

Abstract
Calcium-independent group VIA phospholipase A2 (iPLA2β) is considered to play a role in signal transduction and maintenance of homeostasis or remodeling of membrane phospholipids. A role of iPLA2β has been suggested in various physiological and pathological processes, including immunity, chemotaxis, and cell death, but the details remain unclear. Accordingly, we investigated mice with targeted disruption of the iPLA2β gene. iPLA2β−/− mice developed normally and grew to maturity, but all showed evidence of severe motor dysfunction, including a hindlimb clasping reflex during tail suspension, abnormal gait, and poor performance in the hanging wire grip test. Neuropathological examination of the nervous system revealed widespread degeneration of axons and/or synapses, accompanied by the presence of numerous spheroids (swollen axons) and vacuoles. These findings provide evidence that impairment of iPLA2β causes neuroaxonal degeneration, and indicate that the iPLA2β−/− mouse is an appropriate animal model of human neurodegenerative diseases associated with mutations of the iPLA2β gene, such as infantile neuroaxonal dystrophy and neurodegeneration with brain iron accumulation.
Keywords: phospholipase, neurodegeneration, neuroaxonal dystrophy, knock-out mouse, iPLA2, spheroid
Introduction
The phospholipases A2 (PLA2) comprise a family of esterases that hydrolyze the sn-2 ester bond in phospholipids to yield free fatty acids and lysophospholipids. A number of PLA2 isotypes have been identified in mammals, and depending on their subcellular localization and enzymatic properties, these are divided into three major subfamilies, which are secretory PLA2 (sPLA2), cytosolic Ca2+-dependent PLA2 (cPLA2), and Ca2+-independent PLA2 (iPLA2) (Six and Dennis, 2000; Ma and Turk, 2001). The sPLA2s are extracellular enzymes with a low molecular mass (∼14 kDa) that require millimolar concentrations of Ca2+ for activation. sPLA2s are thought to be potent mediators of inflammation and also show antibacterial activity. In contrast, the cPLA2s are intracellular enzymes that specifically target arachidonic acid at the sn-2 position of phospholipids. Their activity is regulated by submicromolar levels of Ca2+, and these enzymes are believed to play a pivotal role in the production of arachidonic acid metabolites, such as eicosanoids. The iPLA2s do not require Ca2+ for their catalytic activity and are suggested to be involved in remodeling of membrane phospholipids as well as in various cellular signaling processes. In mammals, two iPLA2 genes (iPLA2β and iPLA2γ) have been cloned (Tang et al., 1997; Mancuso et al., 2000).
The physiological role of the PLA2s in the nervous system remains largely unknown in mammals. To our knowledge, there have been no previous reports about neurological dysfunction in PLA2 knock-out mice. The iPLA2β gene is expressed in all regions of the mammalian brain and iPLA2 is the dominant PLA2 activity in rat brain cytosol (Yang et al., 1999; Balboa et al., 2002). Accumulated evidence suggests that changes of phospholipids may be to associated with cell death, including apoptosis and necrosis (caspase-independent cell death) (Shinzawa and Tsujimoto, 2003), and may cause many neurodegenerative diseases, including cerebral infarction and Alzheimer disease (Farooqui et al., 2004). Moreover, it was very recently reported that iPLA2β gene mutations occur in several human neurodegenerative disorders, such as infantile neuraxonal dystrophy (INAD) and neurodegeneration with brain iron accumulation (NBIA) (Khateeb et al., 2006; Morgan et al., 2006), which are all pathologically characterized by widespread development of axonal swellings (spheroids) in the central and peripheral nervous systems, suggesting that impairment of iPLA2β is involved in the pathogenesis of neuroaxonal dystrophy.
To explore the physiological and pathological role of iPLA2β, we generated iPLA2β−/− mice. Here, we report that iPLA2β−/− mice developed motor dysfunction associated with prominent formation of spheroids and vacuoles in axons and synapses throughout the nervous system. These findings provide evidence that impairment of iPLA2β causes neuroaxonal dystrophy.
Materials and Methods
Animal.
The C57BL/6 mice used in this study were obtained from Japan SLC (Shizuoka, Japan). The experimental protocol was approved by the Ethical Review Committee for Animal Experimentation of Osaka University Medical School.
In situ hybridization.
An RNA probe was prepared by in vitro transcription, as described previously (Matsuoka et al., 2002), using digoxigenin (DIG)-UTP and the pGEM-T easy plasmid vector (Promega, Madison, WI) carrying 720 bases (1732–2452) of mouse iPLA2β cDNA.
Brains, spinal cords, and dorsal root ganglia of the mice were dissected, submerged in OCT Tissue-Tek mounting medium (Miles, Elkhart, IN), and rapidly frozen at −40°C in 2-methyl-butane. Sections 7 μm thick were cut on a cryostat and mounted on Matsunami adhesive silane-coated slides (Matsunami, Osaka, Japan). Refixation, acetylation, and hybridization of the sections were performed as described previously (Zhong et al., 2004). After washing three times with 50% formamide and 2× SSC at 60°C, the sections were treated with blocking reagent for 30 min at room temperature, and then incubated with alkaline phosphatase-conjugated anti-DIG antibody for 30 min. After washing three times, the sections were incubated in 100 mm Tris-HCl, pH 9.5, 100 mm NaCl, 0.03% nitro blue tetrazolium, and 0.015% bromochloroindoyl phosphate. Then, color development was stopped by incubation in PBS containing 10 mm EDTA, after which the sections were treated in 100% ethanol and xylene, and then embedded for microscopic observation.
Targeted disruption of theiPLA2β gene.
A vector for disruption of the iPLA2β gene was constructed using genomic DNA derived from 129/Sv embryonic stem (ES) cells and the pMulti-ND-1.0 vector (Inoue et al., 2005). Briefly, a 2.3 kb genomic fragment containing exon 10 and a 7.7 kb genomic fragment containing exons 5–8 were inserted into the PacI site and the ClaI site of the vector, respectively.
ES cells [clone D3 (p13GIRC)] were transfected with the vector, and neomycin-resistant clones were isolated. After screening 288 resistant clones by PCR and subsequently by Southern blot analysis of BglII-digested genomic DNA using a probe A or a probe B, two ES cells lines with disruption of the iPLA2β gene were obtained. These ES cells were injected into C57BL/6 blastocysts to produce chimeric mice, which were crossed with C57BL/6 mice to obtain iPLA2β+/− F1 progeny. The littermates obtained from pairing of iPLA2β+/− F2 mice were pooled and used for this study.
Genotyping of mice by PCR and Southern blot analysis.
Genomic DNA was prepared from mouse tails. To amplify the wild-type and disrupted iPLA2β loci, the following primers were used: wild-type (349 bp), ATGGATCCGTGGTCTTCATCTACCTCCTCG and TGAGCCCAATGCTAGGAATGTCCAATCAGC; disrupted iPLA2β locus (480 bp), TGGCGGACCGCTATCAGGACATAGCGTTGG and AGGTGGAGTGCAGGAACAAGGCCTATCAGG.
The PCR was performed using a Gene Taq (Nippon Gene, Tokyo, Japan), with 32 cycles of 95°C for 30 s, 63°C for 30 s, and 72°C for 1 min. Southern blot analysis was performed using a Gene Images kit (GE Healthcare Bio-Science, Piscataway, NJ). The probes were prepared by PCR using a dNTP mixture containing fluorescein-11-dUTP and the following primes: probe A (423 bp), GCCGGCCTGAACCAGGTAAACAACCAAGGG and TGACCTGCCAAGCAGCAGACTCAAGAGCAG; probe B (777 bp), TCAAGAAGGCGATAGAAGGCGATGCGCTGC and TGAACAAGATGGATTGCACGCAGGTTCTCC. Hybridization, washing, and detection were performed according to the supplier's protocol.
Western blot analysis.
Testes obtained from male mice were homogenized with radioimmunoprecipitation assay buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, and protease inhibitor mixture). As described previously (Shinzawa and Tsujimoto, 2003), immunoblot analysis was performed with a polyclonal anti-iPLA2β antibody (Cayman Chemical, Ann Arbor, MI) or a monoclonal anti-α-tubulin antibody (clone B-5–1-2) (Sigma, St. Louis, MO) and an HRP-conjugated secondary antibody using ECL Western blotting detection reagents (GE Healthcare Bio-Science).
Behavioral analysis.
The hanging wire grip test was performed by placing a mouse on a wire net and then turning the net upside down at a height of ∼20 cm above the cage floor to prevent the animal from easily climbing down. The time that elapsed until the animal fell was recorded three times and the cutoff time was set at 60 s. Footprint patterns were obtained by painting the forepaws and the hindpaws with blue and red ink, respectively.
Histochemical and immunohistochemical analysis.
Mice aged 95–103 weeks (iPLA2β+/+, one male and three females; and iPLA2β−/−, three males and four females), mice aged 56 weeks (iPLA2β+/+, one female; and iPLA2β−/−, four females), mouse aged 32 weeks (iPLA2β−/−, one female), and mice aged 15 weeks (iPLA2β−/−, one male and one female) were used for histological examination. After being treated with an overdose of sodium pentobarbital, each animal was perfused with PBS and then 4% paraformaldehyde. The brain, spinal cord, and sciatic nerve were removed from each mouse, immersed in the same fixative overnight at 4°C, and then dehydrated and embedded in paraffin blocks. Then 4-μm-thick paraffin sections were prepared and stained with hematoxylin and eosin (H&E), Nissl, Luxol fast blue, periodic acid-Shiff (PAS), Bielschowsky, or Berlin blue stain. Small pieces of sciatic nerves from mice aged 95–103 weeks and the cerebral cortex and the dorsal part of the cervical spinal cord from mice aged 56 weeks were fixed with 2.5% glutaraldehyde and processed to Epon blocks as described previously (Sumi et al., 2006). Transverse Epon sections, 1 μm thick, of sciatic nerves were stained with toluidine blue.
To perform immunohistochemistry, deparaffinized sections were processed as described previously (Sumi et al., 2006). The antibodies used were a mouse monoclonal antibody for phosphorylated neurofilament (Covance, Berkeley, CA) and rabbit polyclonal antibody for ubiquitin (Dako, Glostrup, Denmark). Hematoxylin was used to counterstain the cell nuclei.
To estimate the total density of myelinated nerve fibers in the sciatic nerve per unit area, three images of the large nerve fascicles (100× objective, 62,080 μm2) were obtained with a digital camera (VB-7010; Keyence, Osaka, Japan) attached to a light microscope (Eclipse E800; Nikon, Tokyo, Japan), and the number of myelinated fibers in the four images was counted in each mouse. To estimate the area of the sciatic nerves, images of the whole nerve (10× objective) were obtained with a digital camera attached to the light microscope, and the areas of all nerve fascicles were measured using image analysis software (VH-H1A5; Keyence). The total number of myelinated fibers in the sciatic nerve was calculated from the density and the area.
Ultrathin sections of the cerebral cortex and the dorsal horn were cut and stained with uranyl acetate and lead citrate, and were examined by transmission electron microscopy (H-7650; Hitachi, Tokyo, Japan).
Results
Expression of iPLA2β mRNA in the nervous system
As described below, iPLA2β−/− mice suffered from neurodegeneration. Accordingly, we performed detailed examination of the pattern of iPLA2β gene expression in the nervous system by in situ hybridization. Because of unavailability of suitable anti-iPLA2β antibody, we were unable to perform immunohistochemical analysis. As shown in Figure 1A, every layer of the cerebral cortex, except for layer I, was stained by the antisense probe for the iPLA2β gene, but not by the sense probe (Fig. 1B). No signals were detected in the corpus callosum where axons and oligodendrocytes are prominent. In the cerebellum, weak iPLA2β gene expression was seen in the granular layer, whereas there was stronger expression in the Purkinje cells (Fig. 1C). In the spinal cord, both dorsal and ventral horn neurons showed strong expression of the iPLA2β gene (Fig. 1E). Moreover, dorsal root ganglion cells showed somatic expression of iPLA2β, although there was no expression in their axons or in the Schwann cells (Fig. 1G). Together, these results indicated that the iPLA2β gene was widely expressed by neurons.
Figure 1.
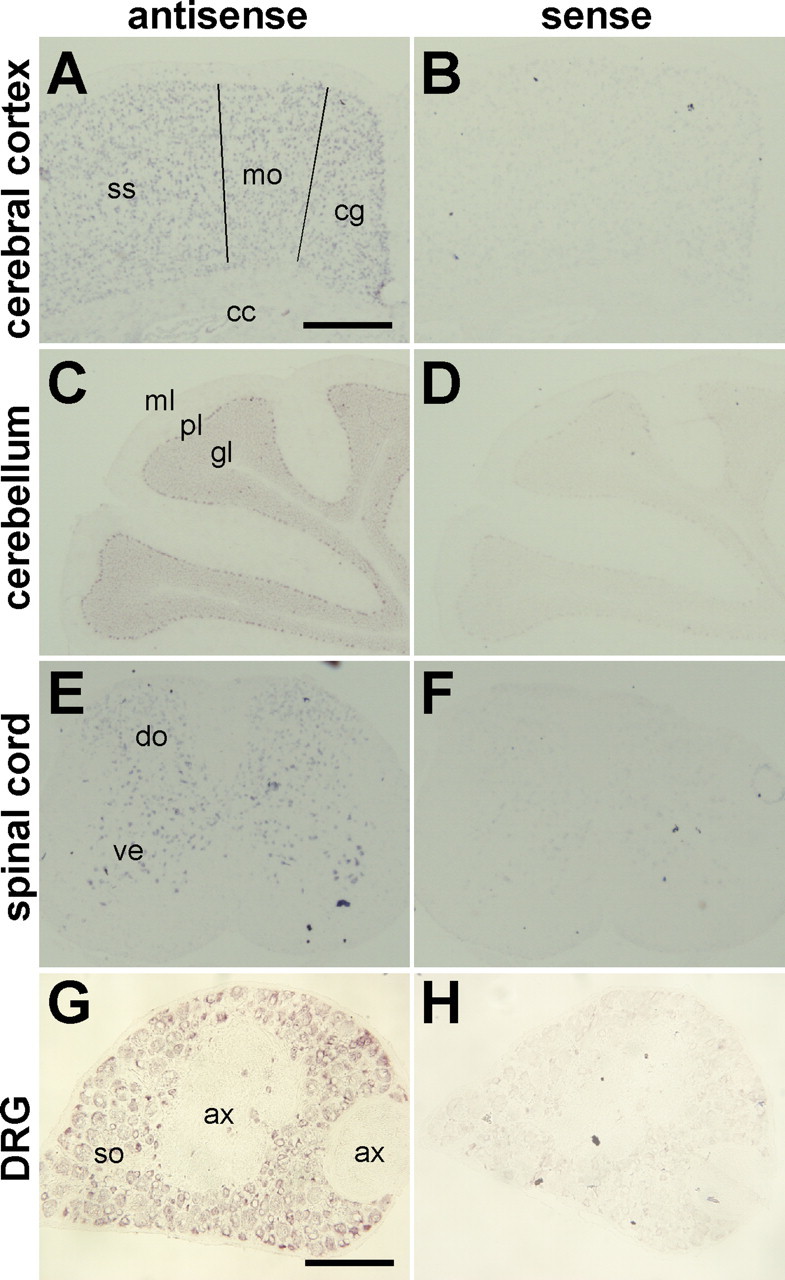
In situ hybridization of iPLA2β mRNA in the nervous system. A–H, The pattern of iPLA2β mRNA expression in the cerebral cortex (A, B), the cerebellum (C, D), the spinal cord (E, F), and the dorsal root ganglion (DRG) (G, H) of 7-week-old female mice. The antisense probe detected signals in all cortical layers, except for layer I, of the cingulate (cg), the motor (mo) cortex, and the somatosensory (ss) cortex, in the Purkinje cell layer (pl) and the granular layer (gl), but not the molecular layer (ml) of the cerebellum, in the dorsal (do) and ventral (ve) horns of the spinal cord, and the soma-rich region of the DRG (so). No signals were detected in the corpus callosum (cc) or in the axon-rich region (ax) that contains mainly axons and oligodendrocytes (cc) or Schwann cells (ax). The sense probe was used as a negative control (B, D, F, H). Scale bars: A–F, 500 μm; G, H, 180 μm.
Generation of iPLA2β−/− mice
To evaluate the physiological role of iPLA2β, we generated mice with targeted disruption of the iPLA2β gene. As described in Figure 2A, a targeting vector was designed, so that exon 9 (which encodes the catalytic domain including the active center amino acid residue, Ser 465) was replaced by a neomycin resistance gene (neo) cassette. Following the standard procedure described in Materials and Methods, two ES cell lines (9E and 12E) with the disrupted allele were obtained (Fig. 2B) and then injected into blastocysts to produce chimeric mice. One of the two cell lines (9E) transmitted the disrupted allele to the germline. iPLA2β+/− mice were interbred to generate iPLA2β−/− mice and their offspring were genotyped by PCR and Southern blot analysis (Fig. 2C). The absence of iPLA2β protein in iPLA2β−/− mice was confirmed by Western blot analysis using testicular lysates (Fig. 2D), indicating that the disrupted allele was a null mutation. The ratio of offspring with each genotype was in accordance with the Mendelian rule (iPLA2β+/+:iPLA2β+/−:iPLA2β−/− = 81:141:67). iPLA2β−/− mice developed normally and grew to maturity. Male mice exhibited reduced fertility (data not shown), confirming previous observations (Bao et al., 2004).
Figure 2.
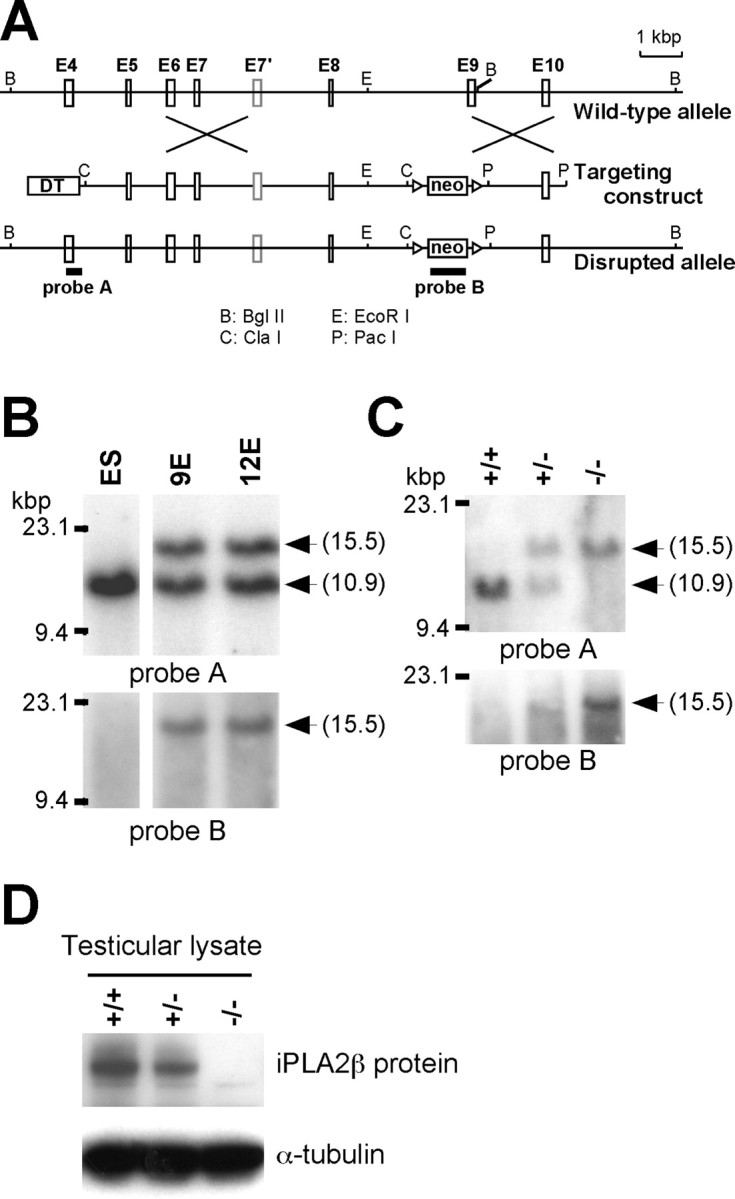
Targeting the iPLA2β gene. A, Diagram of the wild-type iPLA2β gene, the targeting construct, and the disrupted iPLA2β allele. B, C, Southern blot analysis of BglII-digested genomic DNA from ES cell clones (B) and from the tails of iPLA2β+/+, iPLA2β+/−, and iPLA2β−/− mice (C). The DNA probes used for Southern blot analysis were a probe A and B containing exon 4 and neomycin resistance gene (neo), respectively. The iPLA2β+/+ wild-type allele generated a band of 10.9 kbp, whereas the disrupted allele yielded a 15.5 kbp fragment. D, Western blotting of testicular lysates with an anti-iPLA2β antibody (α-tubulin was used as the loading control).
Neurodegeneration and shortened lifespan
Because we could not find any obvious abnormalities of iPLA2β−/− mice by the age of 1 year, except for reduced fertility in males, a substantial number of mice were kept alive to examine the effect on their lifespan and aging. Although at 40–50 weeks no difference of body weight between iPLA2β−/− mice and their littermates (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material), iPLA2β−/− mice gradually lost weight and died earlier than their littermates (Fig. 3A,B). The log-rank test indicated there was a significant survival difference between iPLA2β−/− and iPLA2β+/+ mice (p < 0.02), and between iPLA2β−/− and iPLA2β+/− mice (p < 0.002). The median lifespan of iPLA2β−/− mice was 90 weeks compared with 110–115 weeks for their littermates. By the age of 2 years, all of the iPLA2β−/− mice showed abnormal movement of their hindlimbs. When held by the tail, iPLA2β−/− mice moved their hindlimbs randomly in all directions or stopped moving with feet clasping, and a dangling posture being prominent (Fig. 4Ad,Ae). The mice seemed unable to use their hindlimbs to maintain their balance as they tried to raise their heads. In contrast, iPLA2β+/+ mice extended their hindlimbs out from the trunk to maintain their balance (Fig. 4Aa,Ab), and could easily raise their heads. Moreover, iPLA2β−/− mice dragged their hindlimbs when walking and had an irregular stride (Fig. 4B). In contrast, iPLA2β+/+ and iPLA2β+/− mice did not show any of these changes. When motor function was assessed by the hanging wire grip test, the mice were placed on a wire net that was turned upside down, and the time until the animal fell was recorded three times. Because body weight was predicted to affect the time until falling, the weight was plotted against time (Fig. 4C). All of the iPLA2β−/− mice showed impaired motor function in this test. The iPLA2β−/− mice also tended to adopt a hunched posture (Fig. 4Af), although x-ray radiograms did not reveal any obvious differences of bone structure or density (data not shown). After analysis of 2-year-old mice, the behavioral abnormalities of younger iPLA2β−/− mice (≤1 year old) were carefully monitored. Although iPLA2β−/− mice did not show obvious abnormalities in the footprint and tail suspension tests by the age of 55 weeks (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material) (data not shown), the hanging wire grip test was definitely abnormal. Their time scores decreased gradually from the age of 30 weeks, and by the age of ∼50 weeks all of the iPLA2β−/− mice showed very low time scores (Fig. 4D). These results indicate that the hanging wire grip test may be more sensitive for detecting impaired motor function in iPLA2β−/− mice. All together, these findings indicated that iPLA2β deficiency led to the onset of motor dysfunction and a shorter lifespan.
Figure 3.
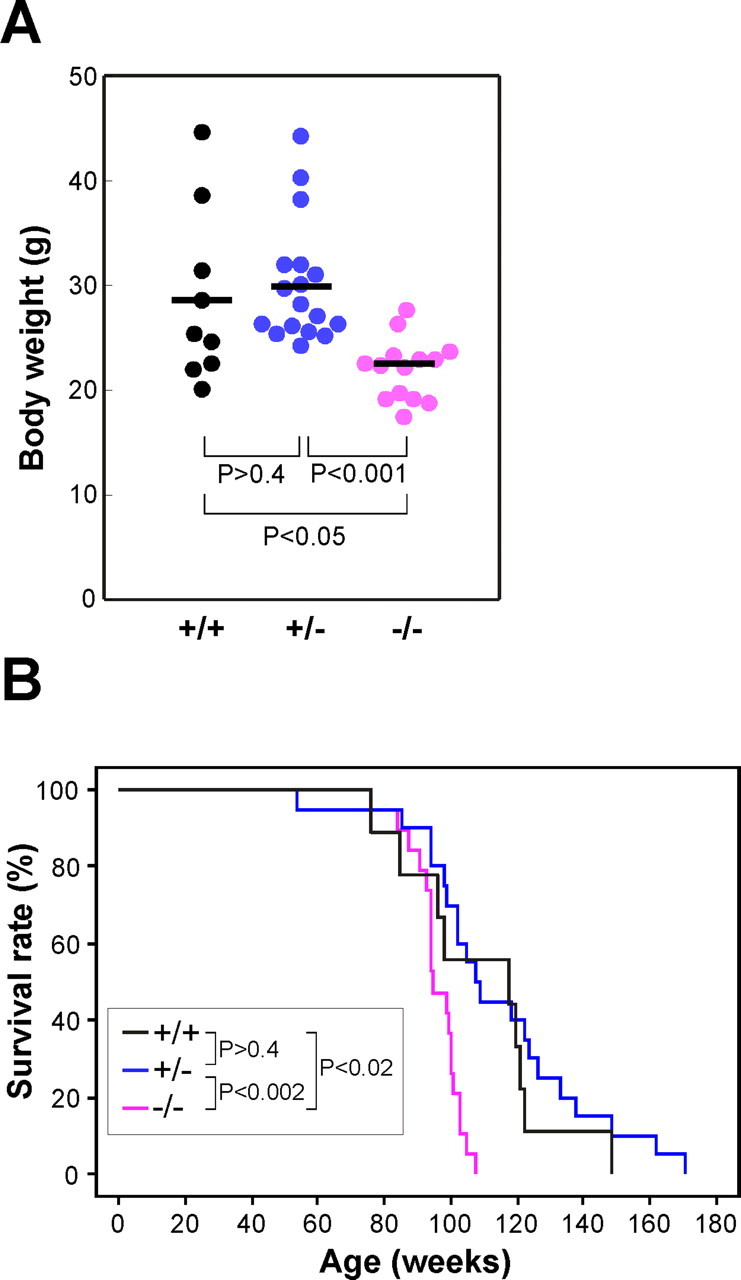
Lower body weight and shorter lifespan of iPLA2β−/− mice. A, Body weight of female iPLA2β+/+ mice (n = 9), iPLA2β+/− mice (n = 17), and iPLA2β−/− mice (n = 14) at 91–100 weeks of age. Bars indicate the mean values. p values were calculated using the Student's t test. B, Survival of female iPLA2β+/+ mice (n = 9), iPLA2β+/− mice (n = 20), and iPLA2β−/− mice (n = 19). p values were calculated using the log-rank test. Results obtained with male mice were virtually identical.
Figure 4.
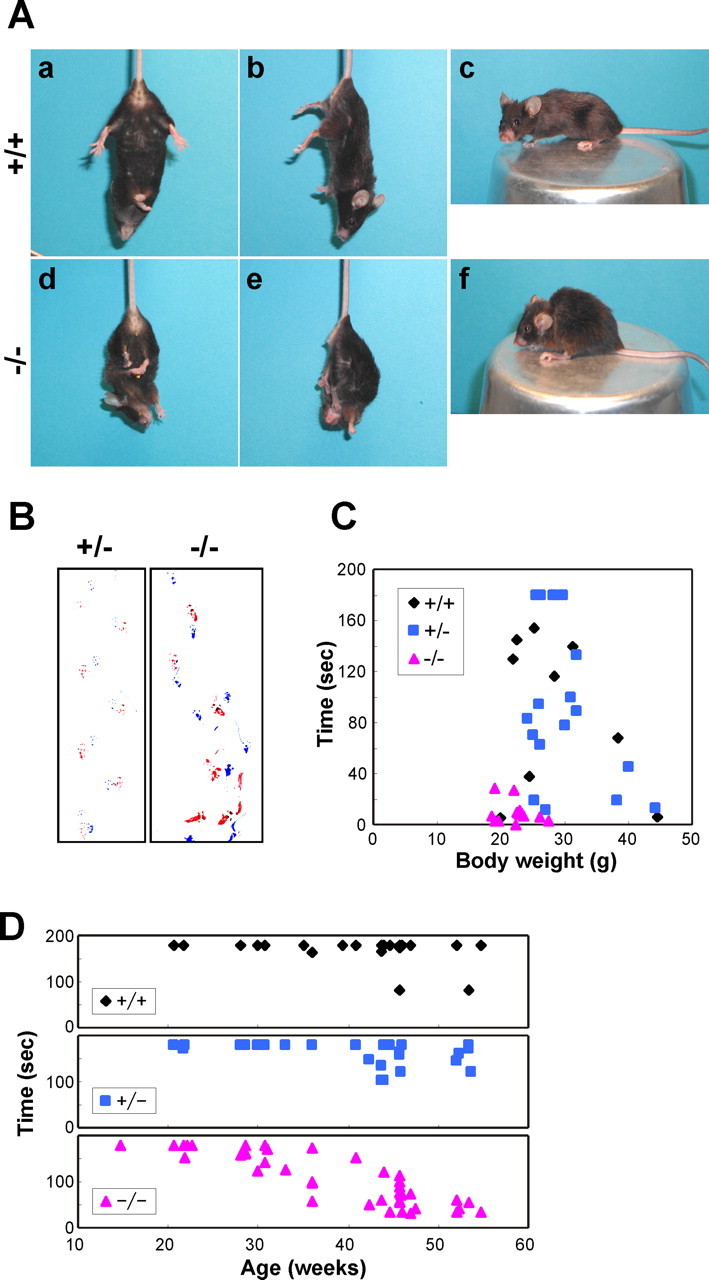
Motor dysfunction in iPLA2β−/− mice. A, Representative photographs of an iPLA2β+/+ mouse (a–c) and an iPLA2β−/− mouse (d–f) at 95 weeks of age. The iPLA2β−/− mouse adopts a feet-clasping posture when suspended by the tail (d, e), whereas the iPLA2β+/+ mouse holds its hindlimbs outward to steady itself (a, b). The iPLA2β−/− mouse has a hunched posture (f). B, Representative footprint patterns of iPLA2β+/− and iPLA2β−/− mice at 90 and 87 weeks of age, respectively. Forepaws and hindpaws were painted with blue and red ink, respectively. The iPLA2β−/− mouse displays an irregular stride and dragging of the hindlimbs. C, D, Hanging wire grip test. C, Female iPLA2β+/+ mice (n = 9), iPLA2β+/− mice (n = 17), and iPLA2β−/− mice (n = 13) (91–100 weeks old) were tested as described in Materials and Methods, and the total time for three trials was plotted against body weight. D, Female iPLA2β+/+ mice (n = 23), iPLA2β+/− mice (n = 25), and iPLA2β−/− mice (n = 42) (15–55 weeks old) were tested. The total time for three trials was plotted against age.
Neuropathological findings in iPLA2β−/− mice
The iPLA2β−/− mice were subjected to neuropathological studies and the changes observed at 2 years are summarized in Table 1. There were numerous spheroids or vacuoles in the axons and neuropil throughout the CNS (Fig. 5) and the peripheral nervous system (PNS) (Fig. 6), indicating the widespread degeneration of axons or synapses. These round or irregular spheroids were 3–90 μm in diameter. The larger spheroids frequently showed vacuolation, fissures, or rarefaction (Figs. 5B,C, 6C). As summarized in Table 1, the spheroids were most prominent in the tegmentum of the medulla (Fig. 5B), the lower pons, and the dorsal horns (Fig. 5C) of the entire spinal cord. In addition to the spheroids, vacuoles of various sizes (3–40 μm in diameter) were observed (Fig. 5B,C,L). Most of the larger vacuoles had a core (Fig. 5B,C). A single irregularly swollen axon that contained both spheroids and a vacuole was detected in a longitudinal section (Fig. 6C). The cerebral cortex contained numerous vacuoles, most of which were rather small (3–5 μm), and there were only a few spheroids (Fig. 5L). Younger iPLA2β−/− mice were also subjected to neuropathological examination and the following data were obtained. At 15 weeks, tiny vacuoles were frequently observed in the molecular layer of the cerebellum (Fig. 5I), which became atrophic at 2 years (Fig. 5H), and spheroids were also found in the dentate nucleus of the cerebellum (Fig. 5J). In other regions, vacuoles and spheroids were only rarely observed. At 32 weeks, some small vacuoles were found in the cerebral cortex, striatum, and hippocampus, and a number of spheroids and large vacuoles were observed in the tegmentum of the lower pons or medulla and in the spinal cord, as well as the cerebellum (data not shown). At 55 weeks, vacuoles or spheroids were distributed throughout the CNS (data not shown). PAS staining revealed many PAS-positive granules, mainly in the normal-looking axons of larger neurons such as anterior horn cells, where very few spheroids or vacuoles were found at 15 weeks (Fig. 5E). At 2 years, PAS-positive granules were also scattered in axons or the neuropil where spheroids were prominent (Fig. 5F). Both spheroids and vacuoles frequently contained PAS-positive granules (Fig. 5C). Although many of the spheroids were positive for PAS to some extent, spheroids were not stained by Berlin blue, which detects iron (data not shown). In iPLA2β+/+ mice, a few spheroids and vacuoles were observed exclusively in the gracile nucleus and part of the cuneate nucleus (data not shown), which might have represented physiological neuroaxonal dystrophy occurring as part of the normal aging process.
Table 1.
Distribution of spheroids and vacuoles in the CNS
| Region | Spheroids | Vacuoles |
|---|---|---|
| Cuneate nucleus | +++ | ++ |
| Gracile nucleus | +++ | ++ |
| Trigeminal nucleus | +++ | ++ |
| Facial nucleus | ++ | + |
| Cochlear nucleus | +++ | ++ |
| Vestibular nucleus | +++ | ++ |
| Hypoglossal nucleus | + | + |
| Spinal cord dorsal horn | +++ | ++ |
| ventral horn | ++ | ++ |
| dorsal funiculus | ++ | ++ |
| ventral funiculus | ++ | ++ |
| Striatum | + | ++ |
| Thalamus | + | ++ |
| Hypothalamus | + | ++ |
| Hippocampus | + | ++ (small)a |
| Amygdala | + | ++ |
| Cerebellum molecular layer | + | ++ (small)a |
| granular layer | + | ++ |
| dentate nucleus | ++ | ++ |
| Cerebral cortex | + / − | +++ (small)a |
Distribution of spheroids and vacuoles in the CNS were assessed and graded as follows: +/−, very few; +, mild degree; ++, moderate degree; +++, marked degree.
aVacuoles of small size were predominantly observed. Mice aged 95–103 weeks were examined.
Figure 5.
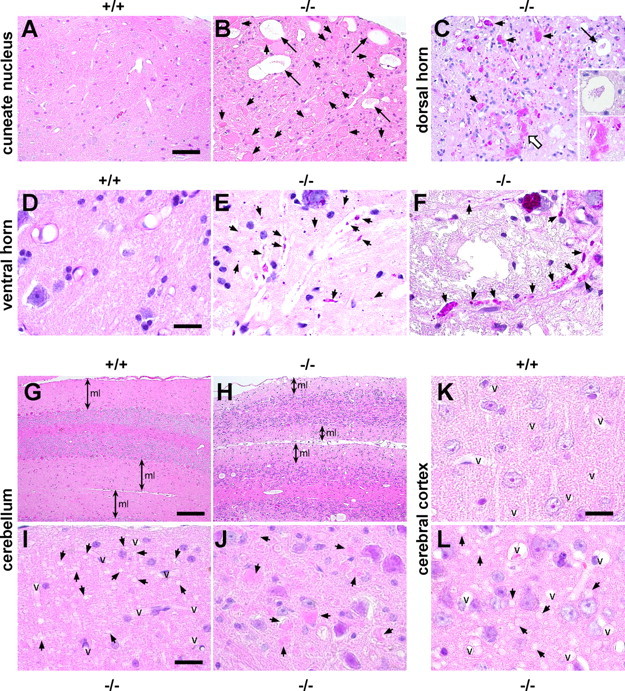
Neuropathological changes in iPLA2β−/− mice. A, B, Cuneate nucleus of an iPLA2β+/+ mouse (A) and an iPLA2β−/− mouse (B) at 2 years (95–103 weeks old). C, Dorsal horn of iPLA2β−/− mouse at 2 years. D–F, Ventral horn of an iPLA2β+/+ mouse at 56 weeks (D) and an iPLA2β−/− mouse at 15 weeks (E) and 2 years (F). G, H, Cerebellum of an iPLA2β+/+ mouse (G) and an iPLA2β−/− mouse (H) at 2 years. I, J, Molecular layer (I) and dentate nucleus (J) of the cerebellum of an iPLA2β−/− mouse at 15 weeks. K, L, Cerebral cortex of an iPLA2β+/+ mouse (K) and an iPLA2β−/− mouse (L) at 2 years. A, B, G–L, H&E stain. C–F, PAS stain. A, No spheroids and vacuoles are detected in the iPLA2β+/+ mouse. B, Many spheroids (arrows) and large vacuoles (large arrows) can be observed. C, Irregular PAS-positive spheroids (small arrows) contain fissures, vacuoles, and PAS-positive granules. The extent of PAS staining is variable. The core of the huge vacuole contains PAS-positive granules (large arrow and open arrow; also shown at a higher magnification). D, No PAS-positive granules are observed in the iPLA2β+/+ mouse. E, Strongly PAS-positive granules can be seen in normal-looking axons or the neuropil. F, PAS-positive granules (arrows) in swollen axons. G, H, The molecular layer (ml) of the cerebellum from the iPLA2β−/− mouse (H) is thinner than that from the iPLA2β+/+ mouse (G). I, Numerous tiny vacuoles (arrows) can be seen in the molecular layer. J, There are only a few spheroids (arrows). K, There are no detectable vacuoles in the iPLA2β+/+ mouse. L, Numerous small vacuoles (arrows) can be observed, but there are no spheroids. Scale bars: A–C, 50 μm; D–F, I–L, 16 μm; G, H, 100 μm. v, Vessels.
Figure 6.
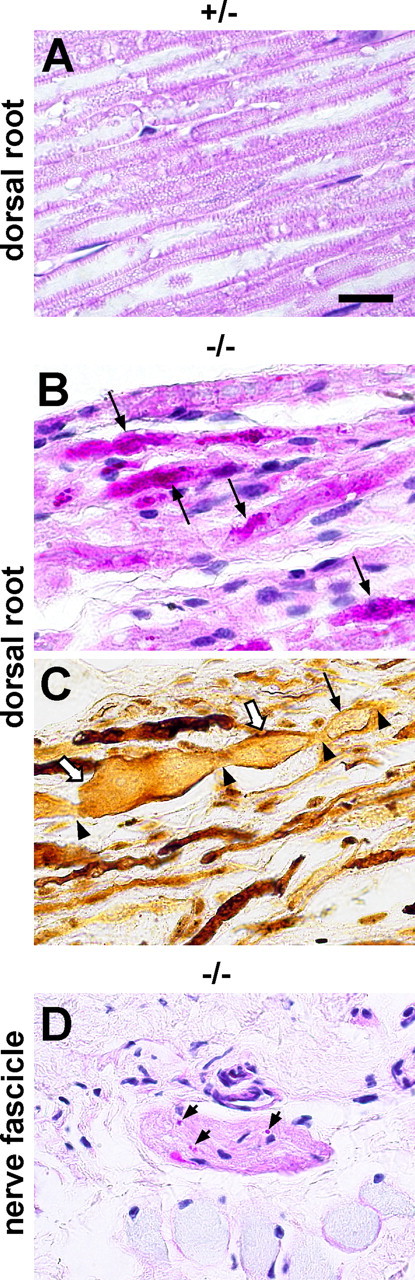
Pathological changes of the PNS. A–C, Dorsal root of an iPLA2β+/− mouse (A) and an iPLA2β−/− mouse (B, C) at 2 years. D, Subcutaneous nerve fascicle of an iPLA2β−/− mouse at 2 years. A, B, and D are PAS stained. C, Bielschowsky stain. Scale bar, 16 μm. A, Axons of the iPLA2β+/− mouse are PAS negative, although the myelin sheath is weakly stained by PAS. B, Swollen axons (arrows) are heterogeneously stained by PAS and contain strongly PAS-positive granules. C, A single swollen axon contains both spheroids (open arrows) and a vacuole (arrow). The central parts of the spheroids (open arrows) are rarefied. Staining of the axonal structures (arrowheads) between the spheroids is weak. D, PAS-positive granules can be seen in the nerve fascicle.
Immunohistochemistry using an anti-phosphorylated neurofilament antibody (SMI31) revealed strong staining of some small spheroids, indicating that these structures originated from axons (Fig. 7A). Most of the large and irregular spheroids that contained narrow clefts or vacuoles were almost completely negative for this antibody, suggesting that severe degeneration had occurred. Some of the spheroids and vacuoles were also stained by anti-ubiquitin antibody, with the cores or fissures of the spheroids and the rims or inner contents of small vacuoles being stained to a varying extent (Fig. 7B,C). These findings indicate that some of the spheroids and vacuoles contained ubiquitinated proteins.
Figure 7.
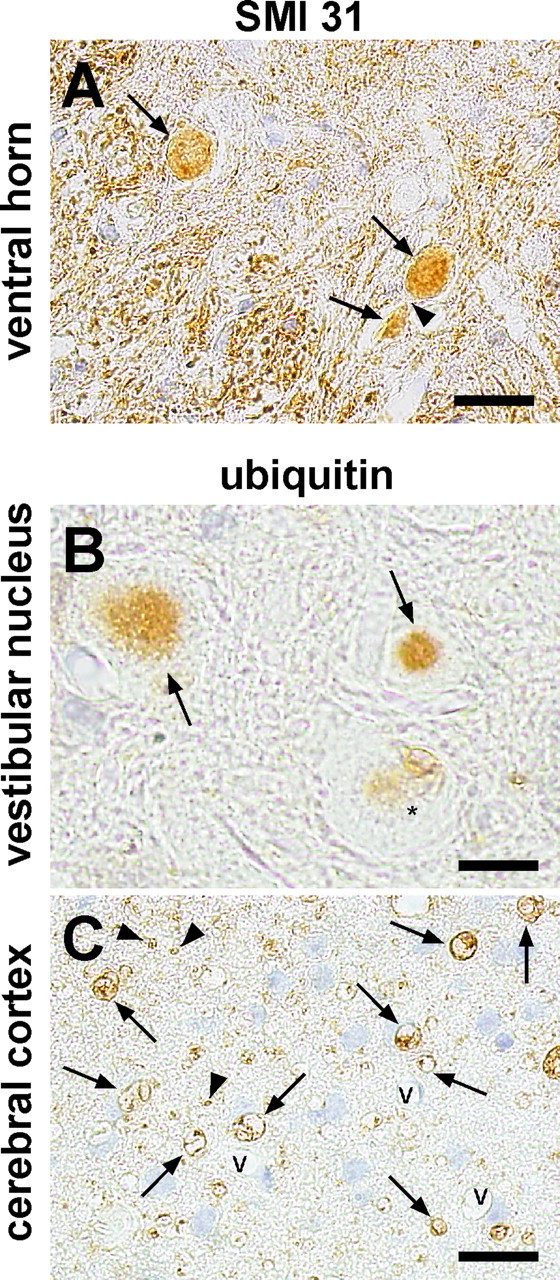
Immunohistochemical analysis of spheroids and vacuoles in iPLA2β−/− mice at 2 years. A, Ventral horn was stained with anti-phosphorylated neurofilament H (SMI31). B, C, Vestibular nucleus (B) and cerebral cortex (C) were stained with anti-ubiquitin antibody. v, Vessels. Scale bars: A, 20 μm; B, 10 μm; C, 16 μm. A, Spheroids (arrows) are positive for SMI31, whereas the axonal portion between the spheroids (arrowhead) is negative. B, Two spheroids (arrows) have positive cores. The other spheroid (asterisk) contains vacuoles that are weakly stained or unstained for ubiquitin. C, Numerous vacuoles (arrows) and tiny circles (arrowheads) that are ubiquitin positive in the neuropil of the cerebral cortex.
Morphological analysis of the sciatic nerve demonstrated that the total number of myelinated fibers (density by area) was significantly reduced in iPLA2β−/− mice (Table 2). The density of myelinated fibers was similar (Table 2, supplemental Fig. 2C,D, available at www.jneurosci.org as supplemental material) in both groups, but the area of the nerve fascicles was significantly smaller in iPLA2β−/− mice (Table 2, supplemental Fig. 2B, available at www.jneurosci.org as supplemental material), indicating that the sciatic nerves of these mice had undergone atrophy because of neuronal degeneration.
Table 2.
Morphological analysis of sciatic nerves between iPLA2β+/+ and iPLA2β−/− mice
| Density of myelinated fibers (per mm2) | Total area of nerve fascicles (mm2) | Number of myelinated fibers (density × area) | |
|---|---|---|---|
| +/+ (n = 4) | 20156 ± 4007 | 0.189 ± 0.037 | 3735 ± 554 |
| −/− (n = 7) | 22922 ± 1695 | 0.136 ± 0.117 | 3115 ± 234 |
| (p < 0.05) | (p < 0.05) |
The number of myelinated fibers per unit area was counted in sciatic nerves. Mice aged 95–103 weeks old were examined. p values were calculated using the Student's t test.
Ultrastructural examination of iPLA2β−/− mice at 55 weeks showed that spheroids in the dorsal funiculus contained tubulovesicular structures, as well as vacuoles, vesicles, and amorphous matrix (Fig. 8). These tubulovesicular structures were also found in the cerebral cortex (data not shown).
Figure 8.
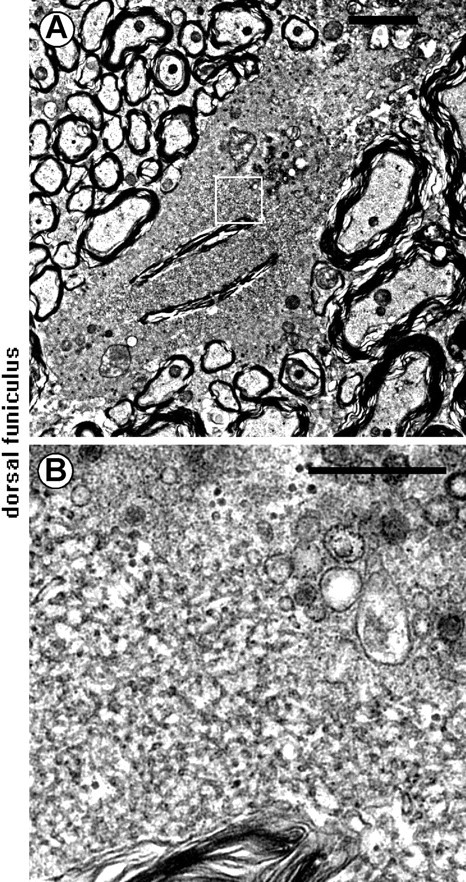
Electron microscopic features of spheroids. A, B, Dorsal funiculus of a iPLA2β−/− mouse at 56 weeks. B shows a higher magnification of the boxed region in A. Tubulovesicular structures can be seen in the spheroid, as well as degenerated vesicles and vacuoles. Scale bars: A, 2 μm; B, 0.5 μm.
Discussion
We showed that iPLA2β deficiency in mice led to motor impairment, which was accompanied by the appearance of axonal swelling (spheroids) and vacuoles throughout the nervous system. Our results provide experimental evidence that iPLA2β plays a critical role in maintaining the structural and functional integrity of axons or synapses, and that iPLA2β dysfunction leads to the occurrence of neuroaxonal dystrophy.
In this study, we found that iPLA2β−/− mice initially developed normally, but began to show significant motor dysfunction from the age of ∼50 weeks (Fig. 4D), which progressed to ataxia and weakness by 2 years. These observations indicate that iPLA2β has an important role in maintaining the function of the nervous system, but not in nervous system development. INAD is caused by mutation of the iPLA2β gene (Khateeb et al., 2006; Morgan et al., 2006) and patients develop the initial symptoms, including hypotonia, areflexia, or weakness < 2 years old. As the disease progresses, dementia, ataxia, blindness, and spasticity occur, and these patients die before puberty (Nardocci et al., 1999). Although some symptoms, such as weakness and ataxia, were common to INAD patients and iPLA2β−/− mice, the clinical course was different. In mice, the onset was late and disease progression was slow. The different clinical pictures could possibly be explained by species differences or compensation for iPLA2β deficiency by other PLA2 isotypes (such as iPLA2γ) in mice.
However, the neuropathological features seen in iPLA2β−/− mice were very similar to those found in patients with INAD. Neuroaxonal dystrophy in INAD and NBIA is characterized by a morphological change of axons in the CNS, which is the development of spheroids (Seitelberger, 1952). Numerous spheroids and vacuoles were distributed throughout the CNS in the iPLA2β−/− mice (Table 1), as is also observed in INAD. As has been reported for INAD (Gonatas et al., 1967), the spheroids were located in the axons and synapses of iPLA2β−/− mice (Figs. 5, 6). In addition, the cerebellum (which is one of the most susceptible regions in INAD) (Nardocci et al., 1999) seemed to be affected quite early among the CNS regions in iPLA2β−/− mice (Fig. 5I,J). Furthermore, examination of the sciatic nerves of iPLA2β−/− mice showed peripheral nerve degeneration (Table 2, supplemental Fig. 2, available at www.jneurosci.org as supplemental material), as also occurs in INAD (Duncan et al., 1970). Moreover, tubulovesicular structures, which are a characteristic finding in INAD patients (Gonatas et al., 1967), were detected in iPLA2β−/− mice by electron microscopy (Fig. 8). Because the pathological changes of the nervous system were almost identical, the iPLA2β−/− mouse seems to be an appropriate model for studying the detailed pathogenesis of neuroaxonal dystrophy in INAD.
The mechanisms leading to the formation of spheroids and vacuoles in the absence of functional iPLA2β remain unclear, but these structures seem to arise from the degeneration of axons or synaptic terminals. Some of the large vacuoles presumably developed from the resorption of spheroids (Cowen and Olmstead, 1963) via a process involving ubiquitination (Fig. 7B). Because numerous PAS-positive granules appeared in the axons or neuropil by 15 weeks, which was before the spheroids and vacuoles formed (Fig. 5E), and because both spheroids and large vacuoles contained PAS-positive granules (Figs. 5C, 6B), these granules might be the key to elucidating the pathogenesis of neuroaxonal degeneration in iPLA2β−/− mice and INAD patients. Because PAS stains materials containing aldehydes as well as staining polysaccharides like glycogen, the PAS-positive material might represent accumulations of polysaccharides, aberrantly glycated proteins, or aldehyde products of lipid peroxides created by an increase of ROS in the iPLA2β-deficient state.
iPLA2β is an important enzyme for maintaining phospholipid membranes through the processes of phospholipid turnover, remodeling, and repair (Ma and Turk, 2001). Therefore, iPLA2β deficiency may alter the phospholipid composition of cellular and subcellular membranes, or may lead to failure to repair oxidative damage to membrane phospholipids, resulting in changes of membrane permeability, fluidity, or ion homeostasis. Why are the axons or presynaptic terminals, and not the cytoplasm, selectively affected in INAD patients and iPLA2β−/− mice? The tubulovesicular structures observed in both INAD patients and iPLA2β−/− mice (Fig. 8) are believed to result from degenerated ER or mitochondria. iPLA2β has been reported to exist in and protect mitochondria (Seleznev et al., 2006), as well as being abundant in neurites and axon terminals (Ong et al., 2005), so its absence would contribute to damage targeting mitochondria in axons and synaptic terminals, leading to degeneration of the axons and synaptic terminals.
Some individuals with NBIA, formerly known as Hallervorden-Spatz syndrome, have a mutation of the pantothenate kinase 2 (PANK2) gene (Zhou et al., 2001). The product of this gene is an enzyme localized to the mitochondria (Kotzbauer et al., 2005) and involved in the biosynthesis of coenzyme A (CoA), which has various roles that include acting as a carrier of free fatty acids. Oxidative damage or impaired fatty acid metabolism resulting from mitochondrial CoA deficiency is thought to be a possible cause of neurodegenerative changes (Johnson et al., 2004). The common mechanism of neuroaxonal dystrophy in iPLA2β deficiency and PANK2 deficiency may be mitochondrial dysfunction affecting the axons and/or axon terminals. Clarification of the pathogenesis of neuroaxonal dystrophy in iPLA2β−/− mice will hopefully lead to the development of treatments for INAD and other neurodegenerative disorders.
Footnotes
This work was supported in part by a grant from the 21st Century Center of Excellence Program, a Scientific Research grant from the Japanese Ministry of Education, Science, Sports, and Culture, and a Comprehensive Research on Aging and Health grant from the Ministry of Health, Labor and Welfare, Japan. We are grateful to Y. Maruyama, A. Kawai, T. Aikawa, K. Ideguchi, and Y. Haseda for technical assistance and Drs. S. Kato and M. Etoh for their helpful advice.
References
- Balboa MA, Varela-Nieto I, Killermann Lucas K, Dennis EA. Expression and function of phospholipase A(2) in brain. FEBS Lett. 2002;531:12–17. doi: 10.1016/s0014-5793(02)03481-6. [DOI] [PubMed] [Google Scholar]
- Bao S, Miller DJ, Ma Z, Wohltmann M, Eng G, Ramanadham S, Moley K, Turk J. Male mice that do not express group VIA phospholipase A2 produce spermatozoa with impaired motility and have greatly reduced fertility. J Biol Chem. 2004;279:38194–38200. doi: 10.1074/jbc.M406489200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowen D, Olmstead EV. Infantile neuroaxonal dystrophy. J Neuropathol Exp Neurol. 1963;22:175–236. doi: 10.1097/00005072-196304000-00001. [DOI] [PubMed] [Google Scholar]
- Duncan C, Strub R, McGarry P, Duncan D. Peripheral nerve biopsy as an aid to diagnosis in infantile neuroaxonal dystrophy. Neurology. 1970;20:1024–1032. doi: 10.1212/wnl.20.10.1024. [DOI] [PubMed] [Google Scholar]
- Farooqui AA, Ong WY, Horrocks LA. Biochemical aspects of neurodegeneration in human brain: involvement of neural membrane phospholipids and phospholipases A2. Neurochem Res. 2004;29:1961–1977. doi: 10.1007/s11064-004-6871-3. [DOI] [PubMed] [Google Scholar]
- Gonatas NK, Evangelista I, Walsh GO. Axonic and synaptic changes in a case of psychomotor retardation: an electron microscopic study. J Neuropathol Exp Neurol. 1967;26:179–199. doi: 10.1097/00005072-196704000-00001. [DOI] [PubMed] [Google Scholar]
- Inoue N, Ikawa M, Isotani A, Okabe M. The immunoglobulin superfamily protein Izumo is required for sperm to fuse with eggs. Nature. 2005;434:234–238. doi: 10.1038/nature03362. [DOI] [PubMed] [Google Scholar]
- Johnson MA, Kuo YM, Westaway SK, Parker SM, Ching KH, Gitschier J, Hayflick SJ. Mitochondrial localization of human PANK2 and hypotheses of secondary iron accumulation in pantothenate kinase-associated neurodegeneration. Ann NY Acad Sci. 2004;1012:282–298. doi: 10.1196/annals.1306.023. [DOI] [PubMed] [Google Scholar]
- Khateeb S, Flusser H, Ofir R, Shelef I, Narkis G, Vardi G, Shorer Z, Levy R, Galil A, Elbedour K, Birk OS. PLA2G6 mutation underlies infantile neuroaxonal dystrophy. Am J Hum Genet. 2006;79:942–948. doi: 10.1086/508572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotzbauer PT, Truax AC, Trojanowski JQ, Lee VM. Altered neuronal mitochondrial coenzyme A synthesis in neurodegeneration with brain iron accumulation caused by abnormal processing, stability, and catalytic activity of mutant pantothenate kinase 2. J Neurosci. 2005;25:689–698. doi: 10.1523/JNEUROSCI.4265-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Turk J. The molecular biology of the group VIA Ca2+-independent phospholipase A2. Prog Nucleic Acid Res Mol Biol. 2001;67:1–33. doi: 10.1016/s0079-6603(01)67023-5. [DOI] [PubMed] [Google Scholar]
- Mancuso DJ, Jenkins CM, Gross RW. The genomic organization, complete mRNA sequence, cloning, and expression of a novel human intracellular membrane-associated calcium-independent phospholipase A(2) J Biol Chem. 2000;275:9937–9945. doi: 10.1074/jbc.275.14.9937. [DOI] [PubMed] [Google Scholar]
- Matsuoka Y, Matsuoka Y, Shibata S, Ban T, Toratani N, Shigekawa M, Ishida H, Yoneda Y. A chromodomain-containing nuclear protein, MRG15 is expressed as a novel type of dendritic mRNA in neurons. Neurosci Res. 2002;42:299–308. doi: 10.1016/s0168-0102(02)00010-x. [DOI] [PubMed] [Google Scholar]
- Morgan NV, Westaway SK, Morton JE, Gregory A, Gissen P, Sonek S, Cangul H, Coryell J, Canham N, Nardocci N, Zorzi G, Pasha S, Rodriguez D, Desguerre I, Mubaidin A, Bertini E, Trembath RC, Simonati A, Schanen C, Johnson CA, et al. PLA2G6, encoding a phospholipase A(2), is mutated in neurodegenerative disorders with high brain iron. Nat Genet. 2006;38:752–754. doi: 10.1038/ng1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardocci N, Zorzi G, Farina L, Binelli S, Scaioli W, Ciano C, Verga L, Angelini L, Savoiardo M, Bugiani O. Infantile neuroaxonal dystrophy: clinical spectrum and diagnostic criteria. Neurology. 1999;52:1472–1478. doi: 10.1212/wnl.52.7.1472. [DOI] [PubMed] [Google Scholar]
- Ong WY, Yeo JF, Ling SF, Farooqui AA. Distribution of calcium-independent phospholipase A2 (iPLA 2) in monkey brain. J Neurocytol. 2005;34:447–458. doi: 10.1007/s11068-006-8730-4. [DOI] [PubMed] [Google Scholar]
- Seitelberger F. Eine unbekannte Form von infantiler Lipoidspeicher-Krankheit des Gehirns. Proc 1st Int Congr Neuropathol. 1952;3:323–333. [Google Scholar]
- Seleznev K, Zhao C, Zhang XH, Song K, Ma ZA. Calcium-independent phospholipase A2 localizes in and protects mitochondria during apoptotic induction by staurosporine. J Biol Chem. 2006;281:22275–22288. doi: 10.1074/jbc.M604330200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinzawa K, Tsujimoto Y. PLA2 activity is required for nuclear shrinkage in caspase-independent cell death. J Cell Biol. 2003;163:1219–1230. doi: 10.1083/jcb.200306159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Six DA, Dennis EA. The expanding superfamily of phospholipase A(2) enzymes: classification and characterization. Biochim Biophys Acta. 2000;1488:1–19. doi: 10.1016/s1388-1981(00)00105-0. [DOI] [PubMed] [Google Scholar]
- Sumi H, Nagano S, Fujimura H, Kato S, Sakoda S. Inverse correlation between the formation of mitochondria-derived vacuoles and Lewy-body-like hyaline inclusions in G93A superoxide-dismutase-transgenic mice. Acta Neuropathol (Berl) 2006;112:52–63. doi: 10.1007/s00401-006-0056-x. [DOI] [PubMed] [Google Scholar]
- Tang J, Kriz RW, Wolfman N, Shaffer M, Seehra J, Jones SS. A novel cytosolic calcium-independent phospholipase A2 contains eight ankyrin motifs. J Biol Chem. 1997;272:8567–8575. doi: 10.1074/jbc.272.13.8567. [DOI] [PubMed] [Google Scholar]
- Yang HC, Mosior M, Ni B, Dennis EA. Regional distribution, ontogeny, purification, and characterization of the Ca2+-independent phospholipase A2 from rat brain. J Neurochem. 1999;73:1278–1287. doi: 10.1046/j.1471-4159.1999.0731278.x. [DOI] [PubMed] [Google Scholar]
- Zhong Y, Takemoto M, Fukuda T, Hattori Y, Murakami F, Nakajima D, Nakayama M, Yamamoto N. Identification of the genes that are expressed in the upper layers of the neocortex. Cereb Cortex. 2004;14:1144–1152. doi: 10.1093/cercor/bhh074. [DOI] [PubMed] [Google Scholar]
- Zhou B, Westaway SK, Levinson B, Johnson MA, Gitschier J, Hayflick SJ. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat Genet. 2001;28:345–349. doi: 10.1038/ng572. [DOI] [PubMed] [Google Scholar]