

Abstract
Li–Fraumeni syndrome (LFS) is a highly penetrant cancer predisposition syndrome caused by heterozygous germline mutations in the TP53 gene. Although more than 200 missense and null TP53 mutations are well established as disease-causing, little is known about the pathogenicity and cancer risks associated with small in-frame deletions. This leads to challenges in variant classification and subsequent difficulty making a molecular diagnosis. We report the genetic testing process for a pediatric patient diagnosed with an undifferentiated high-grade brain tumor following his mother's diagnosis of early-onset bilateral breast cancer. Sequential testing revealed that both harbored a heterozygous three-nucleotide deletion in exon 7 of TP53 (c.764_766delTCA; I255del), which was classified as a variant of uncertain significance. Because the maternal family history was void of any other LFS spectrum tumors, additional information was needed to effectively classify the variant. Targeted TP53 testing of the patient's maternal grandparents confirmed that neither carried the variant; this new de novo data upgraded the variant classification to likely pathogenic. To assess the impact of this mutation on the encoded p53 protein, additional in vitro analyses were performed. Structural modeling predicted that the deletion of isoleucine at codon 255 would disrupt the architecture of the DNA-binding domain, suggesting that it might negatively impact p53 function. Consistent with this notion, the I255del mutant protein exhibited significantly impaired transcriptional activity and greatly reduced growth suppressive properties, similar to more well-characterized LFS-associated p53 mutants. This report illustrates the importance of seeking additional evidence to assign proper pathogenicity classification, which enables optimal genetic counseling and medical management of individuals with LFS and their at-risk relatives.
Keywords: neoplasm of the breast, neoplasm of the central nervous system
INTRODUCTION
Li–Fraumeni syndrome (LFS) is a cancer predisposition syndrome caused by heterozygous germline mutations in TP53 (Malkin et al. 1990). Children and adults with LFS are at high risk to develop cancer, most notably sarcomas, adrenocortical carcinomas, brain tumors, and, in women, early-onset breast cancers (Bougeard et al. 2015; Mai et al. 2016). Although initially considered rare, recent estimates suggest an incidence as high as 1:500 and a new mutation (also known as “de novo”) rate of 7%–20% (Gonzalez et al. 2009; de Andrade et al. 2017). Although LFS is a well-established cancer predisposition syndrome, tailored predictions of exact cancer risk and type remain unavailable. Although the pathogenicity of recurrent variants located within the TP53 DNA-binding domain (DBD) is well established (Kato et al. 2003), limited information is available regarding the clinical and functional implications associated with small in-frame deletions and duplications (Bouaoun et al. 2016), especially when they are encountered for the first time. Here, we report the process of utilizing segregation, de novo status, and structural and functional evidence to establish the pathogenicity of such a variant.
RESULTS
Clinical Presentation and Family History
A previously healthy 2-yr-old Caucasian male presented with progressive tremors of the left extremity and difficulty walking of 2 mo duration. Magnetic resonance imaging (MRI) of the brain demonstrated a large enhancing thalamic mass (Fig. 1). The child underwent a subtotal resection and histologic evaluation showed a high grade poorly differentiated neural tumor (Fig. 2). Immunohistochemical assay showed moderate to strong nuclear staining for p53 in most tumor cells. Under normal conditions, wild-type (wt) p53 is unstable and thus difficult to detect by immunohistochemistry. In contrast, mutant p53 has a longer half-life and accumulates in the nucleus (Yemelyanova et al. 2011). The widespread nuclear immunoreactivity for p53 seen in this case suggested the presence of a mutation in TP53, an important tumor-suppressor gene.
Figure 1.
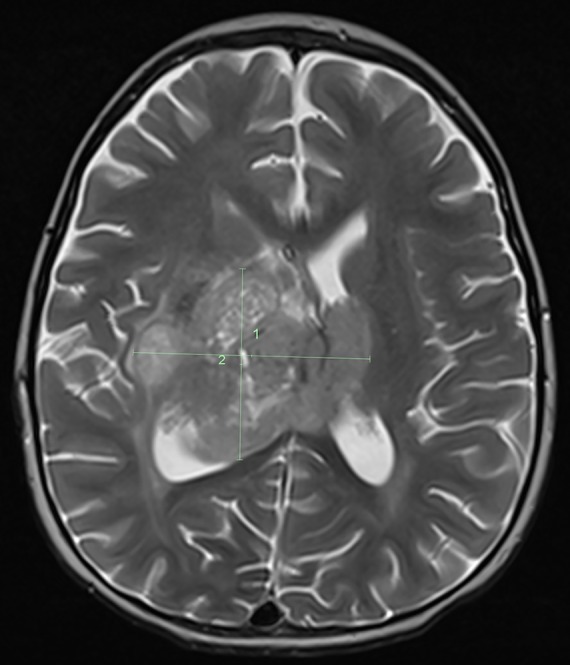
MRI image of the brain. MRI image of the brain shows a large mass involving the right thalamus with extension to the left thalamus and down into the right side of the midbrain.
Figure 2.
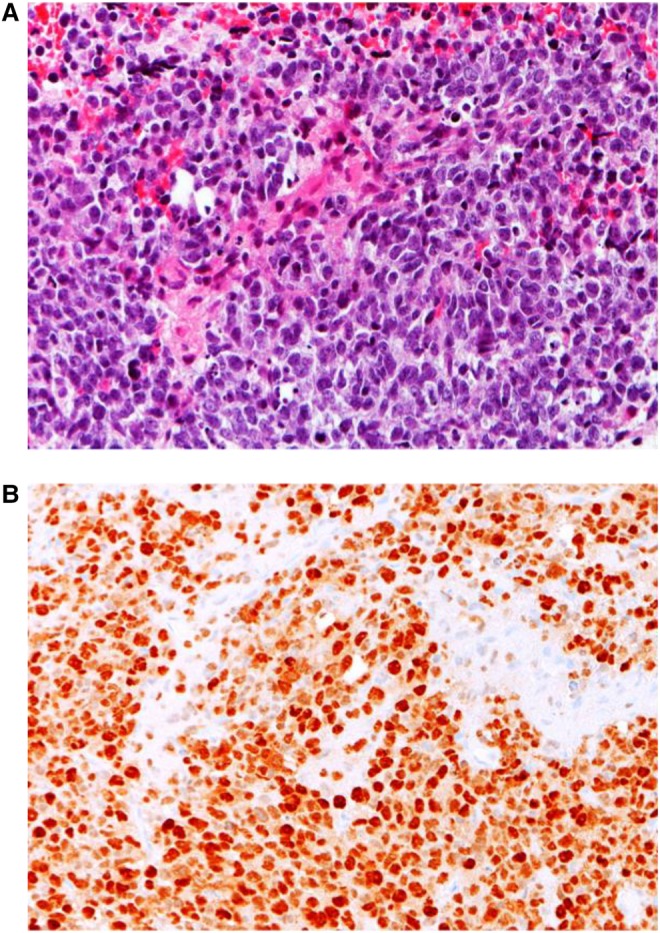
Tumor histopathologic analysis. (A) A high-grade tumor neural tumor is composed of uniform undifferentiated cells with a high nuclear to cytoplasmic ratio and high mitotic count. (B) Almost all tumor cells showed strong nuclear immunoreactivity for p53. Images were taken at 200× magnification.
Upon reviewing the family history, it was learned that the child's mother had been diagnosed with bilateral breast cancer at the age of 32 yr (Fig. 3). The breast tumors tested positive for estrogen and progesterone receptors as well as for human epidermal growth factor receptor 2 (HER2/neu); this “triple-positive” phenotype is a cardinal feature of LFS-associated breast cancer (Masciari et al. 2012). The child's mother shared that she had undergone germline genetic testing for BRCA1 and BRCA2 and no mutations were found. Altogether, the child's brain tumor diagnosis, tumor immunoreactivity for p53, and maternal history of early-onset triple-positive breast cancer with negative BRCA1/2 testing supported the possibility of LFS as the cause for cancer in this family.
Figure 3.
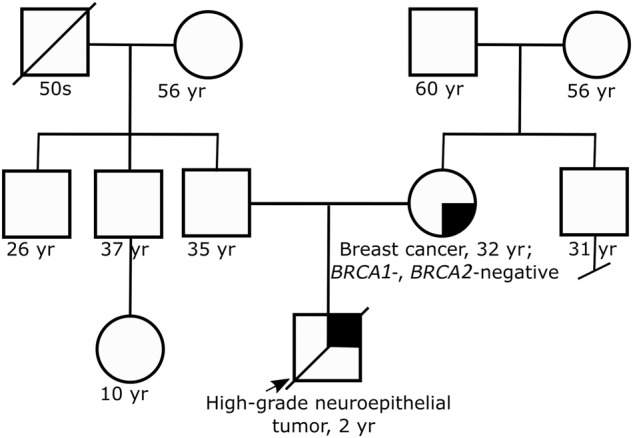
Family pedigree. Family pedigree reveals child with brain tumor and mother with early-onset breast cancer. No other early-onset cancers are reported in the family.
Genomic Analyses
Based on the concern for LFS, the child's mother was offered germline TP53 testing. Results revealed that she carried a heterozygous TP53 variant (c.764_766delTCA; I255del) (Table 1), consisting of a deletion of codon 255 in exon 7, which normally codes for isoleucine (rs1064794309) (Fig. 4). Although this variant was predicted to be deleterious using in silico tools and it was absent from control databases, these criteria alone were not sufficient to call it pathogenic or likely pathogenic according to guidelines from the American College of Medical Genetics (ACMG) and the Association for Molecular Pathology (AMP) (Richards et al. 2015). Therefore the variant was classified as a variant of uncertain significance (VUS).
Table 1.
Variant table
| Gene | Variant | Chr | HGVS DNA Ref. | HGVS protein Ref. | Variant type | Predicted effect | dbSNP ID | Genotype | ClinVar ID | Parent of origin | Observed effect |
|---|---|---|---|---|---|---|---|---|---|---|---|
| TP53 | c.764_766delTCA (p.Ile255del) | 17 | NM_000546.5 | NP_000537.3 | 3-nt in-frame deletion | Deletion | rs1064794309 | Heterozygous | SCV000629866.1 | De novo in proband's mother | Deletion in DBD impairs p53 transcriptional activity and protein folding |
Figure 4.

Schematic of the p53 protein. Schematic of the p53 protein shows the location of the I255del variant within the DNA-binding domain.
Because the family history and tumor types were highly suspicious for LFS, additional evidence of pathogenicity was sought. Approaches to obtain additional information regarding the clinical significance of a VUS are to assess whether it tracks with disease (segregates) in the family and to determine whether it is new (de novo) in an affected individual. In the case of the family presented here, targeted germline testing for the TP53 I255del variant in the child would provide information about segregation, whereas testing both maternal grandparents might make it possible to determine de novo status.
The child expired while his mother was undergoing genetic testing. Therefore, archived germline DNA was sent for TP53 analysis. Through this testing, the child was found to also carry the TP53 I255del variant. Subsequent testing of the maternal grandparents demonstrated that neither carried the variant (Fig. 3), suggesting that this variant occurred as a de novo mutation in the mother. With this additional information, the TP53 I255del variant was reclassified by the testing laboratory as likely pathogenic, thereby confirming the diagnosis of LFS in the deceased child and his mother.
Structural and Functional Analyses
TP53 encodes the p53 protein, a critical transcription factor essential for numerous cellular processes such as DNA repair, cell cycle arrest, apoptosis, and senescence (Chen 2016). The TP53 I255del variant is located within the p53 DBD (Fig. 4), where the majority of LFS-associated mutations are found (Bouaoun et al. 2016). Most TP53 variants affecting this domain are located at residues involved in contacting DNA or supporting the structure of the DNA-binding surface. Accordingly, these mutations result in a protein with impaired ability to bind DNA and altered regulation of downstream target genes. In the case presented here, the three-nucleotide deletion is predicted to lead to loss of a single amino acid with retention of an intact reading frame. Because it was not clear whether such a change would impact the function of the resulting encoded p53 protein, experiments were performed to examine whether the TP53 I255del variant impacted the structure or function of the encoded p53 protein.
Structural modeling showed that deletion of I255 causes a one residue register shift in β-pleated sheet 10 of the p53 DBD, resulting in the burial of glutamate 258 (E258) rather than leucine 257 (L257) in the hydrophobic core (Fig. 5), which would likely dramatically destabilize the protein. To test this hypothesis, we computationally estimated the free energy (ΔG) of folding of the wt-DBD and the I255 deletion mutant (DBD-I255del). Consistent with our hypothesis, the ΔG of folding was predicted to be −8.2 and +2.9 kcal/mol for wt-DBD and DBD-I255del, respectively; the positive value indicates that the I255del p53 DBD mutant is likely to be thermodynamically unstable. We attempted to confirm this experimentally with recombinant proteins expressed in Escherichia coli, but although the DBD-I255del protein bearing well-characterized triple mutations that stabilize the DBD (see Methods) was expressed in bacteria, it was insoluble upon bacterial lysis (data not shown), suggesting that the expressed protein aggregates into inclusion bodies, consistent with the computational prediction that deletion of I255 significantly destabilizes the fold of the DBD. When expressed in p53 null Saos-2 cells, the I255del variant failed to activate transcription of a luciferase reporter under the control of a p53 responsive element. Indeed, p53 I255del was as defective as p53 R175H, the most frequent somatic p53 mutant found in cancer and a known nonfunctional variant (Fig. 6). Furthermore, p53 I255del failed to suppress cell growth, as revealed by the significantly greater number of colonies generated by Saos-2 cells transfected with constructs encoding I255del or R175H as opposed to cells transfected with wt p53 (Fig. 7).
Figure 5.
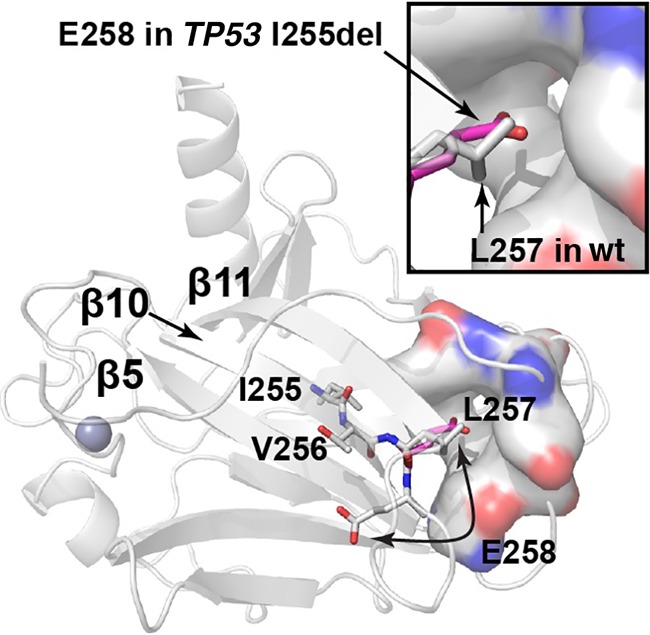
Structural modeling of the TP53 I255del variant. The location of residues in β10 following I255 of the p53 DBD are shown on the crystal structure (PDB 1TUP). Homology models of the I255del mutant show that a register shift in the second half of β10 would result in burial of E258 in a hydrophobic pocket formed by Y107, F109, L145, V147, P151, T155, and Y220 (shown in surface representation). The location of E258 in the I255del mutant model is depicted with the carbon atoms in magenta. The inset shows a zoomed-in view of the L257 pocket. Other portions of the protein are shown in cartoon representation. The zinc atom is shown as a sphere. Nitrogen atoms are colored blue; oxygen atoms are colored red.
Figure 6.
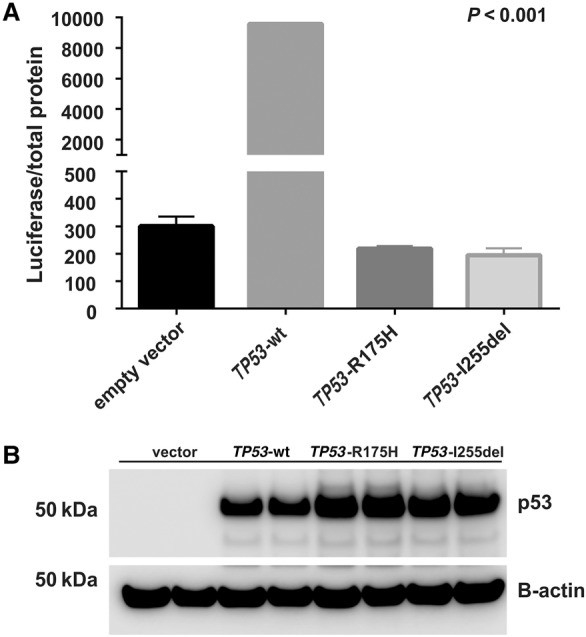
The p53 I255del mutant exhibits reduced transcriptional activity. Constructs encoding p53 wt (TP53-wt), p53-R175H (TP53-R175H), and p53-I255del (TP53-I255del) were transiently transfected into p53-deficient Saos-2 cells. Transcriptional activity was assessed using a promoter–reporter assay. (A) The relative p53 luciferase activity was significantly reduced in cells transfected with TP53-I255del as compared to TP53-wt (P < 0.001) and was like that observed in cells transfected with TP53-R175H. (B) Corresponding p53 protein expression was determined by western blot analysis. Data represent two independent experiments, with each experiment comprising two biologic replicates.
Figure 7.

The p53 I255del mutant fails to suppress colony formation. Saos-2 cells were transfected with constructs encoding p53 wt (TP53-wt), p53-R175H (TP53-R175H), and selected in medium containing G418. (A) TP53-I255del did not suppress colony formation with respect to TP53-wt, with the number of colonies like that produced with the defective TP53-R175H (P = 0.0026). (B) Corresponding colony numbers for each construct are shown with error bars representing the standard deviation. Data represent two independent experiments, with each experiment comprising two biologic replicates.
Collectively, these clinical (segregation, de novo), structural, and functional studies reveal that the TP53 I255del variant is tightly linked with tumor formation and is significantly compromised in its ability to regulate transcriptional activity and suppress tumor cell colony formation. These data provide strong evidence supporting the pathogenicity of the TP53 I255del variant and further confirm its causative role in LFS.
DISCUSSION
In-frame 3-base pair (bp) deletions affecting TP53 have only rarely been reported. A search of the ClinVar database (Landrum et al. 2018) (http://www.ncbi.nlm.nih.gov/clinvar/) downloaded 02.17.19, which contains information on 1390 TP53 variants (Supplemental Table 1a), returns only 10 (0.7%) in-frame 3-bp deletions, including the I255del variant described in this report (Supplemental Table 1b). Except for the I255del variant, each of these nine 3-bp in-frame deletion variants is classified as a VUS. Experimental studies investigating changes in protein structure and function for these nine variants are not yet available; as such, their functional significance remains unknown. Nevertheless, the PROVEAN software tool (Choi and Chan 2015) (http://provean.jcvi.org/index.php) predicts a deleterious effect on protein function for all but two of these variants (Supplemental Table 1b). A similar search of the IARC database (Bouaoun et al. 2016) (R19; http://p53.iarc.fr), the largest comprehensive repository of data on families with germline TP53 mutations and somatic TP53 variations linked to cancer, reveals a total of eight 3-bp in-frame deletions, including I255del (Supplemental Table S2). Of the remaining seven variants, two are reported in ClinVar, both as VUS. By completing additional genetic and functional studies on the p.I255del 3-bp in-frame deletion identified in our family, we were able to show that this TP53 variant significantly impacted the structure and the function of the encoded protein. Therefore, we argue that the other reported in-frame 3-bp deletion variants in ClinVar warrant further investigation as it is possible that they too exert negative effects upon the encoded p53 protein, especially the variants located in the TP53 DBD (Supplemental Table 1b)
As TP53 is increasingly included in multigene panels for cancer susceptibility testing (Rana et al. 2018), it is inevitable that many more TP53 variants including in-frame deletion variants will be discovered. In the absence of supporting clinical or functional information, many of these variants will remain classified as VUS. This is problematic as a VUS may cause undue distress and confusion for the patient and family. With additional evidence, a VUS may eventually be reclassified as pathogenic/likely pathogenic or benign/likely benign. In such cases, it is often the clinical laboratory's position to recontact health-care providers and/or patients with the newly assigned classification. However, it may take years before the desired evidence for precise variant reclassification is obtained. Therefore, it is important that individuals with a VUS, as well as their health-care providers, maintain an ongoing dialogue to ensure proper communication about the possibility of an updated classification and its potential impact on risk assessment or provision of care.
If there are concerns that the VUS in question may be pathogenic (i.e., affects a residue in a conserved domain or occurs in an individual with a relevant tumor type or suspicious family history), it is important to attempt to gather further evidence. Accurate classification is essential as the information gained informs cancer risk and guides clinical management through institution of cancer surveillance and risk reducing measures. Recent reports suggest that surveillance using whole-body MRI and other radiologic and biochemical testing can detect asymptomatic tumors in individuals with LFS, leading to improved survival (Villani et al. 2016). Based on these and other data, the National Comprehensive Cancer Network has recently published surveillance guidelines for adults with LFS and recommends discussion of bilateral mastectomy to reduce the risk of early-onset breast cancer (National Comprehensive Cancer Network; Genetic/Familial High-Risk Assessment: Breast and Ovarian [version 2.2019]).
Although the case presented here highlights LFS and the challenges of interpreting a germline TP53 VUS, the processes encountered are highly pertinent to other genetic conditions, the testing of their respective genes, and interpretation of results. It is important that providers understand that although most VUSs are not considered clinically “actionable,” there are cases that warrant further investigation. In such cases, it may be necessary to interrogate the genetic origin of the VUS within a family or examine its impact on the functions of the encoded protein. Genetic evaluation of family members may be difficult because of death, estrangement, or disinterest in testing by close relatives. Moreover, functional analysis may be problematic because of a lack of appropriate assays or laboratories willing to perform them. Nevertheless, an upgraded classification to pathogenic or likely pathogenic can provide a family with an understanding of disease causality and significantly impact the approach to clinical management.
METHODS
TP53 Genomic Analysis
All diagnostic TP53 analysis was done at Invitae, San Francisco, CA. Genomic DNA obtained from the submitted samples was enriched for targeted regions using a hybridization-based protocol and sequenced using Illumina technology at a read depth of ≥50× (Supplemental Table S3). Reads were aligned to a reference sequence (GRCh37/UCSC hg19), and sequence changes were identified and interpreted in the context of the following TP53 transcript: NM_000546.5. Enrichment and analysis focused on the coding sequence of the transcript with 10 bp of flanking intronic sequence. The TP53 promoter, untranslated regions, and other noncoding regions were not interrogated.
P53 Structural Analysis
SWISS-MODEL (Biasini et al. 2014) was used to construct homology models of the wt and I255del p53 DBD constructs both in the wt and in the stabilized M133L/V213A/N268D (LAD) triple-mutant background (Joerger et al. 2004) from the same template (PDB 1TUP). The free energy of folding was estimated using FoldX (Schymkowitz et al. 2005). The ΔG of folding was predicted to be −6.2 and +1.6 kcal/mol for LAD-DBD and LAD-DBD-I255del, respectively.
The p53-DBD construct bearing the stabilizing M133L/V213A/N268D mutations has been described previously (Follis et al. 2015). The I255 deletion was introduced into the triple mutant construct using QuikChange mutagenesis (Agilent). E. coli cells expressing the DBD construct were grown in LB at 37°C to an OD of ∼0.8 and induced with 0.5 mM IPTG and 50 µM zinc acetate overnight at 16°C. Cells were harvested by centrifugation, resuspended in buffer containing 50 mM tris, 500 mM NaCl, and 0.5 mM TCEP at pH 8.0, and lysed with a microfluidizer. Lysate was spun down at 30,000g and samples were ran on SDS-PAGE before and after lysis.
P53 Functional Analysis
Cell Culture
The human osteosarcoma cell line Saos-2 (TP53-null) was maintained as a monolayer in Dulbecco's modified Eagle's medium with l-Glucose (ThermoFisher Scientific) supplemented with 10% fetal bovine serum, MEM Non-Essential Amino Acids solution (ThermoFisher Scientific), 2.5 mmol/l-Glutamine and penicillin-streptomycin at 37°C under 5% CO2. Cells were seeded at a density of 1 × 106 in 10-cm2 plates and used for luciferase promoter–reporter, western blotting, and colony reduction assays.
Plasmid Construction and Cellular Transfection
The c.764_766delTCA (I255del) mutant TP53 was constructed via site directed mutagenesis using QuikChange II kit (Stratagene) with the forward primer: 5′ CCC ATC CTC ACC ATC ACA CTG GAA GAC TCC AG 3′ and reverse primer: 5′ CTG GAG TCT TCC AGT GTG ATG GTG AGG ATG GG 3′ and pCMV-Neo-Bam expression vector containing wt TP53 cDNA as template. wt and R175H mutant TP53 cDNA were included as controls (West et al. 2006). The entire coding sequence of each plasmid was confirmed by direct sequencing. Saos-2 cells were transiently cotransfected with 250 ng of p50-2 luciferase reporter construct containing consensus p53 element and 1 µg pCMV-Neo-Bam (vector only) or with 1 µg of pCMV-Neo-Bam expressing wt p53 (TP53-wt) or p53-R175H (TP53-R175H) or p53-I255del (TP53-I255del) mutant cDNA by calcium phosphate, as previously described (Zambetti et al. 1992).
Luciferase TP53 Transcriptional Activation Assay
Cells were harvested and lysed in M-PER buffer (Pierce Chemical) containing a complete protease-inhibitor cocktail (Roche Diagnostics Corporation). Protein yield was determined by the Bradford method (Bio-Rad Laboratories). The relative luciferase activity was determined 48 h after transfection using a single Luciferase Assay System (Promega) as per the manufacturer's directions. For the assay, 20 µL of protein lysate was transferred to 100 µL Luciferase Assay Reagent, followed by 2-sec measurement delay followed by a 10-sec measurement read for luciferase activity in an Optocomp I luminometer (MGM Instruments). Relative light units (RLUs) were normalized to total protein (Monti et al. 2011).
Protein Analysis
Total protein extracts (20 µg) were separated on 4% to 12% NuPAGE protein gel (ThermoFisher Scientific) and transferred to nitrocellulose membranes. The membrane was incubated with mouse monoclonal anti-human p53 DO-1 antibody (1:500; Calbiochem) and mouse monoclonal β-Actin antibody (1:4000; Sigma-Aldrich) with correspondent secondary antibodies. Membranes were imaged using a LI-COR Odyssey infrared scanner.
Colony Reduction Assay
For colony reduction assay, cells were maintained and transfected in duplicate. Cells were selected in medium containing 800 ng/mL G418 antibiotic (Invitrogen) 48 h after transfection and maintained for up to 21 d with change of media every 3 d. Cells were washed with Dulbecco's Phosphate Buffered Saline (D-PBS), fixed with 100% methanol, and stained with 1:20 Giemsa stain for 45 min, washed briefly, and air-dried before visualization. The colonies were photographed and counted manually and by using ImageJ software (Schneider et al. 2012). Data were reported as percent reduction relative to empty vector-transfected controls.
ADDITIONAL INFORMATION
Data Deposition and Access
The variant described in this paper has been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) under accession number SCV000629866.1. Genomic sequence data was not deposited because of lack of patient consent.
Ethics Statement
This study was determined to be exempt from review by the St. Jude Children's Research Hospital Institutional Review Board. Written and informed consent was obtained for all genetic testing from either the study participants or, in the case of the child proband, legal guardians.
Acknowledgments
We thank the family for allowing us to share this report.
Author Contributions
E.A.Q., K.E.N., and C.K. wrote the manuscript; J.L.M. reviewed TP53 databases and assisted with manuscript revisions; E.A.Q., K.E.N., C.K., S.A.U., D.B., and M.K. were part of the clinical care team; D.W.E. reviewed tumor pathology; A.H.P. and R.W.K. conducted structural analysis; and E.M.P. and G.P.Z. conducted functional analysis.
Competing Interest Statement
The authors have declared no competing interest.
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Cassarino TG, Bertoni M, Bordoli L, et al. 2014. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42: W252–W258. 10.1093/nar/gku340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouaoun L, Sonkin D, Ardin M, Hollstein M, Byrnes G, Zavadil J, Olivier M. 2016. TP53 variations in human cancers: new lessons from the IARC TP53 database and genomics data. Hum Mutat 37: 865–876. 10.1002/humu.23035 [DOI] [PubMed] [Google Scholar]
- Bougeard G, Renaux-Petel M, Flaman JM, Charbonnier C, Fermey P, Belotti M, Gauthier-Villars M, Stoppa-Lyonnet D, Consolino E, Brugières L, et al. 2015. Revisiting Li–Fraumeni syndrome from TP53 mutation carriers. J Clin Oncol 33: 2345–2352. 10.1200/JCO.2014.59.5728 [DOI] [PubMed] [Google Scholar]
- Chen J. 2016. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb Perspect Med 6: a026104 10.1101/cshperspect.a026104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Chan AP. 2015. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31: 2745–2747. 10.1093/bioinformatics/btv195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Andrade KC, Mirabello L, Stewart DR, Karlins E, Koster R, Wang M, Gapstur SM, Gaudet MM, Freedman ND, Landi MT, et al. 2017. Higher-than-expected population prevalence of potentially pathogenic germline TP53 variants in individuals unselected for cancer history. Hum Mutat 38: 1723–1730. 10.1002/humu.23320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follis AV, Llambi F, Merritt P, Chipuk JE, Green DR, Kriwacki RW. 2015. Pin1-induced proline isomerization in cytosolic p53 mediates BAX activation and apoptosis. Mol Cell 59: 677–684. 10.1016/j.molcel.2015.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez KD, Buzin CH, Noltner KA, Gu D, Li W, Malkin D, Sommer SS. 2009. High frequency of de novo mutations in Li–Fraumeni syndrome. J Med Genet 46: 689–693. 10.1136/jmg.2008.058958 [DOI] [PubMed] [Google Scholar]
- Joerger AC, Allen MD, Fersht AR. 2004. Crystal structure of a superstable mutant of human p53 core domain. Insights into the mechanism of rescuing oncogenic mutations. J Biol Chem 279: 1291–1296. 10.1074/jbc.M309732200 [DOI] [PubMed] [Google Scholar]
- Kato S, Han SY, Liu W, Otsuka K, Shibata H, Kanamaru R, Ishioka C. 2003. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci 100: 8424–8429. 10.1073/pnas.1431692100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, et al. 2018. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res 46: D1062–D1067. 10.1093/nar/gkx1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai PL, Best AF, Peters JA, DeCastro RM, Khincha PP, Loud JT, Bremer RC, Rosenberg PS, Savage SA. 2016. Risks of first and subsequent cancers among TP53 mutation carriers in the National Cancer Institute Li–Fraumeni syndrome cohort. Cancer 122: 3673–3681. 10.1002/cncr.30248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkin D, Li FP, Strong LC, Fraumeni JF Jr, Nelson CE, Kim DH, Kassel J, Gryka MA, Bischoff FZ, Tainsky MA, et al. 1990. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 250: 1233–1238. 10.1126/science.1978757 [DOI] [PubMed] [Google Scholar]
- Masciari S, Dillon DA, Rath M, Robson M, Weitzel JN, Balmana J, Gruber SB, Ford JM, Euhus D, Lebensohn A, et al. 2012. Breast cancer phenotype in women with TP53 germline mutations: a Li–Fraumeni syndrome consortium effort. Breast Cancer Res Treat 133: 1125–1130. 10.1007/s10549-012-1993-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monti P, Perfumo C, Bisio A, Ciribilli Y, Menichini P, Russo D, Umbach DM, Resnick MA, Inga A, Fronza G. 2011. Dominant-negative features of mutant TP53 in germline carriers have limited impact on cancer outcomes. Mol Cancer Res 9: 271–279. 10.1158/1541-7786.MCR-10-0496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana HQ, Gelman R, LaDuca H, McFarland R, Dalton E, Thompson J, Speare V, Dolinsky JS, Chao EC, Garber JE. 2018. Differences in TP53 mutation carrier phenotypes emerge from panel-based testing. J Natl Cancer Inst 110: 863–870. 10.1093/jnci/djy001 [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675. 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. 2005. The FoldX web server: an online force field. Nucleic Acids Res 33: W382–W388. 10.1093/nar/gki387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villani A, Shore A, Wasserman JD, Stephens D, Kim RH, Druker H, Gallinger B, Naumer A, Kohlmann W, Novokmet A. 2016. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li–Fraumeni syndrome: 11 year follow-up of a prospective observational study. Lancet Oncol 17: 1295–1305. 10.1016/S1470-2045(16)30249-2 [DOI] [PubMed] [Google Scholar]
- West AN, Ribeiro RC, Jenkins J, Rodriguez-Galindo C, Figueiredo BC, Kriwacki R, Zambetti GP. 2006. Identification of a novel germ line variant hotspot mutant p53-R175L in pediatric adrenal cortical carcinoma. Cancer Res 66: 5056–5062. 10.1158/0008-5472.CAN-05-4580 [DOI] [PubMed] [Google Scholar]
- Yemelyanova A, Vang R, Kshirsagar M, Lu D, Marks MA, Shih Ie M, Kurman RJ. 2011. Immunohistochemical staining patterns of p53 can serve as a surrogate marker for TP53 mutations in ovarian carcinoma: an immunohistochemical and nucleotide sequencing analysis. Mod Pathol 24: 1248–1253. 10.1038/modpathol.2011.85 [DOI] [PubMed] [Google Scholar]
- Zambetti GP, Bargonetti J, Walker K, Prives C, Levine AJ. 1992. Wild-type p53 mediates positive regulation of gene expression through a specific DNA sequence element. Genes Dev 6: 1143–1152. 10.1101/gad.6.7.1143 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The variant described in this paper has been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) under accession number SCV000629866.1. Genomic sequence data was not deposited because of lack of patient consent.