

Abstract
Huntington disease (HD), caused by CAG expansion in the ubiquitously expressed huntingtin gene, is characterized by early dysfunction and death of striatal medium-sized spiny neurons (MSNs). Previous work has shown MSN-specific alterations in NMDA receptor (NMDAR) expression and cell death signaling. Furthermore, studies in HD human brain tissue and a knock-in mouse model demonstrate increases in calpain activity, which can be stimulated by NMDARs and contribute to excitotoxicity. Here, we report increased calpain activity in MSNs from the yeast artificial chromosome (YAC) transgenic mouse model of HD, expressing human full-length huntingtin with 128 polyglutamine repeats (YAC128), compared with wild type. Moreover, the calpain-cleaved product of NMDAR subunit NR2B is increased early, and NR2B expression levels are reduced, in YAC128 striatum. Although steady-state NMDAR surface expression is similar in wild-type and YAC128 MSNs, the rate of loss of NR2B-containing surface receptors is enhanced in YAC128 MSNs, suggesting that NMDAR forward trafficking to the surface is also faster, as previously reported for YAC72 MSNs. Calpain inhibitor-1 treatment normalized the loss rate of surface NMDARs in YAC128 MSNs to that of wild type, and significantly increased surface NMDAR expression in YAC128, but not in wild type or YAC72. With acute NMDAR overstimulation, the increase in calpain activity correlated with polyglutamine length, and calpain inhibitor treatment reduced NMDA-induced apoptosis in YAC72 and YAC128 MSNs to wild-type levels. Thus, the cumulative effect of increasing huntingtin polyglutamine length is to enhance MSN sensitivity to excitotoxicity at least in part by calpain-mediated cell death signaling.
Keywords: Huntington, NMDA receptor, calpain, calcium, toxicity, trafficking, striatum
Introduction
NMDA glutamate receptors contribute to excitatory neurotransmission (Dingledine et al., 1999), but NMDA receptor (NMDAR) overactivation causes excitotoxic neuronal death (Papadia and Hardingham, 2007). Altered NMDAR activity has been implicated in the pathogenesis of Huntington disease (HD), caused by CAG expansion in the gene for huntingtin (htt), resulting in selective degeneration of striatal medium-sized spiny neurons (MSNs) (The Huntington's Disease Collaborative Research Group, 1993; Vonsattel and DiFiglia, 1998). Similar selective neurodegeneration is reproduced by striatal injection of NMDAR-specific agonists (Beal et al., 1986; Sanberg et al., 1989), and postmortem analysis of human HD brains suggests NMDAR-expressing neurons are particularly vulnerable to degeneration (Young et al., 1988; Albin et al., 1990). Although other mechanisms are also involved in neuronal dysfunction and death in HD (Cowan and Raymond, 2006), excitotoxicity may help explain neuronal selectivity.
Mouse models of HD provide further evidence for the excitotoxicity hypothesis. Yeast artificial chromosome (YAC) transgenic mice express full-length human htt with normal (YAC18) or pathological (YAC72, YAC128) polyQ sizes (Hodgson et al., 1999; Slow et al., 2003). In this model, severity of striatal excitotoxicity parallels expression of htt polyQ expansion-mediated toxicity, since mice expressing caspase-6 cleavage-resistant htt, or particular htt fragments, lack an HD phenotype and exhibit resistance to striatal excitotoxicity despite presence of the polyQ expansion (Slow et al., 2005; Graham et al., 2006). Moreover, NMDA-evoked current density in MSNs increases with htt polyQ length, and the enhanced current observed in YAC72 MSNs can be explained by increased numbers of surface NMDARs, resulting from accelerated forward trafficking (Fan et al., 2007). In YAC72 MSNs, the larger NMDAR current amplitude correlates with enhanced NMDA-induced apoptosis (Shehadeh et al., 2006). In YAC128 MSNs, however, NMDAR current density is similar to WT (Fernandes et al., 2007), whereas NMDA-induced apoptosis is enhanced as in YAC72 MSNs (Graham et al., 2006a; Shehadeh et al., 2006). We hypothesize that the extreme polyQ expansion in YAC128 MSNs produces greater cell stress, resulting in activation of signaling pathways that downregulate NMDAR surface numbers while enhancing sensitivity to apoptosis.
In addition to caspases, calpains, calcium-activated cysteine proteases that are involved in neuronal apoptosis (Harwood et al., 2005), may also play a role in HD. Activated calpains have been detected in striata of HD knock-in mice and postmortem HD human brain (Gafni and Ellerby, 2002; Gafni et al., 2004). The htt protein itself is a substrate for calpains at residues 469 and 536 (Gafni et al., 2004), which may contribute to the toxic gain-of-function theory of htt proteolysis (Goldberg et al., 1996; Wellington and Hayden, 1997). Susceptibility of htt to calpain cleavage in vitro increases with polyQ length (Gafni and Ellerby, 2002), and mutation at the two calpain-cleavage sites eliminates htt aggregation and toxicity (Gafni et al., 2004).
Here we address whether tonic calpain activity is altered in MSNs from YAC mice and what effect this has on NMDAR function and expression. Furthermore, we determine whether calpain activation after NMDA exposure correlates with htt polyQ length and contributes to the enhanced NMDAR-mediated excitotoxicity observed with htt polyQ expansion.
Materials and Methods
Mice
Mice used in all experiments were either wild-type FVB/NJ, or the offspring of crosses between homozygous mice expressing the full-length human htt transgene contained in the yeast artificial chromosome (YAC) on a pure FVB/NJ background. Transgenic mice used were YAC18 (line 212), YAC72 (line 2511), and YAC128 (line 55). Mice were housed and handled according to Canadian Committee on Animal Care guidelines.
MSN primary culture
As previously published (Zeron et al., 2004), striata were dissected from postnatal day 0–1 mice in HBSS on ice; digested in papain solution (4.5% papain, 1 mm CaCl2, 0.2 mg/ml cysteine hydrochloride, 500 μm EDTA in HBSS) at 37°C for 10 min; further dissociated through a series of Pasteur pipettes in 37°C trypsin inhibitor solution (0.25% trypsin inhibitor, 0.25% BSA, 20 μg/ml DNase in Neurobasal media); and plated in serum-free Neurobasal media (Invitrogen) with 2% B27 (Invitrogen), 2 mm l-glutamine and 100 U/ml penicillin/streptomycin at a density of 2 × 105 cells/well on glass coverslips coated with 50 μg/ml poly-d-lysine in 24-well culture plates. Cells were grown at 37°C, 5% CO2 for 9 d.
In vitro excitotoxicity
Methods used to induce NMDAR-mediated apoptosis in cultured MSNs were as previously published (Shehadeh et al., 2006). Primary cultured MSNs at 9 DIV were pretreated (or not) for 1 h at 37°C with 1 μm calpain inhibitor 1 (CI-1) added to conditioned media; then treated for 10 min at 37°C with 500 μm NMDA in BSS (139 mm NaCl, 3.5 mm KCl, 2 mm NaHCO3, 11 mm d-glucose, 50 μm glycine, pH 7.35), in the continued presence or absence of 1 μm CI-1. Controls were treated with DMSO or BSS alone, as appropriate. Cells were then returned to conditioned media, with or without 1 μm CI-1, for 24 h, before being fixed with 4% paraformaldehyde (PFA) in PBS for 15 min at room temperature (for subsequent immunocytochemistry) or for 1 h at 4°C (for analysis of apoptotic death).
Immunocytochemistry
Spectrin.
Cells were treated with 10 mm sodium citrate, pH 6, at 95°C for 15 min; permeabilized in 0.5% Triton X-100 in PBS for 5 min at room temperature; blocked in 5% bovine serum albumin (BSA) in PBS for 30 min at room temperature; incubated in anti-cleaved-spectrin α II (h1186) primary antibody (Santa Cruz, s.c.-23464) at 1:50 in 2% BSA in PBS for 1 h at room temperature; washed 3 times in PBS; incubated in AlexaFluor 488-conjugated anti-goat secondary antibody (Molecular Probes) at 1:400 in 2% BSA in PBS for 1 h at room temperature; washed 3 times in PBS; counterstained with 10 μm Hoechst in PBS for 10 min at room temperature; and mounted with Fluormount (Southern Biotechnology).
Surface/internal NR1.
Sister coverslips of cultured striatal MSNs at 9 DIV were treated with vehicle dimethyl sulfoxide (DMSO) or 1 μm CI-1 dissolved in DMSO for 24 h and subjected to immunostaining of surface and internal NR1 as previously described (Fan et al., 2007). Cells were blocked in 10% normal goat serum (NGS) in PBS for 30 min at room temperature; incubated in anti-NR1 primary antibody (Chemicon, MAB363) at 1:500 in 1% NGS in PBS for 12–18 h at 4°C; washed 3 times in PBS; incubated in AlexaFluor 488-tagged anti-mouse secondary antibody (Molecular Probes) at 1:1000 in 1% NGS in PBS for 40 min at room temperature; washed 3 times in PBS; permeabilized in 0.5% Triton X-100 1% PFA in PBS for 5 min at room temperature; blocked in 10% NGS in PBS for 30 min at room temperature; incubated again in the same anti-NR1 primary antibody for 90 min at room temperature; washed 3 times in PBS; incubated in AlexaFluor 594-labeled anti-mouse secondary antibody (Molecular Probes) at 1:1000 in 1% NGS in PBS for 40 min at room temperature; washed 3 times in PBS; and mounted with Fluormount (Southern Biotechnology).
Analysis of immunocytochemistry
Cells were viewed at 630× magnification with an Axiophot (Zeiss) epifluorescence microscope, with the experimenter blinded to condition. To analyze calpain activity, photographs of MSNs labeled with anti-cleaved spectrin were taken (6 pairs of photographs per coverslip per condition, from n = 3–6 independent batches of cultures, equating to >200 dendrites per data point) keeping exposure time and all other imaging parameters constant. Proximal dendrites of MSNs were then traced using Northern Eclipse Software, and the average fluorescence intensity of each dendrite was measured. To assess the ratio of surface to internal NR1 [as described by Fan et al. (2007)], pairs of photographs were taken in the same way, and ImageJ software was used to stack the red (internal NR1) and green (surface NR1) images, draw around each MSN soma and a section of proximal dendrite, and measure the average fluorescence intensity. An individual background reading was subtracted for each image.
TUNEL
As previously published (Zeron et al., 2002; Shehadeh et al., 2006), cells were incubated in 0.25% Triton X-100 and 0.25% sodium citrate in PBS for 2 min on ice; incubated in 16 μl/ml RNase in PBS for 30 min at room temperature; incubated in fluorescein-conjugated terminal dUTP nick-end labeling (TUNEL) mixture (Roche, as per manufacturer's instructions) for 60 min at 37°C; counterstained with 10 μm propidium iodide or 1 μm Hoechst in PBS for 10–15 min at room temperature; and mounted with Fluormount (Southern Biotechnology). MSNs were scored as apoptotic or nonapoptotic visually at 630× magnification using an epifluorescence microscope (Zeiss Axiophot), with the experimenter blinded to condition. Strict criteria of cellular and nuclear apoptotic morphology in conjunction with TUNEL positivity were used (see Shehadeh et al., 2006). One thousand cells were counted per condition per n; n = 3–7 independent batches of primary cultures.
Preparation of protein samples from tissue and cells
Two-month-old mice were killed by terminal halothane anesthesia followed by decapitation; all procedures were approved by the UBC Animal Care Committee and adhere to Canadian Committee on Animal Care guidelines. Striata were dissected out on ice and homogenized in harvest buffer [1 mm EDTA, 1 mm EGTA, 2 μg/ml aprotinin, 1 mm phenylmethylsulfonyl fluoride, 2 μg/ml leupeptin, 4 μg/ml pepstatin A, 30 mm NaF (pH 7), 40 mm β-glycerophosphate (pH 7), 20 mm sodium pyrophosphate (pH 7), 3.5 mm sodium orthovanadate, in PBS]. The homogenate was spun down briefly at 14,000 rpm, the supernatant discarded, and the pellet resuspended and sonicated in lysis buffer (harvest buffer, 0.2% SDS, 0.8% Triton X-100). Primary MSNs were collected for protein lysates by scraping the cells into chilled harvest buffer, sonicating the cell pellet in lysis buffer in the same way as for tissue, followed by a 30 min rotation at 4°C, 10 min spin at 14,000 rpm, and transfer of supernatant to fresh tube, discarding any pellet. Tissue and cell lysates were stored at −80°C.
Immunoprecipitation
Striatal lysates from 2-month-old mice were prepared as described above. One-tenth volume of the homogenate was reserved for subsequent loading to SDS-PAGE as the lysate (“Lys”) sample. The remainder was incubated with protein A- and protein G-linked beads, rotating for 1 h at 4°C, followed by centrifugation for 5 min at 14,000 rpm. The supernatant was transferred to a fresh tube and incubated with 1 μg of NR2C antibody (A-6475, Invitrogen), rotating 1 h at 4°C, after which protein A- and protein G-linked beads were added with rotation for another 1 h at 4°C. The sample was centrifuged for 3 min at 5000 rpm, and supernatant discarded. Beads were washed 5 times with 500 μl of Tris washing buffer (Tris 50 mm, pH 7.4; NaCl 150 mm; EDTA 1 mm; EGTA 1 mm; 1% Triton X-100), each time centrifuging briefly at 5000 rpm and removing supernatant. Immunoprecipitated proteins were uncoupled from the beads and solubilized in protein sample buffer. All samples in loading buffer were boiled for 3 min at 98°C before loading to SDS-PAGE, as described below.
Western blotting
Protein concentration of tissue or cell lysates was determined by bicinchoninic acid (BCA) protein assay (Pierce); and 20–40 μg of proteins, boiled for 2 min in 3× protein sample buffer (6% SDS, 0.4 mm Tris pH 6.8, 30% glycerol, pyronin Y, 70 mg/ml dithiothreitol), were resolved by 8% SDS-polyacrylamide gel electrophoresis (SDS-PAGE); and transferred by wet electrophoresis onto polyvinylidene difluoride (PVDF) membrane (Bio-Rad). Membranes were wetted in methanol; equilibrated in water and Tris-buffered saline (TBS); blocked for 90 min in 3% BSA 0.5% Tween 20 in TBS; incubated in primary antibody in blocking solution for 12–18 h at 4°C; washed 3 times in 0.05% Tween 20 in TBS (TBS-T); incubated in horseradish peroxidase-coupled secondary antibody (Amersham) at 1:5000 in blocking solution for 1 h at room temperature; washed 3 times in TBS-T; and visualized using enhanced chemiluminescence substrate (Amersham) and exposure to film (Kodak). Primary antibodies used were: anti-calpain 1 (Santa Cruz s.c.-7531 at 1:100); anti-NR1 C-terminal epitope (Upstate, 06-311 at 1 μg/ml); anti-NR2B C-terminal epitopes (Upstate 06-600 at 1:1000; or Transduction Labs, 610417 at 1:250; or Alomone Labs, AGC-003 at 1:1000); anti-NR2B N-terminal epitope (Invitrogen 71–8600 at 1:250); goat anti-actin (Santa Cruz s.c.-1616 at 1:400 to 1:100); and an antibody against the N-terminal 25–130 aa of NR2C (Molecular Probes A-6475 at 1:200). Calpain activity was assessed using a rabbit anti-spectrin antibody recognizing calpain-cleaved spectrin—AB38 1:3000 (Roberts-Lewis et al., 1994) (gift from Dr. David Lynch, University of Pennsylvania, Philadelphia, PA) using the same blotting protocol, except that blocking solution and antibody diluent was 3% milk in TBS-T instead of BSA. Densitometry of resulting bands was performed using Scion Image software.
Biotinlyation analysis of receptor surface expression and degradation rate
Primary cultures of MSNs were prepared in parallel from WT and YAC128 mice. Cells at 9 DIV were washed and precooled to 10°C to halt cellular metabolism, before incubation for 12 min at 10°C with 1.5 mg/ml NHS-SS-Biotin (Pierce) in PBS containing 0.1 mm Ca2+ and 1 mm Mg2+ to biotinylate surface proteins. Following two washes with 0.1% BSA in PBS containing 0.1 mm Ca2+ and 1 mm Mg2+, cells were harvested in PBS supplemented with 1 mm EDTA, 1 mm EGTA, and protease inhibitors, and proteins were solubilized with 1% Triton X-100. Twenty percent of the lysate was reserved for loading to SDS-PAGE (“Lys” sample) and the remaining 80% was incubated with NeutrAvidin biotin-binding resin (Pierce) for 2.5 h at 4°C. Following sedimentation of bound biotin-labeled proteins by centrifugation, one-quarter of the supernatant was saved as the “Sup” sample, while the protein-bound resin was washed 4 times in solubilization buffer before the proteins were eluted by incubation in 3× protein sample buffer (“Biot” sample). Samples were subsequently resolved by SDS-PAGE and immunoblotted for NR1 or NR2B to determine fraction of receptors at the cell surface under basal conditions. Mean band intensities were measured, and the percentage of biotin-labeled receptors relative to total NR1 or NR2B was calculated as “Biot” divided by the sum of “Biot” and four times the intensity of “Sup.”
To analyze the effects of calpain inhibition on receptor degradation rate, cells were pretreated (or not) for 1 h at 37°C with 1 μm CI-1 added to conditioned media. Following the biotinylation step, cells were replenished with conditioned media with or without CI-1 and replaced at 37°C, 5% CO2 for 0, 12, or 24 h. Cell lysates were then collected and processed as before, and NR2B expression measured in eluate and supernatant samples to determine of the rate of receptor loss from the cell surface.
Materials
All reagents are from Sigma except where otherwise stated.
Results
Calpain activity is chronically elevated in the striatum of YAC128 mice
Previous studies indicate that calpain activity is elevated in brains from humans with HD as well as knock-in HD mice (Gafni and Ellerby, 2002; Gafni et al., 2004), and this enzyme is known to contribute to cell death signaling in neurons (Czogalla and Sikorski, 2005). Although NMDAR current amplitudes are similar in YAC128 and WT MSNs (Fernandes et al., 2007), YAC128 MSNs are more sensitive to NMDAR-mediated apoptosis (Graham et al., 2006b) and show a slower recovery following NMDA-induced calcium loads (Fernandes et al., 2007). We hypothesized that an increase in chronic, basal levels of calcium-dependent protease activity may contribute to the increased vulnerability to apoptosis.
To address whether basal protease activity is altered by mutant htt expression, we assessed striatal tissue from 2-month-old WT or YAC mice, which do not show an obvious motor or neuropathological phenotype (Hodgson et al., 1999; Slow et al., 2003). Probing Western blots with an antibody against the calpain protein, we found no difference in levels of calpain between WT, YAC18, YAC72, YAC128 homozygote (line 55) or YAC128 heterozygote (line 53) mice (Fig. 1A,B). However, when we probed similar blots with a neo-epitope antibody specific for spectrin cleaved by calpain (Roberts-Lewis et al., 1994), we found that levels of the 150 kDa spectrin cleavage product were significantly elevated in YAC128 compared with WT striata, and that YAC72 striata showed an intermediate level of calpain activity by this assay (Fig. 1C,D). Thus, even though we found no evidence for increased levels of calpain in the YAC transgenic mice at this stage, cleavage of a calpain substrate was significantly increased in YAC128 striatal tissue, suggesting that calpain activity levels were elevated.
Figure 1.
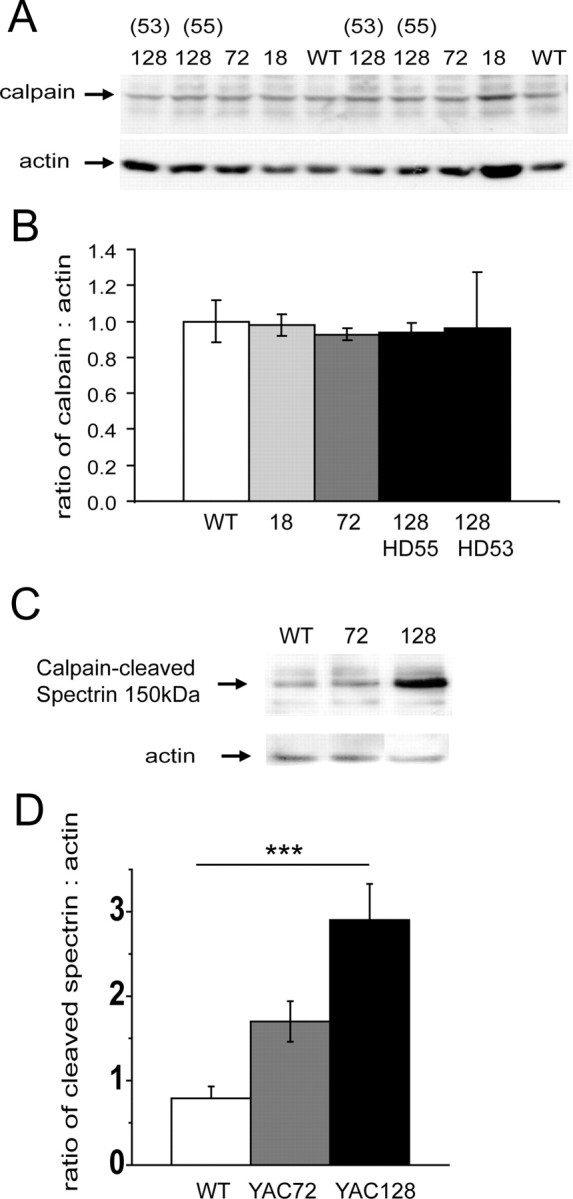
Calpain activity, but not total calpain protein, is increased early and chronically in the striatum of YAC128 mice compared with WT. A, B, Western blot (A) and graph (B) of calpain expression in 2-month-old mouse striatum from WT, 18Q, 72Q, and 128Q (lines 53 and 55) show no difference in the levels of calpain protein between genotypes. However, Western blots of similar tissue probed for the calpain substrate α-spectrin (C, and graph D) show a significant increase in the 150 kDa fragment of α-spectrin produced specifically by calpain-mediated cleavage in striatal tissue from YAC128 over WT mice [**p = 0.0013 by one-way ANOVA for genotype difference among three groups, F(2,12) = 12.07; ***p < 0.001 for YAC128 versus WT by Dunnett's post hoc test; n = 5 independent experiments in which striatal tissue (dissected from different mice for each independent experiment) from each of the three genotypes was run together on the same gel, followed by Western analysis].
Our previous studies measuring NMDAR current and excitotoxicity were done in MSNs cultured from newborn mice, so we repeated the assay for calpain-cleaved spectrin in MSNs under normal culture conditions. As with striatal tissue from 2-month-old mice, we found a trend toward increased levels of the 150 kDa cleaved spectrin product in YAC128 MSNs (cleaved spectrin to actin ratio was: 0.48 ± 0.1, 0.59 ± 0.18, and 1.13 ± 0.57 for WT, YAC72, and YAC128 MSNs, respectively; n = 4 paired experiments; data not shown).
NR2B fragment produced by calpain cleavage is increased in YAC128 striatal tissue
Calpain activation results in cleavage of the C-terminal tail (around amino acid 1030) of the NR2B subunit of NMDARs (Bi et al., 1998a; Guttmann et al., 2001, 2002; Simpkins et al., 2003). We hypothesized that the elevated calpain activity levels we found in YAC128 striatal neurons would be associated with an increased proportion of calpain-cleaved NR2B. To measure this directly, we took advantage of an antibody raised against the N-terminal region of rat full-length NR2C, which in Western blotting has been shown to recognize most NR2 subunit species, including NR2A, NR2C, NR2B, and the C-terminal-truncated fragment of NR2B produced by calpain (Simpkins et al., 2003; Dong et al., 2004).
We probed Western blots of striatal and cerebellar lysates from 2-month-old YAC mice and controls with the NR2C antibody and found that the band corresponding to the 115 kDa calpain-cleaved fragment of NR2B was indeed stronger in YAC128 striatum than striata from other genotypes (Fig. 2A, top panel). This difference was not observed in the cerebellum. As expected from the literature, we also noted that the band density for full-length NR2B was stronger in striatum than cerebellum; NR2C was stronger in cerebellum and undetectable in striatum; and the 115 kDa calpain-cleaved fragment of NR2B was stronger in striatum than cerebellum.
Figure 2.

The 115 kDa fragment of NR2B produced by calpain cleavage is increased in YAC128 tissue, specifically in the striatum. A, Top panel, Western blots showing lysates of striata and cerebella from 2 month-old YAC and WT mice probed with an antibody raised against NR2C (top strip), which recognizes all NR2 subunits. Indicated are the bands representing full-length NR2B/2A, NR2C, and the 115 kDa calpain-cleaved fragment of NR2B. Blots are reprobed with antibodies against NR1 (middle strip) and actin (bottom strip) and are representative of 3 independent experiments. Bottom panel, Control showing that when cultured YAC72 MSNs are treated with 1 μm calpain inhibitor-1 (CI-1) for 24 h before harvesting, the 115 kDa band is markedly reduced, as detected with the anti-NR2C antibody. Blot is representative of n = 7 independent experiments with 4 different culture batches of YAC72 and 3 different batches of WT MSNs. B, NR2 subunits were immunoprecipitated (IP lanes) from 2-month-old WT and YAC128 striatal tissue using the anti-NR2C antibody, and Western blots were probed with an antibody against the N-terminal region of NR2B. Note the bands representing full-length and the 115 kDa fragment of NR2B in both IP and lysate (Lys) lanes, and that the ratio of NR2B fragment to full-length protein is higher in YAC128 than WT striatal tissue. Blot is representative of 4 paired experiments from 4 different mice of each genotype. C, Graph showing the band densities averaged from 3 experiments as shown in A (striatum and cerebellum were dissected from 1–2 mice for each genotype in each independent experiment), expressed as a ratio of 115 kDa NR2B fragment to full-length NR2B. This ratio was significantly higher in YAC128 than in WT striatum (*p < 0.05 by unpaired two-tailed Student's t test). There was no difference between genotypes in the cerebellum. D, Data from the same blots, expressed as a ratio of 115 kDa NR2B fragment to NR1. Again, this ratio was significantly higher in YAC128 striatum than WT striatum (**p < 0.01 by unpaired two-tailed Student's t test), and there was no difference between genotypes in the cerebellum.
To confirm the published reports that the 115 kDa band we detected with the NR2C antibody is indeed the calpain-cleaved fragment of NR2B, we performed additional controls in which cultured MSNs were treated for 24 h with 1 μm CI-1 (Fig. 2A, bottom panel); under these conditions the 115 kDa band density was significantly reduced by 40% (ratio of NR2B 115 kDa fragment to full-length NR2B was 0.69 ± 0.12 and 0.41 ± 0.05 for vehicle-treated versus CI-1-treated MSN cultures; p < 0.05 by paired t test, n = 7 different treatment-paired experiments in either YAC72 or WT MSNs). This long treatment time was chosen because, although calpain should be fully inhibited after 30 min of CI-1 treatment (Adamec et al., 1998), it has been shown that the NR2B fragments are relatively stable for at least that period (Simpkins et al., 2003); however, it is possible that the partial reduction of cleaved NR2B resulted from progressive degradation of CI-1 in the culture medium.
One caveat for interpreting the density of the full-length band is that this could represent either NR2B or NR2A as the antibody does not distinguish between these two with similar molecular weights. To more accurately determine the ratio of NR2B fragment to full-length and also to confirm that the 115 kDa band detected with the anti-NR2C antibody indeed represents a cleaved fragment of NR2B, we used the anti-NR2C antibody to immunoprecipitate NR2 subunits from striatal tissue of WT and YAC128 2-month-old mice, then performed Western blot analysis using an antibody specific for an N-terminal epitope of NR2B. As shown in Figure 2B, this antibody recognized the same 115 kDa band visualized with the anti-NR2C antibody in Figure 2A. Moreover, as with the NR2C antibody, the N-terminal NR2B antibody revealed an increased ratio of 115 kDa fragment to full-length NR2B for YAC128 compared with WT striatal tissue (Fig. 2B) (ratios were 0.85 ± 0.15 and 0.33 ± 0.03, respectively; n = 4 paired experiments from different mice of each genotype; p < 0.05 by two-tailed paired t test)
The experiment shown in Figure 2A was repeated a total of three times using different mice of each genotype for each experiment, and the densities of the 115 kDa NR2B band were expressed as a ratio to full-length NR2B/NR2A (Fig. 2C). Results indicated that there was significantly more calpain-cleaved NR2B in the YAC128 striatum than in striata from other genotypes. To control for the possibility that this may be an artifact resulting from less total NR2B in YAC128 striata (see below), we also expressed the results as a ratio of calpain-cleaved NR2B to NR1. With this analysis, the increase observed in YAC128 striatum was even more significant and also not seen in the cerebellum (Fig. 2D). We also observed a trend toward the same result in primary cultured MSNs (data not shown).
NR2B levels are decreased in YAC128 striatum
Increased levels of calpain-cleaved NR2B would be expected to enhance NMDAR endocytosis and lysosomal degradation (Scott et al., 2004). Although NR2B expression levels are similar for WT and YAC72 striatal tissue from 2-month-old mice (Li et al., 2003), NR2B expression has not been assessed in brains from YAC128 mice. To test whether the observed increase in basal calpain activity in YAC128 MSNs is associated with a decrease in total NR2B protein levels, we performed Western blots on striatal lysates from YAC128 and age-matched WT mice at 2 and 12 months of age. As expected, we found that levels of NR2B protein were significantly decreased in striatal, but not cortical, hippocampal, or cerebellar, tissue from YAC128 compared with WT mice at 12 months of age (shown in Fig. 3A,B for n = 4 different paired experiments of WT and YAC128 tissue). At 2 months, levels of NR2B in YAC128 striata were only 52% of WT, which was not a significant difference for the four paired experiments shown in Figure 3B. However, when the results of an additional four paired experiments were pooled with this data, normalizing band densities for YAC128 to WT in each experiment, the reduction in NR2B expression was significant (the ratio of NR2B to actin band density from YAC128 striatal tissue normalized to that of WT striatal tissue was 0.52 ± 0.12; p < 0.05, n = 8). By 12 months, levels of NR2B in YAC128 striata had further decreased to 29% of WT (p < 0.05 by paired t test, n = 4). At both ages, despite reductions in the striatum, there was no significant difference in NR2B expression levels between YAC128 and WT in the cerebellum, neocortex, or hippocampus; however, there was a trend toward reduced NR2B at 12 months in the YAC128 neocortex. It is also notable that in both genotypes and all brain regions there was a decrease in total levels of NR2B between 2 and 12 months, as previously reported in rats (Magnusson, 2000), and that in both genotypes and both ages there was very little detectable NR2B protein in the cerebellum, as expected (Monyer et al., 1994; Wenzel et al., 1997).
Figure 3.

Levels of NR2B, a calpain substrate, are decreased in YAC128 striatum. A, Representative Western blots of lysates of cerebellum (Cb), cortex (Cx), hippocampus (H), and striatum (S) from 2-month and 12-month mice, probed for NR2B and actin. Blots are representative of results from WT and YAC128 brain tissues that were collected and processed in parallel in 4 independent experiments (brain tissue was dissected from different mice in each independent experiment). B, Bar graphs show the average of 4 independent experiments. Note there is a trend toward a decrease in NR2B in the striatum of YAC128 mice at 2 months, and a statistically significant decrease by 12 months (*p < 0.05). Levels of NR2B are not different between genotype in any other brain region examined. Very little NR2B was detected in the cerebellum.
Accelerated loss of surface NR2B mediated by calpain activity in YAC128 MSNs
Calpain cleavage of the NR2B C terminus would be expected to accelerate receptor loss from the surface by unmasking an AP2 binding site that promotes NR2B endocytosis and trafficking to late endosomes for degradation (Scott et al., 2004). We hypothesized that loss of NMDARs from the surface of YAC128 MSNs would be enhanced as a result of calpain cleavage, and that this might explain the similar NMDAR current densities observed in YAC128 and WT MSNs (Fernandes et al., 2007) while YAC72 MSNs exhibit enhanced NMDAR surface expression and current (Fan et al., 2007).
To test our hypothesis we first determined the proportion of NMDARs expressed at the surface of cultured MSNs, using two different approaches. As shown in Figure 4, A and B, after incubation of MSN cultures with a membrane-impermeable biotinylating agent and isolation of biotinylated (surface) proteins by pull-down with NeutrAvidin beads, the calculated proportion of surface NR1 and NR2B was not significantly different for WT and YAC128 MSNs. As a second approach we used immunocytochemistry, sequentially labeling for NR1 on the cell surface (before membrane permeabilization) and intracellular NR1 (after membrane permeabilization), detected with green and red fluorescent secondary antibodies, respectively, as previously described (Fan et al., 2007). We then assessed the ratio of surface (green) to internal (red) fluorescence intensity (see Materials and Methods). The ratio of surface to internal NR1 in YAC128 MSNs was not significantly different from WT (0.65 ± 0.09 and 0.58 ± 0.08 for YAC128 and WT MSNs, respectively; data from 5 paired experiments on different batches of cultures; p = 0.09 by paired t test). Thus, NMDAR surface expression correlates well with the lack of significant difference between WT and YAC128 MSN NMDAR current densities (Fernandes et al., 2007). Using this immunocytochemical approach, we found that treatment of cultured MSNs with CI-1 for 24 h resulted in a small but significant increase in the surface to internal ratio of NMDAR expression in YAC128 MSNs but not in WT or YAC72 MSNs (Fig. 4C,D), suggesting that excess calpain activity in YAC128 MSNs is associated with downregulation of surface NMDAR expression.
Figure 4.
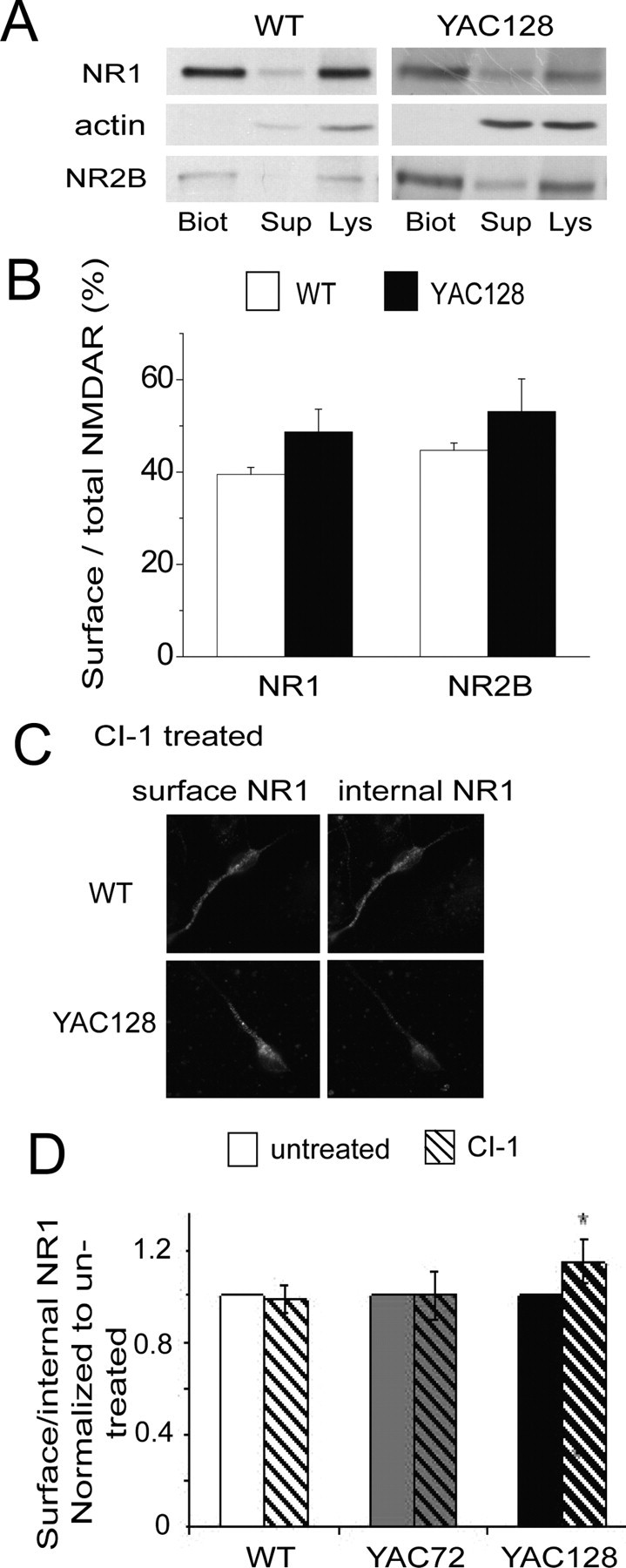
Role of excess calpain activity in regulating NMDAR surface expression in YAC128 versus WT MSNs. A, Representative Western blots probed with antibodies against NR1 or NR2B showing surface [biotinylated (“Biot”)], internal [supernatant from the centrifugation that pulls down biotinylated proteins attached to NeutrAvidin beads (“Sup”)], and total [lysate (“Lys”)] proteins. Note the absence of an actin band in the biotinylated surface protein fraction (Biot), as would be expected. B, Bar graph shows proportion of NR1 and NR2B at the neuronal surface (see Materials and Methods) in untreated WT and YAC128 9 DIV MSNs. NR1: n = 7 and 9 independent experiments for WT and YAC128 MSNs, respectively, from 7 and 9 different culture batches; NR2B: n = 5 and 4 independent experiments for WT and YAC128 MSNs, respectively, from 5 and 4 different cultures batches. C, Representative photomicrographs of surface (left panels) and internal (right panels) NR1 immunostaining in WT compared with YAC128 MSNs. MSN cultures at 9 DIV were incubated with 1 μm CI-1 for 24 h then fixed and incubated with NR1 antibodies before (surface) and after (internal) membrane permeabilization (see Materials and Methods). D, Bar graph shows the ratio of intensities of surface to internal NR1 labeling in WT, YAC72 and YAC128 MSNs treated for 24 h with CI-1, normalized to vehicle-treated sister cultures. Note that calpain inhibition increases NR1 surface to internal ratio only in YAC128 MSNs and not in WT or YAC72. n = 6, 6, and 4 experiments from different culture batches for WT, YAC72, and YAC128, respectively. *p < 0.05, paired two-tailed Student's t test.
Increased cleavage of the NR2B C-terminal region, as shown in Figure 2, might lead to an enhanced rate of loss of surface NR2B. To test this idea we labeled surface NMDARs with biotin and followed their loss over a 24 h period. We chose to use the C-terminal NR2B antibody (that detects full-length NR2B only) for these experiments, as we found it to be more sensitive than the N-terminal NR2B antibody. As shown in Figure 5A, the rate of loss of surface NR2B was significantly faster in YAC128 compared with WT MSNs. Importantly, treatment with 1 μm CI-1 for 1 h before biotinylation, and then for the subsequent 24 h period, abolished the difference between WT and YAC128 MSNs in rate of loss of surface NR2B (Fig. 5B).
Figure 5.
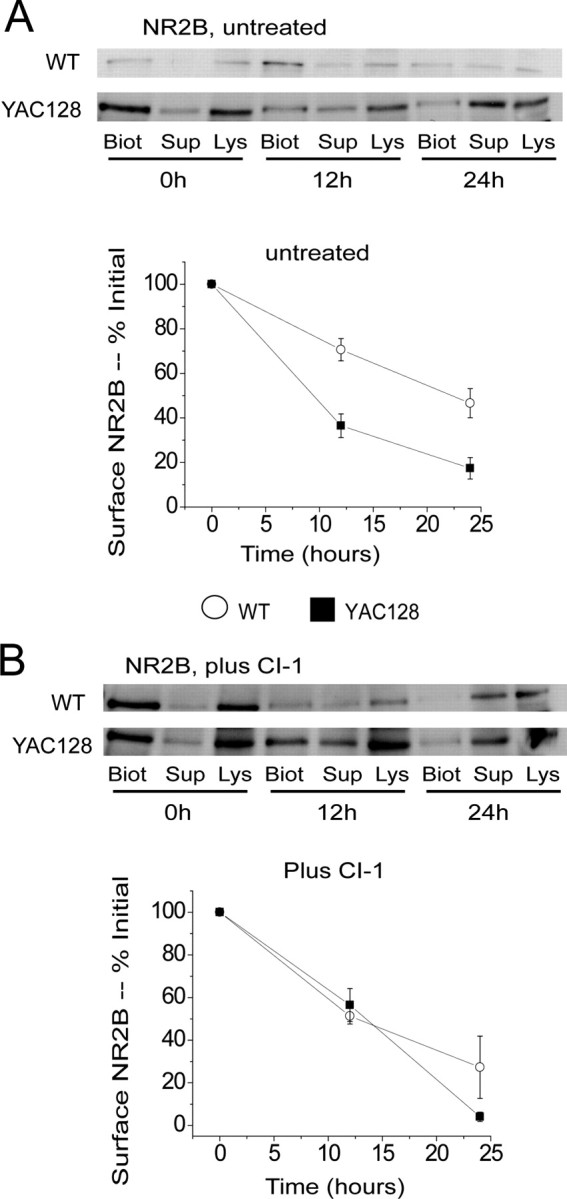
Effect of calpain inhibition on time course of loss of surface NR2B. A, Top panel, Representative Western blots probed with NR2B-specific antibodies after isolation of biotinylated surface proteins (“Biot”) and unbiotinylated internal proteins (“Sup”) as in Figure 4A. Shown is NR2B in WT and YAC128 MSN cultures at 0, 12 and 24 h following treatment of live MSNs with biotinylating reagent. Bottom panel, Plot shows time course of loss of biotinylated, full-length NR2B over 24 h in untreated WT and YAC128 9 DIV MSN cultures. n = 4 independent experiments each from different batches of WT and YAC128 MSN cultures. Difference is significant for time (p < 0.001), genotype (p < 0.001), and interaction between time and genotype (p < 0.01) by two-way ANOVA; **p < 0.01, ***p < 0.001 by Bonferroni posttests. B, Top panel, Representative Western blots probed with NR2B-specific antibodies as in A, except that MSN cultures were pretreated with CI-1 (1 μm) for 1 h before incubation with the biotinylating reagent, and then 1 μm CI-1 was maintained in the medium for the duration of the experiment. Bottom panel, Plot shows time course of loss of biotinylated, full-length NR2B over 24 h in CI-1-treated WT and YAC128 9 DIV MSN cultures. N = 3 and 4 independent experiments from different batches of WT and YAC128 MSN cultures, respectively. No significant difference between genotypes by two-way ANOVA.
NMDA-induced calpain activation correlates with enhanced apoptotic death in cultured YAC72 and YAC128 MSNs
To assess whether calpain activation is involved in NMDA-induced apoptosis of MSNs in the YAC HD mouse model, we again used an antibody specific to the cleaved form of spectrin produced by the enzymatic activity of calpain as a measure of calpain activity, as others have done (Czogalla and Sikorski, 2005). An example of primary MSNs labeled by fluorescence immunocytochemistry for cleaved spectrin is shown (Fig. 6A). We fixed MSNs from the various YAC genotypes at 5 time points from 0 to 6 h after a 10 min treatment with 500 μm NMDA; labeled for anti-calpain-cleaved-spectrin; and assessed the intensity of immunostaining in dendrites using Northern Eclipse (see Materials and Methods). The curves showing calpain activity as a function of time following NMDA exposure were significantly different among genotypes (two-way ANOVA, with genotype and time as variables; p < 0.04, F(3,69) = 2.9 for effect of genotype; p < 0.05 for YAC128 vs YAC18 and for YAC72 vs YAC18 by LSD post hoc test) (Fig. 6B). Notably, calpain activity increased in the first 30 min after NMDA treatment for all genotypes except YAC18, and the magnitude of this change showed a trend toward correlating with polyQ length (Fig. 6B,C). The lack of calpain activation in response to NMDA for YAC18 MSNs is consistent with previous studies showing that striatal neurons from this mouse line are resistant to excitotoxicity both in vitro and in vivo (Leavitt et al., 2006; Shehadeh et al., 2006).
Figure 6.
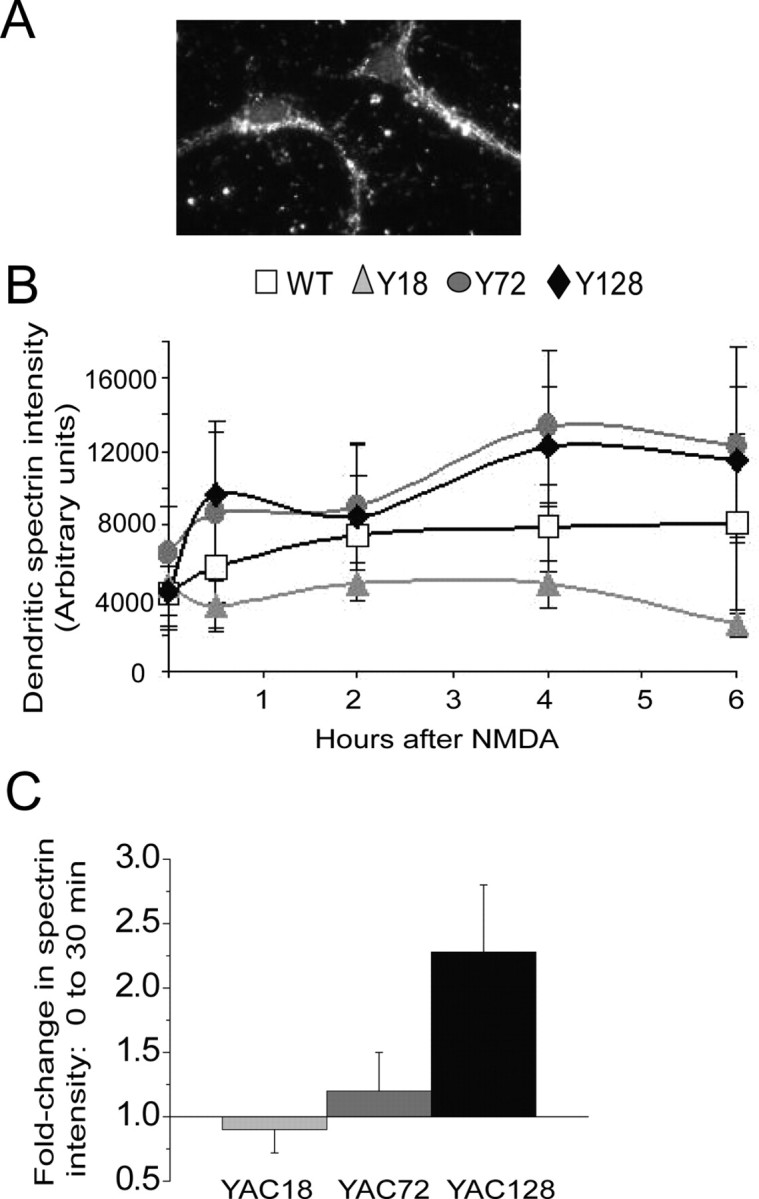
Calpain activity after NMDA insult increases with huntingtin polyQ length. A, Example of NMDA-treated MSNs labeled with anti-calpain-cleaved-spectrin antibody. B, Graph of fluorescence intensity of calpain-cleaved spectrin labeling in MSN dendrites 0–6 h after NMDA insult. n = 4, 3, 5, and 6 independent experiments from different batches of WT, YAC18, YAC72, and YAC128 MSN cultures, respectively. C, Bars represent mean ± SEM fold change in cleaved-spectrin intensity 30 min after NMDA treatment compared with the baseline intensity for experiments shown in B. A trend toward larger magnitude change in cleaved-spectrin intensity was observed with increasing CAG repeat-length.
To assess whether calpain activity is necessary for the increase in NMDA-induced cell death that we have observed in the YAC72 and YAC128 MSNs, we used the in vitro excitotoxicity paradigm as previously described (Shehadeh et al., 2006), in the presence or absence of CI-1. Cells were labeled with TUNEL to detect apoptotic nuclei, and counterstained with Hoechst or propidium iodide to detect all nuclei. We found that treatment with 1 μm CI-1 significantly reduced NMDA-induced apoptosis in both YAC72 and YAC128 MSNs (Fig. 7). Furthermore, the percentage of apoptotic cells seen 24 h after a 10 min NMDA exposure in cultured MSNs treated with CI-1 was similar for all genotypes (Fig. 7B). Therefore calpains contribute to the additional susceptibility to excitotoxicity observed in striatal MSNs from the YAC mice expressing polyQ-expanded htt. Numerous questions, including the temporal and cell specific relationship between caspase and calpain activation, remain to be further explored.
Figure 7.
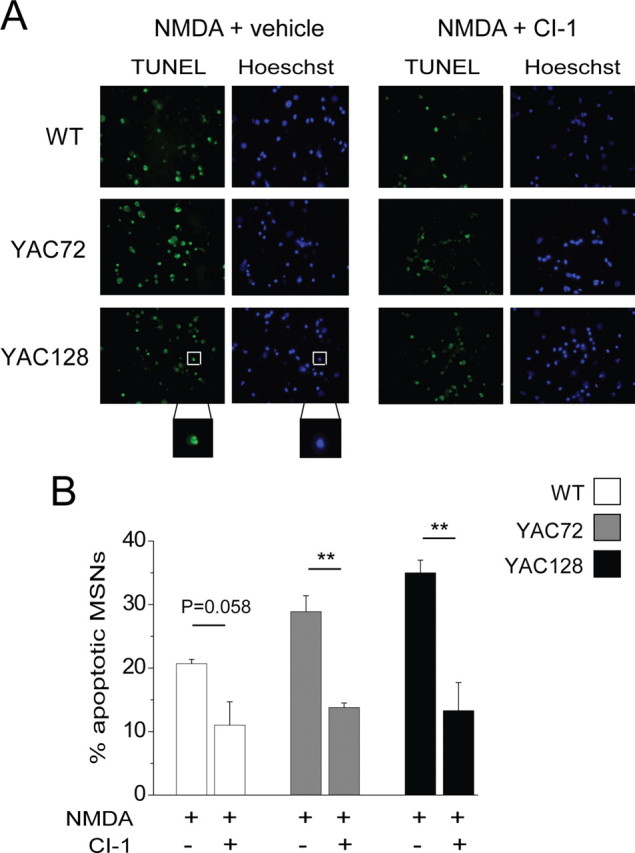
Calpain inhibition reduces NMDAR-mediated apoptosis and abolishes huntingtin polyQ length-dependent differences in excitotoxicity. A, Photomicrographs of MSNs labeled with TUNEL (green, indicating fragmented DNA) and Hoechst (blue, all nuclei). Nuclei were considered apoptotic only if positive for TUNEL and showing condensed, blebbed morphology (see Materials and Methods; typical apoptotic cell is shown in boxed area of bottom left panels). Sister cultures of MSNs were pretreated with vehicle alone (DMSO) or with 1 μm CI-1 for 1 h followed by 10 min exposure to 500 μm NMDA together with vehicle or CI-1, then returned to conditioned medium containing vehicle or CI-1 and fixed 24 h later. Note that MSNs treated with NMDA in conjunction with CI-1 show a reduced proportion of apoptotic nuclei. B, Bar graph quantifying percentage of all nuclei that exhibited apoptotic morphology and TUNEL staining for n = 7, 4 and 4 paired experiments (NMDA plus vehicle vs CI-1) with different batches of cultured MSNs from WT, YAC72, and YAC128 mice, respectively. In each experiment, background apoptosis observed in cultures treated with vehicle or drug treatment alone was subtracted from apoptosis observed in sister cultures treated with NMDA. Statistical significance for CI-1 treatment was determined by paired t test; **p < 0.01.
Discussion
Here we have demonstrated increased tonic calpain activity associated with polyQ-expanded htt in the YAC128 HD mouse model. This result is consistent with previous studies showing elevated calpain activity in human HD autopsy brain tissue and a knock-in HD mouse model (Gafni and Ellerby, 2002; Gafni et al., 2004). The tonically enhanced activity of this protease, which is known to be involved in cell death signaling and also plays an important role in synaptic plasticity (Vanderklish et al., 1996), occurs early in YAC128 striatum and may contribute to the behavioral changes and MSN degeneration observed at ∼2 and 6 months of age, respectively (Slow et al., 2003; Van Raamsdonk et al., 2005).
Although peak NMDAR-mediated current and intracellular Ca2+ responses are similar in YAC128 and WT MSNs, prolonged (5 min) exposure to NMDA triggers a rise in cytosolic Ca2+ that is significantly slower to recover to baseline in YAC128 MSNs (Fernandes et al., 2007), producing a larger cytosolic Ca2+ response integrated over several minutes. Lower levels of excitation that occur repeatedly over time (days to weeks) may also trigger an enhanced, integrated cytosolic Ca2+ load, thereby contributing to the chronically elevated calpain activity in YAC128 MSNs.
Role of calpain in regulating NMDAR surface expression
Calpains have been shown to cleave htt to produce N-terminal fragments, which may accumulate in the nucleus and contribute to cell death and dysfunction as proposed by the toxic fragment hypothesis (Wellington et al., 1997; Gafni and Ellerby, 2002; Gafni et al., 2004). Indeed, mutation of calpain cleavage sites significantly reduces polyQ-expanded htt toxicity in cell lines (Gafni et al., 2004). However, a direct role for calpain-mediated mutant htt cleavage in MSN dysfunction and degeneration has not been established, whereas eliminating caspase-6-mediated cleavage of mutant htt rescues the abnormal behavioral and neurodegeneration observed in YAC128 mice (Graham et al., 2006b).
Our results indicate that chronically augmented calpain activity in the YAC128 striatum results in increased NR2B cleavage and rate of removal/degradation of surface NMDARs. Others have shown that NR2B is cleaved by calpain near the C terminus, removing the region where anchoring protein interactions take place, as well as the Tyr-1472 site which when phosphorylated promotes stability of receptors at the surface, and exposing the 843 Tyr-Trp-Gln-Phe sequence which promotes receptor internalization and subsequent degradation (Kornau et al., 1995; Ehlers et al., 1996; Roche et al., 2001; Scott et al., 2004; Dong et al., 2004). NMDARs with calpain-truncated NR2B initially remain functional and at the plasma membrane, but then are believed to be internalized and degraded rather than recycled (Simpkins et al., 2003; Dong et al., 2004). Indeed, a recent study demonstrates that calpain-cleaved surface NR2B is degraded more quickly than surface full-length NR2B (Yuen et al., 2008), supporting the hypothesis that enhanced calpain-mediated cleavage of NR2B underlies the more rapid degradation rate of surface receptors we found in YAC128 compared with WT cultured MSNs.
We previously reported that NMDAR surface expression and current increases with htt polyQ length in the YAC18, 46 and 72 transgenic mouse models, a result that could be explained by mutant htt-associated accelerated forward trafficking of NMDARs to the neuronal plasma membrane (Fan et al., 2007). The fact that YAC128 MSNs deviate from this trend, exhibiting similar NMDAR current (Fernandes et al., 2007) and surface expression as WT MSNs, may be in part explained by the tonically elevated calpain activity we observed in YAC128 but not YAC72 (or WT) striatal neurons: enhanced calpain-mediated NR2B cleavage would promote more rapid turnover of surface NMDARs that is offset by accelerated NMDAR surface delivery, all resulting from expression of htt with a severely expanded polyQ.
Over time, accelerated calpain cleavage of NR2B might be expected to impact total NR2B expression levels; indeed, we demonstrate 50% reduction of NR2B in the striatum of 2-month-old YAC128 mice that progresses to 70% reduction at 12 months. Although we have not yet determined whether total NR2B expression is reduced in MSNs cultured from P0 YAC128 compared with WT mice, we observed a (nonsignificant) trend toward reduced NMDAR current amplitude (by ∼15%; Fernandes et al., 2007) despite a trend toward increased NR2B surface to internal ratio (by 12–15%) (Fig. 4A, results from immunocytochemical staining) (p = 0.09), suggesting total NR2B is mildly reduced even at this early age.
Downregulation of NMDAR current and surface expression by calpain activation has been demonstrated in other brain regions and disease states. It was first shown that in rat synaptic membrane fractions, treatment with purified calpain decreases full-length NR2B (Bi et al., 1998b). Subsequently, calpain activation by NMDAR-mediated calcium influx was shown to reduce NMDAR current recorded from acutely dissociated or cultured cortical neurons, and in transient forebrain ischemia (Wu et al., 2005; Yuen et al., 2008). Another study reported calpain-dependent reduction in NR2B surface expression 24 h after NMDA insult (Bretin et al., 2006), and there is recent in vivo evidence for calpain proteolysis of NR2B (Araújo et al., 2005). Calpains can also reduce NMDAR function via an indirect mechanism: collapsing response mediator protein (CRMP)-2 is cleaved by calpains activated specifically in response to NMDAR activation, and in turn the cleaved CMRP-2 downregulates NR2B (Bretin et al., 2006).
Enhanced calpain activity and striatal neuronal vulnerability to excitotoxicity
The calpain-mediated downregulation of surface NMDAR expression suggests a protective role of calpains during neuronal overexcitation. On the other hand, calpains cleave a variety of other substrates, including cytoskeletal-associated proteins (Tamada et al., 2005) and calcineurin and its endogenous inhibitor (Kim et al., 2002; Wu et al., 2004), facilitating cell death signaling. Calpains also potentiate the function of proapoptotic Bcl-2-family members, both via transcription-dependent and transcription-independent pathways, and can act in a positive feedback loop with caspase family members (Blomgren et al., 2001). Previous studies indicate that chronically elevated calpain activity contributes to neurodegeneration in Alzheimer's Disease (Siman, 1992; Saito et al., 1993; Nixon et al., 1994). Moreover, calpain cleavage of NR2B to remove the C-terminal region that is required for maintaining NR2B-type NMDARs in the synapse (Prybylowski et al., 2005) is expected to shift the balance of NMDAR signaling away from survival and toward cell death pathways (Wu et al., 2007; Papadia and Hardingham, 2007).
Here, we found that the dominant effect of calpain inhibitor-1 was to protect cultured MSNs against delayed apoptosis mediated by 10 min exposure to NMDA. We observed a rise in calpain activity in MSNs within 30 min of NMDA insult. Early production of the calpain-dependent fragment of spectrin in neurons within 5 min of NMDAR activation has been reported previously (Simpkins et al., 2003). The calpain activity rise we observed showed a trend toward correlation with CAG repeat length in YAC MSNs. Furthermore, treatment with calpain inhibitor-1 (which inhibits calpains 1 and 2, and cathepsins) protected YAC72 and YAC128 MSNs against excessive NMDA-induced in vitro excitotoxicity, reducing apoptosis to levels observed in WT MSNs. Therefore, calpains contribute to the additional susceptibility to excitotoxicity observed in YAC MSNs expressing polyQ-expanded htt, as previously shown for mitochondrial permeability transition (mPT) activation (Zeron et al., 2004). Similar to our results, in a different rodent model of HD, induced by systemic injection of the mitochondrial complex II inhibitor 3-nitropropionic acid, calpain inhibitor treatment also prevented striatal apoptosis (Bizat et al., 2003).
Chronically elevated calpain activity, and the more rapid rise in its NMDA-stimulated activity, in YAC128 MSNs may facilitate cleavage and activation of calcineurin and/or inactivation of its inhibitor (Kim et al., 2002; Wu et al., 2004). Calcineurin dephosphorylates and activates the Bcl-2 family member, Bad (Tan et al., 2006), which is involved in mPT induction and subsequent release of apoptotic factors (Zinkel et al., 2006). Therefore, augmented calpain activity at baseline and with NMDAR stimulation may contribute to increased sensitivity to mPT induction during NMDA application described previously for YAC128 MSNs (Fernandes et al., 2007). Our preliminary results indicate that calcineurin inhibition reduces NMDA-induced apoptosis in YAC128 MSNs with little effect in WT MSNs (CMC and LAR, unpublished data), similar to results reported here for calpain inhibition. Further experiments will determine whether calcineurin acts downstream of calpain activation or if it is involved in a parallel, independent cell death pathway.
Summary
This study underscores the complexity of signaling pathways altered by mutant htt expression, with implications for striatal dysfunction and vulnerability to neuronal loss. The opposing roles of calpain activity in excitotoxicity make it a complex therapeutic target in HD. Recent evidence indicates that NMDARs are protected from calpain cleavage by association with PSD-95 (Wu et al., 2007; Yuen et al., 2008), suggesting that enhanced low-level, chronic calpain activation would preferentially downregulate extrasynaptic NMDARs, which have been implicated in cell death signaling (Hardingham and Bading, 2003). If so, chronic therapy with calpain inhibitors could be detrimental in HD. Moreover, the large number and varied roles of calpain substrates suggest that blanket inhibition of this family of enzymes would be associated with adverse side effects. Therapy targeted to downregulation of enhanced forward trafficking of NMDARs, or else to promoting internalization of extrasynaptic NMDARs in particular without affecting activity of proapoptotic enzymes, may be more fruitful in delaying onset of HD.
Footnotes
This research was funded by operating grants from the Canadian Institutes of Health Research (CIHR) (MOP 12699 to L.A.R. and MOP 84438 to M.R.H.), the HighQ Foundation (L.A.R., M.R.H.), Huntington Disease Society of America (HDSA, to M.R.H.), and a Michael Smith Foundation for Health Research (MSFHR) infrastructure grant. C.M.C. was supported by a Fellowship Award from the HDSA. R.K.G. is supported by an MSFHR award. L.A.R. is a CIHR Investigator and MSFHR Senior Scholar. M.R.H. holds a Canada Research Chair in Human Genetics and is a University Killam Professor. We are grateful to D. Lynch for the anti-spectrin AB38 antibody, A. Milnerwood and C. Gladding for comments and help on the manuscript and experiment, and T. Luo and E. Yu for technical assistance.
References
- Adamec E, Beermann ML, Nixon RA. Calpain I activation in rat hippocampal neurons in culture is NMDA receptor selective and not essential for excitotoxic cell death. Brain Res Mol Brain Res. 1998;54:35–48. doi: 10.1016/s0169-328x(97)00304-5. [DOI] [PubMed] [Google Scholar]
- Albin RL, Young AB, Penney JB, Handelin B, Balfour R, Anderson KD, Markel DS, Tourtellotte WW, Reiner A. Abnormalities of striatal projection neurons and N-methyl-d-aspartate receptors in presymptomatic Huntington's disease. N Engl J Med. 1990;322:1293–1298. doi: 10.1056/NEJM199005033221807. [DOI] [PubMed] [Google Scholar]
- Araújo IM, Xapelli S, Gil JM, Mohapel P, Petersén A, Pinheiro PS, Malva JO, Bahr BA, Brundin P, Carvalho CM. Proteolysis of NR2B by calpain in the hippocampus of epileptic rats. Neuroreport. 2005;16:393–396. doi: 10.1097/00001756-200503150-00017. [DOI] [PubMed] [Google Scholar]
- Beal MF, Kowall NW, Ellison DW, Mazurek MF, Swartz KJ, Martin JB. Replication of the neurochemical characteristics of Huntington's disease by quinolinic acid. Nature. 1986;321:168–171. doi: 10.1038/321168a0. [DOI] [PubMed] [Google Scholar]
- Bi X, Rong Y, Chen J, Dang S, Wang Z, Baudry M. Calpain-mediated regulation of NMDA receptor structure and function. Brain Res. 1998a;790:245–253. doi: 10.1016/s0006-8993(98)00067-5. [DOI] [PubMed] [Google Scholar]
- Bi R, Bi X, Baudry M. Phosphorylation regulates calpain-mediated truncation of glutamate ionotropic receptors. Brain Res. 1998b;797:154–158. doi: 10.1016/s0006-8993(98)00433-8. [DOI] [PubMed] [Google Scholar]
- Bizat N, Hermel JM, Boyer F, Jacquard C, Créminon C, Ouary S, Escartin C, Hantraye P, Kajewski S, Brouillet E. Calpain is a major cell death effector in selective striatal degeneration induced in vivo by 3-nitropropionate: implications for Huntington's disease. J Neurosci. 2003;23:5020–5030. doi: 10.1523/JNEUROSCI.23-12-05020.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomgren K, Zhu C, Wang X, Karlsson JO, Leverin AL, Bahr BA, Mallard C, Hagberg H. Synergistic activation of caspase-3 by m-calpain after neonatal hypoxia-ischemia: a mechanism of “pathological apoptosis”? J Biol Chem. 2001;276:10191–10198. doi: 10.1074/jbc.M007807200. [DOI] [PubMed] [Google Scholar]
- Bretin S, Rogemond V, Marin P, Maus M, Torrens Y, Honnorat J, Glowinski J, Prémont J, Gauchy C. Calpain product of WT-CRMP2 reduces the amount of surface NR2B NMDA receptor subunit. J Neurochem. 2006;98:1252–1265. doi: 10.1111/j.1471-4159.2006.03969.x. [DOI] [PubMed] [Google Scholar]
- Cowan CM, Raymond LA. Selective neuronal degeneration in Huntington's disease. Curr Top Dev Biol. 2006;75:25–71. doi: 10.1016/S0070-2153(06)75002-5. [DOI] [PubMed] [Google Scholar]
- Czogalla A, Sikorski AF. Spectrin and calpain: a ‘target’ and a ‘sniper’ in the pathology of neuronal cells. Cell Mol Life Sci. 2005;62:1913–1924. doi: 10.1007/s00018-005-5097-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- Dong YN, Waxman EA, Lynch DR. Interactions of postsynaptic density-95 and the NMDA receptor 2 subunit control calpain-mediated cleavage of the NMDA receptor. J Neurosci. 2004;24:11035–11045. doi: 10.1523/JNEUROSCI.3722-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD, Mammen AL, Lau LF, Huganir RL. Synaptic targeting of glutamate receptors. Curr Opin Cell Biol. 1996;8:484–489. doi: 10.1016/s0955-0674(96)80024-x. [DOI] [PubMed] [Google Scholar]
- Fan MM, Fernandes HB, Zhang LY, Hayden MR, Raymond LA. Altered NMDA receptor trafficking in a yeast artificial chromosome transgenic mouse model of Huntington's disease. J Neurosci. 2007;27:3768–3779. doi: 10.1523/JNEUROSCI.4356-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes HB, Baimbridge KG, Church J, Hayden MR, Raymond LA. Mitochondrial sensitivity and altered calcium handling underlie enhanced NMDA-induced apoptosis in YAC128 model of Huntington's disease. J Neurosci. 2007;27:13614–13623. doi: 10.1523/JNEUROSCI.3455-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gafni J, Ellerby LM. Calpain activation in Huntington's disease. J Neurosci. 2002;22:4842–4849. doi: 10.1523/JNEUROSCI.22-12-04842.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gafni J, Hermel E, Young JE, Wellington CL, Hayden MR, Ellerby LM. Inhibition of calpain cleavage of huntingtin reduces toxicity: accumulation of calpain/caspase fragments in the nucleus. J Biol Chem. 2004;279:20211–20220. doi: 10.1074/jbc.M401267200. [DOI] [PubMed] [Google Scholar]
- Goldberg YP, Nicholson DW, Rasper DM, Kalchman MA, Koide HB, Graham RK, Bromm M, Kazemi-Esfarjani P, Thornberry NA, Vaillancourt JP, Hayden MR. Cleavage of huntingtin by apopain, a proapoptotic cysteine protease, is modulated by the polyglutamine tract. Nat Genet. 1996;13:442–449. doi: 10.1038/ng0896-442. [DOI] [PubMed] [Google Scholar]
- Graham RK, Slow EJ, Deng Y, Bissada N, Lu G, Pearson J, Shehadeh J, Leavitt BR, Raymond LA, Hayden MR. Levels of mutant huntingtin influence the phenotypic severity of Huntington disease in YAC128 mouse models. Neurobiol Dis. 2006a;21:444–455. doi: 10.1016/j.nbd.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Graham RK, Deng Y, Slow EJ, Haigh B, Bissada N, Lu G, Pearson J, Shehadeh J, Bertram L, Murphy Z, Warby SC, Doty CN, Roy S, Wellington CL, Leavitt BR, Raymond LA, Nicholson DW, Hayden MR. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006b;125:1179–1191. doi: 10.1016/j.cell.2006.04.026. [DOI] [PubMed] [Google Scholar]
- Guttmann RP, Baker DL, Seifert KM, Cohen AS, Coulter DA, Lynch DR. Specific proteolysis of the NR2 subunit at multiple sites by calpain. J Neurochem. 2001;78:1083–1093. doi: 10.1046/j.1471-4159.2001.00493.x. [DOI] [PubMed] [Google Scholar]
- Guttmann RP, Sokol S, Baker DL, Simpkins KL, Dong Y, Lynch DR. Proteolysis of the N-methyl-d-aspartate receptor by calpain in situ. J Pharmacol Exp Ther. 2002;302:1023–1030. doi: 10.1124/jpet.102.036962. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. The Yin and Yang of NMDA receptor signalling. Trends Neurosci. 2003;26:81–89. doi: 10.1016/S0166-2236(02)00040-1. [DOI] [PubMed] [Google Scholar]
- Harwood SM, Yaqoob MM, Allen DA. Caspase and calpain function in cell death: bridging the gap between apoptosis and necrosis. Ann Clin Biochem. 2005;42:415–431. doi: 10.1258/000456305774538238. [DOI] [PubMed] [Google Scholar]
- Hodgson JG, Agopyan N, Gutekunst CA, Leavitt BR, LePiane F, Singaraja R, Smith DJ, Bissada N, McCutcheon K, Nasir J, Jamot L, Li XJ, Stevens ME, Rosemond E, Roder JC, Phillips AG, Rubin EM, Hersch SM, Hayden MR. A YAC mouse model for Huntington's disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 1999;23:181–192. doi: 10.1016/s0896-6273(00)80764-3. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Jo DG, Hong GS, Kim BJ, Lai M, Cho DH, Kim KW, Bandyopadhyay A, Hong YM, Kim DH, Cho C, Liu JO, Snyder SH, Jung YK. Calpain-dependent cleavage of cain/cabin1 activates calcineurin to mediate calcium-triggered cell death. Proc Natl Acad Sci U S A. 2002;99:9870–9875. doi: 10.1073/pnas.152336999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornau HC, Schenker LT, Kennedy MB, Seeburg PH. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science. 1995;269:1737–1740. doi: 10.1126/science.7569905. [DOI] [PubMed] [Google Scholar]
- Leavitt BR, van Raamsdonk JM, Shehadeh J, Fernandes H, Murphy Z, Graham RK, Wellington CL, Raymond LA, Hayden MR. Wild-type huntingtin protects neurons from excitotoxicity. J Neurochem. 2006;96:1121–1129. doi: 10.1111/j.1471-4159.2005.03605.x. [DOI] [PubMed] [Google Scholar]
- Li L, Fan M, Icton CD, Chen N, Leavitt BR, Hayden MR, Murphy TH, Raymond LA. Role of NR2B-type NMDA receptors in selective neurodegeneration in Huntington disease. Neurobiol Aging. 2003;24:1113–1121. doi: 10.1016/j.neurobiolaging.2003.04.003. [DOI] [PubMed] [Google Scholar]
- Magnusson KR. Declines in mRNA expression of different subunits may account for differential effects of aging on agonist and antagonist binding to the NMDA receptor. J Neurosci. 2000;20:1666–1674. doi: 10.1523/JNEUROSCI.20-05-01666.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Saito KI, Grynspan F, Griffin WR, Katayama S, Honda T, Mohan PS, Shea TB, Beermann M. Calcium-activated neutral proteinase (calpain) system in aging and Alzheimer's disease. Ann N Y Acad Sci. 1994;747:77–91. doi: 10.1111/j.1749-6632.1994.tb44402.x. [DOI] [PubMed] [Google Scholar]
- Papadia S, Hardingham GE. The dichotomy of NMDA receptor signaling. Neuroscientist. 2007;13:572–579. doi: 10.1177/10738584070130060401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prybylowski K, Chang K, Sans N, Kan L, Vicini S, Wenthold RJ. The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron. 2005;47:845–857. doi: 10.1016/j.neuron.2005.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts-Lewis JM, Savage MJ, Marcy VR, Pinsker LR, Siman R. Immunolocalization of calpain I-mediated spectrin degradation to vulnerable neurons in the ischemic gerbil brain. J Neurosci. 1994;14:3934–3944. doi: 10.1523/JNEUROSCI.14-06-03934.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche KW, Standley S, McCallum J, Dune Ly C, Ehlers MD, Wenthold RJ. Molecular determinants of NMDA receptor internalization. Nat Neurosci. 2001;4:794–802. doi: 10.1038/90498. [DOI] [PubMed] [Google Scholar]
- Saito K, Elce JS, Hamos JE, Nixon RA. Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: a potential molecular basis for neuronal degeneration. Proc Natl Acad Sci U S A. 1993;90:2628–2632. doi: 10.1073/pnas.90.7.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanberg PR, Calderon SF, Giordano M, Tew JM, Norman AB. The quinolinic acid model of Huntington's disease: locomotor abnormalities. Exp Neurol. 1989;105:45–53. doi: 10.1016/0014-4886(89)90170-2. [DOI] [PubMed] [Google Scholar]
- Scott DB, Michailidis I, Mu Y, Logothetis D, Ehlers MD. Endocytosis and degradative sorting of NMDA receptors by conserved membrane-proximal signals. J Neurosci. 2004;24:7096–7109. doi: 10.1523/JNEUROSCI.0780-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shehadeh J, Fernandes HB, Zeron Mullins MM, Graham RK, Leavitt BR, Hayden MR, Raymond LA. Striatal neuronal apoptosis is preferentially enhanced by NMDA receptor activation in YAC transgenic mouse model of Huntington disease. Neurobiol Dis. 2006;21:392–403. doi: 10.1016/j.nbd.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Siman R. Proteolytic mechanism for the neurodegeneration of Alzheimer's disease. Ann N Y Acad Sci. 1992;674:193–202. doi: 10.1111/j.1749-6632.1992.tb27488.x. [DOI] [PubMed] [Google Scholar]
- Simpkins KL, Guttmann RP, Dong Y, Chen Z, Sokol S, Neumar RW, Lynch DR. Selective activation induced cleavage of the NR2B subunit by calpain. J Neurosci. 2003;23:11322–11331. doi: 10.1523/JNEUROSCI.23-36-11322.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, Oh R, Bissada N, Hossain SM, Yang YZ, Li XJ, Simpson EM, Gutekunst CA, Leavitt BR, Hayden MR. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12:1555–1567. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- Slow EJ, Graham RK, Osmand AP, Devon RS, Lu G, Deng Y, Pearson J, Vaid K, Bissada N, Wetzel R, Leavitt BR, Hayden MR. Absence of behavioral abnormalities and neurodegeneration in vivo despite widespread neuronal huntingtin inclusions. Proc Natl Acad Sci U S A. 2005;102:11402–11407. doi: 10.1073/pnas.0503634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamada Y, Nakajima E, Nakajima T, Shearer TR, Azuma M. Proteolysis of neuronal cytoskeletal proteins by calpain contributes to rat retinal cell death induced by hypoxia. Brain Res. 2005;1050:148–155. doi: 10.1016/j.brainres.2005.05.048. [DOI] [PubMed] [Google Scholar]
- Tan Y, Wu C, De Veyra T, Greer PA. Ubiquitous calpains promote both apoptosis and survival signals in response to different cell death stimuli. J Biol Chem. 2006;281:17689–17698. doi: 10.1074/jbc.M601978200. [DOI] [PubMed] [Google Scholar]
- The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Vanderklish P, Bednarski E, Lynch G. Translational suppression of calpain blocks long- term potentiation. Learn Mem. 1996;3:209–217. doi: 10.1101/lm.3.2-3.209. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Pearson J, Slow EJ, Hossain SM, Leavitt BR, Hayden MR. Cognitive dysfunction precedes neuropathology and motor abnormalities in the YAC128 mouse model of Huntington's disease. J Neurosci. 2005;25:4169–4180. doi: 10.1523/JNEUROSCI.0590-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- Wellington CL, Hayden MR. Of molecular interactions, mice and mechanisms: new insights into Huntington's disease. Curr Opin Neurol. 1997;10:291–298. doi: 10.1097/00019052-199708000-00003. [DOI] [PubMed] [Google Scholar]
- Wenzel A, Fritschy JM, Mohler H, Benke D. NMDA receptor heterogeneity during postnatal development of the rat brain: differential expression of the NR2A, NR2B, and NR2C subunit proteins. J Neurochem. 1997;68:469–478. doi: 10.1046/j.1471-4159.1997.68020469.x. [DOI] [PubMed] [Google Scholar]
- Wu HY, Tomizawa K, Oda Y, Wei FY, Lu YF, Matsushita M, Li ST, Moriwaki A, Matsui H. Critical role of calpain-mediated cleavage of calcineurin in excitotoxic neurodegeneration. J Biol Chem. 2004;279:4929–4940. doi: 10.1074/jbc.M309767200. [DOI] [PubMed] [Google Scholar]
- Wu HY, Yuen EY, Lu YF, Matsushita M, Matsui H, Yan Z, Tomizawa K. Regulation of N-methyl-d-aspartate receptors by calpain in cortical neurons. J Biol Chem. 2005;280:21588–21593. doi: 10.1074/jbc.M501603200. [DOI] [PubMed] [Google Scholar]
- Wu HY, Hsu FC, Gleichman AJ, Baconguis I, Coulter DA, Lynch DR. Fyn-mediated phosphorylation of NR2B Tyr-1336 controls calpain-mediated NR2B cleavage in neurons and heterologous systems. J Biol Chem. 2007;282:20075–20087. doi: 10.1074/jbc.M700624200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young AB, Greenamyre JT, Hollingsworth Z, Albin R, D'Amato C, Shoulson I, Penney JB. NMDA receptor losses in putamen from patients with Huntington's disease. Science. 1988;241:981–983. doi: 10.1126/science.2841762. [DOI] [PubMed] [Google Scholar]
- Yuen EY, Ren Y, Yan Z. Postsynaptic density-95 (PSD-95) and calcineurin control the sensitivity of N-methyl-d-aspartate receptors to calpain cleavage in cortical neurons. Mol Pharmacol. 2008;74:360–370. doi: 10.1124/mol.108.046813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeron MM, Hansson O, Chen N, Wellington CL, Leavitt BR, Brundin P, Hayden MR, Raymond LA. Increased sensitivity to N-methyl-d-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington's disease. Neuron. 2002;33:849–860. doi: 10.1016/s0896-6273(02)00615-3. [DOI] [PubMed] [Google Scholar]
- Zeron MM, Fernandes HB, Krebs C, Shehadeh J, Wellington CL, Leavitt BR, Baimbridge KG, Hayden MR, Raymond LA. Potentiation of NMDA receptor-mediated excitotoxicity linked with intrinsic apoptotic pathway in YAC transgenic mouse model of Huntington's disease. Mol Cell Neurosci. 2004;25:469–479. doi: 10.1016/j.mcn.2003.11.014. [DOI] [PubMed] [Google Scholar]
- Zinkel S, Gross A, Yang E. BCL2 family in DNA damage and cell cycle control. Cell Death Differ. 2006;13:1351–1359. doi: 10.1038/sj.cdd.4401987. [DOI] [PubMed] [Google Scholar]