

Abstract
Osteopontin (OPN) is expressed in many tissues during inflammatory responses. After spinal cord injury, microglia expresses OPN at the site of injury during the early to subacute stages. However, the function of OPN in spinal cord injury is not well understood. This study examines the responses of OPN knock-out (KO) and wild-type (WT) mice to spinal cord contusion injury. KO and WT mice were injured with a modified New York University impactor. Weights of 10 or 5.6 g were dropped 6.25 mm onto the T13 spinal cord under isoflurane anesthesia. At 24 h, homogenized spinal cords were analyzed for total potassium concentration to estimate lesion volumes. Expression of apoptotic genes, proinflammatory cytokines, and nerve growth factors was measured by reverse transcription (RT)-PCR and Western blot. In a series of animals, locomotor recovery was assessed with the Basso mouse scale (BMS) for 6 weeks, and histological analyses was performed to determine tissue preservation. Lesion volume showed no significant differences between KO and WT mice at 24 h. RT-PCR indicated that KO mice had significantly less Bcl-2, tumor necrosis factor-α, interleukin-1β, and interleukin-6 mRNA compared with WT controls. Western blot also showed that KO had significantly less Bcl-2 7 d after spinal cord injury. KO mice had significantly worse BMS locomotor scores than WT at 6 weeks. KO mice also had a significantly reduced area of spared white matter and fewer neuronal-specific nuclear protein-positive neurons in the spinal cord surrounding the impact site. This result supports a potential neuroprotective role for OPN in the inflammatory response to spinal cord injury.
Keywords: osteopontin, BMS, NYU impactor, Bcl-2, knock-out mice, spinal cord injury
Introduction
Osteopontin (OPN) is a widely expressed cytokine and cell adhesion protein that binds to integrins and CD44 variants on the cell surface (Denhardt et al., 2001). It is routinely expressed by injured tissues, for example during myocardial infarction (Komatsubara et al., 2003), autoimmune disease (Petrow et al., 2000), cancer metastasis (Rittling and Chambers, 2004), infectious disease (Nau et al., 2000), stroke (Ellison et al., 1998), and spinal cord injury (Hashimoto et al., 2003). Injury dramatically upregulates OPN in spinal cord microglia by 3 d. OPN upregulation correlates with microglial number and may help attract and stimulate astrocytes around the injury site (Hashimoto et al., 2003). OPN expression after spinal cord injury correlates with cell infiltration after injury. Thus, OPN may accelerate the inflammatory cascade and may create a nonpermissive environment for axonal regeneration.
Studies of OPN knock-out (KO) mice suggest that OPN plays a major role in attracting inflammatory cells to the injury site. For example, OPN-deficient mice show markedly less macrophage infiltration in ischemic kidneys and an enhanced rate of apoptosis during the injury phase of acute renal failure (Persy et al., 2003); they also have reduced tolerance to infectious disease (O'Regan et al., 2001).
OPN expression aggravates autoimmune conditions. OPN-deficient mice are resistant to progressive experimental autoimmune encephalomyelitis (EAE) and have more frequent remissions (Chabas et al., 2001). Myelin-reactive T-cells in OPN −/− mice produce more anti-inflammatory interleukin-10 (IL-10) and less interferon-γ than OPN +/+ mice. OPN −/− mice recover more rapidly without spontaneous relapse in EAE (Jansson et al., 2002). In a rheumatoid arthritis model, the deficiency in OPN protects joints against destruction in anti-type II collagen antibody-induced arthritis in mice (Yumoto et al., 2002). In these autoimmune diseases, the presence of OPN appears to contribute to inflammatory damage of tissues, perhaps by supporting the survival of inflammatory cells.
OPN expression differs considerably between CNS and PNS injuries. Macrophages at crush lesions of the optic nerve strongly express OPN, whereas macrophages in injured sciatic nerve do not, suggesting fundamental differences in the molecular programming of macrophages in these two systems (Kury et al., 2004). OPN expression in the CNS may be related to inability of the CNS to regenerate.
OPN may exacerbate tissue damage after spinal cord injury as it appears to do in autoimmune disease. We hypothesized that the OPN knock-out mice should have less inflammation and tissue damage and better neurological recovery than wild-type (WT) mice. This is consistent with the fact that suppression of inflammation and secondary tissue damage is a clinically effective treatment.
To determine whether OPN expression plays a role in spinal cord injury, we compared gene expression, lesion volume, white and gray matter sparing, and locomotor recovery of OPN knock-out and wild-type mice after a standardized spinal cord contusion injury. We modified an impactor used to produce rat spinal cord contusion injury (Beattie et al., 1997) to produce a suitable injury in mice. This is the first systematic analysis of these outcome measures in a standardized mouse contusion model.
Materials and Methods
Table 1 shows a time line of the present study to identify different procedures to compare KO and WT mice.
Table 1.
Scheme of present study
| 5.6 g rod, 6.25 mm injury | 10 g rod, 6.25 mm injury | |
|---|---|---|
| 24 h | Western blot | Lesion volume, Q-RT-PCR |
| 2 d | BMS | |
| 7 d | BMS, Western blot, immunohistochemistry | |
| 14 d | BMS | |
| 21 d | BMS | |
| 28 d | BMS, corticospinal tract tracing | |
| 35 d | BMS | |
| 42 d | BMS, NeuN cell counts, white matter sparing |
Two types of rods (10 or 5.6g) were used for inflicting spinal cord injury. All experiments were compared between KO and WT mice.
Animal model of spinal cord injury in mice.
We used adult (13–16 weeks old) male 129 strain mice, 49 OPN KO mice, and 49 WT mice for the experiments. The generation and maintenance of the OPN-deficient mice have been described previously (Rittling et al., 1998). All animal use protocols were approved by the Institutional Animal Care and Use Committee of Rutgers University.
Spinal cord contusion injuries were inflicted using a modified impactor as described previously (Young, 2002). The mice were deeply anesthetized by 2% isoflurane in 0.5 L/min oxygen and contused by a 5.6 or 10 g rod with a 1-mm-diameter impact head, dropped a distance of 6.25 mm onto T13 spinal cord exposed by a T10 laminectomy. After injury, the wound was closed with stainless steel clips. Parameter of the impactor was described in supplemental Materials and Methods (available at www.jneurosci.org as supplemental material).
Twenty-four hour lesion volume.
At 24 h after spinal cord injury (10 g rod, 6.25 mm height), the mice were deeply anesthetized by an intraperitoneal dose of pentobarbital (80 mg/kg; Abbott Laboratories, North Chicago, IL) and decapitated. Spinal cord injury sites (1 cm) were rapidly removed, weighed to obtain the wet weight, and homogenized in Trizol (Invitrogen, Carlsbad, CA) to preserve RNA. Trizol contains sodium but no potassium. We measured tissue potassium concentration by atomic absorption spectroscopy to estimate the cell volume fraction and calculate spinal cord lesion volume (Kwo et al., 1989; Constantini and Young, 1994) (supplemental Materials and Methods, available at www.jneurosci.org as supplemental material).
Quantitative reverse transcription-PCR.
Because KO mice had less gray and white matter sparing 6 weeks after spinal cord injury, we examined several cell death-related genes (Bcl-2, Bcl-XL, caspase-3, erythropoietin, and inducible nitric oxide synthase), pro-inflammatory cytokines that can trigger cell death [tumor necrosis factor-α (TNF-α), IL-1β, and IL-6], and nerve growth factors [BDNF, glial cell line-derived neurotrophic factor (GDNF), NGF-β, and neurotrophin-3].
Primers were designed using PrimerExpress software (Applied Biosystems, Foster City, CA) from the same GenBank accession records (Table 2). Levels of quantitative reverse-transcription (Q-RT)-PCR product were measured using SYBR Green fluorescence during real-time PCR on an Applied Biosystems 7900HT. A control cDNA dilution series was created for each gene to establish a standard curve, and all results were within the standard curve range. Each reaction was subjected to melting point analysis to confirm single amplified products. Reactions were run in duplicate, and results were averaged. Glyceraldehyde-3-phosphate dehydrogenase was used to normalize the data to control for variations in the amount of input cDNA.
Table 2.
Oligonucleotide primers for Q-RT-PCR
| GenBank | Description | Forward | Reverse |
|---|---|---|---|
| U34963 | Bcl-xLong | CGTGGAAAGCGTAGACAAGGA | CTTGCAATCCGACTCACCAA |
| NM_016993.1 | Bcl-2 | CTGGGATGCCTTTGTGGAAC | TCAAACAGAGGTCGCATGCT |
| NM_017001 | Erythropoetin | GTCCCACCCTGCTGCTTTTA | GGAGGCCCAGAGGAATCAGTA |
| NM_008366 | IL-2 | GTCAACAGCGCACCCACTT | TGCTTCCGCTGTAGAGCTTG |
| NM_013693 | TNF-α | ATGCTGGGACAGTGACCTGG | CCTTGATGGTGGTGCATGAG |
| NM_008361 | IL-1β | CCAAAAGATGAAGGGCTGCT | TCATCAGGACAGCCCAGGTC |
| NM_031168 | IL-6 | TTCCATCCAGTTGCCTTCTTG | GAAGGCCGTGGTTGTCACC |
| L281117 | NF-κ B | ACGGATTTCCTCCCTACGGT | GATTTCGTGACTCGGGATGG |
| NM_009810 | caspase 3 | GGTTCATCCAGTCCCTTTGC | CTAGCTTGTGCGCGTACAGC |
| NM_010927 | NOS | TGACGGCAAACATGACTTCAG | GCGTACCGGATGAGCTGTG |
| NM_007540 | BDNF | AGGCACTGGAACTCGCAATG | AAGGGCCCGAACATACGATT |
| NM_010275 | GDNF | AAGACCCACGTTTCGCATG | TCCACTTCCCAGTCCTGCAG |
| NM_013609 | NGF-β | GGCAGCTTTTTGGAAACTCCT | TTCACTGGCTTGAGGCACAG |
| NM_008742 | NT-3 | TCCTCAGCCATTGACATTCG | GATCTCCCCCAGCACTGTGA |
| NM_017008.1 | GAPDH | AACTCCCTCAAGATTGTCAGCAA | GGCTAAGCAGTTGGTGGTGC |
Western blot.
At 24 h and 7 d after injury (5.6 g rod, 6.25 mm height), spinal cord samples were homogenized in ice-cold lysis buffer containing 50 mm Tris-HCl, pH 7.6, 150 mm NaCl, 1 mm EDTA, 1% NP-40, and 0.5% Triton X-100, plus a protein inhibitor tablet (Roche Diagnosis, Lewes, UK). Homogenates were centrifuged (15,000 rpm for 10 min), and supernatants were measured using the Bradford reagent (Bio-Rad, Hercules, CA). Aliquots of equal amounts of total protein (50 μg) were electrophoresed on 12% SDS-polyacrylamide gels and transferred onto polyvinylidene difluoride membranes (Hybond-P; Amersham Biosciences, Uppsala, Sweden). The membranes were blocked for 1 h in 5% skim milk and incubated with primary antibodies: rabbit anti-IL-6 (1:500; Millipore, Temecula, CA), rabbit anti-Bcl-2 (1:1500; Millipore), rat anti-rat/mouse TNF-α (1:1000; BD Biosciences, Franklin Lakes, NJ), rabbit anti-mouse IL-1β (1:200; Research Diagnostics, Flanders, NJ), mouse anti-actin (Ab-1, 1:2000; EMD Biosciences, San Diego, CA), and mouse anti-α-tubulin (1:1000; Invitrogen, Carlsbad, CA) for 4°C overnight or 1 h at room temperature. The membranes were washed four times and incubated with secondary antibodies (1:2000, ECL horse radish peroxidase linked whole antibody; Amersham Biosciences) for 45 min. Bands were developed using the ECL+ kit (Amersham Biosciences) and digitalized by Molecular Dynamics (Sunnyvale, CA) Storm 840 version 4.1 and ImageQuant 5.0 (Amersham Biosciences Piscataway, NJ). Band densities were measured by NIH Image software and normalized by actin or α-tubulin and compared between KO and WT mice.
Histology.
After anesthesia with pentobarbital, the mice were transcardially perfused with 4% paraformaldehyde in 0.1 m PBS at 7, 35, and 42 d after injury. Tissue blocks containing lesion center were collected, postfixed in the same fixative overnight at 4°C, and cryoprotected in 20% sucrose in PBS at 4°C for 24 h. Spinal cords were freeze mounted on a holder, cut into 30 μm sections, and placed on poly-l-lysine-coated glass slides. For midsagittal sections, sequential sections of the lesion center (1 cm) were mounted on five slides. Each slide had six to eight sections spaced at 150 μm in each sequential section. For cross sections of the spinal cord, a 3.0-mm-long spinal cord containing the lesion center was dissected and freeze mounted on a holder. Sequential sections were mounted on seven slides with 20–23 sections per slide, spaced in 210 μm intervals.
White matter staining.
One slide per animal was stained with luxol fast blue for myelin. To identify the lesion center, we selected the section containing the least amount of luxol fast blue stain and then assessed that section plus five sections rostral and caudal, a total of 11 sequential sections spanning 2100 μm of spinal cord length. The percentage of spared white matter was calculated from pixels of spared white matter area divided by pixels of total cross-sectional area using Photoshop 5.5 software (Adobe Systems, San Jose, CA). To determine whether OPN affected sparing of white matter, we compared absolute spared white matter pixels for luxol fast blue stain in each of the 11 sections from KO and WT mice. To determine whether the OPN affected spinal cord matter, we compared pixels of total cross section areas.
We also compared uninjured spinal cord, a total of 11 sequential sections spanning 2100 μm of spinal cord length centered at the T13 spinal cord level. Percentage and absolute pixels of white matter were compared between KO and WT mice. Pixels of total cross-sectional areas (absolute spinal cord pixels) were compared within injured KO (KO injury), injured WT (WT injury), uninjured KO (KO normal), and uninjured WT (WT normal) mice.
Immunohistochemistry.
For immunohistochemical stains, we first placed the slides on a slide warmer for 3 h, rinsed the sections with PBS, and blocked the sections with 10% normal goat serum in 0.1 m PBS containing 0.3% Triton X-100 for 2 h at room temperature. Sections were then incubated with anti-neuronal-specific nuclear protein (NeuN) (1:400; Millipore) to stain neurons, rat anti-CD11b (Mac-1; 1:400; Serotec, Oxford, UK) to stain macrophage and microglia, rat anti-CD3 (1:400; Serotec) to stain T-cells, and polyclonal rabbit anti-glial fibrillary acidic protein (GFAP) (1:400; DakoCytomation, Carpinteria, CA) to stain astrocytes at 4°C overnight. After rinsing with PBS, the sections were incubated with biotinylated goat anti-mouse IgG (1:200; Vector Laboratories, Burlingame, CA), Alexa Fluor 488 goat anti-rat IgG (1:800; Invitrogen), and Alexa Fluor 488 goat anti-rabbit IgG (1:800; Invitrogen) for 1 h at room temperature, followed by Vectastain Elite ABC kit (Vector Laboratories) using the protocol of the manufacturer except for the fluorescent secondary antibody. Color was developed by DAB (Vector Laboratories) for 2 min.
NeuN-positive cell counts.
For NeuN staining, a cross-sectional slide from each animal was used. Each section was tile scanned by Axiovert 200 (Zeiss, Thornwood, NY), recorded by AxioVision Release 4.3, and printed out to a letter size paper. All of the NeuN-positive cells of gray matter in one cross section were counted blind. The section that had fewest number of NeuN-positive cells was designated the lesion epicenter and five rostral and five caudal sections in intervals of 210 μm, spanning 2100 μm length of spinal cord centered on injury epicenter. Total neuron counts for each group and for each spinal cord level were compared between KO (KO injury) and WT (WT injury). Uninjured T10 spinal cord sections of KO and WT mice were also stained and tile scanned as above (KO normal and WT normal). Because the staining was more uniform in the uninjured cord than in the injured spinal cord, NeuN-positive cells of gray matter from the uninjured animals were counted by Scion (Frederick, MD) Image.
Quantitative analysis of Mac-1-, CD3-, and GFAP-positive area.
Inflammatory or immune cell accumulation was assessed in spinal cords from KO and WT mice at 7 d after spinal cord contusion. Fluorescent immunohistochemistry of Mac-1, CD3, and GFAP immunoreactivity was assessed in midsagittal sections (Okada et al., 2004). In the case of Mac-1 or CD3 staining, sequential five to six sections per animal were stained as above, and fluorescent signals were captured and tile scanned by Axiovert 200 (AxioVision Release 4.3). For GFAP staining, sequential five sections per animal were stained, and fluorescent signals were captured and tile scanned by Zeiss 510 confocal laser scanning microscope (LSM). Pictures were reopened by LSM510 Workstation (Zeiss), and 1.8 mm length spinal cords for Mac-1 and CD3 or 2.5 mm length spinal cords for GFAP were delineated and extracted. Immunolabeled areas (in square millimeters) were measured and digitalized by using threshold for each antibody (Mac-1, 56; CD3, 98; GFAP, 49). In each animal, five to six sequential sections were averaged and compared between KO and WT mice.
Behavioral analysis.
Motor function of the hindlimbs was evaluated by the locomotor rating test on the Basso mouse scale (BMS) (Engesser-Cesar et al., 2005). This scale was developed specifically for mice because the characteristics of locomotor recovery are different in mice than in rats. Briefly, the numeric ranking system is as follows: 0, no ankle movement; 1–2, slight or extensive ankle movement; 3, planter placing or dorsal stepping; 4, occasional planter stepping; 5, frequent or consistent plantar stepping. There was no animal >6. Subscore was scored if mouse can do frequent plantar stepping. In this study, subscores of all of the animals that can do planter stepping were 0, because they did not have consistent, coordinated, paralleled stepping in addition to tail or trunk instability. More than two examiners participated in all open field tests and were positioned across from each other to observe both sides of mice. Each mouse was assessed for 4 min, and, if the two observers had different scores, then after discussion the lower score was adopted for record. If score was different for the right and left legs, the average was calculated. Three mice have been omitted from this study because leg or legs were contracted. We measured BMS at 2, 7, 14, 21, 28, 35, and 42 d after injury.
Corticospinal tract labeling.
After 4 weeks of recovery from spinal cord injury, two mice (one KO and one WT) were deeply anesthetized by 2% isoflurane in 0.5 L/min oxygen and placed in the stereotaxic device. The left skull over the intended injection sites was first drilled and enlarged with rongeurs around the motor cortex of the left side especially for lower extremities using a surgical microscope (RMS-9210; Zeiss). Dextran amine-conjugated Texas Red (20%, 0.5 μl, molecular weight of 3000; Invitrogen) was injected at the sensorimotor cortex at 10 sites through a 1 μl Hamilton syringe (Hamilton Company, Reno, NV) with a glass tip. After surgery, the mice were housed singly and injected antibiotics for 3 d as describe above. Animals were perfused after 7 d of operation. For triple staining of fluorescent immunohistochemistry, anti-Mac-1 (1:400; Serotec) and GFAP (1:400; DakoCytomation) antibodies were applied for the first antibodies as describe above. Alexa Fluor 488 goat anti-rat IgG (1:800; Invitrogen) and goat anti-rabbit Cy5 (Jackson ImmunoResearch, West Grove, PA) were used for secondary antibody. Representative triple staining was derived by Zeiss 510 confocal LSM.
Statistical analysis.
Change between KO and WT was determined to be significant by Student's t test using a p value of <0.05. In BMS score, NeuN-positive cells counts, and spared white matter, significances were calculated using repeated-measures ANOVA and a Scheffé's post hoc analysis using a p value of <0.05.
Results
Impactor parameters
We created a New York University (NYU) impactor to make a rat spinal cord injury 10 years ago. In the base of experience for technology and acquisition of data, we made a modified NYU impactor for mice. This machine has the sensor for the rod movement, and it is able to obtain impactor data for data collection as described in supplemental Materials and Methods (available at www.jneurosci.org as supplemental material). For data correlation, we always focus on ImpV, Cd, Ct, and Cr (Cd/Ct) (for explanations, refer to supplemental Materials and Methods, available at www.jneurosci.org as supplemental material). Table 3 summarizes the results obtained using the 10 g metal and 5.6 g plastic impactor rods to inflict the spinal cord contusion injury. The 10 g rod was used for lesion volume measurements and Q-RT-PCR study. The 5.6 g rod was used for the other experiments. The ImpV, Cd, and Ct values of the 5.6 g rod were smaller than those of 10 g rod.
Table 3.
Results of impactor parameter at a 6.25 mm height drop
| Rod weight | Rod material | n | ImpV (m/s) | Cd (mm) | Ct (ms) | Cr (m/s) |
|---|---|---|---|---|---|---|
| 10 g | Metal | 33 | 0.347 ± 0.001 | 1.74 ± 0.03 | 6.2 ± 0.09 | 0.281 ± 0.003 |
| 5.6 g | Plastic | 44 | 0.338 ± 0.002 | 1.32 ± 0.03 | 4.77 ± 0.17 | 0.282 ± 0.005 |
We used two types of rods made from different materials and weights. A 10 g weighted rod was used for lesion volume measurement, and a 5.6 g rod was used for the other experiments.
Quantitative study of cell loss 24 h after spinal cord injury
The potassium concentration at the impact site fell by 14–37% after injury. Because >95% of tissue potassium is intracellular, the [K]t is inversely proportional to lesion volume (Kwo et al., 1989; Constantini and Young., 1994). The potassium concentrations of the lesions in the KO and WT mice were 73.72 ± 0.91 and 73.92 ± 0.82 μmol/g, respectively (n = 15 in KO and n = 13 in WT), not statistically different from each other. Lesion volumes calculated from the formula described in supplemental Materials and Methods (available at www.jneurosci.org as supplemental material) were 4.58 ± 0.23 μl in the KO and 4.49 ± 0.29 μl in the WT spinal cord contusion sites, suggesting no significant lesion volume difference at 24 h after spinal cord injury.
Q-RT-PCR
Histological examination of the spinal cords 42 d after spinal cord injury revealed fewer neurons in KO spinal cords compared with WT cords, suggesting that neurons in KO mice may have been more vulnerable than WT neurons to spinal cord contusions. We therefore focused on cell death-related genes, proinflammatory cytokines, and nerve growth factors that could influence neuron survival (Table 2). Of 14 genes analyzed, contused KO spinal cords showed significant downregulation of expression of Bcl-2 and proinflammatory cytokines (TNF-α, IL-1 β, and IL-6) compared with WT (Table 4). Although KO mice showed downregulation of NGF-β and GDNF compared with WT contused spinal cords, t test results suggested that the p value was close to but not <0.05.
Table 4.
Q-RT-PCR results of apoptosis-related genes, proinflammatory cytokines, and nerve growth factor 24 h after spinal cord contusion injury
| GenBank | Description | Ratio of KO/WT | t test p value |
|---|---|---|---|
| U34963 | Bcl-xLong | 0.936 | 0.42 |
| NM_016993.1 | Bcl-2 | 0.697 | p < 0.05 |
| NM_017001 | Erythropoetin | 1.123 | 0.74 |
| NM_008366 | IL-2 | 0.727 | 0.45 |
| NM_013693 | TNF-α | 0.721 | p < 0.05 |
| NM_008361 | IL-1β | 0.569 | p < 0.05 |
| NM_031168 | IL-6 | 0.516 | p < 0.05 |
| L281117 | NF-κ B | 0.851 | 0.35 |
| NM_009810 | caspase 3 | 1.354 | 0.3 |
| NM_010927 | NOS | 1.111 | 0.68 |
| NM_007540 | BDNF | 1.118 | 0.18 |
| NM_010275 | GDNF | 0.693 | 0.0816 |
| NM_013609 | NGF-β | 0.64 | 0.0982 |
| NM_008742 | NT-3 | 1.012 | 0.95 |
Bcl-2, TNF-α, IL-1β, and IL-6 were significantly downregulated in KO by a t test (n = 5–6 in each group).
Western blot
RNA expression does not necessarily reflect protein synthesis or protein level changes. To confirm Q-RT-PCR data, we performed Western blot for Bcl-2, TNF-α, IL-1β, and IL-6 at 24 h and 7 d after injury. Although Bcl-2, IL-1 β, and IL-6 did not show significant difference between KO and WT mice at 24 h after injury (Fig. 1 A–C), membrane-bound TNF-α was upregulated at 24 h in KO spinal cord, contrary to the transcript level (Fig. 1 D, *p < 0.05). Conversely, TNF-α was no longer significantly upregulated after 7 d in the KO (Fig. 1 H, p = 0.5); IL-1beta and IL-6 continued to show no change in protein levels (Fig. 1 F,G). Only Bcl-2 protein was significantly downregulated in KO spinal cord at 7 d after injury (Fig. 1 E, *p < 0.05).
Figure 1.

Changes in protein levels in the injured spinal cord 24 h and 7 d after injury. To confirm Q-RT-PCR data at the protein level, we did Western blot for Bcl-2, IL-1 β, IL-6, and TNF-α at 24 h and 7 d after spinal cord injury. A–D, Protein levels 24 h after spinal cord injury. Although Bcl-2, IL-1 β, and IL-6 mRNA showed significant difference at 24 h after injury in Q-RT-PCR, there were no significant difference in protein level (p = 0.55, p = 0.26, and p = 0.32). Membrane-bound TNF-α was significantly upregulated in the KO mice at 24 h, contrary to Q-RT-PCR data (D, *p < 0.05; n = 3 in KO, n = 4 in WT). E–H, Protein levels 7 d after spinal cord injury. Although IL-1 β (F), IL-6 (G), and TNF-α (H) showed no statistical difference (p = 0.39, p = 0.99, and p = 0.52), Bcl-2 was significantly downregulated in KO 7 d after spinal cord injury (E, *p < 0.05; n = 4 in each group).
BMS locomotor scores
To determine whether OPN deficiency resulted in good or bad locomotor performance after spinal cord contusion injury, we measured BMS locomotor scores for 6 weeks. Expectedly, at 2 d after contusion, the BMS scores were low at 1.31 ± 0.25 in KO and 1.38 ± 0.18 in WT mice. These scores remained lower in KO than WT mice for up to 42 d (Fig. 2). Repeated-measures ANOVA followed by post hoc test showed that KO mice had significantly worse BMS scores than WT mice (p = 0.0332). Post hoc tests of each time point showed that the BMS scores differed significantly at 14 and 42 d after injury (p = 0.0446 and 0.0313, respectively). At 42 d after contusion, the BMS scores of KO mice ranged from 1.5 to 3.5 (average, 2.44 ± 0.24), which meant slight to extensive ankle movement to only occasional planter placing or dorsal stepping. Conversely, BMS scores of WT mice ranged from 1.5 to 4.5 (average, 3.38 ± 0.31). Except for one mouse with a BMS score of 1.5 at 42 d, seven mice achieved planter stepping in at least one leg and maximally frequently planter stepping, without coordination. Although the scores of WT mice continuously increased for 42 d after injury, the KO score of 2.94 ± 0.26 at 28 d after injury was decreased to 2.44 ± 0.24 at 42 d after injury.
Figure 2.

Locomotor recovery after spinal contusion injury in OPN-deficient and WT mice. The BMS scores in KO and WT mice (n = 8 per each group) were determined at 42 d after injury. The BMS scores were consistently greater in WT than KO for 42 d. Repeated-measures ANOVA followed by post hoc test showed that KO mice had significantly worse BMS score than WT (p < 0.05). Post hoc tests at each time point showed statistical significance at 14 and 42 d after injury (*p < 0.05). Although the scores of WT mice increased for 42 d, the scores of KO mice at 28 d were decreased at 42 d. Error bars illustrate the SEM.
White matter sparing
We assessed the amount of spared white matter at the contusion site 42 d after injury to compare KO with WT mice. Representative sections showed little white matter is residual both in KO and WT mice at the injury epicenter (Fig. 3 A,B). Repeated-measures ANOVA and post hoc tests showed no significant difference in the ratio of spared white matter areas between KO injury and WT injury (Fig. 3 C, p > 0.1). We also compared absolute spared white matter pixels (Fig. 3 D) between injured KO mice and injured WT mice. Post hoc tests at each spinal segment showed statistically significant differences at the caudal site (840 μm, p < 0.05; 1050 μm, p < 0.01).
Figure 3.
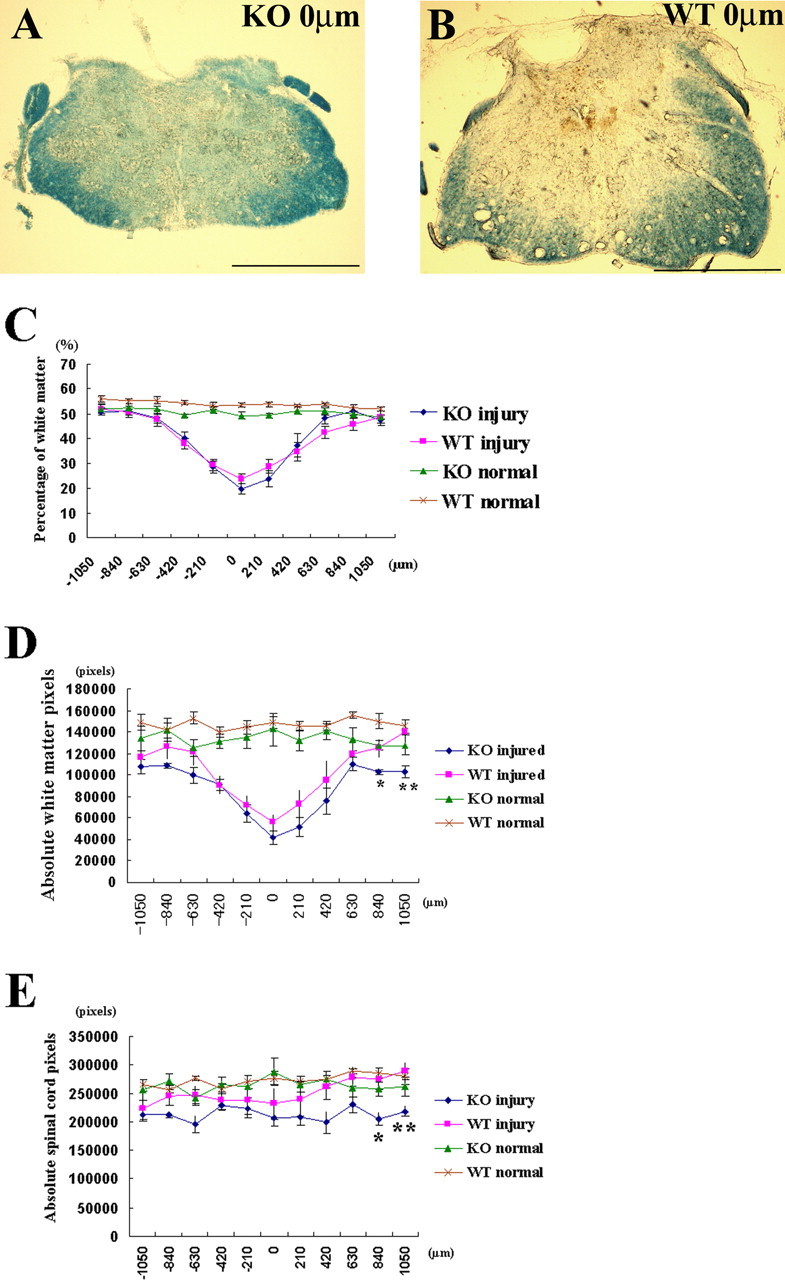
White matter sparing after spinal cord contusion injury in OPN-deficient and WT mice. A, B, Representative spinal cord sections after injury on the lesion epicenter show minimal sparing of white matter in both KO (A) and WT (B) mice. Scale bar, 500 μm. C, Ratio of sparing white matter for 2100 μm length centered on lesion epicenter was compared at 42 d after injury [n = 7 in KO injury (blue ♦) and n = 8 in WT injury (pink ■)]. Uninjured spinal cords of each genotype were also compared in KO (green ▴; n = 5) and WT (brown × symbols; n = 5). Although repeated-measures ANOVA followed by post hoc test showed no significant difference between KO injury and WT injury mice (p = 0.83), there was statistical significance between KO normal and WT normal in ratio of white matter (p = 0.0126). D, Absolute spared white matter pixels in injured animals and absolute white matter pixels in normal spinal cord are shown. Post hoc tests at each spinal segment showed the area of spared white matter was lesser at caudal sites of KO injury than WT injury. E, Absolute spinal cord pixels of KO injury, WT injury, KO normal, and WT normal were plotted at each spinal segment. Repeated-measures ANOVA followed by post hoc test showed statistical significance between KO injury and WT normal (p = 0.0459). Post hoc test for all four groups at each spinal segment showed significant difference between KO injury and WT injury at caudal site of spinal cord. *p < 0.05; **p < 0.01. Error bars illustrate SEM.
We compared uninjured, normal spinal cord between KO and WT. Repeated-measures ANOVA followed by post hoc test showed that the percentage of white matter (Fig. 3 C) as well as the absolute white matter pixels (Fig. 3 D) were both less in uninjured KO mice than uninjured WT mice (p = 0.0136, p = 0.0881 in each). These findings suggest that KO mice have less white matter area than WT mice before spinal cord injury.
To assess changes of spinal cord size before and after spinal cord injury, we compared absolute spinal cord pixels among injured KO mice, injured WT mice, uninjured KO mice, and uninjured WT mice (Fig. 3 E). Repeated-measures ANOVA followed by post hoc test showed significant difference between injured KO mice and uninjured WT mice (p = 0.0459). Post hoc tests comparing all four groups at each spinal segment revealed possible significant difference between injured KO mice and injured WT mice at caudal spinal cord sites (840 μm, p < 0.05; 1050 μm, p < 0.01). These results suggest that injury reduces significantly not only white matter size but also spinal cords sizes in KO compared with WT mice.
NeuN-positive cell counts
To assess numbers of neurons 42 d after spinal cord injury, we stained sections with NeuN to label neurons, counted the number of NeuN-positive neurons, and compared the average counts between KO and WT mice. Representative sections of the injury epicenter showed few spared neuron in both KO and WT mice (Fig. 4 A,B). However, at 630 μm caudal to the lesion epicenter, WT mice had more spared neurons than KO mice (Fig. 4 C,D). Post hoc tests at each spinal segment between injured KO and injured WT mice revealed that NeuN cell counts differed significantly at two caudal sites: +210 μm (p = 0.0267) and +630 μm (p = 0.0311) from lesion center (Fig. 4 E). The mean and SEMs of the NeuN counts at each spinal segment from KO and WT mice were 351 ± 93 and 480 ± 83 cells, respectively, 42 d after spinal cord injury. The cell count difference was, however, not statistically significant on a t test.
Figure 4.
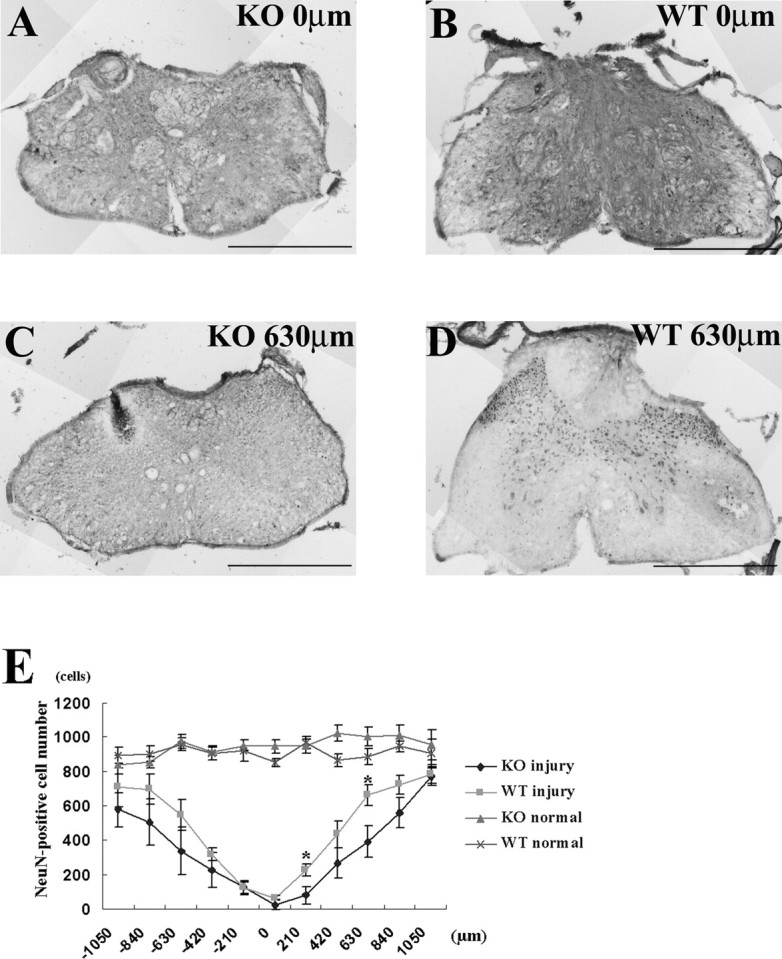
Neuron counts after spinal contusion injury in OPN-deficient and wild-type mice. NeuN-positive cells were counted for 11 sections (2100 μm length) centered on the lesion epicenter, averaged in each group, and compared between KO and WT mice. A, B, Representative sections of injury epicenter show only a few neurons were spared both in KO (A) and WT (B). C, D, At 630 μm caudal from the lesion center there were fewer spared neurons in the KO than in the WT. E, Normal spinal cords of each genotype were also compared (n = 5 in KO and n = 5 in WT). Repeated-measures ANOVA between KO injury and WT injury showed no statistically significant difference in neuron number (p = 0.09), but post hoc tests at each spinal segment showed significant differences between the KO and the WT at 210 and 630 μm caudal from the lesion epicenter (*p < 0.05; n = 7 in KO and n = 6 in WT). Error bars illustrate SEM. Scale bars: 500 μm.
We also counted NeuN-positive cells in uninjured KO and WT mice. Repeated-measures ANOVA and post hoc test between uninjured KO and WT mice revealed no differences around the T13 spinal cord level (2100 μm length centered around T13 spinal cord) (Fig. 4 E). The mean and SEMs of the NeuN counts from KO and WT mice were 946 ± 47 and 910 ± 45 cells, but the difference was not statistically significant on a t test.
Inflammatory and immune cells accumulation after spinal cord injury
At 7 d after spinal cord injury, we used immunohistochemical stain for Mac-1-positive cells and CD3-positive cells. Mac-1 immunoreactivity is higher in KO mice than WT mice, but there was no significant difference between KO and WT (p = 0.56) (data not shown). Conversely, CD-3 immunoreactivity is lighter in KO than WT, but there was no significant difference (p = 0.51) (data not shown).
GFAP-positive astroglial scar accumulation after spinal cord injury
To assess the relationship between OPN deficiency and astroglial scar formation, we measured astroglial scar area and compared between KO and WT mice 7 d after spinal cord injury. Although immunoreactivity of GFAP-positive area was higher in KO than WT, there was no significant difference (p = 0.28) (data not shown).
Corticospinal tract labeling
Although the BMS scores 4 weeks after injury showed no statistical difference between KO and WT mice, they did showed significant differences 6 weeks after injury (Fig. 2). We injected Texas Red-conjugated dextran amine into cerebral cortex at 4 weeks. Low-magnification image (10×) of triple staining of red (corticospinal tract), green (macrophages/microglia), and blue (astrocytes) showed orientation of these colors around the injury sites in KO mouse (Fig. 5 A) and WT mouse (Fig. 5 C). We were unable to see regenerative corticospinal tract or residual tract on the caudal side of GFAP staining area around lesion epicenter. High-magnification image showed that the distance between the caudal end of the corticospinal tract and GFAP staining area of KO (Fig. 5 B) was farther away than that of WT mouse (Fig. 5 D).
Figure 5.

Fluorescent immunohistochemistry at 5 weeks after injury illustrates the relationship between corticospinal tract tracing, macrophages/microglia, and GFAP astrocytes. A, C, Representative 10× triple-labeling images of KO (A) and WT (C) mouse. There were more Mac-1-positive macrophages/microglia and less GFAP-positive astrocytes in KO than that of WT mouse around the injury site. B, D, High-magnification images of A or C (25×) show that corticospinal tract halt before GFAP staining area (B) and in the GFAP staining area (D). There is no visible corticospinal tract staining in caudal spinal cord beyond GFAP staining in KO and WT mouse. Scale bars: A, C, 500 μm; B, D, 200 μm.
Discussion
Spinal cord contusion injury of mice
Several groups have reported spinal cord weight-drop contusion injuries of mice, dropping 1–3 g weights 25–50 mm heights (Kuhn and Wrathall, 1998; Okada et al., 2004). In the present study, we used 10 or 5.6 g rods dropped 6.25 mm. The potential energy (PE) of our model is comparable with those in these other models: PE (10 g rod) = mgh = 10 × g × 6.25 [where m is weight (in kilograms), g is G force (9.8m/s2), and h is height (in meters)] = 0.6125 × 10−3 J. PE (5.6 g rod) = 5.6 × g × 6.25 = 0.343 × 10−3 J.
The average of impact velocity (v) just before impact was 0.348 ± 0.001 m/s in our 24 h lesion volume study. The expected ideal velocity of 6.25 mm weight drop should be 0.350 m/s. The velocity deviated from the ideal by only 0.57%, i.e., (0.350–0.348)/0.348 × 100 = 0.57%. In our 33 impacts, only three were >2 SDs from the mean, but the velocity error of these three impactions was <5% of mean. The average kinetic energy (KE) of impact, i.e., KE = 1/2 mv2, was 0.6053 ± 0.0039 × 10−3 J. The difference between PE and KE (PE − KE) was 11.8% of PE [(PE − KE)/PE × 100], and this difference of energy was lost before impaction. The percentage of energy loss [(PE − KE)/PE × 100] in the 6.25 mm injury was higher than that of 12.5 mm to 50 mm injury in rat (our unpublished data).
The average lesion volume (calculated loss of intracellular volume) in contused mouse spinal cord was 4.67 μl. The lesion volumes produced by 6.25 mm contusions in rat was 6–7 μl (Young, 2000). We also measured spinal cord lesion volumes in mice after 12.5 mm injury but found no significant difference between 6.25 and 12.5 mm. One possible explanation is that the 6.25 mm weight drop is already producing close to the maximum cell loss at the contusion site, because the ratio of intracellular and tissue volume (Vi/Vt) was not significantly different between 6.25 and 12.5 mm injury.
Gene expression difference between KO and WT mice
We used Q-RT-PCR to assess apoptosis-related genes, proinflammatory cytokines, and nerve growth factors. Interestingly, downregulation of Bcl-2, TNF-α, IL-1β, and IL-6 in KO mice was statistically significant in Q-RT-PCR. Bcl-2 protein was still downregulated in KO spinal cords 7 d after spinal cord injury. Some laboratories have reported that OPN plays an important anti-apoptotic role in myeloma cells (Caers et al., 2006), dendritic cells (Kawamura et al., 2005), and cardiac fibroblasts (Zohar et al., 2004). OPN may regulate Bcl-2 upregulation and regulate neuron death after spinal cord injury.
Downregulation of TNF-α in the present study is supported by several studies reporting that OPN deficiency reduces TNF-α expression (Jansson et al., 2002; Morimoto et al., 2004). Similarly, OPN induces Th1-promoting TNF-α (Renkl et al., 2005). These studies support our finding that proinflammatory cytokines are downregulated in injured spinal cords of KO mice. Both TNF-α and IL-1 increase OPN mRNA in both clonal osteoblasts and primary osteoblast-like cells (Jin et al., 1990), suggesting that OPN has functions complementary to proinflammatory cytokines. The downregulation of proinflammatory cytokines in KO mice may result from a deficiency of interaction between OPN and TNF-α, IL-1β, or IL-6.
Another possible explanation of the lower cytokine expression is reduced number and activity of microglia/macrophages, which assemble at the injury site and secrete cytokines in KO mice compared with WT. Microglia and macrophages from injured spinal cord secrete TNF-α (Pearse et al., 2004). Macrophage density increases TNF-α, IL-1, and IL-6 expression (Leskovar et al., 2000). OPN deficiency reduces macrophages and CD4-positive T-cells, as well as TNF-α in hepatic granuloma (Morimoto et al., 2004). OPN is chemotactic for microglia/macrophages, and microglia/macrophages express high levels of OPN (Giachelli et al., 1998). The number of microglia/macrophages in injured spinal cord may have been partially responsible for the difference of gene expression in TNF-α, IL-1, or IL-6 in KO versus WT mice at 24 h after spinal cord injury. Interestingly, Mac-1-positive microglia/macrophages were more accumulated in KO than WT mice at 7 d after spinal cord injury. OPN deficiency may influence the phase shift of inflammatory cell accumulation after spinal cord injury.
Although Q-RT-PCR did not show statistical differences of neurotrophic factor gene expression between injured KO and WT mice, the data suggest that KO mice had less GDNF and NGF-β expression than WT. NGF increases osteopontin in human periodontal ligament cells (Kurihara et al., 2003). OPN also may have complementary function with NGF gene. Increases in the GDNF mRNA levels correlated with the restoration activity of locomotor function (Hashimoto et al., 2005b). Gene expression difference of neurotrophic factor between KO versus WT spinal cord may be partially related with tissue sparing or axonal regeneration.
Locomotor recovery and histology
In the present study, KO mice had less spared white matter measured in pixels than WT. This is one reason why OPN knock-out mice had lower BMS scores than in WT mice during the entire 6 weeks of observed recovery. Note that analysis of uninjured spinal cords indicated that KO mice had less white matter than WT. This is consistent with previous in vitro and in vivo studies indicating that OPN −/− mice contained significantly less MBP than WT mice and that OPN expression is upregulated during demyelination and remyelination (Selvaraju et al., 2004). OPN therefore may play a role in remyelination after spinal cord injury.
NeuN-positive cell counts indicated significant greater neuron loss in KO than WT mice. In addition to anti-apoptotic function as described, OPN improves survival of interstitial and tubular cells (Ophascharoensuk et al., 1999), endothelial cells (Khan et al., 2002), and vascular smooth muscle cells (Weintraub et al., 2000). The deficiency of OPN influenced neuronal loss in KO mice and impaired locomotor recovery after spinal cord injury.
Lower proinflammatory cytokines in OPN KO mice may explain the reason for impaired locomotor recovery than WT, because recombinant IL-1β, TNF-α, and IL-6 treatment of injured spinal cord results in more spared tissue than controls (Klusman and Schwab, 1997). In the present study, we can rather conclude that sufficient upregulation of inflammatory cytokine is essential to attain better neurological recovery after spinal cord injury.
OPN-deficient mice had significantly smaller caudal spinal cords than WT mice after spinal cord injury. Although KO mice had less white matter to begin with, this finding is consistent with the smaller total areas of white matter and counts of NeuN-positive cells in KO mice after spinal cord injury. Exercise prevents regressive changes in the motor neurons and dendrites in transected spinal cords (Gazula et al., 2004). Neuron loss may also be greater in KO mice as a result of greater locomotor impairment in these mice.
Comparison with other KO mice studies
Two other knock-out mice studies compared locomotor scores after spinal cord contusion. One (Abe et al., 2003) studied mice that were deficient in tissue plasminogen activator, whereas the other studied mice that were deficient in matrix metalloproteinase-9 (Noble et al., 2002). These two studies found better locomotor recovery in KO than WT mice. Both of these genes show peak expression at 24 h after spinal cord contusion injury. OPN expression peaks 3–7 d after spinal cord compression in mice (Hashimoto et al., 2005a). The slower time course of OPN expression suggests that OPN may play a role in the recovery.
Several studies compared locomotor scores in knock-out mice after dorsal hemisection. Kerr and Patterson (2004) studied recovery from dorsal hemisection in mice with leukemia inhibitory factor deficiency, and Goldshmit et al. (2004) studied mice that lacked EphA4. Casha et al. (2005) studied the responses of FAS-deficient rats to clip compression injury. In both EphA4- and FAS-deficient mice, locomotor scores after spinal cord lesion were better in KO than WT mice. FAS deficiency rescued more neurons, and EphA4 deficiency prevented glial scar formation.
Our study of OPN-deficient mice showed worse locomotor recovery because of greater neuronal loss and less white matter sparing. Because we did not see astroglial scar difference, increased macrophages/microglia, or CD3-immunoreactive area, we can ascribe worse locomotor recovery to tissue sparing.
In summary, OPN-deficient and wild-type mice were subjected to spinal cord contusion injury using an NYU impactor modified for mice. The impactor delivered consistent impacts to the spinal cords. Lesion volumes calculated from total tissue potassium concentration at 24 h did not differ significantly between KO and WT mice. We selected 14 apoptosis-related genes or neurotrophins for additional analysis by Q-RT-PCR. Bcl-2, TNF-α, IL-1β, and IL-6 mRNAs were downregulated in KO compared with WT mice. Locomotor performance for 6 weeks was assessed by BMS score and found to be significantly lower in KO than WT mice. BMS scores of KO mice never exceeded those of WT mice for 6 weeks after injury. At 6 weeks, histological studies revealed greater NeuN-positive neuron loss and less spared white matter area in KO than WT mice. These data suggest that OPN is beneficial for recovery from spinal cord injury.
Footnotes
This study was sponsored by New Jersey Commission on Spinal Cord Research. We thank Dr. Masashi Yamazaki, Masao Koda, and Yutaka Nishio at Chiba University and Dr. Ron Hart at Rutgers University for useful discussion. We also thank Dr. Bor Tom Ng, Dr. Crista Adamson, Jessica Lum, Dr. Noriko Kane-Goldsmith, Dr. Tsutomu Iseda, and Pui Tom for technical assistance, and Hock Ng and the animal care team at Rutgers University for the animal care in this study.
References
- Abe Y, Nakamura H, Yoshino O, Oya T, Kimura T. Decreased neural damage after spinal cord injury in tPA-deficient mice. J Neurotrauma. 2003;20:43–57. doi: 10.1089/08977150360517173. [DOI] [PubMed] [Google Scholar]
- Beattie MS, Bresnahan JC, Komon J, Tovar CA, Van Meter M, Anderson DK, Faden AI, Hsu CY, Noble LJ, Salzman S, Young W. Endogenous repair after spinal cord contusion injuries in the rat. Exp Neurol. 1997;148:453–463. doi: 10.1006/exnr.1997.6695. [DOI] [PubMed] [Google Scholar]
- Caers J, Gunthert U, Raeve H, Valckenborgh EV, Menu E, Riet IV, Camp BV, Vanderkerken K. The involvement of osteopontin and its receptors in multiple myeloma cell survival, migration and invasion in the murine 5T33MM model. Br J Haematol. 2006;132:469–477. doi: 10.1111/j.1365-2141.2005.05886.x. [DOI] [PubMed] [Google Scholar]
- Casha S, Yu WR, Fehlings MG. MG.FAS deficiency reduces apoptosis, spares axons and improves function after spinal cord injury. Exp Neurol. 2005;196:390–400. doi: 10.1016/j.expneurol.2005.08.020. [DOI] [PubMed] [Google Scholar]
- Chabas D, Baranzini SE, Mitchell D, Bernard CC, Rittling SR, Denhardt DT, Sobel RA, Lock C, Karpuj M, Pedotti R, Heller R, Oksenberg JR, Steinman L. The influence of the proinflammatory cytokine, osteopontin, on autoimmune demyelinating disease. Science. 2001;294:1731–1735. doi: 10.1126/science.1062960. [DOI] [PubMed] [Google Scholar]
- Constantini S, Young W. The effects of methylprednisolone and the ganglioside GM1 on acute spinal cord injury in rats. J Neurosurg. 1994;80:97–111. doi: 10.3171/jns.1994.80.1.0097. [DOI] [PubMed] [Google Scholar]
- Denhardt DT, Noda M, O'Regan AW, Pavlin D, Berman JS. Osteopontin as a means to cope with environmental insults: regulation of inflammation, tissue remodeling, and cell survival. J Clin Invest. 2001;107:1055–1061. doi: 10.1172/JCI12980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison JA, Velier JJ, Spera P, Jonak ZL, Wang X, Barone FC, Feuerstein GZ. Osteopontin and its integrin receptor alpha(v)beta3 are upregulated during formation of the glial scar after focal stroke. Stroke. 1998;29:1698–1706. doi: 10.1161/01.str.29.8.1698. [DOI] [PubMed] [Google Scholar]
- Engesser-Cesar C, Anderson AJ, Basso DM, Edgerton VR, Cotman CW. Voluntary wheel running improves recovery from a moderate spinal cord injury. J Neurotrauma. 2005;22:157–171. doi: 10.1089/neu.2005.22.157. [DOI] [PubMed] [Google Scholar]
- Gazula VR, Roberts M, Luzzio C, Jawad AF, Kalb RG. Effects of limb exercise after spinal cord injury on motor neuron dendrite structure. J Comp Neurol. 2004;476:130–145. doi: 10.1002/cne.20204. [DOI] [PubMed] [Google Scholar]
- Giachelli CM, Lombardi D, Johnson RJ, Murry CE, Almeida M. Evidence for a role of osteopontin in macrophage infiltration in response to pathological stimuli in vivo. Am J Pathol. 1998;152:353–358. [PMC free article] [PubMed] [Google Scholar]
- Goldshmit Y, Galea MP, Wise G, Bartlett PF, Turnley AM. Axonal regeneration and lack of astrocytic gliosis in EphA4-deficient mice. J Neurosci. 2004;24:10064–10073. doi: 10.1523/JNEUROSCI.2981-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto M, Koda M, Ino H, Murakami M, Yamazaki M, Moriya H. Upregulation of osteopontin expression in rat spinal cord microglia after traumatic injury. J Neurotrauma. 2003;20:287–296. doi: 10.1089/089771503321532879. [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Koda M, Ino H, Yoshinaga K, Murata A, Yamazaki M, Kojima K, Chiba K, Mori C, Moriya H. Gene expression profiling of cathepsin D, metallothioneins-1 and -2, osteopontin, and tenascin-C in a mouse spinal cord injury model by cDNA microarray analysis. Acta Neuropathol (Berl) 2005a;109:165–180. doi: 10.1007/s00401-004-0926-z. [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Nitta A, Fukumitsu H, Nomoto H, Shen L, Furukawa S. Inflammation-induced GDNF improves locomotor function after spinal cord injury. NeuroReport. 2005b;16:99–102. doi: 10.1097/00001756-200502080-00004. [DOI] [PubMed] [Google Scholar]
- Jansson M, Panoutsakopoulou V, Baker J, Klein L, Cantor H. Cutting edge: attenuated experimental autoimmune encephalomyelitis in eta-1/osteopontin-deficient mice. J Immunol. 2002;168:2096–2099. doi: 10.4049/jimmunol.168.5.2096. [DOI] [PubMed] [Google Scholar]
- Jin CH, Miyaura C, Ishimi Y, Hong MH, Sato T, Abe E, Suda T. Interleukin 1 regulates the expression of osteopontin mRNA by osteoblasts. Mol Cell Endocrinol. 1990;74:221–228. doi: 10.1016/0303-7207(90)90227-y. [DOI] [PubMed] [Google Scholar]
- Kawamura K, Iyonaga K, Ichiyasu H, Nagano J, Suga M, Sasaki Y. Differentiation, maturation, and survival of dendritic cells by osteopontin regulation. Clin Diagn Lab Immunol. 2005;12:206–212. doi: 10.1128/CDLI.12.1.206-212.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr BJ, Patterson PH. Potent pro-inflammatory actions of leukemia inhibitory factor in the spinal cord of the adult mouse. Exp Neurol. 2004;188:391–407. doi: 10.1016/j.expneurol.2004.04.012. [DOI] [PubMed] [Google Scholar]
- Khan SA, Lopez-Chua CA, Zhang J, Fisher LW, Sorensen ES, Denhardt DT. Soluble osteopontin inhibits apoptosis of adherent endothelial cells deprived of growth factors. J Cell Biochem. 2002;85:728–736. doi: 10.1002/jcb.10170. [DOI] [PubMed] [Google Scholar]
- Klusman I, Schwab ME. Effects of pro-inflammatory cytokines in experimental spinal cord injury. Brain Res. 1997;762:173–184. doi: 10.1016/s0006-8993(97)00381-8. [DOI] [PubMed] [Google Scholar]
- Komatsubara I, Murakami T, Kusachi S, Nakamura K, Hirohata S, Hayashi J, Takemoto S, Suezawa C, Ninomiya Y, Shiratori Y. Spatially and temporally different expression of osteonectin and osteopontin in the infarct zone of experimentally induced myocardial infarction in rats. Cardiovasc Pathol. 2003;12:186–194. doi: 10.1016/s1054-8807(03)00042-5. [DOI] [PubMed] [Google Scholar]
- Kuhn PL, Wrathall JR. A mouse model of graded contusive spinal cord injury. J Neurotrauma. 1998;15:125–140. doi: 10.1089/neu.1998.15.125. [DOI] [PubMed] [Google Scholar]
- Kurihara H, Shinohara H, Yoshino H, Takeda K, Shiba H. Neurotrophins in cultured cells from periodontal tissues. J Periodontol. 2003;74:76–84. doi: 10.1902/jop.2003.74.1.76. [DOI] [PubMed] [Google Scholar]
- Kury P, Zickler P, Stoll G, Hartung HP, Jander S. Osteopontin, a macrophage-derived matricellular glycoprotein, inhibits axon outgrowth. FASEB J. 2004;19:398–400. doi: 10.1096/fj.04-1777fje. [DOI] [PubMed] [Google Scholar]
- Kwo S, Young W, DeCrescito V. Spinal cord sodium, potassium, calcium, and water concentration changes in rats after graded contusion injury. J Neurotrauma. 1989;6:13–24. doi: 10.1089/neu.1989.6.13. [DOI] [PubMed] [Google Scholar]
- Leskovar A, Moriarty LJ, Turek JJ, Schoenlein IA, Borgens RB. The macrophage in acute neural injury: changes in cell numbers over time and levels of cytokine production in mammalian central and peripheral nervous systems. J Exp Biol. 2000;203:1783–1795. doi: 10.1242/jeb.203.12.1783. [DOI] [PubMed] [Google Scholar]
- Morimoto J, Inobe M, Kimura C, Kon S, Diao H, Aoki M, Miyazaki T, Denhardt DT, Rittling S, Uede T. Osteopontin affects the persistence of beta-glucan-induced hepatic granuloma formation and tissue injury through two distinct mechanisms. Int Immunol. 2004;16:477–488. doi: 10.1093/intimm/dxh044. [DOI] [PubMed] [Google Scholar]
- Nau GJ, Chupp GL, Emile JF, Jouanguy E, Berman JS, Casanova JL, Young RA. Osteopontin expression correlates with clinical outcome in patients with mycobacterial infection. Am J Pathol. 2000;157:37–42. doi: 10.1016/S0002-9440(10)64514-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble LJ, Donovan F, Igarashi T, Goussev S, Werb Z. Matrix metalloproteinases limit functional recovery after spinal cord injury by modulation of early vascular events. J Neurosci. 2002;22:7526–7535. doi: 10.1523/JNEUROSCI.22-17-07526.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada S, Nakamura M, Mikami Y, Shimazaki T, Mihara M, Ohsugi Y, Iwamoto Y, Yoshizaki K, Kishimoto T, Toyama Y, Okano H. Blockade of interleukin-6 receptor suppresses reactive astrogliosis and ameliorates functional recovery in experimental spinal cord injury. J Neurosci Res. 2004;76:265–276. doi: 10.1002/jnr.20044. [DOI] [PubMed] [Google Scholar]
- Ophascharoensuk V, Giachelli CM, Gordon K, Hughes J, Pichler R, Brown P, Liaw L, Schmidt R, Shankland SJ, Alpers CE, Couser WG, Johnson RJ. Obstructive uropathy in the mouse: role of osteopontin in interstitial fibrosis and apoptosis. Kidney Int. 1999;56:571–580. doi: 10.1046/j.1523-1755.1999.00580.x. [DOI] [PubMed] [Google Scholar]
- O'Regan AW, Hayden JM, Body S, Liaw L, Mulligan N, Goetschkes M, Berman JS. Abnormal pulmonary granuloma formation in osteopontin-deficient mice. Am J Respir Crit Care Med. 2001;164:2243–2247. doi: 10.1164/ajrccm.164.12.2104139. [DOI] [PubMed] [Google Scholar]
- Pearse DD, Pereira FC, Stolyarova A, Barakat DJ, Bunge MB. Inhibition of tumour necrosis factor-alpha by antisense targeting produces immunophenotypical and morphological changes in injury-activated microglia and macrophages. Eur J Neurosci. 2004;20:3387–3396. doi: 10.1111/j.1460-9568.2004.03799.x. [DOI] [PubMed] [Google Scholar]
- Persy VP, Verhulst A, Ysebaert DK, De Greef KE, De Broe ME. Reduced postischemic macrophage infiltration and interstitial fibrosis in osteopontin knockout mice. Kidney Int. 2003;63:543–553. doi: 10.1046/j.1523-1755.2003.00767.x. [DOI] [PubMed] [Google Scholar]
- Petrow PK, Hummel KM, Schedel J, Franz JK, Klein CL, Muller-Ladner U, Kriegsmann J, Chang PL, Prince CW, Gay RE, Gay S. Expression of osteopontin messenger RNA and protein in rheumatoid arthritis: effects of osteopontin on the release of collagenase 1 from articular chondrocytes and synovial fibroblasts. Arthritis Rheum. 2000;43:1597–1605. doi: 10.1002/1529-0131(200007)43:7<1597::AID-ANR25>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Renkl AC, Wussler J, Ahrens T, Thoma K, Kon S, Uede T, Martin SF, Simon JC, Weiss JM. Osteopontin functionally activates dendritic cells and induces their differentiation toward a Th1-polarizing phenotype. Blood. 2005;106:946–955. doi: 10.1182/blood-2004-08-3228. [DOI] [PubMed] [Google Scholar]
- Rittling SR, Chambers AF. Role of osteopontin in tumour progression. Br J Cancer. 2004;90:1877–1881. doi: 10.1038/sj.bjc.6601839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittling SR, Matsumoto HN, McKee MD, Nanci A, An XR, Novick KE, Kowalski AJ, Noda M, Denhardt DT. Mice lacking osteopontin show normal development and bone structure but display altered osteoclast formation in vitro. J Bone Miner Res. 1998;13:1101–1111. doi: 10.1359/jbmr.1998.13.7.1101. [DOI] [PubMed] [Google Scholar]
- Selvaraju R, Bernasconi L, Losberger C, Graber P, Kadi L, Avellana-Adalid V, Picard-Riera N, Van Evercooren AB, Cirillo R, Kosco-Vilbois M, Feger G, Papoian R, Boschert U. Osteopontin is upregulated during in vivo demyelination and remyelination and enhances myelin formation in vitro. Mol Cell Neurosci. 2004;25:707–721. doi: 10.1016/j.mcn.2003.12.014. [DOI] [PubMed] [Google Scholar]
- Weintraub AS, Schnapp LM, Lin X, Taubman MB. Osteopontin deficiency in rat vascular smooth muscle cells is associated with an inability to adhere to collagen and increased apoptosis. Lab Invest. 2000;80:1603–1615. doi: 10.1038/labinvest.3780171. [DOI] [PubMed] [Google Scholar]
- Young W. Piscataway, NJ: Rutgers University; 2000. Spinal cord injury models and methods, The Spinal Cord Injury Project, W. M. Keck Center for Collaborative Neuroscience, p 7. [Google Scholar]
- Young W. Spinal cord contusion models. Prog Brain Res. 2002;137:231–255. doi: 10.1016/s0079-6123(02)37019-5. [DOI] [PubMed] [Google Scholar]
- Yumoto K, Ishijima M, Rittling SR, Tsuji K, Tsuchiya Y, Kon S, Nifuji A, Uede T, Denhardt DT, Noda M. Osteopontin deficiency protects joints against destruction in anti-type II collagen antibody-induced arthritis in mice. Proc Natl Acad Sci USA. 2002;99:4556–4561. doi: 10.1073/pnas.052523599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zohar R, Zhu B, Liu P, Sodek J, McCulloch CA. Increased cell death in osteopontin-deficient cardiac fibroblasts occurs by a caspase-3-independent pathway. Am J Physiol Heart Circ Physiol. 2004;287:H1730–H1739. doi: 10.1152/ajpheart.00098.2004. [DOI] [PubMed] [Google Scholar]