

Abstract
CNS injury-induced hemorrhage and tissue damage leads to excess iron, which can cause secondary degeneration. The mechanisms that handle this excess iron are not fully understood. We report that spinal cord contusion injury (SCI) in mice induces an “iron homeostatic response” that partially limits iron-catalyzed oxidative damage. We show that ceruloplasmin (Cp), a ferroxidase that oxidizes toxic ferrous iron, is important for this process. SCI in Cp-deficient mice demonstrates that Cp detoxifies and mobilizes iron and reduces secondary tissue degeneration and functional loss. Our results provide new insights into how astrocytes and macrophages handle iron after SCI. Importantly, we show that iron chelator treatment has a delayed effect in improving locomotor recovery between 3 and 6 weeks after SCI. These data reveal important aspects of the molecular control of CNS iron homeostasis after SCI and suggest that iron chelator therapy may improve functional recovery after CNS trauma and hemorrhagic stroke.
Keywords: CNS injury, hemorrhage, macrophage, free radicals, iron chelation, inflammation
Introduction
Free radicals, which can modify and damage DNA, proteins, and lipids, have been implicated as important causative agents in the secondary degeneration that occurs after spinal cord injury (SCI) (Hausmann, 2003). Iron can catalyze the formation of free radicals because of its redox active nature. Ferrous iron (Fe2+) reacts with metabolic hydrogen peroxide to produce harmful hydroxyl radicals, which can then lead to the production of a number of reactive oxygen species (Eaton and Qian, 2002). After injury, iron is released from intracellular reserves and from hemoglobin in red blood cells (RBCs). The latter is an important source of excess iron after SCI. The heme in hemoglobin is broken down via hemoxygenase 1 (HO-1) to ferrous iron and biliverdin. The expression of HO-1 and formation and effects of hydroxyl radicals after SCI have been studied (Mautes et al., 1998; Bao and Liu, 2004; Liu et al., 2004). However, how iron from RBCs and damaged tissue is handled and mobilized out of the spinal cord has not been examined.
Cellular iron homeostasis requires molecules involved in iron import and export across the cell membrane, including transporters and ferroxidases, as well as iron storage proteins. In this work, we examine changes in the expression of these molecules after SCI and focus on the role of the ferroxidase ceruloplasmin, which converts toxic ferrous iron to its nontoxic ferric form. The liver produces a secreted form of ceruloplasmin (Cp) that is abundant in serum but does not cross the blood–brain barrier. We have shown that an alternatively spliced, glycosylphosphatidylinositol (GPI) anchored form of Cp (135 kDa) is expressed by astrocytes and is the major form of Cp in the CNS (Patel and David, 1997; Patel et al., 2000). We also showed that GPI–Cp associates with the iron exporter ferroportin (Fpn) on the cell surface and is required for iron efflux out of astrocytes in vitro (Jeong and David, 2003). Ceruloplasmin, therefore, converts toxic ferrous iron to its nontoxic ferric form and also plays a role in the efflux of iron from cells. Null mutations of the Cp gene in humans (aceruloplasminemia) results in age-dependant iron accumulation in the CNS and neurodegeneration (Miyajima et al., 1987; Hellman and Gitlin, 2002). Cp null mice also show slow iron accumulation in the CNS starting at 12–18 months of age (Patel et al., 2002; Jeong and David, 2006). We now present data on the role of Cp in the iron-homeostatic response after SCI in young adult mice and show that its absence increases secondary damage and functional loss. For this study, we used 2- to 4-month-old Cp−/− mice, an age at which iron accumulation does not occur in the uninjured spinal cord. We also provide evidence that the iron homeostatic response in wild-type mice is inadequate, because iron chelation treatment has a delayed effect in improving locomotor recovery.
Materials and Methods
Animals.
Cp+/+ and Cp−/− mice (litter mates) on a C57BL/6 background between 2 and 4 months of age were used for spinal cord contusion injury (70 kdyn, 600–800 μm displacement) using the Infinite Horizons impactor (Precision Scientific Instrumentation). Mice were deeply anesthetized with a mixture of ketamine, xylazine, and acepromazine, and the contusion injury was performed as described previously (Ghasemlou et al., 2005). Cp−/− mice were generated in our laboratory as described previously (Patel et al., 2002). For the chelator experiments on wild-type mice, contusion injury (60 kdyn, 600–700 μm displacement) was performed on 6- to 8-week-old C57BL/6 mice purchased from Charles River Canada. Using the nine-point Basso mouse scale (BMS) (Basso et al., 2006), two observers trained in the Basso laboratory at Ohio State University scored locomotor behavior independently, and consensus scores were recorded. These analyses were performed by observers blinded to experimental groups. All procedures were approved by the McGill University Animal Care Committee and followed the guidelines of the Canadian Council on Animal Care.
Western blotting.
Approximately 4 mm of the injured spinal cords containing the injury site were dissected out 1, 3, 7, and 21 d post injury (dpi) after intracardiac perfusion with 10 ml of 0.1 m phosphate buffer. Total protein was extracted using radioimmunoprecipitation assay buffer as described previously (Jeong and David, 2003). Twenty micrograms of total protein were loaded onto a 4–12% Novex Bis-Tris gel (Invitrogen) and then transferred to polyvinylidene fluoride membrane (Millipore). Membranes were blocked with 3% nonfat milk in PBS–Tween 20 (0.05%) and incubated at 4°C overnight with anti-ceruloplasmin (1:800; DakoCytomation), anti-ferritin (1:500; DakoCytomation), and anti-DMT1 (divalent metal transporter 1) and anti-Fpn (1:000; Alpha Diagnostics). Blots were washed and incubated with peroxidase-conjugated IgG (1:200,000; Jackson ImmunoResearch) and detected with Western Lightning Chemiluminescence Reagent Plus (PerkinElmer Life and Analytical Sciences). Blots were subsequently reprobed with rabbit anti-actin (1:200; Sigma-Aldrich) to confirm equal loading. Experiments were done in triplicate, and representative blots are shown.
Iron histochemistry.
Cp+/+ and Cp−/− mice were perfused with 0.1 m phosphate buffer, followed by 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.2. Modified Perl's histochemistry was performed on 14 μm cryostat sections of the spinal cord to detect iron accumulation as described previously (Smith et al., 1997). Briefly, sections were incubated with 4% potassium ferrocyanide and 4% HCl, followed by a series of incubations with diaminobenzidine (DAB) and hydrogen peroxide and counterstained with 0.02% methyl green (all from Sigma-Aldrich).
To detect ferrous iron, the modified perfusion–Turnbull's technique was used (Meguro et al., 2003). Animals were perfused with a solution containing 1% HCl, 1% potassium ferricyanide, and 4% paraformaldehyde. Tissue was then removed, cryopreserved, and cryostat sectioned. Sections were incubated with DAB to intensify the stain and counterstained with methyl green.
Flame atomic absorption spectrometry.
Iron content was measured using flame atomic absorption spectrometry (fAAS) as described previously (Santos et al., 2000). Briefly, 4 mm length of spinal cord tissue containing the injury site were removed and then dried for 20 h at 106°C. Samples were weighed and then ashed at 500°C for 17 h, dissolved in 6N HCl, and then diluted to a final concentration of 1.2N HCl. fAAS analysis was done using a Analyst 400 (PerkinElmer Life and Analytical Sciences). Each sample was a pool of two animals, and a total of six samples for each genotype were used. Duplicate readings were obtained on each sample, and a standard curve used to calculate the amount of iron.
Quantitative real-time reverse transcription-PCR.
Total RNA was isolated from 4 mm length of the spinal cord containing the injury site of Cp+/+ and Cp−/− mice at 1, 3, 7, and 21 dpi using the RNeasy Lipid Tissue kit (Qiagen), and quantitative real-time reverse transcription (QRT)-PCR was performed using the Brilliant Probe-based QRT-PCR Reagents and MX4000 (Stratagene), both following the protocols of the manufacturer. Gene-specific primers and Taqman probes were generated using PrimerQuest (Integrated DNA Technology) (Jeong and David, 2006). QRT-PCR for peptidylprolyl isomerase A (PPIA) was performed for use as an internal control (Feroze-Merzoug et al., 2002). For data analysis, the sample Ct values were normalized to that of PPIA and expressed as fold increase. Results are shown as the mean ± SD relative ratio (fold increase) of mRNA from three separate experiments, n = 3 animals per group.
Immunofluorescence, immunohistochemical, and histological analyses.
Double-immunofluorescence labeling was performed as described previously (Jeong and David, 2006). Animals were perfused with 0.1 m phosphate buffer, followed by 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.2, and 14 μm cryostat sections were obtained. Tissue sections were incubated with 0.1 m PBS containing 2% normal goat serum and 1% ovalbumin to block nonspecific binding of antibodies. Tissue was incubated overnight with primary antibodies at the following concentrations: rabbit anti-ceruloplasmin (1:200; DakoCytomation), rat anti-GFAP (1:800; Zymed), rat anti-CD11b/Mac-1 (1:400; Serotec), rabbit anti-Fpn (1:400; Alpha Diagnostics), rabbit anti-DMT1 (1:400; Alpha Diagnostics), rabbit anti-ferritin (1:100; DakoCytomation), rabbit anti-inducible nitric oxide synthase (iNOS) (1:200; BD PharMingen), anti-nitrotyrosine (1:200; Upstate Biotechnology), and anti-4-hydroxynonenal (4-HNE) (1:100; Alpha Diagnostics). For immunofluorescence double labeling, sections were incubated for 1 h at room temperature with fluorescein-conjugated goat anti-rabbit IgG (1:400; Jackson ImmunoResearch) and rhodamine-conjugated goat anti-rat IgG (1:400; Jackson ImmunoResearch). Immunohistochemical stains were visualized by immunoperoxidase labeling using biotinylated secondary antibody and Vectastain Elite ABC kit, followed by color reaction using diaminobenzidine (Sigma-Aldrich). Motor neurons were stained by incubating tissue in 0.1% cresyl violet (Sigma-Aldrich) for 10 min, followed by dehydration through ascending alcohols and Hemo-De (Thermo Fisher Scientific).
Oxyblot.
Protein carbonyl content in Cp+/+ and Cp−/− tissue homogenates was determined using the Oxyblot protein oxidation detection kit (Millipore Bioscience Research Reagents). Briefly, 20 μg of protein was derivatized with 2,4-dinitrophenylhydrazine for 15 min at room temperature. After quenching, equal volumes of all samples were transferred onto nitrocellulose membrane using slot-blot apparatus. After transfer, membranes were blocked with 1% BSA/PBS–Tween 20 and then probed overnight with anti-2,4-dinitrophenol primary antibody at 1:200. The following day, blots were incubated with HRP-conjugated secondary antibody at 1:300, and bands were detected using Western Lightning Chemiluminescence Reagent Plus (PerkinElmer Life and Analytical Sciences) and Hyperfilm (GE Healthcare). Band intensity was quantified using Imagequant (GE Healthcare)
Image analysis and quantification.
All sections were viewed using an Axioskop2 Plus microscope (Zeiss), and images were captured using a QImaging Retiga 1300C camera. Image analysis was performed with BioQuant Nova Prime (BioQuant Image Analysis) to quantify the extent of Perl's, Luxol fast blue, and 4-HNE staining in spinal cord cross sections. Quantification of these stainings was done using the threshold function of the BioQuant Nova Prime tissue analysis software. The threshold was set by selecting all positively stained pixels. This approach eliminates inclusion of weak labeling or background staining from the analysis. The same threshold settings were then applied to every slide analyzed. All analyses were done blind. Percentage myelin spared for each genotype was obtained using the following formula: ratio of myelin to spinal cord (injured)/ratio of myelin to spinal cord (uninjured) × 100. For quantification of iron staining, four sections containing the lesion site were analyzed for pixel counts, per animal.
Systemic iron chelation.
We used a lipophilic iron chelator salicylaldehyde isonicotinoyl hydrazone (SIH), an analog of pyridoxal isonicotinoyl hydrazone that shows high affinity and selectivity for iron (Richardson and Ponka, 1998b). These chelators have been used in a number of in vivo and in vitro models, and the dosage we used was based on previous in vivo work (Richardson and Ponka, 1998a; Klimtova et al., 2003; Simunek et al., 2005, 2008a,b; Sterba et al., 2005, 2006, 2007). An SIH solution was prepared as reported previously (Klimtova et al., 2003). SIH solution was prepared fresh and injected at 1.25 mg/25 g body intraperitoneally.
Statistical analyses.
Data are shown as mean ± SEM. QRT-PCR and histological assessments analyzed using a two-way ANOVA with post hoc Tukey's test. Statistical analyses of the behavioral assessments were performed using two-way repeated measures (RM) ANOVA with post hoc Tukey's test for multiple comparisons or t test for subscores. The t test was also used for other analyses presented as indicated in the figure legends. Differences were considered significant at p < 0.05.
Results
Cp expression is rapidly induced and maintained after SCI
We first assessed the expression of Cp by Western blot analysis of spinal cord tissue after contusion injury. Cp protein is markedly increased at 1 and 3 dpi (approximately sixfold) and remains elevated approximately threefold at 7 dpi. At 21 dpi, Cp expression is increased approximately fourfold above levels in uninjured controls (Fig. 1a). Because Cp is abundant in serum, the sixfold increase in Cp protein seen by Western blot at 1 dpi may be a result of serum Cp present in the spinal cord attributable to tissue damage and hemorrhage. To further assess Cp upregulation after SCI, we performed QRT-PCR analysis. No increase in Cp mRNA was detected at day 1 after injury, further suggesting that the increased Cp protein in tissue at this time may be derived from serum. At later time points, Cp mRNA is increased by approximately twofold at 3 and 7 dpi and reaches approximately fourfold at 21 dpi (Fig. 1b), suggesting de novo expression of Cp after SCI. The expression of Cp in the contused spinal cord was confirmed by double-immunofluorescence labeling. There is a low level of Cp immunoreactivity in astrocytes in the uninjured spinal cord (Fig. 1c–e). In the first few days after injury, Cp staining at the lesion site was diffuse, likely attributable to the continued presence of serum Cp (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). At later time points, Cp immunoreactivity localizes to regions that surround the lesion (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) and is mainly expressed in GFAP+ astrocytes and their processes (Fig. 1f–h), with Mac 1+ macrophages located within the core of the lesion showing only weak Cp immunoreactivity (Fig. 1i–k).
Figure 1.
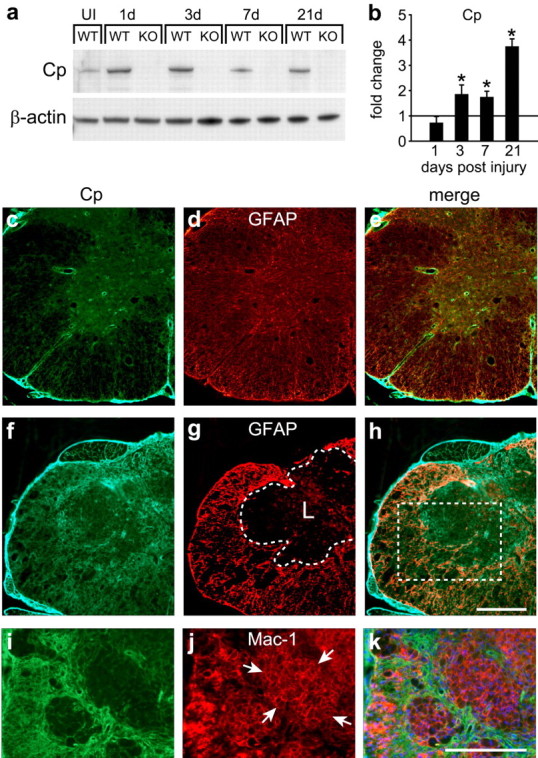
Expression of Cp is increased after SCI. Western blot showing changes in the 135 kDa molecular weight band labeled with anti-Cp antibody (a) and QRT-PCR analysis (b) of spinal cord homogenates of Cp+/+ [wild type (WT)] and Cp−/− [knock-out (KO)] mice. Samples were taken from uninjured (UI) and 1, 3, 7, and 21 d after injury. a, Cp protein levels are high at 1 and 3 d after SCI in Cp+/+ animals. b, Because Cp mRNA is not increased 1 d after SCI, the increase in Cp protein at this time point likely reflects influx of serum Cp attributable to hemorrhage. Cp protein levels remain elevated through 7 and 21 dpi, reflecting the increase in Cp mRNA above control levels at those time points. No protein band is detected in Cp−/− animals. β-Actin serves as loading control (n = 3; *p < 0.05, significantly different from uninjured control, two-way ANOVA, post hoc Tukey's test). In b, the horizontal line at 1 represents the mRNA levels in uninjured control spinal cord. c–e, Double-immunofluorescence labeling of uninjured Cp+/+ spinal cord for Cp (c) and GFAP (d). Note that the expression of Cp in the uninjured spinal cord is restricted to GFAP-positive (GFAP+) astrocytes in the merged image (e). f–k, Cp immunofluorescence staining 21 d after injury in Cp+/+ mice. Cp immunoreactivity is markedly increased (f) in the GFAP+ reactive astrocytes (g, h). The area of the lesion is outlined with the dashed line in g. The lesioned area (L) is essentially devoid of GFAP+ astrocytes (g) but contains Mac-1+ macrophages (j). i–k show a higher magnification of an area similar to the one in the boxed area in h taken from an adjacent section that was double labeled for Cp (i) and Mac-1 (j). Note that the Mac-1+ macrophages (clusters of macrophages labeled with arrows) in the central core of the lesion stain very weakly for Cp (i, k). Note that the Cp labeling, which in this field extends between groups of macrophages, is in astrocytes. Scale bars, 200 μm.
Increased accumulation of iron in the injured spinal cord of Cp−/− mice
We next assessed the role of Cp in mobilizing iron out of the injured spinal cord. Iron accumulation was detected histochemically in the injured spinal cord of Cp+/+ and Cp−/− mice. At 7 dpi, labeling for iron is distributed throughout the gray and some of the white matter (Fig. 2a,b). At earlier time points, RBCs are clearly visible at the lesion site in Epon-embedded sections (data not shown). Subsequently, these cells are phagocytosed by macrophages as seen by electron microscopy (Fig. 2f). The role of macrophages in the phagocytosis and breakdown of RBCs in non-CNS tissue has been described previously (Knutson and Wessling-Resnick, 2003). Quantification of iron levels in the spinal cord at 7 dpi by fAAS showed a 43% increase in Cp−/− mice compared with Cp+/+ mice (Fig. 2g) (p < 0.05). This represents a 3.2-fold increase after injury in wild-type mice and a 4.9-fold increase in Cp−/− mice. There was no difference in the iron levels in uninjured Cp+/+ and Cp−/− mice (Fig. 2g), indicating that iron accumulation does not occur in the normal CNS of Cp−/− mice at the young age used in this study. By 14 d, the iron staining is restricted to the central core of the lesion epicenter (Fig. 2c–e), in which it is contained within large Mac-1+ macrophages (Fig. 2h,i). Based on optical density measurements of histochemically stained tissue, Cp−/− spinal cord sections contained 32% more iron than Cp+/+ mice at day 14 after injury. Perl's histochemistry and fAAS detect both the ferrous and ferric forms of iron. To assess whether there was a selective increase in ferrous iron after SCI, we used a variation of the Turnbull's method that allows for the detection of ferrous iron in tissue (Meguro et al., 2003). This analysis revealed that Cp−/− mice have more ferrous iron at the lesion site than Cp+/+ mice (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Interestingly, by 42 d after injury, there is a noticeable reduction in the intensity of the Perl's staining of macrophages at the lesion epicenter in both Cp+/+ and Cp−/− mice (Fig. 2j,k), suggesting that iron is slowly released from these cells between 21 and 42 dpi. We show later that the viability of these macrophages may be compromised. Furthermore, because these macrophages express low levels of Cp (Fig. 1i–k) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), we speculate based on the literature on the ferroxidase functions of Cp (Bielli and Calabrese, 2002; Hellman and Gitlin, 2002) that the iron released from these macrophages may not be safely oxidized and may therefore remain in the toxic ferrous state.
Figure 2.

Iron accumulation in the injured spinal cord. a, b, Iron staining (dark brown/black) using enhanced Perl's histochemistry is dispersed throughout the lesion site at 7 dpi. Cell nuclei are counterstained with methyl green. c, d, At 14 dpi, iron staining is primarily restricted to the central core region. a and c are wild type (WT), whereas b and d are from Cp−/− mice. Note the greater area of iron accumulation in Cp−/− [knock-out (KO)] compared with Cp+/+ (WT) mice at both 7 and 14 dpi. e, Longitudinal section of the spinal cord of a Cp+/+ mouse 21 dpi stained for iron shows that the iron-labeled cells are located at the epicenter of the lesion. f, Electron micrograph showing a macrophage (outlined with arrows) that has phagocytosed red blood cells (RBC*) at 5 dpi. Note the RBCs that have hemorrhaged into the surrounding spinal cord tissue that have not yet been phagocytosed (RBC). g, Quantitative flame atomic absorption spectrometric analysis of iron levels in the spinal cord of uninjured Cp+/+ (WT) and Cp−/− (KO) mice and of mice 7 d after SCI. Note that there is no difference in the amount of iron in the uninjured spinal cord of Cp+/+ and Cp−/− mice. Note also that, after SCI, the amount of iron is significantly higher in Cp knock-out mice than in wild-type mice (n = 6 per group; *p < 0.05, t test). h, Micrograph at higher magnification showing cells in the central core of the lesion at 14 dpi stained for iron with enhanced Perl's (dark brown/black). Note that some cells are labeled intensely for iron (arrows), whereas others do not contain iron staining (arrowheads) that have phagocytosed other debris. i, An adjacent section stained with the Mac-1 antibody shows that the cells in this central core are macrophages, some of which are indicated with arrows. j, At later time points at day 42 after SCI, the iron staining in cells in the central core of the lesion is markedly diminished (brown staining). k, Staining of an adjacent section with Mac-1 antibody shows that these cells are Mac-1+ and macrophages, some of which are indicated with arrows. Scale bars: a–e, 500 μm; f, 5 μm; h–k, 50 μm.
Impaired functional recovery after SCI in Cp−/− mice is mediated by iron
As shown above (Fig. 2g), there is no iron accumulation in the uninjured spinal cord of Cp−/− mice at the 2–4 months of age at which the mice were used for these experiments. We have reported previously that Cp null mice at 18–24 months of age show impairment on the rotor rod test and loss of dopaminergic neurons in the substantia nigra. We therefore assessed whether such changes occur in 2- to 4-month-old mice used in the present study. Uninjured Cp null mice at this age did not show any deficits in the rotor rod tests (supplemental Fig. 4a,b, available at www.jneurosci.org as supplemental material) and no loss of dopaminergic neurons in the substantia nigra (supplemental Fig. 4c,d, available at www.jneurosci.org as supplemental material).
To evaluate whether the greater iron accumulation in the injured spinal cord of Cp−/− mice has an impact on functional recovery, we assessed locomotor function using the nine-point BMS, which is widely used to evaluate hindlimb motor function in an open field (Basso et al., 2006). Locomotor function was significantly impaired in Cp−/− mice compared with injured wild-type controls beginning at 5 dpi (p < 0.05) (Fig. 3a). The mean score for Cp−/− mice at 28 dpi was 2.2 ± 0.32, whereas that for the Cp+/+ mice was 4.0 ± 0.36. A score of 2 indicates extensive ankle movements but no plantar placement of the paw. A score of 4 indicates consistent weight support and occasional plantar stepping. These data indicate that Cp plays a protective role after SCI, which could likely be via its effects on iron.
Figure 3.

Mice deficient in Cp have greater functional loss and secondary tissue damage after SCI. a, BMS, a scale specifically designed for open-field locomotor analysis of mice after spinal cord contusion injury, was used to assess locomotor recovery. A score of 9 is normal locomotion. Recovery is significantly impaired in Cp−/− [knock-out (KO)] mice (n = 9) compared with Cp+/+ [wild type (WT)] mice (n = 10; two-way RM-ANOVA, *p < 0.001, post hoc Tukey's test). b, Cp−/− injured mice treated with the iron chelator SIH (n = 8) every other day recover significantly better than vehicle-treated Cp−/− mice (n = 7). As before, wild-type injured mice (n = 7) perform significantly better than Cp−/− vehicle-treated mice (two-way RM-ANOVA, *p < 0.05, post hoc Tukey's test). c, d, Micrographs showing the ventral horn of cresyl-violet-stained spinal cord sections of Cp+/+ (c) and Cp−/− (d) mice at 14 dpi. Note the marked loss of motor neurons in Cp−/− mice. Arrows indicate some of the motor neurons. e, Quantification of motor neuron loss from the epicenter (0) to 2 mm rostral and caudal to the lesion using cresyl-violet-stained sections. At 1 mm away from lesion site, there is almost a 50% greater loss of motoneurons in Cp−/− (KO) mice (*p < 0.05, two-way ANOVA, t test). Distance from lesion shown in micrometers. f, g, Micrographs of Luxol fast blue-stained sections of Cp+/+ (f) and Cp−/− (g) mice at 14 dpi. Note the greater myelin loss in Cp−/− mice (g) compared with wild-type mice (f). h, Quantification of the myelin staining shows increased myelin loss in Cp−/− (KO) mice from 250 μm rostral to 500 μm caudal to the lesion epicenter (*p < 0.05, two-way RM-ANOVA, t test). Scale bars: c, d, 100 μm; f, g, 500 μm.
To assess whether the increased locomotor deficit observed after SCI in Cp−/− mice is caused by iron toxicity, Cp−/− mice were treated every other day starting 1 h after contusion injury with SIH, a lipophilic iron chelator that shows high affinity and selectivity for iron (Richardson and Ponka, 1998b; Klimtova et al., 2003). Cp−/− mice treated with SIH showed significantly improved locomotor recovery compared with the vehicle-treated littermate Cp−/− controls (Fig. 3b). The SIH-treated Cp−/− mice reach a score of 3.75 ± 0.21 on day 28, whereas the vehicle-treated Cp−/− mice have a score 2.64 ± 0.21. These results suggest that, after SCI, the lack of Cp causes an early increase in toxic ferrous iron that can be chelated.
Greater motor neuron loss and demyelination in Cp−/− mice
To determine the cellular basis of the functional deficits seen in Cp−/− mice after SCI, we quantified the number of motor neurons in cresyl-violet-stained cross sections of the injured spinal cord and assessed the extent of myelin sparing using Luxol fast blue staining at 14 dpi. Motor neuron loss in Cp−/− mice is almost twofold greater than in Cp+/+ mice at 1 mm rostral and caudal to the lesion epicenter (Fig. 3c–e). There is also a 44% greater loss of myelin in the white matter of Cp−/− mice compared with Cp+/+ mice (Fig. 3f–h). Double labeling of tissue sections to detect myelin and axons showed that axons are present in areas of myelin loss in the dorsal and lateral white matter, indicating greater demyelination in Cp−/− mice (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). These results suggest that the increased accumulation of iron in the absence of Cp may contribute to the neuronal loss and demyelination after SCI and that Cp may normally play a protective role in reducing such damage.
Lack of Cp increases free radical-mediated damage in the injured spinal cord
The protective effects of Cp after SCI are likely attributable to its ability to safely oxidize and thus detoxify ferrous iron. Ferrous iron can catalyze the production of reactive oxygen and nitrogen species, which then deleteriously modify lipids and proteins, leading to cell death and tissue damage. The modifications include the introduction of carbonyl groups into protein side chains. We found significantly greater protein carbonylation in contused spinal cord tissue in Cp−/− mice compared with Cp+/+ mice at early time points after contusion injury (Fig. 4a). Nitric oxide can react with superoxide radicals to produce peroxynitrite (ONOOO−), a reactive oxidant that has been implicated for its cytotoxic role after SCI (Liu et al., 2000). iNOS is upregulated acutely after SCI (Xu et al., 2001). There is also evidence that systemic iron overload induces iNOS expression in the iron-loaded liver (Cornejo et al., 2005). We therefore assessed the expression of iNOS in the injured spinal cord of Cp+/+ and Cp−/− mice. At 14 dpi, iNOS immunoreactivity appears to be mainly localized outside the lesion core that contains macrophages (Fig. 4b,c). The iNOS labeling may be in astrocytes, because the pattern of staining appears similar to that of GFAP (see Fig. 6p). The labeling is more intense in injured tissue of Cp−/− than Cp+/+ mice (Fig. 4b,c). In addition, we also assessed 3-nitrotyrosine immunoreactivity as a measure of nitrosative damage of proteins. Greater 3-nitrotyrosine labeling was detected in Cp−/− mice in both the white and gray matter after SCI (Cp+/+, 13.25 ± 1.25 cells per 0.1 mm2; Cp−/−, 23.75 ± 1.65 cells per 0.1 mm2; p < 0.05) The staining is particularly noticeable in the ventral motor neurons that appear atrophied in Cp−/− mice compared with Cp+/+ mice and also in some of the glial cells in the white matter (Fig. 4d,e). Lipid peroxidation, another hallmark of oxidative damage (Braughler and Hall, 1992), can be assessed by staining for 4-HNE, an end product known to promote cell death (Keller et al., 1999). Greater 4-HNE staining is evident in the white matter adjacent to the lesion in Cp−/− mice compared with Cp+/+ mice (Fig. 4f,g) (optical density values: Cp+/+, 14.4 ± 1.3; Cp−/−, 20.58 ± 0.87; p < 0.05). 4-HNE labeling also localizes to the cell and nuclear membranes of the iron-loaded macrophages at the lesion epicenter (Fig. 4h). 4-HNE has been shown to increase cell permeability (Usatyuk et al., 2006) and may therefore contribute to increased membrane permeability and decreased viability of these macrophages, which may account for the loss of iron from these cells at later time points as noted above (Fig. 2j,k). In addition, RT-PCR analysis at 1 dpi after SCI indicates a marked increase in the mRNA expression of the proinflammatory cytokines interleukin-1β and tumor necrosis factor-α in spinal cord of Cp−/− mice compared with Cp+/+ mice (Fig. 4i), suggesting a greater inflammatory response in Cp−/− mice.
Figure 4.
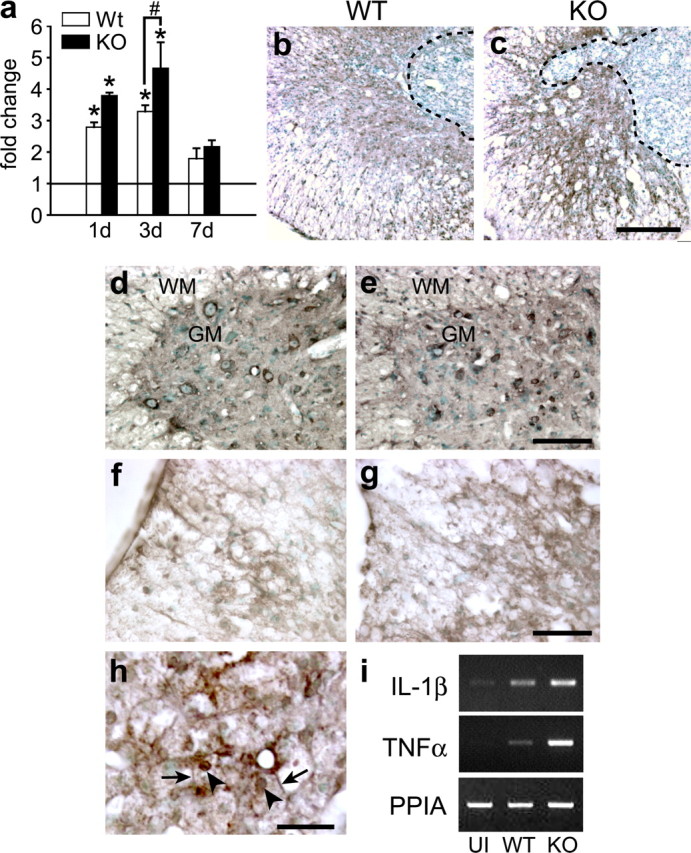
Oxidative and nitrosative damage in the injured spinal cord. a, Densitometric quantification of oxyblots to assess protein oxidation in injured spinal cord of Cp+/+ [wild type (WT)] and Cp−/− [knock-out (KO)] mice at 1, 3, and 7 dpi. Note the increased protein oxidation in Cp−/− mice (n = 4; *p < 0.05, significantly different from uninjured controls; #p < 0.05, significant difference between WT and KO, t test). Horizontal line represents values in normal, uninjured spinal cord. b, c, Immunohistochemical stain for iNOS in the injured spinal cord at 14 dpi. In contrast to the weak staining for iNOS in Cp+/+ mice (b), reactive astrocytes that surround the lesion core in Cp−/− mice (c) stain more intensely. d, e, 3-Nitrotyrosine staining of motoneurons and glia in the gray (GM) and white (WM) matter of Cp+/+ (d) and Cp−/− (e) mice at 14 dpi. Note the atrophied neurons that stain more intensely in Cp−/− than in Cp+/+ tissue. f, g, 4-HNE staining for lipid peroxidation at 14 dpi is more intense in the white matter of Cp−/− mice (g) than Cp+/+ mice (f). h, This micrograph illustrates the strong 4-HNE staining of macrophages in the central core region of the lesion. Note the staining of the nuclear membranes (arrowheads) and cell membranes (arrows) of these macrophages. i, RT-PCR showing the increased mRNA expression of interleukin-1β (IL-1β) and tumor necrosis factor-α (TNFα) in Cp−/− mice 1 d after SCI. UI, Uninjured; WT, wild type; KO, Cp null. Scale bars: c, 200 μm; e, g, 100 μm; h, 25 μm.
Figure 6.
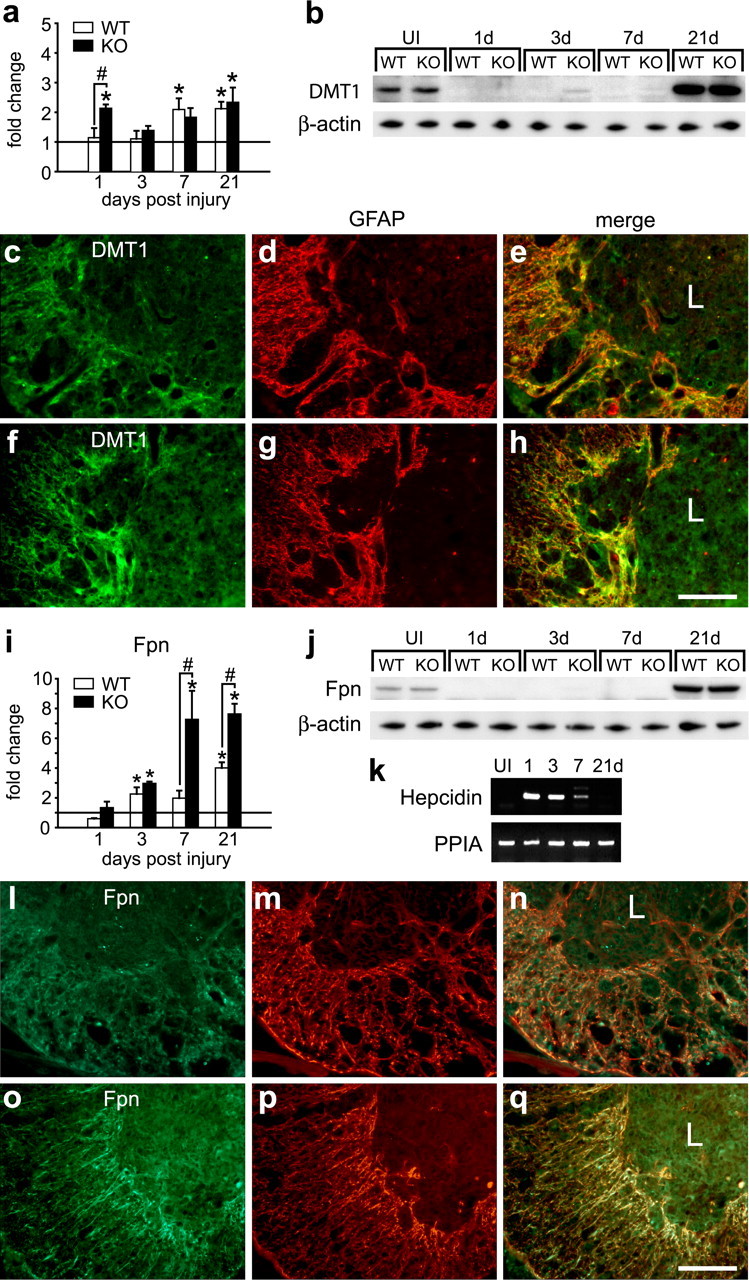
Expression of iron transporters in astrocytes after SCI. a, QRT-PCR analysis of DMT1 mRNA at 1, 3, 7, and 21 dpi. The horizontal line represents expression in uninjured spinal cord. In Cp+/+ mice, the DMT1 mRNA shows a gradual increase with time after injury to reach twofold at 21 dpi. In Cp−/− mice, there is an initial increase at 1 d, which returns to control levels at 3 d, followed by a gradual increase with time to reach approximately twofold at 21 dpi [*p < 0.05, significantly different from uninjured controls (horizontal line)]. b, Western blot showing changes in DMT1 protein at 1, 3, 7, and 21 dpi (65 kDa band labeled with anti-DMT1 antibody). Note the marked reduction in DMT1 protein in the first week after injury. At 21 dpi, the levels are increased approximately fourfold over uninjured spinal cord (UI). c–h, Double-immunofluorescence labeling of Cp+/+ (c–e) and Cp−/− (f–h) mice stained for DMT1 (c, f), and GFAP (d, g). DMT1 is expressed mainly in reactive astrocytes in the area surrounding the central core of the lesion (L). e and h show merged images. i, QRT-PCR analysis of Fpn mRNA at 1, 3, 7, and 21 dpi. The horizontal line represents expression in uninjured spinal cord. Fpn mRNA increases with time after injury, reaching the highest level at 21 dpi in both Cp+/+ [wild type (WT)] and Cp−/− [knock-out (KO)] mice [*p < 0.05, statistically significantly different from uninjured controls (horizontal line); #p < 0.05, significant difference between WT and KO; two-way ANOVA]. j, Western blot showing changes in the 65 kDa Fpn protein band at 1, 3, 7, and 21 dpi. As with DMT1, there is a marked reduction in Fpn protein in the first week after injury. At 21 dpi, the levels are increased ∼2.5-fold over uninjured spinal cord. k, Changes in hepcidin mRNA after SCI detected by RT-PCR. Hepcidin mRNA is not detected in the uninjured spinal cord but is increased at 1, 3, and 7 dpi. l–q, Double-immunofluorescence labeling of Cp+/+ (l–n) and Cp−/− (o–q) mice stained for Fpn (l, o), and GFAP (m, p). Fpn is also expressed mainly in reactive astrocytes in the area surrounding the central core of the lesion (L). n and q show merged images. Scale bars: h, q, 100 μm.
Expression of iron homeostasis proteins in the spinal cord after SCI in Cp+/+ and Cp−/− mice
The increased and prolonged presence of iron in the contused spinal cord of both Cp+/+ and Cp−/− mice suggests a dysregulation of iron homeostatic mechanisms. Because the regulation of iron levels in the CNS is a coordinated response involving several proteins that include influx and efflux transporters and binding proteins, we assessed the expression of these iron homeostasis proteins after SCI.
Macrophages not astrocytes store iron after SCI
Ferritin is the major iron storage protein in cells. An increase in ferritin mRNA and protein are sensitive indicators of increased cellular iron content because ferritin mRNA contains iron response elements (IREs) in the 5′ untranslated region. The IREs are hairpin loop structures that interact with iron regulatory proteins (IRP1 and IRP2) (Rouault, 2006). In the presence of excess iron, IRPs do not bind IREs resulting in increased ferritin mRNA stability and translation. Iron has also been shown to regulate ferritin mRNA transcription (Hintze and Theil, 2006). Our previous work on the aging brain showed that increases in ferritin mRNA and protein are more sensitive indicators of increased iron in the CNS than Perl's histochemistry (Jeong and David, 2006). We therefore assessed ferritin mRNA expression by quantitative real-time RT-PCR. Ferritin-light chain (ferritin-L) (the major storage isoform) mRNA levels are increased ∼10-fold as early as 3 dpi in both genotypes (Fig. 5a). Marked differences in ferritin-L mRNA levels between Cp+/+ and Cp−/− mice are detected by 7 d, being ∼15-fold and 28-fold higher, respectively (Fig. 5a). Its expression remains elevated at ∼10-fold in both genotypes at 21 dpi. Western blot analysis showed a marked increase in ferritin protein in Cp+/+ and Cp−/− mice from 3 d onward. Densitometric measurements showed a 1.5- to 2-fold greater levels in Cp−/− mice compared with wild-type mice (Fig. 5b). Ferritin-heavy chain mRNA levels were significantly higher at 1 dpi in Cp−/− mice compared with Cp+/+ mice. At later time points, the levels in both groups are higher than uninjured controls, but there are no significant differences between the groups at day 21 (data not shown). Immunofluorescence analysis indicates that ferritin is expressed by macrophages within the core of the lesion (Fig. 5c–e) as well as by microglia located around the lesion epicenter and at distances away from the lesion (Fig. 5f–i). Interestingly, increased ferritin immunoreactivity is not observed in reactive astrocytes surrounding the lesion epicenter, in either strain of mice (Fig. 5j–m), indicating that, unlike the aging-related iron accumulation seen in astrocytes in 24-month-old Cp−/− mice, astrocytes are not involved in storing iron after SCI in young mice.
Figure 5.
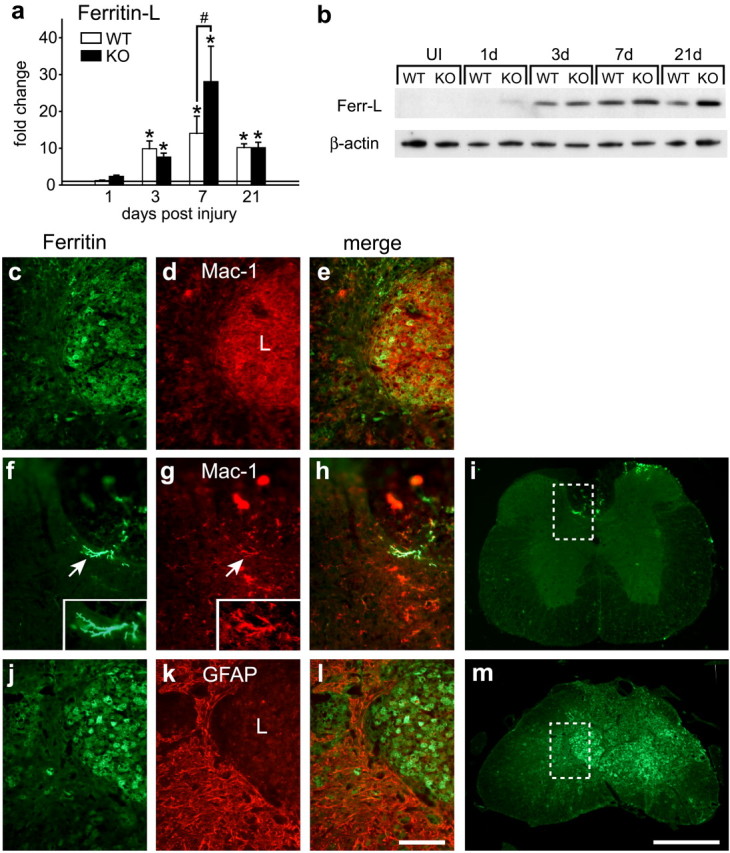
Ferritin expression is induced in macrophages but not in astrocytes after SCI. a, Ferritin-L mRNA expression in the injured spinal cord of Cp+/+ [wild type (WT)] and Cp−/− [knock-out (KO)] mice determined by QRT-PCR [n = 3; *p < 0.05, significantly different from uninjured controls (horizontal line); #p < 0.05, significant difference between WT and KO, two-way ANOVA]. b, Western blot showing changes in the 18 kDa molecular weight band labeled with anti-ferritin antibody. Increase in ferritin protein expression is seen in the injured spinal cord of Cp+/+ (WT) and Cp−/− (KO) mice. Note the significant induction of ferritin protein starting from 3 dpi. β-Actin used as the loading control. c–e, Double immunostaining of a tissue section from Cp+/+ mouse shows that ferritin (c) is mainly in Mac-1+ macrophages (d) at the lesion site 21 dpi. The area shown is similar to that in m. f–i, Cp+/+ tissue section 21 dpi, far from the site of injury, double labeled for ferritin (f) and Mac-1 (g). Note that ferritin (f) is expressed in process-bearing Mac-1+ microglia (g, h) in the dorsal gray matter. i, The area shown in f–h is taken from the boxed area indicated in this low-magnification micrograph. j–m, Labeling of Cp−/− tissue 21 dpi stained for ferritin (j) and GFAP (k). The merged image (l) shows that the reactive astrocytes surrounding the lesion core do not express ferritin. m, The area shown in j–l is indicated by the box in this low-magnification micrograph. Scale bars: c–e, f–h, j–l, 100 μm; i, m, 500 μm.
Astrocytes are critical for iron trafficking after SCI
We also assessed the expression of DMT1 and the iron exporter Fpn in the injured spinal cord. DMT1 is expressed on the astrocyte cell surface in which it plays a role in iron influx (Jeong and David, 2003) as has been shown for other cell types, including enterocytes (Picard et al., 2000). DMT1 therefore contributes to cellular acquisition of divalent ferrous iron. Despite the early increase in DMT1 mRNA 1 d after SCI in Cp−/− mice and the later increase in wild-type and null mice at 7 and 21 d (Fig. 6a), Western blot analysis showed a sharp reduction in DMT1 protein during the first week after SCI and increasing above normal uninjured levels at 21 d (Fig. 6b). This reduction in DMT1 protein during the first week after injury was confirmed by immunofluorescence labeling of tissue sections (supplemental Fig. 6a–f, available at www.jneurosci.org as supplemental material). At 21 d after injury, the increased DMT1 expression is primarily confined to reactive astrocytes at the lesion site in both genotypes (Fig. 6c–h). Because DMT1 specifically transports the ferrous form of iron, these results suggest that the ability of astrocytes to clear toxic ferrous iron from the injury site may be compromised during the first week after SCI.
Astrocytes also express the iron efflux transporter Fpn, the expression of which is regulated at the transcriptional level by iron (Zoller et al., 2002; Knutson and Wessling-Resnick, 2003) and posttranscriptionally through the IREs in the 5′ untranslated region (Lymboussaki et al., 2003). As with DMT1, Western blot analysis showed that Fpn protein levels were markedly reduced in the first week after SCI (Fig. 6j), despite the increases in mRNA levels in wild-type and Cp null mice (Fig. 6i). The Western blot data were further supported by immunofluorescence labeling showing reduced Fpn immunoreactivity at 7 dpi followed by a gradual increase at 14 dpi (supplemental Fig. 6g–l, available at www.jneurosci.org as supplemental material). At 21 dpi, Western blot and immunofluorescence labeling show higher Fpn expression (Fig. 6j,l–q), which appears to be localized mainly to reactive astrocytes at the lesion site (Fig. 6l,q). Recent work has shown that hepcidin, a small peptide, binds to Fpn on the cell surface and causes its internalization and degradation, resulting in a reduction in iron export from cells (Nemeth et al., 2004; Knutson et al., 2005). Interestingly, we found that hepcidin expression is increased in the spinal cord during the period when Fpn protein levels are low during the first week after SCI (Fig. 6k). Therefore, hepcidin-mediated reduction in Fpn during the first week after SCI may compromise the ability of astrocytes to safely export iron out of the CNS.
Previous work has shown that Fpn partners with Cp and that it requires Cp to efflux iron (Jeong and David, 2003) Therefore, the increase in Fpn in astrocytes in the injured spinal cord of Cp null mice is intriguing. Because these astrocytes in the Cp null mice do not show increases in ferritin staining (Fig. 5j–m), it is possible that Fpn is able to efflux iron from these cells in the absence of Cp, possibly via some other extracellular oxidative source, as has been suggested (De Domenico et al., 2007).
Macrophages have also been reported to possess iron efflux mechanisms, namely Fpn (Yang et al., 2002) and Cp (Yang et al., 1986; Mazumder et al., 1997) that underlie the ability of these cells to recycle iron (Knutson and Wessling-Resnick, 2003) in various tissues and organs such as the spleen. However, macrophages in the lesion epicenter that are heavily loaded with iron express very low levels of Fpn, which may explain their prolonged accumulation of iron. Because the amount of iron detected in these macrophages by Perl's histochemistry appears to be markedly reduced between 21 and 42 dpi (Fig. 2h,j), it is possible that the iron released from these cells is likely to be independent of the Fpn/Cp efflux mechanism. We have described above that these macrophages show evidence of lipid peroxidation (Fig. 4h), suggesting that their viability may be compromised. To assess the functional consequences of this delayed loss of iron from these macrophages, we examined the effects of iron chelator after spinal cord contusion injury in wild-type animals.
Systemic iron chelation promotes delayed functional recovery after SCI in wild-type mice
We assessed the effects of chelating iron in wild-type mice after SCI using SIH, a lipophilic iron chelator that shows high affinity and selectivity for iron (Klimtova et al., 2003). SIH was administered twice a week starting 1 h after contusion injury until day 42 after injury. In the first 3 weeks, the chelator-treated mice showed the same degree of motor recovery as the vehicle-treated controls. However, after 21 dpi, the chelator-treated mice exhibited significantly improved locomotor recovery compared with the vehicle-treated control group (Fig. 7a). The vehicle-treated group had a score of 4.57 ± 0.17 at 35 d, whereas the chelator-treated group improved to a score of 5.43 ± 0.20. Animals that show consistent weight support and occasionally plantar step are awarded a score of 4, whereas those with a score of 5 or 6 are able to step frequently or consistently and show some coordination. The differences are even more apparent in the BMS subscores, which rate finer aspects of locomotor control (SIH-treated group, 2.42 ± 0.57; vehicle-treated controls, 0.75 ± 0.49; t test, *p < 0.05). The chelator-treated group was consistently more coordinated and showed more normal paw placement than the vehicle-treated group at 35 dpi. The fact that the locomotor functional improvement is seen after a delay of 21 d suggests that the beneficial effects of SIH may be attributable to chelation of toxic ferrous iron possibly released from macrophages in the core of the lesion between 21 and 42 d after injury. This is further supported by the finding that there is a significant reduction in iron in the injured spinal cord of SIH-treated animals compared with vehicle-treated controls (Fig. 7b,c) (optical density values: vehicle, 62.9 ± 7.3; SIH, 38.2 ± 6.0; p < 0.05).
Figure 7.

Iron chelator treatment promotes functional recovery after SCI. a, Wild-type (WT) injured mice treated with the iron chelator (n = 8) exhibit significant improvement in locomotor recovery compared with vehicle-treated control mice (n = 7; two-way RM-ANOVA, *p < 0.05, post hoc Tukey's test). Functional recovery assessed with the 9-point BMS method. The improvement with the chelator is seen at and after 28 dpi. b, c, Perl's staining of spinal cord 42 d after contusion injury from mice treated with vehicle (b) and iron chelator SIH (c). Scale bar, 500 μm.
Discussion
Trauma to the CNS is generally associated with hemorrhage at the site of injury. We now present data on the iron homeostatic response to spinal cord contusion injury and the role of iron mediated oxidative damage in the pathology that evolves after SCI. The data we present here indicates that expression of Cp is induced after SCI and plays a crucial role in the detoxification and clearance of iron. Mice deficient in Cp exhibit increased iron accumulation, greater secondary damage, and significantly impaired locomotor recovery. This work provides new insights into how astrocytes and macrophages handle iron differently after SCI in wild-type mice. It also highlights the importance of iron-mediated secondary damage several weeks after SCI and demonstrates that treatment with an iron chelator is effective in promoting functional recovery after the acute period.
Ceruloplasmin is a six domain multi-copper oxidase that catalyzes the safe conversion of Fe2+ to Fe3+ with the subsequent reduction of two molecules of oxygen to water (Zaitsev et al., 1999; Bento et al., 2007). The resulting Fe3+ is thought to move to a holding site in ceruloplasmin from where it can be loaded onto iron binding proteins for transport (Zaitsev et al., 1999; Bento et al., 2007). Cp is an acute phase reactant, whose concentration in serum increases with systemic inflammation (Vassiliev et al., 2005). Its properties as an antioxidant are well established, and it is thought to account for most of the antioxidant properties of serum (Gutteridge, 1978; Vassiliev et al., 2005). In the CNS, however, a GPI-anchored variant is the major form of the protein and is expressed by astrocytes (Patel et al., 2000). GPI–Cp on astrocytes partners with Fpn to efflux iron from these cells (Jeong and David, 2003). Soluble serum Cp, which is not normally available to cells in the CNS, can substitute in vitro for the lack of GPI–Cp to efflux iron from astrocytes (Jeong and David, 2003). The results presented here indicate that the expression of ceruloplasmin is substantially increased over several weeks in reactive astrocytes at the injury site. In addition to Cp produced by cells within the injured spinal cord, the extravasation of serum Cp from damaged blood vessels is also likely to play an antioxidant role in the first few days after trauma, detoxifying iron that is released from damaged cells. The absence of this antioxidant activity in Cp−/− mice is likely to contribute to the greater oxidative damage and impaired functional recovery in these mice, which is evident as early as 5 d after injury. Furthermore, the protective effects of the chelator treatment seen as early as 5 d after SCI in Cp−/− mice suggests that Cp plays an early and continuing role in detoxifying iron after SCI. Our data suggest that astrocytes and macrophages play important but distinct roles in the iron homeostatic response after SCI. Astrocytes are thought to play a central role in the efflux and clearance of iron from the normal uninjured CNS (Jeong and David, 2006), likely via the astrocytic end feet that surround 95% of the capillary surface (Nedergaard et al., 2003). They possess the transport mechanisms required for iron import (DMT1) and efflux (Fpn/Cp) (Jeong and David, 2003). Our present data show that, during the first week after SCI, DMT1 and Fpn expression is markedly reduced, suggesting an impaired ability of CNS cells, in particular astrocytes, in clearing potentially toxic divalent ferrous iron from the injured tissue. During this period, macrophages phagocytose RBCs and tissue debris including iron-rich myelin. This is accompanied by an increase in ferritin synthesis in macrophages, suggesting iron storage by these cells. After the first week, there is a gradual increase in the expression of DMT1 and Fpn in reactive astrocytes, suggesting that, in Cp+/+ mice, they are now able to uptake and mobilize iron out of the injured CNS, likely into the circulation via astrocytic end feet. Interestingly, a similar increase in turnover of iron has been proposed recently in studies of lymphocytes from patients with restless legs syndrome (Earley et al., 2008). These studies have shown increased DMT1 and Fpn expression but unchanged levels of ferritin, analogous to the astrocytes around the lesion site that we describe. The initial loss of Fpn protein after SCI may be via hepcidin-mediated internalization and degradation (Nemeth et al., 2004; Knutson et al., 2005), because the timing of hepcidin expression coincides with the period when Fpn protein is reduced. The mechanism mediating the early reduction in DMT1 protein after SCI is currently unknown. We did not detect increased ferritin in astrocytes of Cp−/− mice. Because they express Fpn after the first week after SCI, it suggests that astrocytes in Cp−/− mice are able to efflux iron in the absence of Cp under these conditions, the reasons for which are currently not known. This is in contrast to the accumulation of iron in astrocytes in Cp−/− mice with aging (Jeong and David, 2006). However, in the absence of Cp, ferrous iron that effluxes via Fpn in Cp−/− mice after SCI could react with hydrogen peroxide, found at high levels in injured spinal cord tissue (Liu et al., 1999; Bao and Liu, 2004), and give rise to toxic hydroxyl radicals and superoxides via the Fenton and Haber–Weiss reactions (Eaton and Qian, 2002). Given the reactive nature of ferrous iron, a number of extracellular proteins and lipids could also catalyze this oxidation, albeit in an unsafe and inefficient manner. Therefore, although iron is exported from astrocytes in the injured spinal cord of Cp−/− mice, it may not be mobilized out of the CNS as it would normally and could result in repeated cycles of oxidation and reduction, leading to the high levels of oxidative damage and functional impairment. The altered iron homeostasis that we describe after SCI is likely regulated by iron responsive mechanisms as well as iron-independent mechanisms, particularly inflammation, a known modifier of iron homeostasis (Yang et al., 2002; MacKenzie et al., 2008).
In contrast to astrocytes, macrophages at the lesion site accumulate large quantities of iron derived from the breakdown of hemoglobin in phagocytosed RBCs, as well as tissue debris rich in iron such as myelin. These macrophages store iron in ferritin for a prolonged period of time, because they can be found in the spinal cord for 6 weeks, the longest time point examined. This is in sharp contrast to macrophages in the spleen that phagocytose senescent RBCs but are able to rapidly recycle the iron by exporting it into the circulation (Knutson and Wessling-Resnick, 2003). Macrophages that effectively recycle iron in the spleen (Yang et al., 2002) and in vitro (Knutson et al., 2003) express high levels of Fpn, which in non-CNS tissue can partner with serum Cp to safely efflux iron. In contrast, the iron-laden macrophages in the lesioned spinal cord express low levels of Fpn and cell surface Cp, which may account for their inability to efflux iron efficiently. The impaired cycling of iron from macrophages seen here is reminiscent of the sequestration of iron in peripheral macrophages during systemic inflammatory states (Knutson et al., 2003; Ganz, 2006) and observed in cells treated in vitro with hepcidin (Nemeth et al., 2004). The increased expression of hepcidin in the injured spinal cord may thus underlie the sustained accumulation of iron by macrophages. However, iron is released from these cells between 3 and 6 weeks after injury, as detected by Perl's histochemistry. This release is not likely to be transporter mediated because Fpn is poorly expressed and may thus result from the diffusion of iron across the damaged cell membranes, which show evidence of lipid peroxidation. The very low levels of Cp expressed by these macrophages in wild-type mice suggests that the iron released is likely to remain longer in the toxic, redox-active state and thus capable of generating free radicals and causing secondary tissue damage. This is supported by the evidence that wild-type mice treated with the iron chelator, starting the day after contusion injury, exhibit improvement in locomotor recovery between 3 and 6 weeks after injury that coincides with the period when iron staining is reduced in these macrophages. We cannot rule out the possibility that some of the iron that comes out of these macrophages is safely bound to secreted ferritin (Zhang et al., 2006). Our results with the chelator suggest that at least some of the released iron is redox active and chelatable, which is consistent with reports studying iron release from macrophages after erythrophagocytosis (Moura et al., 1998; Knutson et al., 2005). These effects of the chelator in wild-type mice are likely independent of Cp. Previous work using iron chelators in spinal cord injury include studies on the polyphenolic compound quercetin, which also has antioxidant and other anti-inflammatory effects (Schültke et al., 2003), and studies using a combination of an iron chelator and a mitotic inhibitor for fibroblast proliferation in a model of spinal cord hemisection to reduce the collagen scar induced by meningeal cells (Hermanns et al., 2001; Klapka et al., 2005). In contrast to these studies, our work addresses the issue of iron homeostasis and detoxification, which has not been addressed previously.
Together, these results establish the importance of the iron-homeostatic response in limiting secondary damage after SCI. They provide new insights into the marked differences in the way astrocytes and macrophages handle iron in the injured spinal cord. In wild-type mice, astrocytes may play a protective role in safely cycling iron out of the injured spinal cord. In contrast, macrophages that sequester mainly hemoglobin-derived iron from phagocytosed RBCs appear to contribute to a delayed phase of secondary damage that can be ameliorated by iron chelator therapy. The results presented here also have implications for the treatment of other forms of CNS trauma and hemorrhagic stroke in which iron accumulation occurs.
Footnotes
This work was supported by a grant from the Canadian Institutes of Health Research (CIHR) (S.D.). K.I.R., A.R., and S.Y.J. are recipients of studentships from the CIHR Strategic Training Program in Neuroinflammation, and R.L.V. is the recipient of a CIHR Postdoctoral Fellowship. We thank Ourania Tsatas and Hiba Kazak for technical assistance and Margaret Attiwell for help with preparing the illustrations.
References
- Bao F, Liu D. Hydroxyl radicals generated in the rat spinal cord at the level produced by impact injury induce cell death by necrosis and apoptosis: protection by a metalloporphyrin. Neuroscience. 2004;126:285–295. doi: 10.1016/j.neuroscience.2004.03.054. [DOI] [PubMed] [Google Scholar]
- Basso DM, Fisher LC, Anderson AJ, Jakeman LB, McTigue DM, Popovich PG. Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J Neurotrauma. 2006;23:635–659. doi: 10.1089/neu.2006.23.635. [DOI] [PubMed] [Google Scholar]
- Bento I, Peixoto C, Zaitsev VN, Lindley PF. Ceruloplasmin revisited: structural and functional roles of various metal cation-binding sites. Acta Crystallogr D Biol Crystallogr. 2007;63:240–248. doi: 10.1107/S090744490604947X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielli P, Calabrese L. Structure to function relationships in ceruloplasmin: a “moonlighting” protein. Cell Mol Life Sci. 2002;59:1413–1427. doi: 10.1007/s00018-002-8519-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braughler JM, Hall ED. Involvement of lipid peroxidation in CNS injury. J Neurotrauma. 1992;9(Suppl 1):S1–S7. [PubMed] [Google Scholar]
- Cornejo P, Varela P, Videla LA, Fernández V. Chronic iron overload enhances inducible nitric oxide synthase expression in rat liver. Nitric Oxide. 2005;13:54–61. doi: 10.1016/j.niox.2005.04.009. [DOI] [PubMed] [Google Scholar]
- De Domenico I, Ward DM, di Patti MC, Jeong SY, David S, Musci G, Kaplan J. Ferroxidase activity is required for the stability of cell surface ferroportin in cells expressing GPI-ceruloplasmin. EMBO J. 2007;26:2823–2831. doi: 10.1038/sj.emboj.7601735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley CJ, Ponnuru P, Wang X, Patton SM, Conner JR, Beard JL, Taub DD, Allen RP. Altered iron metabolism in lymphocytes from subjects with restless legs syndrome. Sleep. 2008;31:847–852. doi: 10.1093/sleep/31.6.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton JW, Qian M. Molecular bases of cellular iron toxicity. Free Radic Biol Med. 2002;32:833–840. doi: 10.1016/s0891-5849(02)00772-4. [DOI] [PubMed] [Google Scholar]
- Feroze-Merzoug F, Berquin IM, Dey J, Chen YQ. Peptidylprolyl isomerase A (PPIA) as a preferred internal control over GAPDH and beta-actin in quantitative RNA analyses. Biotechniques. 2002;32:776–778. 780, 782. doi: 10.2144/02324st03. [DOI] [PubMed] [Google Scholar]
- Ganz T. Hepcidin: a peptide hormone at the interface of innate immunity and iron metabolism. Curr Top Microbiol Immunol. 2006;306:183–198. doi: 10.1007/3-540-29916-5_7. [DOI] [PubMed] [Google Scholar]
- Ghasemlou N, Kerr BJ, David S. Tissue displacement and impact force are important contributors to outcome after spinal cord contusion injury. Exp Neurol. 2005;196:9–17. doi: 10.1016/j.expneurol.2005.05.017. [DOI] [PubMed] [Google Scholar]
- Gutteridge JM. Caeruloplasmin: a plasma protein, enzyme, and antioxidant. Ann Clin Biochem. 1978;15:293–296. doi: 10.1177/000456327801500170. [DOI] [PubMed] [Google Scholar]
- Hausmann ON. Post-traumatic inflammation following spinal cord injury. Spinal Cord. 2003;41:369–378. doi: 10.1038/sj.sc.3101483. [DOI] [PubMed] [Google Scholar]
- Hellman NE, Gitlin JD. Ceruloplasmin metabolism and function. Annu Rev Nutr. 2002;22:439–458. doi: 10.1146/annurev.nutr.22.012502.114457. [DOI] [PubMed] [Google Scholar]
- Hermanns S, Reiprich P, Müller HW. A reliable method to reduce collagen scar formation in the lesioned rat spinal cord. J Neurosci Methods. 2001;110:141–146. doi: 10.1016/s0165-0270(01)00427-7. [DOI] [PubMed] [Google Scholar]
- Hintze KJ, Theil EC. Cellular regulation and molecular interactions of the ferritins. Cell Mol Life Sci. 2006;63:591–600. doi: 10.1007/s00018-005-5285-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong SY, David S. Glycosylphosphatidylinositol-anchored ceruloplasmin is required for iron efflux from cells in the central nervous system. J Biol Chem. 2003;278:27144–27148. doi: 10.1074/jbc.M301988200. [DOI] [PubMed] [Google Scholar]
- Jeong SY, David S. Age-related changes in iron homeostasis and cell death in the cerebellum of ceruloplasmin-deficient mice. J Neurosci. 2006;26:9810–9819. doi: 10.1523/JNEUROSCI.2922-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller JN, Hanni KB, Markesbery WR. 4-hydroxynonenal increases neuronal susceptibility to oxidative stress. J Neurosci Res. 1999;58:823–830. [PubMed] [Google Scholar]
- Klapka N, Hermanns S, Straten G, Masanneck C, Duis S, Hamers FP, Müller D, Zuschratter W, Müller HW. Suppression of fibrous scarring in spinal cord injury of rat promotes long-distance regeneration of corticospinal tract axons, rescue of primary motoneurons in somatosensory cortex and significant functional recovery. Eur J Neurosci. 2005;22:3047–3058. doi: 10.1111/j.1460-9568.2005.04495.x. [DOI] [PubMed] [Google Scholar]
- Klimtová I, Simůnek T, Mazurová Y, Kaplanová J, Sterba M, Hrdina R, Gersl V, Adamcová M, Ponka P. A study of potential toxic effects after repeated 10-week administration of a new iron chelator—salicylaldehyde isonicotinoyl hydrazone (SIH) to rabbits. Acta Medica (Hradec Kralove) 2003;46:163–170. [PubMed] [Google Scholar]
- Knutson M, Wessling-Resnick M. Iron metabolism in the reticuloendothelial system. Crit Rev Biochem Mol Biol. 2003;38:61–88. doi: 10.1080/713609210. [DOI] [PubMed] [Google Scholar]
- Knutson MD, Vafa MR, Haile DJ, Wessling-Resnick M. Iron loading and erythrophagocytosis increase ferroportin 1 (FPN1) expression in J774 macrophages. Blood. 2003;102:4191–4197. doi: 10.1182/blood-2003-04-1250. [DOI] [PubMed] [Google Scholar]
- Knutson MD, Oukka M, Koss LM, Aydemir F, Wessling-Resnick M. Iron release from macrophages after erythrophagocytosis is up-regulated by ferroportin 1 overexpression and down-regulated by hepcidin. Proc Natl Acad Sci U S A. 2005;102:1324–1328. doi: 10.1073/pnas.0409409102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Liu J, Wen J. Elevation of hydrogen peroxide after spinal cord injury detected by using the Fenton reaction. Free Radic Biol Med. 1999;27:478–482. doi: 10.1016/s0891-5849(99)00073-8. [DOI] [PubMed] [Google Scholar]
- Liu D, Ling X, Wen J, Liu J. The role of reactive nitrogen species in secondary spinal cord injury: formation of nitric oxide, peroxynitrite, and nitrated protein. J Neurochem. 2000;75:2144–2154. doi: 10.1046/j.1471-4159.2000.0752144.x. [DOI] [PubMed] [Google Scholar]
- Liu D, Liu J, Sun D, Wen J. The time course of hydroxyl radical formation following spinal cord injury: the possible role of the iron-catalyzed Haber-Weiss reaction. J Neurotrauma. 2004;21:805–816. doi: 10.1089/0897715041269650. [DOI] [PubMed] [Google Scholar]
- Lymboussaki A, Pignatti E, Montosi G, Garuti C, Haile DJ, Pietrangelo A. The role of the iron responsive element in the control of ferroportin1/IREG1/MTP1 gene expression. J Hepatol. 2003;39:710–715. doi: 10.1016/s0168-8278(03)00408-2. [DOI] [PubMed] [Google Scholar]
- MacKenzie EL, Iwasaki K, Tsuji Y. Intracellular iron transport and storage: from molecular mechanisms to health implications. Antioxid Redox Signal. 2008;10:997–1030. doi: 10.1089/ars.2007.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mautes AE, Kim DH, Sharp FR, Panter S, Sato M, Maida N, Bergeron M, Guenther K, Noble LJ. Induction of heme oxygenase-1 (HO-1) in the contused spinal cord of the rat. Brain Res. 1998;795:17–24. doi: 10.1016/s0006-8993(98)00230-3. [DOI] [PubMed] [Google Scholar]
- Mazumder B, Mukhopadhyay CK, Prok A, Cathcart MK, Fox PL. Induction of ceruloplasmin synthesis by IFN-gamma in human monocytic cells. J Immunol. 1997;159:1938–1944. [PubMed] [Google Scholar]
- Meguro R, Asano Y, Iwatsuki H, Shoumura K. Perfusion-Perls and -Turnbull methods supplemented by DAB intensification for nonheme iron histochemistry: demonstration of the superior sensitivity of the methods in the liver, spleen, and stomach of the rat. Histochem Cell Biol. 2003;120:73–82. doi: 10.1007/s00418-003-0539-y. [DOI] [PubMed] [Google Scholar]
- Miyajima H, Nishimura Y, Mizoguchi K, Sakamoto M, Shimizu T, Honda N. Familial apoceruloplasmin deficiency associated with blepharospasm and retinal degeneration. Neurology. 1987;37:761–767. doi: 10.1212/wnl.37.5.761. [DOI] [PubMed] [Google Scholar]
- Moura E, Noordermeer MA, Verhoeven N, Verheul AF, Marx JJ. Iron release from human monocytes after erythrophagocytosis in vitro: an investigation in normal subjects and hereditary hemochromatosis patients. Blood. 1998;92:2511–2519. [PubMed] [Google Scholar]
- Nedergaard M, Ransom B, Goldman SA. New roles for astrocytes: redefining the functional architecture of the brain. Trends Neurosci. 2003;26:523–530. doi: 10.1016/j.tins.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- Patel BN, David S. A novel glycosylphosphatidylinositol-anchored form of ceruloplasmin is expressed by mammalian astrocytes. J Biol Chem. 1997;272:20185–20190. doi: 10.1074/jbc.272.32.20185. [DOI] [PubMed] [Google Scholar]
- Patel BN, Dunn RJ, David S. Alternative RNA splicing generates a glycosylphosphatidylinositol-anchored form of ceruloplasmin in mammalian brain. J Biol Chem. 2000;275:4305–4310. doi: 10.1074/jbc.275.6.4305. [DOI] [PubMed] [Google Scholar]
- Patel BN, Dunn RJ, Jeong SY, Zhu Q, Julien JP, David S. Ceruloplasmin regulates iron levels in the CNS and prevents free radical injury. J Neurosci. 2002;22:6578–6586. doi: 10.1523/JNEUROSCI.22-15-06578.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard V, Govoni G, Jabado N, Gros P. Nramp 2 (DCT1/DMT1) expressed at the plasma membrane transports iron and other divalent cations into a calcein-accessible cytoplasmic pool. J Biol Chem. 2000;275:35738–35745. doi: 10.1074/jbc.M005387200. [DOI] [PubMed] [Google Scholar]
- Richardson DR, Ponka P. Orally effective iron chelators for the treatment of iron overload disease: the case for a further look at pyridoxal isonicotinoyl hydrazone and its analogs. J Lab Clin Med. 1998a;132:351–352. doi: 10.1016/s0022-2143(98)90049-x. [DOI] [PubMed] [Google Scholar]
- Richardson DR, Ponka P. Pyridoxal isonicotinoyl hydrazone and its analogs: potential orally effective iron-chelating agents for the treatment of iron overload disease. J Lab Clin Med. 1998b;131:306–315. doi: 10.1016/s0022-2143(98)90180-9. [DOI] [PubMed] [Google Scholar]
- Rouault TA. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat Chem Biol. 2006;2:406–414. doi: 10.1038/nchembio807. [DOI] [PubMed] [Google Scholar]
- Santos MM, de Sousa M, Rademakers LH, Clevers H, Marx JJ, Schilham MW. Iron overload and heart fibrosis in mice deficient for both beta2-microglobulin and Rag1. Am J Pathol. 2000;157:1883–1892. doi: 10.1016/s0002-9440(10)64827-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schültke E, Kendall E, Kamencic H, Ghong Z, Griebel RW, Juurlink BH. Quercetin promotes functional recovery following acute spinal cord injury. J Neurotrauma. 2003;20:583–591. doi: 10.1089/089771503767168500. [DOI] [PubMed] [Google Scholar]
- Simůnek T, Boer C, Bouwman RA, Vlasblom R, Versteilen AM, Sterba M, Gersl V, Hrdina R, Ponka P, de Lange JJ, Paulus WJ, Musters RJ. SIH—a novel lipophilic iron chelator—protects H9c2 cardiomyoblasts from oxidative stress-induced mitochondrial injury and cell death. J Mol Cell Cardiol. 2005;39:345–354. doi: 10.1016/j.yjmcc.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Simunek T, Sterba M, Popelova O, Kaiserova H, Potacova A, Adamcova M, Mazurova Y, Ponka P, Gersl V. Pyridoxal isonicotinoyl hydrazone (PIH) and its analogs as protectants against anthracycline-induced cardiotoxicity. Hemoglobin. 2008a;32:207–215. doi: 10.1080/03630260701680276. [DOI] [PubMed] [Google Scholar]
- Simůnek T, Sterba M, Popelová O, Kaiserová H, Adamcová M, Hroch M, Hasková P, Ponka P, Gersl V. Anthracycline toxicity to cardiomyocytes or cancer cells is differently affected by iron chelation with salicylaldehyde isonicotinoyl hydrazone. Br J Pharmacol. 2008b;155:138–148. doi: 10.1038/bjp.2008.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Harris PL, Sayre LM, Perry G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc Natl Acad Sci U S A. 1997;94:9866–9868. doi: 10.1073/pnas.94.18.9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterba M, Simůnek T, Mazurová Y, Adamcová M, Popelová O, Kaplanova J, Ponka P, Gersl V. Safety and tolerability of repeated administration of pyridoxal 2-chlorobenzoyl hydrazone in rabbits. Hum Exp Toxicol. 2005;24:581–589. doi: 10.1191/0960327105ht571oa. [DOI] [PubMed] [Google Scholar]
- Sterba M, Popelová O, Simunek T, Mazurová Y, Potácová A, Adamcová M, Kaiserová H, Ponka P, Gersl V. Cardioprotective effects of a novel iron chelator, pyridoxal 2-chlorobenzoyl hydrazone, in the rabbit model of daunorubicin-induced cardiotoxicity. J Pharmacol Exp Ther. 2006;319:1336–1347. doi: 10.1124/jpet.106.111468. [DOI] [PubMed] [Google Scholar]
- Sterba M, Popelová O, Simůnek T, Mazurová Y, Potácová A, Adamcová M, Guncová I, Kaiserová H, Palicka V, Ponka P, Gersl V. Iron chelation-afforded cardioprotection against chronic anthracycline cardiotoxicity: a study of salicylaldehyde isonicotinoyl hydrazone (SIH) Toxicology. 2007;235:150–166. doi: 10.1016/j.tox.2007.03.020. [DOI] [PubMed] [Google Scholar]
- Usatyuk PV, Parinandi NL, Natarajan V. Redox regulation of 4-hydroxy-2-nonenal-mediated endothelial barrier dysfunction by focal adhesion, adherens, and tight junction proteins. J Biol Chem. 2006;281:35554–35566. doi: 10.1074/jbc.M607305200. [DOI] [PubMed] [Google Scholar]
- Vassiliev V, Harris ZL, Zatta P. Ceruloplasmin in neurodegenerative diseases. Brain Res Brain Res Rev. 2005;49:633–640. doi: 10.1016/j.brainresrev.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Xu J, Kim GM, Chen S, Yan P, Ahmed SH, Ku G, Beckman JS, Xu XM, Hsu CY. iNOS and nitrotyrosine expression after spinal cord injury. J Neurotrauma. 2001;18:523–532. doi: 10.1089/089771501300227323. [DOI] [PubMed] [Google Scholar]
- Yang F, Liu XB, Quinones M, Melby PC, Ghio A, Haile DJ. Regulation of reticuloendothelial iron transporter MTP1 (Slc11a3) by inflammation. J Biol Chem. 2002;277:39786–39791. doi: 10.1074/jbc.M201485200. [DOI] [PubMed] [Google Scholar]
- Yang F, Naylor SL, Lum JB, Cutshaw S, McCombs JL, Naberhaus KH, McGill JR, Adrian GS, Moore CM, Barnett DR. Characterization, mapping, and expression of the human ceruloplasmin gene. Proc Natl Acad Sci U S A. 1986;83:3257–3261. doi: 10.1073/pnas.83.10.3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaitsev VN, Zaitseva I, Papiz M, Lindley PF. An X-ray crystallographic study of the binding sites of the azide inhibitor and organic substrates to ceruloplasmin, a multi-copper oxidase in the plasma. J Biol Inorg Chem. 1999;4:579–587. doi: 10.1007/s007750050380. [DOI] [PubMed] [Google Scholar]
- Zhang X, Surguladze N, Slagle-Webb B, Cozzi A, Connor JR. Cellular iron status influences the functional relationship between microglia and oligodendrocytes. Glia. 2006;54:795–804. doi: 10.1002/glia.20416. [DOI] [PubMed] [Google Scholar]
- Zoller H, Theurl I, Koch R, Kaser A, Weiss G. Mechanisms of iron mediated regulation of the duodenal iron transporters divalent metal transporter 1 and ferroportin 1. Blood Cells Mol Dis. 2002;29:488–497. doi: 10.1006/bcmd.2002.0587. [DOI] [PubMed] [Google Scholar]