

Abstract
Mutations in the mitochondrial fusion protein mitofusin 2 (MFN2) are the most commonly identified cause of Charcot-Marie-Tooth type 2 (CMT2), a dominantly inherited disease characterized by degeneration of peripheral sensory and motor axons. However, the mechanism by which mutations in this ubiquitously expressed mitochondrial fusion protein lead to neuropathy has not yet been elucidated. To explore how MFN2 mutations lead to degeneration of peripheral axons, we expressed neuropathy-associated forms of MFN2 in cultured dorsal root ganglion neurons, cells preferentially affected in CMT2. Disease-associated MFN2 mutant proteins induced abnormal clustering of small fragmented mitochondria in both neuronal cell bodies and proximal axons. Interestingly, transport of mitochondria in axons was significantly impaired in neurons expressing disease-mutated forms of MFN2. The diminished axonal mitochondrial transport was not attributable to diminished ATP levels in the neurons, and oxidative respiration was normal in mutant MFN2-expressing cells. Additionally, mitochondrial oxidative enzyme activity was normal in muscle mitochondria from a CMT2 patient with an MFN2 mutation, further supporting that abnormal mitochondrial transport in neurons is independent from an energy production defect. This abnormal mitochondrial trafficking provides a likely explanation for the selective susceptibility of the longest peripheral axons to MFN2 mutations, in which proper localization of mitochondria is critical for axonal and synaptic function.
Keywords: axon, axonal transport (axoplasmic transport), genetics, mitochondria, neuropathy, trafficking
Introduction
Charcot-Marie-Tooth disease (CMT) is among the most common inherited disorders of the nervous system, estimated to affect 1:2500 individuals (Berger et al., 2002; Suter and Scherer, 2003; Shy, 2004). CMT syndromes typically involve slowly progressive distal weakness, muscle atrophy, and sensory loss. Additional subcategorization of CMT is based on electrophysiologic studies: CMT type 1 corresponding to slowed nerve conduction velocities (<38 m/s in the median nerve) consistent with primary demyelination, and CMT type 2 corresponding to relative preservation of conduction velocity with diminished compound motor action potentials as evidence of axonal loss (Dyck et al., 1993). In general, genes mutated in CMT1 are structural myelin proteins or those that regulate their expression (Nagarajan et al., 2001; Saifi et al., 2003; Suter and Scherer, 2003). Although less well characterized in general, genes mutated in CMT2 are likely to be important for maintenance of axonal integrity, with their dysfunction leading to primary axonal degeneration (Shy, 2004).
Mutations in mitofusin 2 (MFN2), an outer mitochondrial membrane protein, are the most commonly identified cause of axonal CMT, found in 19–33% of such patients (Zuchner et al., 2004; Kijima et al., 2005; Lawson et al., 2005; Chung et al., 2006; Verhoeven et al., 2006). MFN2 is a dynamin family GTPase believed to primarily be involved in mitochondrial fusion, although it has also been implicated in regulating mitochondrial oxidative function (Pich et al., 2005), and intracellular signaling (Chen et al., 2004). MFN2 and its homolog MFN1 directly participate in docking and tethering of neighboring mitochondria and outer membrane fusion (Koshiba et al., 2004). Optic atrophy 1, or OPA1, an inner membrane-associated GTPase mutated in dominant optic atrophy, likely coordinates inner membrane fusion (Cipolat et al., 2004). Interestingly, the phenotypic spectrum of MFN2 mutations can also include optic atrophy in some severe cases, implicating a role for mitochondrial fusion proteins in optic nerve function (Chung et al., 2006; Zuchner et al., 2006).
Despite MFN2 mutations being the most common cause of axonal CMT (designated CMT2A), the mechanism by which mutations in MFN2 produce selective axonal degeneration has not been investigated. The fact that most CMT disease mutations are missense and all produce a dominant inheritance pattern suggests that mutations in MFN2 lead to a gain of function, altering either normal MFN1 or MFN2 function in a dominant-negative manner, or via another as yet unknown toxic effect. To determine how MFN2 mutations lead to peripheral neuropathy, we expressed disease-mutated MFN2 proteins in cultured dorsal root ganglion (DRG) neurons using lentivirus vectors. MFN2 disease mutants led to marked disruption of axonal mitochondrial transport, providing a possible explanation for the selective susceptibility of distal regions of long peripheral axons in CMT2A. This altered mitochondrial transport occurred in the absence of altered cellular ATP levels, mitochondrial potential, or respiratory chain function. These results suggest that altered mitochondrial transport induced by mutant MFN2 proteins plays a central role in the pathogenesis of CMT2A and indicate an unrecognized role for MFN2 in regulation of mitochondrial transport.
Materials and Methods
Plasmids and MFN2 mutant generation.
A full-length human MFN2 cDNA was obtained from Open Biosystems (Huntsville, AL), and a hemagglutinin tag was added to the N terminus after the initiator ATG using PCR mutagenesis as described previously (Baloh et al., 2000). CMT2A disease-associated point mutants were also generated by PCR mutagenesis, cloned into the phosphorylated enhanced green fluorescent protein–C1 (Clontech, Cambridge, UK), and into CMV-IRES-Venus (CCIV) for packing into lentivirus. CCIV was based on plasmid CSCGW (Sena-Esteves et al., 2004) and is a lentiviral packaging vector with the cytomegalovirus promoter, followed by a multicloning site, an internal ribosomal entry site (IRES), and Venus fluorescent protein. Versions were also made without the IRES–Venus for use in photoactivation experiments. For introduction into neurons, mitochondrially targeted Discosoma red (mito-DsRed2) and mitochondrially targeted photoactivatible GFP (mito-PAGFP) (Karbowski et al., 2004) were cloned into a lentiviral packaging vector downstream of the human ubiquitin promoter.
Primary neuron cultures, immunohistochemistry, and electron microscopy.
Dorsal root ganglion cultures were performed from embryonic rats as described previously (Araki et al., 2004). Timed pregnant Sprague Dawley rats were obtained from Charles River Laboratories (Cambridge, MA). Gestational day 15 embryos were removed, and the dorsal root ganglia were dissected out. Ganglia were dissociated using trypsin/EDTA and plated directly onto 24-well plates coated with laminin and poly-d-lysine. Cultures were maintained in Neurobasal medium with B27 supplement (Invitrogen, Carlsbad, CA), 50 ng/ml NGF, 5 mm glutamine, and antibiotics. Cells were plated in 5-fluorouracil as an antimitotic for the first 3 d in culture. Lentiviral infections were performed on 3–5 d after plating. For activated caspase-3 immunohistochemistry, neurons were fixed in cold 4% paraformaldehyde, blocked using PBS–1% BSA, and incubated with rabbit anti-activated caspase-3 antibody at a 1:50 dilution (Cell Signaling Technology, Beverly, MA) overnight at 4°C. After washing cells with PBS, cells were incubated in anti-rabbit secondary antibody conjugated to cyanine 3 and visualized with fluorescence microscopy. For electron microscopy, human embryonic kidney HEK 293T cells were plated on four-well chamber slides (Nunc, Rochester, NY) and infected with lentivirus encoding wild-type or R94Q mutant MFN2 protein. After 72 h, the cells were fixed and plastic embedded, and standard electron microscopy was performed. EM images were white balanced and processed once with the Sharpen filter in Adobe Photoshop (Adobe Systems, San Jose, CA).
Lentivirus production and infection.
Lentiviruses were produced as described previously (Araki et al., 2004). Briefly, HEK 293T cells were plated onto six-well plates and transfected using Fugene reagent (Roche, Basel, Switzerland) with a packaging vector (Δ8.91), envelope vector (vesicular stomatitis virus-glycoprotein), and transfer vector encoding the gene to be expressed. Supernatants were collected at 24 and 48 h, pooled, and titered on HEK 293T cells. Neuron cultures were infected using ∼107 colony-forming units (CFU)/ml in the final volume, and, in these conditions, >95% of neurons were infected.
Immunoblotting, MFN2 subcellular localization, and GTP-binding assays.
HEK 293T cells were transfected using Fugene (Roche) with either GFP control or the individual wild-type or mutant MFN2 constructs in the CCIV vector. Immunoblotting was performed using standard techniques, with rabbit anti-MFN2 antibody (antibody M6319; Sigma, St. Louis, MO) at 1:1000 dilution of primary antibody. For a loading control, mouse anti-acetylated tubulin antibody (Sigma) was also used at 1:1000 dilution. For confirming subcellular MFN2 localization, HEK 293T cells were transfected with GFP–MFN2 fusion constructs, after 24 h costained with Mitotracker Red dye (Invitrogen), and imaged via fluorescence microscopy.
For GTP-binding studies, HEK 293T cells were transfected with either GFP or GFP–MFN2 fusion constructs and collected in lysis buffer 24 h later [50 mm Tris-HCl, 150 mm NaCl, 1% Triton X-100, 5 mm MgOAc, 0.5 mm PMSF, EDTA-free protease inhibitor cocktail (Roche), and 1 mm DTT]. Cleared lysates were incubated with GTP-agarose beads (Innova Biosciences, Cambridge, UK) for 2 h at room temperature in the presence or absence of 10 mm GTP and subsequently washed three times with lysis buffer. Washed beads were resuspended in lysis buffer and placed in a fluorimeter to measure bound proteins at 485 nm excitation and 538 nm emission.
Mitochondrial mobility measurements.
DRG neurons infected with wild-type and mutant MFN2 constructs and mito-DsRed2 were imaged using a Nikon (Tokyo, Japan) Eclipse TE300 microscope with a 40× Nikon Pan-Fluor lens. Infected neurons were identified by their expression of Venus fluorescent protein, and 10 isolated axon segments for each MFN2 construct were selected for time-lapse imaging. Images were acquired every 3 s for a total of 100 images (5 min) and subjected to kymograph analysis (De Vos et al., 2003; Miller and Sheetz, 2004). Kymographs were generated using MetaMorph Software (Molecular Devices, Sunnyvale, CA) with a width of 11 pixels and using a maximum intensity projection. The number of moving mitochondria in each kymograph was counted, and statistical comparisons of mutant versus wild type were performed using a Student's paired t test.
Mitochondrial photoactivation and confocal microscopy.
DRG neurons were infected with lentiviruses encoding wild-type or mutant MFN2 (without the IRES–Venus), mito-DsRed2 to visualize all mitochondria in the cell, and mito-PAGFP to allow monitoring of individual photoactivated mitochondria. Neuronal cell bodies were imaged using a Zeiss (Oberkochen, Germany) Axiovert 200M inverted laser scanning confocal microscope and a 63×, 1.4 numerical aperture Apochromat objective (Zeiss MicroImaging). Filters used were as follows: DsRed2, excitation at 543 nm, emission long-pass at 560 nm; PAGFP, excitation at 488 nm, emission bandpass at 505–530 nm. Photoactivation of mitochondria in approximately half of the cell was performed using the 405 nm laser, and cells were reimaged immediately after photoactivation and 36 h later using the same settings.
ATP measurements, oxygen electrode experiments, and OXPHOS enzyme complex assays.
For ATP measurements, DRG neurons were placed in culture, infected with lentiviruses after 3–5 d in vitro, and collected 1 week after infection. Cells were lysed in Tris-MES buffer with 4 mm EDTA and 0.5% Triton X-100, and ATP was measured using the CellTiter-Glo reagent (Promega, Madison, WI) at 1:1 volume, with luciferase activity measured using a luminometer. Total protein content was determined from a portion of the samples by the Bio-Rad (Hercules, CA) DC protein assay. Oxidative respiration studies were performed using a Clark oxygen electrode (Oxygraph; Hansatech Instruments, Norfolk, UK) similar to that described previously (Chen et al., 2005). HEK 293T cells were plated on six-well plates and infected with 107 CFU/ml lentivirus encoding wild-type or mutant MFN2 proteins, under which conditions essentially all of the cells are infected. At 72 h later, cells were triturated off the plate and spun down, and 1.5 × 106 cells were resuspended in 300 μl of TD respiration buffer (137 mm NaCl, 5 mm KCl, 0.7 mm Na2HPO4, and 25 mm Tris-HCl, pH 7.5) and placed into the oxygraph chamber. After equilibration for 1 min, the chamber was stoppered, endogenous oxygen consumption was measured for 3 min, and then 2,4-dinitrophenol (Sigma) was added to a final concentration of 400 nm. Oxidative enzyme activity of human muscle tissue was performed as described previously (DiMauro et al., 1987).
Results
Mitofusin 2 disease mutants are produced at normal quantities and have normal stability and GTP-binding activity
The dominant inheritance pattern in CMT2A is consistent with either a dominant-negative, toxic gain of function effect or haploinsufficiency effect of MFN2 mutations. We therefore investigated protein levels and stability of exogenously expressed wild-type and mutant MFN2 in HEK 293T cells, an epithelial cell line in which minimal endogenous MFN2 is present (Fig. 1 A). Transfected wild-type and mutant MFN2 proteins all migrated as a single band at ∼80 kDa (Fig. 1 B, and data not shown). Protein stability was similar for wild-type and disease mutant MFN2, as determined by immunoblotting of transfected cell lysates after the addition of cycloheximide to block protein synthesis (Fig. 1 B). Similar results were seen for all MFN2 disease mutants in this study (data not shown). Wild-type and mutant MFN2 proteins with GFP fused to the N terminus in HEK 293T cells were properly localized to mitochondria, indicating that the CMT2A-associated disease mutants do not disrupt mitochondrial targeting (Fig. 1 C, and data not shown), which is mediated by the C-terminal region (Santel and Fuller, 2001; Rojo et al., 2002). Additionally, because many of the neuropathy-associated MFN2 mutations are near the GTPase domain, we investigated the GTP-binding capability of MFN2 disease mutants (Fig. 2). GFP alone did not bind to GTP-agarose beads, and GFP–MFN2 binding was competed away by incubating in the presence of excess GTP (10 mm), indicating the binding was specific. Duplicate measurements of bound MFN2 mutant proteins normalized to input protein showed no significant differences from wild-type binding. Together, these data support the hypothesis that CMT2A-associated MFN2 mutants do not lead to haploinsufficiency via altering stability or targeting of MFN2 but instead cause disease via a dominant-negative or toxic gain of function mechanism. This hypothesis is also supported by the finding that mice heterozygous for a null MFN2 mutation do not develop a peripheral neuropathy (Chen et al., 2003).
Figure 1.
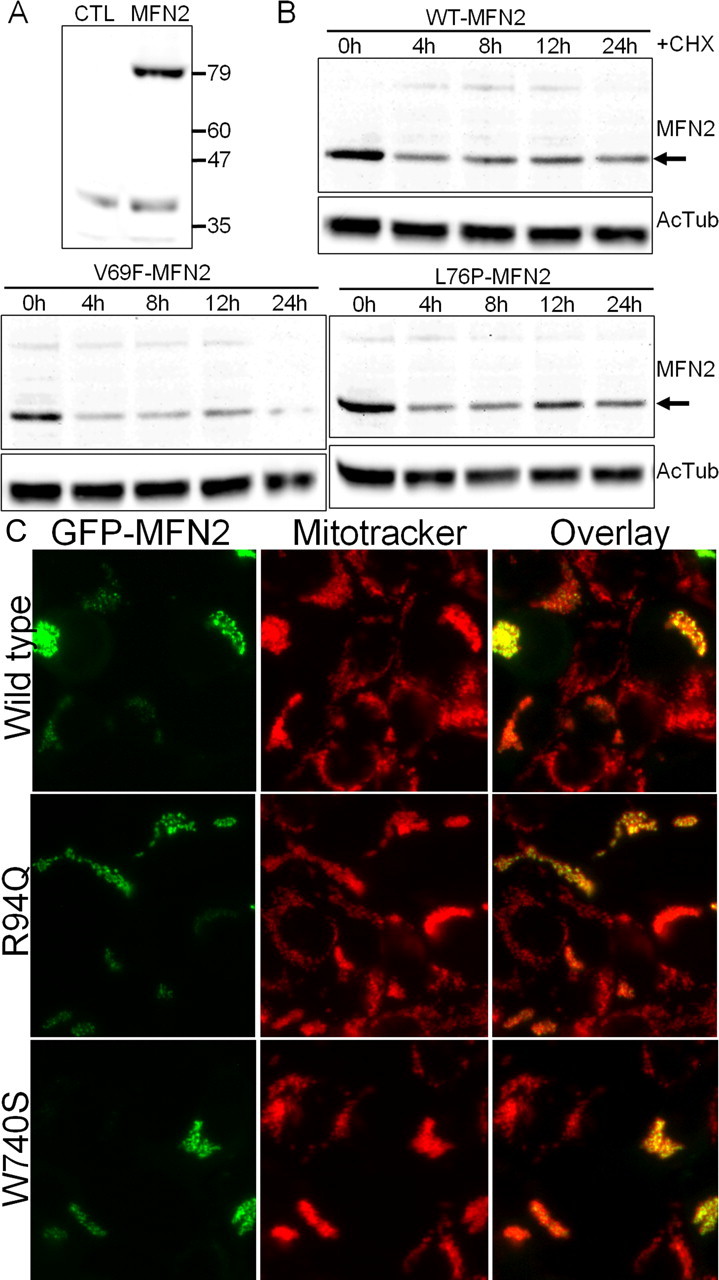
Mutant MFN2 proteins have normal half-lives and are properly localized to mitochondria. A, HEK 293T cells were transfected with an expression vector for GFP (CTL) or wild-type human MFN2 (MFN2), and immunoblotting for MFN2 was performed. Lysate from MFN2-transfected cells showed a single band at ∼80 kDa. Longer exposure did reveal a small amount of endogenous MFN2 present in HEK 293T cells (data not shown). B, HEK 293T cells were transfected with either wild-type (WT-MFN2) or the indicated MFN2 mutant. Cells were lysed at the indicated times after addition of 30 μg/ml cycloheximide (CHX) to inhibit new protein synthesis, and immunoblotting was performed using an anti-MFN2 antibody. Acetylated tubulin (AcTub) immunoblotting is shown on the same samples below as a loading control. Disease mutants ran at identical molecular weight and had a comparable stability to wild-type MFN2. Similar results were obtained for all of the mutants examined in this study (data not shown). C, HEK 293T cells were transfected with wild-type or mutant MFN2 constructs, in which GFP was fused to the N terminus. Cells were treated with Mitotracker Red dye to label mitochondria and imaged with fluorescence microscopy. The overlaid image shows that the GFP-fused MFN2 mutant constructs are properly localized to mitochondria.
Figure 2.

Mitofusin 2 disease mutants have retained GTP-binding activity. Wild-type (WT) and disease mutant MFN2 constructs with GFP fused to the N terminus were transfected into HEK 293T cells, and cell lysates were prepared 24 h later. The lysates were incubated with GTP-agarose beads, and bound GFP-MFN2 protein was measured using a fluorimeter. A, GFP–MFN2 binding to GTP-agarose beads was specific, because there is no background binding of GFP alone, and excess GTP (10 mm) was able to competitively block GFP–MFN2 binding to GTP-agarose beads. B, GTP-agarose binding as above using GFP-MFN2 mutants performed in duplicate demonstrates that all of the MFN2 disease mutants bind GTP to a similar extent.
MFN2 mutants induce aggregation of mitochondria in cultured DRG neurons
To investigate the effect of MFN2 mutants on peripheral neurons, we used recombinant lentiviruses to express wild-type or disease mutant MFN2 proteins in cultured rat dorsal root ganglion neurons. Mitochondria were visualized by coinfection with mito-DsRed2. Neurons infected with GFP alone or wild-type MFN2 showed a normal punctuate distribution of mitochondria in the cell body and axons (Fig. 3). In contrast, neuronal cell bodies expressing MFN2 disease mutants showed varying degrees of mitochondrial aggregation, also with cellular regions devoid of mitochondria (Fig. 3, individual mutants labeled). Aggregation was most pronounced in mutants L76P, R94Q, P251A, and R280H and was present but somewhat less pronounced in mutant V69F. Interesting, mutant W740S, the only disease mutant located near the C-terminal coiled coil domain, did not produce significant mitochondrial aggregation. Electron microscopy of HEK 293T cells infected with MFN2 mutant R94Q confirmed that these represent clusters of individual mitochondria, some of which appear misshapen (Fig. 3). Neurons with mitochondrial aggregates were phase bright, and activated caspase-3 staining was absent, confirming that this was not attributable to induction of apoptosis, a known precipitant of fragmentation of the mitochondrial network (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) (Youle and Karbowski, 2005). Aggregates of mitochondria were also seen in the proximal segments of axons in cells expressing mutant MFN2 proteins (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Low-power images showed that DsRed2-labeled mitochondria are evenly distributed in both proximal and distal axonal segments of DRG neurons expressing wild-type MFN2 (Fig. 4 A). In contrast, neurons expressing mutant MFN2 proteins showed few mito-DsRed2-containing mitochondria in distal axonal segments, accompanied by clustering of mitochondria in the proximal segments (Fig. 4 B,C), suggesting that disease mutant MFN2 proteins produce a mitochondrial trafficking abnormality.
Figure 3.
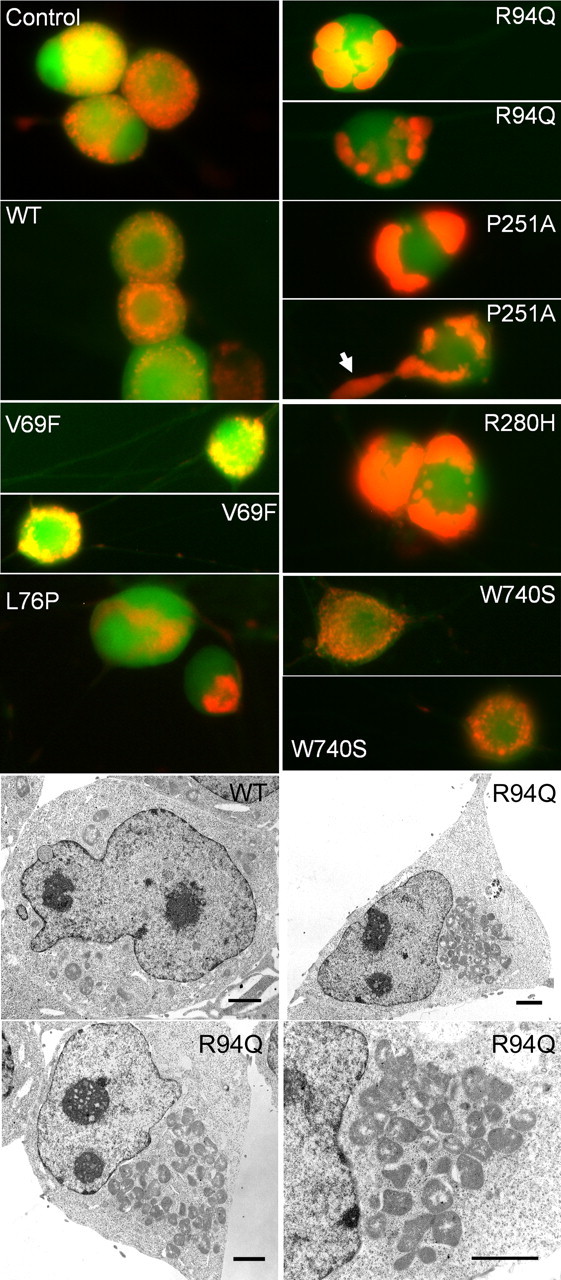
MFN2 mutants induce abnormal mitochondrial clustering and morphology in cell bodies and proximal axons. Embryonic day 15 rat dorsal root ganglion neurons in culture were infected with lentiviruses encoding GFP as a control, wild-type MFN2, or the indicated CMT2 disease mutant followed by an IRES–Venus fluorescent protein to mark infected cells. Subsequently, cells were infected with a second lentivirus encoding a mitochondrially targeted DsRed2, and live cells were imaged. Both GFP infected (GFP) and wild-type MFN2 infected (WT-MFN2) showed a similar pattern of mitochondria in the soma, with a discrete grain-like appearance that was diffuse throughout the cell. In contrast, mutants V69F, L76P, R94Q, P251A, and R280H all led to abnormal clustering of mitochondria, usually in either one or two large clusters, without discrete mitochondria apparent and with cellular regions entirely devoid of mitochondria. This was least pronounced in mutant V69F and not present for mutant W740S. On higher magnification, these appeared as small fragmented mitochondria (see also Figs. 5, 6). Frequently, aggregates of small mitochondria would cluster in the proximal axon (arrow in P251A–MFN2). Electron microscopic images (bottom panels) of HEK 293T cells expressing either wild type or the R94Q mutant MFN2 via lentiviral infection confirmed that the clusters seen in the R94Q mutant represent aggregates of individual mitochondria, many of which appeared misshapen. Scale bars, 2 μm.
Figure 4.

MFN2 disease mutants induce abnormal axonal mitochondrial mobility. Low-power fluorescence images of mito-DsRed2 fluorescence in neurons expressing wild-type (WT) MFN2 (A) or disease mutants R94Q (B) or P251A (C), taken at the same exposure 96 h after infection with mito-DsRed2 lentivirus. In wild-type MFN2-expressing neurons, mitochondria were distributed evenly throughout the distal axonal segments. In contrast, mitochondria in disease mutant MFN2-expressing neurons were found predominantly in the proximal axonal segments and were not seen in distal axonal regions, consistent with a mitochondrial transport defect. To characterize this further, time-lapse imaging was performed over a 5 min period on 10 axons at 40× magnification for each construct, and kymographs were generated from individual axons. D, Representative axon infected with wild-type MFN2, showing mitochondria with normal short tubular morphology, evenly distributed throughout the axon. E, Representative kymograph of time-lapse movie from a wild-type MFN2-expressing axon shows a large number of mobile mitochondria indicated by sloping lines on the kymograph image. The arrow points to an individual axonal mitochondrion moving during the time-lapse movie. F, Representative axon expressing mutant R94Q MFN2 showing clusters of small mitochondria in the proximal axon (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). G, A representative kymograph of an R94Q mutant MFN2-expressing axon showing mostly stationary mitochondria or clusters of mitochondria, with few moving mitochondria during the time lapse (arrow again indicates a moving mitochondrion). H, Quantitative analysis of mobile mitochondria from kymographs. The average number of moving mitochondria per 100 μm axonal segment was significantly lower in the neurons expressing mutant MFN2 constructs compared with those expressing wild-type MFN2 (Student's paired t test, mutant vs wild-type, *p < 0.0001).
Axonal mitochondrial trafficking is significantly disrupted by MFN2 disease mutants
Qualitative changes in mitochondrial mobility has been observed in mouse embryonic fibroblasts deficient in MFN2 (Chen et al., 2003) and in a fibroblast cell line expressing an engineered MFN2 mutant with slowed GTP hydrolysis (Neuspiel et al., 2005). Furthermore, we saw decreased mitochondria movement to the distal axons of cells expressing MFN2 disease mutants (Fig. 4 A–C). Therefore, we investigated whether mitochondrial mobility was altered in neurons expressing disease mutant forms of MFN2 using time-lapse fluorescence imaging and kymograph analysis similar to that described previously (Zhou et al., 2001; De Vos et al., 2003; Miller and Sheetz, 2004). Kymographs represent pixel intensity along a line drawn over the axon of interest as the x-axis and time on the y-axis, generating a two-dimensional image displaying movement along an axon over the entire time-lapse movie. Moving mitochondria appear as sloped lines, changing position (x-axis) over time (y-axis), whereas stationary mitochondria appear as columns. Axons of DRG neurons expressing wild-type MFN2 contained short tubular mitochondria distributed evenly throughout, and kymographs of these axons showed frequent mitochondrial movements (Fig. 4 D,E). In contrast, axons expressing MFN2 mutants frequently showed multiple stationary clusters of mitochondria, with fewer moving mitochondria present on kymograph analysis (Fig. 4 F,G). Whereas the stationary clusters contained multiple mitochondria, moving structures appeared consistent in size with individual mitochondria. The number of moving mitochondria was significantly reduced in axons expressing MFN2 disease mutants compared with wild-type (Fig. 4 H). Of note, a similar reduction in mitochondrial movement was seen in axons expressing disease mutant MFN2 proteins that produced either marked (R94Q, P251A) or minor (V69F, W740S) mitochondrial aggregation.
To further investigate this mobility defect, we generated mito-PAGFP to investigate mitochondrial dynamics in live cells (Patterson and Lippincott-Schwartz, 2002; Karbowski et al., 2004). This method has been used to investigate mitochondrial fusion, by measuring the decrease in fluorescence in a region of interest at time points after GFP photoactivation. Neurons expressing wild-type MFN2 showed complete redistribution of mito-PAGFP 36 h after photoactivation (Fig. 5, top). However, neurons expressing disease mutant MFN2 showed large clusters of small aggregated mitochondria, and, 36 h after photoactivation, these mitochondria had only minimally redistributed throughout the cell. These experiments confirm that disease mutant MFN2 disrupts mitochondrial trafficking in both neuronal cell bodies and axons. Unfortunately, because of the mobility defect, an independent measure of mitochondrial fusion cannot be made because redistribution of mitochondria after photoactivation must be equivalent for fluorescence decrement to be an independent measure of mitochondrial fusion (Karbowski et al., 2004).
Figure 5.
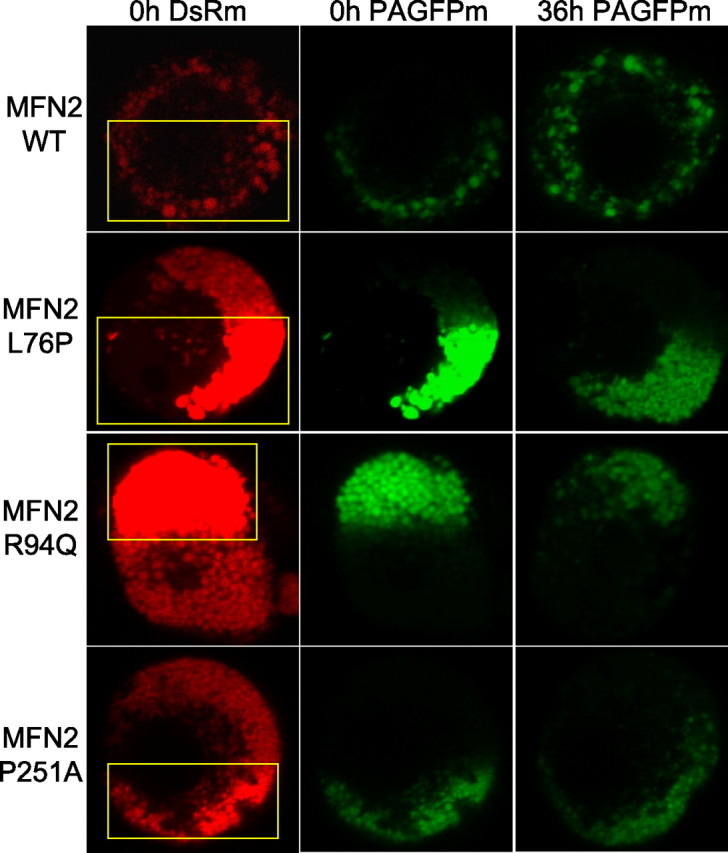
Mitochondrial movement is impaired in neuronal cell bodies expressing mutant MFN2 proteins. DRG neurons were infected with lentiviruses encoding wild-type or disease mutant MFN2 constructs, together with a mitochondrially targeted DsRed2 (DsRm) and photoactivatible GFP (PAGFPm). PAGFP is a variant of GFP and has minimal fluorescence at baseline, but its fluorescence increases ∼100-fold when photoactivated with 405 or 413 nm light (Patterson and Lippincott-Schwartz, 2002). After 10 d in culture, mitochondria in approximately half of the cell body were photoactivated using the 405 nm laser (yellow box indicates region of photoexcitation), and individual cells were reimaged 36 h later. Neurons expressing wild-type MFN2 (WT) showed an even redistribution of photoactivated mitochondria throughout the cell body after 36 h. Neurons expressing mutant MFN2 proteins had clustered, fragmented mitochondria. After 36 h, there was very little movement of mitochondria from the region of photoexcitation. The decrement in the post-photoactivation signal suggests that some mitochondrial fusion still took place, although because of the marked mobility defect, this cannot be measured as an independent variable.
Disrupted axonal mitochondrial transport does not result from diminished ATP production, and neuropathy-associated MFN2 mutations do not alter mitochondrial oxidative function
Axonal mitochondrial transport uses ATP-dependent motors kinesin and dynein to mediate anterograde and retrograde transport, respectively (Hollenbeck, 1996). MFN2 mutants could disrupt axonal mitochondrial transport directly via altering the ability of mitochondria to attach to kinesin or dynein motor proteins. Alternatively, mutant MFN2 could disrupt mitochondrial oxidative function directly, leading to decreased ATP levels and subsequently diminished axonal transport via ATP dependent motor proteins. Therefore, we investigated ATP levels in DRG cultures expressing mutant MFN2 proteins. Whereas control cultures treated with 2-deoxyglucose and oligomycin had near complete depletion of ATP after 12 h, neurons expressing mutant MFN2 proteins had similar ATP levels to those expressing wild-type MFN2, despite the marked abnormality in mitochondrial transport seen in MFN2 mutant-expressing neurons (Fig. 6 A). Furthermore, mitochondrial membrane potential was maintained in neurons expressing mutant MFN2 constructs, as indicated by uptake of Mitotracker Red dye (Fig. 6 B). To further investigate the effect of MFN2 mutants on mitochondrial function, we examined oxidative respiration in HEK 293T cells expressing wild-type or disease mutant MFN2 proteins (Fig. 7). MFN2 disease mutants induced similar mitochondrial clustering in HEK 293T cells to that seen in neurons (Fig. 7 B) (see also Fig. 3). Despite the abnormal clustered mitochondria, both endogenous and maximal rates of oxygen consumption were similar in cells expressing wild-type and disease mutant MFN2 (Fig. 7 C). Furthermore, we found that oxidative enzyme activity was also normal in a muscle biopsy from a patient with CMT2A attributable to a W740S mutation (supplemental table, available at www.jneurosci.org as supplemental material). These data indicate that the mitochondrial transport defect induced by MFN2 mutants is not simply attributable to diminished ATP levels or disruption of mitochondrial oxidative function and suggest instead that MFN2 mutants alter the ability of mitochondria to effectively attach to the microtubule motor apparatus.
Figure 6.

MFN2 mutants do not alter ATP levels or diminish mitochondrial potential in cultured sensory neurons. A, DRG cultures of neurons expressing either wild-type or mutant MFN2 constructs were collected 10 d after infection, and ATP and protein levels were measured. GFP-infected neurons treated with 2-deoxyglucose and oligomycin (2DG/O) for 12 h showed essentially complete depletion of ATP, whereas ATP levels were normal in cells expressing wild-type or disease mutant MFN2 proteins, despite the marked abnormality in ATP-dependent mitochondrial transport (Figs. 3, 4). B, DRG neurons were infected with lentivirus encoding the indicated wild-type (WT) or mutant MFN2 constructs, stained for 30 min with 25 nm Mitotracker Red, and imaged with confocal microscopy. Mitotracker Red staining of neurons expressing wild-type and disease mutant MFN2 showed similar intensity of staining of mitochondria, indicating that the mitochondrial potential in the aggregated mitochondria is maintained. Staining was diminished in the center of the largest mitochondrial clusters, likely attributable to decreased penetration of the Mitotracker dye in the short staining period. The neuron labeled with an asterisk in panel R94Q was uninfected and is shown for comparison with the marked mitochondrial aggregation seen in the neighboring cell. There was no quantitative difference in fluorescence intensity of the Mitotracker dye between WT and mutant infected cells (data not shown), indicating that the mitochondrial membrane potential was not significantly diminished in neurons expressing mutant MFN2 constructs.
Figure 7.

Oxidative respiration is normal in cells expressing MFN2 disease mutants despite mitochondrial clustering. A, Representative oxygen consumption graphs for HEK 293T cells expressing wild-type MFN2 (WT) or disease mutants R94Q or W740S. The arrow indicates the addition of 2,4-dinitrophenol, an uncoupling agent to induce maximal oxidative respiration. The slope of the line indicating the rate of respiration is shown above the curves both before (endogenous respiration) and after (maximal respiration) the addition of the uncoupling agent 2,3-dinitrophenol. B, Fluorescence images of HEK 293T cells expressing wild-type (WT) MFN2 or disease mutant R94Q, together with mito-DsRed2 to visualize mitochondria, showed that these cells form similar mitochondrial aggregates to that seen in primary neurons only in the presence of MFN2 disease mutants. C, Despite the mitochondrial clustering induced by mutant MFN2 proteins, both endogenous and maximal respiration rates were similar in cells expressing wild-type or mutant MFN2 proteins, indicating that mutant MFN2 proteins do not alter mitochondrial oxidative respiration.
Discussion
Our results show that MFN2 mutant proteins consistently lead to a marked disruption of mitochondrial transport in cultured sensory neurons in the absence of a notable energetic effect. Despite this marked transport defect, mutant MFN2 proteins do not lead to a gross defect in ATP production, mitochondrial membrane potential, or oxidative activity in cultured cells or in muscle from a patient with CMT2A. These data have important implications for the relative tissue specificity of MFN2 mutants for distal segments of long motor and sensory axons, because these highly metabolic regions distant from the cell body are likely differentially sensitive to a mitochondrial movement defect compared with other cells. In this way, the pathogenesis of CMT2A may be attributable to inadequate supply of mitochondria to maintain the energy needs of the distal regions of peripheral axons.
Mechanism of mitochondrial trafficking defect from MFN2 mutants
Aggregation of mitochondria has been shown in cell lines expressing mitofusin mutants with altered GTPase activity (Koshiba et al., 2004; Neuspiel et al., 2005). The mechanism of mitochondrial clusters in these cases is hypothesized to be attributable to the formation of tethered intermediates, which are unable to complete mitochondrial fusion, whereby ongoing fission then leads to fragmented-tethered mitochondrial clusters. It is possible that this is also the mechanism by which MFN2 disease mutants induce aggregation of mitochondria. This could lead to a secondary alteration in mitochondrial transport, because “sticky” aggregated mitochondria may not be able to properly attach to the transport machinery.
An alternative hypothesis to explain the abnormal mitochondrial transport is that MFN2 itself could play a regulatory role in the assembly of the molecular complex for attachment to kinesin or dynein motors. This stems from the concept that MFN2 may function as a regulatory GTPase, based on its high GTP affinity and low GTP hydrolysis rate (Ishihara et al., 2004; Zorzano and Pich, 2006). Indeed, one possible explanation for why mutant W740S did not lead to mitochondrial aggregation but did alter axonal mitochondrial transport could be that this region is directly involved in interactions with the mitochondrial transport apparatus. In this way, MFN2 could function similar to other mitochondrial GTPases involved in mitochondrial trafficking, such as Miro (mitochondrial Rho) proteins (Glater et al., 2006). Notably, clustering of mitochondria similar to that observed here is also seen after disruption of mitochondrial transport by altering Miro function (Fransson et al., 2006) and also in cells lacking Kif5b (kinesin family member 5b) of the anterograde transport apparatus (Tanaka et al., 1998).
We should also note that our in vitro neuronal cultures lack myelinating Schwann cells, which may alter axonal transport, and myelinated axons are predominantly affected in CMT2A. Neuron–Schwann cell coculture experiments will be needed to determine whether the presence of myelinating Schwann cells either diminishes or augments the transport defect caused by MFN2 mutant proteins.
Mitochondrial movement and mitochondrial fusion
We were unable to independently measure mitochondrial fusion in neurons using mito-PAGFP, because this technique requires that mitochondrial movement is not altered to be an independent measure of mitochondrial fusion (Karbowski et al., 2004). Furthermore, other cell-based systems, including the use of polyethylene glycol-mediated fusion of cells expressing differentially labeled mitochondria, must also assume that mitochondrial movement is normal within cells to be used as a measure solely of mitochondrial fusion. Interestingly, murine embryonic fibroblasts derived from MFN2 knock-out animals also demonstrate a qualitative defect in mitochondrial movement as seen by time-lapse imaging, in addition to diminished fusion (Chen et al., 2003). Although it is clear that abnormal mitochondrial movement could lead to decreased fusion, it is not obvious why decreased fusion must lead to decreased mitochondrial movement, especially in the absence of overt mitochondrial dysfunction. Future experiments using cell-free systems in which fusion is not dependent on mitochondrial trafficking will be needed to separate a differential effect of MFN2 mutants on mitochondrial fusion and mitochondrial movement.
Mitochondrial trafficking, neuropathy, and neurodegenerative disease
Mitochondria are differentially localized in neurons to sites of high energy demand, including synapses and nodes of Ranvier, and the signals leading to this proper localization are beginning to be understood (Chada and Hollenbeck, 2003; Hollenbeck and Saxton, 2005). Because of the high energy demand at very distant sites from the cell body, the effective trafficking of mitochondria is presumed to be critical for proper neuronal function. This differential sensitivity of neurons in general to proper mitochondrial trafficking has furthermore been proposed to play a role in neurodegenerative diseases (Reynolds et al., 2004). Given that the peripheral sensory and motor axons are the longest in the body, it is likely that these are among the most sensitive to a mitochondrial trafficking insult, and our data support this as a mechanism for the relative tissue specificity for MFN2 mutations to long peripheral neurons. In this way, the distal axons may not be able to adequately supply enough mitochondria to support their energy demands. However, this does not explain why the distal segments of upper motor neurons are only rarely affected (Zhu et al., 2005) or why the optic nerve can be involved in severe cases, in a manner reminiscent of dominant optic atrophy or Leber's hereditary optic neuropathy (Newman, 2005; Zuchner et al., 2006). Perhaps the MFN2 mutations in CMT2A cases in which optic atrophy is involved also produce a direct mitochondrial energy production defect in addition to a mitochondrial movement defect, resulting in optic nerve involvement, the more classic finding in mitochondrial disease. Additional in vitro work looking at genotype–phenotype correlation for MFN2 mutations, effects of mutant MFN2 proteins on other aspects of mitochondrial function, and development of a mouse model of the disease will be helpful in addressing these questions.
Footnotes
R.H.B. was supported by Muscular Dystrophy Association Grant 4152. We especially thank Rati Choksi for performing mitochondrial oxidative enzyme measurements, Drs. Eugene Johnson and David Holtzman for comments on this manuscript, and all members of the Milbrandt laboratory for helpful discussions. We thank Dr. Miguel Sena-Esteves for providing vector CSCGW, which formed the basis of the CCIV construct, and Dr. Richard Youle for the mito-DsRed2 and mito-PAGFP constructs.
References
- Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- Baloh RH, Tansey MG, Johnson EM, Jr, Milbrandt J. Functional mapping of receptor specificity domains of glial cell line- derived neurotrophic factor (GDNF) family ligands and production of GFRalpha1 RET-specific agonists. J Biol Chem. 2000;275:3412–3420. doi: 10.1074/jbc.275.5.3412. [DOI] [PubMed] [Google Scholar]
- Berger P, Young P, Suter U. Molecular cell biology of Charcot-Marie-Tooth disease. Neurogenetics. 2002;4:1–15. doi: 10.1007/s10048-002-0130-z. [DOI] [PubMed] [Google Scholar]
- Chada SR, Hollenbeck PJ. Mitochondrial movement and positioning in axons: the role of growth factor signaling. J Exp Biol. 2003;206:1985–1992. doi: 10.1242/jeb.00263. [DOI] [PubMed] [Google Scholar]
- Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem. 2005;280:26185–26192. doi: 10.1074/jbc.M503062200. [DOI] [PubMed] [Google Scholar]
- Chen KH, Guo X, Ma D, Guo Y, Li Q, Yang D, Li P, Qiu X, Wen S, Xiao RP, Tang J. Dysregulation of HSG triggers vascular proliferative disorders. Nat Cell Biol. 2004;6:872–883. doi: 10.1038/ncb1161. [DOI] [PubMed] [Google Scholar]
- Chung KW, Kim SB, Park KD, Choi KG, Lee JH, Eun HW, Suh JS, Hwang JH, Kim WK, Seo BC, Kim SH, Son IH, Kim SM, Sunwoo IN, Choi BO. Early onset severe and late-onset mild Charcot-Marie-Tooth disease with mitofusin 2 (MFN2) mutations. Brain. 2006;129:2103–2118. doi: 10.1093/brain/awl174. [DOI] [PubMed] [Google Scholar]
- Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci USA. 2004;101:15927–15932. doi: 10.1073/pnas.0407043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos KJ, Sable J, Miller KE, Sheetz MP. Expression of phosphatidylinositol (4,5) bisphosphate-specific pleckstrin homology domains alters direction but not the level of axonal transport of mitochondria. Mol Biol Cell. 2003;14:3636–3649. doi: 10.1091/mbc.E02-10-0638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMauro S, Servidei S, Zeviani M, DiRocco M, DeVivo DC, DiDonato S, Uziel G, Berry K, Hoganson G, Johnsen SD, Johnson PC. Cytochrome c oxidase deficiency in Leigh syndrome. Ann Neurol. 1987;22:498–506. doi: 10.1002/ana.410220409. [DOI] [PubMed] [Google Scholar]
- Dyck PJ, Chance P, Lebo R, Carney JA. Ed 3. Philadelphia: Saunders; 1993. Peripheral neuropathy: hereditary motor and sensory neuropathies. [Google Scholar]
- Fransson S, Ruusala A, Aspenstrom P. The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem Biophys Res Commun. 2006;344:500–510. doi: 10.1016/j.bbrc.2006.03.163. [DOI] [PubMed] [Google Scholar]
- Glater EE, Megeath LJ, Stowers RS, Schwarz TL. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J Cell Biol. 2006;173:545–557. doi: 10.1083/jcb.200601067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck PJ. The pattern and mechanism of mitochondrial transport in axons. Front Biosci. 1996;1:d91–d102. doi: 10.2741/a118. [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ, Saxton WM. The axonal transport of mitochondria. J Cell Sci. 2005;118:5411–5419. doi: 10.1242/jcs.02745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N, Eura Y, Mihara K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J Cell Sci. 2004;117:6535–6546. doi: 10.1242/jcs.01565. [DOI] [PubMed] [Google Scholar]
- Karbowski M, Arnoult D, Chen H, Chan DC, Smith CL, Youle RJ. Quantitation of mitochondrial dynamics by photolabeling of individual organelles shows that mitochondrial fusion is blocked during the Bax activation phase of apoptosis. J Cell Biol. 2004;164:493–499. doi: 10.1083/jcb.200309082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kijima K, Numakura C, Izumino H, Umetsu K, Nezu A, Shiiki T, Ogawa M, Ishizaki Y, Kitamura T, Shozawa Y, Hayasaka K. Mitochondrial GTPase mitofusin 2 mutation in Charcot-Marie-Tooth neuropathy type 2A. Hum Genet. 2005;116:23–27. doi: 10.1007/s00439-004-1199-2. [DOI] [PubMed] [Google Scholar]
- Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC. Structural basis of mitochondrial tethering by mitofusin complexes. Science. 2004;305:858–862. doi: 10.1126/science.1099793. [DOI] [PubMed] [Google Scholar]
- Lawson VH, Graham BV, Flanigan KM. Clinical and electrophysiologic features of CMT2A with mutations in the mitofusin 2 gene. Neurology. 2005;65:197–204. doi: 10.1212/01.wnl.0000168898.76071.70. [DOI] [PubMed] [Google Scholar]
- Miller KE, Sheetz MP. Axonal mitochondrial transport and potential are correlated. J Cell Sci. 2004;117:2791–2804. doi: 10.1242/jcs.01130. [DOI] [PubMed] [Google Scholar]
- Nagarajan R, Svaren J, Le N, Araki T, Watson M, Milbrandt J. EGR2 mutations in inherited neuropathies dominant-negatively inhibit myelin gene expression. Neuron. 2001;30:355–368. doi: 10.1016/s0896-6273(01)00282-3. [DOI] [PubMed] [Google Scholar]
- Neuspiel M, Zunino R, Gangaraju S, Rippstein P, McBride H. Activated mitofusin 2 signals mitochondrial fusion, interferes with Bax activation, and reduces susceptibility to radical induced depolarization. J Biol Chem. 2005;280:25060–25070. doi: 10.1074/jbc.M501599200. [DOI] [PubMed] [Google Scholar]
- Newman NJ. Hereditary optic neuropathies: from the mitochondria to the optic nerve. Am J Ophthalmol. 2005;140:517–523. doi: 10.1016/j.ajo.2005.03.017. [DOI] [PubMed] [Google Scholar]
- Patterson GH, Lippincott-Schwartz J. A photoactivatable GFP for selective photolabeling of proteins and cells. Science. 2002;297:1873–1877. doi: 10.1126/science.1074952. [DOI] [PubMed] [Google Scholar]
- Pich S, Bach D, Briones P, Liesa M, Camps M, Testar X, Palacin M, Zorzano A. The Charcot-Marie-Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum Mol Genet. 2005;14:1405–1415. doi: 10.1093/hmg/ddi149. [DOI] [PubMed] [Google Scholar]
- Reynolds IJ, Malaiyandi LM, Coash M, Rintoul GL. Mitochondrial trafficking in neurons: a key variable in neurodegeneration? J Bioenerg Biomembr. 2004;36:283–286. doi: 10.1023/B:JOBB.0000041754.78313.c2. [DOI] [PubMed] [Google Scholar]
- Rojo M, Legros F, Chateau D, Lombes A. Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J Cell Sci. 2002;115:1663–1674. doi: 10.1242/jcs.115.8.1663. [DOI] [PubMed] [Google Scholar]
- Saifi GM, Szigeti K, Snipes GJ, Garcia CA, Lupski JR. Molecular mechanisms, diagnosis, and rational approaches to management of and therapy for Charcot-Marie-Tooth disease and related peripheral neuropathies. J Investig Med. 2003;51:261–283. doi: 10.1136/jim-51-05-14. [DOI] [PubMed] [Google Scholar]
- Santel A, Fuller MT. Control of mitochondrial morphology by a human mitofusin. J Cell Sci. 2001;114:867–874. doi: 10.1242/jcs.114.5.867. [DOI] [PubMed] [Google Scholar]
- Sena-Esteves M, Tebbets JC, Steffens S, Crombleholme T, Flake AW. Optimized large-scale production of high titer lentivirus vector pseudotypes. J Virol Methods. 2004;122:131–139. doi: 10.1016/j.jviromet.2004.08.017. [DOI] [PubMed] [Google Scholar]
- Shy ME. Charcot-Marie-Tooth disease: an update. Curr Opin Neurol. 2004;17:579–585. doi: 10.1097/00019052-200410000-00008. [DOI] [PubMed] [Google Scholar]
- Suter U, Scherer SS. Disease mechanisms in inherited neuropathies. Nat Rev Neurosci. 2003;4:714–726. doi: 10.1038/nrn1196. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Kanai Y, Okada Y, Nonaka S, Takeda S, Harada A, Hirokawa N. Targeted disruption of mouse conventional kinesin heavy chain, kif5B, results in abnormal perinuclear clustering of mitochondria. Cell. 1998;93:1147–1158. doi: 10.1016/s0092-8674(00)81459-2. [DOI] [PubMed] [Google Scholar]
- Verhoeven K, Claeys KG, Zuchner S, Schroder JM, Weis J, Ceuterick C, Jordanova A, Nelis E, De Vriendt E, Van Hul M, Seeman P, Mazanec R, Saifi GM, Szigeti K, Mancias P, Butler IJ, Kochanski A, Ryniewicz B, De Bleecker J, Van den Bergh P. MFN2 mutation distribution and genotype/phenotype correlation in Charcot-Marie-Tooth type 2. Brain. 2006;129:2093–2102. doi: 10.1093/brain/awl126. [DOI] [PubMed] [Google Scholar]
- Youle RJ, Karbowski M. Mitochondrial fission in apoptosis. Nat Rev Mol Cell Biol. 2005;6:657–663. doi: 10.1038/nrm1697. [DOI] [PubMed] [Google Scholar]
- Zhou HM, Brust-Mascher I, Scholey JM. Direct visualization of the movement of the monomeric axonal transport motor UNC-104 along neuronal processes in living Caenorhabditis elegans . J Neurosci. 2001;21:3749–3755. doi: 10.1523/JNEUROSCI.21-11-03749.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu D, Kennerson ML, Walizada G, Zuchner S, Vance JM, Nicholson GA. Charcot-Marie-Tooth with pyramidal signs is genetically heterogeneous: families with and without MFN2 mutations. Neurology. 2005;65:496–497. doi: 10.1212/01.wnl.0000171345.62270.29. [DOI] [PubMed] [Google Scholar]
- Zorzano A, Pich S. What is the biological significance of the two mitofusin proteins present in the outer mitochondrial membrane of mammalian cells? IUBMB Life. 2006;58:441–443. doi: 10.1080/15216540600644838. [DOI] [PubMed] [Google Scholar]
- Zuchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E, Patitucci A, Senderek J, Parman Y, Evgrafov O, Jonghe PD, Takahashi Y, Tsuji S, Pericak-Vance MA, Quattrone A, Battaloglu E, Polyakov AV, Timmerman V. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004;36:449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
- Zuchner S, De Jonghe P, Jordanova A, Claeys KG, Guergueltcheva V, Cherninkova S, Hamilton SR, Van Stavern G, Krajewski KM, Stajich J, Tournev I, Verhoeven K, Langerhorst CT, de Visser M, Baas F, Bird T, Timmerman V, Shy M, Vance JM. Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2. Ann Neurol. 2006;59:276–281. doi: 10.1002/ana.20797. [DOI] [PubMed] [Google Scholar]