

Abstract
Cannabinoids modulate energy homeostasis and decrease cognitive arousal, possibly by acting on hypothalamic neurons including those that synthesize melanin-concentrating hormone (MCH) or hypocretin/orexin. Using patch-clamp recordings, we compared the actions of cannabinoid agonists and antagonists on identified MCH or hypocretin neurons in green fluorescent protein-expressing transgenic mice. The cannabinoid type-1 receptor (CB1R) agonist R-(+)-[2,3-dihydro-5-methyl-3-(4-morpho linylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone mesylate (WIN55,212,2) depolarized MCH cells and increased spike frequency; in contrast, WIN55,212,2 hyperpolarized and reduced spontaneous firing of the neighboring hypocretin cells, both results consistent with reduced activity seen with intracerebral cannabinoid infusions. These effects were prevented by AM251 [N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide], a CB1R antagonist, and by tetrodotoxin, suggesting no postsynaptic effect on either neuron type. In MCH cells, depolarizing WIN55,212,2 actions were abolished by the GABAA receptor antagonist bicuculline, suggesting that the CB1R-mediated depolarization was attributable to reduced synaptic GABA release. WIN55,212,2 decreased spontaneous IPSCs, reduced the frequency but not amplitude of miniature IPSCs, and reduced electrically evoked synaptic currents in MCH cells. Glutamate microdrop experiments suggest that WIN55,212,2 acted on axons arising from lateral hypothalamus local inhibitory cells that innervate MCH neurons. In hypocretin neurons, the reduced spike frequency induced by WIN55,212,2 was attributable to presynaptic attenuation of glutamate release; CB1R agonists depressed spontaneous and evoked glutamatergic currents and reduced the frequency of miniature EPSCs. Cannabinoid actions on hypocretin neurons were abolished by ionotropic glutamate receptor antagonists. Together, these results show that cannabinoids have opposite effects on MCH and hypocretin neurons. These opposing actions could help explain the increase in feeding and reduction in arousal induced by cannabinoids.
Keywords: hypothalamus, orexin, melanin-concentrating hormone, cannabinoid, feeding, arousal
Introduction
Cannabinoids are the primary psychoactive components of marijuana and hashish. These compounds affect brain function mainly by modulating cannabinoid type-1 receptors (CB1Rs) (Matsuda et al., 1990; Freund et al., 2003; Piomelli, 2005). CB1Rs are widely expressed throughout the CNS, including the cerebellum, hippocampus, and hypothalamus (Herkenham et al., 1991; Matsuda et al., 1993; Tsou et al., 1998). The hypothalamus plays a pivotal role in energy homeostasis and arousal (Harrold, 2004; Leibowitz and Wortley, 2004).
CB1Rs are involved in the regulation of feeding and energy homeostasis (Di Marzo and Matias, 2005; Pagotto et al., 2005; Sharkey and Pittman, 2005). Agonists of CB1Rs increase eating (Williams and Kirkham, 1999; Hao et al., 2000; Jamshidi and Taylor, 2001), whereas CB1R blockade reduces food intake (Arnone et al., 1997; Colombo et al., 1998; Simiand et al., 1998). CB1R-deficient mice consume less food than wild-type littermates, have a leaner and lighter phenotype, and are resistant to diet-induced obesity when given a high-fat diet (Di Marzo et al., 2001; Cota et al., 2003; Ravinet Trillou et al., 2004), underlining the critical role played by CB1Rs in energy homeostasis.
In the hypothalamus, endogenous cannabinoid concentrations have been reported to increase during short-term fasting and decrease during feeding (Kirkham et al., 2002) or after leptin administration (Di Marzo et al., 2001). In addition, CB1R mRNA is found in lateral hypothalamic (LH) melanin-concentrating hormone (MCH) and hypocretin neurons (Cota et al., 2003). MCH neurons play a role in energy homeostasis; injections of MCH peptide into the brain increase food intake (Qu et al., 1996), pharmacological blockade of the MCH receptor reduces body weight (Borowsky et al., 2002), and MCH peptide (Shimada et al., 1998) or receptor (Marsh et al., 2002) knock-out mice eat less and have reduced body weight. Hypocretin-synthesizing neurons have been postulated to play a role in arousal; central administration of hypocretin peptide increases arousal, and the lack of hypocretin or its receptors leads to narcolepsy in mice, dogs, or humans (Chemelli et al., 1999; Lin et al., 1999; Peyron et al., 2000; Thannickal et al., 2000). In addition, hypocretin modulation of arousal states may affect the regulation of energy homeostasis (Yamanaka et al., 2003)
Thus, the observed actions of cannabinoids on energy balance and cognitive arousal may be attributable, in part, to their effects on the activity of MCH and/or hypocretin neurons. Here, using whole-cell voltage- and current-clamp recordings, we studied the cellular effects of CB1R agonists and antagonists on the activity of identified MCH and hypocretin neurons in LH slices from green fluorescent protein (GFP)-expressing transgenic mice.
Materials and Methods
Preparation of hypothalamic slices.
Experiments were performed on hypothalamic slices (250–350 μm thick) obtained from three different lines of transgenic mice. These lines selectively express enhanced GFP in MCH-containing neurons (van den Pol et al., 2004), hypocretin-containing neurons (Li et al., 2002) (provided by Dr. T. Sakurai, University of Tsukuba, Tsukuba, Ibaraki, Japan), or in GABA [glutamic acid decarboxylase 67 (GAD67)-positive]-synthesizing neurons (Tamamaki et al., 2003; Acuna-Goycolea et al., 2005) (provided by Drs. Y. Yanagawa, Gunma University, Gunma, Japan and K. Obata, RIKEN Brain Science Institute, Saitama, Japan). Briefly, 2- to 6-week-old mice maintained in a 12 h light/dark cycle were given an overdose of sodium pentobarbital (100 mg/kg) during the light part of the cycle (11:00 A.M. to 4:00 P.M.). Then, their brains were removed rapidly and placed in an ice-cold oxygenated (95% O2 and 5% CO2) high-sucrose solution that contained (in mm) 220 sucrose, 2.5 KCl, 6 MgCl2, 1 CaCl2, 1.23 NaH2PO4, 26 NaHCO3, and 10 glucose, pH 7.4 (when equilibrated with a mixture of 95% O2 and 5% CO2). A block of tissue containing the hypothalamus was isolated, and coronal slices were cut on a vibratome. After a 1–2 h recovery period, slices were moved to a recording chamber mounted on a BX51WI upright microscope (Olympus, Tokyo, Japan) equipped with video-enhanced infrared-differential interference contrast and fluorescence. Slices were perfused with a continuous flow of gassed artificial CSF (ACSF; 95% O2 and 5% CO2) that contained (in mm) 124 NaCl, 2.5 KCl, 2 MgCl2, 2 CaCl2, 1.23 NaH2PO4, 26 NaHCO3, and 10 glucose, pH 7.4. Bath temperature in the recording chamber was maintained at 35 ± 1°C using a dual-channel heat controller (Warner Instruments, Hamden, CT). Neurons were visualized with an Olympus Optical 40× water-immersion lens. The Yale University Committee on Animal Care and Use approved all procedures used in this study.
Patch-clamp recording.
Whole-cell current- and voltage-clamp recordings were performed using pipettes with 4–6 MΩ resistance after being filled with pipette solution. The pipettes were made of borosilicate glass (World Precision Instruments, Sarasota, FL) using a PP-83 vertical puller (Narishige, Tokyo, Japan). For most recordings, the composition of the pipette solution was as follows (in mm): 145 KMeSO4 (or KCl for IPSCs), 1 MgCl2, 10 HEPES, 1.1 EGTA, 2 Mg-ATP, 0.5 Na2-GTP, pH 7.3, with KOH. Liquid junction potential correction was performed off-line. Slow and fast capacitance compensation was automatically performed using Pulse software (HEKA Elektronik, Lambrecht/Pfalz, Germany). Access resistance was monitored continuously during the experiments. Only those cells in which access resistance was stable (changes <10%) were included in the analysis. An EPC10 amplifier and Pulse software were used for data acquisition (HEKA Elektronik). PulseFit (HEKA Elektronik), Axograph (Molecular Devices, Foster City, CA), and Igor Pro (WaveMetrics, Lake Oswego, OR) software were used for analysis. Both spontaneous EPSCs (sEPSCs) and spontaneous IPSCs (sIPSCs) were detected and measured with an algorithm in Axograph, and only those events with amplitude >5 pA were used, as described in detail previously (Gao and van den Pol, 2001). The frequency of action potentials was measured using Axograph as well. Data are expressed as mean ± SEM. Group statistical significance was assessed using paired Student's t test, for comparison of two groups, and one-way ANOVA followed by a Bonferroni post hoc test, for three or more groups. p < 0.05 was considered statistically significant.
Evoked postsynaptic currents were obtained by using ACSF-filled, theta glass microelectrodes (resistance, 0.2–0.5 MΩ) as stimulation electrodes. The stimulating electrodes were positioned within the lateral hypothalamus. Evoked EPSCs (eEPSCs) were studied with 30 μm bicuculline (Bic) in the bath to block inhibitory GABAA receptor-mediated synaptic transmission. Evoked IPSC (eIPSC) experiments were performed in the presence of dl-2-amino-5-phosphonopentanoic acid (AP-5; 50 μm) and 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; 10 μm) in the external solution to block ionotropic glutamate receptors. Currents were evoked by applying pulses (20–200 μA; duration, 200 μs) through the electrodes. This stimulation protocol was repeated every 6 s. For paired-pulse ratio experiments, the interstimulus interval was 30 ms, and the ratio between the second and the first evoked currents was calculated and averaged for ≥20 trials. To study the effect of locally applied glutamate on the IPSCs recorded in MCH neurons, the following intracellular solution was used (in mm): 130 CsMeSO4, 10 CsCl, 2 MgCl2, 10 HEPES, 0.16 CaCl2, 0.5 EGTA, 4 Na2ATP, 0.4 NaGTP, and 14 Tris-creatine phosphate, adjusted to pH 7.3 with CsOH. AP-5 (50 μm) was added in the bath. Using this internal solution, Cl− reversal potential of recorded cells was approximately −40 mV. Microdrops of glutamate (10–30 mm) were pressure applied (5–8 psi, 5–10 ms) (Picospritzer II; Parker-Hannefin, Fairfield, NJ) to the surface of the lateral hypothalamus (0.5–1 mm away from the recorded cells), using a broken patch pipette of 10–15 μm tip diameter, as described previously (Belousov and van den Pol, 1997; Acuna-Goycolea et al., 2004).
Depolarization-induced suppression of inhibition (DSI) or excitation (DSE) was studied after a 1 or 5 s depolarization pulse (from −70 to 0 mV) every 2–3 min. To quantify DSI or DSE, we calculated the sIPSC or sEPSC waveform integral for 20 s before the depolarization pulse and 20 s after the pulse. The first 2 s after the voltage step were disregarded to allow maximal DSI or DSE to develop. The percentage of suppression was calculated as follows (charge, Q): DSI or DSE = [1 − (Qpost/Qpre)] × 100. To estimate the changes of spontaneous postsynaptic currents in a given cell, data from at least three DSIs or DSEs were averaged.
Immunocytochemistry.
Immunostaining against MCH peptide was performed as described previously (van den Pol et al., 2004). Briefly, GFP–MCH or GAD67–GFP transgenic mice were anesthetized and perfused transcardially with a fixative solution containing 4% paraformaldehyde in 0.1 m phosphate buffer. Sections were then cut on a cryostat at 20–30 μm. After washing in normal buffer containing 0.1% lysine, 1% bovine serum albumin, 1% normal goat serum, and 0.3% Triton X-100, sections were incubated overnight in primary rabbit antiserum against MCH from Drs. P. Sawchenko and W. Vale (Salk Institute, San Diego, CA); the specificity of the antiserum was described in detail previously (Bittencourt et al., 1992). The antisera was used at concentrations of 1:2500 and detected with a secondary donkey anti-rabbit Ig conjugated to Alexa 594. Immunocytochemical verification of selective GFP expression in hypocretin (Li et al., 2002) or GAD67 mRNA-containing GABA neurons (Acuna-Goycolea et al., 2005) has been reported previously.
Effects of CB1R agonist on behavioral activity.
Six- to 10-week-old C57BL/6 mice were stereotaxically implanted with a guide cannula in the lateral ventricle. The surgical procedures and implant coordinates were described previously (Butcher et al., 2002). Cannulated mice were housed individually in cages equipped with running wheels (15 cm diameter); wheel rotation was recorded using Vital View (Minimitter, Bend, OR) data acquisition software. Mice were entrained to a 12 h light/dark cycle for 2 weeks, then dark adapted for a minimum of 2 weeks before infusion. Three hours before the beginning of the active period (subjective nighttime), mice were either infused (500 nl/min) with the CB1R agonist R-(+)-[2,3-dihydro-5-methyl-3-(4-morpho linylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone mesylate (WIN55,212,2; 4 mm, 3 μl, diluted in a DMSO vehicle) or with the same vehicle lacking the CB1R agonist. After infusion, mice were returned to their home cages, and locomotor behavior was monitored. Activity data from the day before drug application was used as the predrug baseline, and the day after drug application was used as the test day.
Drugs and drug application.
AP-5, Bic, and CNQX were purchased from Sigma (St. Louis, MO). Tetrodotoxin (TTX) was obtained from Alomone Labs (Jerusalem, Israel). N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (AM251) and WIN55,212,2 were obtained from Tocris (Ellisville, MO). All drugs were given by large-diameter (500 μm) flow pipette application, directed at the recorded cell, unless noted otherwise. When a drug was not being administered, normal ACSF continuously flowed from the flow pipe. Drug solutions were prepared by diluting the appropriate stock solution with ACSF.
Results
MCH neurons
GFP expression in GFP–MCH transgenic mice
Histological sections from GFP–MCH transgenic mice were examined. GFP-expressing neurons were found in the hypothalamus, with a particularly high concentration in the lateral hypothalamus. The distribution of GFP-labeled cells matches the description of immunoreactive MCH neurons reported previously (Bittencourt et al., 1992). The highest concentration of MCH–GFP cells was found in the LH/perifornical region extending from the posterior paraventricular nucleus caudally to the mammillary recess of the third ventricle and in the zona incerta. Some cells were also seen in the anterior periventricular nucleus, postserolateral to the paraventricular nucleus, and in the dorsomedial hypothalamus. All GFP-positive neurons were immunoreactive with MCH antisera (267 cells examined), whereas 67% (267 of 400) of MCH-immunoreactive cells showed detectable GFP expression. The lack of GFP expression in some immunopositive cells could be attributable to low levels of GFP expression in the negative cells, to aldehyde-mediated quenching of GFP fluorescence, or to cells that do not express GFP. Figure 1A–D showed the expression of GFP and MCH immunostaining (red) in the lateral hypothalamus.
Figure 1.
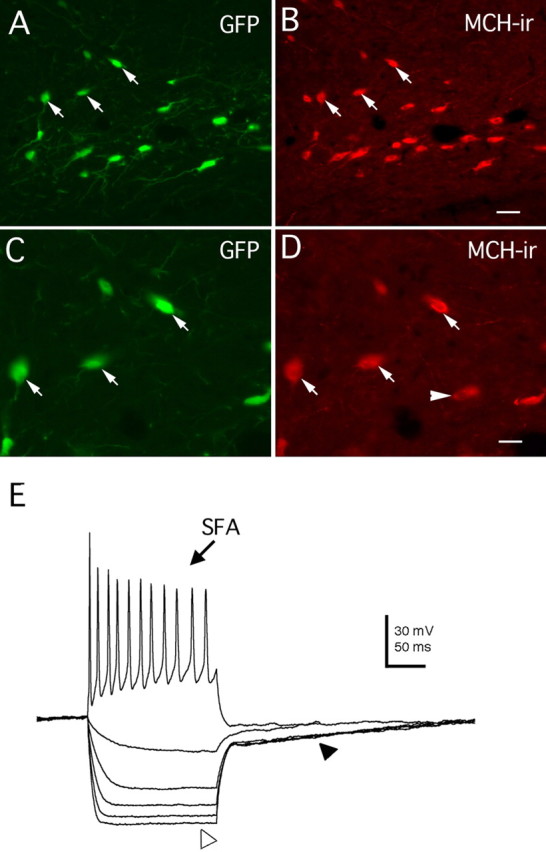
GFP expression restricted to MCH-containing neurons in transgenic mice. A, GFP expression in a hypothalamic section of a GFP–MCH transgenic mouse. Arrows indicate three cells. B, The same three cells (arrows) in A are shown, where antisera reveal immunoreactive MCH (MCH-ir; Texas Red) in the same neurons that express GFP, confirming that GFP expression in this line of transgenic animals is restricted to MCH-synthesizing neurons in the lateral hypothalamus. C, D. The same three cells shown in A and B are depicted at higher magnification. E, Intrinsic membrane properties of MCH neurons identified by GFP expression. SFA, Spike-frequency adaptation (after a 60 pA positive current injection for 200 ms). The open arrowhead indicates the inward rectification induced by −30 to −150 pA current injection for 200 ms, 30 pA steps; the filled arrowhead indicates the outward rectification observed in these hypothalamic cells. Scale bars: A, B, 20 μm; C, D, 10 μm.
As shown before using a recombinant adeno-associated virus (van den Pol et al., 2004), the GFP-expressing MCH neurons are very silent in rest conditions, showing little or no spontaneous activity (firing rate range, 0–0.9 Hz; mean, 0.12 ± 0.1 Hz; n = 41; data not shown). The mean resting potential was −61.7 ± 0.4 mV (n = 41). A positive current step (40–80 pA for 200 ms) induced action potential firing with evident spike-frequency adaptation (Fig. 1E, top, arrow), similar to previous reports (Eggermann et al., 2003; Gao et al., 2003; van den Pol et al., 2004). Hyperpolarizing current injection (from −30 to −150 pA for 200 ms, 30 pA steps) revealed an instantaneous inward rectification for potentials more negative than −85 mV (Fig. 1E, bottom, open arrowhead) and also a transient outward rectification identified as a delayed return to the resting potential after hyperpolarizing current injection (Fig. 1E, bottom, filled arrowhead).
MCH neurons release endocannabinoids
Cannabinoid receptors have been observed in mouse lateral hypothalamus, the region where most MCH neurons are found (Herkenham et al., 1991; Matsuda et al., 1993; Cota et al., 2003). To determine whether endogenous cannabinoids can be released from MCH neurons and modulate their inhibitory synaptic inputs, we recorded the synaptic release of GABA onto MCH cells after stimulation of the postsynaptic cell. Brief depolarizing steps have been shown to effectively evoke postsynaptic release of endocannabinoids in hippocampus (Wilson and Nicoll, 2001), cerebellum (Kreitzer and Regehr, 2001; Yoshida et al., 2002), and other brain areas (Melis et al., 2004; Zhu and Lovinger, 2005). The released endocannabinoids diffuse into the extracellular space and activate presynaptic receptors leading to changes in the synaptic strength (Kreitzer and Regehr, 2002; Wilson and Nicoll, 2002). Brief depolarization (from −70 to 0 mV, 1 s) of MCH cells caused a significant and transient suppression of sIPSCs (called DSI) in six of eight neurons tested (average DSI, 25.0 ± 6.0%; n = 8; p < 0.01, t test; data not shown). The inhibitory effect of depolarizing voltage steps on sIPSCs was not observed when the CB1R antagonist AM251 (5 μm) was present in the bath (averaged DSI under these conditions was 1.8 ± 5.9%; n = 5; p > 0.5, t test). These results suggest that endogenous cannabinoids can be released by postsynaptic depolarization and that they may modulate synaptic transmission in MCH neurons by acting at presynaptic sites. Similar findings have been reported in LH neurons, including some chemically identified MCH neurons (Jo et al., 2005).
CB1Rs modulate MCH neuron activity
To further characterize the physiological role of cannabinoid receptors in modulating MCH neuronal activity, voltage- and current-clamp experiments using selective synthetic agonists/antagonists of CB1R were performed. We first tested whether the CB1R agonist WIN55,212,2 modulated MCH neuron activity in coronal hypothalamic slices in vitro. In current clamp, WIN55,212,2 (1 μm) significantly depolarized GFP-expressing MCH cells by 3.0 ± 0.5 mV (Fig. 2A, trace 2, B,F) (n = 27; p < 0.05, t test). This depolarizing effect was sometimes accompanied by an increase in MCH neuron spike frequency as shown in trace 1 of Figure 2A. WIN55,212,2 actions on both membrane potential and spike frequency were abolished when the slices were preincubated for 5–10 min with the selective CB1R antagonist AM251 (5 μm) (change in membrane potential, 0.3 ± 0.4 mV) (Fig. 2A, trace 3, C,F) (n = 6; p = 0.88, t test), consistent with the view that they were mediated by activation of CB1Rs. The depolarization by WIN55,212,2 could also be blocked by 10–15 min pretreatment with 0.2 or 1 μm AM251 (change in membrane potential was 1.5 ± 0.8 and 0.6 ± 0.5 mV with 0.2 and 1 μm AM251 in the bath, respectively; n = 4–6; p > 0.05; data not shown). With TTX in the external solution, three different doses (1, 5, and 10 μm) of WIN55,212,2 showed no substantive effect on MCH membrane potentials (change by 0.9 ± 0.7, 0.7 ± 0.5, and 0.9 ± 0.7 mV by 1, 5, and 10 μm WIN55,212,2, respectively; n = 4–6; p > 0.05; data not shown), suggesting that the activation of these receptors had little postsynaptic effect. The effect of WIN55,212,2 on neuronal activity was also investigated in older adult 6-week-old mice. WIN55,212,2 (1 μm) significantly depolarized the membrane potential of MCH neurons by 2.8 ± 0.4 mV (n = 9; p < 0.05; data not shown), similar to the effects on MCH neurons from younger mice.
Figure 2.
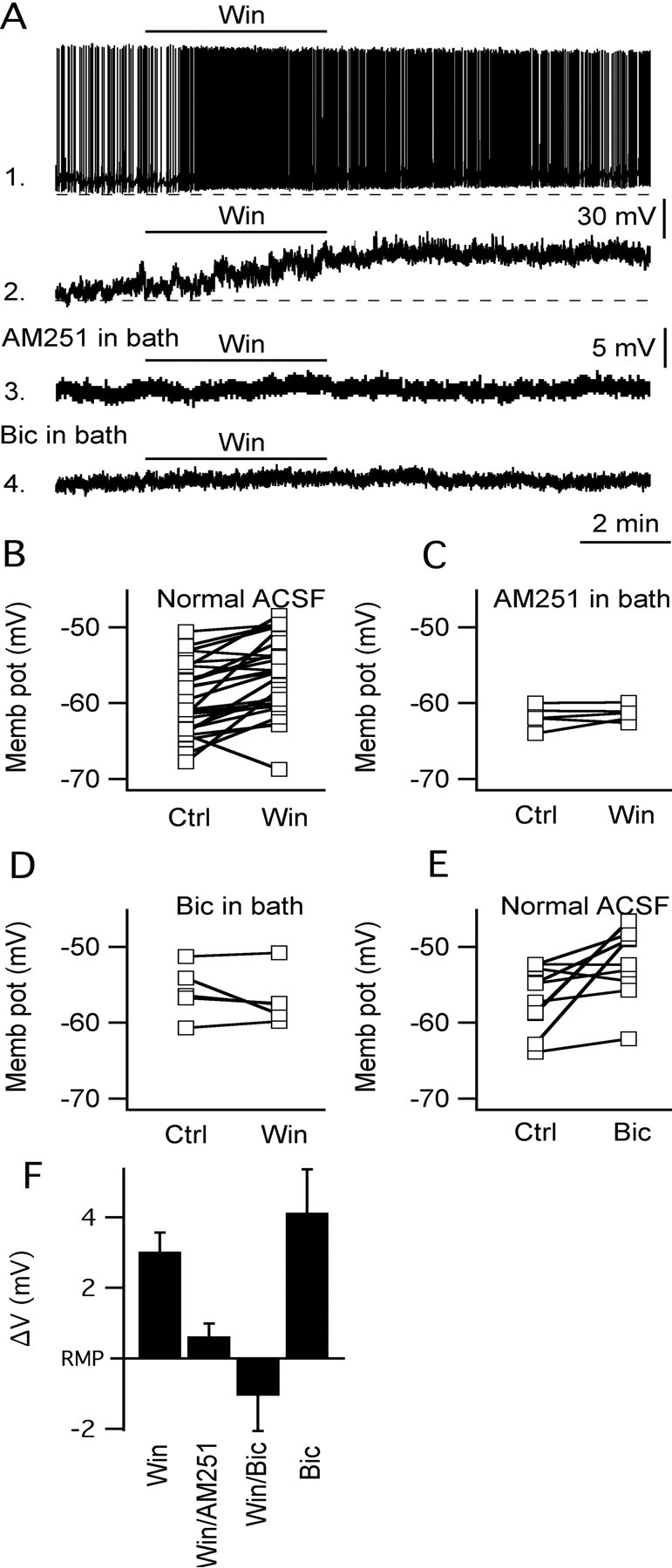
CB1R agonist WIN55,212,2 modulates MCH neuronal activity. A, The application of WIN55,212,2 to the lateral hypothalamus depolarized MCH neurons. Some spontaneously active MCH-expressing cells increased their spike frequency when WIN55,212,2 was applied to the slice [trace 1; resting membrane potential (RMP), −60.5 mV]. B, In silent MCH neurons, WIN55,212,2 usually affected only the membrane potential (trace 2 in A; RMP, −62.8 mV). C, D, The effect of WIN55,212,2 on MCH membrane potential was not observed in the presence of the selective CB1R antagonist AM251 (trace 3 in A; RMP, −63.4 mV; population graph in C) or when the slice was perfused with the GABAA receptor antagonist Bic (trace 4 in A; RMP, −61.8 mV; graph in D). E, Bic application also depolarized MCH cells, suggesting that these hypothalamic neurons are under tonic inhibitory control mediated by GABAA receptor activation. F, Bar graph summarizing the actions of CB1R agonist/antagonist and Bic on MCH membrane potential. Error bars indicate SEM. Win, WIN55,212,2; Ctrl, control; Win/AM251, in the presence of AM251, the change of membrane induced by WIN55,212,2; Win/Bic, with bicuculline in the bath, the change of membrane potential by additional application of WIN55,212,2; Memb pot, membrane potential.
Next, we tested whether some of these CB1R-mediated effects were attributable to modification of intrinsic currents in MCH neurons. First, we studied the action of WIN55,212,2 on whole-cell potassium currents using slow (3 s) voltage-ramp protocols from −140 to +20 mV. This experiment was done with TTX and CdCl2 in the bath. Under these conditions, the application of WIN55,212,2 (1 μm) did not significantly modify the current response in seven cells tested (data not shown), suggesting that potassium current modulations are probably not the primary mechanism by which WIN55,212,2 depolarized MCH neurons. The effect of WIN55,212,2 on voltage-activated calcium currents was also evaluated using CsCH3SO3 pipettes. In these experiments, TEA-Cl (40 mm)-containing ACSF was used as the extracellular solution to inhibit potassium currents, and BaCl2 was substituted for CaCl2 to increase the conductance of the calcium channels. Voltage-dependent sodium currents were blocked by TTX in the bath. A voltage step from −80 to 0 mV for 200 ms was delivered to activate the barium current. Under these conditions, the application of WIN55,212,2 (1 μm) resulted in a statistically nonsignificant depression of barium current (from 517.0 ± 46.0 to 461.0 ± 31.0 pA; n = 8; p > 0.1, t test), suggesting WIN55,212,2 had little modulatory effect on postsynaptic voltage-activated calcium currents.
CB1Rs have been shown to be coupled to G-protein (usually the Gi subtype) and are often located on the presynaptic membrane of inhibitory axons (Tsou et al., 1998; Katona et al., 1999, 2001). We reasoned that the observed WIN55,212,2 actions on MCH cells might be attributable to a presynaptic reduction in GABA release (synaptic disinhibition) onto these hypothalamic neurons. Consistent with this, the addition of Bic alone to the bath mimicked CB1R agonist action in current clamp, leading to a 4.1 ± 1.6 mV depolarization (Fig. 2E,F) (n = 10; p < 0.05, t test). Importantly, with pretreatment with Bic in the external solution, WIN55,212,2 did not change MCH membrane potential (change in membrane potential by WIN55,212,2 with Bic in the bath, 1.1 ± 1.0 mV; n = 5; p > 0.05, t test) (Fig. 2A, trace 4, D,F), suggesting that most of CB1R agonist excitatory actions were indeed attributable to a reduction in GABA release onto MCH neurons.
CB1R agonist modulation of inhibitory activity in MCH neurons
To evaluate the effect of CB1R agonists on MCH inhibitory synaptic inputs, we performed whole-cell voltage-clamp experiments using KCl-based internal solutions. These experiments were done with AP-5 (50 μm) and CNQX (10 μm) in the bath to block ionotropic glutamate receptors. Under these conditions, the sIPSCs were detected as inward currents at −60 mV holding potentials (Fig. 3A). WIN55,212,2 reduced the frequency of sIPSCs by 51.7 ± 6.5% (n = 13; p < 0.05, t test) as shown in Figure 3, A, C, and E. The time course of CB1R agonist actions on the IPSC frequency is presented in Figure 3B. The actions of WIN55,212,2 appeared to be attributable to the activation of CB1Rs, because preincubation of the hypothalamic slices with the selective CB1R antagonist AM251 prevented WIN55,212,2-evoked depression of sIPSC frequency in MCH neurons (change by 2.0 ± 5.1% of control; n = 6; p > 0.5, t test) (Fig. 3D,E). Bar graphs showing the mean effect of WIN55,212,2 on sIPSCs in MCH neurons with and without AMP251 in the bath are presented in Figure 3E. WIN55,212,2 also decreased the frequency of sIPSCs by 38.6 ± 6.1% (n = 8; p < 0.05; data not shown) in MCH neurons from 6-week-old mice. AM251 alone showed no effect on the frequency or amplitude of sIPSCs (change by 6.0 ± 5.9 and 4.0 ± 4.7% of control frequency and amplitude, respectively; n = 5; p > 0.1, t test), or membrane potential of MCH neurons (change by 0.9 ± 0.9 mV; n = 6; p > 0.5), suggesting that, in our experimental conditions, little spontaneous endocannabinoids release occurs from MCH cells.
Figure 3.

Cannabinoids depress GABAergic transmission in MCH neurons. A, Voltage-clamp recording showing the depressing actions of the CB1R agonist WIN55,212,2 on sIPSC in hypothalamic MCH neurons. These recordings were performed with a KCl-based internal solution, and at a −60 mV holding potential, inhibitory currents were detected as inward currents. B, This graph shows the time course of WIN55,212,2 actions on the normalized frequency of IPSCs in 13 MCH neurons tested. C, D, Graph depicting population WIN55,212,2 actions on frequency of IPSCs with normal ACSF as the external solution (C) or in the presence of AM251 in the bath (D). E, Bar graphs summarizing WIN55,212,2 actions on IPSC frequency with or without AM251 in the external solution. *p < 0.05, statistically significant. Error bars indicate SEM. Win, WIN55,212,2; Ctrl, control.
The results presented above support the view that the activation of CB1Rs attenuates the release of GABA onto MCH. To determine whether this WIN55,212,2-induced reduction in GABA release was attributable to selective actions on GABAergic presynaptic terminals innervating MCH neurons, we studied the effect of CB1R agonists on miniature IPSCs (mIPSCs) with 1 μm TTX in the external solution. WIN55,212,2 (1 μm) decreased the frequency of mIPSCs by 47.0 ± 6.0% (Fig. 4A) (n = 9; p < 0.05, t test) with little effect on mIPSC amplitude (change by 5.4 ± 4.3%; n = 9; p = 0.91, t test) (Fig. 4B). In the presence of the CB1R antagonist AM251, WIN55,212,2 showed no significant effect on mIPSCs (change by 3.8 ± 8.9 and 3.0 ± 7.5% of control frequency and amplitude, respectively; n = 5, p > 0.05 t test), indicating that the modulatory effects of WIN55,212,2 on mIPSCs are attributable to the activation of CB1Rs. The bar graphs in Figure 4, C and D, show the mean WIN55,212,2 action on mIPSC frequency (left) and amplitude (right) in slices perfused with normal ACSF (C) or preincubated with the CB1R antagonist AM251 (D). Together, our results are consistent with the idea that activation of CB1Rs in GABA-producing axon terminals leads to a reduction in the release of GABA onto MCH cells, which in turn might relieve these hypothalamic neurons from tonic inhibition mediated by persistent GABAA receptor activation.
Figure 4.

CB1R agonists reduce GABA release by presynaptic mechanisms. A, mIPSC frequency was reduced after WIN55,212,2 application to the slice. B, The amplitude of the mIPSC was not affected by WIN55,212,2 treatment, consistent with a presynaptic mechanism. C, Bar graph showing the averaged actions of WIN55,212,2 on the frequency (left) or the amplitude (right) of mIPSCs in MCH neurons. The asterisk indicates statistical significance. D, No effect of WIN55,212,2 on IPSC frequency (left) or amplitude (right) was observed when slices were preincubated with the CB1R antagonist AM251. Error bars indicate SEM. Win, WIN55,212,2; Ctrl, control.
We further studied WIN55,212,2 actions on eIPSCs in MCH neurons using electrical stimulation of LH neurons with a theta glass microelectrode and KCl-based internal solution. Evoking IPSCs would potentially reveal actions of nonspontaneously active GABA input. Ionotropic glutamate receptors were blocked in these experiments (see above); the stimulating electrode was placed medial or lateral to the recorded cells. eIPSC amplitude was reduced by WIN55,212,2 (1 μm) as shown in the traces of Figure 5A. The amplitude of the eIPSC was reduced by 47.7 ± 7.8% in eight cells tested (p < 0.05, t test) (Fig. 5E, left columns). To determine the synaptic site of action of WIN55,212,2, we examined the effect of WIN55,212,2 on the amplitude ratio of the second IPSC divided by that of the first one (paired-pulse ratio; see Materials and Methods). IPSCs were elicited by two successive stimuli of identical strength at an interval of 30 ms. A change in the paired-pulse ratio is considered to be attributable to a presynaptic change in release probability (Zucker, 1989; Manabe et al., 1993). The eIPSC paired-pulse ratio was consistently increased from 0.8 ± 0.1 to 1.0 ± 0.1 (n = 8; p < 0.05, t test) (Fig. 5B,F, left columns) after WIN55,212,2 application. This result is consistent with our previous observations suggesting a presynaptic site of action of CB1R agonist at GABAergic axons synapsing onto MCH neurons.
Figure 5.

WIN55,212,2 inhibits evoked glutamatergic and GABAergic transmission to MCH neurons. A, C, Traces showing WIN55,212,2 inhibition of the eIPSC (A) and eEPSC (C) amplitude. Thick black traces are the average of seven gray consecutive eIPSCs or eEPSCs. In these and subsequent figures, stimulus artifacts have been truncated for clarity. B, D, Representative paired-pulse traces for eIPSCs (B) and eEPSCs (D) in control (left) or in the presence of WIN55,212,2 (right). E, Bar graphs summarizing WIN55,212,2 actions on the amplitude of eIPSCs and eEPSCs. F, Bar graphs summarizing the effects of WIN55,212,2 on the paired-pulse ratios for eIPSCs and eEPSCs. The asterisks indicate statistical significance. The number of cells is shown in parentheses. Error bars indicate SEM. Win, WIN55,212,2; Ctrl, control.
CB1R agonist and MCH excitatory activity
MCH neurons in slices tend to show low levels of spontaneous excitatory synaptic activity (0.34 ± 0.08 Hz; n = 7) (see also van den Pol et al., 2004), and given the paucity of EPCSs, we were unable to evaluate the actions of WIN55,212,2 on spontaneous glutamatergic currents. To determine whether MCH glutamate-mediated synaptic transmission was affected by CB1R receptor activity, we evaluated the effect of WIN55,212,2 on eEPSCs. These experiments were done in the presence of Bic in the bath to eliminate GABAergic synaptic actions. The application of WIN55,212,2 resulted in a decrease in the amplitude of excitatory currents evoked by electrical stimulation of the lateral hypothalamus (decreased by 27.0 ± 10.1%; n = 7; p < 0.05, t test) (Fig. 5C,E, right columns), suggesting that, in addition to their effects on inhibitory transmission, CB1Rs might also affect the excitatory inputs to MCH neurons. The site of action of WIN55,212,2 on excitatory synapses was then investigated. WIN55,212,2 increased the eEPSC paired-pulse ratio from 1.1 ± 0.1 to 1.5 ± 0.1 (n = 7; p < 0.05, t test) (Fig. 5D,F, right columns), suggesting that the depressing effects of WIN55,212,2 on eEPSCs was caused by presynaptic reduction in glutamate release onto MCH cells.
Cannabinoids inhibit GABA release from local neurons within the lateral hypothalamus
In the absence of any direct effect, one hypothesis that could explain the excitatory actions of cannabinoids is that the CB1R agonists could modulate MCH neuronal activity by attenuating inhibitory transmitter release from axons arising from local GABAergic neurons within the lateral hypothalamus. Many GABA neurons in the LH area express GAD67 mRNA (Rosin et al., 2003), and transgenic mice that selectively express GFP in GAD67-producing neurons show green fluorescence in GABA cells in the perifornical area (Acuna-Goycolea et al., 2005). These LH GAD67-producing neurons do not colocalize with MCH cells as shown in Figure 6A.
Figure 6.

Cannabinoids decrease transmitter release presynaptically from local GABAergic neurons. A, Left, LH GFP expression in GAD67–GFP transgenic mice. Right, Same section immunostained for MCH showing lack of GAD67 fluorescence colocalization (arrows). This shows that the MCH neurons are surrounded by GABAergic neurons. Scale bar, 20 μm. B, With CsMeSO4 in the pipette and holding the membrane potential at 0 mV, sIPSCs are recorded as outward currents (trace 1), and the GABAA antagonist Bic (30 μm) totally blocked the IPSCs (trace 2). With Bic in the bath, a glutamate microdrop (Glu; 10–30 mm) does not directly affect the activity of MCH neurons (trace 2). C, Representative traces showing that in a single MCH neuron, glutamate microapplication (0.5 mm away from the recorded neuron) increased the frequency of IPSCs (trace 1). After washout, WIN55,212,2 decreased the frequency of IPSCs; in the presence of WIN55,212,2, application of glutamate did not increase the frequency of IPSCs (trace 2). D, Graph showing the frequency of IPSCs in 10 MCH neurons tested in the control condition (Ctrl), glutamate microapplication (Glu), WIN55,212,2 application (after washout; Win), and glutamate application in the presence of WIN55,212,2 (Win+Glu). E, Bar graph summarizing the frequency of IPSCs obtained from C. The asterisks indicate statistical significance. n.s., Nonsignificant. Error bars indicate SEM.
Interestingly, in contrast to the electrically silent MCH neurons, we found that LH GABA neurons identified by GFP expression show a high rate of spontaneous action potentials (3.9 ± 0.6 Hz; range, 0–8.5 Hz; n = 12; data not shown), suggesting that they tonically release GABA onto postsynaptic targets. Figure 6A shows that GABAergic neurons surround the MCH cells, raising the possibility that they may be a source of IPSCs recorded in MCH cells.
We tested this possibility by performing glutamate microdrop experiments to stimulate local inhibitory circuits within the lateral hypothalamus. Glutamate (10–30 mm) microdrops were applied 0.5–1.0 mm away from recorded MCH neurons (Belousov and van den Pol, 1997; Acuna-Goycolea et al., 2004). At this distance, the applied glutamate would excite the somatodendritic compartment of GABA neurons in the immediate area of drug microapplication, leading to the generation of action potentials that would facilitate the release of GABA from these neurons onto nearby MCH cells. Excitatory microdrops have the important advantage over electrical stimulation in that they do not stimulate fibers of passage arising from cell bodies elsewhere. We have shown previously that under these experimental conditions, glutamate microdrops induce little direct postsynaptic effects onto recorded neurons. In addition, bath application of TTX completely eliminated the effect of glutamate microdrops on synaptic transmission, further supporting the idea that little direct glutamate actions were induced on presynaptic and/or postsynaptic compartments of recorded cells (Acuna-Goycolea et al., 2004). Our experiments were performed with Cs-based pipettes with a Cl− reversal potential of approximately −40 mV. The NMDA blocker AP-5 (50 μm) was added to the bath throughout the experiment, and sIPSCs were recorded at a 0 mV holding potential to eliminate AMPA-mediated currents (because their reversal potential is expected to be around 0 mV). Under these conditions, sIPSCs were detected as outward currents (Fig. 6B, trace 1); Bic (30 μm) completely eliminated the currents, confirming that they were mediated by GABAA receptor activation (Fig. 6B, trace 2).
Glutamate microdrops did not alter MCH activity in the presence of Bic in the external solution (Fig. 6B, trace 2). With normal ACSF, glutamate microdrops enhanced the frequency of IPSCs in 8 of 10 MCH neurons by 104.0 ± 20.0% (Fig. 6C–E) (n = 10; p < 0.05, ANOVA followed by Bonferroni post hoc procedure), suggesting that activation of local GABA neurons within the lateral hypothalamus led to release of GABA onto MCH cells, which transiently increased the frequency of inhibitory currents. Then, we added WIN55,212,2 to the ACSF and evaluated the effect of glutamate microdrops on IPSCs in the same population of neurons. After WIN55,212,2 application, the frequency of IPSCs was decreased by 37.0 ± 4.5% (n = 10) (Fig. 6C–E), as observed previously. Importantly, with WIN55,212,2 in the bath, glutamate microdrop application failed to significantly enhance the frequency of IPSCs in those MCH neurons [change in IPSCs frequency compared with pre-microdrop condition (WIN55,212,2 alone), 13.7 ± 4.0%; n = 10; p > 0.05, ANOVA followed by Bonferroni post hoc procedure] (Fig. 6C–E). These data suggest that CB1R activation prevented GABA release from local axons arising from LH inhibitory neurons innervating the recorded MCH cells.
Cannabinoids do not alter LH GABA neuron activity
We then tested whether CB1R agonists directly affect the activity of GABAergic cells in the lateral hypothalamus. In these experiments, we used transgenic mice that selectively express GFP in GAD67-synthesing cells (Acuna-Goycolea et al., 2005). WIN55,212,2 (1 μm) did not significantly affect the frequency of spontaneous action potentials of LH GABAergic cells (control, 3.7 ± 0.5 Hz; after, 4.2 ± 0.9 Hz; n = 6; p > 0.2; data not shown). Importantly with TTX (1 μm) in the bath, WIN55,212,2 had no significant effect on membrane potential of GABA cells (change by 0.3 ± 1.0 mV of pretreatment levels; n = 5; p > 0.5, t test; data not shown). These results suggest that WIN55,212,2 has no direct somatic inhibitory effect on GABA neurons and are consistent with the view that cannabinoid agonists inhibit axonal GABA release from local GABA neurons onto MCH neurons by presynaptic mechanisms.
Hypocretin/orexin neurons
Cannabinoids inhibit hypocretin neurons
Next, we focused on another LH cell type, the hypocretin/orexin neuron. As described above, depolarization of MCH cells resulted in a decrease in presynaptic transmitter release, probably because of the actions of postsynaptically released endocannabinoids. Parallel experiments were done on hypocretin cells. Postsynaptic depolarization (from −70 to 0 mV, 5 s) caused a significant and transient suppression of sEPSCs (called DSE) in 9 of 18 neurons tested (average DSE, 21.2 ± 2.7%; n = 18; p < 0.01, t test; data not shown). As in the studies of MCH neurons, application of the CB1R antagonist AM251 (5 μm) blocked this inhibitory effect of depolarization on sEPSCs (DSE: 1.7 ± 2.4%, n = 8 of 8 cells), suggesting that endogenous cannabinoids can be released from hypocretin cells after postsynaptic depolarization and may modulate synaptic transmission in hypocretin neurons by acting at presynaptic sites. We then asked whether DSI might occur in hypocretin neurons. Brief depolarization (from −70 to 0 mV, 5 s) of hypocretin neurons showed no clear effect on the recorded sIPSC in any of the neurons tested (DSI: 2.1 ± 3.0%, n = 20; p > 0.05), suggesting that hypocretin neurons showed no DSI.
We then studied the effect of CB1R activation on hypocretin cell spontaneous activity. When the CB1R agonist WIN55,212,2 (5 μm) was applied to hypothalamic slices from GFP–hypocretin transgenic mice (Li et al., 2002; Yamananka et al., 2003), we observed an 81.0 ± 14.0% decrease in spike frequency accompanied by a 4.0 ± 0.4 mV hyperpolarization under current clamp (n = 7; p < 0.05) (Fig. 7A,C). WIN55,212,2 (1 μm) decreased spike frequency by 28.0 ± 9.4% (n = 15; p < 0.05) and hyperpolarized the membrane potential by 2.1 ± 0.7 mV.
Figure 7.

Cannabinoids inhibit hypocretin neuron activity. A, In a typical cell, WIN55,212,2 (5 μm) blocked spike frequency and hyperpolarized the membrane potential. Resting membrane potential, −57.2 mV. B, The cannabinoid antagonist AM251 (5 μm) increased spike frequency and blocked WIN55,212,2-induced inhibition of spike frequency. C, Bar graph showing the effect of WIN55,212,2, anandamide, and AM251 on spike frequency. D, The CB1R antagonist AM251 reversed the effect of WIN55,212,2 on spike frequency. *p < 0.05, statistical significance. The number of cells is shown in parentheses. Error bars indicate SEM. Win, WIN55,212,2; Ctrl, control; ANA, anandamide; n.s., nonsignificant.
The endogenous cannabinoid agonist anandamide (5 μm) also decreased spike frequency by 58.0 ± 16.0% and hyperpolarized hypocretin cells by 3.6 ± 0.9 mV (n = 5; p < 0.05) (Fig. 7C). In the absence of any exogenously added cannabinoid agonists, the cannabinoid antagonist AM251 (5 μm) increased spike frequency by 22.0 ± 8.0% (n = 6; p < 0.05) (Fig. 7B,C) and depolarized the membrane potential by 2.2 ± 0.7 mV (n = 6; p > 0.05), consistent with an ongoing activation of CB1Rs in hypocretin cells. In the presence of CB1R antagonist AM251 (5 μm), WIN55,212,2 (5 μm) failed to inhibit hypocretin neurons (n = 6; p > 0.05) (Fig. 7B,C). The effect of WIN55,212,2 on spike frequency generally showed only modest recovery. However, after the application of WIN55,212,2 was stopped, subsequent application of AM251 (5 μm) could reverse WIN55,212,2-induced inhibition of spike frequency (to 119.0 ± 16.0% of the control) (Fig. 7D), as reported previously in other regions of the brain (Robbe et al., 2001; Kreitzer et al., 2002). These experiments support a role for CB1R in regulating the spontaneous firing of hypocretin neurons.
Cannabinoids depress excitatory synaptic tone in hypocretin neurons by presynaptic actions
To test whether cannbinoids might inhibit the activity of hypocretin cells by reducing their excitatory input, sEPSCs, eEPSCs, and mEPSCs were examined after application of WIN55,212,2 to hypothalamic slices. WIN55,212,2 (5 μm) decreased sEPSC frequency by 42.0 ± 7.0%, and this recovered after the addition of AM251 (n = 7; p < 0.05) (Fig. 8A,B). In the presence of TTX (0.5 μm), mEPSC frequency was also decreased by 48.0 ± 5.0% (n = 7; p < 0.05) (Fig. 8C,D) after WIN55,212,2 (5 μm) application. WIN55,212,2 did not change the amplitude of mEPSCs (p > 0.05) (Fig. 8E), consistent with the view that it acted on cannabinoid receptors located in presynaptic glutamate terminals innervating hypocretin cells. We also examined WIN55,212,2 and AM251 actions on electrically eEPSCs. Bic (30 μm) was added to the bath to block IPSCs. WIN55,212,2 significantly decreased the amplitude of eEPSCs by 30.7 ± 2.2% (n = 6; ANOVA, p < 0.05) (Fig. 8F,G). AM251 reversed the WIN55,212,2-induced depression of eEPSCs (to 114.4 ± 5.2% of the control) (Fig. 8F,G). The glutamate antagonists AP-5 (50 μm) and CNQX (10 μm) completely blocked the evoked postsynaptic currents (data not shown), confirming their glutamatergic nature. Together, these results indicate that functional CB1Rs exist in glutamatergic terminals innervating hypocretin cells. The activation of these receptors would lead to a reduction in glutamate release onto postsynaptic hypocretin cells.
Figure 8.

WIN55,212,2 inhibits glutamatergic transmission to hypocretin neurons. A, Traces showing the effect of WIN55,212,2 (5 μm) and AM251 (5 μm) on sEPSCs. B, Bar graph showing the effect of WIN55,212,2 and AM251 on sEPSC frequency. C, Traces showing the effect of WIN55,212,2 (5 μm) on mEPSCs in the presence of TTX. D, E, WIN55,212,2 reduces mEPSC frequency (D) but not amplitude (E). F, An example showing the effect of WIN55,212,2 and AM251 on eEPSCs. G, Mean effect of WIN55,212,2 and AM251 on the amplitude of eEPSCs (*p < 0.05, statistically significance). Error bars indicate SEM. Win, WIN55,212,2; Ctrl, control; W/O, washout.
Previous studies have shown that spontaneous discharge of hypocretin neurons is, in part, attributable to the postsynaptic actions of glutamate tonically released in the lateral hypothalamus (Li et al., 2002; Acuna-Goycolea et al., 2004). Thus, we hypothesized that the depressing actions of CB1R agonists on hypocretin spike frequency could be attributable to a reduction in the release of glutamate onto these hypothalamic cells. To test this, we evaluated the effect of WIN55,212,2 on hypocretin neuronal activity in the presence of ionotropic glutamate receptor antagonists AP-5 (50 μm) and CNQX (10 μm). The spike frequency of hypocretin cells was not significantly changed after application of 1 or 5 μm WIN55,212,2 (1 μm, 95.0 ± 6.0%; 5 μm, 90.0 ± 8.0% of control; p > 0.05; n = 6) to hypothalamic slices pretreated with the glutamate antagonists AP-5 (50 μm) and CNQX (10 μm). These findings support the view that CB1R agonist actions on hypocretin spike frequency were dependent on modulation of glutamate release onto these hypothalamic neurons.
Cannabinoid and GABA input to hypocretin neurons
No effect of WIN55,212,2 was found on GABA-mediated IPSCs in hypocretin neurons. After WIN55,212,2 was applied, the frequency of sIPSCs was 95.0 ± 6.0% of control (n = 10; p > 0.05; data not shown), and additional AM251 application did not significantly alter sIPSC frequency (94.0 ± 4.5% of control; n = 10; p > 0.05). Additionally, input resistance was tested under current clamp by injection of current pulses (from −200 to +40 pA for 100 ms with a 20 pA increment, at 2 s intervals). AP-5 (50 μm), CNQX (10 μm), Bic (30 μm), and TTX (0.5 μm) were added to the bath to block synaptic activity. WIN55,212,2 did not change the hypocretin cell input resistance (control, 301.0 ± 36.0 MΩ; WIN55,212,2, 295.0 ± 43.0 MΩ; n = 6; p > 0.05). In addition, with TTX in the bath, WIN55,212,2 had little effect on membrane potential (change by 0.5 ± 0.5 mV; n = 7; p > 0.1). The lack of cannabinoid agonist effects on either membrane input resistance or inhibitory synaptic activity, together with the finding that AP-5 and CNQX block the WIN55,212,2-mediated inhibition, suggests that a reduction in the release of glutamate from presynaptic axons is the primary mechanism by which cannabinoids attenuate hypocretin neuron activity.
Cannabinoids reduce behavioral activity
The lateral hypothalamus has been implicated in behavioral arousal and activity. Both hypocretin and MCH neurons may play a role in regulating activity levels (Hagan et al., 1999; Marsh et al., 2002). In behavioral experiments, we assessed the effects of the CB1R agonist WIN55,212,2 after intracerebral injection into mice. Levels of activity were assessed by monitoring running-wheel activity (Butcher et al., 2005). The activity levels of mice were compared using the day-before WIN55,212,2 administration as the period of normal control activity and the day-after WIN55,212,2 injection as the test day. WIN55,212,2 caused a substantive reduction in activity (>35% decrease) in seven of eight mice, with a mean activity decrease of 63.6 ± 14.2% (n = 8; p < 0.01, paired t test). An example of a typical activity record is shown in Figure 9A, before and after WIN55,212,2 administration. In contrast, intracerebral injection of vehicle caused a similar criterion decrease in only three of seven mice. The mean reduction in control mice given the vehicle was 31.9 ± 13.8%, a nonsignificant effect (n = 7; p > 0.05, paired t test). An example of an activity record from a mouse given control vehicle is seen in Figure 9B.
Figure 9.

Cannabinoids depress behavioral activity. A, A representative activity record of a mouse that received a single infusion of the CB1R agonist WIN55,212,2 (Win) into the lateral ventricle. Each bar represents total activity per 5 min bin; the height of the vertical bars is proportional to the total behavioral activity per binned interval. Each trace shows 18 h of recorded activity. The top trace shows the control day-before intraventricular drug application, whereas the bottom trace shows the day-after drug infusion. B, A representative activity record from a mouse that received a single infusion of drug vehicle.
Discussion
Here, we studied the synaptic actions of CB1R agonists on the activity of GFP-expressing MCH or hypocretin neurons in hypothalamic slices in vitro. Cannabinoids induced a DSI in MCH neurons but a DSE in hypocretin cells. Cannabinoids depolarized and increased spike frequency of MCH neurons by presynaptic attenuation of GABA release from nearby hypothalamic GABA neurons. In contrast, cannabinoids reduced activity of hypocretin neurons by presynaptic attenuation of glutamate release. These results are consistent with the view that both hypocretin and MCH neurons release endocannabinoids but that these cannabinoids have the opposite effect in the two neurons.
Mechanisms of action in MCH and hypocretin neurons
In other brain regions, cannabinoids have been reported to affect neuronal activity by direct modulation of potassium (Deadwyler et al., 1993; Schweitzer, 2000) or calcium (Twitchell et al., 1997) channels. In contrast, we found no detectable postsynaptic effect of CB1R agonists on postsynaptic calcium currents evoked by voltage steps, potassium currents activated by voltage-ramp protocol or input resistance.
Several lines of evidence here suggest that cannabinoids act presynaptically to reduce GABA release, thereby exciting MCH neurons by disinhibition. First, when the GABAA receptor antagonist Bic was included in the external solution, the CB1R agonist WIN55,212,2 failed to depolarize MCH neurons. Second, WIN55,212,2 robustly reduced both the sIPSCs and eIPSCs in these hypothalamic cells. Third, IPSC paired-pulse ratios were increased in the presence of WIN55,212,2. Fourth, the activation of CB1Rs led to a reduction in the frequency of mIPSCs with no changes in their mean amplitude. Fifth, the depolarizing effect of CB1R agonists was mimicked by pharmacological blockade of GABAA receptor with Bic, further suggesting that under our experimental conditions, MCH neurons are tonically inhibited by persistent GABAA receptor activation.
GABAergic (GAD67-expressing) cells are present in the lateral hypothalamus and are interspersed with MCH cells. MCH neurons may contain GABA (Elias et al., 2001); however, the GFP-expressing GABA neurons do not colocalize with MCH-expressing cells found in the same hypothalamic region, suggesting that the GAD67 promoter is only weakly activated or that, for indeterminate reasons, the MCH cells do not express GFP under control of the GAD67 promoter. We used glutamate microdrops within the lateral hypothalamus to directly stimulate the soma of these GABAergic neurons without stimulating axons of passage, while recording inhibitory activity in nearby MCH neurons. We found that in the presence of CB1R agonists, glutamate microdrops were less effective in evoking MCH inhibitory transmission. Somatic whole-cell recording from these identified perifornical GABAergic cells revealed no direct actions of CB1R agonists. More likely, CB1R agonists inhibit the release of GABA from local neurons innervating MCH cells by a presynaptic mechanism.
In contrast to the action of cannabinoids on MCH cells, a reduction in the release of glutamate from excitatory axons innervating hypocretin cells appears to be the primary mechanism mediating cannabinoid effects on these neurons. Both sEPSCs and eEPSCs as well as miniature EPSCs were depressed by cannabinoids. In addition, cannabinoids were unable to change the activity of hypocretin cells in the presence of ionotropic glutamate receptor blockers. Finally, little effect of CB1R agonists on inhibitory input to hypocretin neurons was detected, further suggesting that cannabinoid actions here were primarily because of presynaptic attenuation of glutamate release onto hypocretin cells.
Although the primary action of cannabinoids in MCH cells was excitation attributable to attenuated GABA release, eEPSCs were also depressed by WIN55,212,2, suggesting that CB1Rs are expressed by glutamatergic boutons innervating hypothalamic MCH cells. Thus, in addition to affecting GABA release, the activation of CB1Rs might also modulate MCH neuronal activity by reducing glutamate synaptic input. This might occur selectively during increased periods of glutamate release, because MCH neurons in slices generally show only low levels of spontaneous glutamate synaptic activity compared with robust levels of GABA synaptic activity.
Interestingly, although both MCH and hypocretin neurons are reported to express CB1Rs (Cota et al., 2003), we found little direct effect of cannabinoids on these cell bodies. Similarly, we found little direct cannabinoid action on cell bodies of identified GABA cells in the lateral hypothalamus. The primary actions of cannabinoids on the LH cell types studied here was presynaptic. We cannot exclude the possibility that some of the effects of cannabinoids are caused by CB1Rs that may be expressed by axons of MCH or hypocretin cells, because these axons have been reported to make local neuronal contact within the lateral hypothalamus on MCH and hypocretin cells (Broberger et al., 1998; Elias et al., 1998; Horvath et al., 1999) and both neuron types express mRNA coding for CB1Rs (Cota et al., 2003).
Functional relevance
LH MCH neurons appear to play an important role in energy homeostasis (Williams et al., 2001; Berthoud, 2002). Previous studies have suggested a role for cannabinoids in enhancing food intake (Di Marzo and Matias, 2005; Sharkey and Pittman, 2005). Previous experiments showed that both electrical and chemical stimulation of this hypothalamic region increase food intake (Bernardis and Bellinger, 1996). MCH, which is selectively synthesized by LH area neurons, increases food intake and body weight. The results of the present study show that cannabinoids may depolarize and enhance the activity of MCH neurons, suggesting that some of the reported orexigenic actions of cannabinoids could be explained by their synaptic effects on MCH neuronal activity. This is consistent with the view that MCH cells may integrate signals from both leptins and endocannabinoids (Jo et al., 2005). Another possibility by which cannabinoids might modulate feeding is by inhibiting arcuate nucleus proopiomelanocortin (POMC) neurons that are involved in the repression of food intake; however, cannabinoids reduced GABA synaptic inhibition of POMC cells, potentially raising POMC neuron activity (Hentges et al., 2005). Because POMC neurons are thought to reduce food intake, a CB1R-mediated excitation of these cells is less likely to explain the enhancement of energy stores evoked by cannabinoids in the hypothalamus. Thus, the excitatory effect on MCH cells might be a possible hypothalamic site for the orexigenic action of cannabinoids.
Cannabinoids have been reported to reduce activity in humans. In parallel, injections of a CB1R agonist in mice caused a reduction in activity. Whereas this could be caused by effects of these agonists on several regions of the brain, they are consistent with the electrophysiological effects on both hypocretin and MCH neurons that we report here. The lack of MCH or its receptor leads to a more active phenotype (Marsh et al., 2002). In contrast, injections of hypocretin increase activity and arousal (Hagan et al., 1999). Because hypocretin neuron activity is correlated with cognitive arousal, the reduction in the activity of hypocretin neurons by cannabinoids may, in part, explain the reduction in arousal and increased sleepiness by users of marijuanna and hashish. Interesting, the effects of cannabinoids in increasing the activity of MCH cells but decreasing the activity of hypocretin neurons are both consistent with the reduction in behavioral activity and arousal associated with cannabinoids.
Previous work has shown that hypocretin neurons respond to opiates with changes in transcription and that addiction to opiates, which also reduces arousal and activity, is attenuated in hypocretin knock-out mice (Georgescu et al., 2003). Furthermore, the endogenous opioid dynorphin, similar to cannabinoids, reduces the activity of hypocretin cells (Li and van den Pol, 2006). Our data here showing that, in addition to the inhibitory action of opiates, cannabinoids also modulate and depress the activity of hypocretin cells, suggest that these cells may be a common target for drugs of abuse that depress arousal states.
Footnotes
This work was supported by National Institutes of Health Grants NS34887, 48476, and NS41454. We thank Y. Yang and V. Rogulin for excellent technical assistance with transgenic mice and immunocytochemistry.
References
- Acuna-Goycolea C, Li Y, van den Pol AN. Group 3 metabotropic glutamate receptors maintain tonic inhibition of excitatory synaptic input to hypocretin/orexin neurons. J Neurosci. 2004;24:3013–3022. doi: 10.1523/JNEUROSCI.5416-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acuna-Goycolea C, Tamamaki N, Yanagawa Y, Obata K, van den Pol AN. Mechanisms of neuropeptide Y, peptide YY, and pancreatic polypeptide inhibition of identified green fluorescent protein-expressing GABA neurons in the hypothalamic neuroendocrine arcuate nucleus. J Neurosci. 2005;25:7406–7419. doi: 10.1523/JNEUROSCI.1008-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnone M, Maruani J, Chaperon F, Thiebot MH, Poncelet M, Soubrie P, Le Fur G. Selective inhibition of sucrose and ethanol intake by SR 141716, an antagonist of central cannabinoid (CB1) receptors. Psychopharmacology (Berl) 1997;132:104–106. doi: 10.1007/s002130050326. [DOI] [PubMed] [Google Scholar]
- Belousov AB, van den Pol AN. Local synaptic release of glutamate from neurons in the rat hypothalamic arcuate nucleus. J Physiol (Lond) 1997;499:747–761. doi: 10.1113/jphysiol.1997.sp021966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardis LL, Bellinger LL. The lateral hypothalamic area revisited: ingestive behavior. Neurosci Biobehav Rev. 1996;20:189–287. doi: 10.1016/0149-7634(95)00015-1. [DOI] [PubMed] [Google Scholar]
- Berthoud HR. Multiple neural systems controlling food intake and body weight. Neurosci Biobehav Rev. 2002;26:393–428. doi: 10.1016/s0149-7634(02)00014-3. [DOI] [PubMed] [Google Scholar]
- Bittencourt JC, Presse F, Arias C, Peto C, Vaughan J, Nahon JL, Vale W, Sawchenko PE. The melanin-concentrating hormone system of the rat brain: an immuno- and hybridization histochemical characterization. J Comp Neurol. 1992;319:218–245. doi: 10.1002/cne.903190204. [DOI] [PubMed] [Google Scholar]
- Borowsky B, Durkin MM, Ogozalek K, Marzabadi MR, DeLeon J, Lagu B, Heurich R, Lichtblau H, Shaposhnik Z, Daniewska I, Blackburn TP, Branchek TA, Gerald C, Vaysse PJ, Forray C. Antidepressant, anxiolytic and anorectic effects of a melanin-concentrating hormone-1 receptor antagonist. Nat Med. 2002;8:825–830. doi: 10.1038/nm741. [DOI] [PubMed] [Google Scholar]
- Broberger C, de Lecea L, Sutcliffe JG, Hokfelt T. Hypocretin/orexin- and melanin-concentrating hormone-expressing cells form distinct populations in the rodent lateral hypothalamus: relationship to the neuropeptide Y and agouti gene-related protein systems. J Comp Neurol. 1998;402:460–474. [PubMed] [Google Scholar]
- Butcher GQ, Dziema H, Collamore M, Burgoon PW, Obrietan K. The p42/p44 mitogen-activated protein kinase pathway couples photic input to circadian clock entrainment. J Biol Chem. 2002;277:29519–29525. doi: 10.1074/jbc.M203301200. [DOI] [PubMed] [Google Scholar]
- Butcher GQ, Lee B, Cheng HY, Obrietan K. Light stimulates MSK1 activation in the suprachiasmatic nucleus via a PACAP-ERK/MAP kinase-dependent mechanism. J Neurosci. 2005;25:5305–5313. doi: 10.1523/JNEUROSCI.4361-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C, Richardson JA, Williams SC, Xiong Y, Kisanuki Y, Fitch TE, Nakazato M, Hammer RE, Saper CB, Yanagisawa M. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999;98:437–451. doi: 10.1016/s0092-8674(00)81973-x. [DOI] [PubMed] [Google Scholar]
- Colombo G, Agabio R, Diaz G, Lobina C, Reali R, Gessa GL. Appetite suppression and weight loss after the cannabinoid antagonist SR 141716. Life Sci. 1998;63:PL113–PL117. doi: 10.1016/s0024-3205(98)00322-1. [DOI] [PubMed] [Google Scholar]
- Cota D, Marsicano G, Tschop M, Grubler Y, Flachskamm C, Schubert M, Auer D, Yassouridis A, Thone-Reineke C, Ortmann S, Tomassoni F, Cervino C, Nisoli E, Linthorst AC, Pasquali R, Lutz B, Stalla GK, Pagotto U. The endogenous cannabinoid system affects energy balance via central orexigenic drive and peripheral lipogenesis. J Clin Invest. 2003;112:423–431. doi: 10.1172/JCI17725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deadwyler SA, Hampson RE, Bennett BA, Edwards TA, Mu J, Pacheco MA, Ward SJ, Childers SR. Cannabinoids modulate potassium current in cultured hippocampal neurons. Receptors Channels. 1993;1:121–134. [PubMed] [Google Scholar]
- Di Marzo V, Matias I. Endocannabinoid control of food intake and energy balance. Nat Neurosci. 2005;8:585–589. doi: 10.1038/nn1457. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Goparaju SK, Wang L, Liu J, Batkai S, Jarai Z, Fezza F, Miura GI, Palmiter RD, Sugiura T, Kunos G. Leptin-regulated endocannabinoids are involved in maintaining food intake. Nature. 2001;410:822–825. doi: 10.1038/35071088. [DOI] [PubMed] [Google Scholar]
- Eggermann E, Bayer L, Serafin M, Saint-Mleux B, Bernheim L, Machard D, Jones BE, Muhlethaler M. The wake-promoting hypocretin/orexin neurons are in an intrinsic state of membrane depolarization. J Neurosci. 2003;23:1557–1562. doi: 10.1523/JNEUROSCI.23-05-01557.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias CF, Saper CB, Maratos-Flier E, Tritos NA, Lee C, Kelly J, Tatro JB, Hoffman GE, Ollmann MM, Barsh GS, Sakurai T, Yanagisawa M, Elmquist JK. Chemically defined projections linking the mediobasal hypothalamus and the lateral hypothalamic area. J Comp Neurol. 1998;402:442–459. [PubMed] [Google Scholar]
- Elias CF, Lee CE, Kelly JF, Ahima RS, Kuhar M, Saper CB, Elmquist JK. Characterization of CART neurons in the rat and human hypothalamus. J Comp Neurol. 2001;432:1–19. doi: 10.1002/cne.1085. [DOI] [PubMed] [Google Scholar]
- Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev. 2003;83:1017–1066. doi: 10.1152/physrev.00004.2003. [DOI] [PubMed] [Google Scholar]
- Gao XB, van den Pol AN. Melanin concentrating hormone depresses synaptic activity of glutamate and GABA neurons from rat lateral hypothalamus. J Physiol (Lond) 2001;533:237–252. doi: 10.1111/j.1469-7793.2001.0237b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao XB, Ghosh PK, van den Pol AN. Neurons synthesizing melanin-concentrating hormone identified by selective reporter gene expression after transfection in vitro. J Neurophysiol. 2003;90:3978–3985. doi: 10.1152/jn.00593.2003. [DOI] [PubMed] [Google Scholar]
- Georgescu D, Zachariou V, Barrot M, Mieda M, Willie JT, Eisch AJ, Yanagisawa M, Nestler EJ, DiLeone RJ. Involvement of the lateral hypothalamic peptide orexin in morphine dependence and withdrawal. J Neurosci. 2003;23:3106–3111. doi: 10.1523/JNEUROSCI.23-08-03106.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagan JJ, Leslie RA, Patel S, Evans ML, Wattam TA, Holmes S, Benham CD, Taylor SG, Routledge C, Hemmati P, Munton RP, Ashmeade TE, Shah AS, Hatcher JP, Hatcher PD, Jones DN, Smith MI, Piper DC, Hunter AJ, Porter RA, et al. Orexin A activates locus coeruleus cell firing and increases arousal in the rat. Proc Natl Acad Sci USA. 1999;96:10911–10916. doi: 10.1073/pnas.96.19.10911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao S, Avraham Y, Mechoulam R, Berry EM. Low dose anandamide affects food intake, cognitive function, neurotransmitter and corticosterone levels in diet-restricted mice. Eur J Pharmacol. 2000;392:147–156. doi: 10.1016/s0014-2999(00)00059-5. [DOI] [PubMed] [Google Scholar]
- Harrold JA. Hypothalamic control of energy balance. Curr Drug Targets. 2004;5:207–219. doi: 10.2174/1389450043490460. [DOI] [PubMed] [Google Scholar]
- Hentges ST, Low MJ, Williams JT. Differential regulation of synaptic inputs by constitutively released endocannabinoids and exogenous cannabinoids. J Neurosci. 2005;25:9746–9751. doi: 10.1523/JNEUROSCI.2769-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR, Rice KC. Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J Neurosci. 1991;11:563–583. doi: 10.1523/JNEUROSCI.11-02-00563.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath TL, Diano S, van den Pol AN. Synaptic interaction between hypocretin (orexin) and neuropeptide Y cells in the rodent and primate hypothalamus: a novel circuit implicated in metabolic and endocrine regulations. J Neurosci. 1999;19:1072–1087. doi: 10.1523/JNEUROSCI.19-03-01072.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamshidi N, Taylor DA. Anandamide administration into the ventromedial hypothalamus stimulates appetite in rats. Br J Pharmacol. 2001;134:1151–1154. doi: 10.1038/sj.bjp.0704379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo YH, Chen YJ, Chua SC, Jr, Talmage DA, Role LW. Integration of endocannabinoid and leptin signaling in an appetite-related neural circuit. Neuron. 2005;48:1055–1066. doi: 10.1016/j.neuron.2005.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I, Sperlagh B, Sik A, Kafalvi A, Vizi ES, Mackie K, Freund TF. Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci. 1999;19:4544–4558. doi: 10.1523/JNEUROSCI.19-11-04544.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I, Rancz EA, Acsady L, Ledent C, Mackie K, Hajos N, Freund TF. Distribution of CB1 cannabinoid receptors in the amygdala and their role in the control of GABAergic transmission. J Neurosci. 2001;21:9506–9518. doi: 10.1523/JNEUROSCI.21-23-09506.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkham TC, Williams CM, Fezza F, Di Marzo V. Endocannabinoid levels in rat limbic forebrain and hypothalamus in relation to fasting, feeding and satiation: stimulation of eating by 2-arachidonoyl glycerol. Br J Pharmacol. 2002;136:550–557. doi: 10.1038/sj.bjp.0704767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG. Cerebellar depolarization-induced suppression of inhibition is mediated by endogenous cannabinoids. J Neurosci. 2001;21:RC174. doi: 10.1523/JNEUROSCI.21-20-j0005.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG. Retrograde signaling by endocannabinoids. Curr Opin Neurobiol. 2002;12:324–330. doi: 10.1016/s0959-4388(02)00328-8. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Carter AG, Regehr WG. Inhibition of interneuron firing extends the spread of endocannabinoid signaling in the cerebellum. Neuron. 2002;34:787–796. doi: 10.1016/s0896-6273(02)00695-5. [DOI] [PubMed] [Google Scholar]
- Leibowitz SF, Wortley KE. Hypothalamic control of energy balance: different peptides, different functions. Peptides. 2004;25:473–504. doi: 10.1016/j.peptides.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Li Y, van den Pol AN. Differential target-dependent actions of coexpressed inhibitory dynorphin and excitatory hypocretin/orexin neuropeptides. J Neurosci. 2006;26:13037–13047. doi: 10.1523/JNEUROSCI.3380-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Gao XB, Sakurai T, van den Pol AN. Hypocretin/orexin excites hypocretin neurons via a local glutamate neuron-A potential mechanism for orchestrating the hypothalamic arousal system. Neuron. 2002;36:1169–1181. doi: 10.1016/s0896-6273(02)01132-7. [DOI] [PubMed] [Google Scholar]
- Lin L, Faraco J, Li H, Kadotani R, Rogers W, Lin X, Qui X, deJong PJ, Nishino S, Mignot E. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell. 1999;98:365–376. doi: 10.1016/s0092-8674(00)81965-0. [DOI] [PubMed] [Google Scholar]
- Manabe T, Wyllie DJ, Perkel DJ, Nicoll RA. Modulation of synaptic transmission and long-term potentiation: effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J Neurophysiol. 1993;70:1451–1459. doi: 10.1152/jn.1993.70.4.1451. [DOI] [PubMed] [Google Scholar]
- Marsh DJ, Weingarth DT, Novi DE, Chen HY, Trumbauer ME, Chen AS, Guan XM, Jiang MM, Feng Y, Camacho RE, Shen Z, Frazier EG, Yu H, Metzger JM, Kuca SJ, Shearman LP, Gopal-Truter S, MacNeil DJ, Strack AM, MacIntyre DE, et al. Melanin-concentrating hormone 1 receptor-deficient mice are lean, hyperactive, and hyperphagic and have altered metabolism. Proc Natl Acad Sci USA. 2002;99:3240–3245. doi: 10.1073/pnas.052706899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- Matsuda LA, Bonner TI, Lolait SJ. Localization of cannabinoid receptor mRNA in rat brain. J Comp Neurol. 1993;327:535–550. doi: 10.1002/cne.903270406. [DOI] [PubMed] [Google Scholar]
- Melis M, Pistis M, Perra S, Muntoni AL, Pillolla G, Gessa L. Endocannabinoids mediate presynaptic inhibition of glutamatergic transmission in rat ventral tegmental area dopamine neurons through activation of CB1 receptors. J Neurosci. 2004;24:53–62. doi: 10.1523/JNEUROSCI.4503-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagotto U, Vicennati V, Pasquali R. The endocannabinoid system and the treatment of obesity. Ann Med. 2005;37:270–275. doi: 10.1080/07853890510037419. [DOI] [PubMed] [Google Scholar]
- Peyron C, Faraco J, Rogers W, Ripley B, Overeem S, Charnay Y, Nevsimalova S, Aldrich M, Reynolds D, Albin R, Li R, Hungs M, Pedrazzoli M, Padi-garu M, Kucherlapati M, Fan J, Maki R, Lammers GJ, Bouras C, Kucherlapati R, et al. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med. 2000;6:991–997. doi: 10.1038/79690. [DOI] [PubMed] [Google Scholar]
- Piomelli D. The endocannabinoid system: a drug discovery perspective. Curr Opin Investig Drugs. 2005;6:672–679. [PubMed] [Google Scholar]
- Qu D, Ludwig DS, Gammeltoft S, Piper M, Pelleymounter MA, Cullen MJ, Mathes WF, Przypek J, Kanarek R, Maratos-Flier E. A role of melanin-concentrating hormone in the central regulation of feeding behavior. Nature. 1996;380:243–247. doi: 10.1038/380243a0. [DOI] [PubMed] [Google Scholar]
- Robbe D, Alonso G, Duchamp F, Bockaert J, Manzoni OJ. Localization and mechanisms of action of cannabinoid receptors at the glutamatergic synapses of the mouse nucleus accumbens. J Neurosci. 2001;21:109–116. doi: 10.1523/JNEUROSCI.21-01-00109.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravinet Trillou C, Delgorge C, Menet C, Arnone M, Soubrie P. CB1 cannabinoid receptor knockout in mice leads to leanness, resistance to diet-induced obesity and enhanced leptin sensitivity. Int J Obes Relat Metab Disord. 2004;28:640–648. doi: 10.1038/sj.ijo.0802583. [DOI] [PubMed] [Google Scholar]
- Rosin DL, Weston MC, Sevigny CP, Stornetta RL, Guyenet PG. Hypothalamic orexin (hypocretin) neurons express vesicular glutamate transporters VGLUT1 or VGLUT2. J Comp Neurol. 2003;465:593–603. doi: 10.1002/cne.10860. [DOI] [PubMed] [Google Scholar]
- Schweitzer P. Cannabinoids decrease the K+ M-current in hippocampal CA1 neurons. J Neurosci. 2000;20:51–58. doi: 10.1523/JNEUROSCI.20-01-00051.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharkey KA, Pittman QJ. Central and peripheral signaling mechanisms involved in endocannabinoid regulation of feeding: a perspective on the munchies. Sci STKE. 2005;277:pe15. doi: 10.1126/stke.2772005pe15. [DOI] [PubMed] [Google Scholar]
- Shimada M, Tritos NA, Lowell BB, Flier LS, Maratos-Flier E. Mice lacking melanin-concentrating hormone are hypophagic and lean. Nature. 1998;396:670–674. doi: 10.1038/25341. [DOI] [PubMed] [Google Scholar]
- Simiand J, Keane M, Keane PE, Soubrie P. SR 141716, a CB1 cannabinoid receptor antagonist, selectively reduces sweet food intake in marmoset. Behav Pharmacol. 1998;9:179–181. [PubMed] [Google Scholar]
- Tamamaki N, Yanagawa Y, Tomioka R, Miyazaki J, Obata K, Kaneko T. Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67-GFP knock-in mouse. J Comp Neurol. 2003;467:60–79. doi: 10.1002/cne.10905. [DOI] [PubMed] [Google Scholar]
- Thannickal TC, Moore RY, Nienhuis R, Ramanathan L, Gulyani S, Aldrich M, Cornford M, Siegel JM. Reduced number of hypocretin neurons in human narcolepsy. Neuron. 2000;27:469–474. doi: 10.1016/s0896-6273(00)00058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou K, Brown S, Sanudo-Pena MC, Mackie K, Walker JM. Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience. 1998;83:393–411. doi: 10.1016/s0306-4522(97)00436-3. [DOI] [PubMed] [Google Scholar]
- Twitchell W, Brown S, Mackie K. Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J Neurophysiol. 1997;78:43–50. doi: 10.1152/jn.1997.78.1.43. [DOI] [PubMed] [Google Scholar]
- van den Pol AN, Acuna-Goycolea C, Clark KR, Ghosh PK. Physiological properties of hypothalamic MCH neurons identified with selective expression of reporter gene after recombinant virus infection. Neuron. 2004;42:635–652. doi: 10.1016/s0896-6273(04)00251-x. [DOI] [PubMed] [Google Scholar]
- Williams CM, Kirkham TC. Anandamide induces overeating: mediation by central cannabinoid (CB1) receptors. Psychopharmacology (Berl) 1999;143:315–317. doi: 10.1007/s002130050953. [DOI] [PubMed] [Google Scholar]
- Williams G, Bing C, Cai XJ, Harrold JA, King PJ, Liu XH. The hypothalamus and the control of energy homeostasis: different circuits, different purposes. Physiol Behav. 2001;74:683–701. doi: 10.1016/s0031-9384(01)00612-6. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endocannabinoid signaling in the brain. Science. 2002;296:678–682. doi: 10.1126/science.1063545. [DOI] [PubMed] [Google Scholar]
- Yamanaka A, Beuckmann CT, Willie JT, Hara J, Tsujino N, Mieda M, Tominaga M, Yagami K, Sugiyama F, Goto K, Yanagisawa M, Sakurai T. Hypothalamic orexin neurons regulate arousal according to energy balance in mice. Neuron. 2003;38:701–713. doi: 10.1016/s0896-6273(03)00331-3. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Hashimoto K, Zimmer A, Maejima T, Araishi K, Kano M. The cannabinoid CB1 receptor mediates retrograde signals for depolarization-induced suppression of inhibition in cerebellar Purkinje cells. J Neurosci. 2002;22:1690–1697. doi: 10.1523/JNEUROSCI.22-05-01690.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu PJ, Lovinger DM. Retrograde endocannabinoid signaling in a postsynaptic neuron/synaptic bouton preparation from basolateral amygdala. J Neurosci. 2005;25:6199–6207. doi: 10.1523/JNEUROSCI.1148-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]