

Abstract
A defect in microtubule (MT)-based transport contributes to the neuronal toxicity observed in Huntington's disease (HD). Histone deacetylase (HDAC) inhibitors show neuroprotective effects in this devastating neurodegenerative disorder. We report here that HDAC inhibitors, including trichostatin A (TSA), increase vesicular transport of brain-derived neurotrophic factor (BDNF) by inhibiting HDAC6, thereby increasing acetylation at lysine 40 of α-tubulin. MT acetylation in vitro and in cells causes the recruitment of the molecular motors dynein and kinesin-1 to MTs. In neurons, acetylation at lysine 40 of α-tubulin increases the flux of vesicles and the subsequent release of BDNF. We show that tubulin acetylation is reduced in HD brains and that TSA compensates for the transport- and release-defect phenotypes that are observed in disease. Our findings reveal that HDAC6 inhibition and acetylation at lysine 40 of α-tubulin may be therapeutic targets of interest in disorders such as HD in which intracellular transport is altered.
Keywords: Huntington's disease, polyglutamine, transport, microtubules, BDNF, neuroprotection
Introduction
Huntington's disease (HD), a devastating neurodegenerative disorder characterized by cognitive and motor deficits, is caused by an abnormal polyglutamine (polyQ) expansion in the N-terminal part of the huntingtin protein (htt) (MacDonald et al., 2003). Recent studies have identified an altered microtubule (MT)-dependent transport of organelles in HD (Gunawardena et al., 2003; Szebenyi et al., 2003; Gauthier et al., 2004; Lee et al., 2004; Trushina et al., 2004). Using fast three-dimensional (3D) videomicroscopy and biochemical approaches, we previously demonstrated that htt associates with molecular motors and activates the MT-dependent transport of vesicles containing brain-derived neurotrophic factor (BDNF) (Gauthier et al., 2004). Wild-type (WT) htt enhances the velocity of the vesicles and reduces the amount of time they spend not moving (pausing time). In HD, in which the htt contains the polyQ expansion, the intracellular transport of BDNF-containing vesicles is altered, resulting in reduced trophic support of neurons and their death. We reasoned that it should be possible to use fast 3D videomicroscopy to screen for compounds able to restore intracellular transport.
Currently, there is no treatment available for HD patients, although several studies in yeast, Drosophila, mammalian cells, and mice have identified compounds of therapeutic interest. Among these compounds are histone deacetylase (HDAC) inhibitors such as suberoylanilide hydroxamic acid (SAHA) and trichostatin A (TSA), which have shown neuroprotective effects by inhibiting the HDAC1 enzyme (Butler and Bates, 2006). These drugs are not specific for a given HDAC and act on other HDACs, such as HDAC6 (Haggarty et al., 2003). Unlike other HDACs, HDAC6 is a cytoplasmic enzyme that interacts with and deacetylates MTs in vitro and in vivo (Hubbert et al., 2002; Matsuyama et al., 2002; Zhang et al., 2003). Acetylation is associated with stable MTs, although this relationship is not clear-cut. Some studies have shown that acetylation enhances MT stability (Hubbert et al., 2002; Matsuyama et al., 2002), whereas others have suggested that acetylation occurs only on stable and not dynamic MTs, but the acetylation itself does not stabilize MTs (Haggarty et al., 2003).
Interestingly, a recent report indicates that α-tubulin acetylation at lysine 40 of Tetrahymena thermophila enhances the recruitment of the molecular motor kinesin-1 to MTs and promotes anterograde transport of the kinesin-1 cargo JNK (c-Jun N-terminal kinase)-interacting protein (JIP1) in differentiated neuronal cells (Reed et al., 2006), suggesting a possible link between HDAC6 inhibition and the stimulation of MT-dependent trafficking of vesicles. Given that MT-dependent vesicular transport is altered in HD and that HDAC inhibitors show a neuroprotective effect, we tested whether MT acetylation could regulate vesicular transport and compensate for the observed transport deficit in HD. We report that HDAC inhibitors that selectively enhance tubulin but not histone acetylation lead to the stimulation of MT-dependent transport of BDNF and prevent the alteration observed in HD mutant cells. We show that this effect is specific to HDAC6 inhibition and to the acetylation of α-tubulin at lysine 40. We demonstrate in vitro that purified cytoplasmic dynein and recombinant kinesin-1 bind more effectively to acetylated MTs. Enhancing MT acetylation leads to the recruitment of molecular motors kinesin-1 and cytoplasmic dynein to MTs thereby stimulating anterograde and retrograde transport. We show that this increased transport leads to the enhancement of the anterograde flux of vesicles and the subsequent release of BDNF in the normal and pathological conditions. Our data provide strong evidence for a functional role of MT acetylation on vesicular transport. Furthermore, we identify drugs that could restore the transport deficit in HD.
Materials and Methods
Statistical analyses.
Statview 4.5 software (SAS Institute, Cary, NC) was used for statistical analyses. All data herein described were performed in duplicate or triplicate. Data are expressed as means ± SEM. Statistical analyses may be found in the supplemental material (available at www.jneurosci.org).
DNA constructs.
Constructs encoding BDNF, BDNF-enhanced green fluorescent protein (eGFP), end-binding protein 3 (EB3)-eGFP, HDAC6, inactive HDAC6 (HDAC6m1m2), mCherry-α-tubulin, htt-480–17Q, and htt-480–68Q, were described previously (Haubensak et al., 1998; Saudou et al., 1998; Seigneurin-Berny et al., 2001; Stepanova et al., 2003; Shaner et al., 2004). The mCherry-α-tubulin lysine 40 to alanine mutation (K40A) construct with the mutation at the acetylation site was generated by QuikChange site-directed mutagenesis (Stratagene, La Jolla, CA) using the WT mCherry-α-tubulin plasmid as a nonmutated parental template and complementary oligonucleotides containing the desired mutation (5′-GATGCCAAGTGACGCGACAATTGGGGGAGGAG-3′).
Antibodies.
The polyclonal antibodies used were the following: anti-detyrosinated tubulin (AB3201; Millipore, Bedford, MA), anti-tyrosinated tubulin (Amersham Biosciences, Piscataway, NJ), anti-GFP (Millipore), anti-HDAC6 (Seigneurin-Berny et al., 2001), anti-histone H3, and anti-acetylated histone H3 (Cell Signaling, Boston, MA). The monoclonal antibodies used were the following: anti-htt (2166), anti-kinesin heavy chain (KHC; clone H2), and anti-dynein intermediate chain (DIC; clone 74.1; Millipore); anti-α-tubulin (clone DM1A), anti-α-tubulin (clone DM1A)-FITC conjugated antibodies, anti-acetylated tubulin, anti-β-tubulin cyanine 3 (Cy3)-conjugated antibodies (Sigma, St. Louis, MO); anti-p150Glued, anti-EB1, anti-KIF3A (BD Bioscience, San Jose, CA); and anti-KHC (SUK4; Covance Research Products, Berkeley, CA) (Ingold et al., 1988). The monoclonal antibody anti-acetylated α-tubulin (Zymed, San Francisco, CA) was conjugated to Alexa Fluor 488 using the Alexa Fluor monoclonal antibody labeling kit (Invitrogen, Eugene, OR). Anti-mouse and anti-rabbit secondary antibodies conjugated to Alexa Fluor 488, Alexa Fluor 555, and Alexa Fluor 647 were purchased from Invitrogen. Anti-mouse and anti-rabbit secondary antibodies conjugated to HRP were purchased from Jackson ImmunoResearch (West Grove, PA).
Cytoplasmic dynein purification.
Cytoplasmic dynein from bovine brain tissue was purified and isolated from dynactin as described previously (Bingham et al., 1998), except that the homogenization buffer included 45 nm okadaic acid. The purified dynein was diluted in 45% glycerol and stored at −20°C. Absence of any significant contamination from dynactin was established through SDS-PAGE and Western blotting.
Cell culture, transfection, and drug treatments.
Mouse striatal cells derived from WT htt (WT striatal cells, +/+) mice and from HdhQ109 knock-in (109Q/109Q) mice, HEK293 cells, Cos7 cells, and primary cortical neurons were prepared and cultured as described previously (Xia et al., 1996; Trettel et al., 2000; Humbert et al., 2002; Gauthier et al., 2004). HEK293 cells were transfected by the calcium phosphate technique. Primary cortical neurons were transfected as described previously (Xia et al., 1996) or electroporated with the rat neuron Nucleofector kit according to the supplier's manual (Amaxa Biosystems, Cologne, Germany). Forskolin (10 μm; Sigma) and IBMX (100 μm; Sigma) were added to the culture 5 h after transfection. Neuronal cells were transfected for 4 h with small interfering RNA (siRNA) htt or the corresponding scrambled RNA (19 nucleotides of RNA plus TT) 1 d before BDNF-eGFP transfection. The siRNA sequence targeting mouse htt (siRNA-htt) corresponds to the coding regions 185–206 (GenBank accession number XM 132009). The siRNA sequence targeting mouse or rat KIF5B (kinesin-1) corresponds to the coding region 1215–1233 (GenBank accession number XM 341638). The siRNA sequence targeting human KIF3A (kinesin-2) corresponds to the coding region 1633–1651 (accession number XM 007054). The scrambled RNA htt have the same nucleotide composition as the siRNA-htt but lack a significant sequence homology to any other gene (Gauthier et al., 2004). Transfected or untransfected cells were treated with various HDAC inhibitors: TSA (100 nm, 1 μm, or 5 μm for cell biology and biochemistry, respectively; Sigma), SAHA (2 μm; Coger, Paris, France), MS-275 (HDAC1 inhibitor; 3 μm; Alexis Biochemicals, Lausen, Switzerland), or tubacin (6 μm) (Haggarty et al., 2003) for 4 h; sodium phenylbutyrate (NaPB; 10 mm; Alexis Biochemicals) for 4 or 16 h; and taxol (100 nm; Sigma) for 1 h. Treatment with DMSO (Sigma) was used as a control.
Cell extracts, immunoblotting experiments, and MT preparation.
Cells were lysed 1–4 h after drug treatment with 1% Triton lysis buffer (160 mm NaCl, 50 mm HEPES, 2.5 mm MgCl2, 1.5 mm CaCl2, 2.5 mm KCl, and 1% Triton X-100, pH 7.4, containing 1 mm PMSF, 2 μg/ml aprotinin, 2 μg/ml leupeptin, 2 μg/ml pepstatin). The extracts were sonicated and centrifuged at 11,000 × g (15 min; 4°C). Proteins (1–20 μg) were loaded onto SDS-PAGE and subjected to Western blot analysis.
MTs were depolymerized on ice for 15 min, and the reaction was centrifuged at 10,000 × g for 15 min at 4°C as described previously (Goldstein et al., 1986). MTs in the supernatant (S) were polymerized at 33°C for 30 min by addition of GTP (1 mm final) and taxol (20 μm final; Sigma). Polymerized MTs were pelleted by centrifugation at 10,000 × g for 45 min. After centrifugation, 1 μl of 2% SDS buffer was added to the S, and the pellet (P) was resuspended in 1% SDS buffer, heated 5 min at 100°C, and sonicated before loading. The same volume of each fraction was analyzed by Western blot.
Brain tissues.
Tissues (striatum) were obtained from the Harvard Brain Tissue Resource Center (HBTRC; Belmont, MA): three controls (samples 1–3), one HD grade 3 (HD3; sample 4) patient, and one HD grade 4 (HD4; sample 5) patient. Striatal postmortem samples correspond, respectively, to brain numbers 4741, 4744, 4751, 4797, 4640 as numbered by the HBTRC. Samples were homogenized in NP-40 lysis buffer and cleared by centrifugation at 6000 × g (15 min; 4°C). Western blot analysis was performed on 30 ng (acetylated α-tubulin) or 1 μg (total tubulin) of total extracts.
BDNF immunoenzyme assays.
BDNF assays were performed on cortical neurons 48 h after electroporation with BDNF and WT or polyQ-htt constructs. To measure transport-dependent release, cells were depolarized (treatment for 20 min with DMEM containing high K+ (30 mm CaCl2, 30 mm NaCl, 28 mm KCl), treated 30 min with DMEM, washed, and depolarized again (second depolarization, K2). For TSA treatment, cells were maintained in TSA (1 μm) during all of the steps. The amount of BDNF was measured in S fractions and cell lysates using the BDNF Emax Immunoassay System (Promega, Charbonnieres, France) as described previously (Gauthier et al., 2004).
MT binding assays.
Three micrograms of purified tubulin (Cytoskeleton, Denver, CO) were polymerized in PEM buffer (100 mm PIPES, pH 6.7, 1 mm EGTA, 1 mm MgCl2) in the presence of 200 μm taxol (Sigma) and 1 mm GTP (Sigma) for 30 min at 37°C. MTs were pelleted at 20,000 × g for 30 min, resuspended in PEM buffer containing 20 μm taxol, 1 mm GTP, and 50 mm NaCl, and then sheared with a 25 ga needle (Ligon et al., 2006). MTs were acetylated in vitro by addition of 1% anhydride acetic acid (AA) for 10 min at 37°C (Piperno and Fuller, 1985). Acetylated and unacetylated MTs were then incubated with either 50 ng of recombinant kinesin (Cytoskeleton) or 30 ng dynein for 10 min at 37°C in the presence of 50 μm AMP-PNP (5′-adenyly-limidodiphosphate). MTs were finally pelleted onto a 12 mm coverslip precoated with poly-d-lysine (BD Bioscience) at 27,000 × g for 15 min (Evans et al., 1985), fixed, and visualized by immunofluorescence.
MTs containing the WT mCherry-α-tubulin or K40A construct were purified by two successive rounds of depolymerization/repolymerization from transfected and TSA-treated (1 μm) HEK293 cells as described for MT preparation, except that all of the procedures were performed in the presence of 5 μm TSA to maintain MT acetylation and 10 mm ATP to remove endogenous motors. MTs were resuspended in PEM buffer, polymerized, incubated with recombinant motors, and pelleted as described above. mCherry-α-tubulin was visualized using the anti-GFP antibody. Absence of binding of endogenous kinesin-1 and cytoplasmic dynein on polymerized MTs was verified by immunostaining.
Immunofluorescence experiments.
Neurons and cells were grown on glass coverslips, transfected with various constructs of HDAC6 or of mCherry-α-tubulin, and fixed in anhydrous methanol at −20°C for 5 min and incubated with anti-tubulin, anti-acetylated-tubulin, anti-HDAC6, or anti-acetylated histone H3 antibodies. To visualize molecular motors along MTs, the cell fixation protocol was adapted from Vaughan et al. (1999). Briefly, cells were incubated for 1 min in PBS at room temperature and fixed in anhydrous methanol at −20°C for 5 min and incubated with anti-DIC, anti-KHC, and anti-p150Glued antibodies. In the case of double labeling with two mouse monoclonal antibodies, cells were first fixed and incubated with the primary antibodies anti-kinesin, anti-DIC, or anti-p150Glued antibodies, then cells were washed with 0.1% PBS/BSA and incubated with the anti-mouse secondary antibodies conjugated with Alexa Fluor 488 and 555. Cells then underwent a second fixation (anhydrous methanol at −20°C for 5 min) and were incubated with FITC-conjugated anti-α-tubulin or Cy3-conjugated anti-β-tubulin antibodies. For in vitro experiments, pelleted MTs were fixed with either 4% paraformaldehyde in PHEM buffer for 20 min or anhydrous −20°C methanol for 5 min and incubated, respectively, with anti-KHC or anti-DIC antibodies. Pictures were captured with a 3D deconvolution imaging system as described previously (Gauthier et al., 2004).
Videomicroscopy experiments.
Videomicroscopy experiments were done 2–3 d after transfection. The cells were cotransfected with BDNF-eGFP and various constructs of HDAC6, mCherry-α-tubulin, or the corresponding empty vectors with a DNA ratio of 1:4 and treated as described above. Live videomicroscopy was performed using an imaging system detailed previously (Gauthier et al., 2004). Cells were grown on a glass coverslip that was mounted in a Ludin chamber. The microscope and the chamber were kept at 33°C for striatal cells and 37°C for neurons and Cos7 cells. Stacks of 11 images with a Z step of 0.3 μm were acquired with a 100× PlanApo numerical aperture 1.4 oil immersion objective coupled to a piezo device. Images were collected in stream mode using a CoolSnap HQ camera (Roper Scientific, Trenton, NJ) set at 2 × 2 binning with an exposure time of 50–150 ms (frequency of 2 s). All stacks were treated by automatic batch deconvolution using the point spread function of the optical system. All dynamic parameters of intracellular transport are from data that were typically obtained for each condition from two to three independent transfections with a total of ∼2000–8000 measures in 11–40 independent cells.
Image processing and data analyses.
For videomicroscopy experiments, projections, animations, and analyses (tracking and colocalizations) were generated using ImageJ software (written by W. Rasband at the National Institutes of Health and available at http://rsb.info.nih.gov/ij/). Dynamics were characterized by tracking positions of eGFP vesicles in cells as a function of time with an especially developed plug-in (http://rsb.info.nih.gov/ij/plugins/track/track.html). During tracking, the Cartesian coordinates of the centers of vesicles were used to calculate dynamic parameters (velocity, pausing time, directionality). The threshold for null displacement corresponds to one pixel over the time interval (i.e., 129 nm/2 s = 4 μm/min). For two-color videomicroscopy, an additional midplane on the z-axis was captured in the red channel for every stack completed in the GFP channel. Time projections were generated as follows. After binarization and erosion of the deconvolved images, a composite-projected merge of the 31 frames of the experiment was next generated by continuously substracting the previous frame, thus displaying only the differences between adjacent frames. This time projection allows one to visualize the paths followed by vesicles over the recording time (1 min) (Toomre et al., 1999).
The flux in one direction Φdir was calculated by expressing, for each neuron, the summed individual-traveled distances in this direction di, multiplied by the mean of velocity in the same direction vdir, divided by the number of vesicles moving in this direction ndir, according to the following:
 |
The quantification of the portion of MT-associated proteins (MAPs) signal associated to MTs, was achieved by creating a binary mask from the MT image. First, a Gaussian filter with a five-pixel radius was used to smooth the original image. To get rid of the uneven background, a 3D top-hat filter was then applied, using a 5 × 5 × 2 pixel kernel. The MT image was further binarized using the overall minimum gray level value of the full stack as a threshold. MT gray level value was set to one, whereas the surrounding background pixels were set to zero to obtain the mask image. Finally, the mask was dilated by 5 × 5 pixels to ensure the retrieval of all of the dots of the MAPs associated to MTs in the remainder of the process. The image of MAP signal on MTs was obtained by multiplying the latter and the MAP image. The resultant image was then subtracted to the original MAP image to retrieve the image of the MAP signal excluded from the MTs. Quantifications were expressed as the quotient of the overall MAP signal associated to the MTs and the overall MAP signal. All processes were automated using macros written for ImageJ extended by a homemade plug-in for 3D top-hat filtering (all homemade ImageJ plug-ins are available on request at fabrice.cordelieres@curie.u-psud.fr).
Linescan analyses.
MT intensity profiles were obtained using MT profiler, a homemade plug-in to the open-source software ImageJ. A defined rectangular region of interest was placed around the MT, one of the shortest-bound touching the MT plus tip. The connected maximum intensity path was retrieved in 3D, starting from the tip. Fluorescence signal was then quantified along this path. For quantification of motor recruitment on MT and in vitro experiments, results were expressed as the density of fluorescence (sum of motor intensity along the path divided by the length of MT) and were normalized to control. Ten single MTs per cell (10 cells) were chosen randomly for quantification.
Results
TSA and SAHA increase BDNF intracellular transport and compensate for the trafficking deficit in HD
To test the hypothesis that HDAC inhibitors may regulate vesicular transport, we analyzed the dynamics of BDNF-containing vesicles using fast 3D videomicroscopy followed by deconvolution (Gauthier et al., 2004). Indeed, the control of intracellular dynamics of BDNF-containing vesicles is of particular importance in HD. BDNF is actively transported in corticostriatal-projecting neurons and released in the striatum (Altar et al., 1997) where it acts as a prosurvival factor for the striatal neurons that are particularly vulnerable in HD (Saudou et al., 1998; Baquet et al., 2004). The cellular localization, processing, and secretion of mature BDNF-eGFP are indistinguishable from those of endogenous BDNF (Haubensak et al., 1998; Hartmann et al., 2001; Kohara et al., 2001).
We investigated the effect of HDAC inhibition in the normal and the HD pathological situations by using cell lines derived from striatal precursor cells of knock-in mice in which a CAG expansion was inserted into the endogenous mouse htt gene (Trettel et al., 2000). These cell lines carry either two copies of WT htt (WT striatal cells, +/+) or two copies of mutant htt (homozygous HD mutant cells, 109Q/109Q). We transfected WT and HD mutant cells with BDNF-eGFP and monitored the movement of BDNF-containing vesicles by acquiring 3D time-series images (Fig. 1A). Individual vesicles were then tracked by measuring the x, y, and z coordinates of the vesicles over time. Visualization of the paths of 10 individual vesicles randomly selected in WT and 109Q/109Q cells revealed a significant reduction in the displacement of BDNF vesicles in the HD situation (Fig. 1A). As reported previously (Gauthier et al., 2004), analysis of velocity distribution demonstrated a significant loss of rapidly moving vesicles in HD mutant cells compared with WT cells [32.7% for control (CT) +/+ vs 15.5% for CT 109Q/109Q] (Fig. 1B). We next treated cells with TSA and found that TSA treatment increased the percentage of high-velocity vesicles in the WT cells and restored the distribution of velocities in 109Q/109Q cells (40.8% for TSA +/+ vs 32.7% for CT +/+ and 33.6% for TSA 109Q/109Q vs 15.5% for CT 109Q/109Q) (Fig. 1A,B) (supplemental videos 1–3, available at www.jneurosci.org as supplemental material) back to that of the WT situation, indicating that TSA rescues the alteration of transport in HD cells. We next determined the mean vesicle velocity of moving vesicles between two pauses and the percentage of pausing time of vesicles (time spent by the vesicles without moving). These two parameters are particularly relevant to reflect changes in transport efficiency. In 109Q/109Q cells, the velocity and the pausing time of BDNF vesicles were significantly different than those of +/+ cells (Fig. 1C,D). TSA treatment rescued vesicular transport by significantly increasing the velocity and decreasing the pausing time of BDNF-containing vesicles. We also analyzed the movement of all of the vesicles that are present in cells by calculating the mean percentage of colocalization of vesicles between two successive time points (Manders et al., 1992). A high level of colocalization indicates that vesicles are not moving (static vesicles). The percentage of static vesicles was significantly reduced by TSA treatment both in +/+ and 109Q/109Q cells (Fig. 1E). Finally, TSA treatment did not obviously modify the distribution of BDNF relative to intracellular markers of the secretory pathway, excluding the possibility that BDNF could be transferred to other types of organelles having different dynamics (data not shown). Together, these results demonstrate that TSA stimulates transport of BDNF and rescues the intracellular transport defect in HD mutant cells.
Figure 1.

TSA and SAHA stimulate vesicular transport of BDNF and rescue transport defect in HD Cells. A, B, WT (+/+) and 109Q/109Q cells transfected with BDNF-eGFP were treated for 4 h with DMSO (0.1%, CT), TSA (1 μm), or SAHA (2 μm) and analyzed by videomicroscopy. A, The displacement of BDNF vesicles is reduced in 109Q/109Q compared with +/+ cells as shown by 3D reconstruction of the first time point (T0), time projection of moving structures over the 1 min experiment, and visualization of 10 paths followed by individual vesicles (Tracks). The altered BDNF transport in 109Q/109Q cells (CT) is restored by treating cells with TSA. B, Analysis of the distribution of vesicular velocities shows a marked increase in the number of vesicles that have a low velocity in 109Q/109Q cells compared with +/+ cells. The distribution of velocities in TSA-treated 109Q/109Q cells is similar to the distribution in +/+ cells. Filled bars correspond to nonmoving and low-moving vesicles, and open bars correspond to high-speed moving vesicles. C–E, WT (+/+) and 109Q/109Q cells transfected with BDNF-eGFP were treated for 4 h with DMSO (0.1%, control, CT), TSA (1 μm), SAHA (2 μm), or NaPB (10 mm) and analyzed by videomicroscopy. Dynamics were quantified by the use of three parameters: the mean velocity of vesicles (C), their pausing time (D), and the percentage of static vesicles (E). Dashed lines correspond to the control values. N.S., Not significant. *p < 0.05; **p < 0.01; ***p < 0.001.
We next analyzed the effects of other HDAC inhibitors, such as SAHA and NaPB, on BDNF vesicular dynamics (Fig. 1C–E). Whereas TSA and SAHA significantly increased BDNF transport in +/+ and 109Q/109Q cells, NaPB had no significant effect. Statistical analyses revealed that TSA and SAHA stimulated transport of BDNF and rescued the intracellular transport defect in HD mutant cells to a similar extent (supplemental material, available at www.jneurosci.org).
The stimulatory effect of SAHA and TSA on transport is independent of htt
It has been demonstrated previously by us and other groups that WT htt directly regulates transport. Indeed, increasing htt levels through its overexpression increases transport efficiency, whereas reducing the levels through RNA interference (RNAi) reduces transport efficiency (Gunawardena et al., 2003; Gauthier et al., 2004). To analyze whether TSA and SAHA could act directly on htt to promote vesicular trafficking, we studied the effect of these drugs in cells containing low levels of htt (Fig. 2). Striatal +/+ cells were transfected with BDNF-eGFP and either siRNA directed against htt or the corresponding scrambled RNA. As expected, cells with reduced htt levels (Fig. 2A) showed reduced BDNF vesicle velocity and increased pausing time compared with cells transfected with control RNA (Fig. 2B,C). However, TSA (and SAHA; data not shown) increased BDNF vesicular transport to similar extents in both conditions, showing that the enhanced BDNF transport induced by these drugs does not depend on htt (Fig. 2B,C). Thus, TSA and SAHA stimulate BDNF trafficking through an htt-independent mechanism.
Figure 2.

TSA stimulates vesicular transport of BDNF through an htt-independent mechanism. WT (+/+) cells were transfected with BDNF-eGFP and siRNA directed against mouse htt (siHtt) or scramble RNA (scRNA) 1 d before BDNF-eGFP transfection. A, Total extracts were analyzed by Western blot for htt and α-tubulin levels. B, C, Cells were treated for 4 h with DMSO (0.1%, CT) or TSA (1 μm) and analyzed by videomicroscopy. Dynamics were quantified by the use of the mean velocity of vesicles per cell (B) and their pausing time (C). Dashed lines correspond to the control values. N.S., Not significant. *p < 0.05; **p < 0.01; ***p < 0.001.
SAHA and TSA increase tubulin acetylation
Although HDACs have been widely described to control the acetylation state of histones acting as transcription activators or repressors (Taddei et al., 2005), they also deacetylate cytoplasmic proteins including tubulin (Hubbert et al., 2002; Matsuyama et al., 2002; Zhang et al., 2003). Because BDNF transport is MT dependent (Gauthier et al., 2004), we assessed the acetylation state of α-tubulin in our assay. We treated striatal +/+ cells with TSA, SAHA, and NaPB and used immunofluorescence to analyze the acetylation state of α-tubulin (Fig. 3A). We used histone H3 as a control. The three HDAC inhibitors increased the levels of acetylation of histone H3. However, only TSA and SAHA affected tubulin acetylation. We obtained the same results from immunoblotting extracts of treated WT cells: SAHA and TSA increased histone H3 and tubulin acetylation, whereas even after 16 h of treatment, NaPB increased only the acetylation of histone H3, not the acetylation of α-tubulin (Fig. 3B). In conclusion, these results indicate that only those HDAC inhibitors that increase tubulin acetylation also increase BDNF transport, suggesting that tubulin acetylation may play a role in the regulation of intracellular transport.
Figure 3.
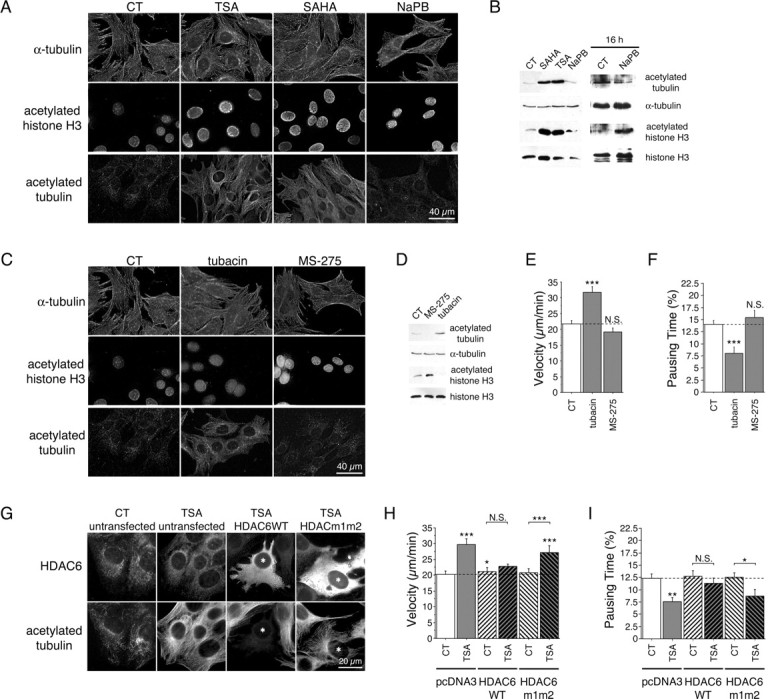
TSA and SAHA acetylate MTs and stimulate vesicular transport of BDNF through an HDAC6-dependent mechanism. A, B, WT (+/+) cells were treated for 4 h with DMSO (0.1%, control, CT), TSA (1 μm), SAHA (2 μm), and NaPB [10 mm (A, B, left) or for 16 h (B, right)] and analyzed by immunofluorescence (A) or Western blot (B) for the presence of acetylated tubulin, α-tubulin, acetylated histone H3, and histone H3. C, WT (+/+) cells were treated for 4 h with DMSO (0.1%, CT), tubacin (6 μm), and MS-275 (3 μm) and stained for α-tubulin, acetylated histone H3, and acetylated tubulin. D, Extracts from WT cells treated for 4 h with DMSO (0.1%, CT), MS-275 (3 μm), or tubacin (6 μm) were processed by Western blot and analyzed for acetylated tubulin, α-tubulin, acetylated histone H3, and histone H3. E, F, WT (+/+) cells transfected with BDNF-eGFP were treated as in D and analyzed by videomicroscopy. G, Untransfected cells and cells transfected with HDAC6WT or HDAC6m1m2 were treated with DMSO (0.1%, CT) and TSA (100 nm) for 4 h and immunostained with anti-HDAC6 and anti-acetylated tubulin antibodies. HDAC6WT or HDAC6m1m2 transfected cells are labeled with an asterisk. H, I, Striatal cells were cotransfected with BDNF-eGFP and with HDAC6WT, HDAC6m1m2, or the corresponding empty vector (pcDNA3), treated with DMSO (0.1%, CT) and TSA (100 nm) for 4 h, and analyzed by videomicroscopy. Dashed lines correspond to the control values. N.S., Not significant. *p < 0.05; **p < 0.01; ***p < 0.001.
TSA increases BDNF intracellular transport and tubulin acetylation through an HDAC6-dependent mechanism
HDACs are divided into three subfamilies: class I, II, and III (Drummond et al., 2005). TSA and SAHA inhibit class I and II HDACs, whereas NaPB only inhibits class I HDACs. Therefore, TSA and SAHA may affect transport through a class II protein. Of the class II HDACs, HDAC6 is unique, being located only in the cytoplasm and having a tubulin deacetylase activity (Hubbert et al., 2002; Matsuyama et al., 2002; Zhang et al., 2003). TSA may thus affect transport specifically through HDAC6 inhibition. Therefore, we analyzed by immunofluorescence (Fig. 3C) and immunoblotting (Fig. 3D) the effects on WT cells of two HDAC inhibitors, tubacin and MS-275, which specifically inhibit HDAC6 and HDAC1, respectively (Haggarty et al., 2003; Hu et al., 2003). Tubacin and MS-275 had opposite effects: tubacin increased the acetylation of tubulin but not of histone H3, whereas MS-275 increased the acetylation of histone H3 but not of tubulin. These results in striatal cells are consistent with previous studies in NIH 3T3 cells (Haggarty et al., 2003), demonstrating that HDAC6 preferentially deacetylates tubulin over histone H3. We next analyzed the effect of tubacin and MS-275 on BDNF transport. We found that tubacin increased the velocity of BDNF vesicles (Fig. 3E) and reduced their pausing time (Fig. 3F), whereas MS-275 had no effect. We conclude that tubacin, which is a specific inhibitor of HDAC6, preferentially increases tubulin acetylation and stimulates intracellular transport.
To further demonstrate the involvement of HDAC6 in the TSA-mediated effect on transport, we overexpressed WT HDAC6 (HDAC6WT) or HDAC6m1m2, an HDAC6 mutant having no deacetylase activity (Seigneurin-Berny et al., 2001), in striatal cells. We next treated the cells with TSA and analyzed by immunofluorescence the level of acetylation of α-tubulin in transfected and untransfected cells. A threshold concentration of 100 nm of TSA was sufficient to enhance significantly the acetylation of α-tubulin (Fig. 3G) and to stimulate vesicular transport (Fig. 3H,I). As described previously (Matsuyama et al., 2002), overexpression of HDAC6WT, but not of HDAC6m1m2, inhibited the acetylation of MTs that is induced by TSA. This also demonstrates that tubulin acetylation induced by TSA is because of the inhibition of the deacetylase activity of HDAC6. In agreement, only high concentrations of TSA are able to overcome the effect of increased HDAC6 activity to induce tubulin acetylation in the HDAC6WT-overexpressing cells (data not shown; Matsuyama et al., 2002). We then compared the dynamics of BDNF vesicles between HDAC6WT and HDAC6m1m2 transfected cells. Strikingly, we found that the stimulatory effect of TSA on BDNF transport was lost in cells expressing HDAC6WT (Fig. 3H,I). However, in cells overexpressing the nonactive HDAC6m1m2 mutant, the TSA-induced increase in BDNF vesicle velocity and decrease in pausing time were not affected compared with untransfected cells. Together, these results show that TSA-induced activation of BDNF transport is mediated, at least in part, through the inhibition of HDAC6 deacetylase activity.
Tubulin acetylation correlates with enhanced transport but not with MT stability
We next considered how TSA, HDAC6 inhibition, and the subsequent increased acetylation of tubulin could contribute to increased intracellular transport. Acetylation has been associated with stable MTs. Therefore, we first analyzed the effect of TSA and tubacin on MT growth in striatal cells using MT EB3 coupled to eGFP. EB3 is a neuron-specific “plus-end” binding protein of the EB1 family, whose dynamics reflect the plus-end MT polymerization rate (Stepanova et al., 2003). As a positive control, we used taxol, a potent stimulator of MT stabilization. Low concentrations of taxol reduced the MT polymerization rate (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). However, neither TSA nor tubacin significantly affected the MT polymerization rate, suggesting that acetylation has no detectable effect on polymerization. Also, TSA had no effect on the localization of endogenous EB1 (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material). Because these approaches give only an indirect value of MT polymerization, we aimed to analyze the effect of increased tubulin acetylation on other MT dynamic parameters. We used Cos7 cells because these cells contain all of the machinery required for tubulin acetylation and, in contrast to neuronal cells, have a large and flattened cytoplasm as well as a low density of MTs. These cells were transfected with mCherry-α-tubulin (Shaner et al., 2004) and treated or not with TSA. Monitoring the dynamics of mCherry-α-tubulin in transfected cells allowed the analysis of the polymerization and depolymerization of MTs at a single MT level as well as the determination of rescue and catastrophe frequencies. These parameters are defined as the transitions between depolymerization and repolymerization phases (rescue) and between repolymerization and depolymerization phases (catastrophe). We tracked MT tips and found that the rates of MT polymerization and depolymerization were not affected by the presence of TSA (Table 1). Also, we observed no significant differences in the rescue and catastrophe frequencies after TSA treatment, suggesting that MT acetylation has little or no effect on MT dynamics.
Table 1.
Effect of HDAC inhibition on microtubule dynamics and stability
| Polymerization rate (μm/min) | Depolymerization rate (μm/min) | Catastrophe frequency (min−1) | Rescue frequency (min−1) | ||
|---|---|---|---|---|---|
| WT | CT (n = 71) | 13.536 ± 0.776 | 13.448 ± 0.811 | 2.77 ± 0.134 | 2.726 ± 0.136 |
| TSA (n = 48) | 12.108 ± 0.862 | 11.776 ± 0.86 | 2.935 ± 0.138 | 2.898 ± 0.138 | |
| K40A | CT (n = 106) | 12.224 ± 0.522 | 14.819 ± 1.027 | 2.937 ± 0.115 | 2.779 ± 0.12 |
MT dynamics are not affected by acetylation. MT dynamic parameters in Cos7 cells transfected with WT mCherry-α-tubulin or mCherry-α-tubulin K40A and treated or not with TSA (1 μm) were determined by tracking the MT tips.
Finally, we also assessed by immunofluorescence and immunoblotting analyses the levels of tubulin detyrosination. Detyrosination occurs after MT assembly and is considered a marker of MT stability (Westermann and Weber, 2003). We found that unlike taxol, TSA and tubacin did not affect tubulin detyrosination (supplemental Fig. 1B,C, available at www.jneurosci.org as supplemental material). Because we did not find any clear association between MT acetylation and stability, we next analyzed the influence of MT stabilization on intracellular transport. We examined this by treating striatal WT cells with TSA or taxol and comparing the dynamics of BDNF vesicles. TSA increased intracellular transport of BDNF, whereas taxol had no significant effect on vesicle velocity and pausing time (supplemental Fig. 1D,E, available at www.jneurosci.org as supplemental material). Together, these results indicate that tubulin acetylation increases transport but has no effect on MT stability and, conversely, that stabilizing MTs has no impact on BDNF intracellular transport.
Inhibition of tubulin deacetylation in cells increases recruitment of dynein/dynactin and kinesin-1 motor complexes to MTs
We have shown previously that BDNF transport is under the control of the retrograde dynein/dynactin complex (Gauthier et al., 2004). However, the molecular motor leading to anterograde transport of BDNF vesicles is unknown. Recent studies have suggested an association between kinesin-1 and HAP1 (Huntingtin-associated protein 1) (McGuire et al., 2006). Using an RNAi approach, we tested the effect of lowering the levels of kinesin-1 and -2 on BDNF trafficking. We found that BDNF transport was reduced in cells treated with siRNA directed against kinesin-1 but not in cells treated with siRNA directed against kinesin-2 (Fig. 4A–C). This suggests that the BDNF vesicular transport depends on the anterograde motor kinesin-1 but not on kinesin-2. Our findings are in agreement with the involvement of conventional kinesin-1 in the anterograde transport of the BDNF receptor TrkB (Yano and Chao, 2004) and with the recent study showing the importance of kinesin-1 in the in vivo transport of BDNF in retinal cells (Butowt and von Bartheld, 2007).
Figure 4.

Inhibition of tubulin deacetylation increases the recruitment of motor complex proteins on MTs. A–C, WT (+/+) cells were transfected with BDNF-eGFP and siRNA directed against scramble RNA (scRNA), kinesin 2 (siKin2), or kinesin 1 (siKin1) 1 d before BDNF-eGFP transfection. A, Total extracts were analyzed by Western blot for kinesin-1 (KHC), kinesin-2 (KIF3A), and α-tubulin. B, C, Cells were analyzed by videomicroscopy. Dynamics were quantified by the use of the mean velocity of vesicles per cell (B) and their pausing time (C). D, MT polymerization experiments were performed from WT cells treated for 4 h with DMSO (0.5%, control, CT) or TSA (5 μm), and analyzed for the presence of kinesin (KHC), dynactin (p150Glued), dynein (DIC), htt, acetylated tubulin, and α-tubulin in S and P fractions using whole-cell extract (WCE). E, Quantitative assessment of the optical density of kinesin (KHC), dynactin (p150Glued), dynein (DIC) expressed as (S/MT P)TSA/(S/MT P)CT ratios. Results were normalized to 1. *p < 0.05; ***p < 0.001. F, WT (+/+) cells were treated for 4 h with DMSO (0.1%, CT) or TSA (1 μm), fixed, and stained for tyrosinated tubulin (green) and dynein (DIC; red) or kinesin (KHC; red). Insets correspond, respectively, to a 1.5× and 3× enlargement of the black and white and color images. G, Representative linescan analyses of the distributions of dynein (DIC) and kinesin (KHC) from the MT tips in CT (blue) and after TSA (1 μm; red) treatment.
Given that acetylation of MTs, and not their stabilization, correlates with increased transport and the observation that the MT-based transport of BDNF involves the dynein/dynactin (Gauthier et al., 2004) and kinesin-1 motor complexes, led us to hypothesize that MT acetylation could regulate the binding of these motor complexes to MTs. Therefore, we analyzed the association of dynein/dynactin and kinesin-1 to MTs under TSA treatment using biochemical approaches. MTs from striatal +/+ cells treated or not with TSA were depolymerized and centrifuged to isolate the soluble fraction containing free tubulin and soluble proteins (Fig. 4D). This fraction was subjected to a subsequent polymerization and sedimentation. We obtained two fractions: the soluble proteins in the S and the polymerized MTs and MAPs in the P. Under TSA treatment, we observed a statistically significant increase in the association of KHC, p150Glued dynactin subunit, and DIC to MTs (Fig. 4D,E).
We next analyzed by immunofluorescence the distribution of DIC and KHC on tyrosinated MTs in the absence or presence of TSA (Fig. 4F). As expected, we observed that KHC was associated with punctate structures along the MTs and that the levels of DIC were higher at the plus ends of polymerizing MTs. When WT cells were treated with TSA, the recruitment of these proteins to MTs was clearly increased (Fig. 4F, insets). Notably, we observed an increased association of these motor proteins along the entire length of the MTs. In support, we also observed an increased recruitment of the p150Glued subunit of dynactin (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material). In contrast, the localization and intensity of EB1, an MT polymerizing plus-end tracking protein, was not affected by TSA treatment. We quantified the recruitment of the motor proteins to MTs by creating a binary mask from the MT image. Analysis of >30 cells (three independent experiments) per condition revealed a statistically significant increase in the association of DIC, KHC, and p150Glued but not EB1 to the MTs (DIC: 122.0% with respect to CT, unpaired t test, t(77) = −2.757, p = 0.008; KHC: 131.6% with respect to CT, unpaired t test, t(75) = −3.311, p = 0.0014) (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material).
As an anti-tyrosinated tubulin antibody stains dynamic MTs, we further confirmed the increased binding of DIC, KHC, and p150Glued to the total pool of MTs using an anti-α-tubulin antibody. We quantified the recruitment of the proteins to the MTs by performing 3D linescan analyses on individual MTs. Single MTs were identified using α-tubulin staining, and a line following the MT structure was generated automatically using an ImageJ plug-in (Fig. 4G). The level of recruitment was expressed as the pixel intensity over the MT length and normalized to control. Again, data from two independent experiments revealed a statistically significant increase in the recruitment of DIC, kinesin, and p150Glued but not EB1 (DIC: 153.2% with respect to CT, unpaired t test, t(154) = −3.226, p = 0.0015; KHC: 231.0% with respect to CT, unpaired t test, t(164) = −4.022, p < 0.0001) (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material). Interestingly, analysis of individual linescans reveals an increase in binding along the entire length of the MTs (Fig. 4G). Altogether, our results show that increasing tubulin acetylation leads to the recruitment of DIC, kinesin, and p150Glued to MTs.
In vitro binding of molecular motors to MTs is enhanced by acetylation
To investigate whether the increased recruitment of cytoplasmic dynein and kinesin-1 depends directly on the acetylation of MTs, we tested the in vitro binding of purified motor components directly on polymerized MTs. Purified tubulin was polymerized into MTs that were next chemically acetylated using AA as described previously (Piperno and Fuller, 1985). Acetylated MTs were pelleted onto coverslips and analyzed by immunostaining. We found that AA treatment induced acetylation at lysine 40 of α-tubulin and had no obvious effect on the length of the MTs or on the level of their detyrosination (supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
Having created a system for the acetylation of MTs in vitro, we assessed the binding of kinesin-1 and cytoplasmic dynein to these acetylated MTs, using methods described previously (Ligon et al., 2006). For kinesin-1, we used a recombinant protein that corresponds to the human KHC motor domain and binds effectively to MTs (Wada et al., 2000). To assess the binding of dynein to MTs, we purified cytoplasmic dynein from bovine brain tissue (Bingham et al., 1998). Absence of any significant contamination from dynactin was established through SDS-PAGE and Western blotting (data not shown). Polymerized MTs were chemically acetylated and incubated with either recombinant KHC or purified dynein and pelleted onto coverslips. Consistent with previous studies (Wada et al., 2000; Mallik et al., 2005), we observed in the control condition a binding of KHC and dynein (Fig. 5). We next determined the binding of both KHC and cytoplasmic dynein after tubulin acetylation and observed a marked increase in the recruitment of motors to the acetylated MTs. We quantified the fluorescence intensity of molecular motors along MTs by performing linescan analyses on 60–150 individual MTs. The increase in the binding of KHC and dynein to acetylated MTs was statistically significant (data from two independent experiments; KHC: 262.5% with respect to CT, unpaired t test, t(134) = −3.394, p = 0.0009; DIC: 358.8% with respect to CT, unpaired t test, t(67) = −3.923, p = 0.0002). We conclude that tubulin acetylation leads to the direct recruitment of the molecular motors cytoplasmic dynein and kinesin-1 on MTs.
Figure 5.
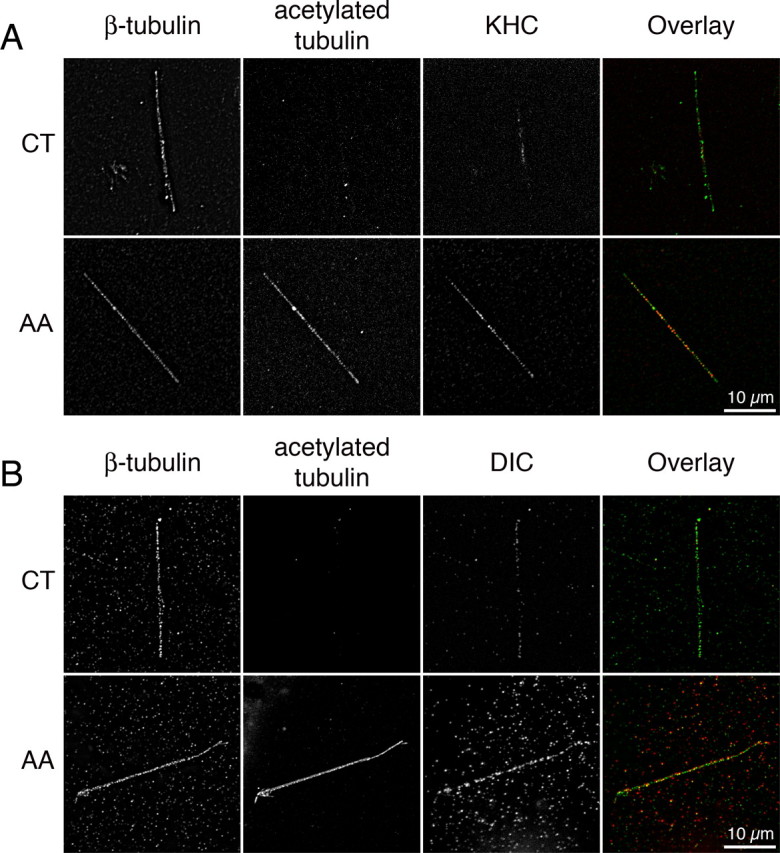
In vitro binding of molecular motors to MTs is enhanced by acetylation. A, B, Recombinant MTs were polymerized and incubated or not with 1% AA for 10 min, incubated with recombinant kinesin (A) or with purified dynein (B), pelleted onto coverslips, fixed, and stained for β-tubulin, acetylated tubulin, and kinesin (KHC) (A) or dynein (DIC) (B).
Mutation of the acetylation site in tubulin inhibits acetylation and reduces recruitment of motor complex proteins and BDNF transport
To unequivocally establish that MT acetylation promotes the attachment of motor complex proteins to MTs and subsequently the efficiency of transport, we generated a K40A mutant in mCherry-α-tubulin because mutation of this lysine of α-tubulin in T. thermophila and in Chlamydomonas reinhardtii results in decreased acetylation of tubulin (Kozminski et al., 1993; Gaertig et al., 1995). This mCherry-α-tubulin K40A mutant was correctly incorporated into the MTs as shown by immunofluorescence (Fig. 6A), and videomicroscopy experiments did not reveal any differences in the MT dynamics parameters compared with the WT mCherry-α-tubulin (Table 1). However, the ability of this mutant to be acetylated after TSA treatment was lost compared with the WT mCherry-α-tubulin as demonstrated by immunofluorescence (Fig. 6A) and Western blotting analyses (Fig. 6B). We next analyzed the consequences of the K40A mutation on the dynamics of BDNF vesicles by performing two-color 3D videomicroscopy on striatal cells transfected with BDNF-eGFP and with the WT or the K40A form of mCherry-α-tubulin (Fig. 6C,D) (supplemental videos 4–7, available at www.jneurosci.org as supplemental material). In control conditions in which cells show a low level of acetylation, we observed a small reduction of BDNF transport in cells expressing the mCherry-α-tubulin K40A construct. However, whereas TSA significantly increased BDNF transport in WT mCherry-α-tubulin-expressing cells, the ability of TSA to increase BDNF transport was lost in mCherry-α-tubulin K40A-expressing cells showing that the stimulatory effect of TSA requires acetylation of tubulin at lysine 40 (supplemental videos 4–7, available at www.jneurosci.org as supplemental material).
Figure 6.

Tubulin acetylation at lysine 40 regulates intracellular transport. A, WT (+/+) cells transfected with WT mCherry-α-tubulin or with mCherry-α-tubulin K40A were treated with DMSO (0.1%, control, CT) or TSA (1 μm) for 4 h, fixed, and immunostained for acetylated tubulin. B, HEK 293 cells were transfected and treated as in A and analyzed by immunoblotting for acetylated- and total-α-tubulin. C, D, WT (+/+) cells were cotransfected with BDNF-eGFP and with WT mCherry-α-tubulin or with mCherry-α-tubulin K40A, treated with DMSO (0.1%, CT) or TSA (1 μm) for 4 h, and analyzed by videomicroscopy. Dashed lines correspond to the control values. N.S., Not significant. *p < 0.05; **p < 0.01.
Because TSA treatment increases the association of dynein/dynactin and kinesin-1 to MTs, we determined in vitro the recruitment of motors to MTs that contain either an intact lysine 40 or a lysine mutated into an alanine. We transfected HEK293 cells with WT mCherry-α-tubulin or K40A constructs, treated the cells with TSA (1 μm), and then purified the MTs by two successive rounds of depolymerization/polymerization in the presence of TSA and high ATP concentration. These conditions led to the removal of most, if not all, motors from the MTs (data not shown). The purified MT fraction was next repolymerized, incubated with either recombinant KHC or purified dynein, and analyzed as above except that quantifications were expressed as a kinesin (or dynein) intensity/mCherry-α-tubulin intensity/MT length ratio. Quantification of motors on MTs revealed a statistically significant reduction in the binding of both KHC and dynein when lysine 40 was mutated (data from two independent experiments; KHC: 35.8% with respect to wild type, unpaired t test, t(161) = 5.214, p < 0.0001; dynein: 33.7% with respect to wild type, unpaired t test, t(171) = 5.416, p < 0.0001). Altogether, our in vitro experiments demonstrate that MT acetylation on lysine 40 increases motor recruitment to MTs.
MT acetylation is altered in HD patients and is enhanced by TSA in neurons
To assess the physiological relevance of our findings to HD, we determined the level of tubulin acetylation in postmortem striatal samples from grade 3 and 4 HD patients. Strikingly, we found a dramatic decrease in the level of acetylated-α-tubulin in HD patients compared with control individuals (Fig. 7A). The total levels of α-tubulin were not changed, further demonstrating a specific decrease of tubulin acetylation in HD.
Figure 7.
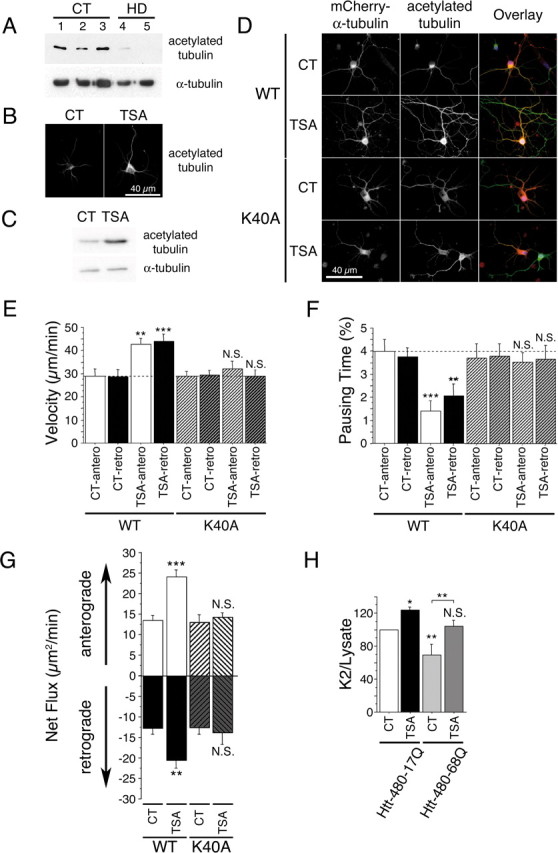
Tubulin acetylation at lysine 40 stimulates bidirectional transport and rescues BDNF release in HD. A, Acetylated tubulin levels are decreased in the brain of HD patients. Protein extracts are prepared from whole striatum of CT (samples 1–3) and HD individuals (HD grade 3, sample 4; HD grade 4, sample 5) and analyzed by immunoblotting for acetylated and total α-tubulin. B, C, Cortical neurons were treated with DMSO (0.1%, CT) or TSA (1 μm) for 4 h and analyzed by immunofluorescence (B) and immunoblotting (C) for the presence of acetylated tubulin. D, Cortical neurons were transfected with WT mCherry-α-tubulin or mCherry-α-tubulin K40A, treated for 4 h with DMSO (0.1%, CT) or TSA (1 μm), and analyzed by immunofluorescence for the presence of mCherry-α-tubulin (red) and acetylated tubulin (green). E, F, Cortical neurons were cotransfected with BDNF-eGFP and either WT mCherry-α-tubulin or mCherry-α-tubulin K40A, treated for 4 h with DMSO (0.1%, CT) or TSA (1 μm), and anterograde and retrograde vesicular movements in neurites were analyzed by videomicroscopy. Dotted lines correspond to the control values. G, TSA treatment increases anterograde and retrograde flux in a lysine 40-dependent manner. H, Cortical neurons were electroporated with BDNF and either htt-480–17Q or htt-480–68Q and treated for 4 h with DMSO (0.1%, CT) or TSA (1 μm). Transport-dependent BDNF release after a second KCl-induced depolarization is expressed as a K2/lysate ratio. N.S., Not significant. *p < 0.05; **p < 0.01; ***p < 0.001.
We next tested whether HDAC inhibitors are able to increase acetylation levels in cortical neurons. Indeed, postmitotic differentiated neurons have stable MTs and a high level of acetylation. However, TSA treatment still led to a significant increase in the acetylation of MTs as confirmed both by immunostaining approaches and by immunoblotting analyses (Fig. 7B,C). TSA had a similar capacity to induce MT acetylation in neurons as in other cell types such as Cos7 cells and WT or mutant striatal cells (supplemental Fig. 4A, available at www.jneurosci.org as supplemental material). We also analyzed the effect of TSA on the distribution of BDNF-eGFP vesicles in cortical neurons and found, as for striatal cells, no significant changes in the localization of BDNF vesicles (supplemental Fig. 4B, available at www.jneurosci.org as supplemental material).
Acetylation at Lys40 of tubulin enhances bidirectional transport in cortical neurons
We next investigated the effect of lysine 40 mutation on both MT acetylation and transport in neurons. Whereas TSA treatment led to a significant increase of tubulin acetylation when neurons express the WT mCherry-α-tubulin, TSA was unable to induce acetylation in neurons that express mCherry-α-tubulin K40A (Fig. 7D). We then analyzed anterograde and retrograde transport of BDNF-eGFP in neurites of cortical neurons, because we observed that TSA induced the recruitment to MTs of both anterograde and retrograde motors in vitro and in cells. TSA treatment significantly increased intracellular transport of vesicles in both directions to the same extent (Fig. 7E,F). Additional analyses revealed that none of the transport values were significantly affected by the overexpression of WT mCherry-α-tubulin (data not shown). Strikingly, we found that the capacity of TSA to increase transport in either direction was lost when α-tubulin cannot be acetylated at lysine 40 (Fig. 7E,F).
We next determined the net flux of BDNF vesicles in both anterograde and retrograde directions. We found that MT acetylation at lysine 40 induced a significant increase in the net flux in both directions. This effect was abolished when α-tubulin cannot be acetylated at lysine 40 (Fig. 7G). Together, we conclude that TSA mediates its stimulatory effect on BDNF transport in neurons through the acetylation of α-tubulin at lysine 40.
MT acetylation in neurons promotes release of BDNF in HD
To analyze the physiological consequences of an increased transport in primary neurons, we electroporated primary cultures of cortical neurons with constructs encoding BDNF and N-terminal fragments of htt that contain the first 480 amino acids with 17Q or 68Q and determined the capacity of neurons to release BDNF when treated with TSA. The 480 N-terminal fragment of htt is as efficient as the full-length htt in promoting intracellular transport (data not shown). We first depolarized neurons to release the pool of vesicles present at the membrane and performed a K2 30 min after the first one to release the vesicles that had been transported to the plasma membrane (Gauthier et al., 2004). The quantity of BDNF released during the K2 normalized to the BDNF content in neuronal lysates reflects transport-dependent release of BDNF. This value was decreased when MTs are depolymerized (Gauthier et al., 2004). As described previously (Gauthier et al., 2004), htt that contains the polyQ expansion released a lower amount of BDNF compared with WT htt (Fig. 7H). However, TSA induced a statistically significant increase in the release of BDNF in neurons expressing either WT or polyQ htt. Our results show that by acetylating MTs at lysine 40, TSA promotes axonal transport and the release of BDNF from neurons. Furthermore, TSA is efficient at restoring the disrupted BDNF transport and release in disease, making it of therapeutic interest in HD.
Discussion
We demonstrate here that HDAC inhibitors increase MT-dependent transport by the inhibition of HDAC6. This inhibition leads to increased acetylation of lysine 40 of α-tubulin and the subsequent recruitment of kinesin-1 and dynein/dynactin to the more acetylated MTs. Our results are in agreement with a recent study by Verhey et al. (Reed et al., 2006) that demonstrated a role for the acetylation at Lys40 of α-tubulin in T. thermophila in the regulation of kinesin-1 binding to MTs. α-Tubulin acetylation at Lys40 was found to enhance kinesin-1 motility along MTs in vitro. In cells, MT acetylation induced an increase in the transport of JIP1. In particular, pharmacological inhibition of HDAC6 in neurons led to the redirection of kinesin-1 transport of JIP1 to the tips of the neurites. Our study shows that, in addition to the recruitment of kinesin-1, MT acetylation also leads to the recruitment of the retrograde motor dynein. Analysis of the dynamic properties of BDNF-containing vesicles revealed that MT acetylation stimulates not only anterograde but also retrograde transport, suggesting a general role for MT acetylation in the stimulation of intracellular dynamics through the recruitment of both anterograde and retrograde motors. A general role for MT acetylation is also supported by the observations that not only transport of cargo proteins such as JIP1 but also of BDNF-containing vesicles is enhanced when MTs are acetylated. In support, we also found that stimulation of vesicular transport was not restricted to BDNF-containing vesicles as intracellular dynamics of lysosomes and of VSV-G, a glycoprotein from the vesicular stomatitis virus, which can be used to follow vesicular transport in the secretory pathway, were also enhanced by TSA treatment (data not shown).
How does acetylated tubulin recruit molecular motors? According to cryoelectron microscopy and 3D reconstruction of intact MTs, lysine 40 is located in the lumen of the MT (Nogales et al., 1998). The increased binding in vitro of dynein and kinesin-1 to acetylated MTs (present study and Reed et al., 2006) suggests that motors are directly recruited to acetylated MTs. Therefore, one possible explanation for how acetylation could lead to an increased binding of motor proteins is that acetylation could change the conformation of MTs to a form with higher affinity for motors. In agreement, several studies focusing on the binding of kinesin-1 to MTs have demonstrated a change in the conformations between plain MTs and MT-kinesin complexes, suggesting that MTs might regulate intracellular transport through modulations of their core structure (Krebs et al., 2004; Skiniotis et al., 2004). A similar mechanism could occur for the dynein/dynactin complex. In support, dynein and kinesin-1 share an overlapping binding site on MTs (Mizuno et al., 2004).
We show that HDAC inhibitors that enhance tubulin acetylation compensate for the transport deficit in striatal cells that contain a pathological polyQ expansion in the htt protein. In particular, using various parameters to assess intracellular transport (mean velocity, pausing time, percentage of static vesicles, and processivity), we show that treatment of HD cells with TSA and SAHA almost completely restore BDNF transport values to the control situation. Although the transport of mitochondria is not altered in HD striatal cells (Gauthier et al., 2004), we observed defects in the transport of small vesicles, such as lysosomes, suggesting an impairment in the transport of other organelles than BDNF in HD (data not shown). Interestingly, the beneficial effect of HDAC6 inhibition is also not restricted to BDNF-containing vesicles because TSA treatment enhances transport of lysosomes and of secretory vesicles such as VSV-G-labeled organelles. Therefore, HDAC6 inhibition, by acting on a wide population of organelles whose transport is altered in disease, could have a larger and more efficient therapeutic effect.
As shown in this study and by Verhey et al. (Reed et al., 2006), this mechanism is relevant in neurons because HDAC6 inhibition in primary cultures of neurons results in an effective MT acetylation and stimulates axonal transport. Indeed, tubacin treatment of hippocampal neurons enhances anterograde transport of JIP1 to the tips of neurites (Reed et al., 2006). We show here that MT acetylation stimulates both anterograde and retrograde transport of BDNF-containing vesicles in the neurites of cortical neurons. Finally, we found that TSA treatment leads to BDNF release in cortical neurons expressing polyQ htt. Together with our previous observations demonstrating an impairment in the axonal transport and subsequent release of BDNF vesicles in cortical neurons expressing polyQ htt, this adds to the rationale that HDAC6 inhibition could be an effective therapeutic strategy in HD.
HDAC6 was reported previously to participate in the clearance of misfolded ubiquitinated proteins in cells by promoting dynein/retrograde dependent transport of such proteins to the centrosome (Kawaguchi et al., 2003). PolyQ-containing proteins, such as htt, also accumulate into aggregates of misfolded proteins that are actively transported to the centrosome to be degraded by the ubiquitin-proteasome system (UPS) or through autophagic degradation when the UPS is impaired (Ravikumar et al., 2002; Ciechanover and Brundin, 2003; Qin et al., 2003; Iwata et al., 2005). Interestingly, efficient autophagic degradation of polyQ-containing peptides is dependent on HDAC6, dynein, and MT-dependent transport (Webb et al., 2004; Iwata et al., 2005; Ravikumar et al., 2005). Here, we dissected the mechanism by which tubulin acetylation enhances intracellular transport along MTs. We believe this HDAC6-dependent mechanism to be distinct from those involved in the clearance of misfolded aggregates for several reasons. First, whereas this transport is a dynein/retrograde-dependent transport to the centrosome, TSA stimulates anterograde and retrograde transport. Second, we show that HDAC6 inhibition increases transport both in WT cells and in primary cultures of neurons. These experiments were performed independently of the pathological context. Third, in experiments using mutant cells that recapitulate the genetic situation observed in HD patients, no microscopically visible aggregates could be detected (data not shown). Finally, we show that HDAC6 inhibition rescues intracellular transport in the pathological situation. These experiments are close to the physiological conditions because full length WT and mutant htt are present in striatal cells at endogenous levels and do not involve the overexpression of short polyQ-htt fragments that are highly susceptible to aggregate formation.
Inhibition of tubulin deacetylases other than HDAC6 could also be of interest in an HD context. In particular, the NAD+-dependent sirtuin 2 (SIRT2) HDAC shows some substrate preference for tubulin peptides compared with histone peptides, and SIRT2 knockdown through siRNA results in tubulin hyperacetylation (North et al., 2003). It will be interesting, when available, to test whether selective SIRT2 inhibitors are able to stimulate intracellular transport.
HDAC inhibitors, such as SAHA, TSA, and NaPB, exhibit neuroprotective effects by inhibiting the HDAC1 enzyme (Butler and Bates, 2006). Here we show that some of these drugs could also protect neurons in HD by promoting the intracellular transport of BDNF through the inhibition of HDAC6 and subsequent tubulin acetylation. Our results support the strategy of combining drugs that allow an effective inhibition of the enzymes of both HDAC1 and HDAC6 families.
Our results validate the use of 3D fast videomicroscopy to screen for compounds able to restore intracellular transport in HD neurons. We show here that the increased acetylation of MTs induced by HDAC6 inhibitors can act as a general mechanism to regulate MT-based transport. Therefore, our finding may also have implications for other neurodegenerative disorders, such as Alzheimer's disease (AD), in which intracellular transport is altered (Kamal et al., 2001; Pigino et al., 2003; Stokin et al., 2005). Indeed, the analysis of postmortem brain samples from AD patients revealed reduced α-tubulin acetylation in neurons with neurofibrillary forms of tau (Hempen and Brion, 1996; Saragoni et al., 2000).
Footnotes
This work was supported by grants from Agence Nationale de la Recherche (MRAR-018-01 to F.S.), Association Française contre les Myopathies (F.S.), Association pour la Recherche sur le Cancer (ARC; 3665 to S.H.), Fondation pour la Recherche Médicale and Fondation BNP Paribas (F.S.), Fédération pour la recherche sur le cerveau (F.S.), HighQ Foundation (F.S., S.H.), Provital P. Chevalier (F.S., S.H.), and National Institutes of Health (NS 48501 to S.J.K.). J.P.D. was supported by the HighQ foundation and currently by Trophos. J.D.G. was supported by a Région Ile de France doctoral fellowship, and B.C.C. was supported by an ARC doctoral fellowship. S.H. is an Institut National de la Santé et de la Recherche Médicale (INSERM) investigator. F.S. is a recipient of a European Molecular Biology Organization Young Investigator award and an INSERM/Assistante Publique-Hôpitaux de Paris investigator. We gratefully acknowledge J. C. Wong, R. Mazitschek, and S. L. Schreiber, who were supported by the Initiative for Chemical Genetics-National Cancer Institute; B. Gilquin, S. Khochbin, V. Lessmann, N. Galjart, M. E. MacDonald, R. Y. Tsien, and M. A. Knossow for reagents and/or discussions; Harvard Brain Tissue Resource Center (Belmont, MA), which is supported in part by PHS Grant MH/NS 31862, for providing human brain tissue; and members of the Saudou/Humbert laboratory for help and comments.
References
- Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, Acheson AL, Lindsay RM, Wiegand SJ. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature. 1997;389:856–860. doi: 10.1038/39885. [DOI] [PubMed] [Google Scholar]
- Baquet ZC, Gorski JA, Jones KR. Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. J Neurosci. 2004;24:4250–4258. doi: 10.1523/JNEUROSCI.3920-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingham JB, King SJ, Schroer TA. Purification of dynactin and dynein from brain tissue. Methods Enzymol. 1998;298:171–184. doi: 10.1016/s0076-6879(98)98017-x. [DOI] [PubMed] [Google Scholar]
- Butler R, Bates GP. Histone deacetylase inhibitors as therapeutics for polyglutamine disorders. Nat Rev Neurosci. 2006;7:784–796. doi: 10.1038/nrn1989. [DOI] [PubMed] [Google Scholar]
- Butowt R, von Bartheld CS. Conventional kinesin-I motors participate in the anterograde axonal transport of neurotrophins in the visual system. J Neurosci Res. 2007 doi: 10.1002/jnr.21165. in press. [DOI] [PubMed] [Google Scholar]
- Ciechanover A, Brundin P. The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron. 2003;40:427–446. doi: 10.1016/s0896-6273(03)00606-8. [DOI] [PubMed] [Google Scholar]
- Drummond DC, Noble CO, Kirpotin DB, Guo Z, Scott GK, Benz CC. Clinical development of histone deacetylase inhibitors as anticancer agents. Annu Rev Pharmacol Toxicol. 2005;45:495–528. doi: 10.1146/annurev.pharmtox.45.120403.095825. [DOI] [PubMed] [Google Scholar]
- Evans L, Mitchison T, Kirschner M. Influence of the centrosome on the structure of nucleated microtubules. J Cell Biol. 1985;100:1185–1191. doi: 10.1083/jcb.100.4.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaertig J, Cruz MA, Bowen J, Gu L, Pennock DG, Gorovsky MA. Acetylation of lysine 40 in alpha-tubulin is not essential in Tetrahymena thermophila. J Cell Biol. 1995;129:1301–1310. doi: 10.1083/jcb.129.5.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier LR, Charrin BC, Borrell-Pages M, Dompierre JP, Rangone H, Cordelieres FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, Saudou F. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Goldstein LS, Laymon RA, McIntosh JR. A microtubule-associated protein in Drosophila melanogaster: identification, characterization, and isolation of coding sequences. J Cell Biol. 1986;102:2076–2087. doi: 10.1083/jcb.102.6.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardena S, Her LS, Brusch RG, Laymon RA, Niesman IR, Gordesky-Gold B, Sintasath L, Bonini NM, Goldstein LS. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron. 2003;40:25–40. doi: 10.1016/s0896-6273(03)00594-4. [DOI] [PubMed] [Google Scholar]
- Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci USA. 2003;100:4389–4394. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann M, Heumann R, Lessmann V. Synaptic secretion of BDNF after high-frequency stimulation of glutamatergic synapses. EMBO J. 2001;20:5887–5897. doi: 10.1093/emboj/20.21.5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haubensak W, Narz F, Heumann R, Lessmann V. BDNF-GFP containing secretory granules are localized in the vicinity of synaptic junctions of cultured cortical neurons. J Cell Sci. 1998;111:1483–1493. doi: 10.1242/jcs.111.11.1483. [DOI] [PubMed] [Google Scholar]
- Hempen B, Brion JP. Reduction of acetylated alpha-tubulin immunoreactivity in neurofibrillary tangle-bearing neurons in Alzheimer's disease. J Neuropathol Exp Neurol. 1996;55:964–972. doi: 10.1097/00005072-199609000-00003. [DOI] [PubMed] [Google Scholar]
- Hu E, Dul E, Sung CM, Chen Z, Kirkpatrick R, Zhang GF, Johanson K, Liu R, Lago A, Hofmann G, Macarron R, de los Frailes M, Perez P, Krawiec J, Winkler J, Jaye M. Identification of novel isoform-selective inhibitors within class I histone deacetylases. J Pharmacol Exp Ther. 2003;307:720–728. doi: 10.1124/jpet.103.055541. [DOI] [PubMed] [Google Scholar]
- Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- Humbert S, Bryson EA, Cordelieres FP, Connors NC, Datta SR, Finkbeiner S, Greenberg ME, Saudou F. The IGF-1/Akt pathway is neuroprotective in Huntington's disease and involves Huntingtin phosphorylation by Akt. Dev Cell. 2002;2:831–837. doi: 10.1016/s1534-5807(02)00188-0. [DOI] [PubMed] [Google Scholar]
- Ingold AL, Cohn SA, Scholey JM. Inhibition of kinesin-driven microtubule motility by monoclonal antibodies to kinesin heavy chains. J Cell Biol. 1988;107:2657–2667. doi: 10.1083/jcb.107.6.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem. 2005;280:40282–40292. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS. Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature. 2001;414:643–648. doi: 10.1038/414643a. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–738. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- Kohara K, Kitamura A, Morishima M, Tsumoto T. Activity-dependent transfer of brain-derived neurotrophic factor to postsynaptic neurons. Science. 2001;291:2419–2423. doi: 10.1126/science.1057415. [DOI] [PubMed] [Google Scholar]
- Kozminski KG, Diener DR, Rosenbaum JL. High level expression of nonacetylatable alpha-tubulin in Chlamydomonas reinhardtii. Cell Motil Cytoskeleton. 1993;25:158–170. doi: 10.1002/cm.970250205. [DOI] [PubMed] [Google Scholar]
- Krebs A, Goldie KN, Hoenger A. Complex formation with kinesin motor domains affects the structure of microtubules. J Mol Biol. 2004;335:139–153. doi: 10.1016/j.jmb.2003.10.039. [DOI] [PubMed] [Google Scholar]
- Lee WC, Yoshihara M, Littleton JT. Cytoplasmic aggregates trap polyglutamine-containing proteins and block axonal transport in a Drosophila model of Huntington's disease. Proc Natl Acad Sci USA. 2004;101:3224–3229. doi: 10.1073/pnas.0400243101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligon LA, Shelly SS, Tokito MK, Holzbaur EL. Microtubule binding proteins CLIP-170, EB1, and p150Glued form distinct plus-end complexes. FEBS Lett. 2006;580:1327–1332. doi: 10.1016/j.febslet.2006.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald ME, Gines S, Gusella JF, Wheeler VC. Huntington's Disease. Neuromolecular Med. 2003;4:7–20. doi: 10.1385/NMM:4:1-2:7. [DOI] [PubMed] [Google Scholar]
- Mallik R, Petrov D, Lex SA, King SJ, Gross SP. Building complexity: an in vitro study of cytoplasmic dynein with in vivo implications. Curr Biol. 2005;15:2075–2085. doi: 10.1016/j.cub.2005.10.039. [DOI] [PubMed] [Google Scholar]
- Manders EM, Stap J, Brakenhoff GJ, van Driel R, Aten JA. Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J Cell Sci. 1992;103:857–862. doi: 10.1242/jcs.103.3.857. [DOI] [PubMed] [Google Scholar]
- Matsuyama A, Shimazu T, Sumida Y, Saito A, Yoshimatsu Y, Seigneurin-Berny D, Osada H, Komatsu Y, Nishino N, Khochbin S, Horinouchi S, Yoshida M. In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J. 2002;21:6820–6831. doi: 10.1093/emboj/cdf682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire JR, Rong J, Li SH, Li XJ. Interaction of Huntingtin-associated protein-1 with kinesin light chain: implications in intracellular trafficking in neurons. J Biol Chem. 2006;281:3552–3559. doi: 10.1074/jbc.M509806200. [DOI] [PubMed] [Google Scholar]
- Mizuno N, Toba S, Edamatsu M, Watai-Nishii J, Hirokawa N, Toyoshima YY, Kikkawa M. Dynein and kinesin share an overlapping microtubule-binding site. EMBO J. 2004;23:2459–2467. doi: 10.1038/sj.emboj.7600240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogales E, Wolf SG, Downing KH. Structure of the alpha beta tubulin dimer by electron crystallography. Nature. 1998;391:199–203. doi: 10.1038/34465. [DOI] [PubMed] [Google Scholar]
- North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell. 2003;11:437–444. doi: 10.1016/s1097-2765(03)00038-8. [DOI] [PubMed] [Google Scholar]
- Pigino G, Morfini G, Pelsman A, Mattson MP, Brady ST, Busciglio J. Alzheimer's presenilin 1 mutations impair kinesin-based axonal transport. J Neurosci. 2003;23:4499–4508. doi: 10.1523/JNEUROSCI.23-11-04499.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piperno G, Fuller MT. Monoclonal antibodies specific for an acetylated form of alpha-tubulin recognize the antigen in cilia and flagella from a variety of organisms. J Cell Biol. 1985;101:2085–2094. doi: 10.1083/jcb.101.6.2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin ZH, Wang Y, Kegel KB, Kazantsev A, Apostol BL, Thompson LM, Yoder J, Aronin N, DiFiglia M. Autophagy regulates the processing of amino terminal huntingtin fragments. Hum Mol Genet. 2003;12:3231–3244. doi: 10.1093/hmg/ddg346. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. 2002;11:1107–1117. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Acevedo-Arozena A, Imarisio S, Berger Z, Vacher C, O'Kane CJ, Brown SD, Rubinsztein DC. Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat Genet. 2005;37:771–776. doi: 10.1038/ng1591. [DOI] [PubMed] [Google Scholar]
- Reed NA, Cai D, Blasius TL, Jih GT, Meyhofer E, Gaertig J, Verhey KJ. Microtubule acetylation promotes kinesin-1 binding and transport. Curr Biol. 2006;16:2166–2172. doi: 10.1016/j.cub.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Saragoni L, Hernandez P, Maccioni RB. Differential association of tau with subsets of microtubules containing posttranslationally-modified tubulin variants in neuroblastoma cells. Neurochem Res. 2000;25:59–70. doi: 10.1023/a:1007587315630. [DOI] [PubMed] [Google Scholar]
- Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- Seigneurin-Berny D, Verdel A, Curtet S, Lemercier C, Garin J, Rousseaux S, Khochbin S. Identification of components of the murine histone deacetylase 6 complex: link between acetylation and ubiquitination signaling pathways. Mol Cell Biol. 2001;21:8035–8044. doi: 10.1128/MCB.21.23.8035-8044.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- Skiniotis G, Cochran JC, Muller J, Mandelkow E, Gilbert SP, Hoenger A. Modulation of kinesin binding by the C-termini of tubulin. EMBO J. 2004;23:989–999. doi: 10.1038/sj.emboj.7600118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepanova T, Slemmer J, Hoogenraad CC, Lansbergen G, Dortland B, De Zeeuw CI, Grosveld F, van Cappellen G, Akhmanova A, Galjart N. Visualization of microtubule growth in cultured neurons via the use of EB3-GFP (end-binding protein 3-green fluorescent protein) J Neurosci. 2003;23:2655–2664. doi: 10.1523/JNEUROSCI.23-07-02655.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LS. Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science. 2005;307:1282–1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- Szebenyi G, Morfini GA, Babcock A, Gould M, Selkoe K, Stenoien DL, Young M, Faber PW, MacDonald ME, McPhaul MJ, Brady ST. Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport. Neuron. 2003;40:41–52. doi: 10.1016/s0896-6273(03)00569-5. [DOI] [PubMed] [Google Scholar]
- Taddei A, Roche D, Bickmore WA, Almouzni G. The effects of histone deacetylase inhibitors on heterochromatin: implications for anticancer therapy? EMBO Rep. 2005;6:520–524. doi: 10.1038/sj.embor.7400441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toomre D, Keller P, White J, Olivo JC, Simons K. Dual-color visualization of trans-Golgi network to plasma membrane traffic along microtubules in living cells. J Cell Sci. 1999;112:21–33. doi: 10.1242/jcs.112.1.21. [DOI] [PubMed] [Google Scholar]
- Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, Cattaneo E, MacDonald ME. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet. 2000;9:2799–2809. doi: 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- Trushina E, Dyer RB, Badger JD, II, Ure D, Eide L, Tran DD, Vrieze BT, Legendre-Guillemin V, McPherson PS, Mandavilli BS, Van Houten B, Zeitlin S, McNiven M, Aebersold R, Hayden M, Parisi JE, Seeberg E, Dragatsis I, Doyle K, Bender A, et al. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol Cell Biol. 2004;24:8195–8209. doi: 10.1128/MCB.24.18.8195-8209.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan KT, Tynan SH, Faulkner NE, Echeverri CJ, Vallee RB. Colocalization of cytoplasmic dynein with dynactin and CLIP-170 at microtubule distal ends. J Cell Sci. 1999;112:1437–1447. doi: 10.1242/jcs.112.10.1437. [DOI] [PubMed] [Google Scholar]
- Wada Y, Hamasaki T, Satir P. Evidence for a novel affinity mechanism of motor-assisted transport along microtubules. Mol Biol Cell. 2000;11:161–169. doi: 10.1091/mbc.11.1.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb JL, Ravikumar B, Rubinsztein DC. Microtubule disruption inhibits autophagosome-lysosome fusion: implications for studying the roles of aggresomes in polyglutamine diseases. Int J Biochem Cell Biol. 2004;36:2541–2550. doi: 10.1016/j.biocel.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Westermann S, Weber K. Post-translational modifications regulate microtubule function. Nat Rev Mol Cell Biol. 2003;4:938–947. doi: 10.1038/nrm1260. [DOI] [PubMed] [Google Scholar]
- Xia Z, Dudek H, Miranti CK, Greenberg ME. Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J Neurosci. 1996;16:5425–5436. doi: 10.1523/JNEUROSCI.16-17-05425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano H, Chao MV. Mechanisms of neurotrophin receptor vesicular transport. J Neurobiol. 2004;58:244–257. doi: 10.1002/neu.10321. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Li N, Caron C, Matthias G, Hess D, Khochbin S, Matthias P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003;22:1168–1179. doi: 10.1093/emboj/cdg115. [DOI] [PMC free article] [PubMed] [Google Scholar]