

Abstract
Auditory fear conditioning, a model for fear learning, is thought to be mediated by synaptic changes in the cortical and thalamic inputs to the lateral amygdala (LA); however, the specific roles of both pathways are still debated. Here, we report that a CaMKII-α-Cre-mediated knock-out (KO) of the rap1a and rap1b genes impaired synaptic plasticity and increased basal synaptic transmission in the cortical but not thalamic input to the LA via presynaptic changes: increases in glutamate release probability and the number of glutamate quanta released by a single action potential. Moreover, KO mice with alterations in the cortico-LA pathway had impaired fear learning, which could be rescued by training with a more aversive unconditional stimulus. These results suggest that Rap1-mediated suppression of synaptic transmission enables plasticity in the cortico-amygdala pathway, which is required for fear learning with a moderately aversive unconditional stimulus.
Keywords: amygdala, fear, LTP, Rap1, cortex, thalamus
Introduction
Auditory fear conditioning, one simple form of fear learning, is mediated by plastic changes in the two major pathways carrying acoustic information to the lateral amygdala (LA): the thalamic pathway, entering the LA via the internal capsule, and the cortical pathway, entering the LA via the external capsule (LeDoux, 1992; McKernan and Shinnick-Gallagher, 1997; Shi and Cassell, 1997; Doron and LeDoux, 1999; Tsvetkov et al., 2002). Previous lesion studies provided evidence that either pathway alone is sufficient for auditory fear conditioning, even when a second input is destroyed before conditioning (Romanski and LeDoux, 1992). Despite these previous findings, the conclusions about the relative contributions of each pathway remain somewhat contradictory. The loss of fear memory after posttraining lesions of cortical input, but not of the thalamic pathway, indicates that the cortical pathway may play a dominant role in fear conditioning when the brain is intact (Campeau and Davis, 1995; Boatman and Kim, 2006). However, more rapid plastic changes in thalamic afferents during fear conditioning suggest that the thalamic pathway plays the primary role (Quirk et al., 1997). The analysis of physiological properties (Quirk et al., 1997; Doyere et al., 2003) and synaptic morphologies (Humeau et al., 2005) revealed differences between the two pathways indicating that each input may have distinct functions. Nevertheless, their specific roles in fear learning remain an open question.
Transgenic lines under control of the calcium/calmodulin-dependent protein kinase II-α (CaMKII-α) promoter preferentially express transgenes in the cortex rather than in the thalamus (Mayford et al., 1996), thus providing a means to examine the specific contribution of cortico-amygdala afferents in fear learning using Cre-loxP technology for preferentially knocking out presynaptically acting molecules in the cortex but not those in the thalamus. Rap1 is a small GTPase that has been found in both presynaptic and postsynaptic terminals (Kim et al., 1990). It has been implicated in regulation of AMPA receptor (AMPAR) trafficking (Zhu et al., 2002), long-term potentiation (LTP) of synaptic transmission in the hippocampus, and spatial learning (Morozov et al., 2003); however, its potential presynaptic role has not yet been explored. Here, we investigated the effects of CaMKII-α-Cre-mediated ablation of rap1a and rap1b genes on the synaptic properties of the thalamic and cortical inputs to the LA and on amygdala-dependent fear learning. The ablation selectively altered synaptic transmission in the cortical but not thalamic inputs to the LA because of the presynaptic but not postsynaptic changes. Moreover, the mutant mice showed impaired auditory fear conditioning, suggesting that sensory inputs from cortex to LA are necessary for fear learning.
Materials and Methods
Generation of mice with restricted knock-out of rap1a and rap1b genes
Rap1a targeting.
Bacterial artificial chromosome (BAC) genomic clones containing rap1a exons 2 and 3 were cloned and mapped using rap1a cDNA as a probe. A targeting construct was made from a 13 kb genomic fragment between the BamHI site upstream of exon 2 and the second NheI site downstream of exon 3 (see Fig. 1A). A single LoxP sequence containing a BamHI site was inserted into the NcoI site upstream of exon 2, and a TK-NEO cassette flanked by the LoxP sequences was inserted into the ApaI site downstream of exon 3. After electroporation into 129SVEV-derived MM13 embryonic stem (ES) cells (obtained from M. Mendelson, Columbia University, New York, NY), DNA from G418 (Geneticin, Invitrogen, Carlsbad, CA)-resistant clones was digested with BamHI and analyzed by Southern blot using a 0.8 kb KpnI/BamHI fragment as a probe. Targeted alleles produced an 8.3 kb BamHI fragment after the insertion of neo gene containing a BamHI site, and the wild-type (WT) allele produced a 14.4 kb band (see Fig. 1A). Transient expression of Cre-recombinase in the targeted clones, followed by the selection with gancyclovir, produced clones with the deletion of the TK-NEO cassette alone or together with the rap1a exons 2 and 3. To identify both types of deletions, DNA from gancyclovir-resistant clones was digested with BamHI and probed with the 0.8 kb KpnI/BamHI fragment. The floxed rap1a allele produced a 10.1 kb fragment, and the complete knock-out (KO) allele yielded a 9 kb fragment. The same strategy was used for mouse genotyping.
Figure 1.

Generation of mice with conditional KO of rap1a and rap1b genes. A, Targeting of rap1a. 1, Targeted locus. 2, Floxed rap1a allele after deletion of the TK-NEO cassette by transient expression of Cre recombinase. 3, The rap1a allele after deletion of coding exons 2 and 3. Rap1a encoding exons 2 and 3 (ex2, ex3), the TK-NEO cassette, and an 0.8 kb BamHI/KpnI fragment used as a probe are shown. BamHI fragments used for identification of mutated rap1a alleles on Southern blot are marked with the horizontal arrows. 4, Southern blot containing DNA from the ES clones after homologous recombination. +/+, WT clone with the WT alleles (14.4 kb); t/+, clone with a targeted rap1a allele (8.3 kb). 5, Southern blot after Cre-mediated excision. f/+, Clone with a floxed allele (10.1 kb); 0/+, clones with a KO allele (9 kb). B, Targeting of rap1b. 1, Targeted locus. 2, Floxed rap1b allele after deletion of the TK-NEO cassette by transient expression of Cre recombinase. 3, The rap1b allele after deletion of the coding exon 1 (ex1). The TK-NEO cassette and 0.5 kb HindIII/HindIII and 1.3 kb BglII/EcoRI fragments used as probes are shown. EcoRI and HindIII fragments used for identification of mutated rap1b alleles on Southern blot are marked with the horizontal arrows. 4, Southern blot containing DNA from the ES clones after homologous recombination. +/+, WT clone with WT alleles (10 kb); t/+, clone with a targeted rap1b allele (11 kb). 5, Southern blot after Cre-mediated excision. 0/+, Clone with the WT (0.5 kb) and KO allele (2.2 kb); +/+, WT contaminating clone; f/+, clone with a floxed allele (2.5 kb). Restriction endonuclease sites ApaI (A), BamHI (B), BglII (Bg), EcoRI (R), HindIII (H), KpnI (K), NcoI (N), NheI (Nh), SalI (S), and XhoI (X) are shown in parentheses when destroyed by cloning. LoxP sites are indicated by triangles. The regions represented with thick lines correspond to the targeting constructs.
Rap1b targeting.
BAC genomic clones containing the rap1b exon 1 were cloned and mapped using rap1b cDNA as a probe (see Fig. 1B). A targeting construct was made from an 11 kb genomic fragment between the BamHI site upstream of the exon 1 and the SalI site inside of the exon 4. A single LoxP site was inserted into the first HindIII site downstream of exon 1, and a TK-NEO cassette flanked by the LoxP sites was inserted into the XhoI site upstream of exon 1. After electroporation into 129SVEV-derived MM13 ES cells, DNA from G418-resistant clones was digested with EcoRI and analyzed by Southern blot using a 1.3 kb BglII/EcoRI fragment as a probe. Targeted alleles produce an 11 kb EcoRI fragment after the insertion of the TK-NEO cassette containing EcoRI sites, and the WT allele produced a 10 kb band (see Fig. 1B). Transient expression of Cre-recombinase in the targeted clones, followed by selection with gancyclovir, produced clones with the deletion of the TK-NEO cassette alone or together with the rap1b exon 1. To identify both types of deletions, DNA from gancyclovir-resistant clones was digested with HindIII and probed with the 0.5 kb HindIII fragment. The floxed rap1b allele produced a 2.5 kb fragment, and the complete KO allele yielded a 2.2 kb fragment. The same strategy was used for mouse genotyping.
To generate mice with the double forebrain rap1a/rap1b KO, the double-floxed homozygous male rap1affrap1bff mice were crossed with double-floxed homozygous females that also contained one transgenic allele expressing Cre-recombinase [the L7Ag13 line (Dragatsis and Zeitlin, 2000)] under the control of the CamKIIα promoter (rap1affrap1bff, Cre mice). All offspring carried double-floxed rap1a and rap1b alleles with or without one cre allele. We refer to the former as Rap1 KO mice and to the latter as Rap1 WT mice. Rap1 double-restricted KOs and their littermates had a mixed 129SVEV/C57 (average, 50%/50%) background.
In situ hybridization
Brain sections (20 μm) were hybridized as described previously (Huang et al., 1999) to [α-35S]-dATP-labeled Rap1A (5′-AGTGTCCAGGATCTCCAGCATGCACTGTTGGCAATCTACCTCGACTTG-3′) or Rap1B (5′-CTTCCCAACGCCTCCTGATCCAAGCACGACGAGCTTATATTCACGCAT-3′) antisense oligonucleotides.
Comparison of excision efficiencies in cortex and thalamus using semiquantitative PCR
The areas of interest were dissected from 0.6 mm brain sections (prepared as described below; see Electrophysiology) obtained from six WT and six KO mice. Tissue was digested in 100 μl of 100 mm Tris-HCl, pH 8.5, 200 mm NaCl, 5 mm EDTA, 0.2% SDS, and 100 μg/ml proteinase K at 56°C for 12 h. One microliter of 1:200 diluted digest was used in a 10 μl PCR containing combinations of primers a1 (5′-CCAAGGCTCTCAGTTGATTTCTA-3′), a2 (5′-TATCTGCACATAATCTGCATGCC-3′), and a3 (5′-TAAAGGTGAAGGCCCTATAGGTA-3′) for detecting rap1a alleles, or primers b1 (5′-CCCTCTCATGCTATTCCTAATGT-3′) and b2 (5′-GCCACCTCAGTAGAAAAGACC-3′) for detecting rap1b alleles. The agarose gel images were analyzed using NIH ImageJ software. The relative intensity of DNA bands representing excised alleles were calculated as the percentage of the cumulative intensity of bands corresponding to both excised and floxed alleles. Statistical significance was determined using an unpaired t test. Because of differences in amplification rates of DNA fragments representing excised and floxed alleles, the values obtained here do not show an actual percentage of excised alleles but allow relative comparison of excision efficiencies between thalamus and cortex.
Rap1 Western blot
The areas of interest were dissected and homogenized (0.15 ml per tissue from one brain) in a buffer containing 50 mm Tris-HCl, pH 7.5, 1 mm EDTA, 1 mm EGTA, 0.5 mm sodium vanadate, 0.1% 2-mercaptoethanol, 1% Triton X-100, 50 mm sodium fluoride, 5 mm sodium phosphate, 10 mm glycerophosphate, and 0.1 mm PMSF by passing the tissue through the 23G1 needle 10 times. The insoluble components were removed by centrifugation at 14,000 × g at 4°C. Protein concentration was determined using Bio-Rad (Hercules, CA) protein assay. Fifteen micrograms of total protein were separated in 15% SDS-polyacrylamide gel transferred on polyvinylidene difluoride membrane, and probed with a 1:500 dilution of Rap1 monoclonal antibody (Transduction Laboratories, Lexington, KY) followed by HRP–anti-mouse conjugate (Cell Signaling Technology, Beverly, MA). All antibody incubations were performed in the manufacturer-recommended blocking solutions. The protein bands were visualized by chemiluminescence using LumiGLO reagent (Cell Signaling Technology). X-ray films were scanned using an Epson scanner (Seiko Epson, Nagano, Japan), and band intensity was quantified using ImageJ software. The percentage deletion was calculated as a percentage decrease in the intensity of the Rap1 band from KO mice relative to those from WT mice and compared using an unpaired t test.
Lentiviral expression of Rap1 in amygdala neurons
cDNA for hemagglutinin epitope (HA)-tagged Rap1B was generated by inserting the HA-linker encoding sequence (annealed oligos: 5′-GATCCGCCATGAGCAGCTACCCATACGACGTGCCAGACTACGCCAGCCTGGGCG-3′ and 5′-GGCCCGCCCAGGCTGGCGTAGTCTGGCACGTCGTATGGGTAGCTGCTCATGGCG-3′) between BamHI and ApaI sites of pBlueScript followed by insertion of the ApaI-digested cDNA fragment of Rap1B obtained by PCR amplification of hippocampal cDNA with oligos 5′-CCAAGG-GCCCTCGAGTCTAGATTAAAGCAGCTGACACGATGA-3′ and 5′-GACCGGGCC-CAGAATGCGTGAATATAAGCTC-3′. HA-Rap1B cDNA was inserted between BamHI and XhoI sites of SIN-W-PGK transfer vector (de Almeida et al., 2002). To generate green fluorescent protein (GFP) transfer vector, enhanced GFP cDNA (Clontech, Mountain View, CA) was PCR amplified and inserted between BamHI and XhoI sites of SIN-W-PGK transfer vector. Lentivirus was prepared as described previously (de Almeida et al., 2002) and concentrated to the titer of 5 × 108 transduction units (TD) per milliliter. A total of 2.5 × 105 TD of HA-Rap1- and 0.5 × 105 TD of GFP-expressing viruses in 1 μl of PBS were coinjected bilaterally in LA of 4-week-old mice at 1.4 mm posterior, 3.4 mm medial from bregma, and 4.1 mm ventral from the brain surface. We then confirmed that 82 ± 11% of GFP-positive cells expressed HA-Rap1B (n = 12 areas) (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). Animals were used for electrophysiology experiments 4–5 weeks after surgery.
Electrophysiology
Amygdala slices were prepared from 8- to 10-week-old mice as described previously (Tsvetkov et al., 2002) with minor modifications. The mice were killed by decapitation, and brains were quickly transferred to ice-cold oxygenated (95% O2/5% CO2) artificial CSF (ACSF) containing (in mm) 119 NaCl, 2.5 KCl, 1 MgSO4, 2.5 CaCl2, 10 glucose, and 26 NaHCO3, pH 7.3. Slices containing the LA were cut with a vibratome and incubated at 35°C for ∼40 min. Later, slices were maintained at room temperature for at least 1 h before recording. The slices were then transferred to a recording chamber continuously superfused with ACSF at a constant rate of ∼60 ml/h. The temperature was held at 30 ± 1°C. Whole-cell recordings of compound EPSPs or EPSCs were obtained from pyramidal cells in the LA under visual guidance (differential interference contrast/infrared optics) with an EPC-10 amplifier and Pulse software (version 8.76; HEKA Elektronik, Lambrecht/Pfalz, Germany). Data were sampled at 10 kHz and filtered at 5 kHz. For “minimal stimulation” experiments, data were filtered at 1 kHz. Compound synaptic responses were evoked by field stimulation of the fibers either in the external capsule (cortical input) or internal capsule (thalamic input) by a 1–3 MΩ glass stimulation electrode (6–8 MΩ for the minimal stimulation) filled with ACSF. In most experiments, the patch pipette was filled with (in mm) 120 K-gluconate, 5 NaCl, 1 MgCl2, 10 HEPES, 0.2 EGTA, 2 ATP-Mg, and 0.1 GTP-Na. The pH was adjusted to 7.3 with KOH, and osmolarity was adjusted to 285 Osm with sucrose. In experiments investigating voltage dependence of EPSC and decay of NMDA receptor (NMDAR) currents in the presence of (+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate (MK-801), Cs+ was used instead of K+ (the pH adjusted to 7.3 by CsOH). The patching pipette resistance was 3–7 MΩ. A concentration of 100 μm picrotoxin was routinely added to the bath solution to block GABAA receptors. All membrane potentials were corrected by a junction potential of 12 mV. Series resistance (Rs) was in the range of 10–20 MΩ and monitored throughout the experiment. If Rs changed >20%, the data were not included in analysis.
In LTP experiments, stimulus intensity was adjusted to produce responses of ∼20–30% of the maximum amplitude. For the spike-timing-dependent plasticity (STD-LTP) protocol, the membrane potential was held at −80 mV for the basal state recordings, and LTP was induced with trains of 10 Hz monosynaptic EPSPs with each EPSP followed by individual action potentials (elicited by 1 nA, 5 ms current steps: EPSP onset to action potential peak delay was no more than 10 ms). This pattern was repeated 15 times at 0.1 Hz. In the pairing protocol, LTP was induced by 80 presynaptic stimuli at 2 Hz while the cell was held at 0 mV. Synaptic strength was measured as the initial slope (first 2–3 ms) of EPSP or the amplitude of EPSC. LTP was quantified by normalizing the data collected in the last 5 min to the mean value of the baseline EPSP(C), recorded at 0.05 Hz for at least 6 min before LTP induction.
For assessing the rate of NMDAR current blockade by MK-801, the recorded neuron was clamped at +30 mV, and the baseline of synaptic responses to 0.1 Hz presynaptic stimulation were recorded for 5 min, followed by the bath application of MK-801 (20 μm) for 10 min without stimulation. Stimulation at 0.1 Hz was then resumed for 20 min. EPSC amplitudes were normalized to the first EPSC. For minimal stimulation, the EPSC amplitudes were measured as the difference between the mean current during the baseline and the mean current over a 1–2 ms window at the peak of EPSC (Bolshakov et al., 1997).
For the minimal stimulation, stimulus intensity was adjusted to just above the threshold: a 10% reduction in the stimulus intensity resulted in all response failures, whereas a 20% increase above the threshold did not alter the average potency of the responses (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The latencies of the responses were invariable throughout repeated stimulations. If the large multiquantal EPSCs in the KO slices were attributable to the recruitment of multiple presynaptic fibers, we would expect that differences in firing latency, conduction, release properties, and electrotonic distance among different fibers would lead to a broadening of the rising phase of EPSCs (Bolshakov et al., 1997). However, we find that the rise time of the large EPSCs is identical to that of the small ones at a given synaptic connection.
For experiments evaluating inhibition of EPSCs by γ-d-glutamylglycine (γ-DGG), only cells showing good recovery of the paired-pulse ratio (PPR) after γ-DGG washout and during perfusion with CNQX (<10% change) were included in the analysis.
Data were expressed as mean ± SEM. Statistical significances were obtained by using paired, or unpaired, or one-group t test. For the analysis of quantal distribution, the Kolmogorov–Smirnov test was used.
Fear conditioning
All experiments were approved by the National Institute of Mental Health Animal Care and Use Committee. The tests were performed on KO and WT male littermates at 3–4 months of age. The experimenter was blind to the genotypes. Conditioning was done as described previously (Herry and Garcia, 2002) with minor modifications; the conditioned stimulus (CS) tone was 12 kHz instead of 2.6 kHz, and long-term fear memory was tested at 48 h instead of 24 h after training, because animals with the 129/C57 mixed background showed stronger conditioning with the 12 kHz tone and less fear generalization from the training to testing context 48 h after training. For the training, the mouse was placed in the conditioning chamber (Med Associates, St. Albans, VT) for 2 min, and five CS–unconditioned stimulus (US) pairings were presented in variable intervals (60–180 s). The CS was a 30 s, 12 kHz, 80 dB tone, and the US was either a 0.5 or 1 s 0.5 mA electrical shock coterminated with the CS. After an additional 30 s in the chamber, mice were returned to their home cages. Cued conditioning was assessed in a different context at 3 or 48 h after the training; the CS (2 min), which had been presented during training, was given after a 1 min habituation period (pre-CS). For all animals tested in cued conditioning, the freezing data before the CS–US pairing were collapsed between the same genotype cohorts receiving identical training. Contextual conditioning was tested on separate groups of animals; it was assessed 48 h after training in the training context (3 min). Freezing was defined as a complete lack of movement measured by a video-based system (Freezeframe; Actimetrics Software, Wilmette, IL). The effect of genotype on freezing was analyzed using a two-way ANOVA.
Water maze experiment
Water maze training and testing was performed essentially as described previously (Bourtchuladze et al., 1994), with the following modifications. After 1 week of handling, mice were trained in a Morris water maze receiving four trials per day with a 20–30 min intertrial interval. During the first 2 d, mice underwent “cued training,” in which a platform was marked by a cue and its position was changed at every trial. During the subsequent 8 d, mice were trained with the hidden platform located in the fixed position throughout the trials. On the 11th day, mice received a 1 min probe trial in the maze without the platform. All trials were analyzed using a video tracking system (HVS Image 2020 Plus). The genotype × day interaction for escape latency and effect of genotype on escape latency were tested using the repeated-measures ANOVA. The time spent by genotypes in the target quadrant during the probe trial was compared using an unpaired two-tailed t test.
Results
Generation of mice with conditional KO of rap1a and rap1b genes
The rap1a and rap1b genes were inactivated by deleting, respectively, exons 2–3 and exon 1 using the three loxP strategy (Fig. 1), and forebrain restriction of the deletions was obtained by crossing transgenic mice expressing Cre-recombinase under the control of the CaMKII-α promoter with the floxed animals (see Materials and Methods). In KO mice, the level rap1a and rap1b mRNA was reduced in the forebrain areas including cortex and hippocampus (Fig. 2A). The efficiencies of rap1a and rap1b gene excision between areas of cortex and thalamus containing neurons projecting to the LA (Fig. 2D) were compared by genomic PCR, which allowed simultaneous detection of excised and nonexcised (floxed) alleles. The relative intensities of bands representing excised alleles in the cortex exceeded those for the thalamus (rap1a: cortex, 0.39 ± 0.05; thalamus, 0.19 ± 0.05; n = 6; p = 0.015; rap1b: cortex, 0.87 ± 0.03; thalamus, 0.67 ± 0.05; n = 6; p = 0.014) (Fig. 2B). A quantification by Western blot (Fig. 2C) revealed an 89.4 ± 3.4% reduction in the total Rap1 protein level in the cortex and a 38.7 ± 14.5% reduction in the thalamus (n = 3; p = 0.027), indicating a more complete cortical ablation of Rap1.
Figure 2.

More efficient deletion of Rap1 genes in cortex compared with thalamus. A, In situ hybridization with antisense rap1a (left) and rap1b (right) probes. B, Comparison of excision efficiencies of the floxed rap1a (left) and rap1b (right) alleles by Cre-recombinase in the cortex (c) and thalamus (t) using semiquantitative PCR. From left to right, Representative agarose gels, length of amplification products in base pairs (bp) corresponding to the floxed and excised alleles, positions of PCR primers sequences (a1, a2, and a3 for rap1a and b1 and b2 for rap1b) on genomic loci, and relative amounts of the PCR products representing excised alleles. *p < 0.05. C, Left, Representative Western blot showing Rap1 total protein expression in the cortex and thalamus; the position of molecular weight standards (numbers indicate kilodaltons) are shown on the left. Arrows point to a 22 kDa Rap1 band (Rap1) and a nonspecific band (ns). Right, Quantification of KO efficiency in the cortex and thalamus by measuring a decrease in the intensity of the Rap1 protein bands from KO relative to WT samples run on same gels. *p < 0.05. D, Dissection scheme for the preparation of genomic DNA and protein extracts.
Rap1 deletion impairs plasticity and increases basal synaptic transmission in the cortical but not thalamic input to LA
To examine the effect of Rap1 KO on LTP of synaptic transmission in the LA inputs, we performed whole-cell recordings from visually identified pyramidal neurons in the LA. We first used a protocol that induces spike-timing-dependent plasticity. This form of LTP results from a temporal association of presynaptic stimulation with an action potential induced in a postsynaptic neuron by current injections through the recording electrode with a short delay from the onset of the EPSP. STD-LTP is considered to be a close physiological model of synaptic modifications that may occur during the integration of several inputs, when the effects of presynaptic stimulation and postsynaptic depolarization interact (Sjostrom et al., 2001).
In the cortico-amygdala pathway, STD-LTP was readily evoked in 8 of 11 cells in slices from WT mice but was substantially reduced in slices from KO animals, in which only 2 of 7 cells showed LTP (mean LTP: WT, 161.2 ± 18.7%, n = 11; KO, 109.9 ± 10.8%, n = 7; p = 0.032) (Fig. 3A,B). In contrast, in thalamic input, STD-LTP could not be induced in slices from mice of either genotype (WT, 96.2 ± 5.8%, n = 9; KO, 105.2 ± 13.4%, n = 10; p = 0.534). Another form of LTP, induced by the pairing protocol, was present in both inputs and was not affected in KO mice (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Thus, Rap1 deletion selectively eliminated one form of plasticity specific to cortical input.
Figure 3.
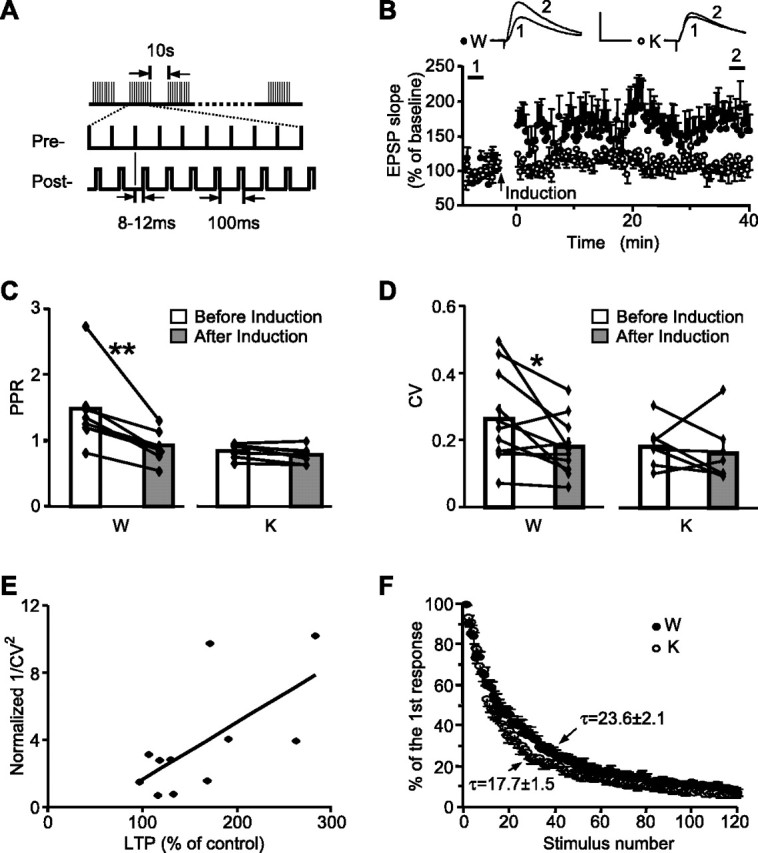
Impaired STD-LTP is associated with elevated Pr in the cortical inputs to LA in Rap1 KO mice. WT (W) and KO (K) slices. A, STD-LTP protocol. B, LTP in cortical input to LA. Insets represent EPSP before (1) and after (2) induction. Calibration: 10 mV, 10 ms. C, PPR before and after LTP induction in cortical input to LA. **p < 0.01. D, CV of EPSP slopes before and after LTP induction in cortical input to LA. *p < 0.05. E, Plot of CV−2 against percentage LTP in slices from WT mice (r = 0.647; n = 11). F, The progressive blockade of NMDA currents by MK-801 during cortical input stimulation. Each point represents averaged normalized currents from eight WT and seven KO neurons.
Plasticity in cortical inputs to the LA correlates with an increase in neurotransmitter release from presynaptic terminals (McKernan and Shinnick-Gallagher, 1997; Tsvetkov et al., 2002). To evaluate possible presynaptic changes in cortical input to LA during STD-LTP, we first looked at paired-pulse facilitation (PPF), a form of short-term plasticity that is primarily a presynaptic process (Zucker and Regehr, 2002). In slices from WT mice, under basal conditions, the PPR was 1.43 ± 0.20, decreased to 0.91 ± 0.08 after the STD-LTP induction protocol (n = 8; p = 0.006) (Fig. 3C), reflecting an increase in the probability of neurotransmitter release (Pr). Consistent with the changes of PPR, the coefficient of variation (CV) of the EPSP slope, an index reversely related to Pr (Malinow and Tsien, 1990), decreased after the induction of STD-LTP (before, 0.26 ± 0.04; after, 0.18 ± 0.02; n = 11; p = 0.028) (Fig. 3D). Together with the positive correlation between STD-LTP and CV−2 (Fig. 3E), these data confirm the presynaptic expression of plasticity in WT slices. In contrast to the WT, slices from KO mice did not show PPF even in the basal state, and neither PPR nor CV changed after the STD-LTP induction protocol (PPR: before, 0.90 ± 0.11; after, 0.83 ± 0.09; n = 7; p = 0.413; CV: before, 0.18 ± 0.02; after, 0.16 ± 0.03; n = 7; p = 0.590) (Fig. 3C,D).
A direct comparison of Pr between mice of two genotypes by measuring the decay of the NMDAR currents during repetitive stimulation of the cortical input in the presence of the noncompetitive NMDAR antagonist MK-801 revealed a 25% faster current decay in slices from KO mice (Fig. 3F) (single exponential fit decay constant; WT: τ = 23.6 ± 2.1, n = 8; KO: t = 17.7 ± 1.5, n = 7; p = 0.045). Together, these data suggest that in slices from Rap1 KO mice the basal Pr is increased at the cortico-amygdala synapses, thus preventing further Pr increases by the STD-LTP induction protocol.
We next examined the effects of Rap1 KO on basal synaptic transmission by recording input–output relationships in two convergent pathways. As expected, Rap 1 ablation did not change synaptic efficacy in thalamic input (the input–output relationship slope; WT: 28.2 ± 3.8 mV/mS · mA, n = 13; KO: 32.4 ± 4.1 mV/mS · mA, n = 11; p = 0.470) but increased it in the cortical input by almost 2.5-fold (WT: 36.8 ± 6.0 mV/mS · mA, n = 13; KO: 90.4 ± 17.1 mV/mS · mA, n = 13; p = 0.007) (Fig. 4A). Because the input–output relationship slope is proportional to the mean efficacy at the individual synaptic connections, which is linearly related to Pr, the 2.5-fold efficacy increase in the KO slices could not be explained solely by a small increase in Pr (estimated to be ∼25%, based on the MK-801 experiment) and therefore was likely caused by increases in the postsynaptic response to a single quantum of transmitter, and/or the number of release sites (Katz, 1969; Gil et al., 1999). There was a possibility that the cortical enhancement of synaptic strength was attributable to the impaired removal of AMPARs from postsynaptic membrane, a process presumably regulated by Rap1 (Zhu et al., 2002). However, the identical current–voltage relationships for AMPAR and NMDAR currents and similar AMPAR/NMDAR current ratios (WT: 2.13 ± 0.27, n = 9; KO: 1.88 ± 0.26, n = 10; p = 0.42) argued against it (Fig. 4B–D). Moreover, both genotypes did not differ in the mean amplitudes (WT: 10.5 ± 1.1pA, n = 7; KO: 10.2 ± 0.6pA, n = 8; p = 0.755) or amplitude distribution (p > 0.9, Kolmogorov–Smirnov test) (Fig. 4E) of the AMPAR-mediated asynchronous EPSC induced in the presence of Sr2+ and thus reflecting release of single quanta of glutamate (Gil et al., 1999). This finding indicates that the enhanced synaptic efficacy was not caused by postsynaptic changes in quantal amplitude.
Figure 4.
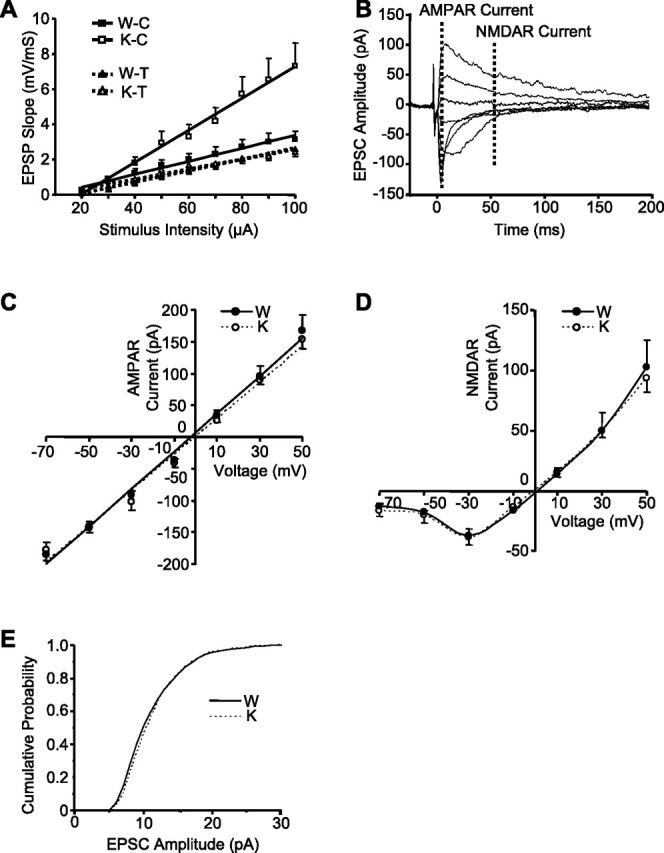
Rap1 KO enhances the efficacy of cortico-LA synapses without altering their postsynaptic properties. A, Input–output curves in the cortical (C) and thalamic (T) inputs to LA. The averaged EPSP slopes are plotted as a function of stimulus intensity. B, Example of EPSCs evoked at holding potentials ranging from −70 to +50 mV at a step of 20 mV. Time points for determination of AMPAR and NMDAR currents are shown with dashed lines. C, D, Averaged current–voltage plots of the AMPAR (C) and NMDAR (D) currents in the WT (filled circles) and KO (open circles) slices. E, Cumulative distributions of the amplitudes of evoked asynchronous EPSCs from seven WT (solid line) and eight KO (dashed line) cells.
An alternative mechanism for the enhanced synaptic transmission could be an increase in the number of quanta released by a single action potential (Kullmann and Siegelbaum, 1995). To test this possibility, we used the minimal stimulation approach, which presumably recruits a single presynaptic fiber, and measured the potencies of successful synaptic responses (Stevens and Wang, 1994). The mean amplitude of the “successes” (potency) was much higher in slices from KO mice (WT: 14.5 ± 2.2pA, n = 10; KO: 34.8 ± 8.0 pA, n = 10; p = 0.025) (Fig. 5A,B), suggesting an ∼2.5-fold rise in the quantal content (given the identical mean amplitude of quantal EPSCs across the genotypes in the experiments when Sr2+ was substituting for external Ca2+). No potency differences between genotypes were found in thalamic input (supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
Figure 5.
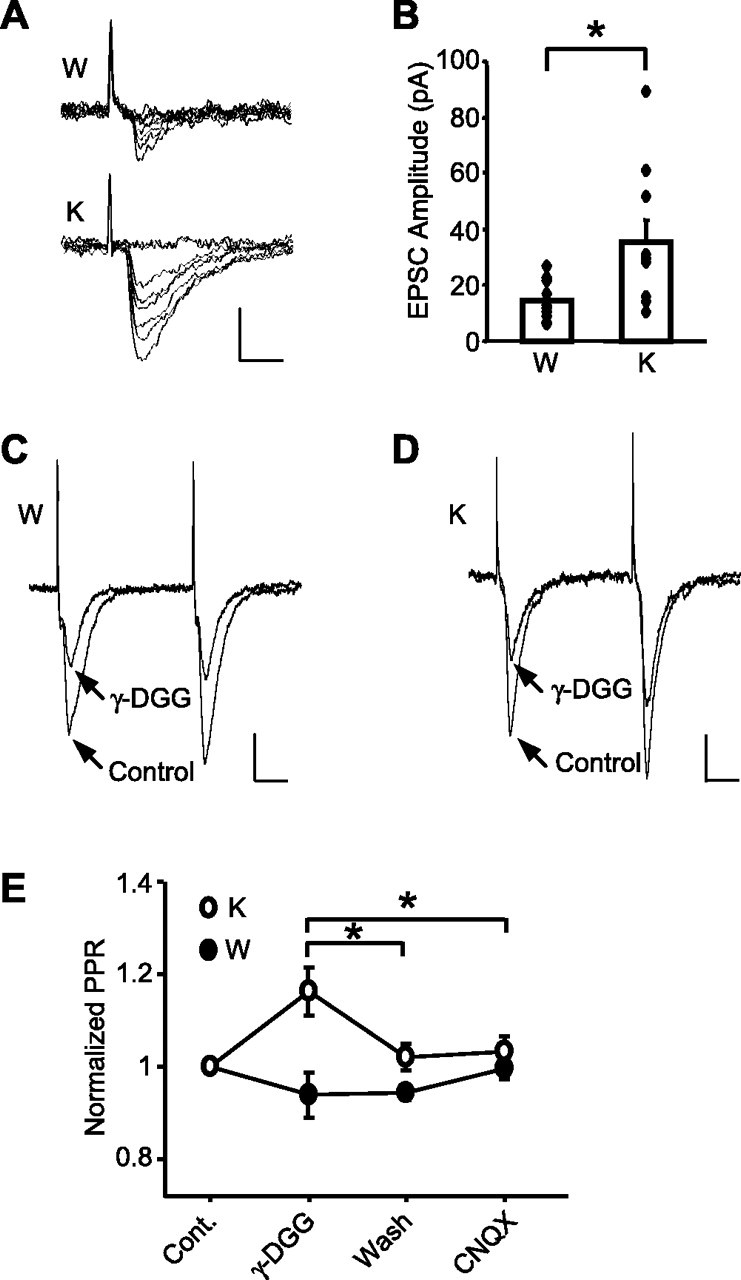
Rap1 KO causes multiquantal release in the cortical input to LA. A, Examples of EPSCs evoked by minimal stimulation in the WT (W) and KO (K) neurons. Calibration: 20 pA, 5 ms. B, Averaged EPSC amplitude of successful responses to minimal stimulation. Diamonds show mean values from individual neurons, and boxes show averaged responses for each genotype. *p < 0.05. C, D, Representative traces of paired EPSCs at an interval of 50 ms before and after γ-DGG application in WT (W) and KO (K) slices. Calibration: 50 pA, 10 ms. E, Summary results showing an increase in the PPR after γ-DGG application in KO but not WT slices. *p < 0.05.
To further confirm that multiquantal release of glutamate may occur in cortical input of KO mice, we tested the effects of γ-DGG, a low-affinity AMPAR antagonist, on EPSCs evoked by paired stimulation. In the case of multivesicular release of glutamate, one would expect a positive correlation between glutamate concentration in the synaptic cleft and Pr, whereas in the case of a single-vesicular release, the glutamate concentration would be independent of Pr (Wadiche and Jahr, 2001; Christie and Jahr, 2006). Subsequently, γ-DGG would produce a relatively weaker inhibition of the AMPA EPSCs arising from the higher concentrations of glutamate. To this end, we examined the relative inhibition of paired EPSCs by γ-DGG during PPF. To obtain PPF, we reduced the external Ca2+ concentration to 1.5 mm when recording from KO slices. In WT slices, γ-DGG (2 mm) similarly inhibited both EPSCs (first EPSC, 52.8 ± 4.2%; second EPSC, 51.8 ± 4.4%; n = 6) with no significant effect on PPR (Fig. 5C,E). In KO slices, γ-DGG inhibited the first EPSC (53.0 ± 3.8%) more strongly than the second EPSC (45.9 ± 4.9%) (n = 6; p = 0.044), leading to a 16.6 ± 5.3% increase in the PPR (Fig. 5D,E). Subsequent perfusion of the same slices with a high-affinity AMPAR antagonist, CNQX (500 nm), after γ-DGG washout resulted in equal inhibition of both EPSCs and did not change the PPR in both genotypes. These data revealed that glutamate concentration in the synaptic cleft was dependent on Pr in KO but not in WT mice, indicating multivesicular release at cortico-amygdala synapses in the absence of Rap1.
The increases in Pr and quantal content suggest presynaptic actions of Rap1; however, postsynaptic absence of Rap1 may also affect presynaptic release via trans-synaptic mechanisms (Macleod and Zinsmaier, 2006). To examine this possibility, we recorded from LA neurons coexpressing Rap1 and GFP in KO slices (supplemental Figs. 4, 5, available at www.jneurosci.org as supplemental material). The input–output relationship (KO: 90.4 ± 17.4 mV/mS · mA, n = 13; KO plus Rap1: 84.3 ± 10.6 mV/mS · mA, n = 10; p = 0.780) and responses to minimal stimulation (KO: 34.8 ± 8.0 pA, n = 10; KO plus Rap1: 27.7 ± 8.1 pA, n = 7; p = 0.557) of the cortical pathway were not altered by postsynaptic reintroduction of Rap1, arguing against a possible trans-synaptic effect of Rap1 KO.
Fear learning is impaired in Rap1 KO mice
We next tested the effects of Rap1 KO on fear conditioning (Fig. 6A). Before training, KO mice did not differ from WT in freezing to a new context (pretraining) (WT: 2.1 ± 0.8%, n = 28; KO: 1.4 ± 0.4%, n = 27; F(1,53) = 0.62; p = 0.434) or CS (CS1) (WT, 4.4 ± 1.3%; KO, 2.5 ± 1.2%; F(1,53) = 1.13; p = 0.292). In a different testing context, 48 h after training, KO mice exhibited a similar amount of freezing as WT animals before the CS (WT: 3.7 ± 2.1%, n = 19; KO: 1.9 ± 1.2%, n = 17; F(1,34) = 0.52; p = 0.477) but less CS-induced freezing (pre-CS freezing subtracted) (WT: 58.5 ± 4.5%, n = 19; KO: 33.9 ± 6.6%, n = 17; F(1,34) = 9.7; p = 0.004), indicating impairment in long-term fear memory. To investigate the KO effect on short-term fear memory, a separate group of mice was tested 3 h after training. In that group, KO mice froze less than WT mice in the testing context before the CS (WT: 9.7 ± 3.6%, n = 9; KO: 0.9 ± 0.6%, n = 10; F(1,17) = 6.6; p = 0.020), showing reduced generalization of context conditioning from the training to the testing chamber. A CS-induced freezing (generalization component subtracted) was also reduced in KO mice (WT: 57.4 ± 6.4%, n = 9; KO: 31.5 ± 9.4%, n = 10; F(1,17) = 5.0; p = 0.040), indicating an impairment of short-term fear memory. When a separate group of animals was tested in the training context 48 h after training, KO mice froze less than WT (WT: 61.0 ± 6.0%, n = 8; KO: 19.6 ± 3.8%, n = 9; F(1,15) = 35.8; p < 0.001), indicating a disruption of contextual fear memory.
Figure 6.

Rap1 KO mice require more aversive US for fear learning. A, Conditioning with the 0.5 s US. Freezing before CS onset (pretraining), freezing during the first CS (CS1), and change in freezing (percentage CS freezing − percentage pre-CS freezing) at 3 and 48 h are shown. *p < 0.05. B, Rescue of fear learning with the 1 s US. Same abbreviations as in A. C, Pain thresholds. Thresholds of current intensities for flinching and running/jumping are shown. *p < 0.05.
The impaired fear learning could be attributable to reduced pain sensitivity or general cognitive deficiencies. To determine pain threshold, animals were exposed to electrical footshocks of increasing intensities (Bourtchuladze et al., 1994). The thresholds for running/jumping in response to the shock did not differ between genotypes (WT: 0.34 ± 0.01, n = 8; KO: 0.32 ± 0.02, n = 6; F(1,12) = 0.81; p = 0.390), whereas the threshold for flinching was even lower in KO mice (WT: 0.28 ± 0.02, n = 8; KO: 0.21 ± 0.02, n = 6; F(1,12) = 8.41; p = 0.013) (Fig. 6C), indicating that the impairments in fear learning were not attributable to nociception deficits. General cognitive deficiencies were also ruled out, because mutation did not affect spatial memory tested in the Morris water maze (supplemental Fig. 6, available at www.jneurosci.org as supplemental material). When mice were trained with a longer and more aversive US (1 s instead of 0.5 s), both genotypes demonstrated similar levels of the CS-induced freezing (WT: 68.9 ± 9.9%, n = 7; KO: 52.4 ± 11.2%, n = 9; F(1,14) = 1.1; p = 0.305) (Fig. 6B, 48 h) and contextual freezing (WT: 57.4 ± 9.4%, n = 9; KO: 49.5 ± 7.6%, n = 11; F(1,18) = 0.43; p = 0.52), suggesting that Rap1 KO only impairs fear learning induced by a moderately aversive US.
Discussion
The aims of the present study were to examine the functions of Rap1 in LA synaptic transmission and to explore the role of cortical input to the LA in fear learning by analyzing mice carrying a predominantly cortical deletion of the rap1a and rap1b genes. The analysis resulted in two findings. First, it revealed a presynaptic role for Rap1, which has thus far only been implicated in postsynaptic neurotransmission (Zhu et al., 2002; Morozov et al., 2003). Second, contrary to the existing view that thalamic and cortical inputs to the LA can fully support auditory fear conditioning independently of one another, we demonstrated that alterations in the cortical pathway could impair auditory fear conditioning even when the thalamic pathway remains functionally intact.
Presynaptic role of Rap1
The elimination of Rap1 impaired synaptic plasticity and enhanced synaptic efficacy in the cortical input to the LA. These phenomena could result from either postsynaptic and/or presynaptic changes. Postsynaptically, the expression of the interfering Rap1 mutant in cultured hippocampal neurons has been shown to inhibit removal of AMPARs from the plasma membrane (Zhu et al., 2002), thus increasing AMPAR density and elevating amplitudes of AMPAR-mediated responses. Three sets of our data, however, argue against such a scenario in the present system. First, the amplitudes of the quantal responses during asynchronous neurotransmitter release in the presence of Sr2+ were not affected by KO, indicating that the AMPAR current generated by a single quantum of glutamate was independent of Rap1 in LA neurons. Second, Rap1 KO did not change the ratio between AMPAR and NMDAR currents evoked in LA neurons by cortical input stimulation. Third, reintroduction of Rap1 into postsynaptic neurons of the KO mice did not reverse the enhanced efficacy of synaptic transmission; it also did not reduce the amplitudes of responses to minimal stimulation. Thus, Rap1 KO did not alter postsynaptic responses in the amygdala but, rather, changed presynaptic release via two mechanisms. First, it increased Pr as reflected by the loss of PPF and a more rapid block of postsynaptic NMDARs during continuous stimulation of the cortical input in the presence of MK-801. Second, it increased the potency of synaptic responses to minimal stimulation by more than twofold. Given invariant quantal amplitudes during asynchronous release in the presence of Sr2+, this change might reflect an increase in the number of synchronously released quanta after activation of a single presynaptic fiber. Accordingly, we found that in KO slices, application of γ-DGG caused stronger inhibition of the first than the second response during paired stimulation. These findings, together with the evidence of Rap1 expression in presynaptic terminals (Kim et al., 1990), suggest a direct involvement of Rap1 in modulation of neurotransmitter release. However, unveiling the molecular mechanism underlying this modulation will require further investigation.
Contribution of cortical input to LA in fear learning
Rap1 KO mice with altered cortico-amygdala but intact thalamo-amygdala transmission were impaired in cued and contextual fear conditioning. Whereas contextual conditioning depends on both amygdala and hippocampus (LeDoux et al., 1990; Bailey et al., 1999; Goosens et al., 2000), the cued conditioning requires amygdala and is considered hippocampus independent (Kim and Fanselow, 1992; Phillips and LeDoux, 1992). Given normal spatial learning by KO mice in the Morris water maze, the deficits in fear conditioning are likely caused by changes in the amygdala-based circuitry. The normal spatial learning by KO mice disagrees with the previously found impairment in transgenic mice expressing the N17Rap1 interfering mutant (Morozov et al., 2003). One explanation for the discrepancy could be compensatory changes mediated by other small GTPases, which may alleviate spatial memory deficits in the KOs but not in the N17Rap1 transgenic mice. The interfering N17Rap1 mutant is believed to act by titrating out Rap1 GDP/GTP-exchange factors (Dupuy et al., 2005) including EPAC (exchange nucleotide protein directly activated by cyclic adenosine-3′,5′-monophosphate), which activates not only Rap1 but several other small GTPases as well (Bos, 2006). Such broader inhibition of small GTPase signaling may block compensatory changes in the N17Rap1 transgenics. However, the ultimate answer to the question would become possible after identification of molecular targets, which mediate Rap1 functions.
Given the forebrain-wide elimination of Rap1, we still cannot completely rule out a possibility that the origin of the impairment of fear learning resides in brain areas outside the thalamo-cortico-amygdala circuit. However, considering numerous evidence that synaptic modifications underlying auditory fear conditioning occur primarily in the thalamic and cortical inputs to the LA (LeDoux, 1992; McKernan and Shinnick-Gallagher, 1997; Shi and Cassell, 1997; Doron and LeDoux, 1999; Tsvetkov et al., 2002), the changes in the cortico-amygdala pathway are the most plausible cause of the fear-learning deficit in the Rap1 KO mice.
Such interpretation appears to be contradictory to the current view that the thalamic pathway can completely support fear conditioning to a simple auditory CS even in the absence of the cortical pathway (Romanski and LeDoux, 1992). The view is based on experiments that demonstrated normal fear learning in rats with pretraining cortical lesions (Romanski and LeDoux, 1992; Campeau and Davis, 1995; Boatman and Kim, 2006). The apparent contradiction appears to originate from differences between fear-conditioning procedures used in the present work versus the previous studies. In our study, the fear-learning deficit in Rap1 KO mice could only be detected with a moderately, but not a highly, aversive US. Notably, the US used in the lesion studies was even more aversive than the most aversive US in this work (compare 1–2.5 mA for 0.5–1 s with 0.5 mA for 1 s footshocks) (Romanski and LeDoux, 1992; Campeau and Davis, 1995; Boatman and Kim, 2006) and produced “normal” fear learning in animals with cortical lesions. Similarly, GluR3 KO mice, which have impaired cortico-amygdala but not thalamo-amygdala plasticity, show normal fear learning (Humeau et al., 2007) when trained with the US comparable to the more aversive US used in this study (0.4 mA for 2 s).
Given the need for cortical input during fear learning with a weak US, the question arises: How can this pathway facilitate fear learning? The simplest explanation could be that the cortical input has more mechanisms for plasticity and undergoes plastic changes more easily than the thalamic pathway. Indeed, it can express both presynaptic and postsynaptic forms of LTP (Huang and Kandel, 1998; Tsvetkov et al., 2002; Humeau et al., 2003, 2005; Tsvetkov et al., 2004; Shin et al., 2006), whereas plasticity in the thalamic pathway is mainly postsynaptic (Humeau et al., 2005, 2007; Rumpel et al., 2005) (but see Apergis-Schoute et al., 2005). Moreover, several LTP protocols, the Poisson-train stimulation of both pathways (Humeau et al., 2003) and STD-LTP in this study, only induce LTP in the cortical but not thalamic input, indicating that the cortical pathway is more plastic and is more likely to undergo changes during a weak training. Despite these findings, a possible involvement of the thalamic pathway in fear conditioning with a weak US cannot be completely ruled out. A recent study demonstrates that a presynaptic form of LTP in the cortical input requires coactivation of the thalamic input during LTP induction (Humeau et al., 2003), indicating that interaction between these two pathways may contribute to facilitation of the cortical input to the LA.
Yet, the deletion of rap1 genes in this study eliminated 90% of Rap1 protein in the cortex, but it also reduced its level in the thalamus by almost 40%, leaving a possibility that some physiological changes in the thalamo-amygdala pathway, not detected by the current techniques, could have contributed to the deficit in fear learning. Therefore, to determine whether the cortico-amygdala pathway is sufficient for fear learning, animals with selective impairment of plasticity in the thalamo-amygdala but not cortico-amygdala pathway would be needed.
Thus, using genetic elimination of Rap1 predominantly in cortical afferents to the LA, we show that changes in cortical LA input can impair fear learning with a moderate, but not a highly aversive US. The underlying mechanisms may include impaired plasticity in the cortico-amygdala pathway that can compromise the CS–US association. Another intriguing possibility is that Rap1 KO may suppress fear learning by enhancing basal release of glutamate, causing overactivation of the local LA inhibitory network (Lang and Pare, 1997) and elevating detection threshold for sensory stimuli arriving in the LA; however, an additional investigation would be required for testing this hypothesis. Currently, the cortico-amygdala pathway is viewed as the main route for emotional learning when more complex stimuli serve as the CS, whereas a simple auditory CS could be processed equally well by the thalamic pathway (Jarrell et al., 1987; Romanski and LeDoux, 1992). Yet, the present study suggests that a complete processing of even a simple auditory CS depends on the cortical pathway.
Footnotes
This work was supported by the National Institute of Mental Health Intramural Research Program and National Institutes of Health (NIH) Grant NS44185 (V.Y.B.). We thank Eric Kandel for support in generating Rap1 knock-out mice, the NIH Fellows Editorial Board and Keri Martinowich for editorial help, and Boris Mirlas and Gennady Merkulov for genotyping.
References
- Apergis-Schoute AM, Debiec J, Doyere V, LeDoux JE, Schafe GE. Auditory fear conditioning and long-term potentiation in the lateral amygdala require ERK/MAP kinase signaling in the auditory thalamus: a role for presynaptic plasticity in the fear system. J Neurosci. 2005;25:5730–5739. doi: 10.1523/JNEUROSCI.0096-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey DJ, Kim JJ, Sun W, Thompson RF, Helmstetter FJ. Acquisition of fear conditioning in rats requires the synthesis of mRNA in the amygdala. Behav Neurosci. 1999;113:276–282. doi: 10.1037//0735-7044.113.2.276. [DOI] [PubMed] [Google Scholar]
- Boatman JA, Kim JJ. A thalamo-cortico-amygdala pathway mediates auditory fear conditioning in the intact brain. Eur J Neurosci. 2006;24:894–900. doi: 10.1111/j.1460-9568.2006.04965.x. [DOI] [PubMed] [Google Scholar]
- Bolshakov VY, Golan H, Kandel ER, Siegelbaum SA. Recruitment of new sites of synaptic transmission during the cAMP-dependent late phase of LTP at CA3-CA1 synapses in the hippocampus. Neuron. 1997;19:635–651. doi: 10.1016/s0896-6273(00)80377-3. [DOI] [PubMed] [Google Scholar]
- Bos JL. Epac proteins: multi-purpose cAMP targets. Trends Biochem Sci. 2006;31:680–686. doi: 10.1016/j.tibs.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell. 1994;79:59–68. doi: 10.1016/0092-8674(94)90400-6. [DOI] [PubMed] [Google Scholar]
- Campeau S, Davis M. Involvement of subcortical and cortical afferents to the lateral nucleus of the amygdala in fear conditioning measured with fear-potentiated startle in rats trained concurrently with auditory and visual conditioned stimuli. J Neurosci. 1995;15:2312–2327. doi: 10.1523/JNEUROSCI.15-03-02312.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie JM, Jahr CE. Multivesicular release at Schaffer collateral–CA1 hippocampal synapses. J Neurosci. 2006;26:210–216. doi: 10.1523/JNEUROSCI.4307-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida LP, Ross CA, Zala D, Aebischer P, Deglon N. Lentiviral-mediated delivery of mutant huntingtin in the striatum of rats induces a selective neuropathology modulated by polyglutamine repeat size, huntingtin expression levels, and protein length. J Neurosci. 2002;22:3473–3483. doi: 10.1523/JNEUROSCI.22-09-03473.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doron NN, LeDoux JE. Organization of projections to the lateral amygdala from auditory and visual areas of the thalamus in the rat. J Comp Neurol. 1999;412:383–409. [PubMed] [Google Scholar]
- Doyere V, Schafe GE, Sigurdsson T, LeDoux JE. Long-term potentiation in freely moving rats reveals asymmetries in thalamic and cortical inputs to the lateral amygdala. Eur J Neurosci. 2003;17:2703–2715. doi: 10.1046/j.1460-9568.2003.02707.x. [DOI] [PubMed] [Google Scholar]
- Dragatsis I, Zeitlin S. CaMKIIalpha-Cre transgene expression and recombination patterns in the mouse brain. Genesis. 2000;26:133–135. doi: 10.1002/(sici)1526-968x(200002)26:2<133::aid-gene10>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Dupuy AG, L'Hoste S, Cherfils J, Camonis J, Gaudriault G, de Gunzburg J. Novel Rap1 dominant-negative mutants interfere selectively with C3G and Epac. Oncogene. 2005;24:4509–4520. doi: 10.1038/sj.onc.1208647. [DOI] [PubMed] [Google Scholar]
- Gil Z, Connors BW, Amitai Y. Efficacy of thalamocortical and intracortical synaptic connections: quanta, innervation, and reliability. Neuron. 1999;23:385–397. doi: 10.1016/s0896-6273(00)80788-6. [DOI] [PubMed] [Google Scholar]
- Goosens KA, Holt W, Maren S. A role for amygdaloid PKA and PKC in the acquisition of long-term conditional fear memories in rats. Behav Brain Res. 2000;114:145–152. doi: 10.1016/s0166-4328(00)00224-2. [DOI] [PubMed] [Google Scholar]
- Herry C, Garcia R. Prefrontal cortex long-term potentiation, but not long-term depression, is associated with the maintenance of extinction of learned fear in mice. J Neurosci. 2002;22:577–583. doi: 10.1523/JNEUROSCI.22-02-00577.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Kandel ER. Postsynaptic induction and PKA-dependent expression of LTP in the lateral amygdala. Neuron. 1998;21:169–178. doi: 10.1016/s0896-6273(00)80524-3. [DOI] [PubMed] [Google Scholar]
- Huang ZJ, Kirkwood A, Pizzorusso T, Porciatti V, Morales B, Bear MF, Maffei L, Tonegawa S. BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell. 1999;98:739–755. doi: 10.1016/s0092-8674(00)81509-3. [DOI] [PubMed] [Google Scholar]
- Humeau Y, Shaban H, Bissiere S, Luthi A. Presynaptic induction of heterosynaptic associative plasticity in the mammalian brain. Nature. 2003;426:841–845. doi: 10.1038/nature02194. [DOI] [PubMed] [Google Scholar]
- Humeau Y, Herry C, Kemp N, Shaban H, Fourcaudot E, Bissiere S, Luthi A. Dendritic spine heterogeneity determines afferent-specific Hebbian plasticity in the amygdala. Neuron. 2005;45:119–131. doi: 10.1016/j.neuron.2004.12.019. [DOI] [PubMed] [Google Scholar]
- Humeau Y, Reisel D, Johnson AW, Borchardt T, Jensen V, Gebhardt C, Bosch V, Gass P, Bannerman DM, Good MA, Hvalby O, Sprengel R, Luthi A. A pathway-specific function for different AMPA receptor subunits in amygdala long-term potentiation and fear conditioning. J Neurosci. 2007;27:10947–10956. doi: 10.1523/JNEUROSCI.2603-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrell TW, Gentile CG, Romanski LM, McCabe PM, Schneiderman N. Involvement of cortical and thalamic auditory regions in retention of differential bradycardiac conditioning to acoustic conditioned stimuli in rabbits. Brain Res. 1987;412:285–294. doi: 10.1016/0006-8993(87)91135-8. [DOI] [PubMed] [Google Scholar]
- Katz B. The release of neural transmitter substance. Liverpool, UK: Liverpool UP; 1969. [Google Scholar]
- Kim JJ, Fanselow MS. Modality-specific retrograde amnesia of fear. Science. 1992;256:675–677. doi: 10.1126/science.1585183. [DOI] [PubMed] [Google Scholar]
- Kim S, Mizoguchi A, Kikuchi A, Takai Y. Tissue and subcellular distributions of the smg-21/rap1/Krev-1 proteins which are partly distinct from those of c-ras p21s. Mol Cell Biol. 1990;10:2645–2652. doi: 10.1128/mcb.10.6.2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann DM, Siegelbaum SA. The site of expression of NMDA receptor-dependent LTP: new fuel for an old fire. Neuron. 1995;15:997–1002. doi: 10.1016/0896-6273(95)90089-6. [DOI] [PubMed] [Google Scholar]
- Lang EJ, Pare D. Similar inhibitory processes dominate the responses of cat lateral amygdaloid projection neurons to their various afferents. J Neurophysiol. 1997;77:341–352. doi: 10.1152/jn.1997.77.1.341. [DOI] [PubMed] [Google Scholar]
- LeDoux JE. Brain mechanisms of emotion and emotional learning. Curr Opin Neurobiol. 1992;2:191–197. doi: 10.1016/0959-4388(92)90011-9. [DOI] [PubMed] [Google Scholar]
- LeDoux JE, Cicchetti P, Xagoraris A, Romanski LM. The lateral amygdaloid nucleus: sensory interface of the amygdala in fear conditioning. J Neurosci. 1990;10:1062–1069. doi: 10.1523/JNEUROSCI.10-04-01062.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macleod GT, Zinsmaier KE. Synaptic homeostasis on the fast track. Neuron. 2006;52:569–571. doi: 10.1016/j.neuron.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Malinow R, Tsien RW. Presynaptic enhancement shown by whole-cell recordings of long-term potentiation in hippocampal slices. Nature. 1990;346:177–180. doi: 10.1038/346177a0. [DOI] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- McKernan MG, Shinnick-Gallagher P. Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature. 1997;390:607–611. doi: 10.1038/37605. [DOI] [PubMed] [Google Scholar]
- Morozov A, Muzzio IA, Bourtchuladze R, Van-Strien N, Lapidus K, Yin D, Winder DG, Adams JP, Sweatt JD, Kandel ER. Rap1 couples cAMP signaling to a distinct pool of p42/44MAPK regulating excitability, synaptic plasticity, learning, and memory. Neuron. 2003;39:309–325. doi: 10.1016/s0896-6273(03)00404-5. [DOI] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106:274–285. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- Quirk GJ, Armony JL, LeDoux JE. Fear conditioning enhances different temporal components of tone-evoked spike trains in auditory cortex and lateral amygdala. Neuron. 1997;19:613–624. doi: 10.1016/s0896-6273(00)80375-x. [DOI] [PubMed] [Google Scholar]
- Romanski LM, LeDoux JE. Equipotentiality of thalamo-amygdala and thalamo-cortico-amygdala circuits in auditory fear conditioning. J Neurosci. 1992;12:4501–4509. doi: 10.1523/JNEUROSCI.12-11-04501.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumpel S, LeDoux J, Zador A, Malinow R. Postsynaptic receptor trafficking underlying a form of associative learning. Science. 2005;308:83–88. doi: 10.1126/science.1103944. [DOI] [PubMed] [Google Scholar]
- Shi CJ, Cassell MD. Cortical, thalamic, and amygdaloid projections of rat temporal cortex. J Comp Neurol. 1997;382:153–175. [PubMed] [Google Scholar]
- Shin RM, Tsvetkov E, Bolshakov VY. Spatiotemporal asymmetry of associative synaptic plasticity in fear conditioning pathways. Neuron. 2006;52:883–896. doi: 10.1016/j.neuron.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjostrom PJ, Turrigiano GG, Nelson SB. Rate, timing, and cooperativity jointly determine cortical synaptic plasticity. Neuron. 2001;32:1149–1164. doi: 10.1016/s0896-6273(01)00542-6. [DOI] [PubMed] [Google Scholar]
- Stevens CF, Wang Y. Changes in reliability of synaptic function as a mechanism for plasticity. Nature. 1994;371:704–707. doi: 10.1038/371704a0. [DOI] [PubMed] [Google Scholar]
- Tsvetkov E, Carlezon WA, Benes FM, Kandel ER, Bolshakov VY. Fear conditioning occludes LTP-induced presynaptic enhancement of synaptic transmission in the cortical pathway to the lateral amygdala. Neuron. 2002;34:289–300. doi: 10.1016/s0896-6273(02)00645-1. [DOI] [PubMed] [Google Scholar]
- Tsvetkov E, Shin RM, Bolshakov VY. Glutamate uptake determines pathway specificity of long-term potentiation in the neural circuitry of fear conditioning. Neuron. 2004;41:139–151. doi: 10.1016/s0896-6273(03)00800-6. [DOI] [PubMed] [Google Scholar]
- Wadiche JI, Jahr CE. Multivesicular release at climbing fiber-Purkinje cell synapses. Neuron. 2001;32:301–313. doi: 10.1016/s0896-6273(01)00488-3. [DOI] [PubMed] [Google Scholar]
- Zhu JJ, Qin Y, Zhao M, Van Aelst L, Malinow R. Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell. 2002;110:443–455. doi: 10.1016/s0092-8674(02)00897-8. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]