

Abstract
Huntington's disease (HD) is an autosomal dominant neurodegenerative disease linked to a polyQ (polyglutamine) expansion in the huntingtin protein. Although general brain atrophy is found in HD patients, the striatum is the most severely affected region. Loss or mutant forms of huntingtin were reported to disrupt fast axonal transport in Drosophila, squid, and mice. However, previous work did not resolve whether mutant huntingtin affects global axonal transport or only a subset of cargoes, nor did it resolve whether striatal neurons are preferentially sensitive to huntingtin-mediated defects. We used amyloid precursor protein (APP)–yellow fluorescent protein and brain-derived neurotrophic factor (BDNF)–mCherry fusion proteins as markers for fast axonal transport when huntingtin is altered. We found that movement of APP and BDNF is impaired in striatal and hippocampal, but not cortical, neurons from presymptomatic homozygous mutant mice carrying 150Q huntingtin knock-in mutations. In addition, loss of huntingtin disrupts APP axonal transport, whereas overexpression of wild-type, but not mutant, huntingtin enhances APP transport in all three types of neurons tested. These data suggest that a loss of wild-type huntingtin function in fast axonal transport plays important roles in the development of cell-type-specific defects in HD.
Keywords: axonal transport, Huntington's disease, BDNF, striatum, cortex, mutant
Introduction
In the nervous system, neurons interact with each other via long axonal and dendritic processes. In axons, macromolecules synthesized in the cell body are transported to synapses by motor proteins, such as kinesin and dynein (Goldstein and Yang, 2000), which move needed materials along microtubules. Defects in axonal transport can lead to neurodegenerative pathology and neurodegenerative diseases, such as Charcot-Marie-Tooth disease and hereditary spastic paraplegia SPG10 (Duncan and Goldstein, 2006). Huntington's disease (HD) is a hereditary dominant neurodegenerative disorder characterized by severe motor and memory dysfunction (Folstein, 1989; Harper, 1996; Ross and Margolis, 2001). Evidence that axonal transport abnormalities might contribute to development of HD comes from examinations of postmortem HD patient brains, which revealed the existence of dystrophic neurites, a possible indication of axonal transport dysfunction (Jackson et al., 1995; DiFiglia et al., 1997; Sapp et al., 1999).
HD is linked genetically to expansion of a polyglutamine (polyQ) repeat within the N terminus of the huntingtin protein (The Huntington's Disease Collaborative Research Group, 1993). Huntingtin associates with microtubules and various vesicles including mitochondria and synaptic vesicles (DiFiglia et al., 1995; Bhide et al., 1996; Velier et al., 1998; Li et al., 2003). Both huntingtin and the huntingtin-associated protein 1 (HAP1) move in axons in both anterograde and retrograde directions (Block-Galarza et al., 1997) and have been reported to associate with both the anterograde and retrograde transport machinery (Engelender et al., 1997; Li et al., 1998; McGuire et al., 2006; Caviston et al., 2007). Several studies suggest that wild-type huntingtin plays roles in axonal transport and that mutant huntingtin interferes with these processes, which may cause neuronal dysfunction and eventually neuronal death (Gunawardena et al., 2003; Szebenyi et al., 2003; Gauthier et al., 2004; Lee et al., 2004; Trushina et al., 2004; Chang et al., 2006; Orr et al., 2008). However, it is controversial whether mutant huntingtin affects axonal transport of all or only a subset of cargoes and whether mutant huntingtin affects axonal transport in all or only certain types of neurons. Thus, we compared directly the effects of loss or polyQ mutation of huntingtin on axonal transport of brain-derived neurotrophic factor (BDNF) and amyloid precursor protein (APP), a cargo of kinesin and dynein (Kamal et al., 2000; Stokin et al., 2005), in primary cortical, striatal, and hippocampal neurons.
Materials and Methods
Mice and genetic crosses.
HdhFlox mice (Dragatsis et al., 2000) and Hdh150Q mice (Lin et al., 2001) were both maintained in a C57BL/6J background according to the Guide for the Care and Use of Laboratory Animals. For endogenous huntingtin depletion experiments, female homozygous HdhFlox mice were crossed with male homozygous HdhFlox mice to produce homozygous HdhFlox mice for primary cortical neuron culture. For experiments comparing the effect of endogenous mutant huntingtin on axonal transport, female heterozygous Hdh150Q mice were crossed with male heterozygous Hdh150Q mice to generate either wild-type Hdh or homozygous Hdh150Q littermates for primary neuron cultures. All animal procedures were reviewed and approved by the University of California, San Diego Institutional Animal Care and Use Committee.
Cell cultures.
Primary cortical, striatal, and hippocampal cells were prepared from postnatal day 1 mice and cultured as described previously with minor modifications (Brewer, 1995, 1997). Neocortex, striatum, and hippocampi were excised in cold HBSS (Invitrogen). For cortical and striatal neuron preparation, neocortex and striatum were digested with a Papain Dissociation kit (Worthington Biochemicals) for 30 min at 37°C. For hippocampal neuron preparation, hippocampi were digested with a 0.22 μm-filtered mixture of 45U papain (Worthington Biochemicals) in PBS (Invitrogen), dl-cysteine HCl (Sigma), bovine serum albumin (Sigma), and d-glucose (Sigma) with 0.05% DNase (Roche Diagnostics) for 30 min at 37°C. After papain digestion, neocortex, striatum, and hippocampi were triturated 20 times in 2 ml of growth medium and plated at 100,000 cells per well of a 24-well dish coated with poly-l-lysine and laminin (Sigma). Cortical and hippocampal neurons were maintained in 0.5 mm glutamine and B27 (Invitrogen) supplemented with Neurobasal medium (Invitrogen) at 37°C in 5% CO2. Striatal neurons were maintained in 1 mm glutamine and B27 (Invitrogen) supplemented with Neurobasal medium at 37°C in 5% CO2.
Transfection.
Primary cortical, striatal, and hippocampal neurons were transfected with Lipofectamine 2000 as recommended (Invitrogen) on 4, 9, and 12 d in vitro (DIV), respectively. The constructions of pCMV-APPYFP, pEBV-HTTQ23F, and pEBV-HTTQ120F plasmids were described previously (Kaether et al., 2000; Zhou et al., 2003). The pCAG-APPYFP and pJPA5-BDNF-mCherry plasmids were gifts from Dr. Z. Yang (University of California, San Diego, San Diego, CA) and Dr. G. Banker (Oregon Health & Sciences University, Portland, OR), respectively.
In vivo imaging.
In vivo movements of APP–yellow fluorescent protein (YFP) and BDNF-mCherry were imaged in primary cultures at 37°C in 5% CO2 on a heating stage (Harvard Apparatus) with an inverted epifluorescent microscope (TE-2000U; Nikon) connected to a Photometrics CoolSNAPHQ-cooled CCD camera (Roper Scientific). Images were captured at a speed of 10 frames per second for 15 s for each stream acquisition stack at 100× magnification and 2 × 2 binning by MetaMorph 6.0 (Universal Imaging Corporation). Image stacks were converted to kymographs and analyzed by MetaMorph (Universal Imaging Corporation). For APP–YFP and BDNF-mCherry movement, images were recorded 16 and 5 h after transfection, respectively. Each experiment was performed twice.
Statistical analysis.
Kymographs of APP–YFP and BDNF-mCherry movements were analyzed manually as described previously (Stokin et al., 2005). Comparisons of average velocity and particle distributions were done using unpaired two-tailed t tests. The data are presented as mean ± SE. Differences between samples were considered significant when p < 0.05 and very significant when p < 0.01.
Tat-cre purification and treatment and viability assay.
Recombinant tat-cre protein was expressed and purified as described previously (Wadia et al., 2004). In brief, the pTat-cre plasmid was transformed into BL21 cells (Novagen). Exponentially growing cells from cultures diluted overnight were induced with 500 μm isopropyl β-d-thiogalactoside for 5 h. Recombinant tat-cre protein was purified by successive steps of the Ni-NTA column (Qiagen) and Source 30S column (Pfizer). Aliquots of purified tat-cre were stored at −70°C in 10% glycerol. For treatment, primary cortical neurons were incubated with 500 μl of 1 mm tat-cre and 5 μm recombinant tat-HA2 peptide in Neurobasal medium for the times stated. After incubation, cells were washed with Neurobasal medium to remove unbound tat-cre. Forty-eight hours after tat-cre treatment, cortical neurons were washed with phosphate-based buffer (PBS) and stained with 15 μg/ml fluorescein diacetate (FDA) and 4.6 μg/ml propidium iodide (PI) in PBS for 15 min at room temperature. After excitation, live cells emit green fluorescence, and dead cells display red fluorescence (Brewer et al., 1993). As a control for dead cells, cortical neurons were treated with 50% methanol in PBS for 10 min at room temperature before the viability test.
Western blot.
Soluble protein extracts were prepared as described previously (Gunawardena et al., 2003). In brief, brains were dounce homogenized in buffer A (in mm: 250 sucrose, 15 Tris-HCl, pH 7.9, 60 KCl, 15 NaCl, 5 EDTA, 1 EGTA, 0.5 DTT) and protease inhibitors (Roche). The lysate was centrifuged at 4000 × g for 10 min, and the supernatant was collected and centrifuged again at 18,000 × g for 10 min. the second supernatant was collected and stored at −20°C for Western blots. The protein concentrations were measured by the DC Protein Assay (Bio-Rad). Equal amounts of protein lysates were loaded on 4–12% Bis-Tris NuPage gels (Invitrogen). After electrophoresis, the samples were transferred to nitrocellulose membrane (Bio-Rad) at 100 mA for 15 h at 4°C (25 mm Tris-HCl, 190 mm glycine, and 20% methanol). After transfer, the membranes were blocked with 5% nonfat milk in TBS-T buffer (50 mm Tris-HCl pH 7.5, 150 mm NaCl, and 0.05% Tween 20) after being washed briefly with TBS-T buffer. The membranes were then incubated with a primary antibody at 4°C overnight. After washing membranes with TBS-T buffer three times with 5 min intervals, membranes were incubated with a corresponding HRP-coupled or fluorescence-coupled secondary antibody at room temperature for 60 min. After washing four times with TBS-T buffer at 5 min intervals, membranes were developed with enhanced chemiluminescence or the Odyssey system as described (GE Healthcare and Li-Cor). Quantization of intensity was done using Odyssey 2.1 software (Li-Cor).
Antibodies.
We used anti-huntingtin (HTT) (MAB2166, 1:1000; Millipore Bioscience Research Reagents), anti-kinesin heavy chain (KHC; MAB1614, 1:1000; Millipore Bioscience Research Reagents), anti-dynein heavy chain (DHC; SC-9115, 1:500; Santa Cruz Biotechnology), anti-BDNF (SC-546, 1:500; Santa Cruz Biotechnology), anti-DARPP32 (AB1656, 1:500; Millipore Bioscience Research Reagents), anti-GAD67 (MAB5406, 1:1000; Millipore Bioscience Research Reagents), anti-APP (MAB348, 1:1000; Millipore Bioscience Research Reagents), anti-p150Glued (671208, 1:250; BD Biosciences), anti-HAP1 (611302, 1:250; BD Biosciences), anti-p50 (611003, 1:250; BD Biosciences), anti-α-tubulin (1:5000; Sigma), and anti-actin (Roche Diagnostics).
Sucrose gradient fractionation.
Sucrose gradient experiments were performed as described previously (LaMonte et al., 2002). In brief, brain extracts were prepared by dounce homogenization in buffer A (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, and 1 mm EDTA) plus protease inhibitors (Roche). The crude extracts were centrifuged at 10,000 × g for 10 min at 4°C. Approximately 100 μl of supernatants (20 mg/ml) was overlaid on a 10.3 ml, 5–20% sucrose gradient and centrifuged at 32,000 × g for 16 h at 4°C in an SW40 rotor (Beckman Coulter). After centrifugation, 800 μl fractions were collected from the top and analyzed by Western blot assays.
Results
Depletion of huntingtin in primary mouse cortical neurons disrupts axonal transport of APP
Previous reports had shown that reduction of endogenous huntingtin in retinoblastoma cells and striatal neurons impairs axonal transport (Gauthier et al., 2004; Trushina et al., 2004). Here we tested whether depletion of endogenous huntingtin also disrupts axonal transport in primary cortical neurons. To completely remove expression of endogenous mouse huntingtin in primary neurons, we used the cre-loxP system. In Hdh floxed mice, exon 1 of the Hdh gene is flanked by two loxP sequences (Dragatsis et al., 2000). In the presence of cre protein, exon 1 of the Hdh gene is removed by recombination of the two loxP sites, thereby completely abolishing the expression of huntingtin. To introduce cre protein, primary cortical neurons (3 or 4 DIV) cultured from homozygous Hdh floxed mice were incubated with recombinant tat-cre protein, a fusion protein between cre and the protein transduction domain of human immunodeficiency virus tat protein that allows proteins to enter cells efficiently (Wadia et al., 2004). After a 60 min treatment with the tat-cre protein, cells were incubated for 48 h, at which time total protein samples were prepared and subjected to Western blot analysis. We found that recombinant tat-cre protein efficiently reduced the level of endogenous huntingtin protein such that <25% of normal levels of huntingtin remained at the time examined (Fig. 1A). Depletion of huntingtin in primary cortical neurons did not affect the level of motor proteins such as KHC and DHC (Fig. 1A).
Figure 1.

Depletion of huntingtin in primary mouse cortical neurons disrupts axonal transport of APP–YFP. A, Depletion of endogenous huntingtin by incubation of cortical neurons isolated from Hdh floxed mice with recombinant tat-cre protein. Con, Control; Tub, tubulin. B, Depletion of endogenous huntingtin from cortical neurons does not cause cell death. C, The velocity of both anterograde and retrograde movement of APP–YFP is reduced in huntingtin-depleted cortical neurons. Anterograde, 0.71 versus 0.45 μm/s, p = 0.002; retrograde 0.53 versus 0.38 μm/s, p < 0.032. D, The number of stationary APP–YFP particles increases whereas the number of anterograde moving particles decreases in huntingtin-depleted cortical neurons. Anterograde, 46 versus 26%, p < 0.05; stationary, 40 versus 60%, p < 0.05. N.S., Not significant. *p < 0.05; **p < 0.01. Error bars indicate SE.
Because complete loss of huntingtin results in embryonic lethal phenotypes in mice (Duyao et al., 1995; Nasir et al., 1995; Zeitlin et al., 1995), we tested whether depletion of huntingtin causes neuronal death. Forty-eight hours after tat-cre treatment, cortical neurons were stained with FDA and PI. After excitation, live cells emit green fluorescence, and dead cells display red fluorescence (Fig. 1B, middle panel). We found that depletion of huntingtin does not affect viability of cortical neurons at the 48 h time point because there are no red PI-positive neurons (Fig. 1B, top and bottom panels).
To evaluate the role of huntingtin in axonal transport, we monitored the kinesin- and dynein-dependent movement of APP–YFP in huntingtin-depleted primary cortical neurons. To differentiate axons from dendrites while imaging, we used the morphology of projections as a guide. In general, axons are long, whereas dendrites are shorter with multiple branches. To confirm these criteria, primary cortical neurons (4 DIV) were transfected with EB3-mCherry, a neuronal microtubule plus-end binding protein (Stepanova et al., 2003). We found that the movement of EB3-mCherry in long projections is uniformly outward (data not shown), a characteristic of axons. Thus, we recorded and analyzed APP–YFP movement in long projections. A total of 304 APP–YFP particles from 23 control cells and 353 APP–YFP particles from 33 tat-cre-treated cells were assessed. As shown (Fig. 1C), anterograde (0.71 vs 0.45 μm/s; p = 0.002) and retrograde (0.53 vs 0.38 μm/s; p = 0.032) movement of APP–YFP was slower in tat-cre-treated cells. In addition, we found that there was an increase in the number of stationary APP–YFP particles in tat-cre-treated cells, apparently at the expense of anterograde particles (Fig. 1D) (anterograde: 46 vs 26%, p < 0.05; stationary: 40 vs 60%, p < 0.05). Thus, depletion of huntingtin disrupts both directions of axonal APP movement in cultured primary cortical neurons.
Overexpression of full-length wild-type huntingtin enhances axonal transport of APP in primary cortical, striatal, and hippocampal neurons
Overexpression of full-length wild-type huntingtin increases transport of BDNF both in the NG108-15 retinoblastoma cells and primary cortical neurons (Gauthier et al., 2004). To test whether wild-type huntingtin enhances axonal transport of cargoes other than BDNF and whether the effect of huntingtin overexpression on axonal transport is limited to certain types of neurons, we monitored APP–YFP transport in cultured primary mouse cortical, striatal, and hippocampal neurons. To verify the cell-type composition of isolated cortical and striatal neurons, total proteins from cultured primary neurons were analyzed by Western blot assays. Consistent with previous reports (Altar et al., 1997), only cortical but not striatal neurons expressed BDNF (Fig. 2A). In contrast, striatal neurons expressed high levels of DARPP32 and GAD67 (Fig. 2A), two proteins that are highly expressed in striatal medium spiny neurons (MSNs) (Reiner et al., 1998). Thus, isolated cortical and striatal neurons express proteins characteristic of each type of neuron. To examine the effects of wild-type huntingtin overexpression on APP axonal transport, primary neurons were cotransfected with plasmids encoding APP–YFP and full-length wild-type human huntingtin. To estimate the level of huntingtin in transfected neurons, total proteins from cortical and striatal neurons were prepared 24 h after transfection and subjected to Western blot assays. The level of total huntingtin is ∼30% higher in cultured neurons cotransfected with APP–YFP and wild-type huntingtin compared with APP–YFP alone (Fig. 2B, compare middle, left panels). Because the transfection efficiency is ∼2–5% of total cells (measured by counting APP–YFP-positive neurons; data not shown), these data suggest that the level of huntingtin is ∼13- to 33-fold higher than endogenous mouse huntingtin in transfected cortical neurons. In transfected striatal neurons, the level of total huntingtin is ∼50% higher in cells cotransfected with APP–YFP and wild-type huntingtin compared with APP–YFP alone (Fig. 2C, compare middle, left panels), suggesting that the expression level of huntingtin is ∼10- to 25-fold higher than endogenous mouse huntingtin in transfected striatal neurons.
Figure 2.

Expression patterns of huntingtin. A, Western blot assays of primary cortical and striatal neurons. BDNF is expressed in cortical but not striatal neurons. Striatal neurons express high level of GAD67 and DARPP32 proteins. B, Expression of wild-type and mutant huntingtin in transfected primary cortical neurons. C, Expression of wild-type and mutant huntingtin in transfected primary striatal neurons. D, Western blot assay of soluble protein levels of wild-type (W.T.) and Hdh150Q/150Q mouse brains. There is no significant difference in the level of huntingtin, p150Glued, DHC, or KHC. The level of BDNF is lower in Hdh150Q/150Q mice. Tub, Tubulin.
To examine the effects of wild-type huntingtin overexpression on APP transport in primary cortical neurons, APP–YFP movement was recorded and analyzed as described previously. A total of 293 APP–YFP particles from 18 cells transfected with APP–YFP alone and 314 APP–YFP particles from 24 cells cotransfected with wild-type huntingtin were analyzed. Overexpression of full-length wild-type huntingtin increases the speed of both anterograde (0.86 vs 1.33 μm/s; p < 0.0001) and retrograde (0.69 vs 1.01 μm/s; p = 0.0003) APP–YFP movements in cortical neurons (Fig. 3A). Overexpression of wild-type huntingtin does not change the number of stationary APP–YFP particles in cortical neurons (Fig. 3B).
Figure 3.

Overexpression of full-length wild-type huntingtin enhances axonal transport of APP in primary cortical, striatal, and hippocampal neurons. A, Overexpression of wild-type huntingtin increases the velocity of both anterograde and retrograde APP–YFP movement in primary cortical neurons. Anterograde, 0.86 versus 1.33 μm/s, p < 0.0001; retrograde, 0.69 versus 1.01 μm/s, p = 0.0003. B, Overexpression of wild-type huntingtin does not change the distribution of moving APP–YFP particles in primary cortical neurons. C, Overexpression of wild-type huntingtin increases the velocity of both anterograde and retrograde APP–YFP movement in primary striatal neurons. Anterograde, 0.72 versus 0.99 μm/s, p < 0.001; retrograde, 0.47 versus 0.82 μm/s, p < 0.001. D, Overexpression of wild-type huntingtin does not change the distribution of moving APP–YFP particles in primary striatal neurons. E, Overexpression of wild-type huntingtin increases the velocity of both anterograde and retrograde APP–YFP movement in primary hippocampal neurons. Anterograde, 0.65 versus 0.85 μm/s, p < 0.001; retrograde, 0.37 versus 0.4 μm/s, p < 0.05. F, Overexpression of wild-type huntingtin does not change the distribution of moving APP–YFP particles in primary hippocampal neurons. N.S., Not significant. *p < 0.05; **p < 0.01. Error bars indicate SE.
In primary striatal neurons, a total of 226 APP–YFP particles from nine cells transfected with APP–YFP alone and 234 APP–YFP particles from 16 cells cotransfected with wild-type huntingtin were evaluated. We found that overexpression of full-length wild-type huntingtin increases the speed of both anterograde (0.72 vs 0.99 μm/s; p < 0.001) and retrograde (0.47 vs 0.82 μm/s; p = 0.003) APP–YFP movements in striatal neurons (Fig. 3C). Overexpression of wild-type huntingtin does not change the number of stationary APP–YFP particles in striatal neurons (Fig. 3D).
In primary mouse hippocampal neurons, a total of 372 APP–YFP particles from 11 cells transfected with APP–YFP alone and 185 APP–YFP particles from 10 cells cotransfected with wild-type huntingtin were analyzed. We found that overexpression of wild-type huntingtin increases the velocity of both anterograde (0.65 vs 0.85 μm/s; p < 0.001) and retrograde (0.37 vs 0.8 μm/s; p < 0.05) movement of APP–YFP (Fig. 3E). Overexpression of wild-type huntingtin does not change the number of stationary APP–YFP particles in hippocampal neurons (Fig. 3F).
Thus, overexpression of wild-type huntingtin accelerates anterograde and retrograde APP axonal transport in both cortical, striatal, and hippocampal neurons.
Overexpression of full-length mutant huntingtin increases the number of stationary APP–YFP particles in primary striatal and hippocampal but not cortical neurons
Because overexpression of wild-type huntingtin accelerates axonal transport of APP in cultured primary cortical and striatal neurons, we asked whether overexpression of mutant huntingtin has comparable effects. After transfection, to estimate the level of mutant huntingtin in transfected neurons, total proteins were prepared 24 h after transfection and subjected to Western blot assays. In transfected cortical neurons, the level of total huntingtin is ∼40% higher in cells cotransfected with mutant huntingtin (Fig. 2B, compare left, right panels). Taking into consideration that the transfection efficiency is ∼2–5%, the expression level of transfected mutant huntingtin is ∼8- to 20-fold higher than endogenous wild-type huntingtin in cortical neurons. In striatal neurons, the level of huntingtin is ∼88% higher in cells cotransfected with mutant huntingtin (Fig. 2C, compare left, right panels), suggesting that the level of transfected mutant huntingtin is ∼18- to 40-fold higher than endogenous wild-type huntingtin in striatal neurons.
In primary cortical neurons, a total of 293 APP–YFP particles from 18 cells transfected with APP–YFP alone and 286 APP–YFP particles from 18 cells cotransfected with mutant huntingtin were analyzed. We found that overexpression of full-length mutant huntingtin does not significantly change the velocity of either anterograde (0.86 vs 0.67 μm/s; p > 0.05) or retrograde (0.69 vs 0.53 μm/s; p > 0.05) APP–YFP movements in cortical neurons (Fig. 4A). Overexpression of full-length mutant huntingtin does not change the number of stationary APP–YFP particles in cortical neurons (Fig. 4B).
Figure 4.
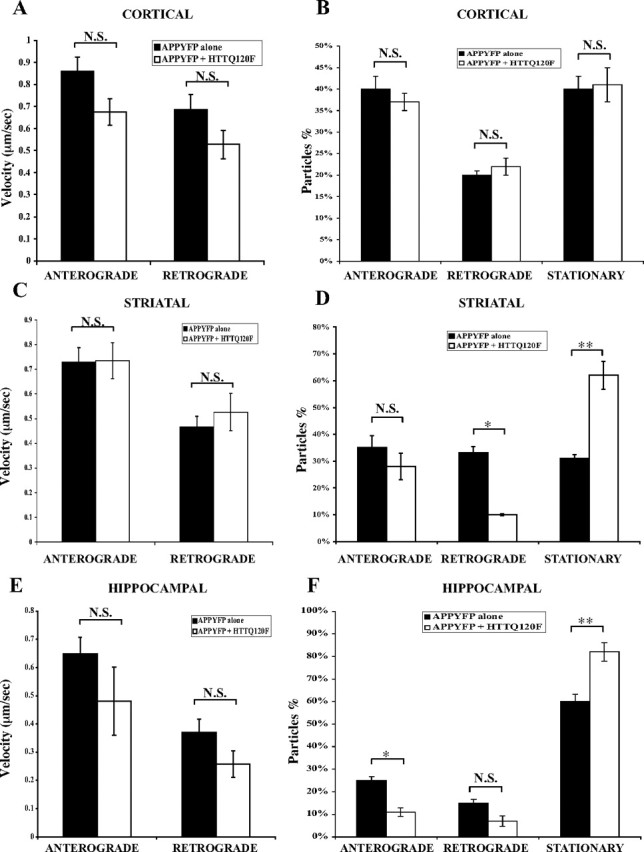
Overexpression of full-length mutant huntingtin increases the number of stationary APP–YFP particles in primary striatal and hippocampal, but not cortical, neurons. A, Overexpression of mutant huntingtin does not affect velocity of either anterograde or retrograde APP–YFP movement in primary cortical neurons. Anterograde, 0.86 versus 0.67 μm/s, p > 0.05; retrograde, 0.69 versus 0.53 μm/s, p > 0.05. B, Overexpression of mutant huntingtin does not change the distribution APP–YFP moving particles in primary cortical neurons. C, Overexpression of mutant huntingtin does not affect velocity of either anterograde or retrograde APP–YFP movement in primary striatal neurons. Anterograde, 0.73 versus 0.74 μm/s, p > 0.05; retrograde, 0.47 versus 0.52 μm/s, p > 0.05. D, Overexpression of mutant huntingtin increases the number of stationary APP–YFP particles whereas it decreases the number of retrograde moving particles in primary striatal neurons. Retrograde, 33 versus 10%, p < 0.05; stationary, 31 versus 62%, p < 0.01. E, Overexpression of mutant huntingtin does not affect velocity of either anterograde or retrograde APP–YFP transport in primary hippocampal neurons. Anterograde, 0.65 versus 0.48 μm/s, p > 0.05; retrograde, 0.37 versus 0.26 μm/s, p > 0.05. F, Overexpression of mutant huntingtin increases the number of stationary APP–YFP particles whereas it decreases the number of anterograde moving particles in primary hippocampal neurons. Anterograde, 25 versus 11%, p < 0.05; stationary, 60 versus 82%, p < 0.01. N.S., Not significant. *p < 0.05; **p < 0.01. Error bars indicate SE.
In primary striatal neurons, a total of 226 APP–YFP particles from 9 cells transfected with APP–YFP alone and 272 APP–YFP particles from 11 cells cotransfected with mutant huntingtin were evaluated. Similar to cortical neurons, overexpression of full-length mutant huntingtin does not significantly change the speed of either anterograde (0.73 vs 0.74 μm/s; p > 0.05) or retrograde (0.47 vs 0.52 μm/s; p > 0.05) APP–YFP movements in striatal neurons (Fig. 4C). However, overexpression of mutant huntingtin increases the number of stationary APP–YFP particles in striatal neurons (Fig. 4D).
In primary mouse hippocampal neurons, a total of 372 APP–YFP particles from 11 cells transfected with APP–YFP alone (controls from previous section) and 392 APP–YFP particles from 14 cells cotransfected with mutant huntingtin were evaluated. Similar to cortical and striatal neurons, overexpression of mutant full-length huntingtin does not significantly change the speed of either anterograde (0.65 vs 0.48 μm/s; p > 0.05) or retrograde (0.37 vs 0.26 μm/s; p > 0.05) APP–YFP movements in hippocampal neurons (Fig. 4E). Similar to the observation in striatal neurons, overexpression of mutant huntingtin increased the number of stationary APP–YFP particles in hippocampal neurons (Fig. 4F).
Thus, overexpression of mutant huntingtin does not affect the velocity of APP–YFP movement significantly in cortical, striatal, or hippocampal neurons. Interestingly, overexpression of mutant full-length huntingtin does increase the number of stationary APP–YFP particles in striatal and hippocampal, but not cortical, neurons, suggesting that there is a differential sensitivity to mutant huntingtin among different neuronal types.
APP transport is disrupted in striatal and hippocampal, but not cortical, neurons of homozygous Hdh150Q mice
Because overexpression of exogenous mutant huntingtin disrupts axonal transport by increasing the number of stationary particles in striatal neurons, we tested whether endogenous mutant huntingtin impairs axonal transport in cortical, striatal, or hippocampal neurons. To do this, APP–YFP movement was recorded and analyzed in primary neurons isolated from either homozygous Hdh150Q knock-in mice or littermate control wild-type mice. To estimate the level of mutant huntingtin in Hdh150Q/150Q knock-in mice and wild-type mice, total brains were analyzed by Western blot assays. The level of mutant huntingtin in Hdh150Q/150Q knock-in mice is comparable to that of wild-type mice (Fig. 2D) (Lin et al., 2001; Orr et al., 2008). In addition, we found that there is no significant difference in the level of motor proteins, such as p150Glued, DHC, and KHC, between wild-type and mutant mouse brains (Fig. 2D).
A total of 197 APP–YFP particles from 12 wild-type cortical neurons and 175 APP–YFP particles from 18 Hdh150Q/150Q cortical neurons were analyzed. Similar to the observation in cortical neurons expressing exogenous mutant huntingtin, there is no difference in the average velocity of APP–YFP movement between cells from wild-type and mutant cortical neurons (anterograde: 0.57 vs 0.58 μm/s, p > 0.05; retrograde: 0.39 vs 0.40 μm/s, p > 0.05) (Fig. 5A). In addition, endogenous mutant huntingtin does not change the number of stationary APP–YFP particles in primary cortical neurons (Fig. 5B).
Figure 5.
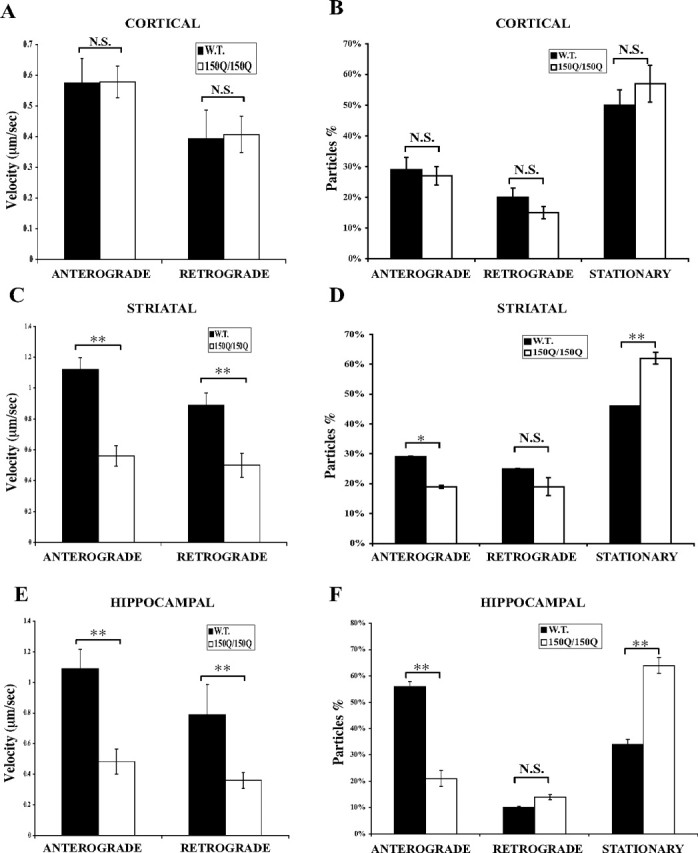
APP transport is disrupted in striatal and hippocampal, but not cortical, neurons of homozygous Hdh150Q mice. A, Endogenous mutant huntingtin does not affect velocity of either anterograde or retrograde APP–YFP movement in primary cortical neurons. Anterograde, 0.57 versus 0.58 μm/s, p > 0.05; retrograde, 0.39 versus 0.40 μm/s, p > 0.05. B, Endogenous mutant huntingtin does not change the distribution of APP–YFP moving particles in primary cortical neurons. C, Endogenous mutant huntingtin reduces the velocity of both anterograde and retrograde APP–YFP movement in primary striatal neurons. Anterograde, 1.12 versus 0.56 μm/s, p < 0.0001; retrograde, 0.89 versus 0.50 μm/s, p = 0.0016. D, Endogenous mutant huntingtin increases the number of stationary APP–YFP particles whereas it decreases the number of anterograde moving APP–YFP particles in primary striatal neurons. Anterograde, 29 versus 19%, p < 0.05; stationary, 46 versus 62%, p < 0.01. E, Endogenous mutant huntingtin reduces the velocity of both anterograde and retrograde APP–YFP movement in primary hippocampal neurons. Anterograde, 1.09 versus 0.48 μm/s, p = 0.0006; retrograde, 0.79 versus 0.36 μm/s, p = 0.0064. F, Endogenous mutant huntingtin increases the number of stationary APP–YFP particles whereas it decreases the number of anterograde moving APP–YFP particles in primary hippocampal neurons. Anterograde, 56 versus 21%, p < 0.01; stationary, 34 versus 64%, p < 0.01. W.T., Wild type; N.S., not significant. *p < 0.05; **p < 0.01. Error bars indicate SE.
In striatal neurons, a total of 330 APP–YFP particles from 7 wild-type striatal neurons and 232 APP–YFP particles from 10 Hdh150Q/150Q striatal neurons were recorded and analyzed. Surprisingly, endogenous mutant huntingtin reduces the velocity of both anterograde and retrograde APP–YFP movement in striatal neurons (anterograde: 1.12 vs 0.56 μm/s, p < 0.0001; retrograde: 0.89 vs 0.50 μm/s, p = 0.0016) (Fig. 5C). Similar to the observation in striatal neurons expressing exogenous mutant huntingtin, endogenous mutant huntingtin also increases the number of stationary APP–YFP particles in primary striatal neurons (Fig. 5D).
In hippocampal neurons, a total of 128 APP–YFP particles from 19 wild-type hippocampal neurons and 207 APP–YFP particles from 19 Hdh150Q/150Q hippocampal neurons were sampled. Similar to observations in striatal neurons, endogenous mutant huntingtin reduces the velocity of both anterograde (1.09 vs 0.48 μm/s; p = 0.0006) and retrograde (0.79 vs 0.36 μm/s; p = 0.0064) movement of APP–YFP (Fig. 5E). As shown in Figure 5F, we found that the number of stationary APP–YFP particles in hippocampal neurons isolated from Hdh150Q/150Q mice increased, apparently at the expense of anterograde particles.
Thus, similar to the results with overexpression of mutant huntingtin, axonal transport of APP–YFP is impaired in striatal and hippocampal, but not cortical, neurons of homozygous Hdh150Q mice, indicating a differential sensitivity to mutant huntingtin between different neuronal types.
BDNF transport is disrupted in striatal but not cortical neurons of homozygous Hdh150Q mice
HD patients experience preferential neuronal loss in the striatum. Survival of striatal neurons requires the growth factor BDNF that is made in the cortex and then transported to the striatum. Because endogenous mutant huntingtin disrupts axonal transport of APP–YFP in striatal but not cortical neurons, we asked whether endogenous mutant huntingtin impairs axonal transport of transfected BDNF in primary cortical and striatal neurons.
To monitor the effect of endogenous mutant huntingtin on transport of BDNF, primary mouse cortical neurons isolated from either wild-type or homozygous Hdh150Q knock-in mice were transfected with plasmid DNA encoding a rat BDNF-mCherry protein. A total of 245 rat BDNF-mCherry particles from 11 wild-type cells and 355 rat BDNF-mCherry particles from 16 Hdh150Q/150Q cells were studied. Rat BDNF-mCherry moves at speeds of 1.23 and 0.72 μm/s in anterograde and retrograde directions, respectively. There is no significant difference in the velocity of rat BDNF-mCherry movement in cortical neurons from wild-type or mutant neurons (Fig. 6A) (anterograde movement: 1.23 vs 1.06 μm/s, p > 0.05; retrograde movement: 0.72 vs 0.72 μm/s, p > 0.05). In addition, there is no difference in the distribution of moving particles of rat BDNF-mCherry between cortical neurons from wild-type and mutant cells (Fig. 6B).
Figure 6.

Retrograde BDNF transport is disrupted in striatal but not cortical neurons of homozygous Hdh150Q mice. A, Endogenous mutant huntingtin does not affect velocity of either anterograde or retrograde BDNF-mCherry movement in primary cortical neurons. Anterograde, 1.23 versus 1.06 μm/s, p > 0.05; retrograde, 0.72 versus 0.2 μm/s, p > 0.05. B, Endogenous mutant huntingtin does not change the distribution BDNF-mCherry moving particles in primary cortical neurons. C, Endogenous mutant huntingtin reduces the velocity of retrograde BDNF-mCherry movement in primary striatal neurons. Anterograde, 0.74 versus 0.64 μm/s, p > 0.05; retrograde, 0.86 versus 0.56 μm/s, p = 0.03. D, Endogenous mutant huntingtin increases the number of stationary BDNF-mCherry particles in primary striatal neurons. Anterograde, 30 versus 15%, p < 0.05; retrograde, 32 versus 10%, p < 0.05; stationary, 38 versus 65%, p < 0.05. W.T., Wild type; N.S., not significant. *p < 0.05. Error bars indicate SE.
In striatal neurons, a total of 216 BDNF-mCherry particles from 12 wild-type striatal neurons and 268 BDNF-mCherry particles from 13 Hdh150Q/150Q striatal neurons were evaluated. Endogenous mutant huntingtin reduces the velocity of retrograde but not anterograde BDNF-mCherry movement in striatal neurons (Fig. 6C) (retrograde: 0.86 vs 0.56 μm/s, p = 0.03; anterograde: 0.74 vs 0.64 μm/s, p > 0.05). Endogenous mutant huntingtin also increases the number of stationary BDNF-mCherry particles in primary striatal neurons (Fig. 6D). Thus, retrograde transport of BDNF is impaired in striatal but not cortical neurons of homozygous Hdh150Q knock-in mice.
Biochemical behavior of huntingtin, kinesin-I, HAP1, and dynactin complexes are not altered in HD model mice
Both loss and mutation of huntingtin result in fast axonal transport defects. Because huntingtin was reported to interact with both the anterograde and retrograde transport machinery, we examined the previously reported associations of huntingtin, HAP1, and motor protein complexes by sucrose gradient fractionation (Li et al., 1998; LaMonte et al., 2002; Gauthier et al., 2004). To do this, total mouse forebrain extracts were fractionated by 5–20% sucrose gradients. As expected, dynein (DHC) and dynactin (p150Glued and p50) migrate as large complexes of 17–19S close to the bottom of the gradient (Fig. 7A, left). Consistent with the finding that HAP1 interacts with p150Glued, HAP1 also migrates to the bottom of the gradient with a pattern similar to that of the dynein and dynactin complexes. In contrast, KHC is found in a smaller complex of ∼8–10S (fractions 5–7). The majority of huntingtin migrates between the kinesin and dynein/dynactin complexes with a size of about 15S. Only a small fraction of huntingtin comigrates with the KHC, dynein, or dynactin complexes.
Figure 7.

Biochemical behavior of huntingtin, kinesin-I, HAP1, and dynactin complexes are not altered in HD model mice. A, Sucrose gradient assay of brain extracts from wild-type and 8-month-old homozygous Hdh150Q knock-in mice. B, Sucrose gradient assay of brain extracts from wild-type and conditional Hdh knock-out mice. W.T., Wild type.
We then tested whether the proposed associations of huntingtin with kinesin-I, dynein, and dynactin complexes are altered in HD model mice. Total forebrain extracts of 8-month-old homozygous Hdh150Q knock-in mouse were subjected to fractionation on 5–20% sucrose gradients. There is no gross change in the pattern of the dynein and dynactin complexes in the Hdh150Q/150Q knock-in mice compared with littermate control mice (Fig. 7A, right). In addition, we found no change in the pattern of kinesin or HAP1 between mutant and control mice.
We also asked whether loss of huntingtin affects the interactions of huntingtin with kinesin and dynein/dynactin complexes. Total forebrain extracts of 6-month-old huntingtin conditional knock-out mice, in which the endogenous Hdh gene is removed by cre expressed from the synapsin promoter, were subjected to 5–20% sucrose gradient fractionation. We found no gross change in the pattern of the dynein and dynactin complexes in the brain extracts of huntingtin conditional knock-out mice compared with littermate control mice (Fig. 7B, right, Hdh floxed, syn-cre). Thus, the biochemical behaviors of huntingtin, kinesin-I, and dynein/dynactin complexes are not obviously altered by either reduction or mutation of huntingtin.
Discussion
Here we have examined the effects of loss of huntingtin and overexpression of both wild-type and mutant huntingtin on axonal transport in different neuronal types, including cortical, striatal, and hippocampal neurons. Summarized in Table 1, these data show that depletion of endogenous huntingtin disrupts both anterograde and retrograde APP axonal transport. In addition, overexpression of wild-type huntingtin increases velocity of APP movements in all three types of neurons tested, whereas overexpression of mutant huntingtin disrupts APP transport in striatal and hippocampal, but not cortical, neurons. Moreover, axonal transport of BDNF is disrupted in striatal but not cortical neurons of homozygous Hdh150Q knock-in mice (Table 2). Finally, although huntingtin is reported to interact with components of both anterograde and retrograde motors, neither reduction nor mutation of endogenous huntingtin appears to disrupt the kinesin-1, dynein, or dynactin complexes.
Table 1.
Summary of the effects of wild-type and mutant huntingtin on APP–YFP movement
| APP-YFP |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cortical |
Striatal |
Hippocampal |
||||||||||
| A |
R |
A |
R |
A |
R |
|||||||
| V | P | V | P | V | P | V | P | V | P | V | P | |
| Hdh knock-out | ↓ | ↓ | ↓ | ↓ | ND | ND | ND | ND | ||||
| ↑ HttQ23F | ↑ | ↔ | ↑ | ↔ | ↑ | ↔ | ↑ | ↔ | ↑ | ↔ | ↑ | ↔ |
| ↑ HttQ120F | ↔ | ↔ | ↔ | ↔ | ↔ | ↓ | ↔ | ↓ | ↔ | ↓ | ↔ | ↓ |
| Hdh150Q/150Q | ↔ | ↔ | ↔ | ↔ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ |
APP–YFP, Primary cultured neurons transfected with plasmid encoding human APP tagged with YFP; Hdh150Q/150Q, primary neurons isolated from Hdh150Q/150Q neonates; HttQ23F, primary neurons isolated from wild-type neonates transfected with plasmid DNA encoding full-length wild-type human huntingtin; HttQ120F, primary neurons isolated from wild-type neonates transfected with plasmid DNA encoding full-length human huntingtin with 120 glutamine residues; A, anterograde transport; R, retrograde transport; V, velocity; P, number of moving vesicles (decreases in P translate to increased numbers of stalled or nonmoving vesicles); ↓, reduced; ↔, no change; ↑, increased; ND, not determined. Note that all experiments are compared with APP–YFP transfections in wild-type cortical, striatal, and hippocampal primary cultures.
Table 2.
Summary of the effects of wild-type and mutant huntingtin on BDNF-mCherry movement
| BDNF-mCherry |
||||||||
|---|---|---|---|---|---|---|---|---|
| Cortical |
Striatal |
|||||||
| A |
R |
A |
R |
|||||
| V | P | V | P | V | P | V | P | |
| Hdh150Q/150Q | ↔ | ↔ | ↔ | ↔ | ↔ | ↓ | ↓ | ↓ |
BDNF-mCherry, Cells transfected with plasmid encoding rat BDNF tagged with mCherry fluorescent protein; Hdh150Q/150Q, primary neurons isolated from Hdh150Q/150Q neonates; A, anterograde transport; R, retrograde transport; V, velocity; P, number of moving vesicles (decreases in P translate to increased numbers of stalled or nonmoving vesicles, in principle lower processivity); ↓, reduced; ↔, no change. Note that all experiments are compared with transfections in wild-type cortical and striatal primary cultures.
Depletion of endogenous huntingtin disrupts axonal transport of APP in primary cortical neurons (Fig. 1). Together with previous findings in Drosophila and mouse neurons (Gunawardena et al., 2003; Gauthier et al., 2004; Trushina et al., 2004), these data establish that endogenous huntingtin is required for axonal transport of a variety of cargoes including APP, BDNF, and mitochondria in various types of neurons. However, the exact role of huntingtin in axonal transport remains unclear. Although huntingtin interacts with dynein intermediate chain, p150Glued, and kinesin light chain, loss of huntingtin does not affect either the level of or complexes of huntingtin, HAP1, dynactin, and kinesin-I (Figs. 2, 7A). Thus, dysfunction of axonal transport in the absence of huntingtin is not attributable to a disruption of motor protein complexes, but instead may result from altered regulation of intact complexes. Moreover, in contrast to published data (Gauthier et al., 2004), no changes in the biochemical behavior and potential interactions of huntingtin with HAP1 and motor protein complexes were observed in Hdh150Q/150Q brain extracts (Fig. 7A). To test whether this discrepancy is caused by differences in the HD model mice used (Hdh150Q/150Q vs Hdh109Q/109Q), age differences, or methods, we performed sucrose gradient fractionation of brain extracts of 14-month-old Hdh109Q/109Q using a 7.5–25% sucrose gradient as described previously (Gauthier et al., 2004). Again, we did not observe any change in the pattern of the kinesin-I, dynein, and dynactin complexes (data not shown). Although endogenous huntingtin is required for axonal transport of various cargoes, it is unclear whether mutant huntingtin disrupts all axonal transport. Mutant huntingtin was reported to impair movement of BDNF but not mitochondria in a cell line derived from the striatum of Hdh109Q/109Q knock-in mice (Gauthier et al., 2004). However, mutant huntingtin disrupts transport of mitochondria and unknown vesicles in striatal and cortical neurons (Trushina et al., 2004; Chang et al., 2006; Orr et al., 2008). Our data in striatal and hippocampal neurons supports the idea that mutant huntingtin disrupts axonal transport of various cargoes including BDNF, APP, and mitochondria. Why only BDNF transport but not mitochondria was altered in some previous experiments is unclear, but we suggest that the immortalized cell line derived from Hdh109Q/109Q mice may have properties different from unaltered striatal neurons.
The mechanism by which mutant huntingtin disrupts axonal transport remains elusive. Mutant huntingtin might form aggregates such that mutant huntingtin binds to cargoes and impairs their movement or physically blocks movement in axons (Chang et al., 2006; Orr et al., 2008). However, this is unlikely in our experiments because no aggregates were observed in the presymptomatic primary neurons used (Lin et al., 2001). Formation of huntingtin aggregates might precipitate motor proteins, thus reducing the soluble pool of motor proteins required for transport, as observed in both HD brain and Drosophila embryos (Gunawardena et al., 2003; Trushina et al., 2004). However, no significant difference in the levels of soluble kinesin-I and dynein were found in brains of HD model mice (Fig. 2D) (Orr et al., 2008)).
Because HD is a hereditary dominant disease, a great deal of effort has focused on gain-of-function phenotypes caused by mutant huntingtin. However, although the cellular functions of wild-type huntingtin are still not clear, it has been suggested that loss of wild-type huntingtin function also contributes to HD pathogenesis (Zuccato et al., 2001; Gauthier et al., 2004). Our findings that huntingtin is required for axonal transport and that mutant huntingtin lacks the ability of wild-type huntingtin to accelerate axonal transport suggest that loss of wild-type huntingtin function in fast axonal transport plays an important role in the development of HD. Overexpression of wild-type huntingtin also protects cells from excitotoxicity and apoptosis (Rigamonti et al., 2000; Ho et al., 2001; Leavitt et al., 2001, 2006; Rigamonti et al., 2001). We suggest that enhanced axonal transport and thus enhanced supply of some materials to synapses might also aid in neuroprotection.
Patients with HD suffer from motor dysfunction and exhibit preferential neuronal loss in the striatum. In striatal neurons, considerable evidence suggests that transport of various cargoes, such as BDNF, APP, and mitochondria, is disrupted (Fig. 4) (Trushina et al., 2004; Orr et al., 2008). These data indicate that transport defects in presymptomatic neurons may be an important contributor to selective neurodegeneration. Disruption of transport could cause mislocalization of mitochondria, leading to apoptosis. Alternatively, defective transport could deplete proteins required for synaptic functions and for their survival, such as BDNF.
Striatal MSNs receive synaptic input and BDNF from the cortex via the corticostriatal pathway (Altar et al., 1997). In addition to the cortex, striatal MSNs also receive BDNF from the substantia nigra, another region that expresses BDNF (Mufson et al., 1994, 1999; Rite et al., 2003). Intriguingly, we found that axonal transport of either APP or BDNF is not affected by mutation of huntingtin in primary mouse cortical neurons (Figs. 4–6, Table 2). Moreover, APP transport is not affected in primary cortical neurons from homozygous Hdh109Q mice either (data not shown). Together with previous data (Gauthier et al., 2004; Chang et al., 2006), these data indicate that axonal transport in cortical neurons is more resistant to polyQ mutation of huntingtin. In contrast, mutant huntingtin affects axonal transport of APP and retrograde transport of BDNF in primary striatal neurons (Figs. 4–6). Thus, we suggest that selective loss of MSN is not attributable to a lack of anterograde BDNF transport in cortical neurons. Instead, our observations raise the possibility that global transport defects, especially reduced retrograde transport of endocytosed BDNF secreted by substantia nigra neurons, might lead to MSN degeneration in HD. Additional experiments are required to examine whether the secretion of BDNF by cortical neurons and uptake of secreted BDNF by striatal neurons are altered by mutation of huntingtin. In addition, it will be crucial to examine the transport of endocytosed BDNF in striatal neurons from HD model mice.
Footnotes
This work was supported by grants from the Hereditary Disease Foundation and the HighQ Foundation to L.S.B.G. L.-S. H. was supported by a fellowship from the Hereditary Disease Foundation. L.S.B.G. is an investigator of the Howard Hughes Medical Institute. We thank members of the Goldstein laboratory, especially Dr. Angels Almenar, Emily A. Davis, Dr. Shauna Yuan, and Dr. Zhaohuai Yang for critical reading and comments. We thank Dr. Carlos Dotti for pCMV-APPYFP plasmid, Dr. Zhaohuai Yang for pCAG-APPYFP plasmid, Dr. Shuo-Chien Ling for pEB3-mCherry plasmid, Dr. Xiao-Jiang Li for pEBV-HttQ23F and pEBV-HttQ120F plasmids, Dr. Gary Banker for pJPA5-BDNF-mCherry plasmid, Dr. Steve F. Dowdy and Dr. Jehangir S. Wadia for pTAT-Cre plasmid and helpful advice, Dr. Scott Zeitlin for HdhFlox mice, and Dr. Peter J. Detloff for Hdh150Q mice.
References
- Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, Acheson AL, Lindsay RM, Wiegand SJ. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature. 1997;389:856–860. doi: 10.1038/39885. [DOI] [PubMed] [Google Scholar]
- Bhide PG, Day M, Sapp E, Schwarz C, Sheth A, Kim J, Young AB, Penney J, Golden J, Aronin N, DiFiglia M. Expression of normal and mutant huntingtin in the developing brain. J Neurosci. 1996;16:5523–5535. doi: 10.1523/JNEUROSCI.16-17-05523.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block-Galarza J, Chase KO, Sapp E, Vaughn KT, Vallee RB, DiFiglia M, Aronin N. Fast transport and retrograde movement of huntingtin and HAP1 in axons. Neuroreport. 1997;8:2247–2251. doi: 10.1097/00001756-199707070-00031. [DOI] [PubMed] [Google Scholar]
- Brewer GJ. Serum-free B27/neurobasal medium supports differentiated growth of neurons from the striatum, substantia nigra, septum, cerebral cortex, cerebellum, and dentate gyrus. J Neurosci Res. 1995;42:674–683. doi: 10.1002/jnr.490420510. [DOI] [PubMed] [Google Scholar]
- Brewer GJ. Isolation and culture of adult rat hippocampal neurons. J Neurosci Methods. 1997;71:143–155. doi: 10.1016/s0165-0270(96)00136-7. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Caviston JP, Ross JL, Antony SM, Tokito M, Holzbaur EL. Huntingtin facilitates dynein/dynactin-mediated vesicle transport. Proc Natl Acad Sci U S A. 2007;104:10045–10050. doi: 10.1073/pnas.0610628104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DT, Rintoul GL, Pandipati S, Reynolds IJ. Mutant huntingtin aggregates impair mitochondrial movement and trafficking in cortical neurons. Neurobiol Dis. 2006;22:388–400. doi: 10.1016/j.nbd.2005.12.007. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase K, Schwarz C, Meloni A, Young C, Martin E, Vonsattel JP, Carraway R, Reeves SA, Boyce FM, Aronin N. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron. 1995;14:1075–1081. doi: 10.1016/0896-6273(95)90346-1. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- Dragatsis I, Levine MS, Zeitlin S. Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat Genet. 2000;26:300–306. doi: 10.1038/81593. [DOI] [PubMed] [Google Scholar]
- Duncan JE, Goldstein LS. The genetics of axonal transport and axonal transport disorders. PLoS Genet. 2006;2:e124. doi: 10.1371/journal.pgen.0020124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyao MP, Auerbach AB, Ryan A, Persichetti F, Barnes GT, McNeil SM, Ge P, Vonsattel JP, Gusella JF, Joyner AL, MacDonald ME, et al. Inactivation of the mouse Huntington's disease gene homolog Hdh. Science. 1995;269:407–410. doi: 10.1126/science.7618107. [DOI] [PubMed] [Google Scholar]
- Engelender S, Sharp AH, Colomer V, Tokito MK, Lanahan A, Worley P, Holzbaur EL, Ross CA. Huntingtin-associated protein 1 (HAP1) interacts with the p150Glued subunit of dynactin. Hum Mol Genet. 1997;6:2205–2212. doi: 10.1093/hmg/6.13.2205. [DOI] [PubMed] [Google Scholar]
- Folstein SE. The Johns Hopkins series in contemporary medicine and public health. Baltimore: Johns Hopkins UP; 1989. Huntington's disease: a disorder of families; p. 251. [Google Scholar]
- Gauthier LR, Charrin BC, Borrell-Pages M, Dompierre JP, Rangone H, Cordelieres FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, Saudou F. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Goldstein LSB, Yang Z. Microtubule-based transport systems in neurons: the roles of kinesins and dyneins. Annu Rev Neurosci. 2000;23:39–71. doi: 10.1146/annurev.neuro.23.1.39. [DOI] [PubMed] [Google Scholar]
- Gunawardena S, Her LS, Brusch RG, Laymon RA, Niesman IR, Gordesky-Gold B, Sintasath L, Bonini NM, Goldstein LS. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron. 2003;40:25–40. doi: 10.1016/s0896-6273(03)00594-4. [DOI] [PubMed] [Google Scholar]
- Harper PS. Huntington's disease. Ed 2. London: Saunders; 1996. [Google Scholar]
- Ho LW, Brown R, Maxwell M, Wyttenbach A, Rubinsztein DC. Wild type Huntingtin reduces the cellular toxicity of mutant Huntingtin in mammalian cell models of Huntington's disease. J Med Genet. 2001;38:450–452. doi: 10.1136/jmg.38.7.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson M, Gentleman S, Lennox G, Ward L, Gray T, Randall K, Morrell K, Lowe J. The cortical neuritic pathology of Huntington's disease. Neuropathol Appl Neurobiol. 1995;21:18–26. doi: 10.1111/j.1365-2990.1995.tb01024.x. [DOI] [PubMed] [Google Scholar]
- Kaether C, Skehel P, Dotti CG. Axonal membrane proteins are transported in distinct carriers: a two-color video microscopy study in cultured hippocampal neurons. Mol Biol Cell. 2000;11:1213–1224. doi: 10.1091/mbc.11.4.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal A, Stokin GB, Yang Z, Xia CH, Goldstein LS. Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron. 2000;28:449–459. doi: 10.1016/s0896-6273(00)00124-0. [DOI] [PubMed] [Google Scholar]
- LaMonte BH, Wallace KE, Holloway BA, Shelly SS, Ascano J, Tokito M, Van Winkle T, Howland DS, Holzbaur EL. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron. 2002;34:715–727. doi: 10.1016/s0896-6273(02)00696-7. [DOI] [PubMed] [Google Scholar]
- Leavitt BR, Guttman JA, Hodgson JG, Kimel GH, Singaraja R, Vogl AW, Hayden MR. Wild-type huntingtin reduces the cellular toxicity of mutant huntingtin in vivo. Am J Hum Genet. 2001;68:313–324. doi: 10.1086/318207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leavitt BR, van Raamsdonk JM, Shehadeh J, Fernandes H, Murphy Z, Graham RK, Wellington CL, Raymond LA, Hayden MR. Wild-type huntingtin protects neurons from excitotoxicity. J Neurochem. 2006;96:1121–1129. doi: 10.1111/j.1471-4159.2005.03605.x. [DOI] [PubMed] [Google Scholar]
- Lee WC, Yoshihara M, Littleton JT. Cytoplasmic aggregates trap polyglutamine-containing proteins and block axonal transport in a Drosophila model of Huntington's disease. Proc Natl Acad Sci U S A. 2004;101:3224–3229. doi: 10.1073/pnas.0400243101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Wyman T, Yu ZX, Li SH, Li XJ. Abnormal association of mutant huntingtin with synaptic vesicles inhibits glutamate release. Hum Mol Genet. 2003;12:2021–2030. doi: 10.1093/hmg/ddg218. [DOI] [PubMed] [Google Scholar]
- Li SH, Gutekunst CA, Hersch SM, Li XJ. Interaction of huntingtin-associated protein with dynactin P150Glued. J Neurosci. 1998;18:1261–1269. doi: 10.1523/JNEUROSCI.18-04-01261.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Tallaksen-Greene S, Chien WM, Cearley JA, Jackson WS, Crouse AB, Ren S, Li XJ, Albin RL, Detloff PJ. Neurological abnormalities in a knock-in mouse model of Huntington's disease. Hum Mol Genet. 2001;10:137–144. doi: 10.1093/hmg/10.2.137. [DOI] [PubMed] [Google Scholar]
- McGuire JR, Rong J, Li SH, Li XJ. Interaction of Huntingtin-associated protein-1 with kinesin light chain: implications in intracellular trafficking in neurons. J Biol Chem. 2006;281:3552–3559. doi: 10.1074/jbc.M509806200. [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Kroin JS, Sobreviela T, Burke MA, Kordower JH, Penn RD, Miller JA. Intrastriatal infusions of brain-derived neurotrophic factor: retrograde transport and colocalization with dopamine containing substantia nigra neurons in rat. Exp Neurol. 1994;129:15–26. doi: 10.1006/exnr.1994.1143. [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Kroin JS, Sendera TJ, Sobreviela T. Distribution and retrograde transport of trophic factors in the central nervous system: functional implications for the treatment of neurodegenerative diseases. Prog Neurobiol. 1999;57:451–484. doi: 10.1016/s0301-0082(98)00059-8. [DOI] [PubMed] [Google Scholar]
- Nasir J, Floresco SB, O'Kusky JR, Diewert VM, Richman JM, Zeisler J, Borowski A, Marth JD, Phillips AG, Hayden MR. Targeted disruption of the Huntington's disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995;81:811–823. doi: 10.1016/0092-8674(95)90542-1. [DOI] [PubMed] [Google Scholar]
- Orr AL, Li S, Wang CE, Li H, Wang J, Rong J, Xu X, Mastroberardino PG, Greenamyre JT, Li XJ. N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J Neurosci. 2008;28:2783–2792. doi: 10.1523/JNEUROSCI.0106-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A, Perera M, Paullus R, Medina L. Immunohistochemical localization of DARPP32 in striatal projection neurons and striatal interneurons in pigeons. J Chem Neuroanat. 1998;16:17–33. doi: 10.1016/s0891-0618(98)00056-8. [DOI] [PubMed] [Google Scholar]
- Rigamonti D, Bauer JH, De-Fraja C, Conti L, Sipione S, Sciorati C, Clementi E, Hackam A, Hayden MR, Li Y, Cooper JK, Ross CA, Govoni S, Vincenz C, Cattaneo E. Wild-type huntingtin protects from apoptosis upstream of caspase-3. J Neurosci. 2000;20:3705–3713. doi: 10.1523/JNEUROSCI.20-10-03705.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigamonti D, Sipione S, Goffredo D, Zuccato C, Fossale E, Cattaneo E. Huntingtin's neuroprotective activity occurs via inhibition of procaspase-9 processing. J Biol Chem. 2001;276:14545–14548. doi: 10.1074/jbc.C100044200. [DOI] [PubMed] [Google Scholar]
- Rite I, Venero JL, Tomas-Camardiel M, Machado A, Cano J. Expression of BDNF mRNA in substantia nigra is dependent on target integrity and independent of neuronal activation. J Neurochem. 2003;87:709–721. doi: 10.1046/j.1471-4159.2003.02041.x. [DOI] [PubMed] [Google Scholar]
- Ross CA, Margolis RL. Huntington's disease. Clin Neurosci Res. 2001;1:142–152. [Google Scholar]
- Sapp E, Penney J, Young A, Aronin N, Vonsattel JP, DiFiglia M. Axonal transport of N-terminal huntingtin suggests early pathology of corticostriatal projections in Huntington disease. J Neuropathol Exp Neurol. 1999;58:165–173. doi: 10.1097/00005072-199902000-00006. [DOI] [PubMed] [Google Scholar]
- Stepanova T, Slemmer J, Hoogenraad CC, Lansbergen G, Dortland B, De Zeeuw CI, Grosveld F, van Cappellen G, Akhmanova A, Galjart N. Visualization of microtubule growth in cultured neurons via the use of EB3-GFP (end-binding protein 3-green fluorescent protein) J Neurosci. 2003;23:2655–2664. doi: 10.1523/JNEUROSCI.23-07-02655.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LS. Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science. 2005;307:1282–1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- Szebenyi G, Morfini GA, Babcock A, Gould M, Selkoe K, Stenoien DL, Young M, Faber PW, MacDonald ME, McPhaul MJ, Brady ST. Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport. Neuron. 2003;40:41–52. doi: 10.1016/s0896-6273(03)00569-5. [DOI] [PubMed] [Google Scholar]
- The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Trushina E, Dyer RB, Badger JD, Ure D, Eide L, Tran DD, Vrieze BT, Legendre-Guillemin V, McPherson PS, Mandavilli BS, Van Houten B, Zeitlin S, McNiven M, Aebersold R, Hayden M, Parisi JE, Seeberg E, Dragatsis I, Doyle K, Bender A, Chacko C, McMurray CT. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol Cell Biol. 2004;24:8195–8209. doi: 10.1128/MCB.24.18.8195-8209.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velier J, Kim M, Schwarz C, Kim TW, Sapp E, Chase K, Aronin N, DiFiglia M. Wild-type and mutant huntingtins function in vesicle trafficking in the secretory and endocytic pathways. Exp Neurol. 1998;152:34–40. doi: 10.1006/exnr.1998.6832. [DOI] [PubMed] [Google Scholar]
- Wadia JS, Stan RV, Dowdy SF. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat Med. 2004;10:310–315. doi: 10.1038/nm996. [DOI] [PubMed] [Google Scholar]
- Zeitlin S, Liu JP, Chapman DL, Papaioannou VE, Efstratiadis A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington's disease gene homologue. Nat Genet. 1995;11:155–163. doi: 10.1038/ng1095-155. [DOI] [PubMed] [Google Scholar]
- Zhou H, Cao F, Wang Z, Yu ZX, Nguyen HP, Evans J, Li SH, Li XJ. Huntingtin forms toxic NH2-terminal fragment complexes that are promoted by the age-dependent decrease in proteasome activity. J Cell Biol. 2003;163:109–118. doi: 10.1083/jcb.200306038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]