

Abstract
Amyotrophic lateral sclerosis (ALS), a fatal and progressive neurodegenerative disorder characterized by weakness, muscle atrophy, and spasticity, is the most common adult-onset motor neuron disease. Although the majority of ALS cases are sporadic, ∼5–10% are familial, including those linked to mutations in SOD1 (Cu/Zn superoxide dismutase). Missense mutations in a dynactin gene (DCTN1) encoding the p150Glued subunit of dynactin have been linked to both familial and sporadic ALS. To determine the molecular mechanism whereby mutant dynactin p150Glued causes selective degeneration of motor neurons, we generated and characterized mice expressing either wild-type or mutant human dynactin p150Glued. Neuronal expression of mutant, but not wild type, dynactin p150Glued causes motor neuron disease in these animals that are characterized by defects in vesicular transport in cell bodies of motor neurons, axonal swelling and axo-terminal degeneration. Importantly, we provide evidence that autophagic cell death is implicated in the pathogenesis of mutant p150Glued mice. This novel mouse model will be instrumental for not only clarifying disease mechanisms in ALS, but also for testing therapeutic strategies to ameliorate this devastating disease.
Keywords: dynactin, p150Glued, ALS, motor neuron disease model, vesicular trafficking, autophagy
Introduction
A family with a slowly progressive autosomal dominant lower motor neuron disease has been linked to a mutation in the p150Glued subunit (G59S) of the dynactin complex (Puls et al., 2003). This disease begins in early adult life with vocal cord paralysis (associated with breathing difficulties), facial weakness, and atrophy of muscles in the hands (Puls et al., 2003, 2005). Subsequently, weakness and atrophy appear in the distal lower extremities. There is evidence of degeneration of motor axons, and reduced numbers of motor neurons in the spinal cord and hypoglossal nucleus (Puls et al., 2005). Significantly, inclusions containing components of dynactin and dynein are evident in motor neurons (Puls et al., 2005). Although no sensory deficits are reported, examinations of skin biopsies disclose abnormalities of axons in the skin. Since this first report, additional mutations in the dynactin gene (DCTN1) have been associated with motor neuron disease (Munch et al., 2004).
Dynactin p150Glued is a critical component of a multiprotein complex that differentially copurifies with dynein (Gill et al., 1991; Schroer and Sheetz, 1991; Schroer, 2004), the molecular motor required for fast retrograde transport of vesicles, organelles, RNAs and proteins along microtubules (Schroer, 2004; Hirokawa and Takemura, 2005; Price et al., 2005). Although dynactin has been postulated to enhance the processivity and efficiency of the dynein motor (LaMonte et al., 2002; Schroer, 2004), previous studies demonstrate in vitro that the dynein-dynactin complexes exhibit bidirectional processive motility (Ross et al., 2006) and in vivo that dynactin is required for coordinated anterograde and retrograde organelle movement (Haghnia et al., 2007). The disruption of the dynactin complex by overexpression of dynamitin (LaMonte et al., 2002) results in motor neuron disease in mice, but the exact pathogenic mechanism remains unknown. Bidirectional transport of vesicles between the endoplasmic reticulum (ER) and Golgi apparatus have also been shown to involve dynein/dynactin mediated transport (Lee et al., 2004). Disruption of dynein function also impairs clearance of aggregate prone proteins by autophagy (Ravikumar et al., 2005), a lysosomal pathway for degrading damaged organelles and aggregated proteins within the cell. Autophagy has been implicated in a number of neurodegenerative disorders, but considerable debate exists regarding the role of autophagy in pathogenesis of specific diseases (Nixon, 2006). The demonstration that genetic deletions of autophagy genes in neurons are sufficient to cause neurodegeneration supports the view that disruption of basal autophagy participates in pathogenesis of some of these illnesses (Hara et al., 2006; Komatsu et al., 2006).
Therefore, given the numerous roles that dynactin plays in intracellular and axonal transport, it is conceivable that alteration in dynactin function could impact on several critical cellular processes within various compartments of the motor neuron. Here, we document that a novel mouse model expressing human mutant p150Glued dynactin exhibits clinical and pathological hallmarks of motor neuron disease. Importantly, we show that motor neuron degeneration in these mutant dynactin mice is associated with abnormalities in intracellular vesicular trafficking, axonal swelling and axo-terminal degeneration. Moreover, autophagic cell death is implicated in the pathogenesis of disease in mutant p150Glued mice.
Materials and Methods
Generation of transgenic mice.
We first engineered the G59S amyotrophic lateral sclerosis (ALS)-linked mutation (Puls et al., 2003) into the human dynactin p150 Glued cDNA by PCR/oligonucleotide primer-directed mutagenesis strategy. In addition, the full-length human cDNA encoding wild-type and mutant dynactin p150Glued were modified to include a C-terminal epitope tag (FLAG) to facilitate identification of the transgenic product. Because previous studies show that the Thy1.2 cassette works well for generation of mice with neuronal expression and was the strategy used for the overexpression of dynactin p50 (LaMonte et al., 2002), the resulting constructs were subcloned into the XhoI site of the mouse Thy1.2 expression cassette to generate expression constructs, one encoding wild-type dynactin p150glued (Thy1.2p150WT), and the other G59S dynactin p150glued (Thy1.2p150G59S). These vectors were initially tested in cultured cells by transient transfection to verify their expression (data not shown). Subsequently, the purified inserts from these vectors were submitted to the Transgenic Facility at Johns Hopkins University School of Medicine for injection into C57BL/6;SJL hybrid mouse embryos. Positive founders were identified by Southern blot analysis and two lines of mutant and three lines of wild-type dynactin p150 Glued were established. F1 offspring, maintained in the hybrid C57BL/6;SJL background were genotyped by Southern blot analysis and subsequent generations were identified using PCR analysis of the tail biopsies using the primer sets: CAA ATG GGT AGG CGT GAT TCT and GTC CCC GCA GTT TGC TAG TC to identify the human p150Glued, and ATG GGT GGG CGT GAT TCT G and ACT GGC GTA CAA AGA TGC CG to identify the endogenous mouse p150Glued. The animal use protocol was approved by the Animal Care and Use Committee of the Johns Hopkins Medical Institutions.
SDS-PAGE and immunoblotting.
Mouse tissues were homogenized in 1:10 PBS containing 1% SDS and protease inhibitor mixture (Roche, Indianapolis, IN). Protein extracts were sonicated before protein assay and protein blot analysis. 20 μg of total protein extracts were loaded onto a 6 or 16% Tris-glycine gel (Invitrogen, San Diego, CA) and transferred to polyvinylidene difluoride membrane (Invitrogen). Immunoblotting was performed using primary antibodies to p150Glued (BD Biosciences, San Jose, CA), FLAG (M2; Sigma, St. Louis, MO), dynamitin (p50; Millipore, Billerica, MA), or LC3 (MBL International, Woburn, MA).
Determination of disease onset and survival.
Mice were regularly monitored to assess the onset and progression of symptoms associated with transgene expression. Observations were made regarding presence of tremor, abnormality of gait or balance, ability to groom as well as weight. Fore and hindlimb grip strength were measured for a cohort of 10 each of transgenic and nontransgenic mice. Measurements were taken using a grip strength meter (San Diego Instruments, San Diego, CA) every 2 weeks from 10 weeks of age. Initial decline in grip strength was considered the onset of motor neuron disease and determined on a Kaplan Meyer graph. Mice were considered at end stage of disease when they were no longer to right themselves within 10 s when placed on their back and lifespan of mice determined on a Kaplan Meyer survival plot.
Immunocytochemistry.
Mice anesthetized with an intraperitoneal injection of 15% chloral hydrate were killed by transcardial perfusion with PBS, pH 7.4, followed by 4% paraformaldehyde in PBS, pH 7.4. Brain, spinal cord, sciatic nerve, and other organs were removed and postfixed in the same fixative. Coronal sections of the brain and cross sections of the spinal cord were embedded in paraffin, sectioned (10 μm), and stained with hematoxylin and eosin (H&E), cresyl violet, and Bielschowski silver. Deparaffinized sections (10 μm) were processed for immunocytochemistry using antibodies recognizing nonphosphorylated neurofilaments (SMI-32; Sternberger Monoclonals, Lutherville, MD), ubiquitin (Dako, High Wycombe, UK), and FLAG (M2 mouse monoclonal; Sigma). The immune reaction was visualized with diaminobenzidine. For neuromuscular junction analysis, sections (50 μm for legs/feet and 100 μm for spines) were incubated overnight at 4°C with primary polyclonal antibody (PGP9.5; Millipore) followed by secondary antibody (goat anti-rabbit biotinylated secondary antibody; Jackson ImmunoResearch, West Grove, PA) and visualized using Vectastain Elite ABC kit (standard; Vector Laboratories, Burlingame, CA).
Electromyography and muscle histology.
Animals were anesthetized by inhalation anesthesia using isoflourane. Needle electromyographic examination for denervation potentials in the form of spontaneous activity (fibrillation potentials) was done in the gastrocnemius muscle.
Fresh skeletal muscle biopsies were obtained before fixation and frozen by immersion in isopentane cooled in liquid nitrogen. Sections (10 μm) were cut and processed for nicotinamide adenine dinucleotide (NADH), ATPase (pH 4.4 and 9.4), Gomori trichrome, and acetlycholinesterase-silver.
Electron microscopy.
Anesthetized mice were perfused with 4% paraformaldehyde in 0.1 m PBS, pH 7.4. Tissues from brain, spinal cord, ventral and dorsal root and sciatic nerve were postfixed with 4% paraformaldehyde with 2% glutaraldehyde in 0.1 m PBS (pH 7.4). Subsequently, tissues were washed again in PBS, dehydrated and embedded in Epon. Thick (1 μm) and thin (100 nm) sections were obtained and stained respectively with toluidine blue and lead citrate/uranyl acetate.
Quantification of motor neurons, ventral roots, and membrane vesicles.
The number of large α-motor neurons (>25 μm) and small γ-motor neurons in the ventral horn of the lumbar spinal cord was quantified using Stereo Investigator software (MicroBrightField, Colchester, VT). Serial sections stained with cresyl violet were taken through the lumbar region and both sides of the cord were counted (nontransgenic, n = 7 animals, 33 sections; mutant dynactin p150Glued, n = 4 animals, 20 sections; and wild-type p150Glued, n = 3 animals, 11 sections). The area was outlined and markers placed to indicate small or large motor neurons, the quick measure tool was used to measure the size of a given motor neuron. One-way ANOVA with Tukey's post hoc test was performed using GraphPad (San Diego, CA) Prism.
Quantification of axonal caliber in the ventral roots was performed using NIH ImageJ software (W. S. Rasband, National Institutes of Health, Bethesda, MD; http://rsb.info.nih.gov/ij/). Each axon was highlighted and the Feret diameter calculated so that a frequency distribution could be plotted (nontransgenic, n = 4 animals, 3532 axons; mutant dynactin p150Glued, n = 5 animals, 2928 axons).
The percentage of autophagosomes in the motor neurons of mutant dynactin p150 Glued mice was counted using electron microscopy images. Images of large motor neurons in the ventral horn of the spinal cord were acquired using a Hitachi (Tokyo, Japan) 7600 transmission electron microscope. Images at 50,000× were obtained from 5-month-old mutant dynactin p150 Glued and 12-month-old wild-type dynactin p150 Glued motor neurons (n = 2 animals for each genotype). Membrane-enclosed structures with empty lumens were scored as dilated ER. Membrane-enclosed structures containing structures were scored as autophagosomes.
Immunofluorescence.
Anesthetized mice were perfused with 4% paraformaldehyde in 0.1 m PBS, pH 7.4. Spinal cords were postfixed for 24 h in 4% paraformaldehyde 0.1 m PBS, pH 7.4, and then for 48 h in 30% sucrose before sectioning on a sliding microtome. Forty micrometer sections were processed for immunofluorescence using antibodies recognizing p115 (BD Biosciences) and GRP94 (Abcam, Cambridge, MA). The secondary antibody used was Alexa Fluor 594 (Invitrogen).
Results
Generation of dynactin p150 Glued transgenic mice
To determine the mechanisms whereby mutant dynactin p150Glued causes selective degeneration of motor neurons, we generated several lines of mice expressing either wild-type or mutant dynactin p150 Glued driven by the Thy1.2 promoter. To distinguish the human from endogenous mouse dynactin p150Glued, the full-length human dynactin p150 Glued cDNAs encoding wild-type or mutant dynactin p150Glued were epitope tagged (via FLAG) at the C-termini (Fig. 1 a). The expression of human wild-type and mutant transgenes was confirmed by examining the accumulation of dynactin p150Glued from spinal cords of nontransgenic, wild-type and mutant dynactin p150 Glued mice. Immunoblot analyses using antisera recognizing the FLAG epitope reveal that the human wild-type and mutant dynactin p150Glued proteins accumulate in spinal cords of wild-type and mutant dynactin p150 Glued mice, respectively (Fig. 1 b). Importantly, antisera that recognize both the human and endogenous mouse dynactin p150Glued protein demonstrated that the level of total p150 protein in spinal cords of nontransgenic mice is similar to that of wild-type or mutant dynactin p150 Glued mice (Fig. 1 b), indicating that the mutant protein is not highly expressed in these transgenic mice.
Figure 1.

Motor neuron disease in mice expressing mutant but not wild-type p150Glued. A, Schematic drawing of the construct used to generate transgenic mice. Human wild-type or mutant p150Glued cDNA with a FLAG tag (indicated by a striped box) were cloned into the Thy1.2 expression cassette at the XhoI site. B, Accumulation of mutant G59S dynactin in mice. Total protein (20 μg) extracted from nontransgenic, wild-type, and mutant dynactin mice were electrophoresed on 6% SDS-PAGE gels and immunoblotted using a monoclonal antibody recognizing the p150Glued (top) or the FLAG (middle) epitope. Identical blot was reprobed with antisera against actin (bottom). C, Kaplan-Meier plot of the onset of tremor for two mutant lines, M1 (gray line) and M2 (black line). The average onset of tremor for M1 is 4.8 ± 1.2 months and for M2 is 6.2 ± 1.3months. D, Onset of the reduction in grip strength for line M1 is plotted on a Kaplan-Meier graph. The black and gray dotted lines denote forelimb and hindlimb grip strength, respectively. E, Kaplan-Meier plot for survival of two mutant lines; line M1 (gray) has a shorter lifespan than line M2 (black).
Mutant dynactin p150 Glued mice display a motor neuron disease phenotype
We have observed clinical and pathological evidence of motor neuron disease in two independent lines of mutant, but not in wild-type dynactin p150 Glued mice. The mutant mice initially show spontaneous tremors that progress with age; gait abnormalities follow. Although the onset of clinical signs occurs at ∼5–6 months of age in both mutant lines, the progression of disease differs significantly between the two lines (Fig. 1 c–e). In mutant dynactin p150 Glued mice, tremor (Fig. 1 c) precedes decline in grip strength, which is observed in both fore and hind limbs (Fig. 1 d). Subsequently, affected mice develop weakness accompanied by muscle wasting in hind limbs, when suspended by the tail these mice fail to extend their hind limbs (supplemental Fig. 1a,b, available at www.jneurosci.org as supplemental material). In the weeks leading up to end-stage of disease, mutant dynactin p150 Glued mice fail to groom and lose weight. Eventually, mice become paralyzed (supplemental Fig. 1c,d, available at www.jneurosci.org as supplemental material). When mice are unable to right themselves after a 10 s interval, they are considered to be at end stage of the disease (Fig. 1 e) and are killed for pathological examination.
Pathological abnormalities in the perikarya and axons of mutant dynactin p150 Glued mice
At end stage of the disease, both mutant dynactin p150 Glued lines exhibit striking cellular abnormalities in motor neurons in lumbar, thoracic and cervical spinal cord and motor neurons in the brainstem (Fig. 2 b,e). Subsets of the motor neurons display chromatolysis, with swollen cell bodies and eccentric nuclei. The cell bodies of motor neurons stain with silver (Fig. 2 h,k), contain ubiquitin positive intracellular inclusions (Fig. 2 n), and also stain positively with anti-FLAG antibody, indicating the presence of the mutant dynactin p150Glued protein (Fig. 2 p). Other members of the dynactin complex including p50 and Arp1 as well as dynein were also found within these inclusions (supplemental Fig. 2b, available at www.jneurosci.org as supplemental material). GFAP staining reveals astrocytic gliosis in the spinal cord (supplemental Fig. 2d, available at www.jneurosci.org as supplemental material). Silver stains also disclose swollen axons, particularly in the ventral root exit zone (Fig. 2 h). In the ventral root exit zone, the swollen axons are evident (Fig. 2 r) and immunocytochemical analysis of neurofilaments revealed that the swollen axons are densely packed with nonphosphorylated neurofilaments (Fig. 2 t). Electron microscopic (EM) analysis confirms this observation and shows massive accumulation of neurofilaments in proximal axons, a pathological hallmark of sporadic ALS (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). At the end stage of disease, the number of large motor neurons in the lumbar region is reduced by 44.3 ± 5% (p < 0.0001) in the mutant dynactin p150 Glued mice compared with that of the wild-type dynactin p150 Glued mice, there was no significant difference between the nontransgenic and wild-type p150Glued mice (Fig. 3 a–e). Some myelinated axons in ventral roots and peripheral nerves of mutant dynactin p150 Glued mice exhibit Wallerian degeneration (Fig. 3 g,i). Moreover, analysis of the ventral roots from the lumbar region of the spinal cord reveals a selective loss of large caliber axons in mutant dynactin p150 Glued mice (Fig. 3 j). Pathology was not present in lines of wild-type p150 Glued mice (Fig. 2 c,f,i,l). No abnormalities were observed in upper motor neurons of the mutant dynactin p150 Glued mice (data not shown).
Figure 2.
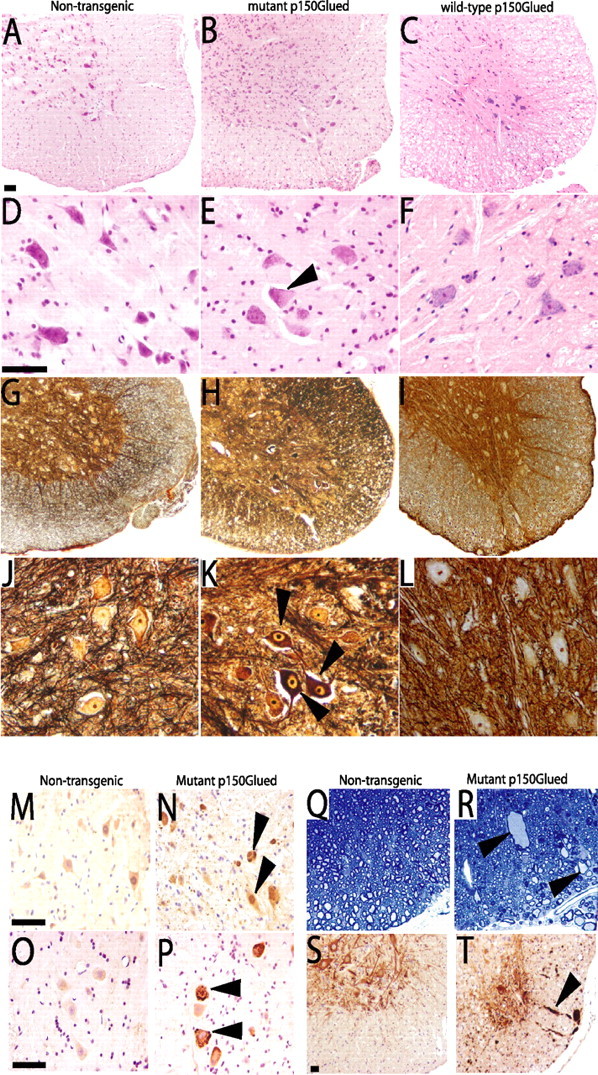
Neuropathological abnormalities in spinal cord of mutant dynactin p150 Glued mice. A–F, Hematoxylin and eosin staining reveals abnormalities in the motor neuron cell bodies of mutant p150Glued mice (B, E). Note subset of swollen motor neuron cell bodies with nuclei pushed to the side (indicated by arrowhead in E). G–L, Silver staining reveals dark staining of motor neuron cell bodies in the spinal cord of mutant mice (H, K, arrowheads), but not of nontransgenic (G, J) or of wild-type p150Glued mice (I, L). Immunohistochemical analysis with ubiquitin (M, N) and FLAG (O, P) shows that the motor neuron cell bodies of mutant mice contain ubiquitin-positive inclusions (N, arrowheads) that also stain positive with FLAG (P, arrowheads), indicating the presence of the mutant dynactin protein. R, Swollen axons are prominent (indicated by arrowheads) in the ventral root exit zone of mutant mice. S, T, Staining with SMI-32 indicates accumulation of nonphosphorylated neurofilaments in the swollen axons (T, arrowhead) in the ventral root exit zone of mutant mice. All figures are representative sections taken from end-stage animals. Scale bars, 50 μm.
Figure 3.
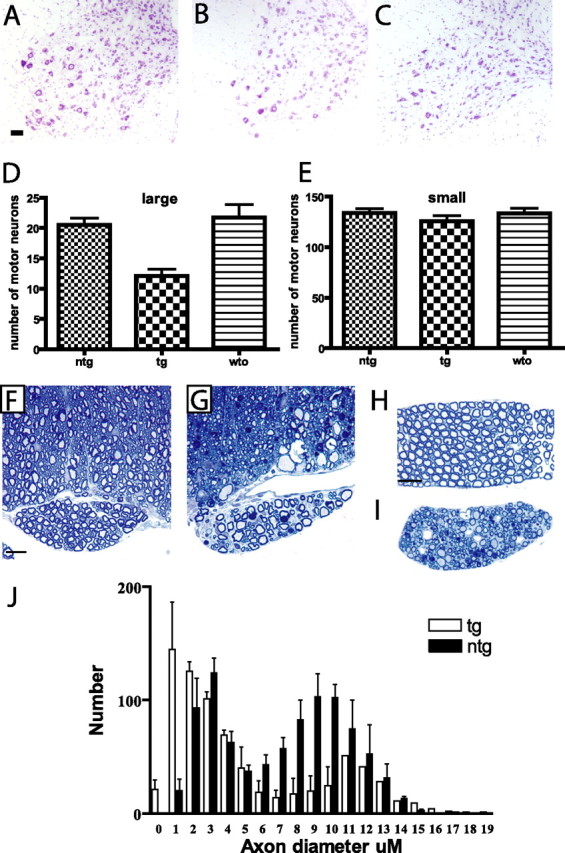
Selective loss of large motor neurons and degeneration of ventral root axons. A–C, Cresyl violet-stained sections from lumbar region of spinal cord from nontransgenic (A), mutant p150Glued mice (B), and wild-type p150Glued mice (C). D, There is a 43% reduction in the number of large motor neurons in these mutant p150Glued mice compared with wild-type p150Glued mice (p < 0.001). E, There is no difference in the number of small motor neurons in the mutant p150Glued mice. F, Ventral root exit zone and root from nontransgenic mouse. G, Ventral root exit zone and root from mutant p150Glued mouse at 5 months of age. Significant degeneration is present. H, Ventral root from nontransgenic mouse. I, Ventral root from mutant p150Glued mouse. A significant loss of large motor axons is observed as well as degeneration including the presence of myelin ovoids and Wallerian degeneration. J, Frequency distribution of axon size in ventral roots from the lumbar region of nontransgenic and mutant p150Glued mice. There is a selective loss of larger motor neurons in mutant p150Glued mice at end stage of disease. Scale bars: (in A) A–C, 50 μm; (in F) F, G, 20 μm; (in H) H, I, 50 μm. Error bars indicate SEM.
Mutant dynactin p150Glued mice exhibit denervation muscle atrophy
Electromyographic analyses reveal spontaneous fibrillations characteristic of denervation (Fig. 4 a) and there is pathological evidence of denervation of muscle and extensive muscle atrophy. At end stage of disease, histochemical stains (e.g., NADH dehydrogenase, ATPase, and Gomori Trichrome) to distinguish between fast (type 1) and slow (type 2), muscle fiber types reveal that the normal random distribution of fiber types is replaced in mutant mice by fiber type grouping (Fig. 4 c,d). Because dynactin is critical for synapse stability at the neuromuscular junction (Eaton et al., 2002) we assessed the integrity of neuromuscular junctions in our mutant mice. Interestingly, when compared with control littermates, mutant p150 Glued mice at symptomatic stage show striking intrajunctional and extrajunctional sprouting (Fig. 4 f). Moreover, this abnormality is accompanied by axoterminal swelling (Fig. 4 g,h) and degeneration (Fig. 4 i) in these mutant animals. Additionally, at the end stage of disease, there is evidence of sensory neuropathy, with mutant p150 Glued mice exhibiting reduced intraepidermal nerve fiber (IENF) densities in the skin (nontransgenic IENF, 19.3/mm; mutant IENF, 3.7/mm) as well as degeneration within the dorsal roots of mutant p150 Glued mice (data not shown). Together, these findings demonstrate that neuronal expression of mutant p150Glued protein in mice is sufficient to cause motor neuron disease, exhibiting pathological features that are observed in sporadic ALS.
Figure 4.
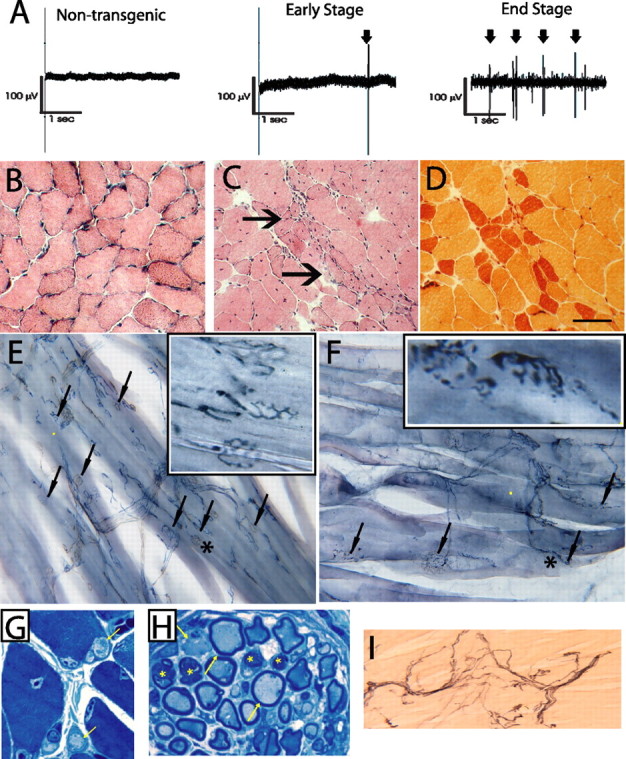
Denervation muscle atrophy in mutant dynactin p150 Glued mice. A, EMG analysis of gastrocnemius muscles from 8-month-old nontransgenic mouse and mutant p150Glued mice at 6 or 8 months of age. Note that rare (arrow) and abundant fibrillation potentials (arrows) are observed during early symptomatic phase (6 months) and at end stage (8 months), respectively. B–D, Examination of gastrocnemius using H&E and esterase histochemistry of muscle fibers from a nontransgenic (B) and end-stage mutant mouse (C, D) reveals group atrophy (arrows). Scale bar, 50 μm. E, F, Neuromuscular junctions were examined using synaptophysin. Typical junctions are highlighted with arrows. Note abnormalities in the neuromuscular junctions of mutant mice (F) including nodules, preterminal accumulations, and intrajunctional and extrajunctional sprouting. Boxes in top right are enlargements of area denoted by an asterisk. G, H, Toluidine blue-stained sections (1 μm) showing axonal swelling in intramuscular nerve of mutant p150Glued mice. Note abnormal intramuscular nerve fibers and axonal swellings as indicated by arrows, respectively, in G and H; asterisks in H indicate degenerating axons. I, p75 staining in the gastrocnemius muscle from mutant p150Glued mice indicates axoterminal degeneration.
Pathological abnormalities occur before the onset of clinical signs of disease
To determine the mechanisms whereby mutant p150Glued protein is toxic, we initially examined the formation of cytoplasmic inclusions in motor neurons at multiple time points during the disease process (Fig. 5). Immunocytochemical analysis using a FLAG antibody that recognizes the wild-type or mutant human p150Glued protein reveals that inclusions appear as early as 2 months of age in motor neurons of mutant mice (Fig. 5 d), a time point before any observable clinical signs. Gradually, these inclusions become larger and by 5 months occupy a significant portion of the cytoplasm (Fig. 5 c,f). In contrast, mice expressing wild-type dynactin p150Glued show a cytoplasmic staining pattern that is consistent with the normal distribution of dynactin throughout the cytoplasm of motor neurons (Fig. 5 b); importantly, we failed to observe large inclusions in these mice, even at 12 months of age (data not shown). The staining patterns are specific to the transgenic products as no FLAG-positive signal is detected in motor neurons of nontransgenic mice (Fig. 5 a).
Figure 5.

Massive accumulation of intracellular inclusions in mutant dynactin p150 Glued mice. A–C, FLAG antibody stained sections from 5-month-old nontransgenic mice (A), wild-type p150Glued mice (B), and mutant p150Glued mice (C). In wild-type mice there is a cytoplasmic staining pattern that is consistent with the normal distribution of dynactin throughout the cytoplasm; however, in mutant p150Glued mice, inclusions positive for the mutant FLAG-tagged protein are evident (C). D, E, Time course, showing sections from 2, 3 and 5 month mutant p150Glued mice. Staining increases from a diffuse pattern at 2 months (D) to numerous dense large inclusions present at 5 months (F). Arrows point to cytoplasmic inclusions in the mutant neurons. Arrowheads in B and C indicate the cells in the insets. Scale bars: (in A) A–C, 20 μm; (in D) D–F, 10 μm.
Alterations in the subcellular architecture of motor neurons of mutant dynactin p150Glued mice
To determine whether these cytoplasmic inclusions comprised of protein aggregates, morphological analysis of motor neurons expressing mutant dynactin p150 Glued were performed. EM analysis of the ventral spinal cord at the lumbar level reveals that the motor neurons of mutant dynactin p150 Glued mice at 3 months of age contain numerous vesicular structures (Fig. 6 c,d). Moreover, by 5 months of age, a massive accumulation of these vesicular structures is evident (Fig. 6 e,f). Some of these vesicles appear to contain subcellular structures (indicated by arrows), indicative that some of these structures are autophagosomes, although the majority are ribosome studded and do not contain any structure (indicated by arrowheads). This accumulation is not observed in the wild-type dynactin p150 Glued mice even at 12 months of age (Fig. 6 g). Quantification of the number of vesicles present in the motor neurons of mutant dynactin p150 Glued mice shows an increased number of dilated endoplasmic reticulum structures and an increase in autophagosomes (Fig. 6 h). Because disruption of the functions of the dynein/dynactin complex impacts basal autophagy (Ravikumar et al., 2005), we asked whether these intracellular inclusions are related to accumulation of autophagosomes. First, we examined the levels of two microtubule associated protein 1 light chain 3 (LC3) species (i.e., cytosolic LC3-I and the autophagosome-associated LC3-II) in the spinal cord of mutant p150 Glued mice. The ratio of LC3-II to LC3-I in mutant p150 Glued mice is significantly increased compared with that of nontransgenic controls (Fig. 6 i), confirming the morphological evidence of an increased accumulation of autophagosomes in motor neurons of mutant p150 Glued mice. This observation is consistent with the idea that mutant dynactin impacts on dynein-dynactin function to alter the regulation of autophagy in motor neurons of mutant dynactin p150 Glued mice. Second, because dynactin plays a role in bidirectional vesicular trafficking between the ER and Golgi, we assessed whether the vesicular compartments that accumulate in the cell body of motor neurons are components of the ER or Golgi. Immunocytochemical analysis using ER (GRP94) and Golgi (P115) markers (Fig. 7 a–f) show that numerous mutant p150Glued-containing inclusions are positively stained, findings that are consistent with the view that vesicular transport are altered leading to abnormal accumulation of membrane compartments within cell bodies of motor neurons of mutant dynactin p150 Glued mice (Fig. 7 b,e). Together with the observation that the G59S mutation destabilizes p150glued and disrupts dynactin function (Lai et al., 2007), our findings indicate that the normal functions of dynactin p150Glued may be compromised by the mutant p150Glued protein in a dominant negative manner.
Figure 6.
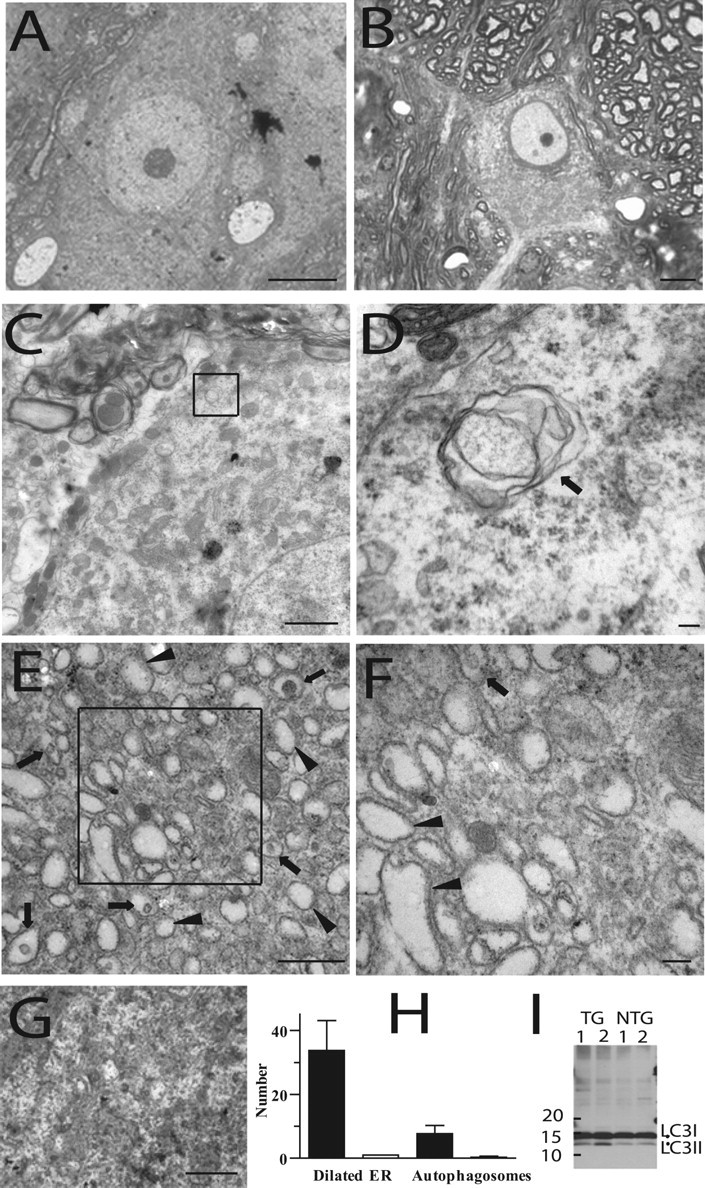
Accumulation of intracellular vesicles in the mutant dynactin p150 Glued mice. A–F, Motor neurons from wild-type p150 Glued (A, G) and mutant p150 Glued mice (B–F). Toluidine blue-stained section (1 μm) shows a motor neuron with eccentric nucleus (B). C–F, EM showed a number of vesicular structures, some of which were distinctive double-membraned autophagosomes in the mutant p150 Glued mice at 3 months of age (C, D). At 5 months of age, there is a massive accumulation of vesicles observed within the cell body of the motor neurons (E, F). Arrows indicate organelle containing vesicles; arrowheads indicate vesicles not containing organelles. Boxes indicate area enlarged in F. No accumulation of vesicles is observed in wild-type p150 Glued mice (12 months of age) (G). H, Quantification of the number of vesicles present in the motor neurons of mutant p150 Glued mice shows an increased number of dilated endoplasmic reticulum structures (mutant, left bar) and an increase in autophagosomes (mutant, left bar) compared with those of wild-type p150 Glued mice. I, Increased level of LC3-II species relative to LC3-I in mutant p150 Glued mice compared with nontransgenic controls. Scale bars: A, B, 10 μm; C, 2 μm; D, 100 nm, E, G, 500 nm; F, 100 nm.
Figure 7.

Some accumulating vesicles in the mutant dynactin p150 Glued mice are components of the ER and Golgi. A–C, Ventral spinal cord of nontransgenic (A) and transgenic (B) mice labeled with GRP94. There is an accumulation of GRP94 labeled structures within a number of the motor neurons. C, Shows FLAG staining in the motor neurons of mutant p150Glued mice. D–F, Ventral spinal cord of nontransgenic (D) and transgenic (E) mice labeled with p115. There is an accumulation of p115 labeled structures in the majority of motor neuron cell bodies in the transgenic mice. F, Shows FLAG staining in the motor neurons of mutant p150Glued mice.
Discussion
Cell culture studies indicated that disease in mutant dynactin can be attributed to a loss and gain of function mechanism (Levy et al., 2006). Although it is plausible that haploinsufficiency can explain disease arising from mutation in dynactin, the finding that heterozygous deletion of Dctn1 in mice (Dctn1 +/− mice) does not result in motor neuron disease (Lai et al., 2007) would not be consistent with this view. Given that homozygous knock-in of a missense mutation in p150Glued (Dctn1 m/m) in mice is embryonically lethal as observed in Dctn1 −/− (Lai et al., 2007), our results indicating that mutant dynactin p150Glued impacts on the normal function of dynactin would be consistent with the idea that disease in dynactin-linked ALS arises through a dominant-negative mechanism. Altering the dose of wild-type p150Glued alleles in mutant dynactin p150 Glued mice through a cross-breeding strategy with either wild-type p150 Glued transgenic mice or p150 Glued knock-out mice should further clarify this issue in the future.
In contrast to the slow, progressive disease observed in distal spinal and bulbar muscular atrophy (Puls et al., 2003), our lines of mutant p150 Glued mice exhibit a rapid disease progression indicating that this mouse model closely mimics the clinical outcomes of sporadic ALS. Moreover, the pathophysiology observed in these mutant p150 Glued mice share many features with those seen in sporadic ALS, including loss of motor neurons, ubiquitin positive inclusions, accumulations of neurofilaments and astrocytic gliosis. Importantly, none of these pathological changes was observed in mice expressing human wild-type p150Glued.
That there is progressive accumulation of intracellular membrane vesicles in motor neurons of mutant mice is consistent with the view that mutant dynactin p150Glued causes aberrant membrane trafficking leading to the “poisoning” of the bidirectional transport of vesicles between the ER and Golgi apparatus within motor neurons of mutant dynactin p150Glued mice. Such membrane trafficking have been shown to involve dynein/dynactin mediated transport (Lee et al., 2004); numerous vesicles are produced from the transitional ER and these coat protein complex II (COPII)-labeled vesicles are relocated to the perinuclear Golgi ribbon along the microtubule cytoskeleton via dynein/dynactin mediated transport (Orci, 1982; Presley et al., 1997; Scales et al., 1997) and the major retrograde vesicular transport from the Golgi to the ER involves COPI-mediated transport (Rothman, 1996; Schekman and Orci, 1996). However, COPI-independent transport can occur via interactions with Bicaudal-D, which binds dynein/dynactin, a mechanism that is regulated by Rab6a (Girod et al., 1999; White et al., 1999; Storrie et al., 2000; Matanis et al., 2002). Thus, abnormal accumulation of membrane vesicles within the motor neuron cell body suggests that mutant p150Glued causes motor neuron degeneration at least in part through disruption and poisoning of intracellular trafficking processes. In this scenario, budding from the ER or Golgi apparatus occurs but the mutant dynactin prevents these vesicles from being transported to their appropriate destination leading to aberrant accumulations of vesicles within the motor neuron.
Our biochemical and morphological evidence strongly indicate that a proportion of the vesicles accumulating in the motor neurons of mutant dynactin mice are autophagosomes. Autophagosome-lysosome fusion is a microtubule dependent process and blockade of this step with microtubule polymerizing drugs results in enlarged autophagosomes and enhanced aggregation of autophagosome substrates (Ravikumar et al., 2005). Moreover, dynein mutations have been shown to impair autophagic clearance of aggregated proteins which has been attributed to disruption of autophagosome-lysosome fusion (Ravikumar et al., 2005). We hypothesize that mutant p150Glued could disrupt intracellular transport of autophagosomes to perinuclear regions where lysosomes are abundant in motor neurons, resulting in accumulation of autophagosomes in these cells. Our observation that inclusions are associated with ubiquitin immunoreactivities suggest that the ubiquitin proteasome system (UPS) may be impacted negatively by mutant dynactin and raise the possibility that autophagy may result from impairment of UPS. The previous findings that conditional deletion of autophagy gene in the CNS causes neurodegeneration characterized by ubiquitin immunoreactive inclusions (Hara et al., 2006; Komatsu et al., 2006) and that autophagy could serve as a compensatory degradation system when the UPS is impaired would support such a notion (Pandey et al., 2007).
In addition to defects in vesicular transport, perturbations in axonal transport have been hypothesized to play a role in the pathogenesis of human diseases, including ALS (Price et al., 1975; Griffin and Price, 1976), and have been shown to occur in various animal models (Price et al., 1975; Griffin and Price, 1976; Griffin et al., 1978; Gold et al., 1985; Collard et al., 1995; Williamson and Cleveland, 1999). In studies of mutant SOD1 (Cu/Zn superoxide dismutase) mice, disruption of transport has been shown to antedate the onset of clinical signs (Williamson and Cleveland, 1999). Although a similar scenario has long been postulated in ALS (Griffin and Price, 1976), the mechanisms leading to impaired axonal transport in models and in human disease are not entirely clear; the most obvious consequences of impaired transport would be the failure of critical proteins synthesized in the cell body and anterogradely delivered to terminals, and/or altered retrograde transport of signals, originating in the periphery, to reach cell bodies (Koliatsos et al., 1993; Ehlers et al., 1995; Grimes et al., 1996, 1997; Riccio et al., 1997; Tsui-Pierchala and Ginty, 1999; Goldstein and Yang, 2000; Terada et al., 2000; Schroer, 2004; Vallee et al., 2004).
Although we have not yet addressed the role of axonal transport in mutant p150 Glued mice, a defect in retrograde transport could be particularly challenging for motor neurons because of the dependence of these cells on trophic support (i.e., BDNF, neurotrophic factor 4,5, and glial cell line-derived neurotrophic factor) provided by distal targets. Impairments in axonal transport could also impact on the autophagy system because peripheral signals are integral to the regulation of this mechanism (Florez-McClure et al., 2004). Moreover, axonal injuries that interrupt trophic signaling and deprive the cell of growth factors have been shown to induce autophagy (Nixon, 2006). Thus, impaired dynactin-dynein mediated transport could reduce retrogradely delivered signaling cargoes to the cell body and possibly lead to an induction of autophagy, stimulating autophagosome formation, while at the same time inhibiting the clearance of these autophagosomes as intracellular transport is also compromised. Identification of defects in axonal transport in mutant dynactin p150 Glued mice will be of great value for clarifying disease mechanisms linked to these processes.
As genetic studies have identified additional missense mutations in dynactin that associate with cases of ALS (Munch et al., 2004), this novel mouse model of motor neuron disease will be a valuable tool to begin to address potentially common pathogenic mechanisms underlying ALS, such as vesicular trafficking, autophagy, as well as axonal transport, and for identification of novel therapeutic strategies to ameliorate degeneration of motor neurons in ALS.
Footnotes
This work was supported in part by The Muscular Dystrophy Association, The Robert Packard Center for Amyotrophic Lateral Sclerosis Research, and National Institute of Neurological Disorder and Stroke Grant RO1 NS40014 (P.C.W.). The Thy1.2 promoter was a gift from P. Caroni. We thank V. Nehus, Y. Pan, D. Prasad, Ashley Moon, and M. Polley for their technical support, and Drs. H. Cai, A. Ludolph, A. Kolodkin, and T. Lloyd for sharing of unpublished results. Our coauthor and friend, Dr. Steven Ackerley, passed away suddenly on January 14, 2008. With deep sorrow and mourning, we dedicate this paper to his memory.
References
- Collard JF, Cote F, Julien JP. Defective axonal transport in a transgenic mouse model of amyotrophic lateral sclerosis. Nature. 1995;375:61–64. doi: 10.1038/375061a0. [DOI] [PubMed] [Google Scholar]
- Eaton BA, Fetter RD, Davis GW. Dynactin is necessary for synapse stabilization. Neuron. 2002;34:729–741. doi: 10.1016/s0896-6273(02)00721-3. [DOI] [PubMed] [Google Scholar]
- Ehlers MD, Kaplan DR, Price DL, Koliatsos VE. NGF-stimulated retrograde transport of trkA in the mammalian nervous system. J Cell Biol. 1995;130:149–156. doi: 10.1083/jcb.130.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florez-McClure ML, Linseman DA, Chu CT, Barker PA, Bouchard RJ, Le SS, Laessig TA, Heidenreich KA. The p75 neurotrophin receptor can induce autophagy and death of cerebellar Purkinje neurons. J Neurosci. 2004;24:4498–4509. doi: 10.1523/JNEUROSCI.5744-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill SR, Schroer TA, Szilak I, Steuer ER, Sheetz MP, Cleveland DW. Dynactin, a conserved, ubiquitously expressed component of an activator of vesicle motility mediated by cytoplasmic dynein. J Cell Biol. 1991;115:1639–1650. doi: 10.1083/jcb.115.6.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girod A, Storrie B, Simpson JC, Johannes L, Goud B, Roberts LM, Lord JM, Nilsson T, Pepperkok R. Evidence for a COP-I-independent transport route from the Golgi complex to the endoplasmic reticulum. Nat Cell Biol. 1999;1:423–430. doi: 10.1038/15658. [DOI] [PubMed] [Google Scholar]
- Gold BG, Griffin JW, Price DL. Slow axonal transport in acrylamide neuropathy: Different abnormalities produced by single-dose and continuous administration. J Neurosci. 1985;5:1755–1768. doi: 10.1523/JNEUROSCI.05-07-01755.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein LS, Yang Z. Microtubule-based transport systems in neurons: the roles of kinesins and dyneins. Annu Rev Neurosci. 2000;23:39–71. doi: 10.1146/annurev.neuro.23.1.39. [DOI] [PubMed] [Google Scholar]
- Griffin JW, Price DL. Axonal transport in motor neuron pathology. In: Andrews J, Johnson R, editors. Amyotrophic lateral sclerosis: recent research trends. New York: Academic; 1976. [Google Scholar]
- Griffin JW, Hoffman PN, Clark AW, Carroll PT, Price DL. Slow axonal transport of neurofilament proteins: Impairment of β,β'-iminodipropionitrile administration. Science. 1978;202:633–635. doi: 10.1126/science.81524. [DOI] [PubMed] [Google Scholar]
- Grimes ML, Zhou J, Beattie EC, Yuen EC, Hall DE, Valletta JS, Topp KS, LaVail JH, Bunnett NW, Mobley WC. Endocytosis of activated TrkA: Evidence that nerve growth factor induces formation of signaling endosomes. J Neurosci. 1996;16:7950–7964. doi: 10.1523/JNEUROSCI.16-24-07950.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimes ML, Beattie E, Mobley WC. A signaling organelle containing the nerve growth factor-activated receptor tyrosine kinase, TrkA. Proc Natl Acad Sci USA. 1997;94:9909–9914. doi: 10.1073/pnas.94.18.9909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghnia M, Cavalli V, Shah SB, Schimmelpfeng K, Brusch R, Yang G, Herrera C, Pilling A, Goldstein LS. Dynactin is required for coordinated bidirectional motility, but not for dynein membrane attachment. Mol Biol Cell. 2007;18:2081–2089. doi: 10.1091/mbc.E06-08-0695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Takemura R. Molecular motors and mechanisms of directional transport in neurons. Nat Rev Neurosci. 2005;6:201–214. doi: 10.1038/nrn1624. [DOI] [PubMed] [Google Scholar]
- Koliatsos VE, Clatterbuck RE, Winslow JW, Cayouette MH, Price DL. Evidence that brain-derived neurotrophic factor is a trophic factor for motor neurons in vivo. Neuron. 1993;10:359–367. doi: 10.1016/0896-6273(93)90326-m. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Lai C, Lin X, Chandran J, Shim H, Yang W-J, Cai H. The G59S mutation in p150glued causes dysfunction of dynactin in mice. J Neurosci. 2007;27:13982–13990. doi: 10.1523/JNEUROSCI.4226-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaMonte BH, Wallace KE, Holloway BA, Shelly SS, Ascano J, Tokito M, Van Winkle T, Howland DS, Holzbaur EL. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron. 2002;34:715–727. doi: 10.1016/s0896-6273(02)00696-7. [DOI] [PubMed] [Google Scholar]
- Lee MC, Miller EA, Goldberg J, Orci L, Schekman R. Bi-directional protein transport between the ER and Golgi. Annu Rev Cell Dev Biol. 2004;20:87–123. doi: 10.1146/annurev.cellbio.20.010403.105307. [DOI] [PubMed] [Google Scholar]
- Levy JR, Sumner CJ, Caviston JP, Tokito MK, Ranganathan S, Ligon LA, Wallace KE, LaMonte BH, Harmison GG, Puls I, Fischbeck KH, Holzbaur EL. A motor neuron disease-associated mutation in p150Glued perturbs dynactin function and induces protein aggregation. J Cell Biol. 2006;172:733–745. doi: 10.1083/jcb.200511068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matanis T, Akhmanova A, Wulf P, Del Nery E, Weide T, Stepanova T, Galjart N, Grosveld F, Goud B, De Zeeuw CI, Barnekow A, Hoogenraad CC. Bicaudal-D regulates COPI-independent Golgi-ER transport by recruiting the dynein-dynactin motor complex. Nat Cell Biol. 2002;4:986–992. doi: 10.1038/ncb891. [DOI] [PubMed] [Google Scholar]
- Munch C, Sedlmeier R, Meyer T, Homberg V, Sperfeld AD, Kurt A, Prudlo J, Peraus G, Hanemann CO, Stumm G, Ludolph AC. Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology. 2004;63:724–726. doi: 10.1212/01.wnl.0000134608.83927.b1. [DOI] [PubMed] [Google Scholar]
- Nixon RA. Autophagy in neurodegenerative disease: friend, foe or turncoat? Trends Neurosci. 2006;29:528–535. doi: 10.1016/j.tins.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Orci L. Macro- and micro-domains in the endocrine pancreas. Diabetes. 1982;31:538–565. doi: 10.2337/diab.31.6.538. [DOI] [PubMed] [Google Scholar]
- Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, Padmanabhan R, Hild M, Berry DL, Garza D, Hubbert CC, Yao TP, Baehrecke EH, Taylor JP. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ, Lippincott-Schwartz J. ER-to-Golgi transport visualized in living cells. Nature. 1997;389:81–85. doi: 10.1038/38001. [DOI] [PubMed] [Google Scholar]
- Price DL, Griffin J, Young A, Peck K, Stocks A. Tetanus toxin: Direct evidence for retrograde intraaxonal transport. Science. 1975;188:945–947. doi: 10.1126/science.49080. [DOI] [PubMed] [Google Scholar]
- Price DL, Ackerly S, Martin LJ, Koliatsos VE, Wong PC. Motor neuron diseases. In: Brady ST, Siegel GJ, Albers RW, Price DL, editors. Basic neurochemistry. Burlington, MA: Elsevier; 2005. [Google Scholar]
- Puls I, Jonnakuty C, LaMonte BH, Holzbaur EL, Tokito M, Mann E, Floeter MK, Bidus K, Drayna D, Oh SJ, Brown RH, Jr, Ludlow CL, Fischbeck KH. Mutant dynactin in motor neuron disease. Nat Genet. 2003;33:455–456. doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- Puls I, Oh SJ, Sumner CJ, Wallace KE, Floeter MK, Mann EA, Kennedy WR, Wendelschafer-Crabb G, Vortmeyer A, Powers R, Finnegan K, Holzbaur EL, Fischbeck KH, Ludlow CL. Distal spinal and bulbar muscular atrophy caused by dynactin mutation. Ann Neurol. 2005;57:687–694. doi: 10.1002/ana.20468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Acevedo-Arozena A, Imarisio S, Berger Z, Vacher C, O'Kane CJ, Brown SD, Rubinsztein DC. Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat Genet. 2005;37:771–776. doi: 10.1038/ng1591. [DOI] [PubMed] [Google Scholar]
- Riccio A, Pierchala BA, Ciarallo CL, Ginty DD. An NGF-TrkA-mediated retrograde signal to transcription factor CREB in sympathetic neurons. Science. 1997;277:1097–1100. doi: 10.1126/science.277.5329.1097. [DOI] [PubMed] [Google Scholar]
- Ross JL, Wallace K, Shuman H, Goldman YE, Holzbaur EL. Processive bidirectional motion of dynein-dynactin complexes in vitro. Nat Cell Biol. 2006;8:562–570. doi: 10.1038/ncb1421. [DOI] [PubMed] [Google Scholar]
- Rothman JE. The protein machinery of vesicle budding and fusion. Protein Sci. 1996;5:185–194. doi: 10.1002/pro.5560050201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scales SJ, Pepperkok R, Kreis TE. Visualization of ER-to-Golgi transport in living cells reveals a sequential mode of action for COPII and COPI. Cell. 1997;90:1137–1148. doi: 10.1016/s0092-8674(00)80379-7. [DOI] [PubMed] [Google Scholar]
- Schekman R, Orci L. Coat proteins and vesicle budding. Science. 1996;271:1526–1533. doi: 10.1126/science.271.5255.1526. [DOI] [PubMed] [Google Scholar]
- Schroer TA. Dynactin. Annu Rev Cell Dev Biol. 2004;20:759–779. doi: 10.1146/annurev.cellbio.20.012103.094623. [DOI] [PubMed] [Google Scholar]
- Schroer TA, Sheetz MP. Functions of microtubule-based motors. Annu Rev Physiol. 1991;53:629–652. doi: 10.1146/annurev.ph.53.030191.003213. [DOI] [PubMed] [Google Scholar]
- Storrie B, Pepperkok R, Nilsson T. Breaking the COPI monopoly on Golgi recycling. Trends Cell Biol. 2000;10:385–391. doi: 10.1016/s0962-8924(00)01818-3. [DOI] [PubMed] [Google Scholar]
- Terada S, Kinjo M, Hirokawa N. Oligomeric tubulin in large transporting complex is transported via kinesin in squid giant axons. Cell. 2000;103:141–155. doi: 10.1016/s0092-8674(00)00094-5. [DOI] [PubMed] [Google Scholar]
- Tsui-Pierchala BA, Ginty DD. Characterization of an NGF-P-TrkA retrograde-signaling complex and age-dependent regulation of TrkA phosphorylation in sympathetic neurons. J Neurosci. 1999;19:8207–8218. doi: 10.1523/JNEUROSCI.19-19-08207.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallee RB, Williams JC, Varma D, Barnhart LE. Dynein: An ancient motor protein involved in multiple modes of transport. J Neurobiol. 2004;58:189–200. doi: 10.1002/neu.10314. [DOI] [PubMed] [Google Scholar]
- White J, Johannes L, Mallard F, Girod A, Grill S, Reinsch S, Keller P, Tzschaschel B, Echard A, Goud B, Stelzer EH. Rab6 coordinates a novel Golgi to ER retrograde transport pathway in live cells. J Cell Biol. 1999;147:743–760. doi: 10.1083/jcb.147.4.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson TL, Cleveland DW. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci. 1999;2:50–56. doi: 10.1038/4553. [DOI] [PubMed] [Google Scholar]