

Abstract
Obstructive sleep apnea is associated with neural injury and dysfunction. Hypoxia/reoxygenation exposures, modeling sleep apnea, injure select populations of neurons, including hypoglossal motoneurons. The mechanisms underlying this motoneuron injury are not understood. We hypothesize that endoplasmic reticulum injury contributes to motoneuron demise. Hypoxia/reoxygenation exposures across 8 weeks in adult mice upregulated the unfolded protein response as evidenced by increased phosphorylation of PERK [PKR-like endoplasmic reticulum (ER) kinase] in facial and hypoglossal motoneurons and persistent upregulation of CCAAT/enhancer-binding protein-homologous protein (CHOP)/growth arrest and DNA damage-inducible protein (GADD153) with nuclear translocation. Long-term hypoxia/reoxygenation also resulted in cleavage and nuclear translocation of caspase-7 and caspase-3 in hypoglossal and facial motoneurons. In contrast, occulomotor and trigeminal motoneurons showed persistent phosphorylation of eIF-2a across hypoxia/reoxygenation, without activations of CHOP/GADD153 or either caspase. Ultrastructural analysis of rough ER in hypoglossal motoneurons revealed hypoxia/reoxygenation-induced luminal swelling and ribosomal detachment. Protection of eIF-2α phosphorylation with systemically administered salubrinal throughout hypoxia/reoxygenation exposure prevented CHOP/GADD153 activation in susceptible motoneurons. Collectively, this work provides evidence that long-term exposure to hypoxia/reoxygenation events, modeling sleep apnea, results in significant endoplasmic reticulum injury in select upper airway motoneurons. Augmentation of eIF-2a phosphorylation minimizes motoneuronal injury in this model. It is anticipated that obstructive sleep apnea results in endoplasmic reticulum injury involving motoneurons, whereas a critical balance of phosphorylated eIF-2a should minimize motoneuronal injury in obstructive sleep apnea.
Keywords: motor neuron, injury, hypoxia/reoxygenation, oxidative, hypoglossal, apoptosis
Introduction
Obstructive sleep apnea syndrome is present in >2% of the adult population and is associated with neural injury (Young et al., 1993). The pathophysiology of this syndrome involves brief sleep state-dependent obstructions of the upper airway that are associated with hypoxemia. Brief arousals from sleep temporarily restore patency of the airway and oxygenation. In severe sleep apnea, these hypoxia/reoxygenation events may cycle every minute or two throughout sleep. Clinical studies have identified neural dysfunction and injury in adults with obstructive sleep apnea. Sensory nerve action potential amplitudes are reduced in individuals with obstructive sleep apnea, and treatment of sleep apnea partially reverses this defect, supporting the concept that sleep apnea contributes to this neural dysfunction (Dziewas et al., 2007). Electromyographic studies of the palatopharyngeus muscle in individuals with sleep apnea show long polyphasic potentials and reduced amplitude at maximum voluntary effort (Svanborg, 2005). Consistent with functional impairment, histological studies have identified demyelination of motoneurons in resected palatal tissue in obstructive sleep apnea (Woodson et al., 1991; Lindman and Stål, 2002; Boyd et al., 2004). The severity of peripheral nerve dysfunction correlates with oxyhemoglobin desaturations (Mayer et al., 1999). Whether obstructive sleep apnea oxygenation events, independent of obesity, diabetes, and other comorbidities, can injure peripheral neurons requires study in animal models.
Exposure to hypoxia/reoxygenation events in rodents, modeling oxygenation patterns in severe sleep apnea, results in neural injury in select groups of neurons including pyramidal, Purkinje, catecholaminergic wake neurons, and upper airway dilator motoneurons (Gozal et al., 2001; Decker et al., 2003, 2005; Veasey et al., 2004a; Kheirandish et al., 2005; Zhan et al., 2005a,b; Zhu et al., 2007). Long-term exposure to hypoxia/reoxygenation impairs hypoglossal whole-nerve responsiveness to both glutamatergic and serotonergic excitation of hypoglossal motoneurons (Veasey et al., 2004b).
There is increasing evidence that the endoplasmic reticulum (ER) plays a central role in both adaptive responses to and injury from ischemia–reperfusion challenges (DeGracia and Montie, 2004; Hayashi et al., 2004, 2005; Benavides et al., 2005). The unfolded protein response (UPR) in the ER represents an adaptive response to minimize accumulation of misfolded proteins that would be toxic to the cell. This is accomplished by reducing overall protein translation, increasing the production of chaperones, upregulating clearance of improperly folded proteins, and increasing antioxidant capacity (Yoshida, 2007). Several components of this protective response are mediated by phosphorylation of eIF-2a. However, when this stress is insurmountable, the ER may take on the role of executioner, activating proapoptotic proteins, including CCAAT/enhancer-binding protein-homologous protein (CHOP)/growth arrest and DNA damage-inducible protein (GADD153) and caspase-7 (Gorlach et al., 2006; Sokka et al., 2007).
Many motoneurons process immense amounts of membrane and secretory proteins that must be properly folded within the ER (Shaw and Eggett, 2000). Consequently, even under basal conditions, select motoneurons evidence an UPR (Yamauchi et al., 2007). We predicted that brainstem motoneurons with a basal UPR would be more susceptible to hypoxia/reoxygenation ER stress injury, and augmentation of the protective ER response, phosphorylated (p)-eIF-2a, should provide protection across hypoxia/reoxygenation. We now show that hypoxia/reoxygenation exposures modeling obstructive sleep apnea manifest in severe ER injury in select brainstem motoneurons, those with UPR evident under basal conditions. Phosphorylation of eIF-2a across hypoxia/reoxygenation exposure provides effective protection from neural injury.
Materials and Methods
Animals.
Young adult male C57BL/6J (The Jackson Laboratory, Bar Harbor, ME), 8 weeks of age at the start of long-term intermittent hypoxia experiments, were studied. Mice were confirmed pathogen free across hypoxia/reoxygenation exposures. The methods and study protocols were approved in full by the Institutional Animal Care and Use Committee of the University of Pennsylvania and conformed with the revised National Institutes of Health Office of Laboratory Animal Welfare Policy. Food and water were provided ad libitum.
Long-term intermittent hypoxia protocol.
Long-term intermittent hypoxia (LTIH) across the sleep-predominant period of a circadian period is an established model of sleep apnea oxygenation (Gozal et al., 2001; Decker et al., 2003; Veasey et al., 2004a,b; Kheirandish et al., 2005; Zhan et al., 2005; Polotsky et al., 2006). An automated nitrogen/oxygen gas delivery system (Oxycycler model A84XOV; Biospherix, Redfield, NY) was used to deliver hypoxia/reoxygenation, using our previously described protocol (Veasey et al., 2004a,b; Zhu et al., 2007). Briefly, this system produces reductions in ambient oxygen level from 21 to 10% for 5 s every 90 s for LTIH and from 21 to 20% for 5 s every 90 s for sham LTIH, resulting in fluctuations in oxyhemoglobin saturation from 94–98% to 76–84% in LTIH and 96–98 to 96–97% in sham LTIH (Veasey et al., 2004). LTIH and sham LTIH conditions were produced for 10 h of the lights-on period. Humidity, ambient CO2, and environmental temperature were held constant within and across exposures. Two durations of intermittent hypoxia (IH) and sham IH (3 d and 8 weeks) were studied to characterize duration-dependent ER stress responses in motoneurons and an intermediate duration (4 weeks) to test the effectiveness of salubrinal therapy.
Western blot.
Western blotting was used to characterize the short-term ER stress protein response to IH in select upper airway motoneurons: occulomotor (III), motor trigeminal (moV), facial (VII), and vagus and hypoglossal (X/XII). After exposure to 3 d IH or sham IH, macropunches of bilateral motor nuclei were obtained and rapidly frozen using our previously detailed protocol (Zhan et al., 2005a,b). Whole-tissue lysate protein content, measured with the Bradford protein assay (Bio-Rad, Hercules, CA), was standardized for each Western assay across IH and sham IH (n = 4–6/nucleus/IH condition). Blots were incubated with primary antibodies to phosphorylated PKR-like ER kinase (p-PERK), p-c-Jun [inositol-requiring enzyme 1 (IRE1) activation], cleaved activating transcription factor-6 (ATF6) (Haze et al., 1999), and CHOP/GADD153; details of primary antibodies are provided in Table 1. Primary antibodies were bound with horseradish peroxidase-conjugated secondary anti-IgG, and were detected with chemiluminescence (SuperSignal Ultra; Pierce, Rockford, IL). Images were analyzed with NIH Image Analysis.
Table 1.
Selected primary antibodies
| Primary antibody | Catalog #/company | IF/EM dilution | WB dilution |
|---|---|---|---|
| ATF-6 | IMG-273/Imgenex (San Diego, CA) | IF 1:100 | WB 1:1000 |
| Caspase-3 | 9661/Cell Signaling (Beverly, MA) | IF 1:500/EM 1:200 | |
| Caspase-7 | 9491/Cell Signaling | IF 1:500 | |
| Choline acetyltransferase | AB144P/Chemicom (Temecula, CA) | IF 1:5000/EM 1:200 | |
| CHOP/GADD153 | sc-575/Santa Cruz (Santa Cruz, CA) | IF 1:500/EM 1:200 | WB 1:200 |
| Dynactin-1 | 612709/BD Transduction (San Diego, CA) | IF 1:500 | |
| MAP-2 | ab-28032/Abcam (Cambridge, MA) | IF 1:200 | |
| p-c-JUN | 231s/Cell Signaling | WB 1:1000 | |
| p-eIF-2a | 44-728G/Biosource (Camarillo, CA) | IF 1:200 | WB 1:1000 |
| p-PERK | SC-32577-R/Santa Cruz | IF 1:500 | WB 1:200 |
IF, Immunofluorescence; WB, Western blot.
Reverse transcription-PCR.
For detection of XBP-1 splicing (as a marker of IRE1 activation), macropunches of the brainstem motor nuclei from mice exposed to IH3d or sham IH3d (n = 5/IH condition) were procured, and RNA was isolated using methods previously described (Zhan et al., 2005b). A total of 0.2 μg of RNA was used for creation of cDNA for NM-013842 primer sets, sense primer: 5′-GAA CCA GGA GTT AAG AAC ACG-3′ and antisense primer: 5′-AGG CAA CAG TGT CAG AGT CC-3′ (Iwawaki et al., 2004) for PCR, 95°C for 9 min followed by 35 cycles at 95°C for 30 s, and then 55°C for 30 s, and then 72°C for 30 s and 72°C for 7 min (2400; Applied Biosystems, Foster City, CA) using 2 and 3.5% gels run 10 cm to identify the 178 bp splice variant and 205 bp unspliced mRNA (Iwawaki et al., 2004).
Immunohistochemistry.
Immunofluorescence was used to determine whether LTIH induces an unfolded protein response in moV, VII, and XII motoneurons and whether this response includes activation of ER proapoptotic proteins in motoneurons. To determine whether the IH persistently activates the UPR in motoneurons, a series of mice (n = 5/group) were exposed to 8 weeks of IH (LTIH) or sham controls and then deeply anesthetized and perfused, as detailed in our previous studies (Zhan et al., 2005a,b). Cryopreserved brains were sectioned 40 mm thick in 1:6 well series, in which wells 1 and 3 were used for UPR studies. Upper airway dilator motor nuclei (moV, VII, and XII) were identified using landmarks in the Franklin and Paxinos (1997) mouse atlas. Within motor nuclei, motoneurons were identified by size >30 mm and immunoreactivity for choline acetyltransferase (ChAT) (Barber et al., 1984; Armstrong et al., 1991). The details for primary antibodies to assess p-AKT, ChAT, p-PERK, p-eIF-2a, p-c-Jun, and N terminus ATF6 are also listed in Table 1. UPR proteins were labeled with Alexa Fluor 594 (red; Invitrogen, Carlsbad, CA), and ChAT was tagged with Alexa Fluor 488 (green). ImagePro Plus software was used to measure immunofluorescence of ER stressor proteins within motoneurons relative to background in each nucleus (Zhu et al., 2007). Mean relative fluorescent intensity per mouse (n = 4–5/group) was used in a two-way ANOVA, Bonferroni corrected for three motor nuclei. To determine whether IH activates ER proapoptotic proteins in upper airway motoneurons, sections from wells 5 and 6 from the same mice as above were used to assess activation of proapoptotic proteins, cleaved caspase-3 and -7 and CHOP/GADD153 in ChAT-labeled motoneurons (Alexa Fluor 488) using primary antibodies and titers as detailed in Table 1, and then labeled with Alexa Fluor 594. To characterize LTIH effects on motoneuronal signaling/transport proteins with established sensitivity to hypoxia, microtubule associated protein-2 (MAP-2), an important dendritic scaffolding protein (Lang-Lazdunski et al., 2000), and dynactin-1/p150glued (Kitano et al., 2004), an axonal motor protein critical for synapse stabilization (Eaton et al., 2002) and implicated in protein misfolding (Levy et al., 2006), were also examined in the three motor nuclei, using primary antibodies in Table 1 on sections from wells 2 and 4 for each mouse (n = 4–5/group).
Electron microscopy.
Immunoelectron microscopy was implemented to determine whether LTIH (8 weeks) results in nuclear translocation of CHOP/GADD153 and altered ultrastructure of ER and ribosomes in the hypoglossal motoneurons. Adult B6 mice were exposed to 8 weeks IH or sham IH. Mice were deeply anesthetized with pentobarbital intraperitoneally and then transcardially perfused with heparin and acrolein (3.8%) in 2% paraformaldehyde in PBS, pH 7.4, and postfixed (Wang et al., 2004). Coronal sections (50 mm) were cut with a vibratome through the pons and medulla, using landmarks as above (Franklin and Paxinos, 1997). Sections were treated with 1% NaBH4 in PBS and rinsed in PBS (Wang et al., 2004), before a Tris-buffered saline rinse and incubation in bovine serum albumin to minimize nonspecific labeling. To assess nuclear translocation of CHOP/GADD153, ChAT primary antibody (Table 1) labeled using biotin-SP-conjugated donkey anti-goat secondary (Jackson ImmunoResearch, West Grove, PA) was detected with DAB, whereas CHOP/GADD153 was labeled using goat anti-rabbit IgG conjugated to 1 nm gold particles (1:50; Amersham Biosciences, Arlington Heights, IL) as described by Aston-Jones et al. (2004). For characterization of ER and polysome ultrastructure, DAB intermediate densities were avoided by labeling ChAT with gold. In both cases, visualization of the gold was enhanced in silver solution (IntenSE M; Amersham Biosciences). After staining, all electron microscopic (EM) sections were postfixed for 1 h in 2% osmium tetroxide in PB, dehydrated through a graded series of alcohols and propylene oxide, and flat-embedded in Epon 812 (Electron Microscopy Science, Fort Washington, PA). Precise location of ChAT-immunoreactive large hypoglossal motoneurons was performed using light microscopy imaging of a semithin (0.35 μm) section or low-power thin section to identify neurons by configuration and adjacent tissue landmarks. Sequential ultrathin (70–80 nm) sections were made using a Leica (Deerfield, IL) Ultracut E ultramicrotome. Sections were collected onto mesh grids, counterstained with Reynold's lead citrate and uranyl acetate for ultrastructural analysis, before imaging with a transmission electron microscope Phillips (Eindhoven, The Netherlands) CM10.
Salubrinal therapy throughout LTIH exposure.
To determine whether eIF-2a phosphorylation protects motoneurons across LTIH exposure, a selective inhibitor of eIF-2a dephosphorylation, salubrinal (Boyce et al., 2005), was administered throughout LTIH exposure and compared with vehicle for effects on UPR and apoptotic responses. Mice were randomized to vehicle or salubrinal pumps (n = 5/group). Salubrinal was solubilized in dimethyl sulfoxide (DMSO) at 10 mg/ml and placed in miniosmotic pumps (1004; Alzet, Cupertino, CA) as 0.52–0.58 mg of salubrinal in 60 ml of DMSO and 40 ml of buffered saline to deliver 1 mg · kg−1 · d−1 subcutaneously for 28 d, using a previously established systemic dose (Sokka et al., 2007). To place pumps, mice were anesthetized with ketamine (80 mg/kg) with xylazine (10 mg/kg) intramuscularly. A 0.5 cm dermal incision between the scapulae was made perpendicular to the spine. Pumps were placed for subcutaneous delivery. The 24 h postop mice were placed in LTIH for 4 weeks. As in the above studies, after LTIH exposure, mice were deeply anesthetized and perfused for immunohistochemical studies, examining p-AKT, ChAT, MAP-2, p-eIF-2a, and CHOP/GADD153.
Results
Short-term intermittent hypoxia elicits an ER stress response in select brainstem motor nuclei
The UPR to 3 d of IH (IH3d) was evaluated by determining activation of the three major ER stress sensors: PERK, IRE-1, and ATF6 (summarized in Fig. 1A). IH3d increased p-PERK levels in the hypoglossal nucleus (t = 3.6; p < 0.01) and in the facial nucleus (t = 2.6; p < 0.05) but not in the motor trigeminal nucleus (t = 1.5; NS) (Fig. 1B). To examine IH induction of IRE-1, we measured phosphorylated c-JUN (p-c-JUN) and XBP-1 mRNA splicing (XBP1S) (Yoshida et al., 2001; Calfon et al., 2002). p-c-JUN protein was evident only in the XII motor nucleus where it was present in both sham and IH3d samples without IH effect on protein. In support, XBP-1 mRNA was unspliced in all samples. Cleaved ATF6 (50 kDa) was present in all XII protein samples (IH3d and sham IH3d), without an effect of IH (IH, 6.3 ± 0.5, vs sham IH, 5.8 ± 1.1; t = 0.5; NS). The 50 kDa band was not detectable in the majority of moV and VII protein samples from either IH or sham IH. In summary, there is evidence for an UPR in the XII motor nucleus, even under sham conditions, as evidenced by phosphorylation of PERK, p-c-JUN, and cleavage of ATF6. However, short-term IH selectively augmented activation of the PERK pathway in some motor nuclei, including hypoglossal and facial without activation on the motor trigeminal.
Figure 1.

Unfolded protein response to intermittent hypoxia, modeling sleep apnea. A, Schematic reveals the general response to unfolded proteins in mammalian cells, including neurons. BiP holds three sensor proteins in an inactive state in the membrane of the ER. In response to improperly folded proteins within the ER lumen, BiP is released from one or more of the three sensors, PERK, IRE1, and ATF6. Release from BiP promotes activation, phosphorylation of PERK and IRE1 (p-PERK and IRE1), and transfer of ATF6 to the Golgi where it is cleaved, before nuclear translocation. All three sensors can activate protective responses to ER stress, including selective translational attenuation and transcriptional upregulation to improve oxidative stress, reduce folding burden, and clear improperly folded proteins from the cell. All three sensors can promote the transcription of CHOP/GADD153, a proapoptotic protein. B, Conditions of short-term intermittent hypoxia (short IH, 3 d) resulted in increased PERK phosphorylation without an IH effect on the other two sensor proteins. Shown in the histograms are average ± SE band densities, with representative hypoglossal control (C) and short IH bands shown above. *p < 0.05.
Longer-term intermittent hypoxia results in sustained activation of PERK in upper airway dilator motoneurons
We next determined whether the UPR observed in the motor nuclei using immunoblots on dissected motor nuclei occurs in motoneurons after longer-term (8 weeks) IH (LTIH) and whether the other ER sensor pathways become activated with this longer IH exposure. Overall, there were highly significant effects of LTIH and motor nucleus site on p-PERK immunoreactivity (p < 0.0001). In sham IH controls, trace p-PERK immunoreactivity was evident in both hypoglossal and facial motoneurons, but not in motor trigeminal neurons. LTIH resulted in significant increases in p-PERK immunoreactivity in motoneurons in the hypoglossal (t = 5.9; p < 0.001) and facial (t = 9.1; p < 0.001) motoneurons, where p-PERK was localized to the perinuclear envelope and nuclei (Fig. 2). p-PERK was evident in moV after LTIH. Cleaved ATF6 immunofluorescence was detected in the cytoplasm in hypoglossal and facial motor nuclei from mice exposed to LTIH without evidence for translocation into motoneuron nuclei. No ATF6 immunoreactivity was detected in moV for either sham IH or LTIH. In summary, long-term intermittent hypoxia results in persistent PERK activation in the motoneurons without evidence for activation of either the IRE1 or ATF6 sensors.
Figure 2.
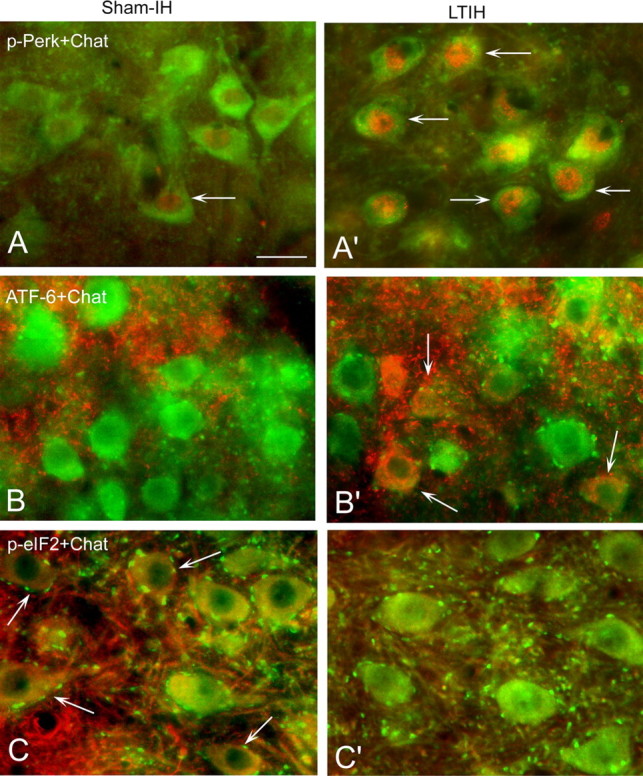
Prolonged hypoxia/reoxygenation exposures, modeling sleep apnea oxygenation, selectively activate the PERK ER sensor protein in hypoglossal motoneurons. A, A′, Under sham-IH conditions, subtle p-PERK immunoreactivity (arrows; Alexa Fluor 594; red color) is evident in ChAT-labeled hypoglossal motoneurons (Alexa Fluor 488; green color); long-term exposure to hypoxia/reoxygenation (LTIH) greatly increases p-PERK immunoreactivity where p-PERK was localized to the perinuclear envelope and nuclei. B, B′, Cleaved ATF6 (immunofluorescence) (arrows; Alexa Fluor 594; red color) was detected in the cytoplasm in ChAT-labeled hypoglossal motor neurons from mice exposed to LTIH, yet without evidence for nuclear translocation. C, C′, Sham IH mice revealed some p-eIF-2a immunoreactivity (arrows; Alexa Fluor 594; red color) in support of baseline activation of the PERK pathway, where p-eIF-2a was observed in the perinuclear envelope in most hypoglossal motoneurons in sham-IH VII and XII. p-eIF-2a is downregulated by LTIH in VII and XII motoneurons. Scale bar, 30 mm.
Intermittent hypoxia activates ER proapoptotic proteins in hypoglossal and facial motoneurons and not in trigeminal
Short-term (3 d) IH increased CHOP/GADD153 protein in X/XII nuclei [10.3 ± 0.9 (immunoblot band density) vs 14.7 ± 0.7; t = 4.7; p < 0.001] and in VII nuclei (10.6 ± 0.7 vs 14.8 ± 0.4; t = 4.3; p < 0.001). In contrast, in moV samples, CHOP/GADD153 was reduced in both sham and IH without an IH effect (4.5 ± 0.3 vs 4.7 ± 0.5; NS). The effects of LTIH (8 weeks) on CHOP/GADD153 and caspase-7 in upper airway motoneurons were next explored. Increased CHOP/GADD153 (expressed as relative to background immunofluorescence) was evident in VII (1.7 ± 0.08 vs 2.2 ± 0.2; t = 2.9; p < 0.5), X (1.8 ± 0.1 vs 2.6 ± 0.3; t = 3.6; p < 0.01), and XII, (1.7 ± 0.06 vs 2.5 ± 0.3; t = 4.7; p < 0.01). In moV, LTIH had no effect on CHOP immunofluorescent intensity (1.2 ± 0.02 vs 1.2 ± 0.03; NS). Semithin sections prepared for electron microscopic imaging and analysis with DAB-labeled ChAT and silver-enhanced gold-labeled CHOP/GADD were then examined with light microscopy in which a striking LTIH increase in CHOP/GADD153 immunoreactivity was evident in both facial and hypoglossal motoneurons, throughout somata, dendrites, and in the majority of nuclei (Fig. 3). In contrast, minimal CHOP/GADD153 was evident in motor trigeminal neurons. Electron microscopy revealed CHOP/GADD153 silver-labeled particle density increased in the soma and dendrites by fourfold to fivefold (soma, t = 7.0, p < 0.001; dendrites, t = 6.9, p < 0.001). Consistent with light microscopy findings, CHOP/GADD153-labeled gold particles were clearly evident in all LTIH nuclei examined (n = 35) and throughout dendrites (n = 122). The density of CHOP-immunoreactive silver particles within nuclei of ChAT peroxidase-labeled motoneurons was markedly increased by LTIH (17.7 ± 1.3 particles/mm2 vs 1.4 ± 0.1 particles/mm2; t = 67; p < 0.0001) (Fig. 4). Cleaved caspase-7 (cC-7) was not evident in any of the studied brainstem motoneurons in sham IH control mice. In contrast, cC-7 was evident in the majority of facial and hypoglossal motoneurons in mice exposed to LTIH (Fig. 5). No cC-7 immunoreactivity was detected in trigeminal motoneurons. Although cC-7 was detected in most LTIH VII and XII motoneurons, only 8–10 ± 3–4% of LTIH VII and XII motoneurons showed nuclear translocation of caspase-7. Cleaved caspase-3 showed a similar pattern of immunoreactivity response to LTIH as observed for cC-7 (Fig. 6). Thus, CHOP/GADD153 protein increases and translocates to the nuclei markedly in hypoglossal and facial motoneurons, whereas caspases-3 and -7 are cleaved in response to LTIH with nuclear translocation in a small percentage of motoneurons.
Figure 3.
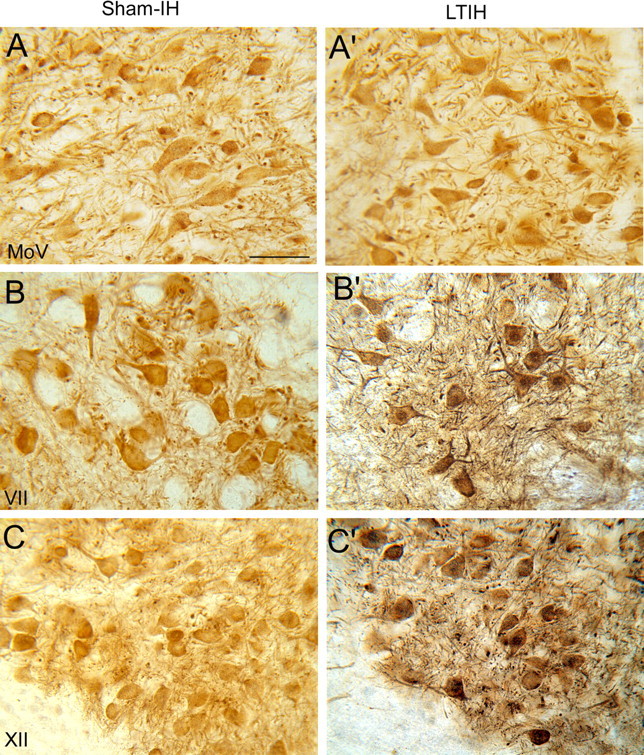
LTIH increase in CHOP/GADD153 immunoreactivity in both facial and hypoglossal but not in motor trigeminal neurons. The 50 μm vibratome sections, prepared for electron microscopic imaging and analysis with DAB-labeled ChAT and silver-enhanced gold-labeled CHOP/GADD, were first examined with light microscopy in which a striking LTIH increase in CHOP/GADD153 immunoreactivity was evident in both facial (B′) and hypoglossal motoneurons (C′), throughout somata, dendrites, and in the majority of nuclei. In contrast, minimal CHOP/GADD153 was observed in motor trigeminal neurons (A′) and in all three motor nuclei under sham IH conditions (A–C). Scale bar, 50 mm.
Figure 4.
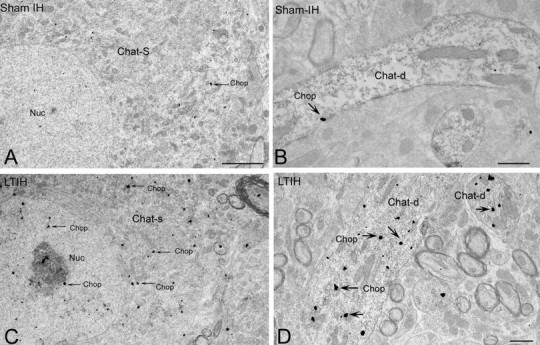
Ultrastructural localization of CHOP/GADD153 immunoreactivity in hypoglossal neurons. A–D, EM photomicrograph reveals CHOP/GADD153 silver-labeled particles (arrow) were rarely evident in ChAT-labeled somata (A; ChAT-S) or dendrites (B; ChAT-d) in sham LTIH mice. In contrast, exposure to LTIH (8 weeks) was associated with increased CHOP/GADD153-labeled particles within nuclei and throughout the soma and dendrites of ChAT peroxidase-labeled motoneurons (C, arrows). Within the nucleus (nuc) and nucleolus, CHOP/GADD153 colocalizes within subregions of increased electron density. Within dendrites of mice exposed to LTIH, CHOP/GADD immunoreactivity increased significantly. Scale bars: A, C, 2 mm; B, D, 0.5 mm.
Figure 5.

cC-7 immunoreactivity response to LTIH for 8 weeks. Fluorescence photomicrographs of ChAT-immunoreactive (ir) facial (VII) motoneurons. Note cC-7 (Alexa Fluor 594; red color) was localized to the perinuclear envelope in the majority of ChAT-ir neurons (Alexa Fluor 488; green color), and 10 ± 3% of motoneurons showed nuclear translocation of caspase-7 (indicated by arrows) in mice exposed to LTIH for 8 weeks (B, B′). In contrast, no cC-7 ir was detected in sham-IH mice (A, A′). Scale bar, 50 mm.
Figure 6.
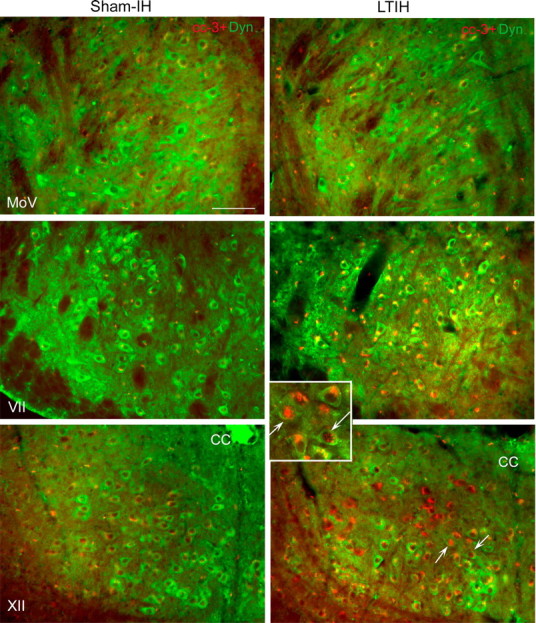
LTIH increase in cC-3 immunoreactivity in both facial and hypoglossal but not in motor trigeminal neurons. Low-power fluorescence photomicrographs of cC-3 (Alexa Fluor 594; red color) with dynactin-1/p150glued (Alexa Fluor 488; green color) in facial, hypoglossal, and trigeminal neurons are shown to illustrate heterogeneities in cC-3 immunoreactivities across motor nuclei groups and within motor nuclei. Overall, cC-3 immunoreactivity followed the LTIH cC-7 response (as shown in Fig. 5) with the majority of cC-3 localized to the perinuclear envelope in hypoglossal and facial motoneurons, and only rare nuclear translocation. A small percentage of hypoglossal and facial motoneurons revealed nuclear translocation of cC-3 in response to LTIH (arrows, inset box for LTIH XII). Minimal cC-3 was observed in sham-LTIH motoneurons in all three motor nuclei studied and motor trigeminal neurons in mice exposed to LTIH. Scale bar, 100 mm.
Activation of AKT has been proposed as a protective mechanism in hippocampal neurons after exposure to short-term IH and sustained hypoxia, where tissue homogenates show increased p-AKT in response to 6 h IH (Klein et al., 2005). Thus, in parallel with characterization of caspase activation, we explored the response of AKT activation to LTIH across vulnerable and resistant motoneuron groups. p-AKT was high at baseline in III and moV but only in non-neuronal tissue, and did not change with LTIH (supplemental figure, available at www.jneurosci.org as supplemental material). LTIH increased p-AKT immunoreactivity in motoneurons only in VII and XII motoneurons, in which some p-AKT was evident at baseline (supplemental figure, available at www.jneurosci.org as supplemental material).
LTIH results in a sustained increases in eIF-2a phosphorylation in trigeminal motoneurons but not hypoglossal or facial
Activation of PERK phosphorylates eIF-2a that triggers a cascade that serves to minimize protein misfolding. However, increased CHOP/GADD153 will increase GADD34 that in turn serves to enhance dephosphorylation of p-eIF-2a, as illustrated in Figure 1A. Thus, we next determined whether the protective component of PERK activation, phosphorylation of eIF-2a, is lost under conditions of LTIH in any of the upper airway motoneuronal groups. Sham IH mice revealed some p-eIF-2a immunoreactivity in the perinuclear envelope in most motoneurons (Fig. 2) with higher levels in moV motoneurons relative to VII and XII for sham IH condition (p < 0.05). An increase in p-eIF-2a in response to LTIH was evident only in moV motoneurons (moV sham, 1.5 ± 0.03, vs IH, 1.7 ± 0.02; t = 3.2; p < 0.05). VII and motoneurons showed no effect across IH conditions (VII, 1.2 ± 0.02 vs 1.1 ± 0.02, NS; XII, 1.2 ± 0.02 vs 1.2 ± 0.02, NS). Thus, eIF-2a phosphorylation is higher at baseline in the motoneurons resistant to LTIH injury and is maintained across LTIH. The increased p-eIF-2a in sham IH motor trigeminal neurons relative to hypoglossal and facial was confirmed with Western blot (sham IH moV, 3.68 ± 0.3, VII, 1.8 ± 0.2, and X/XII, 1.8 ± 0.3; F = 21; p < 0.001 for both moV vs VII and moV vs X/XII comparisons).
Long-term intermittent hypoxia reduces expression of signaling proteins
Overall, IH had a significant effect on ChAT immunofluorescent relative intensity in motoneurons (p < 0.0001) (Fig. 7). The effect was dependent on motor nucleus, with IH8wk reducing ChAT in facial (t = 5.1; p < 0.01) and hypoglossal motoneurons (t = 4.2; p < 0.01), whereas no change was evident in motor trigeminal motoneurons. Within the hypoglossal nucleus, central motoneurons showed greater reductions in ChAT, the same subregion with greatest cleaved caspase-3 immunoreactivity (Fig. 6, bottom panels). Dynactin-1 (p150glued) was reduced also in facial (t = 3.8; p < 0.001) and hypoglossal (t = 4.4; p < 0.001) motoneurons (Fig. 7). Similarly, MAP-2 was reduced in facial (t = 2.6; p < 0.05) and hypoglossal (t = 3.0; p < 0.05), without effect in trigeminal. Thus, all three proteins were reduced in similar magnitude in facial and hypoglossal motoneurons with an apparent sparing of effect in trigeminal motoneurons.
Figure 7.

LTIH suppresses protein levels in hypoglossal motoneurons. Average immunofluorescent signaling, relative to adjacent background, ± SE using techniques as previously described (Zhu et al., 2007), reveals decreased levels of all three motoneuronal proteins assayed. Asterisks denote p < 0.05, relative to sham IH. Note all three proteins are only partially reduced, and to a similar degree.
LTIH disrupts ER ultrastructure
Electron microscopy revealed significant alterations in ER ultrastructure within hypoglossal motoneurons of mice exposed to LTIH. Specifically, disaggregation of ribosomes and degranulation of rough ER was evident in almost all LTIH XII somata, as illustrated in Figure 8. Well formed polyribosomes were evident throughout the cytoplasm of XII motoneurons in sham-treated mice (Fig. 8C,C′) but were rarely identified in LTIH XII motoneurons (Fig. 8D,D′). Parallel rows or rough ER were typical in sham LTIH XII motoneurons. In contrast, LTIH ER was swollen, distorted, and less frequently parallel, as illustrated in Figure 8, D′, E, and F. Medium densities within the ER lumen were consistent with protein aggregation (Fig. 8D′,F). LTIH effects were not limited to the ER. Swelling was also evident in Golgi cisternae and mitochondria, where vacuoles within mitochondria were observed and cristae were less evident (Fig. 8G). Golgi cisternae were also fragmented (Fig. 8D, perinuclear). The plasma membrane appeared intact, whereas the nuclear membrane evidenced irregular swelling with chromatin condensation near regions of membrane irregularities. Irregular chromatin condensation was present to a lesser extent throughout the karyoplasm. However, darkened cytoplasm and pyknotic nuclei were not observed, suggesting that advanced apoptosis was not present in the vast majority of motoneurons.
Figure 8.
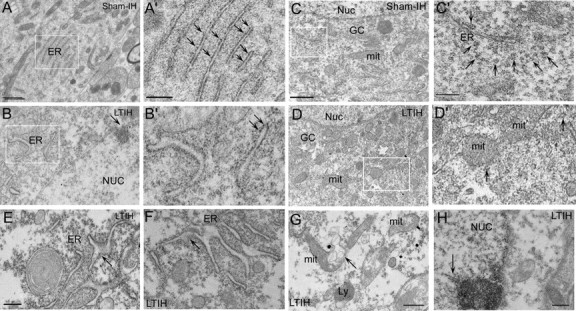
Significant alterations in ER ultrastructure after 8 weeks of hypoxia/reoxygenation exposure. Electron microscopy revealed significant alterations in ER ultrastructure within ChAT peroxidase-labeled hypoglossal motoneurons of mice exposed to LTIH 8 weeks. Most perinuclear ER formed parallel sheets in sham LTIH XII motoneurons (A, A′). In contrast, LTIH ER was swollen, distorted, and less frequently parallel (B, B′). Medium densities within the ER lumen were consistent with protein aggregation (B, B′, E, F). Disaggregation of polyribosomes (D, D′) and degranulation of rough ER (A′, B′) were evident in almost all LTIH XII somata. Mitochondria in LTIH showed increased size and occasionally vacuoles (G, arrow). Plasma membranes of motoneurons appeared intact, whereas the nuclear membranes evidenced irregular swelling with chromatin condensation near regions of membrane irregularities (H). However, darkened cytoplasm and pyknotic nuclei were not observed. Nuc, Nucleus; ER, rough endoplasmic reticulum; mit, mitochondria; Ly, lysosomes; GC, Golgi complex. Scale bars: A–D, 1 mm; A′–D′, 0.25 mm; E–H, 0.50 mm.
Salubrinal increases phosphorylation of eIF-2a and prevents LTIH upregulation of CHOP/GADD153 and protects select motoneuron signaling proteins
Salubrinal increased p-eIF-2a immunoreactivity relative to background in both the VII (vehicle, 1.3 ± 0.03, vs salubrinal, 1.5 ± 0.05; t = 4.5; p < 0.001) and the XII nuclei (vehicle, 1.2 ± 0.03, vs salubrinal, 1.5 ± 0.04; t = 5.1; p < 0.001). Representative photomicrographs are presented in Figure 9A. Whereas salubrinal increased p-eIF-2a, CHOP/GADD153 immunoreactivity was reduced in salubrinal-treated mice exposed to LTIH for both hypoglossal and facial (XII: salubrinal, 1.4 ± 0.04, XII: DMSO, 1.7 ± 0.04, t = 4.4, p < 0.001; VII: salubrinal, 1.3 ± 0.04, vs VII: DMSO, 1.5 ± 0.06, t = 2.5, p < 0.05). A typical salubrinal effect is shown in Figure 9B. Moreover, salubrinal increased MAP-2 under conditions of LTIH in both VII (vehicle, 1.4 ± 0.04, vs salubrinal, 1.6 ± 0.05; t = 2.8; p < 0.05) and XII (vehicle, 1.3 ± 0.02, vs salubrinal, 1.6 ± 0.05; t = 4.3; p < 0.001) (Fig. 10). A three-dimensional reconstruction of a confocal z-stack movie is shown in the supplemental material (available at www.jneurosci.org), in which preservation of cholinergic dendritic projections with salubrinal is readily evident in comparison with vehicle-treated mice. Whereas salubrinal protected motoneurons from LTIH caspase activation, p-AKT activation was less evident in XII and VII motoneurons in salubrinal-treated mice (data shown in supplemental material, available at www.jneurosci.org).
Figure 9.

Salubrinal increases phosphorylation of eIF-2a and prevents LTIH upregulation of CHOP/GADD153. A, Fluorescence photomicrograph of p-eIF-2a immunoreactivity (Alexa Fluor 594; red color) and ChAT immunoreactivity (Alexa Fluor 488; green color) in facial neurons. Salubrinal increased p-eIF-2a immunoreactivity relative to background in both the VII (vehicle, 1.3 ± 0.03, vs salubrinal, 1.5 ± 0.05; t = 4.5; p < 0.001) and the XII nuclei (vehicle, 1.2 ± 0.03, vs salubrinal, 1.5 ± 0.04; t = 5.1; p < 0.001). B, Fluorescence photomicrograph of CHOP/GADD153 immunoreactivity (Alexa Fluor 594; red color) and ChAT immunoreactivity (Alexa Fluor 488; green color) in hypoglossal neuron. CHOP/GADD153 immunoreactivity as suppressed in salubrinal-treated mice exposed to LTIH. Scale bar, 50 mm.
Figure 10.

Salubrinal protects select motoneuron signaling proteins. Confocal photomicrograph of facial motoneurons. Preservation of cholinergic dendritic (Alexa Fluor 594; red color) projections with salubrinal (A, A′, A″) is readily evident in comparison with vehicle-treated mice (B, B′, B″). Moreover, salubrinal increased MAP-2 (Alexa Fluor 488; green color) under conditions of LTIH in both VII (vehicle, 1.4 ± 0.04, vs salubrinal, 1.6 ± 0.05; t = 2.8; p < 0.05) and XII (vehicle, 1.3 ± 0.02, vs salubrinal, 1.6 ± 0.05; t = 4.3; p < 0.001). An arrow highlights a dendrite with colocalization of ChAT (A′′, B′′) and MAP-2 (A′, B′). The supplemental material (available at www.jneurosci.org) provides a three-dimensional reconstruction movie of the z-stacks from which the images were obtained. Scale bar, 30 mm.
Discussion
The present collection of studies suggests that exposure to the oxygenation pattern commonly observed in severe obstructive sleep apnea (1% of the adult population) injures select populations of motoneurons innervating the critical upper airway dilator muscles. By contrasting responses in upper airway motoneuronal groups with differential susceptibility to hypoxia/reoxygenation injury and by characterizing the effects of a specific protein phosphatase inhibitor on responses in these motoneurons, this work provides insight into the mechanisms of injury and protection from repeated hypoxia/reoxygenation events modeling sleep apnea oxygenation.
Physiological studies have demonstrated impaired hypoglossal nerve function after long-term intermittent hypoxia exposure, but mechanisms by which impairment occurs were not known. Long-term intermittent hypoxia was shown to shift hypoglossal nerve excitation response to serotonin and glutamate microinjection into the hypoglossal nucleus by several log doses rightward without affecting receptor density (Veasey et al., 2004b). The present studies have identified an important mechanism by which upper airway nerve function can be impaired by LTIH. Intermittent hypoxia appears to selectively activate the PERK ER sensor. At this point, the fate of a neuron can follow two divergent pathways (summarized in our proposed model in Fig. 11). Neurons with basal CHOP/GADD153 are expected to have high GADD34, which promotes dephosphorylation of p-eIF-2a (Oyadomari and Mori, 2004). Thus, from the first insult of intermittent hypoxia, these neurons are unable to mount an appropriate homeostatic ER stress response. In contrast, motor trigeminal neurons and occulomotor neurons do not have significant CHOP/GADD153 at baseline, and can mount a protective eIF-2a response. This hypothesis is strongly supported by the collective findings in this study. First, motoneurons with basal CHOP/GADD153 within the nucleus had lower p-eIF-2a immunoreactivity and did not increase p-eIF-2a with long-term intermittent hypoxia; whereas motoneurons without detectable CHOP/GADD153 at baseline were able to increase p-eIF-2a within motoneurons, and neurons in these motor nuclei did not evidence CHOP/GADD153 even with long-term exposures to intermittent hypoxia. Finally, salubrinal, a protein phosphatase inhibitor selective for eIF-2a, was shown in our studies to alter the phenotype from susceptible to resistant motoneurons, preventing CHOP and caspase activation. Thus, eIF-2a appears essential for an appropriate homeostatic response to this stressor.
Figure 11.

Proposed model of hypoxia/reoxygenation ER injury and protection. Motoneurons may be considered susceptible or resistant to injury from hypoxia/reoxygenation. Susceptible neurons have at baseline an unfolded protein response and activation of CHOP/GADD153, a proapoptotic protein. Under conditions of hypoxia/reoxygenation, CHOP/GADD153 is markedly increased and activated in susceptible motoneurons. This activation parallels an increase in activation of caspase-7 (ER specific) and caspase-3. In contrast, resistant motoneurons are able to phosphorylated eIF-2a and circumvent an activation of CHOP/GADD153 or caspases. Salubrinal, a specific protein phosphatase inhibitor, maintains phosphorylation of eIF-2a in susceptible motoneurons and converts the phenotype from susceptible to resistant to hypoxia/reoxygenation injury.
Hypoxia/reoxygenation may either augment or impair motoneuronal function. Severe ischemia followed by reperfusion markedly impairs neural function and can result in substantial loss of motoneurons (Sakurai et al., 2003). Intriguingly, neuronal loss is preceded by upregulation of the unfolded protein response and caspase-12, a murine marker for ER stress-related apoptosis, suggesting ER injury contributes to motoneuron demise (Sakurai et al., 2003, 2005; Yamauchi et al., 2007). On the other end of the spectrum, several short bursts of mild hypoxia result in long-term facilitation of phrenic and hypoglossal motoneuronal activity (Millhorn et al., 1980; Bach and Mitchell, 1996). The intermittent hypoxia used in the present study is designed to model a very common form of hypoxia/reoxygenation, obstructive sleep apnea. Although substantial loss of motoneurons in our model was not evident, this hypoxia/reoxygenation exposure is clearly injurious to select brainstem motoneurons, as evidenced by increased CHOP/GADD153 and cleaved caspases-3 and -7. Whether LTIH for longer exposure durations and whether humans with obstructive sleep apnea develop irreversible injury to motoneurons should be explored. Moreover, it will be important to discern the susceptibility of spinal motoneurons to this hypoxia/reoxygenation injury.
A differential susceptibility across motoneuronal groups is consistent with findings in most murine models of motoneuronal diseases. Established murine models of a human motoneuron disease (e.g., wobbler, progressive motor neuropathy, and the G93A mutation of superoxide dismutase-1 models) each show unique temporal and severity patterns of injury across occulomotor, trigeminal, facial, and hypoglossal motoneurons, as well as across spinal motoneuronal pools. In most studies, the injury is classified as motoneuronal loss rather than ER injury. In the wobbler mouse, motoneuronal loss is early in the hypoglossal nucleus (Haenggeli and Kato, 2002). In the SOD G93A mutant mouse, there is a significant loss of motoneurons in the hypoglossal and trigeminal nuclei before spinal motor involvement (Haenggeli and Kato, 2002). A similar overall susceptibility hierarchy is evident in our model of sleep apnea oxygenation. In the case of LTIH injury to facial and hypoglossal motoneurons with sparing of occulomotor and motor trigeminal neurons, we observed several important differences in ER responses at baseline and in response to LTIH. First, trigeminal motoneurons did not demonstrate appreciable basal ER stress. In contrast, the ER in hypoglossal and facial neurons was challenged under basal conditions with evidence of basal p-PERK and CHOP/GADD153. Second, moV motoneurons responded to LTIH with persistent phosphorylation of eIF-2a. As described, this response is expected to facilitate ER homeostasis by favoring translation of proteins that should facilitate resolution of protein misfolding. Third, although LTIH increased p-PERK, trigeminal, as well as occulomotor, motoneurons did not develop CHOP activation, as observed in hypoglossal and facial motoneurons. Our findings in moV motoneurons exposed to LTIH resulting in high p-eIF-2a, yet absent CHOP/GADD153 levels support the concept that CHOP/GADD153 must be readily degraded in these healthier neurons (Ohoka et al., 2007), and that increasing p-eIF-2a in vulnerable neurons should promote an effective adaptive response (upregulation of chaperones and antioxidants) without the injurious response (CHOP/GADD153 activation). p-AKT was higher in resistant motor nuclei III and moV, yet it was clearly not evident in motoneurons and appeared to be in glia. Thus, there may be baseline differences in the glial cell AKT activation that contribute to differential neuronal susceptibility to ER injury. This intriguing observation deserves future analysis.
Hypoglossal motoneurons have been shown highly susceptible to oxidative injury in several animal models. The Cu/Zn transgenic murine model of amyotrophic lateral sclerosis develops high nitrotyrosine immunoreactivity in hypoglossal motoneurons before loss of motoneurons (Cha et al., 2000). LTIH results in carbonylation of proteins in the hypoglossal nucleus to a greater extent than surrounding neurons, yet hypoglossal motoneuronal function may be maintained, and carbonylation minimized if animals are treated with a superoxide dismutase mimetic across LTIH (Veasey et al., 2004b). The rescue from LTIH motoneuronal injury by either preventing eIF-2a inactivation or supplementation with a superoxide dismutase mimetic supports the concept of important cross talk between oxidative and endoplasmic reticulum stress in motoneuronal injury. Effective prevention of neural injury in sleep apnea may require addressing both oxidative and ER injury pathways.
Hypoxia has been shown to activate the ER stress response, particularly the PERK pathway (Koumenis et al., 2002). Of interest, sustained hypoxia used to generate an ER response is typically severe [e.g., ≤0.02% ambient oxygen for at least 1 h for in vitro studies or cardiac arrest for 5 min or ligation of the middle cerebral artery for >5 min in vivo (Koumenis et al., 2002)]. In the present model of sleep apnea oxygenation, hypoxia is relatively mild and each episode is extremely brief. The model was selected to represent oxygenation fluctuations in a significant proportion of individuals with sleep apnea. Rarely, individuals with sleep apnea have longer and more profound hypoxemic episodes, particularly in individuals with obesity hypoventilation. Whether longer exposure durations or more severe hypoxia would result in more profound injury, including phrenic and other spinal motoneurons, should now be examined.
Long-term intermittent hypoxia reduced the levels of selected signaling and cytoskeletal proteins (ChAT, MAP-2, and dynactin-1) in motoneurons susceptible to ER injury. Preservation of all three proteins in moV motoneurons highlights the heterogeneity in protein responses to intermittent hypoxia. Activation of eIF-2a can cause temporary translation attenuation. However, in our studies, the neurons with high p-eIF-2a were the neurons with protected protein levels, whereas neurons without p-eIF-2a response to LTIH showed reduced protein levels by immunoreactivity. Thus, p-eIF-2a is not the source of reduced cytoskeletal and signaling proteins. Rather, the substantial loss of integrity of polyribosomes and rough ER are likely to contribute to reduced translation and improper protein folding. The increased densities within the ER lumen are consistent with protein aggregation. Whether the reduced signaling/cytoskeletal proteins are reversible will be important to discern.
In summary, hypoxia/reoxygenation events modeling obstructive sleep apnea oxygenation patterns injure select populations of brainstem motoneurons through an impaired ER stress homeostatic response involving an absence of eIF-2a phosphorylation. Augmentation of eIF-2a phosphorylation with salubrinal converts susceptible motoneurons into motoneurons resistant to hypoxia/reoxygenation injury, at least temporarily. It will be important to discern whether the injury we observe in the murine model occurs in humans with obstructive sleep apnea and whether this injury can be prevented, reversed, or minimized with therapies increasing ER homeostatic efforts.
Footnotes
This work was supported in part by National Institutes of Health Grants HL080492 and HL079555.
References
- Armstrong DM, Brady R, Hersh LB, Hayes RC, Wiley RG. Expression of choline acetyltransferase and nerve growth factor receptor within hypoglossal motoneurons following nerve injury. J Comp Neurol. 1991;304:596–607. doi: 10.1002/cne.903040407. [DOI] [PubMed] [Google Scholar]
- Aston-Jones G, Zhu Y, Card JP. Numerous GABAergic afferents to locus ceruleus in the pericerulear dendritic zone: possible interneuronal pool. J Neurosci. 2004;24:2313–2321. doi: 10.1523/JNEUROSCI.5339-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach KB, Mitchell GS. Hypoxia-induced long-term facilitation of respiratory activity is serotonin dependent. Respir Physiol. 1996;104:251–260. doi: 10.1016/0034-5687(96)00017-5. [DOI] [PubMed] [Google Scholar]
- Barber RP, Phelps PE, Houser CR, Crawford GD, Salvaterra PM, Vaughn JE. The morphology and distribution of neurons containing choline acetyltransferase in the adult rat spinal cord: an immunocytochemical study. J Comp Neurol. 1984;229:329–346. doi: 10.1002/cne.902290305. [DOI] [PubMed] [Google Scholar]
- Benavides A, Pastor D, Santos P, Tranque P, Calvo S. CHOP plays a pivotal role in the astrocyte death induced by oxygen and glucose deprivation. Glia. 2005;52:261–275. doi: 10.1002/glia.20242. [DOI] [PubMed] [Google Scholar]
- Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D, Yuan J. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005;307:935–939. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- Boyd JH, Petrof BJ, Hamid Q, Fraser R, Kimoff RJ. Upper airway muscle inflammation and denervation changes in obstructive sleep apnea. Am J Respir Crit Care Med. 2004;170:541–546. doi: 10.1164/rccm.200308-1100OC. [DOI] [PubMed] [Google Scholar]
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- Cha CI, Chung YH, Shin CM, Shin DH, Kim YS, Gurney ME, Lee KW. Immunocytochemical study on the distribution of nitrotyrosine in the brain of the transgenic mice expressing a human Cu/Zn SOD mutation. Brain Res. 2000;853:156–161. doi: 10.1016/s0006-8993(99)02302-1. [DOI] [PubMed] [Google Scholar]
- Decker MJ, Hue GE, Caudle WM, Miller GW, Keating GL, Rye DB. Episodic neonatal hypoxia evokes executive dysfunction and regionally specific alterations in markers of dopamine signaling. Neuroscience. 2003;117:417–425. doi: 10.1016/s0306-4522(02)00805-9. [DOI] [PubMed] [Google Scholar]
- Decker MJ, Jones KA, Solomon IG, Keating GL, Rye DB. Reduced extracellular dopamine and increased responsiveness to novelty: neurochemical and behavioral sequelae of intermittent hypoxia. Sleep. 2005;28:169–176. doi: 10.1093/sleep/28.2.169. [DOI] [PubMed] [Google Scholar]
- DeGracia DJ, Montie HL. Cerebral ischemia and the unfolded protein response. J Neurochem. 2004;91:1–8. doi: 10.1111/j.1471-4159.2004.02703.x. [DOI] [PubMed] [Google Scholar]
- Dziewas R, Schilling M, Engel P, Boentert M, Hor H, Okegwo A, Ludemann P, Ringelstein EB, Young P. Treatment for obstructive sleep apnoea: effect on peripheral nerve function. J Neurol Neurosurg Psychiatry. 2007;78:295–297. doi: 10.1136/jnnp.2006.102806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton BA, Fetter RD, Davis GW. Dynactin is necessary for synapse stabilization. Neuron. 2002;34:729–741. doi: 10.1016/s0896-6273(02)00721-3. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. San Diego: Academic; 1997. [Google Scholar]
- Gorlach A, Klappa P, Kietzmann T. The endoplasmic reticulum: folding, calcium homeostasis, signaling, and redox control. Antioxid Redox Signal. 2006;8:1391–1418. doi: 10.1089/ars.2006.8.1391. [DOI] [PubMed] [Google Scholar]
- Gozal D, Daniel JM, Dohanich GP. Behavioral and anatomical correlates of chronic episodic hypoxia during sleep in the rat. J Neurosci. 2001;21:2442–2450. doi: 10.1523/JNEUROSCI.21-07-02442.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haenggeli C, Kato AC. Differential vulnerability of cranial motoneurons in mouse models with motor neuron degeneration. Neurosci Lett. 2002;335:39–43. doi: 10.1016/s0304-3940(02)01140-0. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Saito A, Okuno S, Ferrand-Drake M, Dodd RL, Chan PH. Oxidative injury to the endoplasmic reticulum in mouse brains after transient focal ischemia. Neurobiol Dis. 2004;15:229–239. doi: 10.1016/j.nbd.2003.10.005. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Saito A, Okuno S, Ferrand-Drake M, Dodd RL, Chan PH. Damage to the endoplasmic reticulum and activation of apoptotic machinery by oxidative stress in ischemic neurons. J Cereb Blood Flow Metab. 2005;25:41–53. doi: 10.1038/sj.jcbfm.9600005. [DOI] [PubMed] [Google Scholar]
- Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787–3799. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwawaki T, Akai R, Kohno K, Miura M. A transgenic mouse model for monitoring endoplasmic reticulum stress. Nat Med. 2004;10:98–102. doi: 10.1038/nm970. [DOI] [PubMed] [Google Scholar]
- Kheirandish L, Gozal D, Pequignot JM, Pequignot J, Row BW. Intermittent hypoxia during development induces long-term alterations in spatial working memory, monoamines, and dendritic branching in rat frontal cortex. Pediatr Res. 2005;58:594–599. doi: 10.1203/01.pdr.0000176915.19287.e2. [DOI] [PubMed] [Google Scholar]
- Kitano H, Nishimura H, Tachibana H, Yoshikawa H, Matsuyama T. ORP150 ameliorates ischemia/reperfusion injury from middle cerebral artery occlusion in mouse brain. Brain Res. 2004;1015:122–128. doi: 10.1016/j.brainres.2004.04.058. [DOI] [PubMed] [Google Scholar]
- Klein JB, Barati MT, Wu R, Gozal D, Sachleben LR, Jr, Kausar H, Trent JO, Gozal E, Rane MJ. Akt-mediated valosin-containing protein 97 phosphorylation regulates its association with ubiquitinated proteins. J Biol Chem. 2005;280:31870–31881. doi: 10.1074/jbc.M501802200. [DOI] [PubMed] [Google Scholar]
- Koumenis C, Naczki C, Koritzinsky M, Rastani S, Diehl A, Sonenberg N, Koromilas A, Wouters BG. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor elF2alpha. Mol Cell Biol. 2002;22:7405–7416. doi: 10.1128/MCB.22.21.7405-7416.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang-Lazdunski L, Heurteaux C, Mignon A, Mantz J, Widmann C, Desmonts J, Lazdunski M. Ischemic spinal cord injury induced by aortic cross-clamping: prevention by riluzole. Eur J Cardiothorac Surg. 2000;18:174–181. doi: 10.1016/s1010-7940(00)00430-9. [DOI] [PubMed] [Google Scholar]
- Levy JR, Sumner CJ, Caviston JP, Tokito MK, Ranganathan S, Ligon LA, Wallace KE, LaMonte BH, Harmison GG, Puls I, Fischbeck KH, Holzbaur EL. A motor neuron disease-associated mutation in p150Glued perturbs dynactin function and induces protein aggregation. J Cell Biol. 2006;172:733–745. doi: 10.1083/jcb.200511068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindman R, Stål PS. Abnormal palatopharyngeal muscle morphology in sleep-disordered breathing. J Neurol Sci. 2002;195:11–23. doi: 10.1016/s0022-510x(01)00676-1. [DOI] [PubMed] [Google Scholar]
- Mayer P, Dematteis M, Pepin JL, Wuyam B, Veale D, Vila A, Levy P. Peripheral neuropathy in sleep apnea. A tissue marker of the severity of nocturnal desaturation. Am J Respir Crit Care Med. 1999;159:213–219. doi: 10.1164/ajrccm.159.1.9709051. [DOI] [PubMed] [Google Scholar]
- Millhorn DE, Eldridge FL, Waldrop TG. Prolonged stimulation of respiration by a new central neural mechanism. Respir Physiol. 1980;41:87–103. doi: 10.1016/0034-5687(80)90025-0. [DOI] [PubMed] [Google Scholar]
- Ohoka N, Hattori T, Kitagawa M, Onozaki K, Hayashi H. Critical and functional regulation of CHOP (C/EBP homologous protein) through the N-terminal portion. J Biol Chem. 2007;282:35687–35694. doi: 10.1074/jbc.M703735200. [DOI] [PubMed] [Google Scholar]
- Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- Polotsky VY, Rubin AE, Balbir A, Dean T, Smith PL, Schwartz AR, O'Donnell CP. Intermittent hypoxia causes REM sleep deficits and decreases EEG delta power in NREM sleep in the C57BL/6J mouse. Sleep Med. 2006;7:7–16. doi: 10.1016/j.sleep.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Sakurai M, Nagata T, Abe K, Horinouchi T, Itoyama Y, Tabayashi K. Survival and death-promoting events after transient spinal cord ischemia in rabbits: induction of Akt and caspase3 in motor neurons. J Thorac Cardiovasc Surg. 2003;125:370–377. doi: 10.1067/mtc.2003.112. [DOI] [PubMed] [Google Scholar]
- Sakurai M, Takahashi G, Abe K, Horinouchi T, Itoyama Y, Tabayashi K. Endoplasmic reticulum stress induced in motor neurons by transient spinal cord ischemia in rabbits. J Thorac Cardiovasc Surg. 2005;130:640–645. doi: 10.1016/j.jtcvs.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Shaw PJ, Eggett CJ. Molecular factors underlying selective vulnerability of motor neurons to neurodegeneration in amyotrophic lateral sclerosis. J Neurol. 2000;247(Suppl 1):I17–I27. doi: 10.1007/BF03161151. [DOI] [PubMed] [Google Scholar]
- Sokka AL, Putkonen N, Mudo G, Pryazhnikov E, Reijonen S, Khiroug L, Belluardo N, Lindholm D, Korhonen L. Endoplasmic reticulum stress inhibition protects against excitotoxic neuronal injury in the rat brain. J Neurosci. 2007;27:901–908. doi: 10.1523/JNEUROSCI.4289-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svanborg E. Impact of obstructive apnea syndrome on upper airway respiratory muscles. Respir Physiol Neurobiol. 2005;147:263–272. doi: 10.1016/j.resp.2005.06.012. [DOI] [PubMed] [Google Scholar]
- Veasey SC, Davis C, Zhan G, Hsu YJ, Fenik P, Pratico D, Gow AJ. Intermittent hypoxia in mice: protracted hypersomnolence and basal forebrain redox alterations. Sleep. 2004a;27:194–201. doi: 10.1093/sleep/27.2.194. [DOI] [PubMed] [Google Scholar]
- Veasey SC, Zhan G, Fenik P, Pratico D. Long-term intermittent hypoxia: reduced excitatory hypoglossal nerve output. Am J Respir Crit Care Med. 2004b;170:665–672. doi: 10.1164/rccm.200403-261OC. [DOI] [PubMed] [Google Scholar]
- Wang G, Anrather J, Huang J, Speth RC, Pickel VM, Iadecola C. NADPH oxidase contributes to angiotensin II signaling in the nucleus tractus solitarius. J Neurosci. 2004;24:5516–5524. doi: 10.1523/JNEUROSCI.1176-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodson BT, Garancis JC, Toohill RJ. Histopathologic changes in snoring and obstructive sleep apnea syndrome. Laryngoscope. 1991;101:1318–1322. doi: 10.1002/lary.5541011211. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Sakurai M, Abe K, Matsumiya G, Sawa Y. Impact of the endoplasmic reticulum stress response in spinal cord after transient ischemia. Brain Res. 2007;1169:24–33. doi: 10.1016/j.brainres.2007.06.093. [DOI] [PubMed] [Google Scholar]
- Yoshida H. ER stress and diseases. FEBS J. 2007;274:630–658. doi: 10.1111/j.1742-4658.2007.05639.x. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med. 1993;328:1230–1235. doi: 10.1056/NEJM199304293281704. [DOI] [PubMed] [Google Scholar]
- Zhan G, Fenik P, Pratico D, Veasey SC. Inducible nitric oxide synthase in long-term intermittent hypoxia: hypersomnolence and brain injury. Am J Respir Crit Care Med. 2005a;171:1414–1420. doi: 10.1164/rccm.200411-1564OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan G, Serrano F, Fenik P, Hsu R, Kong L, Pratico D, Klann E, Veasey SC. NADPH oxidase mediates hypersomnolence and brain oxidative injury in a murine model of sleep apnea. Am J Respir Crit Care Med. 2005b;172:921–929. doi: 10.1164/rccm.200504-581OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Fenik P, Zhan G, Mazza E, Kelz M, Aston-Jones G, Veasey SC. Selective loss of catecholaminergic wake active neurons in a murine sleep apnea model. J Neurosci. 2007;27:10060–10071. doi: 10.1523/JNEUROSCI.0857-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]