatum and midbrain (Fig. 3A–G).
Translocation of p53 to the nucleus after MPTP exposure
p53 can be phosphorylated by p38 MAP kinase, which facilitates its accumulation in the nucleus. We, therefore, examined the levels of both p53 and phospho-p53 (Ser15) in the nuclear compartment and found that they were increased indicating that increased p53 mediated transcription could potentially take place (Fig. 4A). MPTP also increased the levels of total p53 in the postnuclear supernatant and specifically in the cytosol (Fig. 4B). This was validated by immunohistochemistry wherein we observed the translocation of p53 into the nucleus of the surviving tyrosine hydroxylase positive neurons in SNpc after 8 d of MPTP treatment (Fig. 4C). We then quantitated the mRNA levels of Noxa and Puma, which are transcribed through p53 dependent mechanism using qRT-PCR and found that a single dose of MPTP resulted in significant increase in the levels of Puma but not Noxa (Fig. 4D) 12 h after MPTP treatment. In addition, in the postnuclear supernatant, Bax, another target gene of p53 was also increased in a sustained manner after a single dose of MPTP (Fig. 4E).
Figure 4.

MPTP administration up-regulates p53 and induces p53 mediated transactivation, in vivo. Animals were treated with a single dose of vehicle or MPTP and killed 12 and 24 h later or with a daily dose of MPTP for 8 d and killed on the ninth day. A, pp53 (Ser15) accumulates in the nucleus 12 h after a single dose of MPTP and after subchronic exposure to MPTP for 8 d. Representative immunoblots of nuclear p53 and phospho-p53 Ser15 with their densitometric analyses are depicted. Values are mean ± SD (n = 7 animals). B, Representative immunoblot of extranuclear and cytosolic p53 in ventral midbrain (MB) and densitometric analyses of the post nuclear supernatant (PNS) and cytosolic p53 show the upregulation after MPTP exposure. Values are mean ± SD (n = 6 animals). C, Quantitative assessment of the expression of Noxa and Puma in ventral midbrain after MPTP treatment. qRT-PCR analysis using mouse ventral midbrain RNA shows relatively higher expression of Noxa and Puma in mouse ventral midbrain after MPTP treatment for 12 h. All samples were normalized using 18S rRNA expression. Values are mean ± SD (n = 6 animals). D, Representative immunoblot for Bax from ventral midbrain (MB; n = 5) of animals and densitometric analyses are shown. E, Animals were treated with a single dose of vehicle (3% DMSO) or MPTP and killed 12 h later. Some animals also received a single dose of SB239063 intrathecally. E–G, Representative blots from ventral midbrain of animals treated with DMSO (lane 1), MPTP (lane 2), SB239063 (lane 3), and SB239063 + MPTP (lane 4) depicting the protein levels of phospho-p38, p38, and Bax (G) in the extranuclear compartment and pp53 Ser15 and p53 (F) in the nuclear compartment as determined by immunoblot are presented. pp38/p38 ratio represents p38 activation in ventral midbrain. Densitometric analysis of the immunoblots representing the relative intensity of the immunoreactive bands are shown. Values are mean ± SD (n = 4 animals). β-Tubulin levels were measured as loading control for extranuclear compartment and lamin/histone levels were measured as loading control for nuclear compartment. Asterisks indicate values significantly different from corresponding control (p < 0.05). Statistical analysis using t test for A–E and repeated measures of ANOVA followed by Dunnet's test was performed for F–I.
MPTP mediated phosphorylation and translocation of p53 to the nucleus is abolished by p38 inhibitor SB239063, in vivo in mice
We examined the effect of pretreatment with SB239063, a selective inhibitor of p38 on the phosphorylation and translocation of p53 to the nucleus after MPTP exposure because p38 is known to phosphorylate and stabilize p53 (Shieh et al., 1997; Vitale et al., 2008). Pretreatment with SB239063 attenuated MPTP mediated phosphorylation of p38 MAP kinase as expected (Fig. 4F,G). Further, the downstream phosphorylation and translocation of p53 was also abolished (Fig. 4I). p53 mediated transcription of Bax was attenuated but not abolished (Fig. 4H) indicating that transcription of Bax could occur through alternate pathways (Vila et al., 2001; Melino et al., 2004).
p38 inhibitor and pifithrin-α afford protection against MPP+ induced cell death in primary neurons derived from human CNS progenitor cells
Primary cultures of human CNS progenitor cells were differentiated into neurons and characterized for the dopaminergic phenotype by studying the expression of tyrosine hydroxylase (TH) and neuronal marker β-III-tubulin (Tuj1) (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). On average, 89.9% cells were found to be positive for Tuj1 and 87.7% cells were found to be positive for tyrosine hydroxylase staining (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). Glial population was negligible, only 2% of the cells were found to be positive for GFAP (supplemental Fig. 1D, available at www.jneurosci.org as supplemental material). Cells were responsive to MPP+ mediated toxicity as examined by TUNEL assay and dose-dependent increase in TUNEL positive neurons was observed after exposure to increasing concentration of MPP+(supplemental Fig. 1E, available at www.jneurosci.org as supplemental material). Both p38 inhibitor SB239063 (1 μm) (Fig. 5A) and p53 inhibitor pifithrin-α (250 nm) (Fig. 5B) afforded complete protection against MPP+ mediated cell death.
Figure 5.
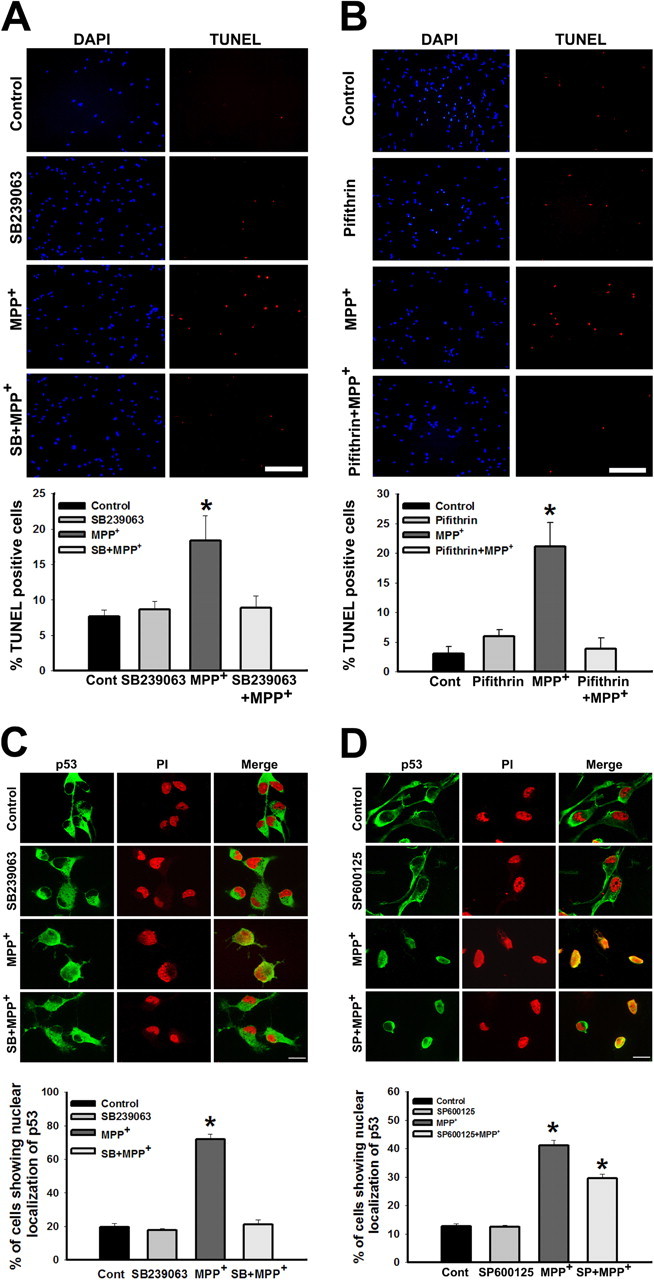
p38 inhibitor confers protection against MPP+ mediated cell death by preventing p53 translocation into the nucleus. A, Cells treated with SB239063 (1 μm) for 60 min before exposure to MPP+ (10 μm) for 24 h were protected from MPP+ mediated cell death. Scale bar, 200 μm. Cells were treated with either vehicle or SB239063 before the treatment with MPP+, quantification represents percentage of cells positive for TUNEL staining per total number of cells as represented by DAPI staining. B, p53 inhibitor pifithrin-α confers protection against MPP+ mediated cell death. Cells treated with pifithrin-α (250 nm) for 60 min before exposure to MPP+ (10 μm) for 24 h were not affected by MPP+ mediated cell death. Scale bar, 200 μm. Cells were treated with either vehicle or pifithrin-α before the treatment with MPP+, quantification represents, percentage of cells positive for TUNEL staining per total number of cells as represented by DAPI count. C, p53, localized in the cytoplasm in primary neurons under control condition translocated into the nucleus in response to MPP+ exposure. The nuclear translocation was prevented by pretreating the cells with p38 inhibitor, SB239063. D, Nuclear translocation was partially prevented by pretreating the cells with JNK inhibitor, SP600125 (panel 4). Total number of cells is represented by propidium iodide (PI) staining. Data are represented as mean ± SD of four independent experiments. Asterisks indicate values significantly different from controls (p < 0.05).
MPP+ mediated nuclear translocation of p53 is abolished by p38 inhibitor but not by JNK inhibitor
p53 is normally localized in the cytoplasm of primary neurons derived from the human CNS progenitor cells (Fig. 5C). Exposure to MPP+ (10 μm) for 24 h caused nuclear translocation of p53, which was prevented by pretreating the cells with p38 inhibitor, SB239063 (Fig. 5C), indicating the role of p38 in the translocation of p53. However, pretreatment of cells with JNK inhibitor, SP600125 attenuated the nuclear translocation of p53 minimally (Fig. 5D).
pp38 and pJNK in the substantia nigra of PD patients
Phospho-p38 and p-JNK are expressed in tyrosine hydroxylase positive neurons of human substantia nigra (Fig. 6A,B). In the control brain from traffic accident victims both pp38 and pJNK immunostaining appeared to co localize within the tyrosine hydroxylase positive neurons. In PD patients, the surviving neurons from substantia nigra appeared shrunken with decreased tyrosine hydroxylase immunostaining (Fig. 6A,B) and pp38 immunostaining was very prominent throughout the surviving tyrosine hydroxylase positive neuron (Fig. 6A). In PD brains, pJNK immunostaining was not evident in the neuronal cells in contrast to control (Fig. 6B); however, pJNK positive cells colocalized with Iba 1 positive glia indicating an enhanced gliosis in PD brains (Fig. 6C).
Figure 6.

pp38 and pJNK in the substantia nigra of PD patients and schematic representation of the role of p38 in MPTP mediated toxicity. A, Immunohistochemical colocalization of phospho-p38 (green) in Tyrosine hydroxylase (red) positive neurons of control (traffic accident victims) and PD substantia nigra. B, Immunohistochemical colocalization of phospho-JNK (green) in Tyrosine hydroxylase (red) positive neurons of control (traffic accident victims) and PD substantia nigra. C, pJNK immunopositive cells colocalizes with Iba-1 positive cells of substantia nigra (green) in PD brain. The bottom depicts a magnified microglial cell indicating the colocalization of pJNK with Iba-1. Negative control was performed for each set of experiment. Scale bars: (B, C) 10 μm; (A) 25 μm. D, MPP+ is taken up by VMAT and concentrated in the synaptic vesicle generating ROS. MPP+ the toxic metabolite of MPTP causes increased production of ROS and mitochondrial dysfunction in dopaminergic neurons by inhibiting complex I of the electron transport chain. Mitochondrial complex I inhibition further contributes to ROS generation which presumably activates p38 MAP kinase pathway. As a consequence, the downstream target p53 is phosphorylated. p53 subsequently translocates to the nucleus transcriptionally activating the expression of Puma, Noxa and Bax. Bax translocates to the mitochondria ultimately leading to cell death. Approaches aimed at inhibiting p38 activation can help terminate this cascade and attenuate MPTP-induced neurodegeneration. DAT, Dopamine transporter; N, nucleus; VMAT, vesicular monoamine transporter.
Discussion
p53 knock-out mice or mice pretreated with pfithrin, a p53 inhibitor are protected from dopaminergic cell death in SNpc after MPTP (Trimmer. et al. 1996; Duan et al., 2002; Perier et al., 2007). However, the upstream signaling pathways have not been clearly defined.
The failure of the clinical trials using the JNK inhibitor CEP1347 that does not affect ERK1/2 or p38 (Gallo and Johnson, 2002) raises the question about the role of JNK in the demise of dopaminergic cells in SNpc. By blocking mixed lineage kinases (MLK) of MAPKKK superfamily, CEP1347 is also known to modulate the microglial activation of JNK/p38 under certain stressful conditions (Lund et al., 2005). However, by only inhibiting the MLK family, the capability of other MAPKKK to activate p38 and MAPKK-independent activation of p38 may still be preserved (Ge et al., 2002). We now report that selective activation of p38 occurs in the dopaminergic neurons of SNpc in mice treated with MPTP, whereas JNK phosphorylation is predominant in microglia. Thus, the partial protection seen with CEP1347 in animal models of PD could be due to its effect on microglia mediated inflammatory response.
To discern whether the activation of p38 was indeed linked to the demise of the neurons, we examined the effect of p38 inhibitor on MPP+ mediated toxicity, both in vitro in dopaminergic neurons derived from human neural progenitor cells (Fig. 5A) and in vivo, in mice (Fig. 4). We observed that p38 inhibitor; SB239063, which inhibits both p38 α and β, prevented the translocation of p53 to the nucleus and afforded complete protection against cell death, in vitro. In vivo, in the ventral midbrain, treatment with p38 inhibitor prevented the downstream phosphorylation of p53 and its translocation to the nucleus. p38 inhibitor SB239063 does not cross the blood brain barrier (Cao et al., 2007), and therefore we administered it intrathecally.
p38 is known to phosphorylate p53. This post-translational modification facilitates p53 accumulation in the nucleus (Shieh et al., 1997; Vitale et al., 2008). Phosphorylation of p53 within or close to the N-terminal MDM2 binding domain, i.e., at Ser15 increases the stability of p53 by interfering with its ability to bind MDM2 and by decreasing ubiquitin-dependent degradation (Shieh et al., 1997). We saw increased phosphorylation of p53 in the nuclear compartment of ventral midbrain of mice treated with MPTP, which was abolished by pretreatment with p38 inhibitor (Fig. 4I). Thus, our results point to the selective activation of p38 in the neurons of SNpc and its role in the downstream phosphorylation of p53 leading to increased p53 mediated transcription of Bax and Puma, but not Noxa. It needs to be examined whether other targets of p53 are transcriptionally activated in addition to the above. p38 inhibitor did not abolish but attenuated the increase in Bax levels (Fig. 4H). This indicates that transcription of Bax can occur through alternate pathways also (Vila et al., 2001; Melino et al., 2004).
MK2 is a downstream target of p38 and MK2 knock-out mice are protected from MPTP toxicity (Thomas et al., 2008). This has been attributed to the attenuation of inflammatory response mediated through p38 in the microglia. The lack of coherence between our observations could be due to the fact that MK2 can be activated by upstream kinases other than p38. Further, the dosing paradigm for MPTP involved 4 doses in 24 h resulting in a massive insult rather than the chronic toxicity paradigm used in our experiments wherein a single dose of MPTP was given daily for 8 d.
A protective role for p38 dependent phosphorylation has been recently reported (Plun-Favreau et al., 2007). Activated p38 phosphorylates HtrA2 (Omi) in a PINK1 dependent manner which may contribute to increased resistance of cells to mitochondrial stress and further Omi decreased in brain of PD patients. Although the above report suggests an important function for p38 under normal conditions in the mitochondria, it is to be determined whether aberrant activation of p38 as seen in the present study plays a dysfunctional role within the mitochondria also.
Increased pp38 immunostaining was observed in the surviving tyrosine hydroxylase positive neurons in the SNpc from patients with Parkinson's disease, which was not observed in control brain from traffic accident victims (Fig. 6A). Further, pJNK staining seemed to be restricted to the glial cells which colocalized with Iba1 staining indicating that JNK was phosphorylated in the microglia (Fig. 6C). The surviving neurons in SNpc did not show predominant JNK activation (Fig. 6B). Although the above observation is similar to that seen in the MPTP mouse model, the data need to be interpreted carefully. The animal models do not replicate the human disease entirely. Further, there is greater redundancy in human brain such that additional pathways can be involved and the cross talk between the pathways could potentially add to the complexity of the pathogenic process. Nevertheless, the studies present herein point to the cell-specific activation of death signaling cascade with similarities between the mouse model and the human disease.
Although the present study points to selective activation of p38 within the dopaminergic neurons of SNpc and its role in p53 mediated cell death, the upstream mediators of p38 activation remain to be identified. p38 is activated by the MAPKKs, which in turn are activated by MAPKKK. We observed the activation of pMKK6 (a MAPKK) in the ventral midbrain after MPTP exposure (data not shown). Apoptosis signal regulating kinase (ASK1, a MAPKKK) is activated in the ventral midbrain however, it is yet to be determined whether ASK1 activation (Karunakaran et al., 2007) is indeed essential for the p38 activation. Potentially, the presence of Iba1 positive activated microglia seen in the SNpc signifying the inflammatory response (Fig. 3) could lead to generation of ROS, which in turn could activate ASK1 (Matsuzawa and Ichijo, 2008; Noguchi et al., 2008), thus triggering the aberrant MAPK cascade resulting in p38 activation. However, the evidence for the above needs to be established.
Earlier studies on the role of JNK activation have been performed largely on cultured cells, whereas signaling, in vivo is considerably more complex. For example, in cultured cells exposed to MPP+ or paraquat, JNK is phosphorylated and JNK inhibitors afford neuroprotection (Saporito et al., 1999; Wang et al., 2004; Peng et al., 2004). However, in vivo cell-specific activation of signaling cascades ensues as seen in the present study with p38 and JNK phosphorylation occurring in neuronal and glial cells respectively. This difference may be attributed to the use of cultured/primary cells; the toxin used and the dosage paradigm, wherein chronic exposure involves administering several doses on a single day versus a single dose for several days.
The dopaminergic neurons derived from the human neural progenitors offer an excellent in vitro model for discerning molecular mechanisms after a neurotoxic insult. These primary neurons express tyrosine hydroxylase and most of the Tuj1 positive neurons also express tyrosine hydroxylase (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material) indicating the predominant dopaminergic phenotype of these cells (Mishra et al., 2008).
One of the compelling questions in understanding the pathogenesis of neurodegenerative diseases in general and PD in particular is the vulnerability of selective cell population to neurodegeneration. The results described herein demonstrating the selective activation of JNK and p38 in distinct cell populations within the SNpc, namely the microglia and the dopaminergic neurons, is a case in point (Figs. 2, 3). Identification of cell-specific signaling pathways is vital for discovering and developing therapeutic strategies that will go beyond global neuroprotection and hold promise as disease modifying agents that can slow down the degeneration of vulnerable cell population. It is to be determined whether selective inhibitors of p38 would help preserve the surviving neurons in PD and slow down the disease progression, which the JNK inhibitor CEP1347 failed to do in human trials (Parkinson Study Group PRECEPT Investigators, 2007). We acknowledge that from a global perspective several mechanisms could contribute to the neurodegeneration seen in diseases like PD, nevertheless the cell-specific activation of death signaling cascade as seen in this study indicates that activation of p38 is potentially an important contributor to the disease process leading to the degeneration of dopaminergic neurons of the SNpc in PD. CEP1347 only attenuated but could not completely abolish MPTP mediated degeneration of striatal dopaminergic nerve terminals and cell bodies in the nigra (Saporito et al., 2000). In this scenario, a combinatorial approach involving JNK and p38 inhibitors, which would target pathogenic mechanisms in distinct cell populations would enhance neuroprotection in a region specific manner and beyond.
Footnotes
We thank Prof. S. K. Shankar for providing the human brain samples through the Human Brain Tissue Repository at Department of Neuropathology, National Institute of Mental Health and Neuro Sciences, Bangalore. We also thank Neha Sehgal for her help with some experiments and Ajit Ray for proofreading this manuscript. The laboratory assistance of P. Manish is acknowledged.
References
- Barden H. The oxidative generation of sulfonic acid groups in neuromelanin and lipofuscin in the human brain. J Histochem Cytochem. 1984;32:329–336. doi: 10.1177/32.3.6198360. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Cao FL, Liu MG, Hao J, Li Z, Lu ZM, Chen J. Different roles of spinal p38 and c-Jun N-terminal kinase pathways in bee venom-induced multiple pain-related behaviors. Neurosci Lett. 2007;427:50–54. doi: 10.1016/j.neulet.2007.09.005. [DOI] [PubMed] [Google Scholar]
- Duan W, Zhu X, Ladenheim B, Yu QS, Guo Z, Oyler J, Cutler RG, Cadet JL, Greig NH, Mattson MP. p53 inhibitors preserve dopamine neurons and motor function in experimental parkinsonism. Ann Neurol. 2002;52:597–606. doi: 10.1002/ana.10350. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Blanco R, Carmona M, Puig B, Barrachina M, Gómez C, Ambrosio S. Active, phosphorylation-dependent mitogen-activated protein kinase (MAPK/ERK), stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK), and p38 kinase expression in Parkinson's disease and Dementia with Lewy bodies. J Neural Transm. 2001;108:1383–1396. doi: 10.1007/s007020100015. [DOI] [PubMed] [Google Scholar]
- Gallo KA, Johnson GL. Mixed-lineage kinase control of JNK and p38 MAPK pathways. Nat Rev Mol Cell Biol. 2002;3:663–672. doi: 10.1038/nrm906. [DOI] [PubMed] [Google Scholar]
- Ge B, Gram H, Di Padova F, Huang B, New L, Ulevitch RJ, Luo Y, Han J. MAPKK-independent activation of p38alpha mediated by TAB1-dependent autophosphorylation of p38alpha. Science. 2002;295:1291–1294. doi: 10.1126/science.1067289. [DOI] [PubMed] [Google Scholar]
- Guan Z, Kim JH, Lomvardas S, Holick K, Xu S, Kandel ER, Schwartz JH. p38 MAP kinase mediates both short-term and long-term synaptic depression in aplysia. J Neurosci. 2003;23:7317–7325. doi: 10.1523/JNEUROSCI.23-19-07317.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann A, Michel PP, Troadec JD, Mouatt-Prigent A, Faucheux BA, Ruberg M, Agid Y, Hirsch EC. Is Bax a mitochondrial mediator in apoptotic death of dopaminergic neurons in Parkinson's disease? J Neurochem. 2001;76:1785–1793. doi: 10.1046/j.1471-4159.2001.00160.x. [DOI] [PubMed] [Google Scholar]
- Karunakaran S, Diwakar L, Saeed U, Agarwal V, Ramakrishnan S, Iyengar S, Ravindranath V. Activation of apoptosis signal regulating kinase 1 (ASK1) and translocation of death-associated protein, Daxx, in substantia nigra pars compacta in a mouse model of Parkinson's disease: protection by alpha-lipoic acid. FASEB J. 2007;21:2226–2236. doi: 10.1096/fj.06-7580com. [DOI] [PubMed] [Google Scholar]
- Klintworth H, Newhouse K, Li T, Choi WS, Faigle R, Xia Z. Activation of c-Jun N-terminal protein kinase is a common mechanism underlying paraquat- and rotenone-induced dopaminergic cell apoptosis. Toxicol Sci. 2007;97:149–162. doi: 10.1093/toxsci/kfm029. [DOI] [PubMed] [Google Scholar]
- Korner M, Rattner A, Mauxion F, Sen R, Citri Y. A brain-specific transcription activator. Neuron. 1989;3:563–572. doi: 10.1016/0896-6273(89)90266-3. [DOI] [PubMed] [Google Scholar]
- Lund S, Porzgen P, Mortensen AL, Hasseldam H, Bozyczko-Coyne D, Morath S, Hartung T, Bianchi M, Ghezzi P, Bsibsi M, Dijkstra S, Leist M. Inhibition of microglial inflammation by the MLK inhibitor CEP-1347. J Neurochem. 2005;92:1439–1451. doi: 10.1111/j.1471-4159.2005.03014.x. [DOI] [PubMed] [Google Scholar]
- Mancuso C, Scapagini G, Currò D, Giuffrida Stella AM, De Marco C, Butterfield DA, Calabrese V. Mitochondrial dysfunction, free radical generation and cellular stress response in neurodegenerative disorders. Front Biosci. 2007 Jan;1(12):1107–1123. doi: 10.2741/2130. [DOI] [PubMed] [Google Scholar]
- Matsuzawa A, Ichijo H. Redox control of cell fate by MAP kinase: physiological roles of ASK1-MAP kinase pathway in stress signaling. Biochim Biophys Acta. 2008;1780:1325–1336. doi: 10.1016/j.bbagen.2007.12.011. [DOI] [PubMed] [Google Scholar]
- Melino G, Bernassola F, Ranalli M, Yee K, Zong WX, Corazzari M, Knight RA, Green DR, Thompson C, Vousden KH. p73 Induces apoptosis via PUMA transactivation and Bax mitochondrial translocation. J Biol Chem. 2004;279:8076–8083. doi: 10.1074/jbc.M307469200. [DOI] [PubMed] [Google Scholar]
- Mishra M, Vetrivel S, Siddappa NB, Ranga U, Seth P. Clade-specific differences in neurotoxicity of human immunodeficiency virus-1 B and C Tat of human neurons: significance of dicysteine C30C31 motif. Ann Neurol. 2008;63:366–376. doi: 10.1002/ana.21292. [DOI] [PubMed] [Google Scholar]
- Mizuno Y, Ikebe S, Hattori N, Nakagawa-Hattori Y, Mochizuki H, Tanaka M, Ozawa T. Role of mitochondria in the etiology and pathogenesis of Parkinson's disease. Biochim Biophys Acta. 1995;1271:265–274. doi: 10.1016/0925-4439(95)00038-6. [DOI] [PubMed] [Google Scholar]
- Noguchi T, Ishii K, Fukutomi H, Naguro I, Matsuzawa A, Takeda K, Ichijo H. Requirement of Reactive Oxygen Species-dependent Activation of ASK1–p38 MAPK Pathway for Extracellular ATP-induced Apoptosis in Macrophage. J Biol Chem. 2008;283:7657–7665. doi: 10.1074/jbc.M708402200. [DOI] [PubMed] [Google Scholar]
- Olanow CW. The pathogenesis of cell death in Parkinson's disease–2007. Mov Disord. 2007;17(22) Suppl:S335–S342. doi: 10.1002/mds.21675. [DOI] [PubMed] [Google Scholar]
- Parker WD, Jr, Boyson SJ, Parks JK. Abnormalities of the electron transport chain in idiopathic Parkinson's disease. Ann Neurol. 1989;26:719–723. doi: 10.1002/ana.410260606. [DOI] [PubMed] [Google Scholar]
- Parkinson Study Group PRECEPT Investigators. Mixed lineage kinase inhibitor CEP-1347 fails to delay disability in early Parkinson disease. Neurology. 2007;69:1480–1490. doi: 10.1212/01.wnl.0000277648.63931.c0. [DOI] [PubMed] [Google Scholar]
- Peng J, Mao XO, Stevenson FF, Hsu M, Andersen JK. The herbicide paraquat induces dopaminergic nigral apoptosis through sustained activation of the JNK pathway. J Biol Chem. 2004;279:32626–32632. doi: 10.1074/jbc.M404596200. [DOI] [PubMed] [Google Scholar]
- Perier C, Tieu K, Guégan C, Caspersen C, Jackson-Lewis V, Carelli V, Martinuzzi A, Hirano M, Przedborski S, Vila M. Complex I deficiency primes Bax-dependent neuronal apoptosis through mitochondrial oxidative damage. Proc Natl Acad Sci U S A. 2005;102:19126–19131. doi: 10.1073/pnas.0508215102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perier C, Bové J, Wu DC, Dehay B, Choi DK, Jackson-Lewis V, Rathke-Hartlieb S, Bouillet P, Strasser A, Schulz JB, Przedborski S, Vila M. Two molecular pathways initiate mitochondria-dependent dopaminergic neurodegeneration in experimental Parkinson's disease. Proc Natl Acad Sci U S A. 2007;104:8161–8166. doi: 10.1073/pnas.0609874104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plun-Favreau H, Klupsch K, Moisoi N, Gandhi S, Kjaer S, Frith D, Harvey K, Deas E, Harvey RJ, McDonald N, Wood NW, Martins LM, Downward J. The mitochondrial protease HtrA2 is regulated by Parkinson's disease-associated kinase PINK1. Nat Cell Biol. 2007;9:1243–1252. doi: 10.1038/ncb1644. [DOI] [PubMed] [Google Scholar]
- Saporito MS, Brown EM, Miller MS, Carswell S. CEP-1347/KT-7515, an inhibitor of c-jun N-terminal kinase activation, attenuates the 1-methyl-4-phenyl tetrahydropyridine-mediated loss of nigrostriatal dopaminergic neurons In vivo. J Pharmacol Exp Ther. 1999;288:421–427. [PubMed] [Google Scholar]
- Saporito MS, Thomas BA, Scott RW. MPTP activates c-Jun NH(2)-terminal kinase (JNK) and its upstream regulatory kinase MKK4 in nigrostriatal neurons in vivo. J Neurochem. 2000;75:1200–1208. doi: 10.1046/j.1471-4159.2000.0751200.x. [DOI] [PubMed] [Google Scholar]
- Schnell SA, Staines WA, Wessendorf MW. Reduction of lipofuscin-like autofluorescence in fluorescently labeled tissue. J Histochem Cytochem. 1999;47:719–730. doi: 10.1177/002215549904700601. [DOI] [PubMed] [Google Scholar]
- Sharma SK, Sherff CM, Stough S, Hsuan V, Carew TJ. A tropomyosin-related kinase B ligand is required for ERK activation, long-term synaptic facilitation, and long-term memory in Aplysia. Proc Natl Acad Sci U S A. 2006;103:14206–14210. doi: 10.1073/pnas.0603412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91:325–334. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- Thomas T, Timmer M, Cesnulevicius K, Hitti E, Kotlyarov A, Gaestel M. MAPKAP kinase 2-deficiency prevents neurons from cell death by reducing neuroinflammation–relevance in a mouse model of Parkinson's disease. J Neurochem. 2008;105:2039–2052. doi: 10.1111/j.1471-4159.2008.05310.x. [DOI] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimmer PA, Smith TS, Jung AB, Bennett JP., Jr Dopamine neurons from transgenic mice with a knockout of the p53 gene resist MPTP neurotoxicity. Neurodegeneration. 1996;5:233–239. doi: 10.1006/neur.1996.0031. [DOI] [PubMed] [Google Scholar]
- Vila M, Jackson-Lewis V, Vukosavic S, Djaldetti R, Liberatore G, Offen D, Korsmeyer SJ, Przedborski S. Bax ablation prevents dopaminergic neurodegeneration in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson's disease. Proc Natl Acad Sci U S A. 2001;98:2837–2842. doi: 10.1073/pnas.051633998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale I, Senovilla L, Galluzzi L, Criollo A, Vivet S, Castedo M, Kroemer G. Chk1 inhibition activates p53 through p38 MAPK in tetraploid cancer cells. Cell Cycle. 2008;13:1956–1961. doi: 10.4161/cc.7.13.6073. [DOI] [PubMed] [Google Scholar]
- Wang W, Shi L, Xie Y, Ma C, Li W, Su X, Huang S, Chen R, Zhu Z, Mao Z, Han Y, Li M. SP600125, a new JNK inhibitor, protects dopaminergic neurons in the MPTP model of Parkinson's disease. Neurosci Res. 2004;48:195–202. doi: 10.1016/j.neures.2003.10.012. [DOI] [PubMed] [Google Scholar]