

Abstract
Alcoholism is a complex and debilitating syndrome affecting ∼140 million people worldwide. However, not everyone who consumes ethanol develops abuse, raising the possibility that some individuals have a protective mechanism that inhibits elevated alcohol consumption. We tested the hypothesis that the δ-opioid receptor (DOR) plays such a protective role. Here we show that DOR activity in the ventral tegmental area (VTA) robustly decreases ethanol consumption in rats and that these effects depend on baseline ethanol consumption. Intra-VTA microinjection of the DOR agonist DPDPE decreases drinking, particularly in low-drinking animals. Furthermore, VTA microinjection of the DOR selective antagonist TIPP-Ψ increases drinking in low, but not high, drinkers and this increase is blocked by comicroinjection of the GABAA antagonist bicuculline. Using electrophysiological techniques we found that in VTA brain slices from drinking rats DPDPE presynaptically inhibits GABAA receptor mediated IPSCs in low drinkers, but not in high drinkers or naive animals, most likely through activation of DORs on GABA terminals. This DOR-mediated inhibition of IPSCs also correlates inversely with behavioral correlates of anxiety measured in the elevated plus maze. In contrast, presynaptic inhibition of VTA GABAA IPSCs by the μ- opioid receptor agonist DAMGO is significantly reduced in both high- and low-drinking rats (<30%) compared with age-matched nondrinking controls (>70%). Together, our findings demonstrate the protective nature of VTA DORs and identify an important new target for therapeutic intervention for alcoholism.
Keywords: ethanol, ventral tegmental area, δ-opioid, alcoholism, drinking, consumption
Introduction
The δ-opioid receptor (DOR) plays a protective role in cerebral ischemia, hypoxia, cardiac dysfunction, skeletal muscle damage, peripheral organ survival, and vulnerability to stress (Borlongan et al., 2004; Hebb et al., 2005; Hong et al., 2005; Saitoh et al., 2005; Chao et al., 2007; Förster et al., 2007). Interestingly, CNS expression of DORs is often dynamically induced following physiological challenge (Commons, 2003; Hack et al., 2005; Cahill et al., 2007) and trafficking to the plasma membrane is regulated through a variety of mechanisms, which are dependent on stimulus type and duration (Cahill et al., 2007). Although previous studies have identified a role of the DOR in opioid tolerance and dependence (Gomes et al., 2004; Zhang et al., 2006), and have suggested that DOR activity modulates both food consumption and drug reward (Duvauchelle et al., 1996; Kelley et al., 1996; Ragnauth et al., 1997; Suzuki et al., 1997; Lamonte et al., 2002; Khaimova et al., 2004), to date no one has specifically investigated the possible protective effects of the DOR on drug or ethanol (EtOH) consumption.
The ventral tegmental area (VTA) has been implicated in the motivational actions of opioids (Bozarth and Wise, 1984) and EtOH (McBride et al., 1991): injection of a μ-opioid receptor (MOR) agonist into the VTA is reinforcing (Bals-Kubik et al., 1993; Olmstead and Franklin, 1997; Zangen et al., 2002; Terashvili et al., 2004) while MOR antagonists in the VTA block both opioid and EtOH place preference (Phillips and LePiane, 1980; Bechtholt and Cunningham, 2005). The major reinforcing process in the VTA is thought to be excitation of dopamine (DA) neurons (Schultz, 2007) and MOR agonist-mediated reinforcement in the VTA is thought to be due to inhibition of GABA release onto DA neurons (Johnson and North, 1992). Indeed, MOR agonists in the VTA increase DA levels in multiple VTA projection targets (Spanagel et al., 1992; Devine et al., 1993b; Yoshida et al., 1993) and it has been suggested that endogenous opioid release in the VTA in response to EtOH consumption is accessing the same mechanism to produce reinforcement (Nylander et al., 1994; Bechtholt and Cunningham, 2005).
Although many studies have reported a role for DORs in regulating EtOH intake, no consistent picture of its actions has emerged. DOR knock-out mice consume more EtOH than wild types (Roberts et al., 2001), yet pharmacologically blocking DOR signaling either attenuates EtOH consumption (Lê et al., 1993; Froehlich, 1995; Krishnan-Sarin et al., 1995a,b; June et al., 1999; Hyytiä and Kiianmaa, 2001; Ciccocioppo et al., 2002) or has no effect (Hyytiä, 1993; Ingman et al., 2003). The reasons for this variability are not known; however, one possibility is that DOR efficacy changes dynamically during EtOH exposure. To determine whether dynamic regulation of the DOR affects EtOH intake in chronic drinkers, we studied the effect of DOR ligands on EtOH consumption in high- and low-drinking rats following long-term voluntary EtOH consumption. We performed this investigation in the VTA because opioid signaling in this brain region is sufficient to modulate EtOH reinforcement (Bechtholt and Cunningham, 2005).
Materials and Methods
Animals.
Fifty-three male Lewis rats (Harlan Laboratories) weighing between 275 and 300 g on arrival were housed individually in a temperature controlled colony room (21°C) on a 12 h reversed light/dark cycle (lights off at 10 A.M.). All experiments were performed during the dark portion of the cycle. Rat chow, water, and 10% EtOH (Gold Shield) were available ad libitum. Experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health) and were approved by the Ernest Gallo Clinic and Research Center Committee on Animal Research. Animals were never food or water deprived.
EtOH self-administration.
EtOH was administered via a two-bottle continuous access, free-choice paradigm in which one bottle contained 10% EtOH (v/v) and the other bottle contained water. Sucrose was never added to the EtOH solution. The amount of EtOH and water consumed was measured at the same time daily (10 A.M.). Stable self-administration was achieved in 12–14 weeks (Fig. 1). Experiments began after a stable level of EtOH self-administration was achieved for all animals. Animals were weighed daily. No significant differences or changes in weight were observed between high and low drinkers or between any treatment groups. Bottles were identical and their positions were counterbalanced and rotated daily.
Figure 1.

Drinking habits develop and stabilize over months. A, Cumulative EtOH consumption for individual high (black; n = 12) and low (red; n = 12) drinking animals over the course of 5 months on a two-bottle free choice, continuous access paradigm. Drinking patterns are clearly established by the end of the fourth month. B, Stable EtOH consumption plateau in high and low drinkers over the last 2 weeks before manipulation. Note that there is no crossover between high (black) and low (red) drinking populations. Broken horizontal lines indicate mean consumption over the 12 measurement points and shaded regions ±SEM for each group.
VTA cannulations.
Animals were anesthetized and maintained on isoflurane (0.5 l/min) as needed for the duration of surgery. Animals were placed in a stereotaxic frame and were implanted with bilateral 26-gauge stainless steel chronic guide cannulas (Plastics One) into the VTA (AP, −5.8; ML, ±0.5; DV, −7.0) based on the atlas of Paxinos and Watson (1997). Cannulas were secured to the skull with dental cement. At the end of the surgical procedure, animals were treated with penicillin and topical antibiotics. A stainless steel dummy cannula (Plastics One) was inserted into each guide cannula and remained in place when the guide cannulas were not in use. Animals were allowed a 1 week recovery period before behavioral testing.
VTA microinjections.
Each injection was made using a 1 μl syringe (Hamilton) attached to 20 cm of PE 50 tubing connected to a 33-gauge injection cannula (Plastics One). Microinjections of 0.5 μl volumes were given at a rate of 0.5 μl/min using a syringe pump (kd Scientific) into each side of the VTA, except for bicuculline injections, where 0.25 μl was injected per side. Injection cannulas extended 2 mm beyond guide cannulas and were left in place for 1 min following microinjections to minimize the backflow of drug solution. In addition to drug microinjections, physiological saline microinjections were made in every rat to measure the effect of the injection manipulation alone on drinking. Drug injections were randomized and counterbalanced. Change in drinking due to drug microinjections was calculated by comparing to both the previous day's consumption (“baseline”) and to saline injection. At the conclusion of the experiment, animals were anesthetized with pentobarbital and perfused intracardially through the ascending aorta with 0.1 m phosphate buffered saline followed by 10% formalin. Brains were sectioned coronally at 50 μm, mounted and stained with cresyl violet. Only animals with confirmed injection sites within the VTA were included (see supplemental figure, available at www.jneurosci.org, for placements). No behavioral differences were observed between animals with anterior compared with posterior cannula placements.
Elevated plus-maze.
Before electrophysiological slice recordings, animals were run in an Elevated Plus-Maze (Med Associates) for 5 min. The main platform was elevated 75 cm from the ground and consisted of four arms (50 × 10 cm each) joined by a central platform (10 × 10 cm) with floor lines delineating the entrance to each arm. Enclosed arm walls were 40 cm high. Amount of time in open and closed arms, and number of rearings in open and closed arms were recorded for analysis. Animals began testing when placed in the central hub with their head facing toward a closed arm.
Slice preparation and electrophysiology.
Lewis rats were maintained on 2 bottle choice, as described for the behavioral experiments, until their drinking stabilized. Electrophysiological experiments were completed blind to EtOH consumption levels. Recordings were made throughout the VTA. To commence electrophysiological experiments, rats were anesthetized with isoflurane and their brains were removed. Horizontal brain slices (200 μm thick) containing the VTA were prepared using a vibratome (Leica Microsystems). Slices were submerged in artificial CSF solution containing (in mm): 126 NaCl, 2.5 KCl, 1.2 MgCl, 1.4 NaH2PO4, 2.5 CaCl2, 25 NaHCO3, and 11 glucose saturated with 95% O2– 5% CO2 and allowed to recover at 32°C for at least 1 h. Individual slices were visualized using a Zeiss Axioskop microscope with differential interference contrast optics and infrared illumination. Whole-cell patch-clamp recordings were made at 32°C using 2.5–5 MΩ pipettes containing (in mm) 128 KCl, 20 NaCl, 1 MgCl2, 1 EGTA, 0.3 CaCl2, 10 HEPES, 2 MgATP, and 0.3 Na3GTP (pH 7.2, osmolarity adjusted to 275), plus 0.1% biocytin or lucifer yellow to label the recorded neuron. Signals were amplified using a Multiclamp 700B amplifier (Axon Instruments), filtered at 2 kHz, and collected at 5 kHz using IGOR Pro (Wavemetric). Ih was measured by voltage clamping cells and stepping from −60 to −40, −50, −70, −80, −90, −100, and −120 mV. Cells were recorded in voltage-clamp mode (V = −70 mV). Series resistance and input resistance were sampled throughout the experiment with 4 mV, 200 ms hyperpolarizing steps. GABAA IPSCs were pharmacologically isolated with 6,7-dinitroquinoxaline-2,3(1H,4H)-dione (DNQX: 10 μm), strychnine (1 μm), and sulpiride (10 μm). Picrotoxin (100 μm) was added at the end of some recordings to confirm the remaining signal was due to GABAA receptor activation. Stimulating electrodes were placed 80–250 μm away from the soma. To measure drug effects on evoked IPSCs, paired pulses (50-ms interval) were delivered once every 10 s. The IPSC amplitude was calculated by comparing a 2 ms period around the peak to a 2 ms interval just before stimulation. The paired-pulse ratio (PPR) was calculated by dividing the amplitude of the second IPSC by that of the first, after averaging together 8 consecutive trials. Spontaneous events were detected by searching the smoothed first derivative of the data trace for values that exceeded a set threshold, and these events were confirmed visually. Dose–response experiments were completed in the presence of a MOR antagonist (1 μm CTAP) to insure the observed effect remained DOR selective at the 10 μm dose of DPDPE.
All neurons were identified as Ih(+) or Ih(−). Wherever possible, Ih(+) neurons were immunocytochemically processed for tyrosine hydroxylase (TH) content as a marker for DA neurons (see below). However, since no results differed across any of these cell groups, the data were grouped together for analysis.
Immunocytochemistry.
Immediately after electrophysiological recording, slices were fixed in 4% formaldehyde for 2 h and then stored at 4°C in PBS. Slices were preblocked for 2 h in PBS plus 0.3% (v/v) Tween, 0.2% BSA and 5% normal goat serum and then incubated at 4°C with a rabbit anti-tyrosine hydroxylase polyclonal antibody (1:100). The slices were then washed thoroughly in PBS with 0.3% Tween and 0.2% (w/v) BSA before being agitated overnight at 4°C with Cy5 or FITC anti-rabbit secondary antibody (1:100). For cells filled with biocytin, fluorescein (DTAF)-conjugated streptavidin (3.25 μl/ml) was also added during this step. Sections were rinsed and mounted on slides using Bio-Rad Fluoroguard Antifade Reagent mounting media and visualized under a Zeiss LSM 510 META microscope. Primary antibodies were obtained from Chemicon International, secondary antibodies from Jackson ImmunoResearch Laboratories, and all other reagents from Sigma Chemical.
Drugs and doses.
For behavioral experiments, DPDPE (10 mm; Sigma), TIPP-Ψ (5 μm; NIDA), Bicuculline (1 mm; Sigma), DAMGO (0.2 mm; Sigma), and CTOP (10 mm; Tocris) were prepared in physiological saline for microinjection into the VTA. For electrophysiology, all drugs were applied by bath perfusion. Stock solutions were made and diluted in artificial CSF immediately before application. All chemicals were obtained from Sigma or Tocris except TIPP-Ψ, which was acquired from NIDA.
Data analysis.
Results are presented as mean ± SEM where appropriate. For behavioral data, drinking was analyzed using 24 h time points. Raw drinking data were used for paired Student's t test comparisons probing drug effects on drinking compared with baseline or saline injections, a more conservative comparison than analyzing normalized data. Drinking comparisons and regression analyses were completed in Excel (v.11.4.1; Microsoft). Because TIPP-Ψ induced a long-lasting, robust increase in EtOH consumption in low-drinking animals (see below), cross-over saline data could not be obtained for three animals that received the TIPP-Ψ injection before saline. Baseline data were substituted for these three animals for benefit of statistical comparison. For electrophysiology, the analyzed data were composed of the 4 min of baseline just preceding drug application and minutes 4–7 of drug application. Comparisons across electrophysiology groups were made with one-way ANOVA followed by the Student–Newman–Keuls (SNK) method for multiple comparisons where appropriate using SigmaStat software (SPSS). p < 0.05 was required for significance in all experiments.
Results
DOR activation in the VTA attenuates EtOH consumption
We initially investigated the effects of DOR activation on EtOH consumption by microinjecting DOR selective compounds into the VTA of chronically drinking Lewis rats. The DOR selective agonist DPDPE (10 mm) decreased drinking across all animals compared with drinking the day before treatment (n = 15, t = 2.14, p = 0.008) (Fig. 2A). This effect was also evident compared with control saline microinjections in the same rats, and was particularly prominent in low drinkers (n = 7, t = 2.45, p = 0.02) (Fig. 2B). This DPDPE effect on drinking was only at trend level in high drinkers (n = 8, t = 2.36, p = 0.059) (Fig. 2B). Additionally, there was an inverse correlation between baseline EtOH consumption and % baseline drinking following DPDPE administration (n = 15, F = 6.35, r = 0.57, p = 0.02), which demonstrated that the lowest drinking animals were the most affected by DPDPE. Consistent with this result, the DOR selective antagonist TIPP-Ψ (5 μm) microinjected into the VTA increased drinking across all animals compared with EtOH consumption the day preceding treatment (n = 14, t = 2.16, p = 0.006) (Fig. 3A). This was also the case compared with saline injections, and a median split of the data revealed that this effect was again driven by the low drinkers (n = 7, t = 2.45, p = 0.0002 for low drinkers vs n = 7, t = 2.45, p = 0.32 for high drinkers) (Fig. 3B). These data suggest that DOR activation by endogenous opioids released in the VTA normally suppresses EtOH intake. Moreover, some animals appear to lack this protective mechanism, resulting in increased EtOH intake.
Figure 2.

Chronically drinking rats decreased their EtOH consumption following DOR activation in the VTA. A, EtOH drinking decreased across the whole population of rats when the DOR agonist DPDPE (10 mm) was microinjected bilaterally into their VTA compared with the preceding 24 h. B, This decrease in drinking was also evident when compared with control saline injection effects and was particularly pronounced in low-drinking animals. *p < 0.05; **p < 0.01.
Figure 3.

Chronically drinking rats increased their EtOH consumption when DOR signaling was blocked in the VTA. A, Microinjecting the DOR selective antagonist TIPP-Ψ (5 μm) into the VTA increased drinking compared with the preceding 24 h of consumption. B, This increase in drinking was also apparent compared with saline injections and was driven by low-drinking animals. *p < 0.05.
DOR activation decreases GABA release in the VTA in chronically drinking animals
Our behavioral observations indicate that DOR activation in the VTA is interacting with EtOH reinforcement. One possibility is that in low drinkers EtOH is reinforcing at a lower concentration. Since attenuation of GABAA signaling in the VTA is reinforcing (Ikemoto et al., 1997) and presynaptic opioid receptor activation can inhibit neurotransmitter release (Williams et al., 2001), we examined whether DOR activation decreases GABA release onto VTA neurons in drinking animals. We measured pharmacologically isolated evoked and spontaneous GABAA IPSCs in brain slices from animals with drinking histories similar to those used in the behavioral experiments. We confirmed that the evoked and spontaneous IPSCs were due to GABAA receptor activation, as the GABAA receptor selective antagonist picrotoxin (100 μm) completely blocked both evoked and spontaneous IPSCs (n = 4, data not shown).
DPDPE (1 μm) significantly inhibited both evoked (Fig. 4) and spontaneous (Fig. 5) IPSCs in VTA neurons from drinking animals. In contrast, in age-matched, ethanol-naive, control animals there was no effect of DPDPE on GABAA IPSCs (Figs. 4A,C,D, 5B,C). The DPDPE effect in drinking animals was blocked by the DOR selective antagonist TIPP-Ψ (1 μm), indicating that the agonist was acting through the DOR (Fig. 4C). Application of the antagonist alone had no effect on either evoked IPSC amplitude (6.8 ± 9.0% decrease from baseline, n = 3) or spontaneous IPSCs (8.6 ± 2.4% decrease in frequency and 2.6 ± 1.6% decrease in amplitude, n = 2), suggesting that there is no tonic activation of DOR in this slice preparation. Importantly, for both evoked IPSCs and spontaneous IPSCs there was an inverse correlation between DPDPE induced inhibition and amount of EtOH consumed, and this relationship was particularly strong for spontaneous IPSC frequency (Fig. 6). We also collected dose–response data to probe whether elevated EtOH consumption shifted the IC50 of this DOR-mediated effect or whether the maximal effect was smaller in high-drinking and EtOH naive animals. We found that in all animal groups 1 μm DPDPE was a saturating dose with no apparent shift in the IC50 between groups (Fig. 4D).
Figure 4.

DOR agonists inhibit evoked GABAA signaling in the VTA of drinking rats. A, Example traces of evoked GABAA IPSCs from VTA neurons from EtOH naive, low and high drinkers. Only in the cell from a low drinker did the selective DOR agonist DPDPE (1 μm) decrease the amplitude of the evoked IPSC. Calibration: 50 pA, 5 ms. B, Time course of the DPDPE effects in VTA neurons from drinking animals. C, The effect of DPDPE in low drinkers (n = 4) was significantly larger than that in naive (n = 3) or high drinkers (n = 6). The effect in low drinkers was blocked by the DOR selective antagonist TIPP-Ψ (n = 3). D, Dose–response data were collected from each group, 2–4 neurons per data point.
Figure 5.
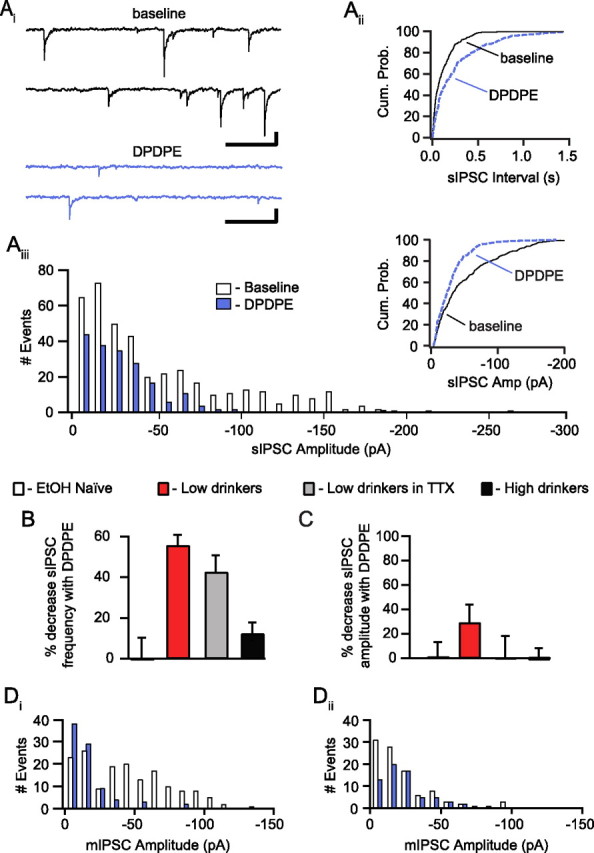
DOR agonists inhibit spontaneous GABAA signaling in the VTA of drinking rats. Ai, Example traces from the baseline period and DPDPE (1 μm) application from a cell from a low-drinking animal showing spontaneous IPSCs. Calibration: 50 pA, 200 ms. Aii, The cumulative probability plot of event interval from the same neuron shows a rightward shift in interval length in the presence of DPDPE in this case. Aiii, A histogram of the event amplitudes shows a decrease in the frequency of the events of all amplitudes with DPDPE. Inset, The cumulative probability plot of the event amplitudes shows a shift to the left with DPDPE indicating a decrease in event amplitudes in this cell. B, There was an overall greater DPDPE-induced inhibition of sIPSC frequency in neurons from low-drinking animals (n = 5) than in naive (n = 4) or high drinkers (n = 7), and this effect was also present in neurons from low drinkers when the experiment was conducted in the presence of TTX (500 nm; n = 5). C, There was a nonsignificant trend for DPDPE to decrease the sIPSC amplitude in low-drinking animals. D, Example event amplitude histograms from mIPSC experiments collected in the presence of TTX (500 nm). Very large events persist in the presence of TTX (Di), and DPDPE can either decrease (Di) or increase (Dii) the mean amplitude of mIPSCs within a particular neuron.
Figure 6.

DOR-mediated inhibition of GABAA signaling correlates with EtOH consumption. A, The magnitude of the DPDPE inhibition of evoked IPSCs was inversely correlated with EtOH consumption. B, There was also an inverse correlation between the extent of the DPDPE inhibition of sIPSC frequency and the amount of EtOH the animal consumed.
To confirm that these DOR-mediated effects were mediated by presynaptic receptors located on GABAergic terminals synapsing onto recorded neurons, we examined additional properties of the DPDPE effect in EtOH consuming animals. An increase in PPR of evoked postsynaptic currents suggests a decrease in the probability of release of neurotransmitter (Manabe et al., 1993). There was a significant increase in the PPR of evoked IPSCs following DPDPE bath application (baseline: 0.85 ± 0.08; DPDPE: 1.18 ± 0.1; p = 0.03). DPDPE also inhibited spontaneous IPSC frequency but not amplitude, across neurons (Fig. 5B,C). Moreover, the change in spontaneous IPSC frequency correlated with the inhibition of evoked IPSCs (slope = 0.75, intercept = 3.0; F = 7.17, r = 0.74, p = 0.04). We also measured miniature IPSCs (mIPSCs) recorded in the presence of tetrodotoxin (TTX; 500 nm) to block possible action potential driven events. DPDPE decreased the frequency of mIPSCs and sIPSCs similarly in neurons from low-drinking animals (Fig. 5B). Also, as with the spontaneous events, across cells there was no change in the amplitude of mIPSCs with DPDPE (Fig. 5C). We therefore conclude that these DPDPE effects on IPSCs are due to inhibition of GABA release by presynaptic DORs on GABA terminals.
While there was no significant change in the amplitude of spontaneous IPSCs following DPDPE application, a subset of neurons showed a decrease in spontaneous IPSC amplitude. This change was driven by a loss of the largest events (see example in Fig. 5A), which could result from events onto or very close to the soma or from simultaneous release of multiple vesicles. Since these large events persist in the presence of TTX (Fig. 5Di), they are not action potential dependent. At some synapses the amplitude distributions of IPSC events are sensitive to changes in the probability of release (Behrends and ten Bruggencate, 1998). Another alternative is that DORs may be expressed on a subset of presynaptic terminals. Consistent with this possibility are the example distributions of mIPSC event amplitudes showing that DPDPE selectively inhibits the frequency of large events in one cell (Fig. 5Di) and small events in a different cell (Fig. 5Dii).
DOR mediates EtOH consumption through GABAA signaling
If presynaptic inhibition of GABA release plays a role in our observed intra-VTA DOR modulation of EtOH consumption, then the TIPP-Ψ induced increase in drinking should be blocked by coinjection of a GABAA receptor antagonist. In fact, when the GABAA receptor antagonist bicuculline (1 mm) was coinjected with TIPP-Ψ (5 μm) into the VTA, it completely blocked the TIPP-Ψ induced increase in EtOH consumption in low-drinking animals (Fig. 7). Bicuculline also produced a small overall decrease in drinking across all animals following coadministration (n = 15, p = 0.035). However, a median split revealed that this effect was carried by high-drinking animals (n = 7, t = 2.45, p = 0.017) (Fig. 7) in whom TIPP-Ψ alone had no significant effect. Furthermore, TIPP-Ψ and bicuculline cotreatment had no effect on EtOH consumption in low-drinking animals (n = 8, t = 2.36, p = 0.79) (Fig. 7). Therefore, VTA GABA neurotransmission plays a critical role in DOR mediated control of EtOH consumption in low-drinking animals.
Figure 7.

DOR mediates drinking in the VTA through a GABAA mechanism. Microinjection of the GABAA antagonist bicuculline (1 mm) into the VTA does not alter drinking when compared with baseline consumption. Coinjection of bicuculline with TIPP-Ψ (5 μm) blocks the TIPP-Ψ induced increase in drinking.
DOR modulation of GABA in the VTA correlates with anxiety-like behaviors
Because anxiety has been linked to EtOH consumption (Boyd et al., 1989) we also measured behavioral correlates of anxiety using an elevated plus maze prior to electrophysiological experiments. More time spent in the open arms and more open arm rearings suggest an animal is less anxious. We found that the animals that spent more time in the open arms also expressed greater DPDPE-induced inhibition of GABAA sIPSC frequency (Fig. 8A). There was also a significant correlation between the number of rearings in the open arms and DPDPE inhibition of sIPSC frequency (Fig. 8B). These correlations suggest that animals that are less anxious have more functional DORs on GABA terminals in the VTA during chronic drinking, and raise the possibility that activation of these DORs is anxiolytic.
Figure 8.

DOR agonist effects on spontaneous IPSC frequency correlate with anxiety measures in drinking animals. Before recordings, animals were tested in the elevated plus maze. The behavioral measures of time in the open arm (A) and number of rearings (B) correlated with the DOR mediated inhibition of spontaneous GABAA IPSC frequency.
MOR antagonists in the VTA cause a decrease in EtOH consumption
Naltrexone's therapeutic effects on drinking have been proposed to be mediated through the MOR (Ghozland et al., 2005; Weerts et al., 2008), and blocking opioid signaling in the VTA diminishes EtOH reinforcement (Bechtholt and Cunningham, 2005). To evaluate the role of VTA MORs in drinking and to compare with our DOR findings, we completed similar behavioral and electrophysiological experiments with MOR ligands. In contrast to DOR, microinjection of the MOR agonist DAMGO (0.2 mm) into the VTA did not affect drinking (Fig. 9A). However, the MOR selective antagonist CTOP (10 mm) significantly decreased drinking (Fig. 9B). In contrast to the DOR antagonist mediated enhancement of ethanol consumption in low drinkers, this MOR antagonist mediated decrease was similar in high- and low- drinking animals.
Figure 9.

MOR antagonists in the VTA decrease drinking. A, The MOR agonist DAMGO (0.2 mm) microinjected into the VTA does not change EtOH consumption. B, The MOR antagonist CTOP (10 mm) microinjected into the VTA decreases EtOH consumption. *p < 0.05; **p < 0.01.
MOR agonist effects on GABA release are diminished in drinking animals
Electrophysiologically we found that, compared with EtOH naive animals, DAMGO (1 μm) had a relatively small effect on evoked and spontaneous GABAA signaling in drinking animals (27 ± 10% in drinkers versus 74 ± 5% decrease in evoked GABAA amplitude in naive animals) (Fig. 10A). However, there was great variability among these effects (Fig. 10B) and there was no apparent relationship between DAMGO effect and either EtOH consumption or cell type. The DAMGO effects on evoked IPSC amplitude and spontaneous IPSC frequency were correlated within each cell (Fig. 10B), suggesting a presynaptic site of MOR agonist action on GABAA signaling.
Figure 10.

Chronic EtOH consumption decreases presynaptic MOR inhibition at VTA GABA terminals. A, Mean DAMGO (500 nm) effects on evoked IPSC amplitude by treatment group. EtOH naive animals (n = 5) showed a greater inhibition of evoked IPSC amplitude with DAMGO than either high (n = 4) or low (n = 8) drinking animals. B, There is a correlation between the effects of DAMGO on evoked IPSC amplitude and spontaneous IPSC frequency but not drinking behavior.
Baseline GABAA signaling following chronic EtOH
Melis et al. (2002) reported an augmentation of probability of release of GABA onto VTA Ih(+) neurons 24 h after EtOH injection in mice. We examined our data to see whether such changes persist following chronic EtOH consumption. We found no difference in baseline spontaneous IPSC frequency in Ih(+) neurons between drinking rats (3.0 ± 0.6; n = 23 neurons) compared with EtOH naive rats (2.3 ± 0.4; n = 11 neurons; p = 0.45). Furthermore, the baseline PPR was similar in Ih(+) neurons from drinking (0.91 ± 0.10; n = 21) compared with EtOH naive (0.97 ± 0.09; n = 12; p = 0.70) rats. We also observed no difference in the mean amplitude of spontaneous IPSCs between drinking (32 ± 4 pA; n = 23 neurons) and EtOH naive rats (25 ± 3 pA; n = 11 neurons; p = 0.23). Therefore, the changes in GABA signaling in the VTA reported in response to acute EtOH administration appear to not persist following prolonged EtOH consumption.
Discussion
Our results demonstrate that DOR activation in the VTA inhibits EtOH consumption in actively drinking rats. In low-drinking rats, VTA microinjection of the DOR agonist DPDPE significantly decreases EtOH consumption, while the DOR selective antagonist TIPP-Ψ significantly increases drinking. These effects were not seen in high-drinking rats. Our data implicate presynaptic modulation of GABA release by DOR in these behavioral effects since DPDPE inhibits evoked, spontaneous, and miniature GABAA IPSCs in low-drinking animals, but does not change evoked or spontaneous GABAA IPSCs in high-drinking or naive animals, and also because the TIPP-Ψ induced increase in EtOH consumption in low drinkers is blocked by comicroinjection of the GABAA antagonist bicuculline.
The emergence of functional DORs following exposure to drugs of abuse and other physiological challenges is a prevalent theme in DOR research. Ultrastructural studies (Cahill et al., 2007) show that in many brain regions DORs have a primarily cytoplasmic location. These cytoplasmic DORs only become available to bind most ligands when they translocate to the cell surface. Such translocation may underlie the emergence of functional DOR on GABA terminals in the periaqueductal gray following 48 h of exposure to morphine (Hack et al., 2005). Translocation of DORs to the cell membrane also occurs in the nucleus accumbens following exposure to psychostimulants (Ambrose-Lanci et al., 2008). With respect to EtOH, we show here an increase in DOR function on GABA terminals in the VTA following chronic drinking, and this increase in DOR appears to protect against elevated EtOH consumption. This is consistent with the protective effects of DORs in other systems.
Previous research in EtOH preferring and/or high-drinking rats indicates that a DOR selective antagonist can either have no effect on EtOH consumption (Stromberg et al., 1998; Ingman et al., 2003) or can decrease EtOH consumption (Krishnan-Sarin et al., 1995a,b; June et al., 1999). The present data are consistent with the former findings. However, our current data demonstrate that, in contrast, there is significant DOR action in low-drinking animals. These results highlight the importance of considering individual differences in behavior relative to specific brain functions and offer a possible explanation for the previously reported heterogeneous effects of DOR manipulation on EtOH consumption. Furthermore, since stress and pain also cause increases in functional DORs (Besse et al., 1992; Hao et al., 1998; Ploj et al., 2003), previous experiences may influence DOR ligand modulation of drinking.
Although the anxiolytic properties of DOR selective agonists have been reported previously (Saitoh et al., 2004; Perrine et al., 2006; Jutkiewicz, 2007), the potential electrophysiological mechanisms by which these effects occur have yet to be investigated. Behaviorally, DOR knock-out mice display greater measures of anxiety than matched controls (Filliol et al., 2000), and selective δ antagonists can attenuate the anxiolytic actions of benzodiazepines (Primeaux et al., 2006). However, to our knowledge, ours is the first examination of an electrophysiological process linking anxiety-like behaviors to specific synaptic actions of DOR activation. We demonstrate that EtOH consuming animals with lower levels of anxiety have greater DOR function on GABA terminals in the VTA, raising the possibility that DOR activation in the VTA is anxiolytic. Furthermore, since less DOR inhibition of GABA release in the VTA correlates with both heightened drinking and higher behavioral anxiety scores, DOR expression in the VTA may be a potential physiological link between anxiety behaviors and EtOH consumption. These data are also intriguing in light of recent results showing an interaction between corticotrophin releasing factor (CRF), a physiological indicator of stress, and EtOH consumption, such that blocking CRF receptor activation decreases EtOH consumption (Heilig and Koob, 2007; Marinelli et al., 2007; Lowery et al., 2008). Additional studies are necessary to investigate the relationship between different types of stress, anxiety, EtOH consumption, and DOR function and to determine the time course of these effects.
Behaviorally, we found that MOR and DOR selective antagonists have opposing effects on EtOH self-administration: intra-VTA microinjection of the MOR selective antagonist CTOP decreases consumption while the highly selective DOR antagonist TIPP-Ψ increases consumption. One possible explanation for this difference is that MORs and DORs regulate EtOH consumption through actions at distinct cellular sites within the VTA. Unlike DORs that emerge with EtOH exposure, EtOH downregulates MORs in the VTA (Mendez et al., 2001), in particular diminishing the ability of MOR agonists to decrease GABAA signaling. Therefore the dominant effect of MOR activation in drinking animals may be shifted to postsynaptic cell bodies and/or presynaptic glutamate terminals. Microdialysis studies in naive rats show that microinjecting either the MOR agonist DAMGO or the MOR antagonist CTOP into the VTA increases DA levels in the nucleus accumbens (Devine et al., 1993a), suggesting a highly complex function of VTA MORs. In naive animals, MORs also modulate glutamate release and postsynaptically inhibit subsets of both DA and non-DA VTA neurons (Cameron et al., 1997; Bonci and Malenka, 1999; Manzoni and Williams, 1999; Margolis et al., 2003, 2005). Thus, MOR modulation of EtOH consumption may be due to a combination of these different synaptic effects in the VTA.
GABAA signaling in the VTA participates in a circuit involved in motivation and reinforcement (Ikemoto et al., 1997). Although VTA GABAA signaling is required for the DOR antagonist mediated increase in EtOH consumption, the downstream consequences of DOR activation are unclear since DOR-mediated inhibition of GABA release is similar onto DA and non-DA neurons. Therefore, in vivo activation of DORs on GABA terminals could disinhibit of all classes of VTA neurons, including glutamate, GABA, and DA neurons (Dahlstroem and Fuxe, 1964; Van Bockstaele and Pickel, 1995; Yamaguchi et al., 2007). Since there are DA-independent outputs of the VTA that can contribute to behavioral reinforcement (Nader and van der Kooy, 1997; Laviolette et al., 2004), it is possible that projecting glutamate or GABA VTA neurons are responsible for the modulation of drinking reported here. DA may also be involved in this modulation, since DA agonists and antagonists alter drinking (Pfeffer and Samson, 1988; Weiss et al., 1990; Rassnick et al., 1992, 1993; Cohen et al., 1998, 1999). If DA does play a role in DOR-mediated EtOH consumption, our data are consistent with an inverted u-shaped relationship between DA level and reward value. According to this model, animals self administer drugs or consume EtOH to achieve an optimal level of DA release. Therefore, animals with low basal DA or systems that are more resistant to processes that increase DA release tend to self administer larger quantities of drugs or EtOH, while animals with higher basal DA or more responsive DA systems tend to self administer less (Weissenborn et al., 1996; Volkow et al., 1999; Caine et al., 2000; Mattay et al., 2003; Meyer-Lindenberg et al., 2005; Cohen et al., 2007). Our data are consistent with this model in that endogenous opioids released when the animal is drinking should disinhibit DA neurons by activating DORs on GABA terminals, thereby increasing DA release and attenuating EtOH consumption in low-drinking animals. Blocking the DOR with TIPP-Ψ increases GABA release, thereby decreasing DA neural activity in low-drinking animals, which drives these animals to consume more EtOH to achieve optimal DA release. High-drinking animals, in which these DOR effects are minimal, may drink more because their basal DA levels are lower (Murphy et al., 1983, 1987; Bustamante et al., 2008) or because higher EtOH concentrations are required to increase DA release.
The nonspecific opioid antagonist naltrexone (Revia) is the most widely prescribed FDA approved treatment for alcohol abuse. However, its actions at multiple receptor types are likely a drawback to its use as a therapeutic for alcoholism. Understanding the contribution of each opioid receptor to EtOH consumption could lead to the development of a more effective therapeutic by permitting more selective targeting of the appropriate receptor(s). For example, MOR selective antagonists reduce EtOH intake (Krishnan-Sarin et al., 1998) while KOR selective antagonists can increase EtOH intake (Mitchell et al., 2005; but see Walker and Koob, 2008) in rats. Therefore, antagonizing all opioid receptors simultaneously is not an optimal treatment strategy. We show here that activating, not antagonizing, DORs may be most appropriate for decreasing EtOH consumption. Therefore an improved therapeutic efficacy would likely result from a compound that acts as both a DOR agonist and MOR antagonist. Together, our findings demonstrate a protective action of the VTA DOR on alcohol consumption and identify an important new target for therapeutic intervention.
Footnotes
This work was supported by U. S. Army Awards W81XWH-08-1-0017 and W81XWH-07-1-043; the awarding and administering acquisition office is The U. S. Army Medical Research Acquisition Activity, 820 Chandler Street, Fort Detrick, MD 21702-5014. The content of the information does not necessarily reflect the position or the policy of the Government, and no official endorsement should be inferred. This work was also supported by the State of California for medical research on alcohol and substance abuse through the University of California, San Francisco. We thank Leah Grossman and Joe Driscoll for their technical assistance.
References
- Ambrose-Lanci LM, Peiris NB, Unterwald EM, Van Bockstaele EJ. Cocaine withdrawal-induced trafficking of delta-opioid receptors in rat nucleus accumbens. Brain Res. 2008;1210:92–102. doi: 10.1016/j.brainres.2008.02.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bals-Kubik R, Ableitner A, Herz A, Shippenberg TS. Neuroanatomical sites mediating the motivational effects of opioids as mapped by the conditioned place preference paradigm in rats. J Pharmacol Exp Ther. 1993;264:489–495. [PubMed] [Google Scholar]
- Bechtholt AJ, Cunningham CL. Ethanol-induced conditioned place preference is expressed through a ventral tegmental area dependent mechanism. Behav Neurosci. 2005;119:213–223. doi: 10.1037/0735-7044.119.1.213. [DOI] [PubMed] [Google Scholar]
- Behrends JC, ten Bruggencate G. Changes in quantal size distributions upon experimental variations in the probability of release at striatal inhibitory synapses. J Neurophysiol. 1998;79:2999–3011. doi: 10.1152/jn.1998.79.6.2999. [DOI] [PubMed] [Google Scholar]
- Besse D, Weil-Fugazza J, Lombard MC, Butler SH, Besson JM. Monoarthritis induces complex changes in mu-, delta- and kappa-opioid binding sites in the superficial dorsal horn of the rat spinal cord. Eur J Pharmacol. 1992;223:123–131. doi: 10.1016/0014-2999(92)94830-o. [DOI] [PubMed] [Google Scholar]
- Bonci A, Malenka RC. Properties and plasticity of excitatory synapses on dopaminergic and GABAergic cells in the ventral tegmental area. J Neurosci. 1999;19:3723–3730. doi: 10.1523/JNEUROSCI.19-10-03723.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borlongan CV, Wang Y, Su TP. Delta opioid peptide (D-Ala 2, D-Leu 5) enkephalin: linking hibernation and neuroprotection. Front Biosci. 2004;9:3392–3398. doi: 10.2741/1490. [DOI] [PubMed] [Google Scholar]
- Boyd TL, Callen EJ, House WJ. The effects of post-stress exposure to alcohol upon the development of alcohol consumption in rats. Behav Res Ther. 1989;27:35–41. doi: 10.1016/0005-7967(89)90117-4. [DOI] [PubMed] [Google Scholar]
- Bozarth MA, Wise RA. Anatomically distinct opiate receptor fields mediate reward and physical dependence. Science. 1984;224:516–517. doi: 10.1126/science.6324347. [DOI] [PubMed] [Google Scholar]
- Bustamante D, Quintanilla ME, Tampier L, Gonzalez-Lira V, Israel Y, Herrera-Marschitz M. Ethanol induces stronger dopamine release in nucleus accumbens (shell) of alcohol-preferring (bibulous) than in alcohol-avoiding (abstainer) rats. Eur J Pharmacol. 2008;591:153–158. doi: 10.1016/j.ejphar.2008.06.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill CM, Holdridge SV, Morinville A. Trafficking of delta-opioid receptors and other G-protein-coupled receptors: implications for pain and analgesia. Trends Pharmacol Sci. 2007;28:23–31. doi: 10.1016/j.tips.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Caine SB, Negus SS, Mello NK. Effects of dopamine D(1-like) and D(2-like) agonists on cocaine self-administration in rhesus monkeys: rapid assessment of cocaine dose-effect functions. Psychopharmacology (Berl) 2000;148:41–51. doi: 10.1007/s002130050023. [DOI] [PubMed] [Google Scholar]
- Cameron DL, Wessendorf MW, Williams JT. A subset of ventral tegmental area neurons is inhibited by dopamine, 5-hydroxytryptamine and opioids. Neuroscience. 1997;77:155–166. doi: 10.1016/s0306-4522(96)00444-7. [DOI] [PubMed] [Google Scholar]
- Chao D, Bazzy-Asaad A, Balboni G, Xia Y. delta-, but not mu-, opioid receptor stabilizes K(+) homeostasis by reducing Ca(2+) influx in the cortex during acute hypoxia. J Cell Physiol. 2007;212:60–67. doi: 10.1002/jcp.21000. [DOI] [PubMed] [Google Scholar]
- Ciccocioppo R, Martin-Fardon R, Weiss F. Effect of selective blockade of mu(1) or delta opioid receptors on reinstatement of alcohol-seeking behavior by drug-associated stimuli in rats. Neuropsychopharmacology. 2002;27:391–399. doi: 10.1016/S0893-133X(02)00302-0. [DOI] [PubMed] [Google Scholar]
- Cohen C, Perrault G, Sanger DJ. Preferential involvement of D3 versus D2 dopamine receptors in the effects of dopamine receptor ligands on oral ethanol self-administration in rats. Psychopharmacology (Berl) 1998;140:478–485. doi: 10.1007/s002130050792. [DOI] [PubMed] [Google Scholar]
- Cohen C, Perrault G, Sanger DJ. Effects of D1 dopamine receptor agonists on oral ethanol self-administration in rats: comparison with their efficacy to produce grooming and hyperactivity. Psychopharmacology (Berl) 1999;142:102–110. doi: 10.1007/s002130050868. [DOI] [PubMed] [Google Scholar]
- Cohen MX, Krohn-Grimberghe A, Elger CE, Weber B. Dopamine gene predicts the brain's response to dopaminergic drug. Eur J Neurosci. 2007;26:3652–3660. doi: 10.1111/j.1460-9568.2007.05947.x. [DOI] [PubMed] [Google Scholar]
- Commons KG. Translocation of presynaptic delta opioid receptors in the ventrolateral periaqueductal gray after swim stress. J Comp Neurol. 2003;464:197–207. doi: 10.1002/cne.10788. [DOI] [PubMed] [Google Scholar]
- Dahlstroem A, Fuxe K. Evidence for the existence of monoamine-containing neurons in the central nervous system. I. Demonstration of monoamines in the cell bodies of brain stem neurons. Acta Physiol Scand Suppl. 1964;232:1–55. [PubMed] [Google Scholar]
- Devine DP, Leone P, Wise RA. Mesolimbic dopamine neurotransmission is increased by administration of mu-opioid receptor antagonists. Eur J Pharmacol. 1993a;243:55–64. doi: 10.1016/0014-2999(93)90167-g. [DOI] [PubMed] [Google Scholar]
- Devine DP, Leone P, Pocock D, Wise RA. Differential involvement of ventral tegmental mu, delta and kappa opioid receptors in modulation of basal mesolimbic dopamine release: in vivo microdialysis studies. J Pharmacol Exp Ther. 1993b;266:1236–1246. [PubMed] [Google Scholar]
- Duvauchelle CL, Fleming SM, Kornetsky C. Involvement of delta- and mu-opioid receptors in the potentiation of brain-stimulation reward. Eur J Pharmacol. 1996;316:137–143. doi: 10.1016/s0014-2999(96)00674-7. [DOI] [PubMed] [Google Scholar]
- Filliol D, Ghozland S, Chluba J, Martin M, Matthes HW, Simonin F, Befort K, Gavériaux-Ruff C, Dierich A, LeMeur M, Valverde O, Maldonado R, Kieffer BL. Mice deficient for delta- and mu-opioid receptors exhibit opposing alterations of emotional responses. Nat Genet. 2000;25:195–200. doi: 10.1038/76061. [DOI] [PubMed] [Google Scholar]
- Förster K, Kuno A, Solenkova N, Felix SB, Krieg T. The delta-opioid receptor agonist DADLE at reperfusion protects the heart through activation of pro-survival kinases via EGF receptor transactivation. Am J Physiol Heart Circ Physiol. 2007;293:H1604–H1608. doi: 10.1152/ajpheart.00418.2007. [DOI] [PubMed] [Google Scholar]
- Froehlich JC. Genetic factors in alcohol self-administration. J Clin Psychiatry. 1995;56(Suppl 7):15–23. [PubMed] [Google Scholar]
- Ghozland S, Chu K, Kieffer BL, Roberts AJ. Lack of stimulant and anxiolytic-like effects of ethanol and accelerated development of ethanol dependence in mu-opioid receptor knockout mice. Neuropharmacology. 2005;49:493–501. doi: 10.1016/j.neuropharm.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Gomes I, Gupta A, Filipovska J, Szeto HH, Pintar JE, Devi LA. A role for heterodimerization of mu and delta opiate receptors in enhancing morphine analgesia. Proc Natl Acad Sci U S A. 2004;101:5135–5139. doi: 10.1073/pnas.0307601101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hack SP, Bagley EE, Chieng BC, Christie MJ. Induction of delta-opioid receptor function in the midbrain after chronic morphine treatment. J Neurosci. 2005;25:3192–3198. doi: 10.1523/JNEUROSCI.4585-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao JX, Yu W, Xu XJ. Evidence that spinal endogenous opioidergic systems control the expression of chronic pain-related behaviors in spinally injured rats. Exp Brain Res. 1998;118:259–268. doi: 10.1007/s002210050280. [DOI] [PubMed] [Google Scholar]
- Hebb AL, Drolet G, Mendella PD, Roach SP, Gauthier MS, Zacharko RM. Intracerebroventricular D-Pen2, D-Pen5-enkephalin administration soon after stressor imposition influences behavioral responsivity to a subsequent stressor encounter in CD-1 mice. Pharmacol Biochem Behav. 2005;82:453–469. doi: 10.1016/j.pbb.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Heilig M, Koob GF. A key role for corticotropin-releasing factor in alcohol dependence. Trends Neurosci. 2007;30:399–406. doi: 10.1016/j.tins.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong J, Sigg DC, Coles JA, Jr, Oeltgen PR, Harlow HJ, Soule CL, Iaizzo PA. Hibernation induction trigger reduces hypoxic damage of swine skeletal muscle. Muscle Nerve. 2005;32:200–207. doi: 10.1002/mus.20354. [DOI] [PubMed] [Google Scholar]
- Hyytiä P. Involvement of mu-opioid receptors in alcohol drinking by alcohol-preferring AA rats. Pharmacol Biochem Behav. 1993;45:697–701. doi: 10.1016/0091-3057(93)90527-z. [DOI] [PubMed] [Google Scholar]
- Hyytiä P, Kiianmaa K. Suppression of ethanol responding by centrally administered CTOP and naltrindole in AA and Wistar rats. Alcohol Clin Exp Res. 2001;25:25–33. doi: 10.1111/j.1530-0277.2001.tb02123.x. [DOI] [PubMed] [Google Scholar]
- Ikemoto S, Murphy JM, McBride WJ. Self-infusion of GABA(A) antagonists directly into the ventral tegmental area and adjacent regions. Behav Neurosci. 1997;111:369–380. doi: 10.1037//0735-7044.111.2.369. [DOI] [PubMed] [Google Scholar]
- Ingman K, Salvadori S, Lazarus L, Korpi ER, Honkanen A. Selective delta-opioid receptor antagonist N,N(CH3)2-Dmt-Tic-OH does not reduce ethanol intake in alcohol-preferring AA rats. Addict Biol. 2003;8:173–179. doi: 10.1080/1355621031000117400. [DOI] [PubMed] [Google Scholar]
- Johnson SW, North RA. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci. 1992;12:483–488. doi: 10.1523/JNEUROSCI.12-02-00483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- June HL, McCane SR, Zink RW, Portoghese PS, Li TK, Froehlich JC. The delta 2-opioid receptor antagonist naltriben reduces motivated responding for ethanol. Psychopharmacology (Berl) 1999;147:81–89. doi: 10.1007/s002130051145. [DOI] [PubMed] [Google Scholar]
- Jutkiewicz EM. RB101-mediated protection of endogenous opioids: potential therapeutic utility? CNS Drug Rev. 2007;13:192–205. doi: 10.1111/j.1527-3458.2007.00011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley AE, Bless EP, Swanson CJ. Investigation of the effects of opiate antagonists infused into the nucleus accumbens on feeding and sucrose drinking in rats. J Pharmacol Exp Ther. 1996;278:1499–1507. [PubMed] [Google Scholar]
- Khaimova E, Kandov Y, Israel Y, Cataldo G, Hadjimarkou MM, Bodnar RJ. Opioid receptor subtype antagonists differentially alter GABA agonist-induced feeding elicited from either the nucleus accumbens shell or ventral tegmental area regions in rats. Brain Res. 2004;1026:284–294. doi: 10.1016/j.brainres.2004.08.032. [DOI] [PubMed] [Google Scholar]
- Krishnan-Sarin S, Portoghese PS, Li TK, Froehlich JC. The delta 2-opioid receptor antagonist naltriben selectively attenuates alcohol intake in rats bred for alcohol preference. Pharmacol Biochem Behav. 1995a;52:153–159. doi: 10.1016/0091-3057(95)00080-g. [DOI] [PubMed] [Google Scholar]
- Krishnan-Sarin S, Jing SL, Kurtz DL, Zweifel M, Portoghese PS, Li TK, Froehlich JC. The delta opioid receptor antagonist naltrindole attenuates both alcohol and saccharin intake in rats selectively bred for alcohol preference. Psychopharmacology (Berl) 1995b;120:177–185. doi: 10.1007/BF02246191. [DOI] [PubMed] [Google Scholar]
- Krishnan-Sarin S, Wand GS, Li XW, Portoghese PS, Froehlich JC. Effect of mu opioid receptor blockade on alcohol intake in rats bred for high alcohol drinking. Pharmacol Biochem Behav. 1998;59:627–635. doi: 10.1016/s0091-3057(97)00474-7. [DOI] [PubMed] [Google Scholar]
- Lamonte N, Echo JA, Ackerman TF, Christian G, Bodnar RJ. Analysis of opioid receptor subtype antagonist effects upon mu opioid agonist-induced feeding elicited from the ventral tegmental area of rats. Brain Res. 2002;929:96–100. doi: 10.1016/s0006-8993(01)03382-0. [DOI] [PubMed] [Google Scholar]
- Laviolette SR, Gallegos RA, Henriksen SJ, van der Kooy D. Opiate state controls bi-directional reward signaling via GABAA receptors in the ventral tegmental area. Nat Neurosci. 2004;7:160–169. doi: 10.1038/nn1182. [DOI] [PubMed] [Google Scholar]
- Lê AD, Poulos CX, Quan B, Chow S. The effects of selective blockade of delta and mu opiate receptors on ethanol consumption by C57BL/6 mice in a restricted access paradigm. Brain Res. 1993;630:330–332. doi: 10.1016/0006-8993(93)90672-a. [DOI] [PubMed] [Google Scholar]
- Lowery EG, Sparrow AM, Breese GR, Knapp DJ, Thiele TE. The CRF-1 receptor antagonist, CP-154,526, attenuates stress-induced increases in ethanol consumption by BALB/cJ mice. Alcohol Clin Exp Res. 2008;32:240–248. doi: 10.1111/j.1530-0277.2007.00573.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manabe T, Wyllie DJ, Perkel DJ, Nicoll RA. Modulation of synaptic transmission and long-term potentiation: effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J Neurophysiol. 1993;70:1451–1459. doi: 10.1152/jn.1993.70.4.1451. [DOI] [PubMed] [Google Scholar]
- Manzoni OJ, Williams JT. Presynaptic regulation of glutamate release in the ventral tegmental area during morphine withdrawal. J Neurosci. 1999;19:6629–6636. doi: 10.1523/JNEUROSCI.19-15-06629.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Hjelmstad GO, Bonci A, Fields HL. Kappa-opioid agonists directly inhibit midbrain dopaminergic neurons. J Neurosci. 2003;23:9981–9986. doi: 10.1523/JNEUROSCI.23-31-09981.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Hjelmstad GO, Bonci A, Fields HL. Both kappa and mu opioid agonists inhibit glutamatergic input to ventral tegmental area neurons. J Neurophysiol. 2005;93:3086–3093. doi: 10.1152/jn.00855.2004. [DOI] [PubMed] [Google Scholar]
- Marinelli PW, Funk D, Juzytsch W, Harding S, Rice KC, Shaham Y, Lê AD. The CRF1 receptor antagonist antalarmin attenuates yohimbine-induced increases in operant alcohol self-administration and reinstatement of alcohol seeking in rats. Psychopharmacology (Berl) 2007;195:345–355. doi: 10.1007/s00213-007-0905-x. [DOI] [PubMed] [Google Scholar]
- Mattay VS, Goldberg TE, Fera F, Hariri AR, Tessitore A, Egan MF, Kolachana B, Callicott JH, Weinberger DR. Catechol O-methyltransferase val158-met genotype and individual variation in the brain response to amphetamine. Proc Natl Acad Sci U S A. 2003;100:6186–6191. doi: 10.1073/pnas.0931309100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride WJ, Murphy JM, Gatto GJ, Levy AD, Lumeng L, Li TK. Serotonin and dopamine systems regulating alcohol intake. Alcohol Alcohol Suppl. 1991;1:411–416. [PubMed] [Google Scholar]
- Melis M, Camarini R, Ungless MA, Bonci A. Long-lasting potentiation of GABAergic synapses in dopamine neurons after a single in vivo ethanol exposure. J Neurosci. 2002;22:2074–2082. doi: 10.1523/JNEUROSCI.22-06-02074.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez M, Leriche M, Calva JC. Acute ethanol administration differentially modulates mu opioid receptors in the rat meso-accumbens and mesocortical pathways. Brain Res Mol Brain Res. 2001;94:148–156. doi: 10.1016/s0169-328x(01)00232-7. [DOI] [PubMed] [Google Scholar]
- Meyer-Lindenberg A, Kohn PD, Kolachana B, Kippenhan S, McInerney-Leo A, Nussbaum R, Weinberger DR, Berman KF. Midbrain dopamine and prefrontal function in humans: interaction and modulation by COMT genotype. Nat Neurosci. 2005;8:594–596. doi: 10.1038/nn1438. [DOI] [PubMed] [Google Scholar]
- Mitchell JM, Liang MT, Fields HL. A single injection of the kappa opioid antagonist norbinaltorphimine increases ethanol consumption in rats. Psychopharmacology (Berl) 2005;182:384–392. doi: 10.1007/s00213-005-0067-7. [DOI] [PubMed] [Google Scholar]
- Murphy JM, McBride WJ, Lumeng L, Li TK. Monoamine and metabolite levels in CNS regions of the P line of alcohol-preferring rats after acute and chronic ethanol treatment. Pharmacol Biochem Behav. 1983;19:849–856. doi: 10.1016/0091-3057(83)90092-8. [DOI] [PubMed] [Google Scholar]
- Murphy JM, McBride WJ, Lumeng L, Li TK. Contents of monoamines in forebrain regions of alcohol-preferring (P) and -nonpreferring (NP) lines of rats. Pharmacol Biochem Behav. 1987;26:389–392. doi: 10.1016/0091-3057(87)90134-1. [DOI] [PubMed] [Google Scholar]
- Nader K, van der Kooy D. Deprivation state switches the neurobiological substrates mediating opiate reward in the ventral tegmental area. J Neurosci. 1997;17:383–390. doi: 10.1523/JNEUROSCI.17-01-00383.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nylander I, Hyytiä P, Forsander O, Terenius L. Differences between alcohol-preferring (AA) and alcohol-avoiding (ANA) rats in the prodynorphin and proenkephalin systems. Alcohol Clin Exp Res. 1994;18:1272–1279. doi: 10.1111/j.1530-0277.1994.tb00118.x. [DOI] [PubMed] [Google Scholar]
- Olmstead MC, Franklin KB. The development of a conditioned place preference to morphine: effects of microinjections into various CNS sites. Behav Neurosci. 1997;111:1324–1334. doi: 10.1037//0735-7044.111.6.1324. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates, compact. Ed 3. San Diego: Academic; 1997. [Google Scholar]
- Perrine SA, Hoshaw BA, Unterwald EM. Delta opioid receptor ligands modulate anxiety-like behaviors in the rat. Br J Pharmacol. 2006;147:864–872. doi: 10.1038/sj.bjp.0706686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer AO, Samson HH. Haloperidol and apomorphine effects on ethanol reinforcement in free feeding rats. Pharmacol Biochem Behav. 1988;29:343–350. doi: 10.1016/0091-3057(88)90167-0. [DOI] [PubMed] [Google Scholar]
- Phillips AG, LePiane FG. Reinforcing effects of morphine microinjection into the ventral tegmental area. Pharmacol Biochem Behav. 1980;12:965–968. doi: 10.1016/0091-3057(80)90460-8. [DOI] [PubMed] [Google Scholar]
- Ploj K, Roman E, Nylander I. Long-term effects of maternal separation on ethanol intake and brain opioid and dopamine receptors in male Wistar rats. Neuroscience. 2003;121:787–799. doi: 10.1016/s0306-4522(03)00499-8. [DOI] [PubMed] [Google Scholar]
- Primeaux SD, Wilson SP, McDonald AJ, Mascagni F, Wilson MA. The role of delta opioid receptors in the anxiolytic actions of benzodiazepines. Pharmacol Biochem Behav. 2006;85:545–554. doi: 10.1016/j.pbb.2006.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragnauth A, Ruegg H, Bodnar RJ. Evaluation of opioid receptor subtype antagonist effects in the ventral tegmental area upon food intake under deprivation, glucoprivic and palatable conditions. Brain Res. 1997;767:8–16. doi: 10.1016/s0006-8993(97)00539-8. [DOI] [PubMed] [Google Scholar]
- Rassnick S, Pulvirenti L, Koob GF. Oral ethanol self-administration in rats is reduced by the administration of dopamine and glutamate receptor antagonists into the nucleus accumbens. Psychopharmacology (Berl) 1992;109:92–98. doi: 10.1007/BF02245485. [DOI] [PubMed] [Google Scholar]
- Rassnick S, Pulvirenti L, Koob GF. SDZ-205,152, a novel dopamine receptor agonist, reduces oral ethanol self-administration in rats. Alcohol. 1993;10:127–132. doi: 10.1016/0741-8329(93)90091-2. [DOI] [PubMed] [Google Scholar]
- Roberts AJ, Gold LH, Polis I, McDonald JS, Filliol D, Kieffer BL, Koob GF. Increased ethanol self-administration in delta-opioid receptor knockout mice. Alcohol Clin Exp Res. 2001;25:1249–1256. [PubMed] [Google Scholar]
- Saitoh A, Kimura Y, Suzuki T, Kawai K, Nagase H, Kamei J. Potential anxiolytic and antidepressant-like activities of SNC80, a selective delta-opioid agonist, in behavioral models in rodents. J Pharmacol Sci. 2004;95:374–380. doi: 10.1254/jphs.fpj04014x. [DOI] [PubMed] [Google Scholar]
- Saitoh A, Yoshikawa Y, Onodera K, Kamei J. Role of delta-opioid receptor subtypes in anxiety-related behaviors in the elevated plus-maze in rats. Psychopharmacology (Berl) 2005;182:327–334. doi: 10.1007/s00213-005-0112-6. [DOI] [PubMed] [Google Scholar]
- Schultz W. Behavioral dopamine signals. Trends Neurosci. 2007;30:203–210. doi: 10.1016/j.tins.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Spanagel R, Herz A, Shippenberg TS. Opposing tonically active endogenous opioid systems modulate the mesolimbic dopaminergic pathway. Proc Natl Acad Sci U S A. 1992;89:2046–2050. doi: 10.1073/pnas.89.6.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromberg MF, Casale M, Volpicelli L, Volpicelli JR, O'Brien CP. A comparison of the effects of the opioid antagonists naltrexone, naltrindole, and beta-funaltrexamine on ethanol consumption in the rat. Alcohol. 1998;15:281–289. doi: 10.1016/s0741-8329(97)00131-6. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Tsuji M, Ikeda H, Misawa M, Narita M, Tseng LF. Antisense oligodeoxynucleotide to delta opioid receptors blocks cocaine-induced place preference in mice. Life Sci. 1997;60:PL 283–288. doi: 10.1016/s0024-3205(97)00143-4. [DOI] [PubMed] [Google Scholar]
- Terashvili M, Wu HE, Leitermann RJ, Hung KC, Clithero AD, Schwasinger ET, Tseng LF. Differential conditioned place preference responses to endomorphin-1 and endomorphin-2 microinjected into the posterior nucleus accumbens shell and ventral tegmental area in the rat. J Pharmacol Exp Ther. 2004;309:816–824. doi: 10.1124/jpet.103.059287. [DOI] [PubMed] [Google Scholar]
- Van Bockstaele EJ, Pickel VM. GABA-containing neurons in the ventral tegmental area project to the nucleus accumbens in rat brain. Brain Res. 1995;682:215–221. doi: 10.1016/0006-8993(95)00334-m. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Fowler JS, Logan J, Gatley SJ, Gifford A, Hitzemann R, Ding YS, Pappas N. Prediction of reinforcing responses to psychostimulants in humans by brain dopamine D2 receptor levels. Am J Psychiatry. 1999;156:1440–1443. doi: 10.1176/ajp.156.9.1440. [DOI] [PubMed] [Google Scholar]
- Walker BM, Koob GF. Pharmacological evidence for a motivational role of kappa-opioid systems in ethanol dependence. Neuropsychopharmacology. 2008;33:643–652. doi: 10.1038/sj.npp.1301438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weerts EM, Kim YK, Wand GS, Dannals RF, Lee JS, Frost JJ, McCaul ME. Differences in delta- and mu-opioid receptor blockade measured by positron emission tomography in naltrexone-treated recently abstinent alcohol-dependent subjects. Neuropsychopharmacology. 2008;33:653–665. doi: 10.1038/sj.npp.1301440. [DOI] [PubMed] [Google Scholar]
- Weiss F, Mitchiner M, Bloom FE, Koob GF. Free-choice responding for ethanol versus water in alcohol preferring (P) and unselected Wistar rats is differentially modified by naloxone, bromocriptine, and methysergide. Psychopharmacology (Berl) 1990;101:178–186. doi: 10.1007/BF02244123. [DOI] [PubMed] [Google Scholar]
- Weissenborn R, Deroche V, Koob GF, Weiss F. Effects of dopamine agonists and antagonists on cocaine-induced operant responding for a cocaine-associated stimulus. Psychopharmacology (Berl) 1996;126:311–322. doi: 10.1007/BF02247382. [DOI] [PubMed] [Google Scholar]
- Williams JT, Christie MJ, Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev. 2001;81:299–343. doi: 10.1152/physrev.2001.81.1.299. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Sheen W, Morales M. Glutamatergic neurons are present in the rat ventral tegmental area. Eur J Neurosci. 2007;25:106–118. doi: 10.1111/j.1460-9568.2006.05263.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M, Yokoo H, Tanaka T, Mizoguchi K, Emoto H, Ishii H, Tanaka M. Facilitatory modulation of mesolimbic dopamine neuronal activity by a mu-opioid agonist and nicotine as examined with in vivo microdialysis. Brain Res. 1993;624:277–280. doi: 10.1016/0006-8993(93)90087-4. [DOI] [PubMed] [Google Scholar]
- Zangen A, Ikemoto S, Zadina JE, Wise RA. Rewarding and psychomotor stimulant effects of endomorphin-1: anteroposterior differences within the ventral tegmental area and lack of effect in nucleus accumbens. J Neurosci. 2002;22:7225–7233. doi: 10.1523/JNEUROSCI.22-16-07225.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Bao L, Guan JS. Role of delivery and trafficking of delta-opioid peptide receptors in opioid analgesia and tolerance. Trends Pharmacol Sci. 2006;27:324–329. doi: 10.1016/j.tips.2006.04.005. [DOI] [PubMed] [Google Scholar]