

Abstract
Here, we describe the metagenome composition of a microbial community in a hot spring sediment as well as a sequence-based and function-based screening of the metagenome for identification of novel xylanases. The sediment was collected from the Lobios Hot Spring located in the province of Ourense (Spain). Environmental DNA was extracted and sequenced using Illumina technology, and a total of 3.6 Gbp of clean paired reads was produced. A taxonomic classification that was obtained by comparison to the NCBI protein nr database revealed a dominance of Bacteria (93%), followed by Archaea (6%). The most abundant bacterial phylum was Acidobacteria (25%), while Thaumarchaeota (5%) was the main archaeal phylum. Reads were assembled into contigs. Open reading frames (ORFs) predicted on these contigs were searched by BLAST against the CAZy database to retrieve xylanase encoding ORFs. A metagenomic fosmid library of approximately 150,000 clones was constructed to identify functional genes encoding thermostable xylanase enzymes. Function-based screening revealed a novel xylanase-encoding gene (XynA3), which was successfully expressed in E. coli BL21. The resulting protein (41 kDa), a member of glycoside hydrolase family 11 was purified and biochemically characterized. The highest activity was measured at 80 °C and pH 6.5. The protein was extremely thermostable and showed 94% remaining activity after incubation at 60 °C for 24 h and over 70% remaining activity after incubation at 70 °C for 24 h. Xylanolytic activity of the XynA3 enzyme was stimulated in the presence of β-mercaptoethanol, dithiothreitol and Fe3+ ions. HPLC analysis showed that XynA3 hydrolyzes xylan forming xylobiose with lower proportion of xylotriose and xylose. Specific activity of the enzyme was 9080 U/mg for oat arabinoxylan and 5080 U/mg for beechwood xylan, respectively, without cellulase activity.
Subject terms: Metagenomics, Hydrolases
Introduction
Xylans, the second most abundant group of polysaccharides in nature and the major component of plant cell walls, are made of a xylose backbone linked by β-1,4-glycosidic bonds and side chains1. Xylanases catalyze the hydrolysis of 1,4‐β‐d‐xylosidic linkages in xylan and the hydrolysis products consist of monomers D-xylose and xylooligosaccharides of different sizes2. Enzymes degrading xylan are found in microbes, plants, seeds, snails and insects. Glycoside hydrolase families containing enzymes with a demonstrated endo-1,4-β-xylanase activity on xylan have been classified into six GH families 5, 7, 8, 10, 11 and 43 in CAZY database2. Among these, families 5, 10 and 11 are known to contain thermostable xylanases2,3.
Thermostable xylanases have been recognized as very useful biocatalyst in many industrial applications because they can withstand the harsh conditions of industrial processing without denaturation, like high temperature, alkaline or acidic pretreatment, or the use of solvents. Xylanases are of interest in the food and feed industries, paper and pulp technology, as well as in textile and biofuel production2. Hot springs are an interesting source of novel enzymes with potential biotechnological applications because extremely hot environments are inhabited by specialized microorganisms adapted to high temperatures and extreme pHs4. Many hot spring-derived xylanases have been recovered from cultured microorganisms5,6. However, most bacteria in any given environment cannot be cultured using conventional methods7, therefore biotechnological potential from uncultivable hot spring microorganisms remains untapped. The culture-independent metagenomic approaches provide access to the collective nucleic acids from all microorganisms present in an environmental sample8,9. Direct sequencing of environmental DNA and homology analysis in comparison with sequences already present in the databases9 has led to the identification of many novel enzymes10. Another approach is functional metagenomics based on the direct isolation of DNA from environmental samples and generation of metagenomic libraries from the isolated DNA. Screening of the constructed libraries for metabolic activity has led to the identification and characterization of a variety of novel xylanases from soil (Hu and others 2008), compost‐soil11, termites12 and rumen13.
Galicia, a region in the north-west of Spain, is rich in geothermal springs. One of these springs, Lobios, is characterized by high water temperature (76 °C) and alkaline pH (8.2). Our previous study showed that this environment is an attractive source of novel thermostable and alkaline-tolerant enzymes with biotechnological applications14. In this study, a sequence-based metagenomic approach was applied to the hot spring sediment sample using the next-generation sequencing to identify candidate genes coding for xylan-degrading enzymes. Moreover, metagenomic DNA sequences were also analyzed to assess the taxonomic composition of the hot spring sediment metagenome and to evaluate the microbial diversity related to GH families of predicted xylanases. Finally, a functional-based metagenomic approach was used to screen for novel xylanase enzymes from constructed metagenomic fosmid library. A thermostable xylanase showing remarkable characteristics for industrial use has been identified, purified and characterized.
Materials and Methods
Sample collection and DNA extraction
A sediment sample was collected from an alkaline hot spring located (GPS 41.86113, −8.1062) in Lobios, Ourense, Galicia, Spain, in January 2014. Approximately 400 grams of wet sediment was collected from a borehole into sterile container and transported to the laboratory. The pH of sediment was determined by using pH meter. Briefly, sediment was mixed with distilled water at a ratio (sediment/water) of 1:2.5 and pH was measured after 10 min. Metagenomic DNA was extracted using two independent extractions (20 g of sediment was processed for each extraction) using the PowerMax Soil DNA Isolation kit (Mobio Laboratories Inc.), and a modified protocol, in which cell lysis was achieved by shaking the sediment suspension at 500 rpm in a 65 °C incubator for 45 min.
Metagenomic sequencing on the Illumina HiSeq platform
Approximately 1 µg of the extracted DNA was sequenced using Pair-end Illumina HiSeq. 1500 platform at Health in Code (A Coruña, Spain). A total of 387,524,268 reads with a read size of 100 bp were generated.
Read processing and annotation
Paired reads were joined on the overlapping ends using the fastq-join15 with a minimum overlap of 8 bp and a maximum difference of 10%. Unpaired reads were discarded. Reads with ambiguous bases (“Ns”), sequence duplicates, minimum quality score of 10 and low-complexity sequences with DUST score < 7 were removed using PRINSEQ (Schmieder and Edwards, 2011). Paired reads were annotated by BLASTX using DIAMOND16 against the NCBI protein (nr) database using an e-value 1e-03 and one best match was retained. BLAST output files (in xml format) were imported into MEGAN (MEtaGenome ANalyzer) software (version 5.4.0) to perform taxonomic analysis using a Min score of 50, Top percent value 10%, Min support percent of 0 and Min support of 117. KronaTools-2.7 was used to visualize the BLAST results18.
Read assembly and ORFs prediction
For the assembly, raw reads with ambiguous bases (“Ns”), sequence duplicates, minimum quality score of 25 and low-complexity sequences with DUST score < 7 were removed using PRINSEQ.19. Filtered reads were then assembled using IDBA-UD version 1.0.920. The putative Open Reading Frames (ORFs) were predicted from contigs using MetaGeneMark Heuristic Approach21.
Xylanase sequences in metagenomic contigs
CAZYdb (Carbohydrate-Active enZYmes Database) (updated on 20 July 2017) was downloaded from dbCAN22,23 (http://csbl.bmb.uga.edu/dbCAN/download.php). The predicted ORFs were used for BLASTP (with e-value of 1e-5 and minimum bit-score of 50) search against the CAZy database using BLAST + (version 2.6.0) for identification of carbohydrate-active enzymes. Sequences classified within the glycosyl hydrolase (GH) families 5, 7, 8, 10, 11 and 43, known to contain enzymes with endo-1,4-β-xylanase activity2, were retrieved and further annotated using BLASTP against NCBI nr database.
Sequence deposition
The raw sequencing reads and the assembled metagenome dataset have been deposited at the NCBI Short Read Archive under BioProject ID PRJNA540576 and accession number SRR9001935. Identified xylanase XynA3 sequence were submitted to GenBank under accession number MK878879.
Metagenomic library construction
Metagenomic library construction was performed using the CopyControl™ HTP Fosmid Library Production Kit (Epicentre) and the pCT3FK fosmid vector24. Approximately 6 µg of high-molecular-weight (HMW) DNA was end-repaired and separated on 1% low melting point agarose gel electrophoresis for 12 h at 40 V. DNA fragments with size ranging from 30 to 40 kb were isolated and recovered from the gel with GELase (Epicentre Technologies). A fosmid library of purified end-repaired 30–40 kb metagenomic DNA was constructed using the CopyControl Fosmid Library Production kit (Epicentre Biotechnologies). Approximately 300 ng of DNA was ligated to 500 ng of the linearized fosmid pCT3FK vector, packaged into the lambda phage and used to infect E. coli EPI300-T1R cells. The library size was determined by dilution titering on LB (Luria-Bertani) agar plates containing 12.5 μg/ml chloramphenicol (LB/Cm). The resulting fosmid library was plated onto 76 LB/Cm plates at a density of ≈2000 bacterial clones per plate. Colonies from each plate were resuspended in 2 ml of LB containing 20% glycerol and a 150 μl aliquot was transferred to a 96-well plate; each well contained ≈2000 independent clones. The library was stored at −80 °C. The average DNA insert size was estimated by isolation and purification of 20 randomly chosen fosmids.
Functional screening
20 µl aliquot from metagenomic fosmid library was transferred to a new 96-well plate containing 130 µl of liquid LB/Cm and incubated overnight at 37 °C. Subsequently, the overnight culture was replica plated using a 48-pin replicator stamp onto LB/Cm plates containing 0.02% arabinose and 0.05% insoluble AZCL-HE-xylan (Megazyme) (powder has been transferred in 500 µl of absolute ethanol and added into the medium after autoclaving)25,26. The plates were incubated at 37 °C for 7 days. Xylanase activity was identified by the formation of a blue halo around the stamped colonies. The fosmid that showed xylanolitic activity was isolated, digested with DraI and PvuII restriction enzymes and sub-cloned into pJET1.2. The positive sub-clone was sequenced using primer walking and the ORF of the putative xylanase gene was identified using ORFfinder available at https://www.ncbi.nlm.nih.gov/orffinder/.
XynA3 sequence analysis
The nucleotide sequence of the identified XynA3 gene was searched against the NCBI nr database using BLASTX and protein sequences of top 7 BLAST hits were retrieved and used to construct a multiple sequence alignment using Clustal Omega27. Conserved regions were determined as described previously28. The theoretical pI (isoelectric point) and Mw (molecular weight) molecular mass of the XynA3 protein sequence was determined using ExPASy’s Compute pI/mw tool29. A potential ribosome-binding site was identified by visual inspection. In order to identify longer sequence in the sequenced hot spring sediment metagenome that matched the identified xylanase, the nucleotide sequence of XynA3 was searched against the metagenomic reads, ORFs and contigs using BLASTN and e-value of 1e-3.
Cloning, expression and purification of XynA3
The ORF of the putative xylanase gene was PCR amplified with the following primers: XynA3F 5′-AAAATTCATATGAAAAACAGAAAAGGATGGCTG- 3′ and XynA3R 5′-ATTATTGTCTCGAGTTTGATCTCCAGATAATCGAG-3′. The underlined bases indicate NdeI and XhoI restriction sites incorporated into primers. The purified PCR products were digested with NdeI and XhoI and cloned into pET21a expression vector digested with the same restriction enzymes. The resulting recombinant plasmid (pET21a-xynA3) was transformed into E. coli XL1-Blue and then BL21. Transformed BL21 cells were grown in four flasks containing 1 L of LB medium with ampicillin (100 µg/ml) at 37 °C until an OD600 nm of 0.6–0.8 was reached. The cells were induced by addition of IPTG (isopropyl β-D-1-thiogalactopyranoside) to a final concentration 0.2 mM, and the flasks were further incubated at 37 °C for 2 h. Induced cells were harvested by centrifugation, resuspended in 30 ml of binding buffer (150 mM NaCl, 50 mM Tris-HCl, pH 7.3) and put to −80 °C. Subsequently, the cells were sonicated on ice at 100% amplitude for 15 min consisting of 2 sec pulse-on and 8 sec pulse-off intervals. Insoluble material was removed by centrifugation (20 minutes, 14,000 rpm, 4 °C) and filtration through a 0.45 um syringe filter. Clarified lysate was passed through a 5 ml HiTrap Q HP column (GE Healthcare Life Sciences) that had been equilibrated with the same binding buffer. The sample was eluted with an elution buffer (150 mM NaCl, 500 mM imidazole, 50 mM Tris-HCl, pH 7.3), with a linear imidazole gradient of 30−500 mM at a flow rate of 3.5 ml/min. The eluted fractions showing xylanase activity were combined, concentrated, and further purified by anion exchange using a 5 ml HiTrap SP HP column (GE Healthcare Life Sciences) with a binding buffer composed of 30 mM NaCl, 5 mM β-Mercaptoethanol, 10 mM Tris-HCl, pH 6.8, and eluted with a linear gradient of 0–500 mM NaCl in 5 mM β-Mercaptoethanol, and 50 mM Tris-HCl, pH 6.8. Eluted fractions were pooled, and the proteins were exchanged into a protein storage buffer (50 mM Tris-HCl, 150 mM NaCl, pH 7.3) using Amicon Ultra-15 centrifugal filter unit with Ultracel-30 membrane (Millipore) down to ~1 ml and stored in 100 µl aliquots at −80 °C until use. All purification steps were performed using an ÄKTA Prime purification system (GE Healthcare Life Sciences). SDS-PAGE was used to analyze the purity and the molecular weight of the purified protein. The protein concentration was analyzed using Eppendorf BioPhotometer D30 and Bradford assay.
Enzyme assays
Enzyme activity was measured using dinitrosalicylic acid (DNS) assay30 with xylose (Sigma) as the standard. The composition of DNS reagent was as follows: 8 g of sodium hydroxide, 5 g of 3,5-dinitrosalicylic acid, 150 g of potassium sodium tartrate in 500 ml dH2O. The xylanase activity assay was carried out using 1% arabinoxylan (Megazyme) substrate in citrate-phosphate (McIlvaine) buffer31. The pH values for buffer solutions were adjusted at room temperature. The optimum pH for xylanase activity was obtained by assaying the purified enzyme at different pH (3.0–8.0) using citrate-phosphate buffer at 60 °C. The optimum temperature was determined by measuring the purified enzyme activity at different temperatures (40–100 °C) at pH 6.5. The assay consisted of 20 μl of appropriately diluted enzyme pre-heated for 30 sec and mixed with 180 μl of pre-heated for 2 min 1% arabinoxylan or beechwood xylan and incubated at respective temperature for 10 min. To stop the reaction, 300 μl DNS was added to the solution, and the mixture was immediately boiled for 5 min, then cooled on ice for 5 min. The absorbance was determined spectrophotometrically at 540 nm. Cellulolytic activity was tested on carboxymethyl cellulose (SIGMA) and AZCL-HE-cellulose (Megazyme). The thermostability of the enzyme was examined by incubating the enzyme in citrate-phosphate buffer (pH 6.5) at 60 °C, 70 °C and 80 °C for 24 h, respectively. The aliquots were withdrawn at different time intervals (10 min, 30 min, 1 h, 2 h, 3 h, 4 h, and 24 h) and the residual activities were measured by the method as described above. One unit of enzyme activity (U) was defined as the amount of the enzyme that will produce 1 μmol of reducing sugar (measured as xylose) from xylan per minute at pH 6.5 at 80 °C.
Effect of metal ions on xylanase activity
Effects of metal ions and other chemicals on xylanase activity was determined using 1 mM and 10 mM solutions of CaCl2, FeCl3, CuSO4, MnCl2, MgCl2, or KCl and EDTA, and 0.1% and 1% solutions of SDS, 2-Mercaptoethanol, DTT, N-Bromosuccinimide, Tween 20, Tritone X-100. The xylanase activity was assayed using the DNS method described above. Reaction mixture without any additive was considered as the control and its xylanase activity was designated as 100%.
Xylose and xylooligosaccharide quantification
The 2% beechwood xylan solution in citrate-phosphate buffer (pH 6.5) was incubated with 20 U of purified XynA3 at 60 °C for 24 h and boiled for 5 min. Samples and standard XOs (xylose, xylobiose and xylotriose) were analysed using a Waters HPLC at 90 °C using a Sugar-PakTM I column and refractive index detector (Waters 2414). Distilled water was used as the mobile phase with a flow rate of 0.5 ml/min, and injection volume of 15 µl and retention time 14 min.
Homology modelling
The models of XynA3 and 1,4-beta-xylanase from Paenibacillaceae bacterium JTherm (GenBank accession number: PDO11710) were made with the fully automated protein structure homology-modelling server Swiss-Model (https://swissmodel.expasy.org/)32 and the figures were generated with PyMOL (The PyMOL Molecular Graphics System, Version 2.1.1 Schrödinger, LLC).
Results and Discussion
Taxonomic classification of the sediment prokaryotic community
A sediment sample was collected from the Lobios Hot Spring in January 2014. The hot spring sediment pH was acidic (pH 5.9), in contrast to the alkaline pH of water (pH 8.2). The water temperature of this hot spring was 76 °C14. The Illumina HiSeq sequencing of the environmental DNA generated 38.8 Gbp of sequence reads. Pairs of overlapping fragments were combined and unpaired reads were discarded. After the quality control, the remaining reads (21,681,238 reads with an average read length of 165 bp and total length of 3.6 Gbp) were used for phylogenetic annotation using DIAMOND against the nr protein database using BLASTX. Taxonomy read counts were exported from MEGAN (File 1 in E-supplementary data) and visualized using Krona (Fig. 1). Of a total of 21.7 M reads, 62% were annotated and classified mainly within the Bacteria and Archaea domains. Bacteria were the most abundant prokaryotic domain, constituting 93% of all annotated reads, whereas archaeal sequences represented only 6% of all annotated reads. The phylum Acidobacteria (25%) was the most abundant among the annotated reads. This is in agreement with culture independent studies which showed that Acidobacteria are widespread in soil, sediments and hot spring environments33. The majority of the sequences that fell into that phylum were similar to sequences identified using other metagenomic studies, to the uncultured Acidobacteria bacterium found in a water stream in Japan34 and environmental Acidobacteria present in soil samples35. This may be due to the fact that Acidobacteria are rarely cultured therefore cultured isolates may be underrepresented in the database36. The closest cultured representatives were Pyrinomonas methylaliphatogenes and Chloracidobacterium thermophilum, bacteria found in geothermal soils and hot springs37,38. Second most abundant phylum was Chloroflexi (20%). This phylum was mainly represented by uncultured Chloroflexi bacteria from sediment metagenomic surveys39,40 and non-photosynthetic Thermomicrobium roseum isolated from a hot spring41. Remaining sequences were affiliated with other phyla: Proteobacteria (10%), Firmicutes (7%), Armatimonadetes (6%), Actinobacteria (4%), Bacteroidetes (2%), Deinococcus-Thermus (1%), Planctomycetes (1%), Cyanobacteria (1%), Gemmatimonadetes (1%) and other with abundance less than 1%. Archaeal reads (6%) showed that almost all fell into the phylum Thaumarchaeota (5%), which was predominantly affiliated with Candidatus Caldiarchaeum subterraneum (4%) previously isolated from the geothermal water42. The most abundant phyla identified in the sediment sample of the Lobios Hot Spring were also observed in the water sample from the same hot spring as determined in our previous study14, however the relative abundances of these taxa was different. In the water, sequences related to Deinococcus-Thermus were the most abundant (21%), whereas in the sediment sample their abundance was low (1%). Acidobacteria (25%) and Chloroflexi (20%) were the most abundant in the sediment, and their proportions in the water were less abundant (9% and 7%, respectively).
Figure 1.

Krona chart representation of taxonomic classification of the hot spring sediment metagenome.
Identification of xylan-degrading enzymes by sequence-based approach
Reads were assembled into 117,315 contigs (totaling 147.8 Mbp). The ORFs (216,305) predicted from these contigs were queried against the CAZy database using a BLAST search, leading to the identification of 15,863 ORFs with significant BLAST hits (E value < 10−5). Most abundant CAZy-associated genes identified in the hot spring metagenome were related to glycosyl transferase families GT2 (2,955 ORFs) and GT4 (2,331 ORFs). These large families include diverse proteins involved in biosynthesis of cellulose, chitin, sucrose and N‐glycosylation. These families were also the most abundant in the microbial community decomposing poplar wood chips43. Twenty-three putative xylanase-encoding ORFs were identified and classified within the family of GH5 (7 ORFs), GH8 (1 ORF) and GH10 (15 ORFs) (Fig. 2). Five of them were complete or nearly complete. Annotation of these ORFs using the NCBI nr database revealed that they were most similar to bacteria belonging to Paenibacillaceae family (5 ORFs), Armatimonadetes (4 ORFs) and Myxococcales (3 ORFs) (Fig. 2), proteins derived from a hot spring, sediment, compost and soil environment40,44,45. The number of putative ORFs involved in degradation of xylan found in this study was lower than that detected in metagenomes from samples with high lignocellulose-degrading ability, such as lignocellulosic biomass microbiota10, crop-eating snail microbiome46 and yak rumen microbiome47. These findings are not surprising as hot spring environment does not generally contain high plant biomas.
Figure 2.

Taxonomic classification of endo‐1,4‐β‐xylanases found in the hot spring sediment metagenome belonging to glycosyl hydrolase (GH) family 5, 8 and 10.
Identification of xylan-degrading enzymes by function-based approach
A metagenomic fosmid library containing approximately 150,000 E. coli clones, was constructed from microbial DNA extracted from a hot spring sediment sample. The insert size ranged from 22 kb to 59 kb (average size of 43 kb), covering approximately 6.5 Gbp of DNA in the library. To facilitate expression screening and positive clone recovery efforts, the library was arrayed into 76 wells of 96-well plate at a density of 2,000 clones per well. The library subsequently was screened for xylan active enzymes. Functional screening using azurine-cross linked (AZCL) xylan as a substrate resulted in the detection of a fosmid, named pCT3FK-XynA3, forming a blue halo on the indicator plate (Fig. 3A). The positive hit rate from hot spring sediment was much lower in comparison with rumen metagenome known to have high lignocellulose-degrading ability (52 positive clones per 14,000 fosmid clones screened)48.
Figure 3.

Xylanase identification and purification. (A) LB plate containing insoluble AZCL-xylan. The positive stamped clone is indicated by a red arrow. (B) SDS-PAGE analysis of recombinant xylanase XynA3 purified from E. coli (pET21a-xynA3). Lane 1, crude extract; lane 2, crude extract induced with IPTG; lane 3, after Ni-NTA affinity chromatography; lane 4, after ion exchange chromatography; M, NZYcolour Protein Marker II (NZYTech).
Characterization of XynA3 gene
The DNA insert of fosmid pCT3FK-XynA3 was partially digested with DraI and PvuII and subcloned. One subclone expressing xylanase activity was sequenced and an ORF encoding for a putative xylanase gene (named XynA3) was identified. The XynA3 ORF encodes a predicted protein of 371 amino acid residues, with a calculated molecular mass of 39,965 Da and a pI of 8.25. A potential ribosome-binding site (SD) sequence, 5′-AGGAGA-3′, was located 11 bp upstream of the translation start codon (ATG). BLASTX analysis revealed that the XynA3 displayed the highest identity (92%) with thermostable endo-1,4-xylanase from Paenibacillaceae bacterium JTherm (GenBank accession number: PDO11710) derived from the compost metagenome, and other endo-1,4-xylanases, members of glycosyl hydrolase family 11. Alignment of the amino acid sequence of XynA3 with those of other endo-β-1,4-xylanases showed presence of four conserved regions (black boxes highlighted in Fig. 4) and two conserved glutamic acid residues forming the catalytic site (green color highlighted in Fig. 4), typically observed in family 11 xylanases28. A highly-conserved asparagine (N) is found in position 73 (shown in bold in Fig. 4). The N is conserved in the alkaline xylanase group, and is responsible for the pH adaptation28.
Figure 4.

Alignment of the predicted amino acid sequence of XynA3 and its closest relatives from the GenBank databases. The closest sequences shown are: 1,4-beta-xylanase from Paenibacillaceae bacterium JTherm derived from compost metagenome (PDO11710); Paenibacillus ihumii (WP_055108594); Paenibacillus kribbensis (WP_094156067); Bacillus pseudalcaliphilus (KMK76771); Paenibacillus campinasensis (WP_095266106 and AEI54132) and synthetic construct (ACJ67896). Gaps are indicated by dashes. Asterisks indicate identical amino acids. The black boxes indicate conserved regions and the secondary structure elements are selected by black lines. Asparagine (N) present in alkaline xylanases is shown in bold. The glutamic acid residues corresponding to Glu 122 and 212 of XynA3, essential for the catalytic activity, are highlighted in green.
Search for XynA3 in sediment metagenomic dataset
A search of XynA3 nucleotide sequence against the sequenced sediment metagenome revealed that only 8 reads, one short contig (414 bp) and one short ORF (87 bp) aligned to the XynA3 sequence. This suggests that the organism that harbors this enzyme is present in this environment at low abundance. A higher sequencing coverage would be required to identify this enzyme using the sequence-based approach. Moreover, the identified short ORF that matched the XynA3 sequence was not annotated as endo-1,4-β-xylanase in the CAZy database, most likely due to short sequence length or insufficient homology. Despite 23 putative xylanase sequences were identified using the sequence-based approach, the activity-based approach identified only one xylanase. The major drawback of the function-based screening is that the gene expression from uncultured organisms and proper folding of the expressed protein is not always achieved in E. coli host49. Both sequence- and function-based screening approaches therefore have their advantages and disadvantages. A combined analysis using both methods may be complementary and provide the better chance of novel enzyme discovery.
Purification and biochemical characterization of XynA3
Identified xylanase-encoding gene was cloned, expressed and characterized in E. coli. Recombinant enzyme was purified in two steps, Ni-NTA affinity chromatography and ion exchange chromatography. SDS-PAGE revealed that the purified protein showed single band with a molecular mass of 41 kDa (Fig. 3B), higher than the typical molecular mass of GH11 xylanases (<30 kDa)50. The molecular weight of the recombinant xylanases corresponds to the calculated molecular mass of the mature peptide, suggesting that the recombinant enzyme is not glycosylated. The specific activity of the enzyme preparation was 9080 U/mg on arabinoxylan and 5060 U/mg on beechwood xylan. There was no activity towards carboxymethyl cellulose (CMC) and cellulose (data not shown), indicating that the enzyme can be useful in paper production. The enzyme exhibited >80% of its maximal activity in the temperature ranging from 65 to 85 °C, with the highest activity recorded at 80 °C (Fig. 5A), and >80% of its maximal activity in the pH range of 5.5 to 6.8, with the highest activity at a pH of approximately 6.5 (Fig. 5B).
Figure 5.
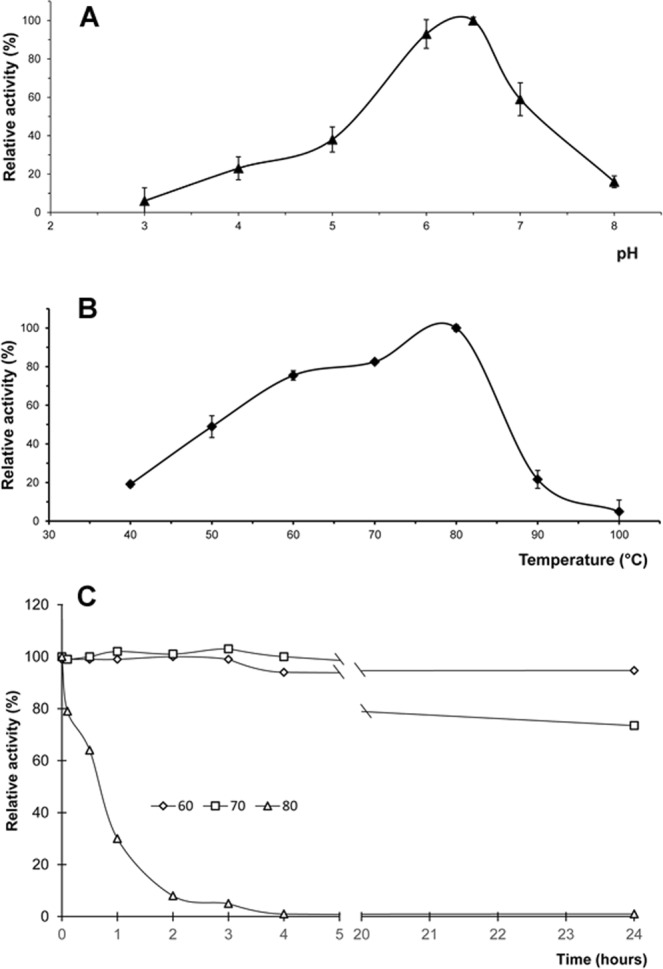
pH (A) and temperature (B) optima and thermostability (C) of XynA3 from E. coli (pET21a-xynA3). (A) Effect of pH on the activity. Enzyme activity was assayed in a pH range of 3.0–8.0. (B) Effect of temperature on the activity. Enzyme activity was assayed at various temperatures of 40–100 °C. (C) Thermal stability of purified XynA3 at pH 6.5 in the absence of xylan. Residual activity was monitored at various times (10 min, 30 min, 1 h, 2 h, 3 h, 4 h, and 24 h) after incubation at 60 °C (◊), 70 °C (□), and 80 °C (∆). The initial activity was defined as 100%.
The xylanase was stable at temperature up to 70 °C. It maintained 94% of the original activity at 60 °C after incubation for 24 h and 73% of the original activity at 70 °C after incubation for 24 h (Fig. 5C). It lost its activity at temperature 80 °C after incubation for 4 h.
The effect of metallic ions and inhibitors on the purified xylanase
At low concertation (1 mM), Fe3+ ions, dithiothreitol (DTT) and β-mercaptoethanol were found to increase the xylanase activity by 16%, 17%, and 144%, respectively. At high concertation (10 mM), β-mercaptoethanol increased the xylanase activity by 174%. Ca2+, K+ ions, EDTA, Tween 20 slightly inhibited xylanase activity by 9–19%, while Mn2+, Mg2+, Cu+, N-Bromosuccinimide, SDS and Tritone X-100 strongly inhibited its activity by 33–97% (Table 1). The presence of FeCl3 increased the activity while EDTA slightly inhibited the activity of the enzyme suggesting the enzyme is a metalloenzyme. Interestingly, the addition of β-mercaptoethanol enhanced the catalytic efficiency of the enzyme by 1.7-fold. The disulfide bonds reducing agents, β-mercaptoethanol and DTT, have been reported previously to stimulate activity of other xylanases and indicate that cysteine residues are a part of catalytic site in the xylanase51.
Table 1.
Effect of different substances on relative activity (%) of purified XynA3 xylanase.
| Substance | Concentration | |
|---|---|---|
| 1 mM | 10 mM | |
| Control | 100 | 100 |
| Ca2+ | 90 | 84 |
| Fe3+ | 116 | 86 |
| Mn2+ | 59 | 40 |
| Mg2+ | 67 | 99 |
| K+ | 86 | 93 |
| Cu+ | 80 | 62 |
| EDTA | 93 | 81 |
| DTT | 117 | 72 |
| 2-Mercaptoethanol | 244 | 274 |
| N-Bromosuccinimide | 91 | 3 |
| Substance | Concentration | |
| 0.1% | 1% | |
| SDS | ND | 40 |
| Tween 20 | 94 | 91 |
| Tritone X-100 | 12 | 44 |
Production of xylose and xylooligosaccharides by purified xylanase
HPLC analysis revealed that the recombinant xylanase released mainly xylobiose and to a lower extent of xylotriose and xylose as main products of beechwood xylan hydrolysis (Fig. 6). This characteristic is observed in GH11 xylanases, which produce xylobiose and xylotriose as main end reaction products from xylans52.
Figure 6.
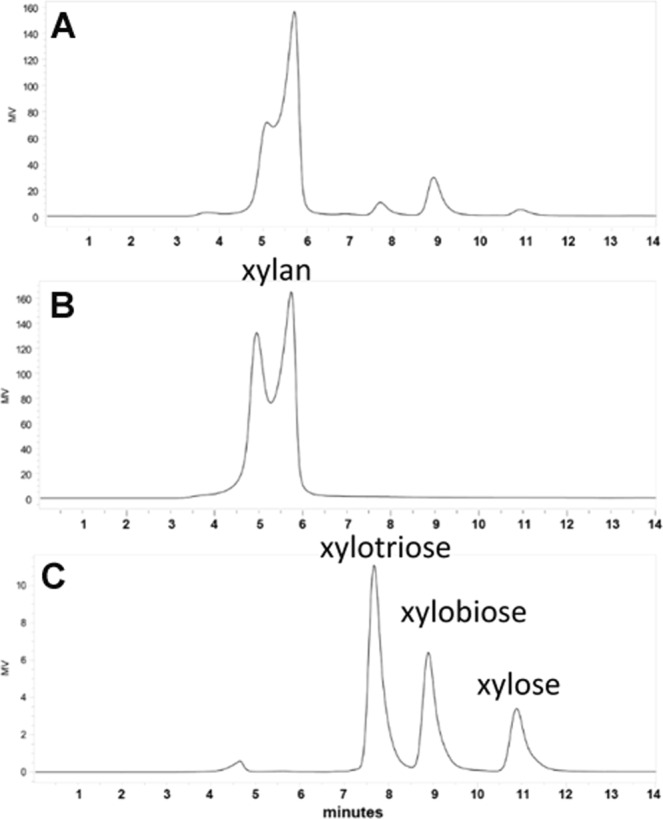
HPLC analysis of xylooligosaccharides produced by XynA3 enzyme from Beechwood xylan. (A) Products released after incubation of 2% Beechwood xylan with XynA3. (B) Beechwood xylan (substrate). (C) Xylose, xylobiose and xylotriose (standards).
Protein model analysis
A prediction of the tertiary structure of the XynA3 and the 1,4-beta-xylanase from Paenibacillaceae bacterium JTherm was made using Swiss-Model32. In both cases, the structure of the alkaliphilic XynJ from Bacillus sp. 41M-1 (Protein Data Bank code 2dcj.1.A) was used as a template to build the models. XynA3 and the 1,4-beta-xylanase from P. bacterium JTherm showed 74.7% and 75.0% sequence identity, respectively, with the xylanase XynJ. Both proteins, XynA3 and the 1,4-beta-xylanase from P. bacterium JTherm, showed a similar structure, with two well-differentiated domains: a glycoside hydrolase (GH) family 11 catalytic domain at the N-terminus and a carbohydrate binding module (CBM) of the family 6 at the C-terminus. Joining the two domains there is a linker sequence that presents differences in both proteins (Fig. 7). This linker sequence protrudes in opposite directions in both models and corresponds to a GSG insertion in the XynA3 sequence not present in the 1,4-beta-xylanase from P. bacterium JTherm. It is an area rich in small and polar amino acids such as G, S and T (GGSGGGSGGGTST) that is characteristic of flexible linkers connecting domains that require a certain degree of movement or interaction53. Although it is difficult to predict its importance at a biological level, it could facilitate the affinity or selection of certain ligands for the CBM domain and could determine the difference between the two proteins.
Figure 7.
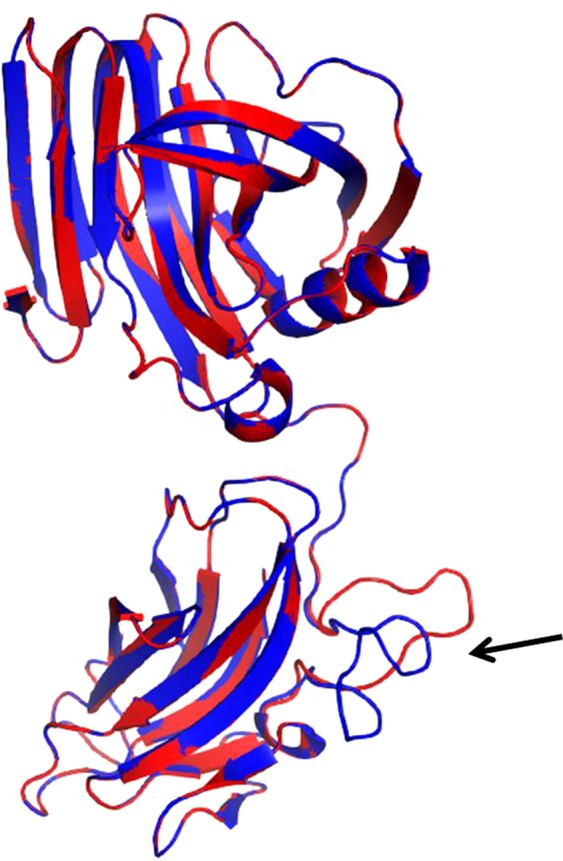
Ribbon representation. Ribbon diagram corresponding to the prediction of the tertiary structure of XynA3 (blue) using the Swiss-Model program superimposed with the prediction of the tertiary structure of the 1,4-beta-xylanase from P. bacterium JTherm (red). The black arrow shows the linker sequence between domains.
Conclusions
Metagenomic sequencing of the DNA extracted from the Lobios hot spring sediment (Ourense, Spain) demonstrated the predominance of Bacteria (93%) over Archaea (7%), being Acidobacteria the most abundant phylum. Sequence-based analysis showed that the hot spring metagenome is a potential source of novel microbial xylanases. A novel GH11 family xylanase, XynA3, was isolated by screening of the fosmid metagenomic library. The purified enzyme is low molecular weight (41 kDa), cellulase-free, thermostable (stability at high temperature of 60–70 °C), active at mildly acidic pH (pH 5.5 to 6.8) and producing xylobiose and xylotriose, suggesting that the enzyme is potentially useful for various industrial purposes, for example for pulp bleaching process or prebiotics production.
Supplementary information
Acknowledgements
This work was supported by the European Union Seventh Framework Programme (FP7/2007–2013) [grant number 324439]; and the Xunta de Galicia (Consolidación D.O.G. X-12-2016) co-financed by FEDER [grant number 2016/012]. We thank Ángel Vizoso Vázquez and Agustín Rico-Díaz for help and advices on protein purification.
Author Contributions
Kamila Knapik designed and performed the experiments and wrote the manuscript. María-Isabel González-Siso conceived the experiments. Manuel Becerra performed the HPLC analysis, protein structure homology-modelling and wrote the appropriate sections of the manuscript. All authors revised and approved the final version of the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-47637-z.
References
- 1.Beg QK, Kapoor M, Mahajan L, Hoondal GS. Microbial xylanases and their industrial applications: a review. Appl Microbiol Biotechnol. 2001;56:326–338. doi: 10.1007/s002530100704. [DOI] [PubMed] [Google Scholar]
- 2.Collins T, Gerday C, Feller G. Xylanases, xylanase families and extremophilic xylanases. FEMS Microbiol Rev. 2005;29:3–23. doi: 10.1016/j.femsre.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 3.Gallardo O, et al. Characterization of a family GH5 xylanase with activity on neutral oligosaccharides and evaluation as a pulp bleaching aid. Appl Environ Microbiol. 2010;76:6290–6294. doi: 10.1128/AEM.00871-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Escuder-Rodríguez Juan-José, DeCastro María-Eugenia, Cerdán María-Esperanza, Rodríguez-Belmonte Esther, Becerra Manuel, González-Siso María-Isabel. Cellulases from Thermophiles Found by Metagenomics. Microorganisms. 2018;6(3):66. doi: 10.3390/microorganisms6030066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu B, et al. Characterization of a recombinant thermostable xylanase from hot spring thermophilic Geobacillus sp. TC-W7. J Microbiol Biotechnol. 2012;22:1388–1394. doi: 10.4014/jmb.1203.03045. [DOI] [PubMed] [Google Scholar]
- 6.Sunna A, Prowe SG, Stoffregen T, Antranikian G. Characterization of the xylanases from the new isolated thermophilic xylan-degrading Bacillus thermoleovorans strain K-3d and Bacillus flavothermus strain LB3A. FEMS Microbiol Lett. 1997;148:209–216. doi: 10.1111/j.1574-6968.1997.tb10290.x. [DOI] [PubMed] [Google Scholar]
- 7.Cowan D, et al. Metagenomic gene discovery: past, present and future. Trends Biotechnol. 2005;23:321–329. doi: 10.1016/j.tibtech.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 8.Handelsman J. Metagenomics: application of genomics to uncultured microorganisms. Microbiol Mol Biol Rev. 2004;68:669–685. doi: 10.1128/MMBR.68.4.669-685.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simon C, Daniel R. Metagenomic analyses: past and future trends. Appl Environ Microbiol. 2011;77:1153–1161. doi: 10.1128/AEM.02345-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Montella S, et al. Discovery of genes coding for carbohydrate-active enzyme by metagenomic analysis of lignocellulosic biomasses. Sci Rep. 2017;7:42623. doi: 10.1038/srep42623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Verma D, Kawarabayasi Y, Miyazaki K, Satyanarayana T. Cloning, expression and characteristics of a novel alkalistable and thermostable xylanase encoding gene (Mxyl) retrieved from compost-soil metagenome. PLoS One. 2013;8:e52459. doi: 10.1371/journal.pone.0052459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Warnecke F, et al. Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature. 2007;450:560–565. doi: 10.1038/nature06269. [DOI] [PubMed] [Google Scholar]
- 13.Cheng F, et al. Novel xylanase from a holstein cattle rumen metagenomic library and its application in xylooligosaccharide and ferulic Acid production from wheat straw. J Agric Food Chem. 2012;60:12516–12524. doi: 10.1021/jf302337w. [DOI] [PubMed] [Google Scholar]
- 14.Lopez-Lopez O, Knapik K, Cerdan ME, Gonzalez-Siso MI. Metagenomics of an Alkaline Hot Spring in Galicia (Spain): Microbial Diversity Analysis and Screening for Novel Lipolytic Enzymes. Front Microbiol. 2015;6:1291. doi: 10.3389/fmicb.2015.01291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aronesty E. Comparison of Sequencing Utility Programs. The Open Bioinformatics Journal. 2013;7:1–8. doi: 10.2174/1875036201307010001. [DOI] [Google Scholar]
- 16.Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12:59–60. doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- 17.Huson DH, Auch AF, Qi J, Schuster SC. MEGAN analysis of metagenomic data. Genome Res. 2007;17:377–386. doi: 10.1101/gr.5969107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ondov BD, Bergman NH, Phillippy AM. Interactive metagenomic visualization in a Web browser. BMC Bioinformatics. 2011;12:385. doi: 10.1186/1471-2105-12-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27:863–864. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peng Y, Leung HC, Yiu SM, Chin FY. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. 2012;28:1420–1428. doi: 10.1093/bioinformatics/bts174. [DOI] [PubMed] [Google Scholar]
- 21.Zhu W, Lomsadze A, Borodovsky M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010;38:e132. doi: 10.1093/nar/gkq275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yin Y, et al. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012;40:W445–451. doi: 10.1093/nar/gks479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42:D490–495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Angelov A, Mientus M, Liebl S, Liebl W. A two-host fosmid system for functional screening of (meta)genomic libraries from extreme thermophiles. Syst Appl Microbiol. 2009;32:177–185. doi: 10.1016/j.syapm.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 25.Martínez, A. & Osburne, M. S. in Methods in Enzymology Vol. 531 (ed E. F. DeLong) 123–142 (Academic Press, 2013). [DOI] [PubMed]
- 26.Coman G, Cotarlet M, Bahrim G, Stougaard P. Increasing the eficiency of screening Streptomycetes able to produce glucanases by using insoluble chromogenic substrates. Romanian Biotechnological Letters. 2008;13:20–25. [Google Scholar]
- 27.Chojnacki S, Cowley A, Lee J, Foix A, Lopez R. Programmatic access to bioinformatics tools from EMBL-EBI update: 2017. Nucleic Acids Res. 2017;45:W550–W553. doi: 10.1093/nar/gkx273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sapag A, et al. The endoxylanases from family 11: computer analysis of protein sequences reveals important structural and phylogenetic relationships. J Biotechnol. 2002;95:109–131. doi: 10.1016/S0168-1656(02)00002-0. [DOI] [PubMed] [Google Scholar]
- 29.Wilkins MR, et al. Protein identification and analysis tools in the ExPASy server. Methods Mol Biol. 1999;112:531–552. doi: 10.1385/1-59259-584-7:531. [DOI] [PubMed] [Google Scholar]
- 30.Miller GL. Use of Dinitrosalicylic Acid Reagent for Determination of Reducing Sugar. Anal. Chem. 1959;31:426–428. doi: 10.1021/ac60147a030. [DOI] [Google Scholar]
- 31.McIlvaine TC. A buffer solution for colorimetric comparison. J. Biol. Chem. 1921;49:183–186. [Google Scholar]
- 32.Waterhouse A, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barns SM, Takala SL, Kuske CR. Wide distribution and diversity of members of the bacterial kingdom Acidobacterium in the environment. Appl Environ Microbiol. 1999;65:1731–1737. doi: 10.1128/aem.65.4.1731-1737.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nunoura T, et al. Genetic and functional properties of uncultivated thermophilic crenarchaeotes from a subsurface gold mine as revealed by analysis of genome fragments. Environ Microbiol. 2005;7:1967–1984. doi: 10.1111/j.1462-2920.2005.00881.x. [DOI] [PubMed] [Google Scholar]
- 35.Butterfield CN, et al. Proteogenomic analyses indicate bacterial methylotrophy and archaeal heterotrophy are prevalent below the grass root zone. PeerJ. 2016;4:e2687. doi: 10.7717/peerj.2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hugenholtz P, Pitulle C, Hershberger KL, Pace NR. Novel division level bacterial diversity in a Yellowstone hot spring. J Bacteriol. 1998;180:366–376. doi: 10.1128/jb.180.2.366-376.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bryant DA, et al. Candidatus Chloracidobacterium thermophilum: an aerobic phototrophic Acidobacterium. Science. 2007;317:523–526. doi: 10.1126/science.1143236. [DOI] [PubMed] [Google Scholar]
- 38.Crowe MA, et al. Pyrinomonas methylaliphatogenes gen. nov., sp. nov., a novel group 4 thermophilic member of the phylum Acidobacteria from geothermal soils. Int J Syst Evol Microbiol. 2014;64:220–227. doi: 10.1099/ijs.0.055079-0. [DOI] [PubMed] [Google Scholar]
- 39.Anantharaman K, et al. Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat Commun. 2016;7:13219. doi: 10.1038/ncomms13219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baker BJ, Lazar CS, Teske AP, Dick GJ. Genomic resolution of linkages in carbon, nitrogen, and sulfur cycling among widespread estuary sediment bacteria. Microbiome. 2015;3:14. doi: 10.1186/s40168-015-0077-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu D, et al. Complete genome sequence of the aerobic CO-oxidizing thermophile Thermomicrobium roseum. PLoS One. 2009;4:e4207. doi: 10.1371/journal.pone.0004207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nunoura T, et al. Insights into the evolution of Archaea and eukaryotic protein modifier systems revealed by the genome of a novel archaeal group. Nucleic Acids Research. 2011;39:3204–3223. doi: 10.1093/nar/gkq1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van der Lelie D, et al. The metagenome of an anaerobic microbial community decomposing poplar wood chips. PLoS One. 2012;7:e36740. doi: 10.1371/journal.pone.0036740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim DY, et al. Novel intracellular GH10 xylanase from Cohnella laeviribosi HY-21: biocatalytic properties and alterations of substrate specificities by site-directed mutagenesis of Trp residues. Bioresour Technol. 2010;101:8814–8821. doi: 10.1016/j.biortech.2010.06.023. [DOI] [PubMed] [Google Scholar]
- 45.Barabote RD, et al. Complete genome of the cellulolytic thermophile Acidothermus cellulolyticus 11B provides insights into its ecophysiological and evolutionary adaptations. Genome Res. 2009;19:1033–1043. doi: 10.1101/gr.084848.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cardoso AM, et al. Metagenomic analysis of the microbiota from the crop of an invasive snail reveals a rich reservoir of novel genes. PLoS One. 2012;7:e48505. doi: 10.1371/journal.pone.0048505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dai X, et al. Metagenomic insights into the fibrolytic microbiome in yak rumen. PLoS One. 2012;7:e40430. doi: 10.1371/journal.pone.0040430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang L, Hatem A, Catalyurek UV, Morrison M, Yu Z. Metagenomic insights into the carbohydrate-active enzymes carried by the microorganisms adhering to solid digesta in the rumen of cows. PLoS One. 2013;8:e78507. doi: 10.1371/journal.pone.0078507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gabor EM, Alkema WB, Janssen DB. Quantifying the accessibility of the metagenome by random expression cloning techniques. Environ Microbiol. 2004;6:879–886. doi: 10.1111/j.1462-2920.2004.00640.x. [DOI] [PubMed] [Google Scholar]
- 50.Linares-Pasten JA, Aronsson A, Karlsson EN. Structural Considerations on the Use of Endo-Xylanases for the Production of prebiotic Xylooligosaccharides from Biomass. Curr Protein Pept Sci. 2018;19:48–67. doi: 10.2174/1389203717666160923155209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gupta S, Bhushan B, Hoondal GS. Isolation, purification and characterization of xylanasefrom Staphylococcus sp. SG-13 and its application in biobleaching of kraft pulp. J Appl Microbiol. 2000;88:325–334. doi: 10.1046/j.1365-2672.2000.00974.x. [DOI] [PubMed] [Google Scholar]
- 52.Pollet A, Delcour JA, Courtin CM. Structural determinants of the substrate specificities of xylanases from different glycoside hydrolase families. Crit Rev Biotechnol. 2010;30:176–191. doi: 10.3109/07388551003645599. [DOI] [PubMed] [Google Scholar]
- 53.Chen X, Zaro JL, Shen WC. Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev. 2013;65:1357–1369. doi: 10.1016/j.addr.2012.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.