

Abstract
After an ischemic stroke, neurons in the core are rapidly committed to die, whereas neuron death in the slowly developing penumbra is more amenable to therapeutic intervention. Microglia activation contributes to delayed inflammation, but because neurotoxic mechanisms in the penumbra are not well understood, we developed an in vitro model of microglia activation and propagated neuron killing. To recapitulate inflammatory triggers in the core, microglia were exposed to oxygen glucose-deprived neurons and astrocytes. To model the developing penumbra, the microglia were washed and allowed to interact with healthy naive neurons and astrocytes. We found that oxygen–glucose deprivation (OGD)-stressed neurons released glutamate, which activated microglia through their group II metabotropic glutamate receptors (mGluRs). Microglia activation involved nuclear factor κB (NF-κB), a transcription factor that promotes their proinflammatory functions. The activated microglia became neurotoxic, killing naive neurons through an apoptotic mechanism that was mediated by tumor necrosis factor-α (TNF-α), and involved activation of both caspase-8 and caspase-3. In contrast to some earlier models (e.g., microglia activation by lipopolysaccharide), neurotoxicity was not decreased by an inducible nitric oxide synthase (iNOS) inhibitor (S-methylisothiourea) or a peroxynitrite scavenger [5,10,15,20-tetrakis(N-methyl-4′-pyridyl)porphinato iron (III) chloride], and did not require p38 mitogen-activated protein kinase (MAPK) activation. The same microglia neurotoxic behavior was evoked without exposure to OGD-stressed neurons, by directly activating microglial group II mGluRs with (2S,2′R,3′R)-2-(2′3′-dicarboxycyclopropyl) glycine or glutamate, which stimulated production of TNF-α (not nitric oxide) and mediated TNF-α-dependent neurotoxicity through activation of NF-κB (not p38 MAPK). Together, these results support potential therapeutic strategies that target microglial group II mGluRs, TNFα overproduction, and NF-κB activation to reduce neuron death in the ischemic penumbra.
Keywords: microglia activation, metabotropic glutamate receptors, neurodegeneration, neuroinflammation, neuron death, TNF-α
Introduction
An ischemic stroke results in an initial area of neuron death, the core, surrounded by an area vulnerable to further damage: the penumbra. When blood flow in the core is reduced, initial neuron death results from reduced ATP levels and protein synthesis, ionic imbalances, and glutamate-mediated excitotoxicity (Sharp et al., 2000). Because of this rapid commitment to cell death and the delay to hospitalization, it is not feasible to target therapies to rescue neurons in the core (Barone and Feuerstein, 1999). Over time, damage can propagate into the penumbra, where delayed neuron death by apoptosis and necrosis can extend for days to weeks (Zheng et al., 2003), a time frame that renders these cells more amenable to rescue. Thus, it is essential to understand events that occur in the penumbra.
After an ischemic stroke, a prominent inflammatory response develops, propagates, and lasts for many days (Schilling et al., 2003), and is widely believed to exacerbate neuron death (Siao and Tsirka, 2002; Bramlett and Dietrich, 2004). Although there is evidence that inflammation in the penumbra is orchestrated by brain-resident immune cells (microglia) (Wood, 1995), the actions of these cells are not fully understood. Microglial activation is observed within 1 d after a stroke in vivo (Schilling et al., 2003), but it has been difficult to identify the molecule(s) that activate the microglia. A prevalent theory is that damaged neurons release ATP (Volonte et al., 2003) and glutamate (Fujimoto et al., 2004), which activate microglial purinergic (Haynes et al., 2006) and glutamate (Taylor et al., 2002, 2005) receptors. Much of our understanding of these events comes from in vitro models using neuron cultures or organotypic brain slice cultures, and although it is clear that such models cannot exactly replicate in vivo conditions, it is important to refine the models to better represent events occurring in the penumbra. Some stimuli used to activate the microglia in vitro are of questionable relevance to the stroke penumbra; e.g., many studies apply the bacterial endotoxin, lipopolysaccharide (LPS), an ATP analog (e.g., benzoyl-ATP), or tumor necrosis factor-α (TNF-α). Clearly, LPS is not a model of stroke, and although ATP and TNF-α are relevant, it is very unlikely that brain tissue subjected to an ischemic stress will produce only a single molecule. A more appropriate in vitro stroke model is one that subjects isolated neurons or brain slices to oxygen–glucose deprivation (OGD), and in doing so, evokes neuron death through mitochondrial dysfunction and apoptosis (Iijima et al., 2003; Cho et al., 2004). However, it can be argued that this is a better model of the ischemic core than the penumbra.
The aim of this study was to develop an in vitro model that is more relevant to the in vivo penumbra, to investigate (1) whether ischemic neurons activate microglia and, if so, the molecules and signaling pathways involved; and (2) whether microglia activated in this manner can propagate the damage by killing healthy naive neurons and, if they do, the signals involved.
Materials and Methods
Culturing microglia and embryonic rat neurons
All animal handling was in accordance with guidelines from the Canadian Council on Animal Care. Highly purified microglia cultures were prepared from brains of 1- to 2-d-old Wistar rat pups (Charles River, St. Constant, Quebec, Canada) as previously described (Xie et al., 2002; Fordyce et al., 2005). Briefly, rat pups were killed by cervical dislocation, and the whole cortex was dissected out. Dissociated brain tissue was minced in cold minimal essential medium (MEM; Invitrogen, Carlsbad, CA) and centrifuged (1000 rpm for 10 min). The pelleted cells were resuspended and seeded in tissue-culture flasks containing MEM supplemented with 5% horse serum and 5% fetal bovine serum. The medium was changed once, after 2 d. After 10–12 d, the flasks were shaken on an orbital shaker for 4–6 h at 72 rpm in the incubator to dislodge the microglia from the underlying bed of astrocytes. At this stage, the cultures were 99–100% microglia, as judged by labeling with isolectin B4, tomato lectin, and antibodies to complement receptor 3 (CR3) or ionized calcium-binding adaptor molecule 1 (Iba1), and by real-time reverse transcription-PCR for CR3 versus other brain cell markers (Kaushal et al., 2007). Microglia were harvested, centrifuged for 10 min at 1000 rpm, resuspended in Neurobasal A/B27 without antioxidants (as above), and seeded at 106 cells/well onto the upper inserts of Transwell chambers, which have 3 μm diameter pores (BD Biosciences, Mississauga, Ontario, Canada). The Transwell chambers were placed in 24-well plates in a humidified incubator (95% O2, 5% CO2, 37°C).
Neuronal cultures were prepared as previously described (Fordyce et al., 2005), using embryonic day 18 Wistar rats (Charles River). Briefly, the cortex was dissected out and incubated in 2 mg/ml papain (Worthington Biochemical, Lakewood, NJ) for 30 min at 37°C. The dissociated tissue was triturated and resuspended in serum-free culture medium (Neurobasal A/B27 medium), which contained Neurobasal A and 2% B27 supplement with antioxidants, 0.05 mg/ml Gentamycin, and 0.5 mm l-glutamine (all from Invitrogen). The cultured cortical neurons were seeded onto poly-l-ornithine-treated (Sigma, St. Louis, MO) German coverslips (Bellco Glass, Vineland, NJ) at 3 × 104 cells/well and grown for 7–10 d to increase the proportion of mature neurons. Every 4 d, 50% of the medium was replaced with fresh Neurobasal A/B27 medium without antioxidants. After 7 d, the culture purity was determined by staining mature neurons with microtubule-associated protein 2 (MAP2) and astrocytes with GFAP: ∼70% of the cells were neurons, and ∼30% were astrocytes (for data, see Kaushal et al., 2007).
The in vitro penumbra model
There were three stages in the experimental paradigm (Fig. 1).
Figure 1.

The in vitro penumbra model: basic experimental paradigm (see Materials and Methods and Results for further details). Microglia were seeded onto the upper permeable insert of a Transwell chamber, and neuron cultures (with ∼30% astrocytes) were grown on coverslips, which were placed in the lower well. Importantly, because the upper and lower parts of the chamber can be separated, the microglia and neurons can be treated separately. Stage 1, Neurons in the bottom well of a Transwell chamber were subjected to OGD, and two of their responses were assessed: apoptosis (see Fig. 3B) and glutamate release (see Fig. 5A). Stage 2, Microglia growing in the porous upper insert were exposed to these OGD-SNs in a Transwell chamber for varying times and then assessed for activation of p38 MAPK and NF-κB and production of TNF-α and nitric oxide (see Fig. 2). Stage 3, Alternatively, microglia were exposed to OGD-SNs for 24 h, washed thoroughly, and then placed in a new Transwell chamber above healthy target (“naive”) neurons (with ∼30% astrocytes), which were then removed and assessed for activation of caspase-3 and caspase-8 after 24 h (see Fig. 3C) or TUNEL after 48 h (see Fig. 3A,B).
Stage 1.
Neuron/astrocyte (∼70:30%) cultures were subjected to OGD by growing them on coverslips in 24-well plates and placing them in a Modular incubator chamber (Billups-Rothenberg, Del Mar, CA), where the Neurobasal A/B27 medium (without antioxidants) was replaced with oxygen-glucose free DMEM containing 5 mm HEPES (Invitrogen). The chamber was flushed with 5% CO2 and 95% N2 for 5 min and then sealed and incubated at 37°C for the desired time. Oxygen–glucose-free DMEM was prepared by bubbling glucose-free DMEM with 5% CO2 and 95% N2 for 5 min, and incubating overnight in a sealed 15 ml Falcon tube. After OGD, the neuron-containing plates were removed from the chamber, and the DMEM was replaced with normal Neurobasal A/B27 medium without antioxidants. Having determined that 60 min OGD caused ∼40% of the neurons to become terminal deoxyuridine-triphosphate nick-end labeling (TUNEL) positive by 24 h, this duration was used for all further experimentation, except when time course experiments were conducted. Longer periods of OGD can evoke necrosis, particularly if the neurons are isolated from more OGD-sensitive regions (e.g., hippocampus) (Jiang et al., 2004; Xu et al., 2005).
Stage 2.
Because the inflammatory response develops over many hours (see Introduction), this stage was designed to represent the expanding penumbra, wherein molecules released from damaged neurons in the core of the infarct can diffuse to, and activate, nearby microglia. This was accomplished by allowing the OGD-stressed neurons (“OGD-SNs”) in the lower well to chemically communicate with microglia in the porous upper insert of the Transwell chamber for 24 h (95% O2, 5% CO2, 37°C). The 3-μm-diameter pores allow molecules to diffuse between the neurons and microglia, which are separated by ∼1 mm, without permitting cell–cell contact. Note that the microglia were never exposed to OGD, because it is not known how ischemia would affect their activation.
Stage 3.
This stage was designed to ask whether the stimulated microglia develop a neurotoxic phenotype and, if so, what diffusible molecules are responsible for microglial activation and their ability to kill healthy neurons. Because it was important to avoid carryover of molecules secreted by OGD-stressed neurons or compounds used to treat the microglia, the microglia-bearing inserts were thoroughly washed with Neurobasal A/B27 medium (without antioxidants) before they were placed in new Transwell chambers above healthy naive neurons. Importantly, these test neurons were never exposed to OGD.
Treatments
For selected experiments, microglia alone were treated with the following: an inhibitor of inducible nitric oxide synthase, 100 μm S-methylisothiourea (SMT; EMD Biosciences, San Diego, CA); 50 ng/ml soluble tumor necrosis factor-α receptor 1 protein (sTNFR1; R & D Systems, Minneapolis, MN); the peroxynitrite scavenger, 2 μm 5,10,15,20-tetrakis(N-methyl-4′-pyridyl)porphinato iron (III) chloride (FeTmPyP; EMD Biosciences); the p38 mitogen-activated protein kinase (MAPK) inhibitor, 2 μm 4-(4-fluorophenyl)-2-(4-hydroxyphenyl)-5-(4-pyridyl)-1H-imidazole (SB 202190; EMD Biosciences); 50 ng/ml of an IκB kinase inhibitor peptide (EMD Biosciences); or an agonist of group II metabotropic glutamate receptors (mGluRIIs), 250 nm (2S,2′R,3′R)-2-(2′3′-dicarboxycyclopropyl) glycine (DCG-IV; Tocris, Avonmouth, UK). For some experiments, microglia and neurons/astrocytes in Transwell chambers (stage 2) were exposed to an antagonist of group II mGluRs or to a caspase inhibitor. To inhibit mGluRs, we used either 250 μm 2S-α-ethylglutamic acid (EGLU; Tocris), which blocks mGluR2 (Kd, 35 μm) and mGluR3 (Kd, 4 μm), or 500 nm 2S-2-amino-2-(1S,2S-2-carboxycycloprop-1-yl)-3-(xanth-9-yl) propanoic acid (LY341495; Tocris), which blocks mGluR2 (Kd, 10 nm) and mGluR3 (Kd, 6 nM) (Pin and Acher, 2002). To inhibit caspase-3, we used 100 μm DEVD-CHO (also called caspase-3 inhibitor I or CPP32/apopain inhibitor) (Nicholson et al., 1995), which has long been used to inhibit neuronal apoptosis in culture (Du et al., 1997). We compared the degree of neuronal rescue with the broad-spectrum caspase peptide inhibitor, 100 μm Boc-d-FMK [also called BOC-Asp(OMe)-fluoromethyl ketone or caspase inhibitor III] (Thornberry and Lazebnik, 1998), which inhibits neuronal apoptosis (Hayashi et al., 2007). Stock solutions were made in distilled water, except LY341495, DEVD-CHO, and Boc-d-FMK (both from EMD Biosciences), which were dissolved in DMSO and compared with DMSO controls. The final dilutions were made in the experimental solution: Neurobasal A/B27 medium without antioxidants. Control experiments included exposing neuron/astrocyte cultures to the group II mGluR antagonists (EGLU, LY341495), because the OGD-stressed cultures were exposed to these drugs.
Plate-reader assays
Neurons were grown on coverslips, and microglia were grown on the porous upper inserts of Transwell chambers (as above). After experimentation, the cells were washed, fixed, permeabilized, placed in 24-well black-walled plates (PerkinElmer, Waltham, MA), and stained (described below). Then, a spectrofluorometer (SPECTRAmax Gemini EM; Molecular Devices, Sunnyvale CA) was used to measure the average fluorescence intensity in each well. Signals were standardized as relative fluorescence units (RFU) per milligram of protein in each well, where the protein concentration was measured in each well with a Bio-Rad colorimetric protein assay and BSA standards (Bio-Rad Laboratories, Hercules, CA), using a colorimetric plate reader (model EL311SX, Bio-Tek Instruments, Winooski, VT). For every experiment, the baseline signal was calculated as the average from three wells containing neuron/astrocyte cultures that had previously been exposed to unstimulated microglia but no other reagents. Then, for each treatment, an individual experimental value was obtained by averaging the RFU/mg protein from three wells of cells from the same culture, after subtracting the baseline signal. Multiple n values were obtained using cell cultures from different animals. Because the plate-reader assays depend on appropriate TUNEL or antibody labeling, we verified staining and monitored negative controls before using the plate reader.
Assessing neuron damage and apoptosis.
Neuronal DNA damage was quantified by TUNEL, e.g., in target neurons that had been exposed to microglia that had been preexposed to separate OGD-stressed neurons (stage 3 of the standard experimental protocol shown in Fig. 1). In this case, neuron-bearing coverslips were removed from the Transwell chamber after 48 h, washed with PBS, fixed (30 min, 4% paraformaldehyde), permeabilized with 0.01% Triton X-100 (Sigma) in PBS containing 1% sodium citrate (2 min on ice); and then incubated (30 min, 37°C) with the TUNEL reaction mixture according to the manufacturer's protocol (Roche Applied Science, Laval, Quebec, Canada). The fluorescence signal was then detected using FITC-conjugated streptavidin (1:500; Invitrogen) and the fluorescence plate reader. To demonstrate the validity of this higher-throughput plate-reader assay, TUNEL-positive neurons were counted as a percentage of all cells, where all nuclei were stained by a 5 min treatment with 4′,6′-diamidino-2-phenylindole (DAPI; 1:2000; Sigma). The coverslips were washed in PBS (three times, 5 min each), incubated for 30 min with FITC-conjugated streptavidin, washed with PBS (three times, 5 min each), and mounted on glass slides with 1:1 glycerol:PBS. Cells were counted in five separate fields using a 10× objective, a Zeiss Axioplan 2 epifluorescence microscope (Zeiss, Thornwood, NY), and Zeiss Axiocam digital camera. The TUNEL-positive cells were confirmed to be neurons, by double labeling with anti-NeuN antibody (1:200; Millipore, Billerica, MA) (see Fig. 3A).
Figure 3.
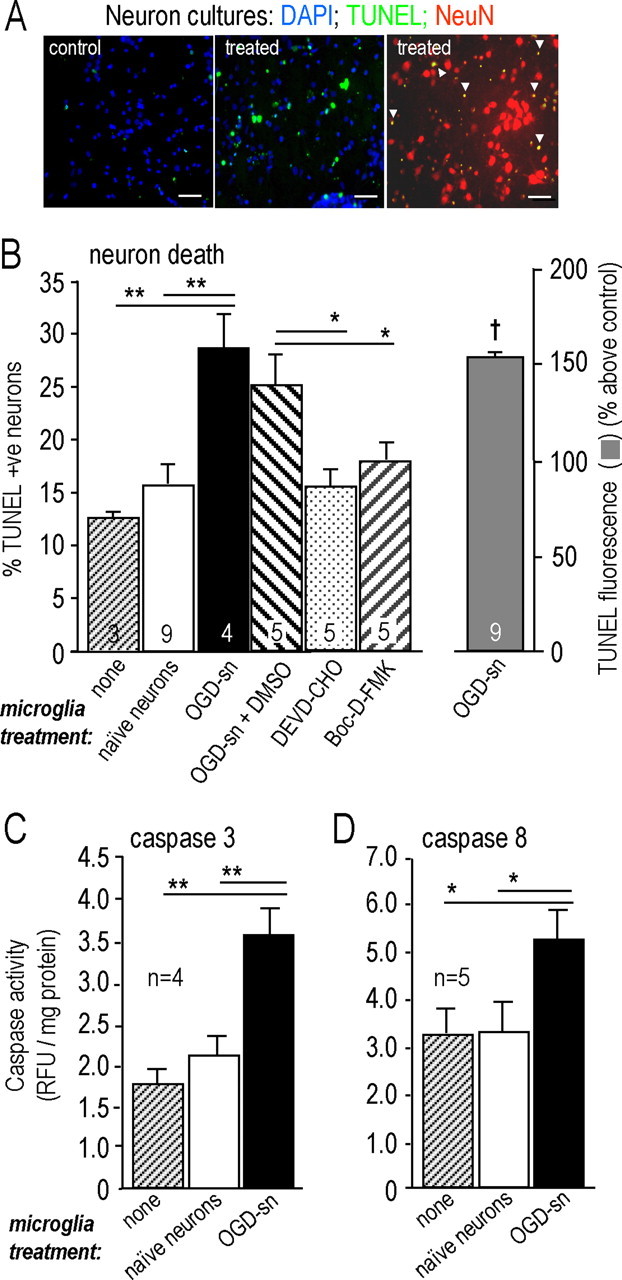
OGD-SNs increase the neurotoxic activity of microglia. A, Representative photomicrographs of target neurons in the lower wells of a Transwell chamber after 48 h of exposure to microglia. The microglia were either untreated or exposed to OGD-SNs for 24 h (“treated”). Cell nuclei were labeled with DAPI (blue), TUNEL (green), or an antibody against NeuN (red). Scale bars, 10 μm. The remaining panels show similar experiments, with outcomes in target neurons in the lower wells expressed as mean ± SEM for the number of separate cell cultures indicated. The label below each bar indicates the pretreatment of the neurons, before they were used to stimulate the microglia. B, Treating microglia with OGD-SNs evokes neurotoxic behavior. Bars 1–6 show TUNEL-positive nuclei counted after 48 h and expressed as a percentage of all DAPI-stained nuclei (*p < 0.05; **p < 0.01). The right bar shows the corresponding increase in the fluorescence signal from TUNEL, measured using the plate reader and compared with neurons exposed to untreated microglia (†p < 0.05). Either the caspase-3 inhibitor, 100 μm DEVD-CHO, or the broad-spectrum caspase inhibitor, 100 μm Boc-d-FMK, was added at the beginning of the 48 h incubation. C, D, Microglia that had been exposed to OGD-SNs evoked activation of both caspase-3 (C) and caspase-8 (D) in naive target neurons. Fluorogenic substrates and the fluorescence plate reader (see Materials and Methods) were used to measure caspase activity in target neurons 24 h after exposure to microglia. *p < 0.05; **p < 0.01.
Neuron levels of activated caspase-3 and caspase-8 were monitored after 24 h and expressed as RFU standardized to protein content, as above. For caspase-3, neurons were scraped off the coverslips and suspended in 200 μl of caspase extraction buffer (50 mm KCl, 5 mm EGTA, 1 mm MgCl2, 5 mm DTT, 0.2% CHAPS, 5% sucrose, and 50 mm HEPES, pH 7.5). The extract was centrifuged (1 min at 20,000 × g), the pellet was discarded, and the supernatant (50 μl) was combined with Ac-DEVD-AMC (5 μl) and 2× reaction mixture (50 μl), which contained 50 mm HEPES, pH 7.4, 100 mm NaCl, 0.1% CHAPS, 1 mm EDTA, 10% glycerol, and 10 mm DTT. Caspase-3 activity was measured with a spectrofluorometer (Molecular Devices) with excitation at 365 nm and emission at 430 nm. For caspase-8, a purchased kit was used, according to the manufacturer's instructions (EMD Biosciences). Neurons were scraped off coverslips, suspended in PBS, centrifuged (5 min, 500 × g), and the supernatant was discarded. Caspase-8 activity in the cell pellet was measured with a fluorescence plate reader (Model Victor3; PerkinElmer) at 345 nm excitation and 480 nm emission.
Microglial NF-κB and p38 MAPK activation.
Microglia activation was monitored as activation of p38 MAPK and nuclear factor κB (NF-κB) after 30 min of exposure to OGD-SNs. Phosphorylated p38 MAPK was monitored, and a standard approach was used to measure NF-κB activation, i.e., degradation of IκB-α (Nikodemova et al., 2006). The microglia-bearing porous Transwell insets were cut out, placed in 24-well black-walled plates, washed with PBS (three times, 5 min each), fixed for 30 min in 4% paraformaldehyde at room temperature, and permeabilized for 2 min on ice with 0.01% Triton X-100. After further washing (three times, 5 min each) the microglia were labeled (18 h, 4°C) with a rabbit polyclonal antibody against phospho-p38 MAPK (1:50; Cell Signaling Technology, Beverley, MA) or IκB-α (1:100; Santa Cruz Biotechnology, Santa Cruz, CA). After washing in PBS (three times, 5 min each), immunoreactivity was detected using a secondary antibody (2 h, room temperature) conjugated to Cy3 (1:500; Jackson ImmunoResearch Laboratories, West Grove, PA). The relative fluorescence signal was quantified using the spectrofluorometer, as above.
Neuronal glutamate release.
Neurons were exposed for 24 h to group II mGluR antagonists or control medium, and then the supernatants were harvested. Glutamate levels were determined with an Amplex Red glutamic acid assay, according to the manufacturer's instructions (Invitrogen). A black-walled, clear-bottom 96-well plate was loaded with triplicates of each sample and read (excitation 545 nm, emission 590 nm) using the spectrofluorometer. Relative fluorescence units were standardized to protein content using the BSA assay (as above), and glutamate levels were quantified by interpolation on a standard curve.
Microglial NO production.
Microglia were grown on coverslips in 24-well plates, exposed for 24 h to OGD-stressed neurons, and then washed and incubated for 48 h. Nitric oxide production was measured as nitrite, using the colorimetric Griess assay (Invitrogen), as follows. Supernatant (150 μl) from each well was mixed with 10 μl of N-(1-naphthyl) ethylenediamine dihydrochloride, 10 μl of 1% sulfanilic acid and 130 μl of distilled H2O (30 min, room temperature), and absorbance at 570 nm was quantified using the colorimetric plate reader.
Microglial TNF-α production.
TNF-α production was quantified using an ELISA kit, according to the manufacturer's instructions (R & D Systems). After microglia growing on Transwell inserts were exposed to OGD-stressed neurons for 24 h, the inserts were washed and incubated for a further 48 h. Because TNF-α has never been examined with this stimulus, we quantified both the amount secreted and that remaining in the microglia. That is, after sampling the supernatant, the cells were washed, and TNF-α remaining in the adherent microglia was measured by lysing them with 1% Triton X-100 in PBS. Total TNF-α was calculated as the sum of the two values. Alternatively, after microglia were stimulated with the mGluRII agonist (DCG-IV) for 24 h, we measured the amount of secreted TNF-α (from 50,000/cells per well in a 96-well plate) for comparison with an earlier study (Taylor et al., 2005). For all TNF-α assays, absorbance was read at 405 nm using the colorimetric plate reader (as above), and each treatment was averaged from three wells and compared with a standard curve to quantify the concentration of TNF-α.
Statistical analyses
All data are presented as the mean of each treatment group ± SEM, with the number of experiments indicated. Each n value represents a separate cell culture from a different rat litter. The statistical significance of differences between experimental groups was analyzed using Origin 7.0 software (OriginLab, Northampton, MA) from one-way ANOVAs, followed by Bonferroni post hoc tests unless otherwise specified.
Results
OGD-stressed neurons activate microglia
NF-κB and p38 MAPK are key molecules in pathways involved in activating the proinflammatory properties of microglia (Zhang and Stanimirovic, 2002; Pawate et al., 2004). We monitored activation of these molecules by quantifying levels of IκB-α and phosphorylated p38 MAPK (see Materials and Methods), a method we previously validated for microglia treated with the commonly used stimulus, lipopolysaccharide (Fordyce et al., 2005; Kaushal et al., 2007). After microglia were exposed for 30 min to healthy naive neurons or to OGD-SNs, their phospho-p38 MAPK levels did not differ from microglia that had not been exposed to neurons (“untreated”) (Fig. 2A). As a positive control, a 30 min exposure to lipopolysaccharide evoked a more than threefold increase in phospho-p38 MAPK, as previously seen (Kaushal et al., 2007). In contrast to p38 MAPK, 30 min of exposure to OGD-stressed neurons resulted in activation of NF-κB in microglia, as seen from the ∼80% reduction in IκB-α (p < 0.05) (Fig. 2B).
Figure 2.

OGD-SNs activate microglia. Comparisons were made between untreated microglia, microglia exposed for 30 min to naive neurons, and microglia exposed for 30 min to OGD-SNs. A, OGD-stressed neurons do not activate p38 MAPK, as judged by monitoring phospho-p38 MAPK using a rabbit polyclonal antibody (1:50; Cell Signaling Technology). A 30 min exposure to lipopolysaccharide, which is included as a positive control, evoked a more than threefold increase in phospho-p38 MAPK (*p < 0.05). B, OGD-stressed neurons activate NF-κB (*p < 0.05), monitored as degradation of IκB-α using a rabbit polyclonal antibody against IκB-α (1:100; Santa Cruz Biotechnology). C, Microglial TNF-α production is elicited by neurons that had been exposed to increasing durations of OGD (from 0 to 120 min; *p < 0.05). Microglia on Transwell inserts were exposed to the OGD-SNs for 24 h and then washed and incubated for a further 48 h. The total amount of TNF-α was measured by ELISA (see Materials and Methods for details). D, Exposure of microglia to neurons that were OGD stressed for 0–120 min did not stimulate nitric oxide production. NO was quantified with the Griess reagent, as nitrite in the medium.
Next, we examined two potential outcomes of microglia activation, production of nitric oxide and TNF-α, because both have been implicated in neurotoxicity after ischemic stroke. To assess the outcome of increasing the severity of the challenge to neurons, the OGD duration was increased, after which microglia were exposed to the neurons for 24 h. After exposure to OGD-stressed neurons, isolated microglia on inserts were washed and incubated for 48 h, and then TNF-α and NO were measured. Total TNF-α (i.e., secreted plus remaining in the microglia) increased from 94 ± 149 pg/ml (untreated microglia) to 349 ± 122 pg/ml for microglia exposed to naive neurons for 30 min (Fig. 2C). Microglial TNF-α production increased with longer durations of neuron exposure to OGD, reaching 1068 ± 341 pg/ml for a 120 min exposure; however, microglial NO production was not affected by naive or OGD-stressed neurons (Fig. 2D). Some spontaneous microglial activation is anticipated in culture, especially in the presence of naive neurons, a few of which spontaneously die; however, this was greatly reduced by conducting all experiments in Neurobasal A/B27 medium. Dramatic activation of microglia exposed to OGD-stressed neurons is evident from the observed increases in NF-κB activation and TNF-α production. Interestingly, microglial responses in the present model differed from LPS-stimulated microglia, which showed p38 MAPK activation and increased NO production (Kaushal et al., 2007).
OGD-stressed neurons increase the neurotoxic activity of microglia
Because microglial activation and propagating neuron death are implicated in development of the stroke penumbra (see Introduction), stage 3 experiments (see Materials and Methods) (Fig. 1) were designed to test whether the neurotoxic potential of microglia is increased by preexposing them to OGD-SNs. Unless otherwise indicated, neurons alone were exposed to OGD for 60 min, and then coincubated in Transwell chambers with microglia for 24 h. Next, the microglia were thoroughly washed and placed in a new Transwell chamber above naive healthy neurons. These target neurons were collected at 48 h to monitor TUNEL, by counting cells or using the fluorescence plate reader after verifying the appropriate fluorescence labeling. Examples of staining are shown in Figure 3A, and the results of counting TUNEL-positive nuclei are compared with those using the fluorescence plate reader (Fig. 3B). Approximately 4% of the target neurons spontaneously became TUNEL positive by 48 h, which increased to 12 ± 0.1% when they were exposed to untreated microglia for 24 h (Fig. 3B). TUNEL-positive cells were identified as neurons by colabeling their nuclei with DAPI and a neuron-specific antibody against NeuN (Fig. 3A). When microglia were preexposed to OGD-stressed neurons, their killing of target neurons was increased by a similar degree whether measured by cell counting (an increase to 28 ± 3%; p < 0.01) or the plate reader (Fig. 3B). Because TUNEL does not discriminate well between apoptotic and necrotic cell death (Labat-Moleur et al., 1998), to further determine whether neuron death was through apoptosis, changes in activity of caspase-3 were monitored in the neuron layer (Fig. 3C,D). Preexposure to untreated neurons did not increase the neurotoxic potential of the microglia; 15 ± 2% of neurons became TUNEL positive. However, when the microglia had been preexposed to OGD-stressed neurons, caspase-3 activation in the target neurons nearly doubled (p < 0.01), indicating that an apoptotic pathway was activated in the neurons. Caspase 3 activation was required for neuron death (Fig. 3B), because the caspase-3 inhibitor, DEVD-CHO (Nicholson et al., 1995), and the broad spectrum caspase inhibitor, Boc-d-FMK (Thornberry and Lazebnik, 1998), reduced neuron death to the background level (i.e., to 15 ± 1% and 17 ± 1%, respectively).
Group II mGluRs increase microglia neurotoxic activity via TNF-α
It has been proposed that the signal that propagates from dying neurons to more distant microglia in the stroke penumbra is neuron-derived glutamate acting on microglial group II mGlu receptors (Taylor et al., 2002, 2005). Microglia express mGluR2 and mGluR3 receptors within the group II type (Geurts et al., 2003). Thus, the next step was to determine whether activation of mGluRIIs in microglia promotes their neurotoxic activity in our model, and if so, to identify the effector molecule(s). Microglia were stimulated for 24 h with the mGluRII agonist, DCG-IV, and then washed and incubated with naive target neurons for 48 h in the Transwell chamber. As a consequence (Fig. 4A), there was a significant increase in TUNEL in the target neurons compared with neurons exposed to untreated microglia (p < 0.05), and this increased toxicity was inhibited ∼70% by the mGluRII antagonist, EGLU (p < 0.05). Neuron killing involved TNF-α, but not NO, because the increase in TUNEL was reduced ∼85% by adding the soluble TNF-α receptor (p < 0.05), and unaffected by inhibiting microglia iNOS (with SMT) or scavenging peroxynitrite (with FeTmPyP). This observation prompted us to examine whether microglial activation via mGluRII receptors increased production of TNF-α or NO. When microglia were activated with DCG-IV for 24 h, there was more than double the TNF-α production (from 192 ± 23 to 414 ± 124 pg/ml; p < 0.05), but no increase in NO production (Fig. 4B). This is very similar to the effect of OGD-stressed neurons on microglia (compare with Fig. 2B,C).
Figure 4.
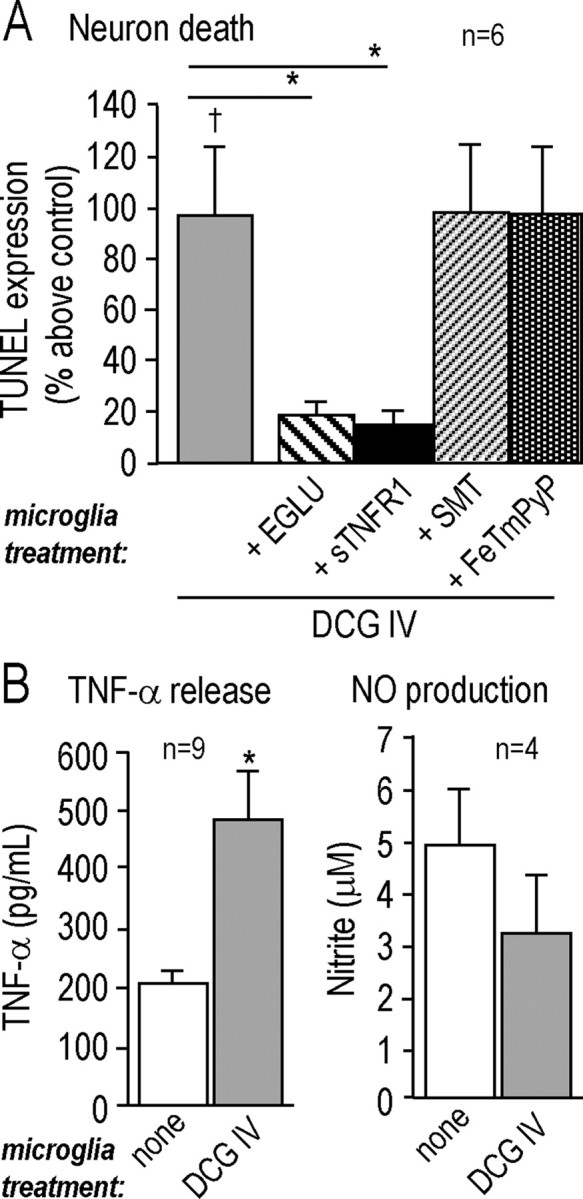
Group II mGluRs increase microglia neurotoxic activity via TNF-α. A, Microglial neurotoxic activity is stimulated by a specific agonist of group II metabotropic glutamate receptors and inhibited by antagonists of mGluRII or TNF-α. Microglia were treated for 24 h with the mGluRII agonist (250 nm DCG-IV), without (control) or with a drug added during a specific stage of the experiment (see Fig. 1), as follows. The mGluRII antagonist (250 μm EGLU) or iNOS inhibitor (100 μm SMT) was added during stage 2, and then after washing, the microglia were placed in the Transwell chamber above naive target neurons. The soluble TNF-α receptor 1 (50 ng/ml sTNFR1) or the peroxynitrite scavenger (2 μm FeTmPyP) was added during stage 3. Killing of naive target neurons was monitored by TUNEL after 48 h and normalized to the signal from control neurons (as in Fig. 3). All values are mean ± SEM for the number of separate cell cultures indicated. Significant differences are shown between DCG-IV-treated and untreated microglia (†p < 0.05), and for DCG-IV-treatment with or without a drug (*p < 0.05). B, The mGluRII agonist evokes TNF-α (but not nitric oxide) production from microglia. Microglia were treated for 24 h with 250 nm DCG-IV, and then TNF-α released into the supernatant was measured by ELISA (*p < 0.05), and NO (nitrite) production was monitored with the Griess assay.
OGD-stressed neurons promote microglia mGluRII-dependent neurotoxic activity through TNF-α and NF-κB
Above, we showed that microglia are activated by OGD-SNs to produce TNF-α (Fig. 2), and that engaging microglial mGluRII increases their neurotoxic behavior and TNF-α production (Fig. 4). It was then important to determine whether the same molecules are involved in neuron killing by microglia in the penumbra model and to identify the pathways used. In order for microglia mGluRII to be involved in their activation by OGD-stressed neurons, it is essential that sufficient glutamate be produced; i.e., the affinity (Kd) of group II mGluRs is 5.9 μm for mGluR2 and 0.34 μm for mGluR3 (Pin and Acher, 2002). When cultured neurons were exposed to OGD for 60 min, they released significantly more glutamate than untreated neurons (Fig. 5A) (p < 0.05), and the concentration in the supernatant rose from 7 ± 2 to 15 ± 2 μm, an appropriate range for increasing mGluRII activation. We confirmed that the microglial mGluRII was involved in the penumbra model, because subsequent killing of naive target neurons was markedly reduced when the microglia were treated with mGluRII antagonists. That is, Figure 5B shows that TUNEL increased in target neurons exposed to microglia that had been pretreated with OGD-stressed neurons (p < 0.05), and this excess killing was reduced 72–80% by EGLU or LY341495 (p < 0.05). Because both microglia and neurons were exposed to the drugs, an important finding was that neuronal glutamate release was not affected by EGLU, LY341495, or its solvent, DMSO (Fig. 5A).
Figure 5.

OGD-SNs increase microglia neurotoxic activity via mGluRII: involvement of NF-κB activation and TNF-α release. A, Neuronal glutamate release was stimulated by OGD and unaffected by mGluRII inhibitors. Glutamate was measured in the supernatant of control neurons or neurons that had been exposed to 60 min of OGD (*p < 0.05), with or without an mGluRII antagonist, i.e., 250 μm EGLU or 500 nm LY341495. Note that LY341495 was compared with its solvent, DMSO. Values were normalized to the amount of cell protein in each well and plotted as mean ± SEM for the number of separate cell cultures indicated. B–D, Microglia on upper inserts of Transwell chambers were exposed to OGD-SNs for 24 h (B, C) or to glutamate for 24 h (15 μm; D) with or without drugs and then washed and placed above naive target neurons in the lower chamber for 48 h, after which neuron death was assessed by TUNEL. B, Evidence for involvement of microglia mGluRII in neuron killing. OGD-SNs increased microglia neurotoxic activity compared with untreated microglia (†p < 0.05), and this was inhibited by the mGluRII antagonists, 250 μm EGLU or 500 nm LY341495 (*p < 0.05). C, TNF-α contributes to neuron killing. OGD-SNs increased microglia neurotoxic activity (†p < 0.05), and this was inhibited by the soluble TNF-α receptor 1 (50 ng/ml sTNFR1; *p < 0.05). There was no effect of the peroxynitrite scavenger (2 μm FeTmPyP) or the iNOS inhibitor (100 μm SMT). D, Glutamate stimulates microglial neurotoxic activity through mGluRII and NF-κB. Significant differences are shown for untreated versus glutamate-treated microglia (**p < 0.01) and glutamate-treated microglia with or without the mGluRII antagonist (250 μm EGLU) or the IκB inhibitor peptide (50 ng/ml) (*p < 0.05). Note that the p38 MAPK inhibitor (2 μm SB 202190) is compared with its vehicle, DMSO.
Because we had observed an NF-κB-dependent activation of microglia and TNF-α production (Fig. 2), we examined whether these two molecules were involved in the glutamate- and mGluRII-dependent activation of microglia by OGD-stressed neurons, and consequent killing of naive neurons. As before, microglia that were activated by OGD-stressed neurons killed naive neurons (p < 0.05) (Fig. 5C). This increased killing was prevented by adding sTNFR1 (p < 0.05) at approximately a 50-fold higher concentration than the TNF-α produced by microglia (Fig. 2B). In contrast, neither the peroxynitrite scavenger (FeTmPyP) nor the iNOS inhibitor (SMT) affected neuron killing. Our observation that caspase-8 activation increased ∼40% in the target neurons (Fig. 3D) (p < 0.05) is entirely consistent with neuronal damage through the low-affinity TNF-α receptor, p55/TNFR1 (Feuerstein et al., 1998; Gorman et al., 1998).
Finally, specific inhibitors were used to examine potential roles of microglial NF-κB and p38 MAPK in neuron killing in this penumbra model. To avoid possible effects of these inhibitors on neurons, microglia were directly activated with 15 μm glutamate (Fig. 5D), the same concentration as measured in the neuron supernatant after 60 min of exposure to OGD (see above). As expected, glutamate activated the microglia through mGluRII and significantly increased their neurotoxic activity; i.e., the prevalence of TUNEL-positive target neurons increased to 23 ± 1% compared with 12 ± 1% after exposure to control (untreated) microglia (p < 0.01). This glutamate-dependent killing was prevented by blocking the microglial mGluRII with EGLU (p < 0.05) or by inhibiting NF-κB activation, using a peptide that prevents dissociation and activation of the NF-κB complex (p < 0.05). In contrast, the p38 MAPK inhibitor had no effect. Note that the target neurons were not exposed to any of the inhibitors.
Together, these results point to involvement of glutamate from OGD-stressed neurons in activating microglial mGluRII receptors and increasing their neurotoxic functions through activation of NF-κB and production of TNF-α.
Discussion
By developing a new in vitro model of the ischemic penumbra, which differs from the infarct core, we were able to dissect mechanisms underlying inflammation-mediated neurotoxicity with a degree of precision not previously possible. Figure 6 summarizes our in vitro results and relates them to postulated events, including microglial morphological changes as they progressively activate after a stroke in vivo (Fig. 6B). Microglia are well positioned to respond to chemical cues because they are in close proximity to neurons (Fig. 6A) and can rapidly extend their processes toward damaged neurons (Davalos et al., 2005; Nimmerjahn et al., 2005). After ischemia, these chemicals include glutamate, released from damaged neurons and by reversal of glutamate transporters in astrocytes. We found that microglial activation was caused by glutamate released from the OGD-stressed neuron/astrocyte cultures. Glutamate activated mGluRIIs on the “resting” microglia, which activated the transcription factor, NF-κB. Subsequently, the activated microglia released TNF-α, which killed naive neurons by an apoptotic pathway that was prevented by inhibiting caspase-3. Direct activation of microglial mGluRII did not stimulate nitric oxide production; thus, it is not surprising that the neurotoxicity did not involve iNOS, nitric oxide, or peroxynitrite. The same level of neurotoxicity was evident whether microglia were activated by OGD-stressed neurons, or the same concentration of glutamate they released, or by an mGluRII agonist. This is apparently the first study showing that microglia become neurotoxic after being exposed to OGD-stressed neurons, to elucidate the molecular events occurring in the microglia, or to identify the neurotoxic moiety.
Figure 6.
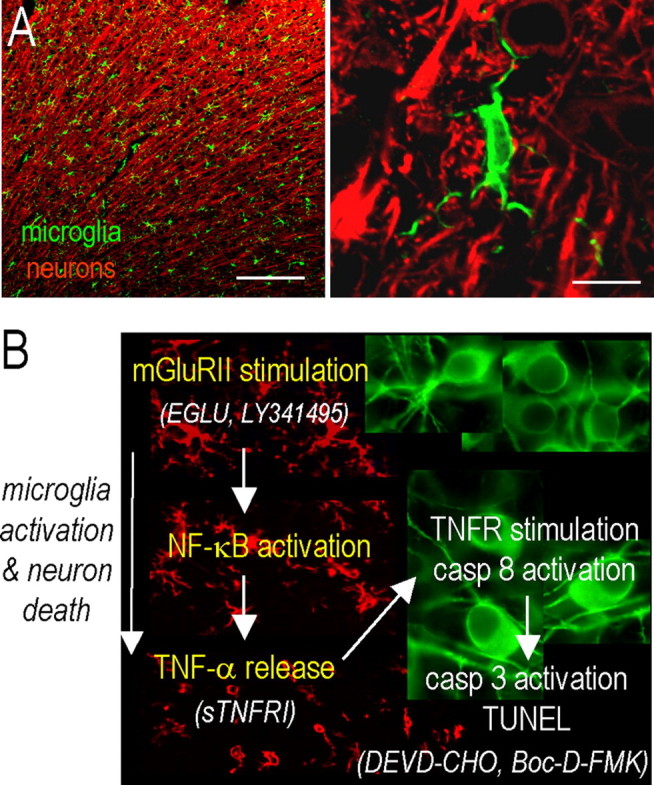
Summary of findings and model of the ischemic penumbra. Neurons subjected to OGD in the infarct core die by apoptosis and necrosis. Transient ischemia was modeled by exposing cortical neuron/astrocyte cultures to OGD for 60 min and then to a normal solution to represent reperfusion. By 24 h later, they had secreted substantial amounts of glutamate but were not yet TUNEL positive (∼40% became TUNEL positive by 48 h). A, Representative confocal micrographs of the healthy rat cortex, showing the dense network of ramified microglia whose processes are in close apposition to the neurons. Scale bars: left, 200 μm; right, 10 μm. A higher-magnification image shows a typical microglial cell with processes abutting the neuronal cell body (near top) and wrapping around the axonal processes. Microglia are labeled with a rabbit polyclonal antibody against a membrane protein, Iba1 (Wako, Osaka, Japan), and an Alexa 488-conjugated goat anti-rabbit secondary antibody (green; Jackson ImmunoResearch Laboratories). Neurons are labeled with a mouse monoclonal MAP2 antibody (Millipore) and Cy3-conjugated goat anti-mouse secondary antibody (red; Jackson ImmunoResearch Laboratories). B, After a stroke, an inflammatory response develops over time, microglia become activated (their processes retract and they migrate to the damage site), and neuronal damage propagates into the surrounding tissue, the ischemic “penumbra.” Note that the colors are reversed from A; i.e., microglia (red) are labeled with Iba1 antibody and a Cy3-conjugated secondary antibody, whereas rat cortical neurons (green) are labeled with MAP2 and a FITC-conjugated secondary antibody. In these representative confocal micrographs from the rat brain after a stroke, microglia are ramified and densely distributed in the uninjured contralateral hemisphere (top image), but retract their processes and eventually become rounded up as they progressively activate on the damaged ipsilateral side (bottom two images). In this study, using an in vitro model of the stroke penumbra, we identified several events. (1) Glutamate, released by the OGD-stressed neuron-astrocyte cultures, reached sufficient concentrations to stimulate microglial mGluRIIs; this stimulation was blocked by the selective mGluRII antagonists, EGLU or LY341495. (2) Microglial activation was accompanied by activation of NF-κB, not p38 MAPK. (3) Activated microglia produced and released TNF-α. (4) TNF-α interacted with the target neurons, activating their p55/TNFR1 receptors, as judged by activation of caspase-8 (Gorman et al., 1998). Neurotoxicity was reduced by scavenging TNF-α with soluble TNF-α receptor 1 (sTNFR1). (5) The target neurons died by apoptosis, as judged by caspase-3 activation and TUNEL. (6) Caspase 3 activation was required for excess neuron killing, because it was prevented by the caspase-3 inhibitor, DEVD-CHO, and by the broad-spectrum caspase inhibitor Boc-d-FMK.
Neurons are extremely susceptible to changes in oxygen or blood flow, and clinical studies have shown impaired neuron function after as little as 10 min of ischemia (Oechmichen and Meissner, 2006). In animal models of ischemic stroke, neurons in the core rapidly become committed to die, because of changes in ion homeostasis and mitochondrial dysfunction (Beilharz et al., 1995); thus, it is not surprising that therapeutic strategies to rescue these neurons have failed. As a consequence, attention has increasingly focused on the surrounding penumbra, where neuron death can extend for days to weeks (Zheng et al., 2003). To develop effective therapies, it is crucial to understand the neurotoxic mechanisms occurring in the penumbra. In such studies, a key question is how well the in vitro models represent the penumbra. Neuron necrosis in the core is thought to trigger microglia activation (Gehrmann et al., 1992), with increased MHC (major histocompatibility complex) class II expression and production of proinflammatory cytokines (Lehrmann et al., 1997; Block et al., 2000). Activated microglia can increase neuron death in animal models of ischemia (Block et al., 2000) and propagate neuron death in the penumbra (Sharp et al., 2000). To our knowledge, no previous in vitro studies have mimicked events thought to occur in the penumbra. Instead, in vitro studies of neurotoxicity have often used coculture models, in which microglia stimulated with exogenous chemical activators (e.g., lipopolysaccharide) are used to kill neurons (Xie et al., 2002; Fordyce et al., 2005). One study showed that microglia treated with neuron-conditioned medium became more activated, changed their morphology, and increased production of reactive oxygen species (Sudo et al., 1998). Microglia activation after neuron damage has been examined in the more complex organotypic brain slice cultures (Pringle et al., 1997; Skibo et al., 2000), and some of these studies support a protective role for microglia after OGD, through production of neurotrophic factors (Neumann et al., 2006). Overall, the protective versus detrimental effects of microglia after an ischemic stroke remain controversial. We designed the present penumbra model to elucidate communication mechanisms (particularly through soluble factors) between microglia, neurons, and astrocytes. It should prove useful for testing potential therapeutic approaches in an in vitro model that is much more relevant to the ischemic penumbra than previous models. An obvious limitation, compared with in vivo models, is that it does not account for the influence of infiltrating immune cells or the blood–brain barrier.
After a stroke, glutamate (Sharp et al., 2000; Bruhn et al., 2003) and other soluble factors released by neurons in the infarct core can diffuse to nearby cells and propagate the damage (for review, see Dirnagl et al., 1999). Astrocytes, which normally help remove glutamate through uptake transporters, can potentially exacerbate the stroke damage when the transporters reverse to release glutamate (Kosugi and Kawahara 2006). Glutamate apparently activates microglia in several CNS diseases (Matute et al., 2006) and in cerebrocortical slice cultures (De Cristobal et al., 2002; Fujimoto et al., 2004), consistent with in vitro studies showing that direct activation of microglial mGluRIIs increases their neurotoxic behavior (Taylor et al., 2002, 2005; present study). Here, we showed that OGD-stressed neurons activated the neurotoxic functions of microglia through their group II mGluRs, and we demonstrated neuroprotection by mGluRII antagonists that block mGluR2 and mGluR3. Based on the concentrations of glutamate produced by naive (∼7 μm) and OGD-stressed neurons (∼15 μm), these results are consistent with the lower-affinity mGluR2 receptor being the target. An advantage of the present experimental paradigm is that the role of mGluRs in microglia can be distinguished from other cells. In vivo studies using direct, chemical activation of group II mGluRs have produced conflicting results. After focal ischemia, in which microglia are thought to orchestrate damage in the penumbra (Block et al., 2000), activating group II mGluRs had no effect (Lam et al., 1998; Bond et al., 1999), whereas, this approach was neuroprotective after traumatic brain injury (Allen et al., 1999; Kingston et al., 1999) or global ischemia (Bond et al., 1999). Some possible explanations include the increased tissue complexity and comparisons between quite different damage models; that only global effects were studied, rather than site-specific effects in the penumbra; and possibly that agonist concentrations were used that activated different subtypes of group II mGluRs (i.e., mGluR2 vs GluR3), and on other cell types.
Specific molecules thought to trigger neuron death in the penumbra after a stroke include TNF-α, IL-1β, and reactive oxygen and nitrogen species (Sharp et al., 2000). Although studies of a single molecule cannot rule out involvement of multiple pathways, high TNF-α concentrations cause neuron apoptosis through a caspase-8-mediated pathway (Feuerstein et al., 1998; Gorman et al., 1998). TNF-α production increases after ischemia in vivo (Gabriel et al., 1999), and depleting TNF-α receptors can reduce neuron damage (Sriram et al., 2006). In the present in vitro penumbra model, we found that microglia activated by OGD-stressed neurons produced excess TNF-α, which contributed to killing naive, healthy neurons. In principle, TNF-α can act in an autocrine manner on microglia (Kuno et al., 2005; Takeuchi et al., 2006) and a paracrine manner on neurons; however, increased caspase-8 activity in the target neurons demonstrates involvement of neuronal TNF-α receptors (Feuerstein et al., 1998; Gorman et al., 1998). Our observation that microglia activated by the mGluRII agonist, DCG-IV, secreted TNF-α (but not nitric oxide), and that TNF-α was required for neuron killing, is consistent with earlier studies using the same stimulus (Taylor et al., 2002, 2005). Some studies found that inhibiting iNOS was neuroprotective (Galea et al., 1998; Zhang and Stanimirovic, 2002) and reduced infarct size in vivo after focal ischemia (Zhu et al., 2002). We found that iNOS and peroxynitrite were not required for neurotoxicity, an important observation because this model is more relevant to stroke than previous in vitro models of microglia activation. Clearly, the stimulus used can affect the neurotoxic molecules produced; i.e., LPS-activated microglia produce abundant nitric oxide, and peroxynitrite contributes to their neurotoxic behavior (Fordyce et al., 2005; Kaushal et al., 2007).
Two primary signaling molecules in microglia, p38 MAPK and NF-κB, and the consequent production of proinflammatory molecules (Pawate et al., 2004) have been linked to neuron death in vitro and in vivo [for review, see Zhang and Stanimirovic (2002) and Pawate et al. (2004)]. We found that OGD-stressed neurons evoke NF-κB activation in microglia, through a glutamate- and mGluRII-dependent mechanism, and that the resulting neurotoxic activity of the microglia requires NF-κB activation. p38 MAPK was neither activated nor required. This is the first study linking microglial mGluRII with NF-κB signaling, and is especially important because NF-κB stimulates microglial production of TNF-α (Ginis et al., 2000). Of note, the penumbra model differs from microglia stimulation by LPS, which activates both p38 MAPK and NF-κB (Zhang and Stanimirovic, 2002; Kaushal et al., 2007); however, LPS, from Gram-negative bacteria, is clearly a poor model of ischemic stroke. More relevant is that p38 MAPK and NF-κB activation have been observed in the penumbra in vivo after focal ischemia (Seegers et al., 2000; Krupinski et al., 2003), their inhibition is protective (Zhang and Stanimirovic, 2002), and both have been implicated in inflammation-induced neuron injury (Gabriel et al., 1999; Schneider et al., 1999; Sharp et al., 2000; Ferrer et al., 2003). Despite the likelihood of additional mechanisms in vivo, the present in vitro study supports targeting microglial group II metabotropic receptors, TNF-α overproduction, and NF-κB for reducing inflammation-mediated neurotoxicity after ischemic stroke.
Footnotes
This work was supported by operating grants to L.C.S. from the Canadian Institutes for Health Research (MT-13657) and the Heart and Stroke Foundation of Canada (T4670, T5782). We are grateful for valuable comments on this manuscript to Drs. J. Eubanks, L. Mills, and L. Ohana and to J. Wasserman and V. Kena-Cohen. We are also grateful to Jason Wasserman for the confocal images of microglia and neurons in the rat brain in vivo.
References
- Allen JW, Ivanova SA, Fan L, Espey MG, Basile AS, Faden AI. Group II metabotropic glutamate receptor activation attenuates traumatic neuronal injury and improves neurological recovery after traumatic brain injury. J Pharmacol Exp Ther. 1999;290:112–120. [PubMed] [Google Scholar]
- Barone FC, Feuerstein GZ. Inflammatory mediators and stroke: new opportunities for novel therapeutics. J Cereb Blood Flow Metab. 1999;19:819–834. doi: 10.1097/00004647-199908000-00001. [DOI] [PubMed] [Google Scholar]
- Beilharz EJ, Williams CE, Dragunow M, Sirimanne ES, Gluckman PD. Mechanisms of delayed cell death following hypoxic-ischemic injury in the immature rat: evidence for apoptosis during selective neuronal loss. Brain Res Mol Brain Res. 1995;29:1–14. doi: 10.1016/0169-328x(94)00217-3. [DOI] [PubMed] [Google Scholar]
- Block F, Peters M, Nolden-Koch M. Expression of IL-6 in the ischemic penumbra. NeuroReport. 2000;11:963–967. doi: 10.1097/00001756-200004070-00013. [DOI] [PubMed] [Google Scholar]
- Bond A, Ragumoorthy N, Monn JA, Hicks CA, Ward MA, Lodge D, O'Neill MJ. LY379268, a potent and selective group II metabotropic glutamate receptor agonist, is neuroprotective in gerbil global, but not focal, cerebral ischaemia. Neurosci Lett. 1999;273:191–194. doi: 10.1016/s0304-3940(99)00663-1. [DOI] [PubMed] [Google Scholar]
- Bramlett HM, Dietrich WD. Pathophysiology of cerebral ischemia and brain trauma: similarities and differences. J Cereb Blood Flow Metab. 2004;24:133–150. doi: 10.1097/01.WCB.0000111614.19196.04. [DOI] [PubMed] [Google Scholar]
- Bruhn T, Christensen T, Diemer NH. Uptake of glutamate is impaired in the cortical penumbra of the rat following middle cerebral artery occlusion: an in vivo microdialysis extraction study. J Neurosci Res. 2003;71:551–558. doi: 10.1002/jnr.10492. [DOI] [PubMed] [Google Scholar]
- Cho S, Liu D, Fairman D, Li P, Jenkins L, McGonigle P, Wood A. Spatiotemporal evidence of apoptosis-mediated ischemic injury in organotypic hippocampal slice cultures. Neurochem Int. 2004;45:117–127. doi: 10.1016/j.neuint.2003.11.012. [DOI] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- De Cristobal J, Cardenas A, Lizasoain I, Leza JC, Fernandez-Tome P, Lorenzo P, Moro MA. Inhibition of glutamate release via recovery of ATP levels accounts for a neuroprotective effect of aspirin in rat cortical neurons exposed to oxygen-glucose deprivation. Stroke. 2002;33:261–267. doi: 10.1161/hs0102.101299. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Du Y, Bales KR, Dodel RC, Hamilton-Byrd E, Horn JW, Czilli DL, Simmons LK, Ni B, Paul SM. Activation of a caspase 3-related cysteine protease is required for glutamate-mediated apoptosis of cultured cerebellar granule neurons. Proc Natl Acad Sci USA. 1997;94:11657–11662. doi: 10.1073/pnas.94.21.11657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Friguls B, Dalfo E, Planas AM. Early modifications in the expression of mitogen-activated protein kinase (MAPK/ERK), stress-activated kinases SAPK/JNK and p38, and their phosphorylated substrates following focal cerebral ischemia. Acta Neuropathol (Berl) 2003;105:425–437. doi: 10.1007/s00401-002-0661-2. [DOI] [PubMed] [Google Scholar]
- Feuerstein G, Wang X, Barone FC. Cytokines in brain ischemia—the role of TNF-α. Cell Mol Neurobiol. 1998;18:695–701. doi: 10.1023/A:1020690004094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fordyce CB, Jagasia R, Zhu X, Schlichter LC. Microglia Kv1.3 channels contribute to their ability to kill neurons. J Neurosci. 2005;25:7139–7149. doi: 10.1523/JNEUROSCI.1251-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto S, Katsuki H, Kume T, Kaneko S, Akaike A. Mechanisms of oxygen glucose deprivation-induced glutamate release from cerebrocortical slice cultures. Neurosci Res. 2004;50:179–187. doi: 10.1016/j.neures.2004.06.013. [DOI] [PubMed] [Google Scholar]
- Gabriel C, Justicia C, Camins A, Planas AM. Activation of nuclear factor-κB in the rat brain after transient focal ischemia. Brain Res Mol Brain Res. 1999;65:61–69. doi: 10.1016/s0169-328x(98)00330-1. [DOI] [PubMed] [Google Scholar]
- Galea E, Golanov EV, Feinstein DL, Kobylarz KA, Glickstein SB, Reis DJ. Cerebellar stimulation reduces inducible nitric oxide synthase expression and protects brain from ischemia. Am J Physiol. 1998;274:H2035–H2045. doi: 10.1152/ajpheart.1998.274.6.H2035. [DOI] [PubMed] [Google Scholar]
- Gehrmann J, Bonnekoh P, Miyazawa T, Hossmann KA, Kreutzberg GW. Immunocytochemical study of an early microglial activation in ischemia. J Cereb Blood Flow Metab. 1992;12:257–269. doi: 10.1038/jcbfm.1992.36. [DOI] [PubMed] [Google Scholar]
- Geurts JJ, Wolswijk G, Bo L, van der Valk P, Polman CH, Troost D, Aronica E. Altered expression patterns of group I and II metabotropic glutamate receptors in multiple sclerosis. Brain. 2003;126:1755–1766. doi: 10.1093/brain/awg179. [DOI] [PubMed] [Google Scholar]
- Ginis I, Hallenbeck JM, Liu J, Spatz M, Jaiswal R, Shohami E. Tumor necrosis factor and reactive oxygen species cooperative cytotoxicity is mediated via inhibition of NF-κB. Mol Med. 2000;6:1028–1041. [PMC free article] [PubMed] [Google Scholar]
- Gorman AM, Orrenius S, Ceccatelli S. Apoptosis in neuronal cells: role of caspases. NeuroReport. 1998;9:R49–R55. doi: 10.1097/00001756-199807130-00001. [DOI] [PubMed] [Google Scholar]
- Hayashi H, Campenot RB, Vance DE, Vance JE. Apolipoprotein E-containing lipoproteins protect neurons from apoptosis via a signaling pathway involving low-density lipoprotein receptor-related protein-1. J Neurosci. 2007;27:1933–1941. doi: 10.1523/JNEUROSCI.5471-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan WB, Julius D. The P2Y(12) receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci. 2006;9:1512–1519. doi: 10.1038/nn1805. [DOI] [PubMed] [Google Scholar]
- Iijima T, Mishima T, Akagawa K, Iwao Y. Mitochondrial hyperpolarization after transient oxygen-glucose deprivation and subsequent apoptosis in cultured rat hippocampal neurons. Brain Res. 2003;993:140–145. doi: 10.1016/j.brainres.2003.09.041. [DOI] [PubMed] [Google Scholar]
- Jiang X, Mu D, Manabat C, Koshy AA, Christen S, Tauber MG, Vexler ZS, Ferriero DM. Differential vulnerability of immature murine neurons to oxygen-glucose deprivation. Exp Neurol. 2004;190:224–232. doi: 10.1016/j.expneurol.2004.07.010. [DOI] [PubMed] [Google Scholar]
- Kaushal V, Koeberle PD, Wang Y, Schlichter LC. The Ca2+-activated K+ channel KCNN4/KCa3.1 contributes to microglia activation and nitric oxide-dependent neurodegeneration. J Neurosci. 2007;27:234–244. doi: 10.1523/JNEUROSCI.3593-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingston AE, O'Neill MJ, Bond A, Bruno V, Battaglia G, Nicoletti F, Harris JR, Clark BP, Monn JA, Lodge D, Schoepp DD. Neuroprotective actions of novel and potent ligands of group I and group II metabotropic glutamate receptors. Ann NY Acad Sci. 1999;890:438–449. doi: 10.1111/j.1749-6632.1999.tb08022.x. [DOI] [PubMed] [Google Scholar]
- Kosugi T, Kawahara K. Reversed astrocytic GLT-1 during ischemia is crucial to excitotoxic death of neurons, but contributes to the survival of astrocytes themselves. Neurochem Res. 2006;31:933–943. doi: 10.1007/s11064-006-9099-6. [DOI] [PubMed] [Google Scholar]
- Krupinski J, Slevin M, Marti E, Catena E, Rubio F, Gaffney J. Time-course phosphorylation of the mitogen activated protein (MAP) kinase group of signalling proteins and related molecules following middle cerebral artery occlusion (MCAO) in rats. Neuropathol Appl Neurobiol. 2003;29:144–158. doi: 10.1046/j.1365-2990.2003.00454.x. [DOI] [PubMed] [Google Scholar]
- Kuno R, Wang J, Kawanokuchi J, Takeuchi H, Mizuno T, Suzumura A. Autocrine activation of microglia by tumor necrosis factor-α. J Neuroimmunol. 2005;162:89–96. doi: 10.1016/j.jneuroim.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Labat-Moleur F, Guillermet C, Lorimier P, Robert C, Lantuejoul S, Brambilla E, Negoescu A. TUNEL apoptotic cell detection in tissue sections: critical evaluation and improvement critical evaluation and improvement. J Histochem Cytochem. 1998;46:327–334. doi: 10.1177/002215549804600306. [DOI] [PubMed] [Google Scholar]
- Lam AG, Soriano MA, Monn JA, Schoepp DD, Lodge D, McCulloch J. Effects of the selective metabotropic glutamate agonist LY354740 in a rat model of permanent ischaemia. Neurosci Lett. 1998;254:121–123. doi: 10.1016/s0304-3940(98)00651-x. [DOI] [PubMed] [Google Scholar]
- Lehrmann E, Christensen T, Zimmer J, Diemer NH, Finsen B. Microglial and macrophage reactions mark progressive changes and define the penumbra in the rat neocortex and striatum after transient middle cerebral artery occlusion. J Comp Neurol. 1997;386:461–476. [PubMed] [Google Scholar]
- Matute C, Domercq M, Sanchez-Gomez MV. Glutamate-mediated glial injury: mechanisms and clinical importance. Glia. 2006;53:212–224. doi: 10.1002/glia.20275. [DOI] [PubMed] [Google Scholar]
- Neumann J, Gunzer M, Gutzeit HO, Ullrich O, Reymann KG, Dinkel K. Microglia provide neuroprotection after ischemia. FASEB J. 2006;20:714–716. doi: 10.1096/fj.05-4882fje. [DOI] [PubMed] [Google Scholar]
- Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, Munday NA, Raju SM, Smulson ME, Yamin T-T, Yu VL, Miller DK. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- Nikodemova M, Duncan ID, Watters JJ. Minocycline exerts inhibitory effects on multiple mitogen-activated protein kinases and IκBα degradation in a stimulus-specific manner in microglia. J Neurochem. 2006;96:314–323. doi: 10.1111/j.1471-4159.2005.03520.x. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Oechmichen M, Meissner C. Cerebral hypoxia and ischemia: the forensic point of view: a review. J Forensic Sci. 2006;51:880–887. doi: 10.1111/j.1556-4029.2006.00174.x. [DOI] [PubMed] [Google Scholar]
- Pawate S, Shen Q, Fan F, Bhat NR. Redox regulation of glial inflammatory response to lipopolysaccharide and interferongamma. J Neurosci Res. 2004;77:540–551. doi: 10.1002/jnr.20180. [DOI] [PubMed] [Google Scholar]
- Pin JP, Acher F. The metabotropic glutamate receptors: structure, activation mechanism and pharmacology. Curr Drug Targets CNS Neurol Disord. 2002;1:297–317. doi: 10.2174/1568007023339328. [DOI] [PubMed] [Google Scholar]
- Pringle AK, Angunawela R, Wilde GJ, Mepham JA, Sundstrom LE, Iannotti F. Induction of 72 kDa heat-shock protein following sub-lethal oxygen deprivation in organotypic hippocampal slice cultures. Neuropathol Appl Neurobiol. 1997;23:289–298. [PubMed] [Google Scholar]
- Schilling M, Besselmann M, Leonhard C, Mueller M, Ringelstein EB, Kiefer R. Microglial activation precedes and predominates over macrophage infiltration in transient focal cerebral ischemia: a study in green fluorescent protein transgenic bone marrow chimeric mice. Exp Neurol. 2003;183:25–33. doi: 10.1016/s0014-4886(03)00082-7. [DOI] [PubMed] [Google Scholar]
- Schneider A, Martin-Villalba A, Weih F, Vogel J, Wirth T, Schwaninger M. NF-κB is activated and promotes cell death in focal cerebral ischemia. Nat Med. 1999;5:554–559. doi: 10.1038/8432. [DOI] [PubMed] [Google Scholar]
- Seegers H, Grillon E, Trioullier Y, Vath A, Verna JM, Blum D. NF-κB activation in permanent intraluminal focal cerebral ischemia in the rat. Neurosci Lett. 2000;288:241–245. doi: 10.1016/s0304-3940(00)01245-3. [DOI] [PubMed] [Google Scholar]
- Sharp FR, Lu A, Tang Y, Millhorn DE. Multiple molecular penumbras after focal cerebral ischemia. J Cereb Blood Flow Metab. 2000;20:1011–1032. doi: 10.1097/00004647-200007000-00001. [DOI] [PubMed] [Google Scholar]
- Siao CJ, Tsirka SE. Extracellular proteases and neuronal cell death. Cell Mol Biol (Noisy-le-grand) 2002;48:151–161. [PubMed] [Google Scholar]
- Skibo GG, Nikonenko IR, Savchenko VL, McKanna JA. Microglia in organotypic hippocampal slice culture and effects of hypoxia: ultrastructure and lipocortin-1 immunoreactivity. Neuroscience. 2000;96:427–438. doi: 10.1016/s0306-4522(99)00562-x. [DOI] [PubMed] [Google Scholar]
- Sriram K, Matheson JM, Benkovic SA, Miller DB, Luster MI, O'Callaghan JP. Deficiency of TNF receptors suppresses microglial activation and alters the susceptibility of brain regions to MPTP-induced neurotoxicity: role of TNF-α. FASEB J. 2006;20:670–682. doi: 10.1096/fj.05-5106com. [DOI] [PubMed] [Google Scholar]
- Sudo S, Tanaka J, Toku K, Desaki J, Matsuda S, Arai T, Sakanaka M, Maeda N. Neurons induce the activation of microglial cells in vitro. Exp Neurol. 1998;154:499–510. doi: 10.1006/exnr.1998.6911. [DOI] [PubMed] [Google Scholar]
- Takeuchi H, Jin S, Wang J, Zhang G, Kawanokuchi J, Kuno R, Sonobe Y, Mizuno T, Suzumura A. Tumor necrosis factor-α induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem. 2006;281:21362–21368. doi: 10.1074/jbc.M600504200. [DOI] [PubMed] [Google Scholar]
- Taylor DL, Diemel LT, Cuzner ML, Pocock JM. Activation of group II metabotropic glutamate receptors underlies microglial reactivity and neurotoxicity following stimulation with chromogranin A, a peptide up-regulated in Alzheimer's disease. J Neurochem. 2002;82:1179–1191. doi: 10.1046/j.1471-4159.2002.01062.x. [DOI] [PubMed] [Google Scholar]
- Taylor DL, Jones F, Kubota ES, Pocock JM. Stimulation of microglial metabotropic glutamate receptor mGlu2 triggers tumor necrosis factor α-induced neurotoxicity in concert with microglial-derived Fas ligand. J Neurosci. 2005;25:2952–2964. doi: 10.1523/JNEUROSCI.4456-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- Volonte C, Amadio S, Cavaliere F, D'Ambrosi N, Vacca F, Bernardi G. Extracellular ATP and neurodegeneration. Curr Drug Targets CNS Neurol Disord. 2003;2:403–412. doi: 10.2174/1568007033482643. [DOI] [PubMed] [Google Scholar]
- Wood PL. Microglia as a unique cellular target in the treatment of stroke: potential neurotoxic mediators produced by activated microglia. Neurol Res. 1995;17:242–248. doi: 10.1080/01616412.1995.11740321. [DOI] [PubMed] [Google Scholar]
- Xie Z, Wei M, Morgan TE, Fabrizio P, Han D, Finch CE, Longo VD. Peroxynitrite mediates neurotoxicity of amyloid β-peptide 1–42 and lipopolysaccharide-activated microglia. J Neurosci. 2002;22:3484–3492. doi: 10.1523/JNEUROSCI.22-09-03484.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Xu RX, Liu BS, Jiang XD, Huang T, Ding LS, Yuan J. Time window characteristics of cultured rat hippocampal neurons subjected to ischemia and reperfusion. Chin J Traumatol. 2005;8:179–182. [PubMed] [Google Scholar]
- Zhang W, Stanimirovic D. Current and future therapeutic strategies to target inflammation in stroke. Curr Drug Targets Inflamm Allergy. 2002;1:151–166. doi: 10.2174/1568010023344689. [DOI] [PubMed] [Google Scholar]
- Zheng Z, Zhao H, Steinberg GK, Yenari MA. Cellular and molecular events underlying ischemia-induced neuronal apoptosis. Drug News Perspect. 2003;16:497–503. doi: 10.1358/dnp.2003.16.8.829348. [DOI] [PubMed] [Google Scholar]
- Zhu DY, Deng Q, Yao HH, Wang DC, Deng Y, Liu GQ. Inducible nitric oxide synthase expression in the ischemic core and penumbra after transient focal cerebral ischemia in mice. Life Sci. 2002;71:1985–1996. doi: 10.1016/s0024-3205(02)01970-7. [DOI] [PubMed] [Google Scholar]