

Abstract
Localization of Trk neurotrophin receptors is an important factor in directing cellular communication in developing and mature neurons. One potential site of action is in lipid raft membrane microdomains. Although Trk receptors have been localized to lipid rafts, little is known about how these neurotrophin receptors are directed there or how localization to these membrane microdomains regulates Trk signaling. Here, we report that the TrkB brain-derived neurotrophic factor (BDNF) receptor specifically localized to intracellular lipid rafts in cortical and hippocampal membranes in response to BDNF and that this process was critically dependent on the tyrosine kinase Fyn. BDNF-induced TrkB accumulation at lipid rafts was prevented by blocking the internalization of TrkB. BDNF stimulation also resulted in the association between endogenous TrkB and Fyn. Moreover, in neurons derived from Fyn knock-out mice, the translocation of TrkB to lipid rafts in response to BDNF was compromised, whereas the corticohippocampal region of Fyn mutants displayed lower amounts of TrkB in lipid rafts in vivo. In support of a role for lipid rafts in neurotrophin signaling, inhibiting TrkB translocation to lipid rafts, either by using Fyn knock-out neurons or lipid raft-disturbing agents, prevented the full activation of TrkB and of downstream phospholipase C-γ. These results indicate that the lipid raft localization of TrkB receptors is regulated by Fyn and represents an important factor in determining the outcome of BDNF signaling in neurons.
Keywords: TrkB, Fyn, PLCγ, lipid rafts, intracellular, rat
Introduction
Neurotrophins activate Trk tyrosine kinase receptors, whose signaling properties determine neuronal survival and differentiation decisions and numerous synaptic functions (Poo, 2001; Ginty and Segal, 2002; Chao, 2003). Each neurotrophin, nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), and NT-4, is capable of binding to a specific Trk receptor tyrosine kinase (TrkA, TrkB, and TrkC) and to the p75 neurotrophin receptor (Chao, 1992; Huang and Reichardt, 2001). Neurotrophin binding to cognate Trk receptors results in the activation of phospholipase Cγ (PLCγ) and in the recruitment of a number of proteins that are involved in the activation of extracellular signal-regulated kinases 1 and 2 (ERK1/2), phosphatidylinositol 3-kinase (PI3K) and its downstream target Akt. Whereas ERK1/2 mediate neurotrophin-induced neuronal survival and differentiation (Huang and Reichardt, 2003), PI3K activity is responsible for retrograde survival signaling (Kuruvilla et al., 2000). The activity of PLCγ has been implicated in the ability of TrkB receptors to modulate synaptic transmission and long-term potentiation (LTP) (Matsumoto et al., 2001; Minichiello et al., 2002).
The localization of Trk receptors in specific neuronal subdomains has been suggested to be crucial for determining the biological functions of neurotrophins. For example, recent studies have indicated that TrkB at synaptic sites may mediate enhancement of glutamate release (Yano et al., 2006), as well as enhance NMDA receptor function (Lu, 2003). The endocytosis of Trk receptors after ligand binding, which occurs by both clathrin-dependent and -independent mechanisms, as well as pinocytotic pathways (Shao et al., 2002), is a crucial factor for the cellular localization of activated Trk. The internalized neurotrophin–Trk complex is sorted to an endosomal compartment and then enters into recycling, retrograde transport, or degradation pathways (Howe et al., 2001). Clearly, identification of membrane compartments carrying Trk is important to understand the regulation and the components of Trk trafficking vesicles and how they affect Trk signaling and function.
Lipid rafts are membrane microdomains rich in cholesterol and sphingolipids that have been proposed as important regions for cellular signaling, by bringing together transmembrane receptors with adaptor and signaling proteins (Simons and Toomre, 2000; Tsui-Pierchala et al., 2002). Both Trk and p75 neurotrophin receptors are known to be localized in these membrane domains (Wu et al., 1997; Huang et al., 1999; Peiro et al., 2000; Higuchi et al., 2003; Suzuki et al., 2004), suggesting a role for lipid rafts in neurotrophin signaling. However, the mechanism whereby Trk is localized to lipid rafts and the importance of Trk localization in these lipid domains to downstream signaling have not been investigated.
Here, we report isolation of lipid raft membrane fractions in primary neurons enriched with activated TrkB receptors after BDNF treatment. In contrast to a number of downstream receptor-interacting proteins, we provide genetic evidence that TrkB receptors are selectively recruited to lipid raft membranes in intracellular locations through a mechanism that involves Fyn.
Materials and Methods
Materials.
The solutions used for cell culture, such as the HBSS, the minimal essential medium (MEM), the Neurobasal medium, the B-27 supplement, and the fetal bovine serum (FBS), as well as the Lipofectamine, the Amplex Red Cholesterol Assay Kit, and the Alexa 488-coupled anti-mouse antibody and cholera toxin subunit B were purchased from Invitrogen (Carlsbad, CA). BDNF was obtained from Peprotech (Rocky Hill, NJ). The Bradford reagent was from Bio-Rad (Hercules, CA). Protein A-Sepharose and the ECL reagent were purchased from GE Healthcare (Little Chalfont, UK). The sulfosuccinimidyl 2-(biotinamido)-ethyl-1,3-dithiopropionate (sulfo-NHS-SS-biotin) and the agarose-coupled streptavidin were from Pierce Biotechnology (Rockford, IL). The Optiprep was from Axis-Shield (Oslo, Norway). The rabbit anti-Trk (C-14) and anti-EphA3 antibodies, the normal rabbit IgG, and the mouse anti-Fyn (15), anti-Akt (B1), and anti-PLCγ1 (E12) antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). The mouse anti-phospho-ERK1/2 antibody, the HRP-coupled goat anti-rabbit antibody and the rabbit anti-phospho-Akt (Ser473), anti-phospho-PLCγ1 (Tyr783), anti-phospho-TrkA (Tyr490), and anti-ERK1/2 antibodies were purchased from Cell Signaling Technology (Danvers, MA). The mouse anti-flotillin-1 and anti-TrkB antibodies were obtained from BD Biosciences (San Jose, CA). The mouse anti-transferrin receptor antibody was from Zymed Laboratories (San Francisco, CA). The mouse anti-Flag antibody was purchased from Sigma (St. Louis, MO). The HRP-coupled goat anti-mouse antibody and the cyanine 3 (Cy3)-coupled anti-rabbit and anti-chicken antibodies were from Jackson ImmunoResearch (West Grove, PA). The chicken anti-TrkB antibody was a kind gift from Dr. Louis Reichardt (University of California, San Francisco, CA). The Vectashield mounting medium was from Vector Laboratories (Burlingame, CA). The SW40 centrifuge tubes were from Beckman (Palo Alto, CA). The Microcon YM-30 centrifugal filter tubes and the polyvinylidene difluoride (PVDF) membranes were from Millipore (Bedford, MA). Fyn knock-out founder mice (Fyntm1Sor) (Stein et al., 1992), of B6, 129 mixed background, were purchased from The Jackson Laboratory (Bar Harbor, ME), and the primers used for genotyping the mouse colony were obtained from Operon Biotechnologies (Huntsville, AL). All other reagents were from Sigma.
Cell culture and preparation of lysates.
Rat primary cortical neuron cultures were prepared as described previously (Kim et al., 2002), with some modifications. The cortices of embryonic day 18 (E18) Sprague Dawley rat embryos were dissected in Ca2+- and Mg2+-free HBSS supplemented with 0.37% glucose and digested in the same medium supplemented with 0.05% trypsin. Cortical cells were mechanically dissociated with fire-polished Pasteur pipettes in plating medium (MEM containing 2 mm glutamine, supplemented with 10% FBS, 1 mm pyruvate, 0.37% glucose, and 25 U/ml penicillin/streptomycin), plated in poly-l-lysine-coated dishes at 85–115 × 103 cells/cm2 for biochemical experiments and at 20–30 × 103 cells/cm2 for immunocytochemistry, and kept in a humidified incubator at 37°C and 5% CO2. Cells were maintained in serum-free Neurobasal medium with B-27 supplement, 0.5 mm glutamine, 25 μm glutamate, and 25 U/ml penicillin/streptomycin, and at day in vitro 3 (DIV 3), 5-fluoro-2-deoxyuridine was added. At DIV 7–11, cells were starved for 4 h in MEM with 2 mm glutamine, 0.37% glucose, and 10 μm MK-801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate] (starving medium) and then stimulated with 50 ng/ml BDNF. For the experiments depicted in Figure 8, cells were also treated with 3 mm methyl-β-cyclodextrin (MCD) (added just for 10 min before the treatment with BDNF) or 100 μm GM1 (present for 30 min before and also during stimulation with BDNF). For biochemical experiments, cells were then washed in cold PBS and lysed in radioimmunoprecipitation assay buffer (150 mm NaCl, 50 mm Tris, pH 8.0, 5 mm EDTA, 1% Triton X-100, 0.5% deoxycholic acid, and 0.1% SDS) containing protease and phosphatase inhibitors (2 μg/ml leupeptin, 2 μg/ml aprotinin, 1 mm sodium orthovanadate, 10 mm sodium fluoride, and 1 mm phenylmethylsulfonyl fluoride), unless otherwise indicated. After [protein] measurement by the Bradford method (Bradford, 1976), 5× sample buffer (350 mm Tris, pH 6.8, 30% glycerol, 10% SDS, 600 mm dithiothreitol, and 0.012% bromophenol blue) was added, and samples were denatured at 100°C for 10 min.
Figure 8.

The lipid raft-disturbing compounds MCD and ganglioside GM1 inhibit BDNF-induced TrkB and PLCγ phosphorylation. A–D, DIV 9 rat cortical neurons were treated with MCD (A, B) and ganglioside GM1 (C, D) before stimulation with BDNF for 40 min. Optiprep gradient fractions (A, C) and whole lysates (B, D) of these cells were probed with antibodies against TrkB and Fyn (A, C) or against the phosphorylated and total forms of Trk and of the signaling proteins described in the legend of Figure 7 (B, D). Quantification is presented in B and D as mean percentage of total ± SEM for n = 3–4. Statistically significant differences between BDNF-stimulated conditions are indicated (*p < 0.05; **p < 0.01; ***p < 0.001). Ctr, Control.
Mouse primary corticohippocampal neuron cultures were prepared using E15.5–16.5 mouse embryos obtained from the crossing of Fyn mutant heterozygous mice. Brains were kept in dissection medium, on ice, while each embryo was genotyped by PCR, using tail DNA, with the primers fyn i2F3 (5′-GTCCCTCTTCCCACTCTTC-3′), neo F4 (5′-CGCCTTCTATCGCCTTCTT-3′), and fyn i2R3 (5′-TACTCCCAACGCTCACTAA-3′) described at www.fhcrc.org/science/labs/soriano/protocols/pcrgen.html (Stein et al., 1992). Wild-type, heterozygous, and Fyn knock-out brains were then pooled together, and their cortex and hippocampus were dissected. Cell dissociation, plating, and maintenance were performed as described for the rat cultures. At DIV 8–10, cells were starved, treated with BDNF, and lysed as described above.
Preparation of mouse brain extracts.
The cortex and hippocampi of littermate Fyn knock-out, heterozygous, and wild-type mice of 3 weeks of age were dissected and homogenized in detergent-free TNE buffer (150 mm NaCl, 50 mm Tris, pH 8.0, and 2 mm EDTA) containing protease and phosphatase inhibitors (as above), using a motor-driven tissue grinder (Omni TH Homogenizer; Omni International, Warrenton, VA) at low speed. Triton X-100 was then added to a concentration of 0.5%, and, after a 1 h incubation, [protein] was measured by the Bradford method. The whole procedure was performed at 4°C.
Lipid raft isolation.
Lipid rafts were isolated as described previously, with some modifications (Bruckner et al., 1999). DIV 9–11 rat cortical neurons and DIV 8–10 mouse corticohippocampal neurons were lysed in TNE buffer containing 0.5% Triton X-100 (TXNE). The lysates were then brought up to the same protein concentration. These samples, as well as the extracts obtained from 3-week-old mouse brain, were loaded on the bottom of discontinuous Optiprep gradients by mixing 750 μl of sample with 1050 μl of a 60% Optiprep (36% Iodixanol) TXNE solution, in SW40 centrifuge tubes. The resulting 35% Optiprep solution was overlayered sequentially with 8 ml of 30% Optiprep in TXNE and 3.5 ml of TXNE. Protease and phosphatase inhibitors were present in all layers. The gradients were spun for 6 h at 36,000 rpm in a Beckman XL-70 ultracentrifuge, using an SW40 rotor. Eight fractions of 1.5 ml were then collected from the top, after discarding the first 1.1 ml. The whole procedure was conducted at 4°C. For Western blot analysis, equal volumes of each fraction were loaded on SDS-polyacrylamide gels. In some cases, the samples were concentrated by centrifugation on Microcon YM-30 centrifugal filter tubes, to allow the loading of higher amounts of material.
Immunoprecipitation and immunoblotting.
BDNF-treated rat cortical neurons were lysed in protease- and phosphatase-containing TNE with 1% NP-40 and 60 mm β-octylglucoside, to efficiently extract proteins from lipid rafts. After bringing all of the samples to the same concentration, 5 μg/ml rabbit anti-Trk (C-14) antibody or of normal rabbit IgG were added for an overnight incubation at 4°C, which was followed by a 2 h incubation with protein A-Sepharose. After extensive washing, the precipitates were eluted from the Sepharose beads by boiling in 2× sample buffer for 10 min. Precipitated proteins were separated by 12% SDS-PAGE and transferred to PVDF membranes. These were then blocked for 1 h in TBS with 0.1% Tween 20 (TBS-T) with 5% low-fat milk. The incubation with the primary as well as with the HRP-coupled secondary antibodies was performed overnight at 4°C in TBS-T with 3% BSA or 1% low-fat milk, respectively. Immunoreactive proteins were visualized by ECL detection and film autoradiography. Band intensity was quantified using ImageJ. The total lysates and lipid raft samples obtained as described above and the biotinylation samples that we describe below were run on 8–12% SDS-PAGE and immunoblotted in a similar manner to that described for the immunoprecipitation samples. The primary antibodies used are indicated in the legends of the corresponding figures.
Biotinylation experiments.
Plasma membrane proteins were biotinylated essentially as described previously (Mammen et al., 1997). After BDNF treatment, cells were washed once with ice-cold PBS/Ca2+/Mg2+ (PBS with 1 mm CaCl2 and 0.5 mm MgCl2) and then incubated for 30 min at 4°C in PBS/Ca2+/Mg2+ containing 1 mg/ml sulfo-NHS-SS-biotin. To remove the excess of biotin, cultures were washed three times in PBS/Ca2+/Mg2+ with 100 mm glycine and once more in PBS/Ca2+/Mg2+. For the experiments described in Figure 2A–C, cell lysates were then prepared and processed for density gradient fractionation as described above in Lipid raft isolation. β-Octylglucoside was added to 500 μl of fractions 2, 5, and 8, to a final concentration of 60 mm, to solubilize lipid raft and other membrane proteins. The biotinylated proteins present in these samples were then precipitated twice with 50 μl of a 50% slurry of agarose-coupled streptavidin. A third streptavidin precipitation was performed to confirm that all biotinylated proteins were precipitated in the first two precipitations. Trk was then immunoprecipitated from the final supernatant, as described above in Immunoprecipitation and immunoblotting. For the experiments described in Figure 3, A and B, cells were incubated in starving medium supplemented with 50 mm HEPES, pH 7.4, at 37 or 12°C, in the presence or absence of 50 ng/ml BDNF. They were then subjected to the biotinylation procedure described above. Biotinylated proteins present in the whole cell lysates were precipitated with agarose-coupled streptavidin, as described for the biotinylated lipid raft samples.
Figure 2.
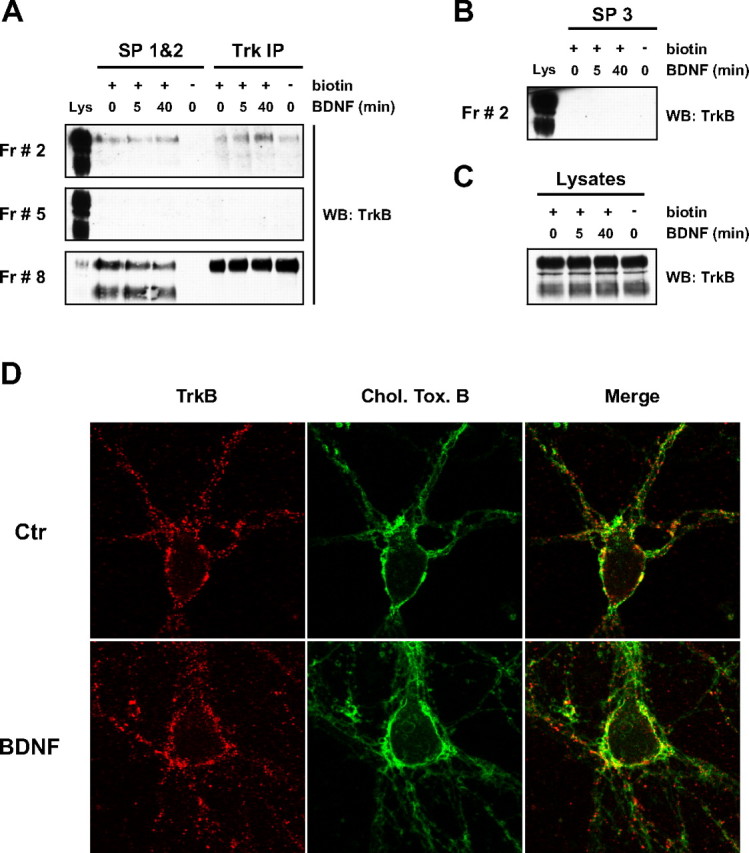
The BDNF-induced recruitment of TrkB to lipid rafts occurs at intracellular membranes. A–C, DIV 9 rat cortical neurons were stimulated with BDNF, incubated with sulfo-NHS-SS-biotin, lysed, and processed for Optiprep gradient fractionation. Three sequential streptavidin precipitations were performed on samples from fractions (Fr) 2, 5, and 8, and the final supernatant was immunoprecipitated with an anti-panTrk antibody (anti-Trk C-14). The precipitates from the first two streptavidin precipitations (SP 1&2) were combined for probing with an anti-TrkB antibody by Western blotting (WB). This is shown in A, together with the results obtained for the precipitates from the Trk immunoprecipitation (Trk IP). The exposure of the fraction 8 panel is lower than the exposure used for fractions 2 and 5 to allow clear visualization of the protein bands. Only the full-length isoform is detected in the Trk IP precipitates, because the antibody used to immunoprecipitate Trk was raised against its intracellular domain, not recognizing truncated TrkB. As indicated, a control in which no biotin was added to the cells was included, and a sample of whole lysate (Lys) was run with the precipitates from each fraction. B, An immunoblot against TrkB is shown for fraction 2 precipitates from the third streptavidin precipitation (SP 3). C, TrkB levels present in the total lysates of all conditions. The results shown in A–C are representative of two independent experiments. D, Copatching of cell surface lipid rafts and TrkB receptors was performed as described in Materials and Methods, on control (Ctr) and BDNF-stimulated DIV 7–8 rat cortical neurons. These images, which were used for the quantification described in Results, were taken at a Z plane close to the cell bottom, where both the cell body and neurites were visible and where most of the lipid raft patches accumulated, for the majority of the neurons. They are representative of three independent experiments in which an average of 10 cells per condition per experiment were imaged. Chol. Tox. B, Cholera toxin subunit B.
Figure 3.

Blocking TrkB internalization impairs BDNF-induced TrkB recruitment to lipid rafts. A–D, DIV 9 rat cortical neurons were stimulated with BDNF for 40 min, at 37 or 12°C, and used either for cell surface biotinylation (A, B) or for lipid raft fractionation studies (C, D). A, Biotinylated proteins were precipitated with agarose-coupled streptavidin as described in the legend of Figure 2. The combined precipitates from the first two streptavidin precipitations (SP 1&2) were probed with anti-TrkB and anti-EphA3 antibodies. SP3 did not pull down a significant amount of biotinylated TrkB (data not shown). As a loading control, the TrkB and EphA3 levels present in the input lysates are shown on the right. B, Quantification of biotinylated TrkB (SP TrkB) in control and BDNF-treated conditions, at 37 and 12°C, was performed by dividing the intensity of each TrkB-immunoreactive band by the intensity of the corresponding EphA3 band (SP EphA3). Results are expressed as mean percentage of total (the sum of the intensities of all of the bands analyzed) ± SEM for n = 3. C, For lipid raft fractionation studies, the cells were lysed and processed for Optiprep gradient fractionation. Gradient fractions (Fr) were then probed with the mouse anti-TrkB and anti-Fyn antibodies. D, Quantification of lipid raft TrkB in the different conditions was performed by dividing the intensity of each fraction 2 (f#2) TrkB-immunoreactive band by the corresponding Fyn band intensity. Results are expressed as mean percentage of total ± SEM of four independent experiments. The indicated conditions are statistically different from the unstimulated control at 37°C (*p < 0.05; ***p < 0.001).
Cell transfection, patching experiments, and immunocytochemistry.
For Trk–Fyn colocalization experiments, cells were transfected at DIV 6 with Flag–Fyn (1 μg/ml pFlagCMV2-Fyn) at a 1:2.5 ratio with Lipofectamine, according to the manufacturer's instructions. At DIV 9, cells were starved and stimulated with 50 ng/ml BDNF for 40 min. After fixation in PBS with 4% paraformaldehyde for 10 min at room temperature, cells were permeabilized for 10 min in PBS with 0.3% Triton X-100 at 4°C and blocked in PBS with 0.1% Tween 20 (PBS-T) with 5% normal goat serum (NGS) for 1 h at room temperature. The incubation with the primary antibodies [rabbit anti-Trk (C-14), 1:100; mouse anti-Flag, 1:10,000] was performed overnight at 4°C in PBS-T with 1.5% NGS. After washing in PBS-T, cells were incubated with the secondary antibodies (anti-rabbit Cy3, 1:300; anti-mouse Alexa 488, 1:500) in PBS-T with 1.5% NGS for 2 h at room temperature, washed again in PBS-T, and mounted with Vectashield mounting medium.
For TrkB and lipid raft copatching experiments, the protocol described by Guirland et al. (2004) was followed with minor alterations. Briefly, DIV 7–8 neurons were starved, treated with BDNF as above, and incubated for 1 h at 12°C with chicken anti-TrkB antibody (1:200) and Alexa 488-coupled cholera toxin subunit B at 1 μg/ml in MEM with 50 mm HEPES and 2 mg/ml BSA at pH 7.3. After washing, cells were incubated for 45 min at 12°C with Cy3-coupled anti-chicken IgY, at 1:200, in the same medium. Fixation was performed in PBS with 4% paraformaldehyde for 4 min followed by 5 min in cold methanol. Mounting was performed as above. Images were acquired with a Plan-Apochromat 63×/1.40 oil objective on a Zeiss (Thornwood, NY) LSM 510 confocal microscope, using the multitrack configuration and pinhole settings adjusted for similar optical slices for the green and red channels. Quantification was performed using the Zeiss LSM software as described in the corresponding figure legends.
Analysis of [cholesterol] and ganglioside GM1 distribution.
The concentration of cholesterol was measured using the Amplex Red Cholesterol Assay Kit, according to the manufacturer's instructions. Ganglioside GM1 levels were determined by dot blot with cholera toxin subunit B–HRP at 1:10,000.
Statistical analysis.
Results are presented as means ± SEM of the indicated number of experiments. Statistical significance was determined using one-way ANOVA with Newman–Keuls multiple-comparison test, unless otherwise indicated.
Results
BDNF induces TrkB translocation to intracellular lipid rafts
To characterize Trk-containing membranes in rat primary cortical neurons in culture, we performed a series of subcellular biochemical fractionation experiments. Lipid rafts are characterized by their insolubility in Triton X-100 at 4°C and low buoyant density, allowing their isolation in discontinuous density gradients. After cell lysis in 0.5% Triton X-100-containing buffer and centrifugation on Optiprep discontinuous gradients, fractions were collected and analyzed by Western blotting (Fig. 1). The lipid raft membranes were recovered in fraction 2, which was enriched in the lipid markers cholesterol and ganglioside GM1 (Fig. 1A) and in the protein markers Fyn (Fig. 1A) and flotillin (see Fig. 4A). Nonraft membranes containing the transferrin receptor were localized in fraction 8 (Fig. 1A).
Figure 1.

BDNF induces the recruitment of TrkB to lipid rafts, in cultured cortical neurons. DIV 9–11 rat cortical neurons were lysed in 0.5% Triton X-100-containing TNE buffer and centrifuged in Optiprep density gradients to isolate lipid rafts, as described in Materials and Methods. A, A diagram of the density gradient fractions (Fr) shows that the band present in the 0–30% Optiprep solution interface is totally recovered in fraction 2. Also shown is a representative dot blot of each of these fractions, probed for ganglioside GM1, and a representative plot of the [cholesterol] in each of them. These fractions were also probed for Fyn [anti-Fyn (15) at 1:100] and the transferrin receptor (TfR; anti-TfR at 1:500), by immunoblotting. B, Equal volumes of each of the density gradient fractions, obtained from control neurons or neurons stimulated with 50 ng/ml BDNF for the indicated periods of time (t), were immunoblotted and probed with anti-phospho-TrkA (Tyr490) (pTrk; 1:750), mouse anti-TrkB (1:150), and anti-Fyn (15) antibodies. These immunoblots are representative of three to four independent experiments.
Figure 4.
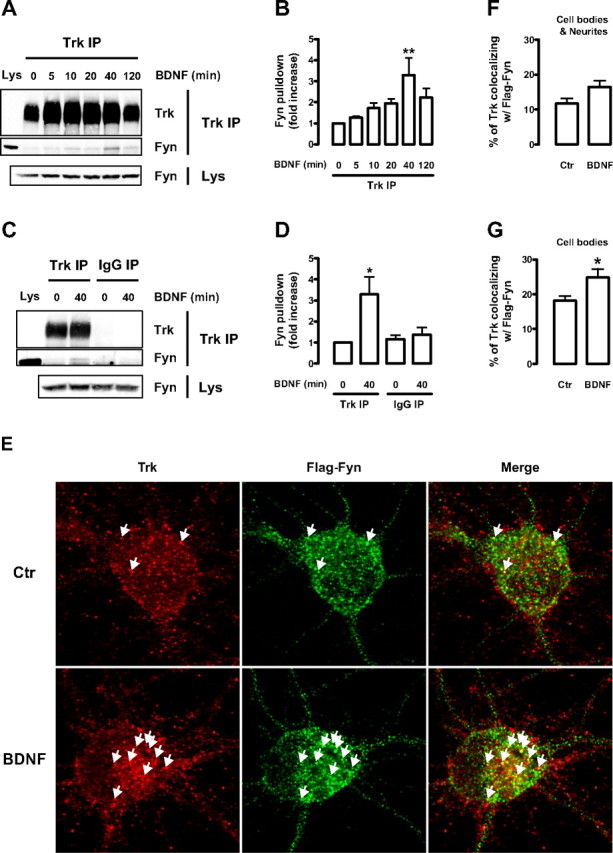
BDNF induces endogenous Trk–Fyn association and increases the colocalization of Trk and Flag–Fyn in cultured cortical neurons. A–D, Lysates obtained from DIV 9 rat cortical neurons, stimulated with BDNF for the indicated periods of time, were immunoprecipitated with anti-Trk (C-14) antibody (Trk IP) (A) or with normal rabbit IgG (IgG IP) (B). The immunoprecipitates were probed with anti-Trk (C-14) (1:100) and anti-Fyn (15) antibodies. A sample of whole lysate (Lys) was run side by side with the immunoprecipitates. As a loading control, the amount of Fyn present in the whole lysates is shown at the bottom. Quantitative analysis of coimmunoprecipitated Fyn from the experiments depicted in A and B are shown in C and D, respectively. Data are presented as mean fold increase ± SEM of three to five independent experiments. The indicated conditions are statistically different from the unstimulated control (*p < 0.05; **p < 0.01). E, Immunocytochemistry of control (Ctr) and BDNF-stimulated DIV 9 rat cortical neurons transfected with Flag–Fyn at DIV 6. Cells were double stained with anti-Trk (C-14) and anti-Flag antibodies. Arrows depict colocalization between green and red puncta. F, G, Quantification of the percentage of Trk that colocalizes with Flag–Fyn. Colocalization was assessed in a region of interest that contained the whole cell (cell bodies and neurites; F) or just the cell body (G) of Flag–Fyn-positive neurons. Results are expressed as mean percentage ± SEM for six independent experiments, in which ∼8–12 cells per condition per experiment were imaged. The indicated condition is statistically different from the control as determined by unpaired two-tailed t test (*p < 0.05).
TrkB receptors were abundantly observed in the bottom fractions of the density gradient, as detected with an antibody raised against an extracellular epitope of TrkB that recognizes the full-length and truncated isoforms (Fig. 1B). However, low levels of full-length TrkB could also be detected in fraction 2. Treatment with 50 ng/ml BDNF increased full-length TrkB protein levels in the lipid raft fraction, without affecting the distribution of the truncated TrkB isoform (Fig. 1B). The translocation of TrkB to lipid rafts was maximally observed at later time points of BDNF treatment (20 min and 1 h), whereas a smaller effect was visible at the 5 min time point. TrkB phosphorylation in response to BDNF was also assessed with an antibody that recognizes the phosphorylated Shc-binding site that is present on all Trk receptors [anti-phospho-TrkA (Tyr490)]. As depicted in Figure 1B, most of the BDNF-induced phospho-TrkB receptors were present in fraction 8, but a significant amount were also seen in the lipid raft fraction. Immunoblots were performed on Fyn to control for equal protein loading and to confirm the distribution of this lipid raft marker in the gradient (Fig. 1B).
The slow kinetics of TrkB translocation to lipid rafts in response to BDNF led us to question whether this translocation occurs at the plasma membrane or at an intracellular location after TrkB internalization and intracellular trafficking. This was investigated by biotinylation experiments with sulfo-NHS-SS-biotin to label all extracellular proteins at the cell surface. After BDNF treatment, cells were biotinylated and processed for Optiprep density gradient fractionation. Biotinylated proteins were recovered by two sequential agarose-coupled streptavidin precipitations, whose precipitates were combined for Western blotting. Full-length TrkB was detected in plasma membrane lipid rafts in control conditions (Fig. 2A, Fr 2, SP 1&2). However, BDNF stimulation for 5 or 40 min failed to increase TrkB levels at lipid rafts present at the cell surface (Fig. 2A, Fr 2, SP 1&2). Nonbiotinylated and thus intracellular Trk was recovered by immunoprecipitating the supernatant obtained from the streptavidin precipitations with an anti-panTrk antibody [anti-Trk (C-14)]. Interestingly, BDNF caused an increase in the amount of TrkB receptors in intracellular lipid rafts (Fig. 2A, Fr 2, Trk IP). This response followed a time course similar to the one described for total TrkB (Fig. 1B).
A number of controls were performed to verify the extent and specificity of the streptavidin precipitation. A third additional streptavidin precipitation was performed on the supernatant of fraction 2 samples to confirm that all lipid raft biotinylated TrkB was pulled down in the first two streptavidin precipitations. As depicted in Figure 2B, no TrkB signal was detected in the precipitates of the third streptavidin precipitation. Second, TrkB was not observed in precipitates obtained from nonbiotinylated cell lysates (Fig. 2A, SP 1&2). A similar procedure conducted for the nonraft fraction 5, in which TrkB is below detection levels, did not produce any biotinylated or nonbiotinylated TrkB receptors (Fig. 2A, Fr 5). On the contrary, the nonraft fraction that contains the highest levels of TrkB (i.e., fraction 8) showed high amounts of biotinylated and nonbiotinylated receptor (Fig. 2A, Fr 8). Therefore, nonraft TrkB was distributed between intracellular compartments and the plasma membrane. As expected, a reduction in the levels of full-length TrkB present at nonraft plasma membrane was seen after treatment with BDNF (Fig. 2A, Fr 8, SP 1&2), as a result of ligand-induced internalization. Finally, an immunoblot of the whole lysate samples corresponding to the conditions tested in Figure 2, A and B, demonstrated equal levels of TrkB in all conditions (Fig. 2C).
As an alternative approach to study TrkB localization in lipid rafts, we performed lipid raft and TrkB receptor copatching experiments at the surface of living cells (Fig. 2D). This method relies on the coalescence of cross-linked raft elements, mediated by their common lipid environments, into patches that are big enough to be resolved by fluorescence microscopy (Harder et al., 1998). After BDNF treatment of rat cortical neurons, cells were brought down to 12°C, and surface TrkB receptors were cross-linked with a chicken anti-TrkB antibody, which recognizes the extracellular portion of the receptor. Lipid raft ganglioside GM1 was cross-linked with Alexa 488-coupled cholera toxin subunit B. In control conditions, most of the cell surface TrkB receptor patches were localized outside lipid rafts domains (Fig. 2D). However, some colocalization was observed between surface TrkB and lipid raft patches, which occurred mostly at the cell body but also in discrete locations along processes (Fig. 2D). BDNF treatment did not increase the amount of TrkB that was present in lipid rafts at the cell surface (Fig. 2D). This was confirmed by quantifying the percentage of surface TrkB in lipid rafts, which was not significantly different between control and BDNF-stimulated conditions (30.4 ± 0.5% vs 26.9 ± 2.0%, respectively, for n = 3, of an average of 10 cells imaged per condition per experiment; p = 0.170 as determined by unpaired two-tailed t test). These results used a different approach to support our previous biochemical observation that the BDNF-induced recruitment of TrkB to lipid rafts did not occur at the cell surface.
Overall, these data suggest that BDNF induces the translocation of TrkB receptors to lipid rafts located in intracellular membranes. The slow kinetics of this translocation may reflect the time required by TrkB to be internalized and reach the appropriate subcellular location.
Inhibition of TrkB internalization impairs BDNF-induced TrkB translocation to lipid rafts
To determine whether TrkB internalization is necessary for relocalization of the receptor to lipid rafts, we stimulated rat cortical neuron cultures with BDNF at 12°C to block internalization. Cell surface biotinylation of untreated cells and cells stimulated with BDNF for 40 min showed that whereas BDNF treatment led to a sharp reduction in TrkB surface levels at 37°C, TrkB internalization by BDNF was abolished at 12°C (Fig. 3A,B). Although BDNF was not able to further decrease biotinylated TrkB levels at 12°C (Fig. 3B), a small but significant decrease in surface TrkB levels was seen at 12°C in control conditions, compared with the control at 37°C (Fig. 3B). The levels of biotinylated EphA3, an unrelated receptor tyrosine kinase, were found to be unchanged by BDNF at either temperature (Fig. 3A).
The effect of BDNF on lipid raft TrkB levels was then investigated at 37 and 12°C. Stimulation of rat cortical neuron cultures with BDNF for 40 min at 12°C failed to induce a significant increase in the localization of TrkB receptors in lipid rafts, in contrast to what was observed for BDNF stimulation at 37°C (Fig. 3C,D). The quantification of these results, shown in Figure 3D, was performed by normalizing the amount of lipid raft TrkB to the corresponding levels of the lipid raft marker Fyn.
These results demonstrate that the inhibition of TrkB receptor internalization abolished TrkB translocation to lipid rafts in response to BDNF, confirming that the accumulation of this receptor in lipid rafts occurs at intracellular membranes.
BDNF induces the association and colocalization of TrkB with the lipid raft-resident protein Fyn
The mechanism by which TrkB receptors are directed to and retained in lipid rafts is unknown. It was previously reported that BDNF can induce the association of TrkB with the lipid raft marker Fyn, in cultured cortical neurons (Iwasaki et al., 1998). We hypothesized that the association of TrkB with Fyn could represent a putative mechanism to account for TrkB translocation to lipid rafts. Therefore, we first aimed to investigate whether BDNF could induce the endogenous association of TrkB and Fyn in primary cultures of rat cortical neurons.
BDNF stimulation increased the amount of Fyn that coimmunoprecipitated with Trk in a time-dependent manner (Fig. 4A,B). A small amount of Fyn was associated with Trk in unstimulated conditions, but after 5 min of BDNF treatment, there was a small increase in Trk–Fyn association that became more pronounced at 10 and 20 min, peaking at ∼40 min (Fig. 4A,B). After 2 h, the amount of Fyn that coimmunoprecipitated with Trk was still consistently higher than the amount associated without BDNF treatment (Fig. 4A,B). The increase in Trk–Fyn interaction in response to BDNF was not observed when the lysates were immunoprecipitated with normal rabbit IgG (Fig. 4C,D). Therefore, the BDNF-induced Trk–Fyn association seen in neuronal membranes was specific in nature.
To investigate the subcellular localization of Trk and Fyn in response to BDNF, an immunocytochemistry study was performed. Because the available Fyn antibodies displayed nonspecific staining in Fyn knock-out mouse neurons (data not shown), we transfected rat cortical neurons with a Flag–Fyn construct as an alternative approach. In unstimulated conditions, Trk was found to colocalize with Flag–Fyn mostly at the cell body but also in neurites at discrete regions (Fig. 4E, data not shown). BDNF treatment for 40 min induced a small but reproducible increase in the amount of Trk that colocalized with Flag–Fyn (Fig. 4E–G). This increase was statistically significant for colocalization of Trk and Flag–Fyn at the cell body. There was also colocalization when the neurites were included; however, it was not statistically significant (Fig. 4F,G). Several of the Trk puncta that colocalized with Flag–Fyn at the cell body appeared to be present at intracellular locations after analysis of Z stacks that were obtained for some of the cells imaged (data not shown).
These data indicate that BDNF induces endogenous TrkB–Fyn association in rat cultured cortical neurons, with a time course similar to the induction of TrkB translocation to lipid rafts, and it places a fraction of Trk receptors in the same intracellular location as transfected Flag–Fyn.
BDNF-induced TrkB translocation to lipid rafts is impaired in Fyn knock-out neurons
To address whether Fyn is involved in BDNF-mediated TrkB translocation to lipid rafts, we made use of corticohippocampal cultures isolated from Fyn wild-type, heterozygous, and knock-out mouse littermates. These animals were obtained by crossing Fyn heterozygous mice derived from Fyn knock-out animals that were previously generated (Stein et al., 1992). After stimulation with BDNF for 40 min, Optiprep density gradient fractions were obtained and analyzed for their content in TrkB and in the lipid raft marker flotillin, a glycosylphosphatidylinositol-linked protein. Flotillin is highly enriched in lipid rafts, although it is also present in fractions 7 and 8 (Fig. 4A) (see also, for example, Dermine et al., 2001).
BDNF increased the levels of TrkB in lipid rafts of wild-type mouse corticohippocampal cultures (Fig. 5A,B), in similarity to what was observed using rat cortical cultures (Fig. 1B). BDNF-induced TrkB translocation to lipid rafts was significantly reduced in Fyn heterozygous mouse neuronal cultures and nearly abolished in Fyn knock-out neurons (Fig. 5A,B). This inhibition of BDNF-induced TrkB translocation to lipid rafts was not attributable to impaired TrkB internalization, because BDNF treatment resulted in similar levels of TrkB internalization in Fyn wild-type and knock-out neurons (data not shown).
Figure 5.

Fyn mutant mouse neurons have impaired localization of TrkB in lipid rafts. A, DIV 8–10 mouse corticohippocampal neurons, obtained from Fyn wild-type (Fyn +/+), heterozygous (Fyn +/−), and knock-out (Fyn −/−) littermates, were stimulated with BDNF for 40 min. Density gradient fractions (Fr) were probed with anti-TrkB and anti-flotillin (1:150) antibodies. B, TrkB levels in the lipid raft fractions were quantified by dividing the intensity of fraction 2 TrkB-immunoreactive bands by the intensity of the corresponding flotillin band. This allowed the normalization of TrkB levels to the amount of lipid raft material loaded for each condition. Results are shown as mean percentage of total ± SEM of three independent experiments. Unless otherwise indicated, the statistical analysis refers to a comparison of the indicated condition with the corresponding unstimulated control of the same genotype (*p < 0.05; **p < 0.01; ***p < 0.001). C, The cortex and hippocampus of 3-week-old Fyn +/+, +/−, and −/− mouse littermates were processed for lipid raft isolation. The gradient fractions were immunoblotted and probed with anti-TrkB and anti-flotillin antibodies. To clearly detect TrkB in lipid rafts from brain extracts, high volumes of concentrated samples were used, resulting in a high concentration of protein and Optiprep in the bottom fractions. Therefore, some proteins, such as flotillin, did not run at their expected molecular weight in those fractions. The migration of TrkB was not affected. D, The quantification of these results was performed as described in B. Data are presented as mean percentage of total ± SEM of four to six independent experiments. The indicated condition is statistically different from the wild-type condition (*p < 0.05). Arrows indicate the lipid raft fraction for each panel. Ctr, Control.
The involvement of Fyn in the lipid raft localization of TrkB receptors was also investigated in vivo by analyzing the levels of TrkB in the lipid rafts of brain extracts obtained from 3-week-old Fyn wild-type, heterozygous, and knock-out mouse littermates. The cortex and hippocampi of these animals were dissected and lysed, and Optiprep density gradient fractionation was performed. As depicted in Figure 5C, Fyn wild-type mice had a small amount of full-length TrkB in lipid rafts obtained from their cortex and hippocampi (Fig. 5C), which was not significantly altered in Fyn heterozygous animals (Fig. 5C,D). Fyn knock-out mice, however, had ∼40% less TrkB in their corticohippocampal lipid rafts (Fig. 5C,D).
Our results suggest that BDNF increases TrkB levels in lipid rafts by inducing the association of this receptor with the lipid raft-resident protein Fyn.
BDNF-induced TrkB, Akt, and PLCγ phosphorylation is affected in Fyn knock-out neurons
To establish the effects of lipid raft translocation on BDNF receptor function, we first analyzed the distribution of TrkB downstream signaling proteins using Optiprep density gradient analysis. Proteins are considered enriched in lipid rafts when their amount in the lipid raft fraction is higher than in the neighboring nonraft fractions (3–6). Many proteins involved in TrkB signaling were found to be enriched in lipid rafts. This included PLCγ (Fig. 6A), Shc, ARMS, Ras, Rap1, and PI3K (data not shown). However, several key proteins, such as Akt and ERK (Fig. 6A), were not enriched in lipid rafts. Also, unlike TrkB, the primary signaling molecules downstream of this receptor were not actively recruited to lipid rafts in response to BDNF (Fig. 6A, data not shown).
Figure 6.

TrkB downstream signaling proteins are differentially distributed on Optiprep density gradients. DIV 9–11 rat cortical neurons were stimulated with BDNF and processed for lipid raft isolation. A, B, The density gradient fractions (Fr) were immunoblotted and probed with anti-Akt (B1) (1:200), anti-ERK1/2 (1:500), and anti-PLCγ1 (E12) (1:100) antibodies (A) or with antibodies against the phosphorylated form of these proteins [anti-phospho-Akt (Ser473) (pAkt), 1:500; anti-phospho-ERK1/2 (pERK), 1:500; anti-phospho-PLCγ (Tyr783) (pPLCγ), 1:250; B]. Arrows indicate the lipid raft fraction for each set of immunoblot panels. These results are representative of two to three independent experiments. t, Time.
We then analyzed the distribution of BDNF-induced phosphorylated forms of Akt, ERK, and PLCγ in the Optiprep density gradient fractions. Interestingly, although phospho-PLCγ was enriched in lipid rafts, phospho-Akt and phospho-ERK were not enriched in these membrane subdomains (Fig. 6B). In fact, if we compare the amount of the phosphorylated form of Akt and ERK in the lipid raft fraction with the amount present in the nonraft neighboring fractions, phospho-Akt and phospho-ERK were mostly excluded from lipid rafts. These results suggest that PLCγ is more represented in neuronal lipid rafts than the ERK or Akt enzymes.
Because Fyn was involved in TrkB translocation to lipid rafts, we next sought to determine the effects of Fyn on BDNF-induced phosphorylation of TrkB and downstream signaling proteins. For these studies, we used the corticohippocampal cultures obtained from Fyn wild-type, heterozygous, or knock-out mice. After BDNF treatment for 40 min, the activation of TrkB was reduced in Fyn heterozygous and knock-out neurons (Fig. 7A,B). Quantitation indicated a reduction of ∼15 and 25%, respectively (Fig. 7B). At the same time, the levels of both phospho-Akt and phospho-PLCγ were also decreased in Fyn knock-out neurons (Fig. 7A,B). Phospho-Akt was reduced by 40% in Fyn knock-out neurons, whereas PLCγ phosphorylation was inhibited by ∼15 and 20% in Fyn heterozygous and knock-out neuronal cultures, respectively (Fig. 7A,B). Interestingly, BDNF-induced ERK phosphorylation was not significantly affected in corticohippocampal neurons that lacked Fyn (Fig. 7A,B).
Figure 7.

BDNF is unable to induce full activation of TrkB and of downstream Akt and PLCγ in Fyn-deficient neurons. A, DIV 8–10 mouse corticohippocampal neurons, obtained from Fyn wild-type (+/+), heterozygous (+/−), and knock-out (−/−) littermates, were stimulated with BDNF for 40 min. Whole lysates from these cells were probed with the anti-Trk (C14), anti-phospho-TrkA (Tyr490), and anti-Fyn (15) antibodies, in addition to the antibodies against the TrkB signaling proteins described in the legend of Figure 6. B, Quantification was performed by dividing the intensity of the phosphorylated form by the corresponding intensity of the total form of each protein. Data are presented as mean percentage of total ± SEM for n = 8. The indicated conditions are statistically different from the corresponding wild-type condition (**p < 0.01; ***p < 0.001). Ctr, Control.
Of note, the Fyn protein levels expressed in the heterozygous mice were markedly reduced compared with the levels present in their wild-type littermates (Fig. 7A). The differences in Fyn protein between heterozygous and knock-out neurons suggest that minimal amounts of Fyn can rescue the inhibition of TrkB translocation to lipid rafts and downstream signaling.
Together, these results indicate that neurons lacking Fyn display a detectable effect on the activation of TrkB, PLCγ, and Akt in response to BDNF. This supports a role for Fyn, not only in the translocation of TrkB to lipid rafts, but also in the efficient engagement of selective BDNF signal transduction pathways.
BDNF-induced TrkB and PLCγ phosphorylation is decreased by lipid raft-disturbing drugs
The altered TrkB signaling properties observed in Fyn-deficient mice may be a result of changes in TrkB signaling caused by developmental defects, impaired TrkB translocation to lipid rafts, or direct effects of Fyn on TrkB activation. To address which of the TrkB signaling deficits shown by the Fyn knock-out neurons were attributable to the inhibition of TrkB recruitment to lipid rafts, we examined the effect of lipid raft-disturbing drugs on BDNF-induced signaling.
MCD is a cholesterol-binding drug that disturbs lipid rafts by removing this lipid from the cell membranes (Simons and Toomre, 2000). Ganglioside GM1 is a normal component of lipid rafts, and exogenous application of this sphingolipid to the cells affects the stability and protein composition of lipid rafts (Simons et al., 1999). Both compounds have been used previously to disturb Trk localization at lipid rafts in PC12 cells and rat cortical neurons (Peiro et al., 2000; Nishio et al., 2004; Suzuki et al., 2004). MCD, at a concentration of 3 mm, and ganglioside GM1, at 100 μm, abolished the TrkB recruitment to lipid rafts in response to BDNF (Fig. 8A,C), without affecting cell morphology (data not shown). We next studied the effect of MCD and ganglioside GM1 on BDNF-induced signaling, using these same drug concentrations. The phosphorylation of TrkB in response to BDNF was inhibited by MCD and ganglioside GM1 by ∼30 and 65%, respectively (Fig. 8B,D). BDNF-induced ERK phosphorylation was slightly affected by both drugs, although in opposite ways. MCD increased phospho-ERK levels by ∼35%, whereas ganglioside GM1 decreased ERK phosphorylation by 20% (Fig. 8B,D). The lipid raft-disturbing drugs also showed different effects on Akt phosphorylation in response to BDNF. Ganglioside GM1 reduced phospho-Akt by close to 40%, whereas MCD had no effect on the phosphorylation of Akt (Fig. 8B,D). However, BDNF-induced PLCγ phosphorylation was inhibited by both drugs. MCD reduced phospho-PLCγ levels by ∼35%, whereas ganglioside GM1 reduced PLCγ phosphorylation by ∼65% (Fig. 8B,D). The inhibitory effect of MCD on BDNF-induced signaling was therefore restricted to TrkB and PLCγ, which were also most extensively affected by the GM1 ganglioside. GM1 also inhibited Akt and ERK, although to much lower extents.
These data indicate that BDNF-induced TrkB translocation to lipid rafts is necessary for full activation of TrkB and PLCγ. Together with our previous results, it also suggests that the lack of Fyn leads to a decrease in the phosphorylation of TrkB and PLCγ in response to BDNF, by inhibiting TrkB translocation to lipid rafts.
Discussion
In this study, we have demonstrated a novel role for Fyn in mediating the BDNF-induced recruitment of the TrkB receptor to lipid rafts located in intracellular compartments. We observed that BDNF promotes the association between TrkB and Fyn with a time course similar to that seen for the translocation of TrkB to lipid rafts. Moreover, BDNF was unable to direct TrkB to these membrane domains in Fyn knock-out neurons. The full activation of TrkB and of selective downstream signaling proteins was compromised by the absence of Fyn. Interestingly, BDNF-induced TrkB and PLCγ phosphorylation were reduced both by lipid raft-disturbing drugs, which abolish TrkB recruitment to lipid rafts, and by the lack of Fyn. This suggests that Fyn is essential for the proper distribution of TrkB among intracellular membrane subdomains after ligand binding, which in turn is crucial for full TrkB activation and efficient engagement of the PLCγ pathway.
TrkB recruitment to lipid rafts in response to BDNF was previously shown in rat cortical neurons (Suzuki et al., 2004), with a similar time course to the one that we obtained. In agreement with a requirement for TrkB activation for the translocation to take place (Suzuki et al., 2004), we observed BDNF-induced translocation of full-length TrkB but not of the truncated isoform to lipid rafts. Additionally, we found that the TrkB recruitment to lipid rafts in response to BDNF occurs intracellularly, because (1) cell surface biotinylation experiments showed that, in contrast to biotinylated TrkB, nonbiotinylated and hence intracellular TrkB accumulated in lipid rafts in response to the neurotrophin, (2) copatching of lipid rafts and TrkB receptors at the surface of living cells did not reveal an increase of colocalizing TrkB and lipid raft patches after BDNF treatment, and (3) blocking TrkB internalization by reducing the temperature to 12°C abolished BDNF-induced TrkB translocation to lipid rafts. The observation that the increase of TrkB levels in lipid rafts occurred intracellularly is very interesting, because the intracellular membranous localization of phosphorylated Trk in primary CNS neurons is critical in determining neurotrophin actions (Riccio et al., 1997; Drake et al., 1999; Ye et al., 2003; Rajagopal et al., 2004; Yano and Chao, 2004).
Previous studies have shown a BDNF-induced recruitment of surface TrkB receptors to lipid rafts, in Xenopus spinal neuron growth cones, by the same copatching method that we followed on Figure 2D (Guirland et al., 2004). We attribute the discrepancies between these results and ours to differences in cellular context (DIV 1 Xenopus spinal neurons vs DIV 7–9 rat cortical neurons) and in the subcellular localization of the TrkB receptors studied (growth cone vs cell body and neurites).
The mechanisms responsible for ligand-induced Trk translocation to lipid rafts have so far remained elusive. The lipid raft resident protein Fyn was previously reported to coimmunoprecipitate with Trk in response to BDNF in rat cortical neuron cultures (Iwasaki et al., 1998), appearing to be a good candidate to mediate TrkB localization to lipid rafts. We studied the kinetics of the TrkB–Fyn association in response to BDNF and found that this neurotrophin increased the interaction between endogenous Fyn and TrkB with a time course similar to that in which it induced TrkB recruitment to lipid rafts. We also found that BDNF induced an increase in Trk colocalization with transfected Flag–Fyn at intracellular locations in the cell body. Finally, using Fyn-deficient mice, we have demonstrated that TrkB translocation into lipid rafts in primary neurons in culture was dependent on the presence of Fyn.
In support of a role for Fyn in the distribution of TrkB among different membrane subdomains in vivo, we found that the brains of Fyn knock-out mice exhibited lower levels of TrkB in lipid rafts. The TrkB receptor detected in lipid rafts extracted from the brain of these animals may be of neuronal or glial origin. However, full-length TrkB appears to be restricted to neurons, whereas CNS glial cells, such as astrocytes and oligodendrocytes, only express the truncated TrkB.T1 isoform (Armanini et al., 1995; Wetmore and Olson, 1995; Ohira et al., 2005). This suggests that most of the full-length TrkB present at lipid rafts from brain extracts is neuron-derived.
The BDNF-induced association between TrkB and Fyn may be direct or indirect in nature. A direct interaction between the SH2 domain of Fyn with BDNF-induced phosphorylated TrkB, obtained from cortical neuron lysates, was proposed previously (Iwasaki et al., 1998). An indirect interaction mediated by the SH3 domain of Fyn and the TrkA juxtamembrane region was also recently demonstrated by in vitro approaches (Iwasaki et al., 1998; Rajagopal and Chao, 2006).
A role for Src-family kinases in Trk receptor signaling has been suggested, although it is far from being understood. Direct phosphorylation of TrkA by Fyn and Src was previously demonstrated in vitro (Tsuruda et al., 2004; Rajagopal and Chao, 2006). Direct TrkB phosphorylation by lipid raft-located Fyn could explain why, in the absence of Fyn and after treatment with lipid raft-disturbing drugs, BDNF did not induce full TrkB activation.
Although our biochemical fractionation experiments suggest that only a small fraction of TrkB was directed to lipid rafts by BDNF, this event plays an important role in BDNF-induced signaling. In fact, preventing TrkB recruitment to lipid rafts led to a marked decrease in the phosphorylation of TrkB and downstream proteins. Several possibilities may account for this apparent discrepancy. First, the small amount of TrkB in lipid rafts reflects a window of a highly dynamic process of protein movement within the plane of the membrane. Indeed, it was previously suggested that raft proteins rapidly partition into and out of lipid rafts (Kenworthy et al., 2004). Therefore, although a bigger fraction of TrkB may traffic through lipid rafts, only a small fraction may be present there at a given moment. Second, the stringent Triton X-100-based extraction of lipid rafts may solubilize a good portion of lipid raft-located proteins, including TrkB, which are not strongly associated with these membrane domains. The fact that the amount of surface TrkB patches that colocalized with lipid rafts was much higher than that revealed by our biochemical fractionation procedures (Fig. 2D; see Results), argues in favor of the latter possibility.
A role for lipid rafts in Trk receptor signaling was previously addressed in PC12 cells (Huang et al., 1999; Peiro et al., 2000; Nishio et al., 2004). TrkA was shown to be constitutively present in caveolae, a specific type of lipid rafts (Simons and Toomre, 2000), whereas lipid raft-disrupting compounds markedly inhibited NGF-induced ERK phosphorylation (Peiro et al., 2000; Nishio et al., 2004). However, the presence of caveolae in CNS neurons remains controversial. Using primary cortical neurons, we found that ERK phosphorylation was slightly affected by lipid raft-disturbing drugs and unaffected by the lack of Fyn. On the other hand, TrkB and PLCγ were consistently inhibited by preventing TrkB translocation to lipid rafts. Akt phosphorylation was also reduced by ganglioside GM1 and by the absence of Fyn. However, preventing the translocation of TrkB with MCD did not affect the phospho-Akt levels, suggesting that the effect of GM1 and Fyn on Akt does not depend on the presence of TrkB in lipid rafts. This would be consistent with the selective enrichment of PLCγ in cortical neuron lipid rafts, which was not observed for ERK or Akt. Altogether, the presence of Trk receptors in lipid rafts (constitutive vs ligand-induced), and the exact role of these membrane microdomains in Trk signaling, seems to depend on cell context and/or receptor subtype.
We should note that previous reports have suggested that ganglioside GM1 induces Trk phosphorylation by itself and that it potentiates NGF-induced TrkA phosphorylation (Ferrari et al., 1995; Mutoh et al., 1995; Farooqui et al., 1997). We were able to detect a modest increase in TrkB phosphorylation in response to ganglioside GM1 only after high exposure of the autoradiography films (data not shown). This phosphorylation was several orders of magnitude below that obtained in response to BDNF, and, as shown in Figure 8, it inhibited instead of potentiating BDNF-induced TrkB phosphorylation. We interpret these discrepancies as possible differences in cell context and sensitivity of the different Trk isoforms to ganglioside GM1. In fact, this lipid was shown to be less effective at inducing TrkB phosphorylation than at inducing phosphorylation of TrkA or TrkC (Duchemin et al., 2002).
While we provide evidence that the integrity of lipid rafts is essential for efficient TrkB signaling, previous reports demonstrate that it is also crucial for certain BDNF physiological functions. Preventing BDNF-induced TrkB translocation to lipid rafts dramatically inhibits BDNF potentiation of neurotransmitter release and BDNF-induced growth cone turning of rat cortical and Xenopus spinal neurons in culture, respectively (Guirland et al., 2004; Suzuki et al., 2004). Interestingly, the pathway that was most convincingly associated with these effects of BDNF was the PLCγ pathway (Matsumoto et al., 2001; Li et al., 2005). This is the same pathway that we found to be consistently affected by lipid raft-disturbing drugs and by the lack of Fyn.
Deficiency in Fyn kinase is known to result in prominent neurological defects (Grant, 1996), such as a deficit in LTP at CA1 synapses in the hippocampus (Grant et al., 1992; Kojima et al., 1997). This defect in LTP in Fyn knock-out mice is very similar to the impaired LTP phenotype shown by mice that lack either BDNF or functional TrkB (Korte et al., 1996; Patterson et al., 1996; Minichiello et al., 1999). This suggests that Fyn and Trk may converge on similar signaling pathways that regulate LTP in the hippocampus. In fact, the pathway that was genetically demonstrated to be responsible for the TrkB-mediated LTP in the hippocampus was again the PLCγ pathway (Minichiello et al., 2002). Our results provide a link between Fyn and BDNF-induced PLCγ activation, through the recruitment of TrkB to specific intracellular membrane domains, which may explain the similarity of synaptic defects elicited by the lack of these molecules.
In conclusion, we propose the following model for BDNF-induced TrkB translocation to lipid rafts. After binding to BDNF, TrkB is internalized and reaches intracellular compartments in which Fyn is located. Because proteins can rapidly partition into and out of lipid rafts, TrkB may increase the amount of time spent in rafts by interacting with Fyn. This can facilitate direct phosphorylation of this receptor by Fyn, which will lead to full activation of TrkB and enhance the engagement to downstream PLCγ, which is enriched in lipid rafts. Our findings bring additional support to the idea that internalized Trk receptors signal in a selective manner in segregated compartments to regulate specific fundamental neuronal responses.
Footnotes
This work was supported by the National Institutes of Health Grants NS21072 and HD23315. D.B.P. was supported by a postdoctoral fellowship from Fundação Portuguesa para a Ciência e a Tecnologia. We thank Toby Starr and Dr. Michael Dustin for help with the [cholesterol] measurements, Dr. Paul Stein for providing Fyn knock-out animals for initial studies, Dr. Louis Reichardt for the chicken anti-TrkB antibody, and Drs. Marilyn Resh and Michael Lisanti for advice and reagents. We would also like to thank all of the members of the Chao laboratory, and in particular, Juan Carlos Arevalo, Hiroko Yano, Rithwick Rajagopal, and Katrin Deinhardt for advice and Mercedes Beyna for technical assistance.
References
- Armanini MP, McMahon SB, Sutherland J, Shelton DL, Phillips HS. Truncated and catalytic isoforms of trkB are co-expressed in neurons of rat and mouse CNS. Eur J Neurosci. 1995;7:1403–1409. doi: 10.1111/j.1460-9568.1995.tb01132.x. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Bruckner K, Pablo Labrador J, Scheiffele P, Herb A, Seeburg PH, Klein R. EphrinB ligands recruit GRIP family PDZ adaptor proteins into raft membrane microdomains. Neuron. 1999;22:511–524. doi: 10.1016/s0896-6273(00)80706-0. [DOI] [PubMed] [Google Scholar]
- Chao MV. Neurotrophin receptors: a window into neuronal differentiation. Neuron. 1992;9:583–593. doi: 10.1016/0896-6273(92)90023-7. [DOI] [PubMed] [Google Scholar]
- Chao MV. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci. 2003;4:299–309. doi: 10.1038/nrn1078. [DOI] [PubMed] [Google Scholar]
- Dermine JF, Duclos S, Garin J, St-Louis F, Rea S, Parton RG, Desjardins M. Flotillin-1-enriched lipid raft domains accumulate on maturing phagosomes. J Biol Chem. 2001;276:18507–18512. doi: 10.1074/jbc.M101113200. [DOI] [PubMed] [Google Scholar]
- Drake CT, Milner TA, Patterson SL. Ultrastructural localization of full-length trkB immunoreactivity in rat hippocampus suggests multiple roles in modulating activity-dependent synaptic plasticity. J Neurosci. 1999;19:8009–8026. doi: 10.1523/JNEUROSCI.19-18-08009.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchemin AM, Ren Q, Mo L, Neff NH, Hadjiconstantinou M. GM1 ganglioside induces phosphorylation and activation of Trk and Erk in brain. J Neurochem. 2002;81:696–707. doi: 10.1046/j.1471-4159.2002.00831.x. [DOI] [PubMed] [Google Scholar]
- Farooqui T, Franklin T, Pearl DK, Yates AJ. Ganglioside GM1 enhances induction by nerve growth factor of a putative dimer of TrkA. J Neurochem. 1997;68:2348–2355. doi: 10.1046/j.1471-4159.1997.68062348.x. [DOI] [PubMed] [Google Scholar]
- Ferrari G, Anderson BL, Stephens RM, Kaplan DR, Greene LA. Prevention of apoptotic neuronal death by GM1 ganglioside. Involvement of Trk neurotrophin receptors. J Biol Chem. 1995;270:3074–3080. doi: 10.1074/jbc.270.7.3074. [DOI] [PubMed] [Google Scholar]
- Ginty DD, Segal RA. Retrograde neurotrophin signaling: Trk-ing along the axon. Curr Opin Neurobiol. 2002;12:268–274. doi: 10.1016/s0959-4388(02)00326-4. [DOI] [PubMed] [Google Scholar]
- Grant SG. Analysis of NMDA receptor mediated synaptic plasticity using gene targeting: roles of Fyn and FAK non-receptor tyrosine kinases. J Physiol Paris. 1996;90:337–338. doi: 10.1016/s0928-4257(97)87914-4. [DOI] [PubMed] [Google Scholar]
- Grant SG, O'Dell TJ, Karl KA, Stein PL, Soriano P, Kandel ER. Impaired long-term potentiation, spatial learning, and hippocampal development in fyn mutant mice. Science. 1992;258:1903–1910. doi: 10.1126/science.1361685. [DOI] [PubMed] [Google Scholar]
- Guirland C, Suzuki S, Kojima M, Lu B, Zheng JQ. Lipid rafts mediate chemotropic guidance of nerve growth cones. Neuron. 2004;42:51–62. doi: 10.1016/s0896-6273(04)00157-6. [DOI] [PubMed] [Google Scholar]
- Harder T, Scheiffele P, Verkade P, Simons K. Lipid domain structure of the plasma membrane revealed by patching of membrane components. J Cell Biol. 1998;141:929–942. doi: 10.1083/jcb.141.4.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi H, Yamashita T, Yoshikawa H, Tohyama M. PKA phosphorylates the p75 receptor and regulates its localization to lipid rafts. EMBO J. 2003;22:1790–1800. doi: 10.1093/emboj/cdg177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe CL, Valletta JS, Rusnak AS, Mobley WC. NGF signaling from clathrin-coated vesicles: evidence that signaling endosomes serve as a platform for the Ras-MAPK pathway. Neuron. 2001;32:801–814. doi: 10.1016/s0896-6273(01)00526-8. [DOI] [PubMed] [Google Scholar]
- Huang CS, Zhou J, Feng AK, Lynch CC, Klumperman J, DeArmond SJ, Mobley WC. Nerve growth factor signaling in caveolae-like domains at the plasma membrane. J Biol Chem. 1999;274:36707–36714. doi: 10.1074/jbc.274.51.36707. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Iwasaki Y, Gay B, Wada K, Koizumi S. Association of the Src family tyrosine kinase Fyn with TrkB. J Neurochem. 1998;71:106–111. doi: 10.1046/j.1471-4159.1998.71010106.x. [DOI] [PubMed] [Google Scholar]
- Kenworthy AK, Nichols BJ, Remmert CL, Hendrix GM, Kumar M, Zimmerberg J, Lippincott-Schwartz J. Dynamics of putative raft-associated proteins at the cell surface. J Cell Biol. 2004;165:735–746. doi: 10.1083/jcb.200312170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim AH, Yano H, Cho H, Meyer D, Monks B, Margolis B, Birnbaum MJ, Chao MV. Akt1 regulates a JNK scaffold during excitotoxic apoptosis. Neuron. 2002;35:697–709. doi: 10.1016/s0896-6273(02)00821-8. [DOI] [PubMed] [Google Scholar]
- Kojima N, Wang J, Mansuy IM, Grant SG, Mayford M, Kandel ER. Rescuing impairment of long-term potentiation in fyn-deficient mice by introducing Fyn transgene. Proc Natl Acad Sci USA. 1997;94:4761–4765. doi: 10.1073/pnas.94.9.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte M, Griesbeck O, Gravel C, Carroll P, Staiger V, Thoenen H, Bonhoeffer T. Virus-mediated gene transfer into hippocampal CA1 region restores long-term potentiation in brain-derived neurotrophic factor mutant mice. Proc Natl Acad Sci USA. 1996;93:12547–12552. doi: 10.1073/pnas.93.22.12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuruvilla R, Ye H, Ginty DD. Spatially and functionally distinct roles of the PI3-K effector pathway during NGF signaling in sympathetic neurons. Neuron. 2000;27:499–512. doi: 10.1016/s0896-6273(00)00061-1. [DOI] [PubMed] [Google Scholar]
- Li Y, Jia YC, Cui K, Li N, Zheng ZY, Wang YZ, Yuan XB. Essential role of TRPC channels in the guidance of nerve growth cones by brain-derived neurotrophic factor. Nature. 2005;434:894–898. doi: 10.1038/nature03477. [DOI] [PubMed] [Google Scholar]
- Lu B. BDNF and activity-dependent synaptic modulation. Learn Mem. 2003;10:86–98. doi: 10.1101/lm.54603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammen AL, Huganir RL, O'Brien RJ. Redistribution and stabilization of cell surface glutamate receptors during synapse formation. J Neurosci. 1997;17:7351–7358. doi: 10.1523/JNEUROSCI.17-19-07351.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto T, Numakawa T, Adachi N, Yokomaku D, Yamagishi S, Takei N, Hatanaka H. Brain-derived neurotrophic factor enhances depolarization-evoked glutamate release in cultured cortical neurons. J Neurochem. 2001;79:522–530. doi: 10.1046/j.1471-4159.2001.00591.x. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Korte M, Wolfer D, Kuhn R, Unsicker K, Cestari V, Rossi-Arnaud C, Lipp HP, Bonhoeffer T, Klein R. Essential role for TrkB receptors in hippocampus-mediated learning. Neuron. 1999;24:401–414. doi: 10.1016/s0896-6273(00)80853-3. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Calella AM, Medina DL, Bonhoeffer T, Klein R, Korte M. Mechanism of TrkB-mediated hippocampal long-term potentiation. Neuron. 2002;36:121–137. doi: 10.1016/s0896-6273(02)00942-x. [DOI] [PubMed] [Google Scholar]
- Mutoh T, Tokuda A, Miyadai T, Hamaguchi M, Fujiki N. Ganglioside GM1 binds to the Trk protein and regulates receptor function. Proc Natl Acad Sci USA. 1995;92:5087–5091. doi: 10.1073/pnas.92.11.5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishio M, Fukumoto S, Furukawa K, Ichimura A, Miyazaki H, Kusunoki S, Urano T, Furukawa K. Overexpressed GM1 suppresses nerve growth factor (NGF) signals by modulating the intracellular localization of NGF receptors and membrane fluidity in PC12 cells. J Biol Chem. 2004;279:33368–33378. doi: 10.1074/jbc.M403816200. [DOI] [PubMed] [Google Scholar]
- Ohira K, Kumanogoh H, Sahara Y, Homma KJ, Hirai H, Nakamura S, Hayashi M. A truncated tropomyosin-related kinase B receptor, T1, regulates glial cell morphology via Rho GDP dissociation inhibitor 1. J Neurosci. 2005;25:1343–1353. doi: 10.1523/JNEUROSCI.4436-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- Peiro S, Comella JX, Enrich C, Martin-Zanca D, Rocamora N. PC12 cells have caveolae that contain TrkA. Caveolae-disrupting drugs inhibit nerve growth factor-induced, but not epidermal growth factor-induced, MAPK phosphorylation. J Biol Chem. 2000;275:37846–37852. doi: 10.1074/jbc.M000487200. [DOI] [PubMed] [Google Scholar]
- Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- Rajagopal R, Chao MV. A role for Fyn in Trk receptor transactivation by G-protein-coupled receptor signaling. Mol Cell Neurosci. 2006;33:36–46. doi: 10.1016/j.mcn.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Rajagopal R, Chen ZY, Lee FS, Chao MV. Transactivation of Trk neurotrophin receptors by G-protein-coupled receptor ligands occurs on intracellular membranes. J Neurosci. 2004;24:6650–6658. doi: 10.1523/JNEUROSCI.0010-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccio A, Pierchala BA, Ciarallo CL, Ginty DD. An NGF-TrkA-mediated retrograde signal to transcription factor CREB in sympathetic neurons. Science. 1997;277:1097–1100. doi: 10.1126/science.277.5329.1097. [DOI] [PubMed] [Google Scholar]
- Shao Y, Akmentin W, Toledo-Aral JJ, Rosenbaum J, Valdez G, Cabot JB, Hilbush BS, Halegoua S. Pincher, a pinocytic chaperone for nerve growth factor/TrkA signaling endosomes. J Cell Biol. 2002;157:679–691. doi: 10.1083/jcb.200201063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1:31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- Simons M, Friedrichson T, Schulz JB, Pitto M, Masserini M, Kurzchalia TV. Exogenous administration of gangliosides displaces GPI-anchored proteins from lipid microdomains in living cells. Mol Biol Cell. 1999;10:3187–3196. doi: 10.1091/mbc.10.10.3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein PL, Lee HM, Rich S, Soriano P. pp59fyn mutant mice display differential signaling in thymocytes and peripheral T cells. Cell. 1992;70:741–750. doi: 10.1016/0092-8674(92)90308-y. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Numakawa T, Shimazu K, Koshimizu H, Hara T, Hatanaka H, Mei L, Lu B, Kojima M. BDNF-induced recruitment of TrkB receptor into neuronal lipid rafts: roles in synaptic modulation. J Cell Biol. 2004;167:1205–1215. doi: 10.1083/jcb.200404106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui-Pierchala BA, Encinas M, Milbrandt J, Johnson EM., Jr Lipid rafts in neuronal signaling and function. Trends Neurosci. 2002;25:412–417. doi: 10.1016/s0166-2236(02)02215-4. [DOI] [PubMed] [Google Scholar]
- Tsuruda A, Suzuki S, Maekawa T, Oka S. Constitutively active Src facilitates NGF-induced phosphorylation of TrkA and causes enhancement of the MAPK signaling in SK-N-MC cells. FEBS Lett. 2004;560:215–220. doi: 10.1016/S0014-5793(04)00115-2. [DOI] [PubMed] [Google Scholar]
- Wetmore C, Olson L. Neuronal and nonneuronal expression of neurotrophins and their receptors in sensory and sympathetic ganglia suggest new intercellular trophic interactions. J Comp Neurol. 1995;353:143–159. doi: 10.1002/cne.903530113. [DOI] [PubMed] [Google Scholar]
- Wu C, Butz S, Ying Y, Anderson RG. Tyrosine kinase receptors concentrated in caveolae-like domains from neuronal plasma membrane. J Biol Chem. 1997;272:3554–3559. doi: 10.1074/jbc.272.6.3554. [DOI] [PubMed] [Google Scholar]
- Yano H, Chao MV. Mechanisms of neurotrophin receptor vesicular transport. J Neurobiol. 2004;58:244–257. doi: 10.1002/neu.10321. [DOI] [PubMed] [Google Scholar]
- Yano H, Ninan I, Zhang H, Milner TA, Arancio O, Chao MV. BDNF-mediated neurotransmission relies upon a myosin VI motor complex. Nat Neurosci. 2006;9:1009–1018. doi: 10.1038/nn1730. [DOI] [PubMed] [Google Scholar]
- Ye H, Kuruvilla R, Zweifel LS, Ginty DD. Evidence in support of signaling endosome-based retrograde survival of sympathetic neurons. Neuron. 2003;39:57–68. doi: 10.1016/s0896-6273(03)00266-6. [DOI] [PubMed] [Google Scholar]