

Abstract
Postsynaptic nitric oxide (NO) production affects synaptic plasticity and neuronal cell death. Ca2+ fluxes through the NMDA receptor (NMDAR) stimulate the production of NO by neuronal nitric oxide synthase (nNOS). However, the mechanisms by which nNOS activity is regulated are poorly understood. We evaluated the effect of neuronal stimulation with glutamate on the phosphorylation of nNOS. We show that, in cortical neurons, a low glutamate concentration (30 μm) induces rapid and transient NMDAR-dependent phosphorylation of S1412 by Akt, followed by sustained phosphorylation of S847 by CaMKII (calcium-calmodulin-dependent kinase II). We demonstrate that phosphorylation of S1412 by Akt is necessary for activation of nNOS by the NMDAR. nNOS mutagenesis confirms that these phosphorylations respectively activate and inhibit nNOS and, thus, transiently activate NO production. A constitutively active (S1412D), but not a constitutively repressed (S847D) nNOS mutant elevated surface glutamate receptor 2 levels, demonstrating that these phosphorylations can control AMPA receptor trafficking via NO. Notably, an excitotoxic stimulus (150 μm glutamate) induced S1412, but not S847 phosphorylation, leading to deregulated nNOS activation. S1412D did not kill neurons; however, it enhanced the excitotoxicity of a concomitant glutamate stimulus. We propose a swinging domain model for the regulation of nNOS: S1412 phosphorylation facilitates electron flow within the reductase module of nNOS, increasing nNOS sensitivity to Ca2+-calmodulin. These findings suggest a critical role for a kinetically complex and novel series of regulatory nNOS phosphorylations induced by the NMDA receptor for the in vivo control of nNOS.
Keywords: neuronal nitric oxide synthase, nNOS, NMDA receptor, AMPA receptor, excitotoxicity, phosphorylation, dephosphorylation
Introduction
Signaling by the NMDA receptor (NMDAR) plays a major role in mechanisms of synaptic plasticity and cell death (Vanhoutte and Bading, 2003). In the case of NMDAR control of the novel second messenger, nitric oxide (NO), the enzyme neuronal nitric oxide synthase (nNOS), which catalyzes NO synthesis, is tethered to the NMDAR by the scaffolding protein postsynaptic density-95 (PSD-95). In this complex, NMDAR-mediated Ca2+ influxes regulate nNOS activity and NO production (Bredt and Snyder, 1989; Garthwaite et al., 1989; Kornau et al., 1997; Christopherson et al., 1999).
NO function has been implicated in the modification of neuronal circuitry in numerous brain systems, including those functioning in learning and memory (Rose, 2000), auditory fear conditioning (Schafe et al., 2005), drug addiction (Balda et al., 2006), and visual adaptation to light (Sekaran et al., 2005). Changes in synaptic strength, such as LTP or LTD, are thought to involve the rapid movement of AMPA receptors (AMPARs) into and out of synapses (Malinow and Malenka, 2002). NO exerts its effects by activating the cGMP/protein kinase G pathway and by S-nitrosylation of proteins (Bredt, 2003). S-nitrosylation of N-ethylmaleimide-sensitive factor (NSF) increases the surface expression of the glutamate receptor 2 (GluR2) subunit of AMPARs (Huang et al., 2005). A pathway dependent on NO, cGMP-dependent kinase, RhoA, and actin increases GluR1 in synaptic puncta (Huang et al., 2005).
Under pathological circumstances, overproduction of NO and increased levels of reactive oxygen species can lead to the formation of peroxynitrite, a reactive agent implicated in mechanisms of excitotoxicity after stroke (Keynes and Garthwaite, 2004). S-nitrosylation and nitration of proteins by peroxynitrite also exacerbate neurological disorders such as dementia (Giasson et al., 2000), Parkinson's disease (Chung et al., 2004), and Alzheimer's disease (Uehara et al., 2006).
The control of NO production by the NMDAR at neural synapses involves regulatory phosphorylations of nNOS (Rameau et al., 2003, 2004). However, it is not known how these phosphorylations are induced in neurons and the mechanism by which they regulate nNOS activity. Calcium-calmodulin-dependent kinase II (CaMKII), which phosphorylates S847 of nNOS (Rameau et al., 2004) binds to the C-terminal domain of the NR2B subunit (Robison et al., 2005) in proximity to nNOS tethered by PSD-95. Also the C-terminal tail of nNOS contains a putative site for phosphorylation at S1412 (Adak et al., 2001a). Phosphatidylinositol-3 (PI3) kinase binds to tyrosine phosphorylated NR2B leading to phosphorylation of phosphoinositides and the recruitment and activation of Akt at the plasma membrane, which could phosphorylate nNOS (Hisatsune et al., 1999; Waxman and Lynch, 2005).
Here, we examine how NMDAR stimulation by glutamate regulates the phosphorylation of nNOS at S1412 and S847. Because functions of the NMDAR depend on the intensity of the glutamate stimulus (Hardingham and Bading, 2003), we determined how changes in the concentration of the glutamate stimulus affect nNOS phosphorylation. Our findings show that by regulating nNOS phosphorylations at S1412 and S847, the NMDAR tightly couples its own activity to nNOS biochemical functions. We interpret the regulation through a molecular model of nNOS and show that these phosphorylations can induce changes in surface AMPAR abundance and mediate glutamate excitotoxicity by controlling NO production.
Materials and Methods
Cell culture and pharmacology.
Cultured embryonic day 18 (E18) rat cortical and hippocampal neurons were prepared as described previously (Rameau et al., 2000). Except when used for Western blots, the cultures were treated with cytosine β-d-arabinofuranoside 3 d after plating and until non-neuronal cells were absent at 15–21 d in vitro (DIV). For Western blots, pharmacological treatments were performed at 10–13 DIV. Comparisons were between treated and untreated cells from the same cultures and three plates were pooled to make lysates.
Western blots and immunocytochemistry.
The preparation of lysates for Western blots was described previously (Rameau et al., 2004). Each experiment was performed in triplicate and repeated at least three times. Equal amounts of proteins were fractionated on an SDS-PAGE gel and densitometric analysis of Western blots was performed with NIH Image software. For immunostaining, cultures were fixed and blocked as described previously (Rameau et al., 2003). For surface staining, incubations with anti-GluR1 (N-terminal antibody, 1:250 dilution; Millipore, Temecula, CA) and anti-GluR2 (N-terminal antibody, 1:100 dilution; Millipore) were performed before cell permeabilization overnight at room temperature and were labeled after permeabilization with 0.2% Triton X-100. Incubations with anti-S1412-PO4-nNOS, anti-S847-PO4-nNOS, and anti-PSD-95 monoclonal antibody (Upstate, Lake Placid, NY) were also performed overnight. All secondary antibodies were applied for 1 h at room temperature and were conjugated to either Alexa Fluorescein or Alexa Rhodamine-Red-X (Invitrogen, Eugene, OR). Images were acquired using a Nikon (Tokyo, Japan) PCM2000 confocal laser-scanning microscope. Synaptophysin-positive areas were selected as the regions of interest and the total intensity of synaptophysin, anti-S1412-PO4-nNOS, or anti-S847-PO4-nNOS was measured in these areas. Synapses were isolated along primary dendritic branches within a box of 6.7 × 25.91 mm per cell; only individual synapses that could be distinguished clearly were included and analysis was performed using Image J software. For surface GluR2 and GluR1, the intensity of the antibody-conjugated fluorochromes was measured for primary dendrites within boxes of identical size for images acquired using identical settings using C-Imaging Systems software. Signal from an average of 20 neurons per coverslip was averaged to obtain a population mean (presented as mean ± SEM). Statistical significance of differences between means was calculated using Student's t test.
Antibodies.
Anti-S1412-PO4 nNOS serum was generated by immunizing rabbits with a synthetic phosphopeptide synthesized by the Tufts University Core Facility (Boston, MA) corresponding to 12 amino acids in nNOS: NH2-CRLRSE(S-PO4)IAFIE-COOH. Antibodies were generated and affinity-purified by Covance (Princeton, NJ). Anti-PSD-95 monoclonal antibody was purchased from Upstate, monoclonal anti-synaptophysin from Sigma (St. Louis, MO), and polyclonal anti-microtubule-associated protein 2 (MAP-2) from Santa Cruz Biotechnology (Santa Cruz, CA).
cGMP assays.
Cultured cortical neurons were grown 10 DIV, lysed, and cGMP levels were assayed using a cGMP Direct Biotrak Enzyme Immunoassay kit (GE Healthcare, Pittsburgh, PA). Where indicated, cells were preincubated with inhibitors (Akt inhibitor, 50 μm and MK-801, 15 μm, both from Calbiochem, La Jolla, CA) for 5 min before glutamate or NMDA stimulation.
TUNEL assays.
Cortical neurons were grown 15 DIV, infected for 8 h with Sindbis virus expressing S1412D, wild-type nNOS, or green fluorescent protein (GFP), and were treated for 10 min with glutamate 30 or 150 μm, added directly to the growth medium. The cells were washed once and returned to the growth medium for 3 h of incubation, after which they were fixed and permeabilized. The neurons were stained for terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) using the in situ death detection kit (Boehringer Mannheim, Indianapolis, IN). The cells were costained with a polyclonal antibody to the Myc epitope tag (Santa Cruz) and a monoclonal antibody to MAP-2, a positive marker for neurons. Detection was performed using a goat anti-rabbit antibody conjugated to Rhodamine (Invitrogen) and a goat anti-mouse antibody conjugated to Cy5 (blue channel) (Jackson ImmunoResearch, Bar Harbor, ME). Representative field of cells were systematically scanned at 20× magnification, with an average of 15 infected cells per field. We scanned 20 fields for an average of 300 infected neurons per experimental condition and the experiment was repeated three times. Neuroprotection or cell death induced by the various nNOS mutants tested was derived from the following equation: TUNEL(glutamate) − TUNEL(no glutamate)/Total(no glutamate) infected cells.
Cloning.
The rat nNOS cDNA in pBluescript (SK(−)) was a kind gift from Dr. Solomon Snyder, Johns Hopkins University, Baltimore, MD. The cDNA was excised by PCR with a pair of primers containing an XbaI restriction site plus the sequence for the C-myc epitope tag (upstream, 5′GCTCTAGAGCCACCATGGAACAAAAACTCATCTCAGAAGAGGATCTGGAAGAGAACACGTTTGGGGTTCAGCAGATCCAACCCAAT; downstream, 5′GTTCTAGACAGTTAGG AGCTGAAAACCTCATCTGCGTCTTTTTTGCTCTCTTCGAT) using high-fidelity polymerase (Roche Molecular Biochemicals, Indianapolis, IN), and subcloned into pcDNA3 vector and into pSinrep5 vector. The putative Akt phosphorylation and the CaMKII sites in nNOS were mutated using the Quick Change site-directed mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer instructions. All mutants were verified by DNA sequencing.
Statistical analysis.
Data are presented as means ± SEM. Statistical analysis was performed using the unpaired Student's t test and ANOVA, and Bonferroni/Dunn post hoc test using Stat View (Abacus Concepts, Berkeley, CA).
Results
Detection of phosphorylation of nNOS at S1412 with a phosphopeptide antibody specific for S1412
nNOS contains a potential site for phosphorylation at S1412 at its C-terminal tail that is analogous to the established Akt site found in endothelial nitric oxide synthase (Dimmeler et al., 1999; Fulton et al., 1999; Gallis et al., 1999; Michell et al., 1999) (Fig. 1A). A second phosphorylation site at S847 resides in an α-helix of the autoinhibitory (AI) domain of NOS reductase (NOSred) (Hayashi et al., 1999; Rameau et al., 2004) (Fig. 1A). To determine whether Akt phosphorylates nNOS, purified nNOS expressed in Escherichia coli was incubated with the Akt enzyme in an in vitro 32PO4-ATP kinase assay. Robust labeling confirmed that the nNOS protein is an Akt substrate (Fig. 1B). We prepared an affinity-purified anti-phosphopeptide antibody specific for nNOS S1412-PO4. This antiserum recognized bacterially expressed nNOS after in vitro Akt phosphorylation, but not an unphosphorylated nNOS control (Fig. 1C), confirming that Akt phosphorylates nNOS at S1412 and demonstrating the specificity of the antibody. This antibody also recognized nNOS in rat whole-brain lysates (Fig. 1D), indicating that basal levels of S1412 phosphorylation exist in the brain.
Figure 1.

Detection of phosphorylation of nNOS at S1412 with a phosphopeptide antiserum specific for S1412. A, Schematic diagram of nNOS showing the NOSox and NOSred modules, the CaM-binding linker, and the location of the phosphorylation sites at S1412 and S847. The AI insert, the C-terminal tail (CT), and the FMN domain of NOS are also illustrated. B, Autoradiogram of SDS-PAGE of 32P-nNOS labeled in the presence of γ32P ATP and Akt. C, Western blots showing the specificity of the antibody against S1412-PO4 for nNOS phosphorylated by Akt. NOS was incubated with Akt plus nonradioactive ATP, or without any enzyme as a control. Lane 1, No enzyme, control; lane 2, Akt. A Western blot for total nNOS showing equivalent levels of nNOS protein loading. The specificity of the antibody to S1412-PO4 for phosphorylated nNOS was demonstrated further using extracts from an adult rat brain. D, Western blots of a rat whole-brain lysate, fractionated by 10% SDS-PAGE, with antibody to S1412-PO4 and polyclonal nNOS antibody (Transduction Laboratories, Lexington, KY).
NMDAR-mediated pathways differentially regulate S1412 and S847 phosphorylation
The regulation of phosphorylation of endogenous nNOS at S1412 by the NMDAR has not been investigated, nor have the kinetics of phosphorylation at this site relative to phosphorylation of S847. To address these issues, we used the phosphospecific S1412-PO4 nNOS antibody and a previously reported S847-PO4 nNOS antibody (Rameau et al., 2004) to analyze nNOS phosphorylation induced by the NMDAR in cultured primary embryonic neurons.
Primary cultured cortical neurons 13–15 DIV were treated with 30 μm glutamate, a concentration that mimics a physiologic stimulus (McBain and Mayer, 1994), and were then analyzed for S1412-PO4 and S847-PO4 by Western blotting. S1412-PO4 phosphorylation increased rapidly after stimulation, peaking at a fivefold increase over basal levels in 5 min, followed by a decline of the signal to below basal levels (Fig. 2A,C). This finding indicates rapid sequential actions of a kinase and a phosphatase. In contrast, phosphorylation at S847 increased slowly from 15 to 45 min peaking at eightfold over the basal levels (Fig. 2B,C). This differential phosphorylation indicates that glutamate induces NMDAR-dependent, self-limiting phosphorylations of nNOS at S847 and S1412.
Figure 2.

Glutamate regulates phosphorylation of nNOS at S847 and S1412 at synapses and Akt phosphorylation at S1412. A, Time course of increase of phosphorylation at S1412. Lane 1, No glutamate, control; lane 2, 30 μm glutamate, 5 min; lane 3, 30 μm glutamate, 15 min; lane 4, 30 μm glutamate, 30 min; lane 5, 30 μm glutamate, 45 min. Western blot for total nNOS is shown below. B, Time course of increase of phosphorylation at S847. Lane 1, No glutamate, control; lane 2, 30 μm glutamate, 5 min; lane 3, 30 μm glutamate, 15 min; lane 4, 30 μm glutamate, 30 min; lane 5, 30 μm glutamate, 45 min. Western blot for total nNOS is shown below. C, Quantitation of A and B was performed by taking the ratio of phospho-nNOS to total nNOS.
Phosphorylation of nNOS at S1412 by Akt and regulation of nNOS by phosphorylation takes place at synapses
PSD-95 tethers nNOS to the NMDAR at synapses, thus providing a basis for close coupling of nNOS to the NMDAR (Kornau et al., 1995). This tethering also suggests preferential induction of phosphorylation of nNOS in dendritic spines. S1412-PO4 nNOS colocalized with the synaptic marker synaptophysin in unstimulated neurons, and the synaptic S1412-PO4 signals increased in cells stimulated for 5 min with 30 μm glutamate (Fig. 3A,B). This demonstrates NMDAR-dependent S1412 phosphorylation of the synaptic population of nNOS and confirms the S1412-PO4 increase detected by Western blot. The synaptic S847-PO4 signal also increased after 30 μm glutamate treatment for 30 min (Fig. 3C,D). Although a proportion of S1412 and S847 phospho-nNOS was cytoplasmic, the most significant accumulations of S1412-PO4 and S847-PO4 nNOS were in synaptic puncta (Fig. 3A,C). Western blots show that the NMDAR antagonist APV and the PI3 kinase inhibitor LY294002 [2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one] each blocked the S1412-PO4 increase (Fig. 3E,F). This supports the conclusion that NMDAR activation induces phosphorylation of S1412 by Akt, a kinase whose activity is regulated by PI3 kinase.
Figure 3.
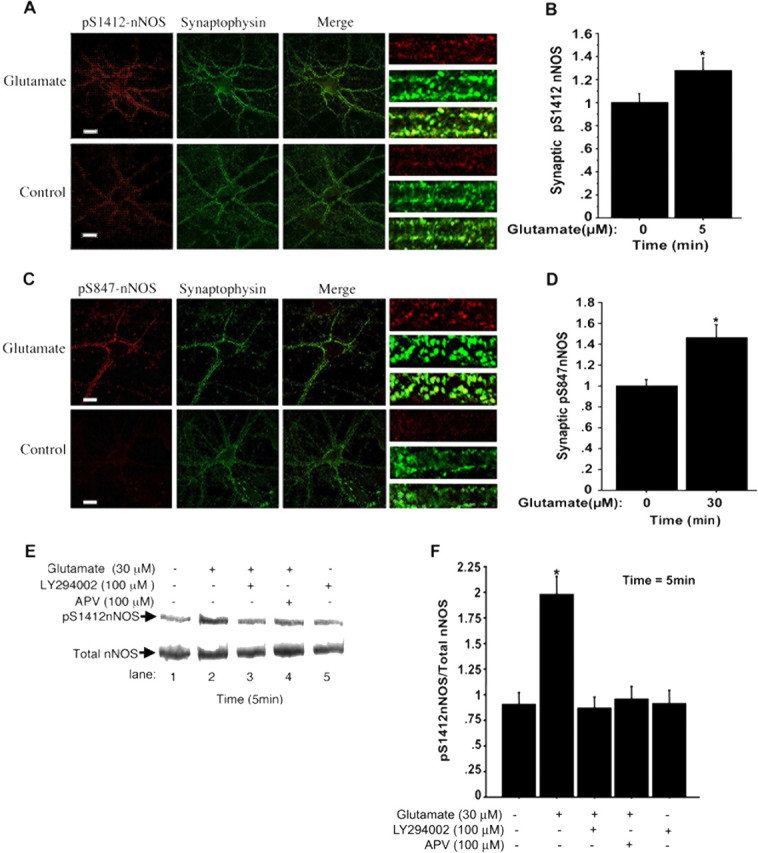
Glutamate treatment leads to increased synaptic S1412-PO4 and S847-PO4 nNOS. A, C, Representative images showing S1412-PO4 and S847-PO4 (red) and synaptophysin (green) in glutamate-treated cells and in no-drug control cells. On the right side are given magnifications of segments of primary proximal dendrites; the bottom image (merged; yellow) shows an example of synaptic regions identified by synaptophysin. B, Treatment with 30 μm glutamate for 5 min significantly increases the level of S1412-PO4 at synaptophysin containing puncta by ∼30% relative to control (n = 3; *p ≤ 0.006). D, Treatment with 30 μm glutamate for 30 min also significantly increases S847-PO4 levels at synaptophysin containing puncta by ∼50% relative to control (no glutamate; n = 3; *p ≤ 0.0001). Glutamate regulates Akt phosphorylation of nNOS at S1412. E, Phosphorylation at S1412 induced by stimulation with glutamate for 5 min is blocked by LY294002. Lane 1, No glutamate, control; lane 2, 30 μm glutamate; lane 3, 30 μm glutamate administered after preincubation with 100 μm LY294002; lane 4, 30 μm glutamate plus 100 μm APV; lane 5, 100 μm LY294002. F, Quantification of E, which shows that the PI3-kinase inhibitor LY294002 and the NMDAR antagonist APV significantly blocked NMDAR-induced phosphorylation at S1412 (n = 4; *p ≤ 0.002). Scale bars, 20 μm.
NMDAR-induced phosphorylation of nNOS by Akt is required to increase neuronal nNOS activity.
The effects of phosphorylation of S1412 on endogenous nNOS activity in neurons and the impact of this phosphorylation on cellular function are not known. We use a pharmacological approach to determine how nNOS phosphorylation and nNOS activity are coupled in neurons. The activity of endogenous nNOS was measured by assaying cGMP production, after an application of 30 or 150 μm glutamate for 2 min, using an ELISA-based assay. Glutamate induces cGMP production in a dose-dependent manner, with 1.5-fold and threefold increases observed after 30 and 150 μm treatments, respectively (n = 5; p ≤ 0.005 and 0.0001) (Fig. 4A). Production of cGMP was dependent on NOS and NMDAR activity because the increase in cGMP was blocked by the NOS inhibitor Nω-nitro-l-arginine (NA) and by the NMDAR antagonist MK801 (Fig. 4A). Akt phosphorylation of nNOS is essential for the increase in nNOS activity after NMDAR stimulation because Akt Inhibitor, a phosphatidylinositol ether analog that selectively inhibits Akt, blocked both NMDA and glutamate-induced elevation of cGMP (Fig. 4B,C). This dependence on Akt is also consistent with nNOS S1412 phosphorylation increasing the activity of the enzyme.
Figure 4.

Stimulation of NMDAR induces Akt activity required for nNOS activity. cGMP levels were measured after treatments with glutamate or NMDA and in the presence or absence of inhibitors of NMDAR or Akt. A, cGMP levels were increased 1.7-fold by 30 μm glutamate and 2.5-fold by 150 μm glutamate relative to the control with no agonist. Increase in cGMP levels by 150 μm glutamate stimulation was blocked by NA and by 150 and 15 μm MK801. B, Increase in cGMP by 150 μm glutamate was blocked by Akt inhibitor, 10 μm. C, NMDA, 50 μm, leads to a twofold increase in cGMP levels. Akt inhibitor, 10 μm, blocked the NMDAR-mediated increase in cGMP, as did 15 μm MK801. *p ≤ 0.005; **p ≤ 0.0001.
To analyze the function of nNOS phosphorylation, we mutated the regulatory serines of nNOS to alanines (to block phosphorylation) or aspartates (which act as phosphomimetic mutations). Western blots showed that cultured hippocampal neurons at 15–17 DIV expressed similar levels of wild-type, phosphomimetic, and phospho-blocking Myc-tagged-nNOS mutants when infected with Sindbis virus vectors expressing the different constructs (Fig. 5E). The expression of the nNOS mutants and wild-types were detected by anti-Myc antibody and their phenotypes were assayed in situ using an anti-cGMP antibody to label cGMP. The levels of cGMP were measured in individual neurons by quantifying the fluorescence intensity of images obtained by confocal microscopy, as described previously (Rameau et al., 2004). In the absence of glutamate stimulation, neurons expressing nNOS S1412D, which mimics the Akt phosphorylated form of nNOS, exhibited a twofold increase in cGMP (n = 4; p ≤ 0.0001) (Fig. 5A,F) relative to cells expressing wild-type nNOS (Fig. 5A,F). This confirms biochemical studies that suggest that phosphorylation at this site increases activity of the enzyme (Adak et al., 2001b). In cells expressing S847D nNOS (Fig. 5B,F), which mimics the CaMKII phosphorylated nNOS, cGMP levels were reduced to one-half (n = 4; p ≤ 0.026) of those detected in cells expressing wild-type nNOS (Fig. 5C,F), although the levels were still higher than in control cells expressing GFP (Fig. 5D,F). This demonstrates that phosphorylation of S847 reduces nNOS activity. Neither the S1412A nor the S847A mutant activity differed significantly from that of wild-type nNOS (Fig. 5F). This is consistent with the proposed roles of phosphorylation in the regulation of nNOS activity. These results also support the idea that NMDAR induction of sequential phosphorylations at S1412 and S847 facilitates transient nNOS activation.
Figure 5.

nNOS regulation analyzed by phosphorylation site mutagenesis. A–D, Representative confocal images of neurons show cGMP levels (red) and myc-epitope-tagged nNOS (green) for neurons expressing (from Sindbis virus vectors) phosphomimetic mutants S1412D (A) and S847D (B), wild-type nNOS (C), and GFP (D) as a control. On the left side of each panel is a segment of a proximal dendrite, in boxed regions of identical size, that illustrates the levels of cGMP in cells expressing the phosphomimetics S1412D and S847D, and wild-type nNOS relative to cells expressing GFP as a control. E, Western blot showing the expression of the phosphomimetic and phospho-blocking nNOS mutants relative to wild-type nNOS. Graph showing nNOS activity from cells expressing mutants or wild-type nNOS relative to cells expressing cGMP as a control. F, Relative to cells expressing wild-type nNOS, cells expressing S1412D nNOS show a twofold increase in cGMP levels (n = 4; **p ≤ 0.0001), whereas cells expressing nNOS mutant S847D show a 1.5-fold to twofold decrease in cGMP levels (n = 4; *p ≤ 0.026). Expression of the S1412A and S847A nonphosphorylatable nNOS mutants did not change cGMP levels appreciably relative to wild-type nNOS. Scale bars, 20 μm.
nNOS phosphorylation differentially regulates AMPA receptor trafficking
Previous studies have shown that NO regulates GluR2 surface expression (Huang et al., 2005). Because phosphorylation of nNOS regulates the production of NO, we asked whether the phosphomimetic and phospho-blocking mutants at S1412 and S847 had differential effects on GluR2 trafficking. Surface GluR2 levels were determined by labeling the cells with anti-GluR2 antibodies that recognize the GluR2 extracellular domain, and receptor surface levels were measured by quantitative confocal microscopy. Expression of the S1412D mutant increased the surface GluR2 signal 2.5-fold relative to cells expressing GFP (n = 4; p ≤ 0.0002), whereas wild-type nNOS increased surface GluR2 1.5-fold compared with GFP (p ≤ 0.0068) (Fig. 6A–C,G). The S847D mutant did not significantly change surface GluR2 levels relative to control neurons expressing GFP (Fig. 6G). The S1412A and S847A mutants increase surface GluR2 to the same extent as the wild-type nNOS expression (Fig. 6G). These results are in agreement with a requirement for phosphorylation at these sites to control nNOS activity. The elevation of surface GluR2 induced by the S1412D mutant cannot be attributed to an increase in GluR2 production, because total GluR2 levels were similar in cells expressing S1412D or wild-type nNOS relative to the control, GFP (Fig. 6I). We next examined GluR1. The wild-type nNOS and all of the mutants increased surface GluR1 relative to GFP, but these increases were similar to one another (Fig. 6H). As with GluR2, the expression of wild-type or mutant nNOS did not significantly change the intracellular levels of GluR1 relative to cells expressing GFP (Fig. 6I), indicating that the effects of nNOS were at the level of trafficking. The lack of difference in GluR1 surface expression induced by wild-type nNOS and the mutants suggests that the production of NO has in each case saturated the machinery required for GluR1 trafficking. Additional studies are required to determine the mechanism by which cGMP contributes to trafficking of GluR1-containing AMPARs. We conclude that nNOS mutations that mimic the stimulatory and inhibitory phosphorylations at S1412 and S847, respectively, control GluR2-containing AMPAR trafficking, as predicted from the regulatory properties of these phosphorylations.
Figure 6.

Phosphomimetic nNOS mutations S1412D and S847D regulate AMPAR surface expression. Wild-type and nNOS mutants were expressed in cultured hippocampal neurons (21 DIV). Cells were immunostained for surface GluR2 (red) followed by permeabilization and staining for Myc-epitope-tagged nNOS. A–F, Representative images of hippocampal neurons expressing S1412D, wild-type nNOS, or GFP (green), respectively, from Sindbis virus. Scale bars, 20 μm. To the left of each panel, a segment of primary dendrite displays the levels of surface GluR2 or GluR1. A total of 240 boxed regions containing proximal dendrites were analyzed to quantify surface GluR2. G, Results of the quantification of surface GluR2 from cells expressing S1412D, S1412A, S847D, S847A, wild-type nNOS, and GFP as a control. We quantify surface GluR1 by using the same strategy used for determining surface GluR2. H, Results of the quantification of surface GluR1 from cells expressing S1412D, S1412A, S847D, S847A, wild-type nNOS, and GFP as a control. I, Cells were immunostained for total GluR1 and GluR2 by permeabilization and staining with antibody to GluR1 and GluR2. A total of 40 boxed regions containing proximal dendrites were analyzed for each experiment to quantify GluR2 and GluR1. For GluR2 surface expression, the experiments were repeated four times (n = 4; *p ≤ 0.0068; **p ≤ 0.0002) as indicated, and for GluR1 the experiments were repeated three times (n = 3; *p ≤ 0.0001).
Phosphorylation of nNOS is regulated by glutamatergic regulation of the kinase and phosphatase balance
Protein phosphorylation is regulated by the relative activities of kinases and phosphatases, which are in turn regulated by extracellular cues. In neurons, the differential stimulation of the NMDAR is thought to affect the magnitudes of NMDAR Ca2+ fluxes, and the relative activation of Ca2-regulated kinases and phosphatases (Dell'Acqua et al., 2006). Therefore, we investigated how the strength of the glutamate stimulus influences the kinetics of phosphorylation of nNOS. When neurons were stimulated with varying concentrations of glutamate for 5 min, phosphorylation of S1412 was highly dependent on glutamate stimulus concentration and was maximal at 30 μm glutamate, which gave a fivefold higher level than the control (n = 4; p ≤ 0.0001) (Fig. 7A,B). We also assayed phosphorylation kinetics over a 60 min time course, after treatment with 150 μm glutamate, an excitotoxic stimulus. At 0–15 min after such a stimulus, S847-PO4 levels declined progressively and this phosphorylation was not detected thereafter (Fig. 7G). In contrast, a 30 μm glutamate treatment increased S847-PO4 over the basal level from 15 to 45 min post stimulation. Excitotoxic treatment also altered S1412 phosphorylation kinetics. Treatment with 150 μm glutamate induced a transient increase that was more rapid than the increase after 30 μm glutamate; it peaked at 1 min and declined to below basal levels after 2.5 min (Fig. 7C). The S847 levels did not change during this interval (Fig. 7D). We conclude that an elevated glutamate stimulus (150 μm) induces transient S1412 phosphorylation with a more rapid rate of onset and decay than the lower glutamate stimulus (30 μm), but promotes S847 dephosphorylation (Figs. 2A, 7E). This indicates that high glutamate treatment induces the stimulatory, but not the inhibitory modification of nNOS.
Figure 7.

Deregulation of nNOS activity by an excitotoxic glutamate stimulus. The magnitude of the glutamate stimulus modifies the phosphorylation of nNOS at S1412. The levels of S1412 were determined by Western blots after 5 min of neuron stimulation with glutamate. A, Lane 1, No glutamate; lane 2, 5 μm glutamate; lane 3, 10 μm glutamate; lane 4, 30 μm glutamate; lane 5, 75 μm glutamate; lane 6, 100 μm glutamate; lane 7, 250 μm glutamate. B, Quantification of A from measurements that were done in triplicate for each experiment, with experiments repeated three times with similar results. C, D, Extended stimulation with excitotoxic glutamate decreases phosphorylation of S847. Lane 1, No glutamate, control; lane 2, 150 μm glutamate, 5 min; lane 3, 150 μm glutamate, 15 min; lane 4, 150 μm glutamate, 30 min; lane 5, 150 μm glutamate, 45 min; lane 6, 150 μm glutamate, 60 min. Treatment with excitotoxic glutamate increases the rate of nNOS phosphorylation at S1412. E, F, Levels of nNOS phosphorylation at S1412 (E) and S847 (F) are shown. Lane 1, No glutamate, control; lane 2, 150 μm glutamate, 30 s; lane 3, 150 μm glutamate, 1 min; lane 4, 150 μm glutamate, 2.5 min; lane 5, 150 μm glutamate, 5 min. Western blot for total nNOS for each sample is shown below. G, Quantification of C and D. p ≤ 0.002; **p ≤ 0.0001.
The finding that phosphorylations at the two nNOS sites are differentially regulated raised the possibility that multiple pathways may contribute to nNOS dephosphorylation. At glutamatergic synapses, non-NMDA glutamate-gated ion channels and voltage-sensitive ion channels may cooperate with the NMDAR to transduce the glutamate stimulus into electrical and biochemical responses. The activity of these receptors could modify the NMDAR signals that regulate nNOS phosphorylation. Therefore, we examined the role of AMPAR and L-type voltage sensitive channels in regulating NMDAR-induced phosphorylation of nNOS. We found that S1412-PO4 dephosphorylation after 150 μm glutamate treatment for 15 min (Fig. 8A, lane 2) was blocked by the L-type voltage-gated calcium channel (VGCC) blocker nifedipine and by CNQX, an AMPAR antagonist (Fig. 8A, lanes 3, 4), relative to a control with no glutamate (Fig. 8A, lane 1). Indeed, despite treatments with high glutamate, nNOS phosphorylation at S1412 increased by approximately twofold in the presence of nifedipine and CNQX. Under these conditions, however, nifedipine and CNQX did not block glutamate-induced dephosphorylation at S847 (Fig. 8A, lanes 3, 4). Interestingly, nifedipine and CNQX, in the absence of glutamate stimulation, strongly elevated the basal steady-state levels of S1412-PO4 by fivefold and sixfold, respectively, and of basal S847-PO4 by 2.5-fold within 15 min (Fig. 8A, lanes 5, 6). These results are consistent with the existence of a distinct AMPAR and L-type VGCC-dependent pathway that activates phosphatases responsible for nNOS dephosphorylation. To investigate further the pathways of nNOS dephosphorylation at S1412-PO4 and S847-PO4, we determined whether inhibitors of protein phosphatase 2B (PP2B)/calcineurin and PP1 block NMDAR-induced nNOS dephosphorylation. Treatments with glutamate, 30 and 150 μm, for 15 min depressed the level of phosphorylation at S1412 (Fig. 8D, lanes 2, 5) and the extent of dephosphorylation was reduced by 2 μm ascomycin, a PP2B inhibitor (Fig. 8D, lanes 3, 6) or 0.5 μm okadaic acid, which inhibits PP1 (Fig. 8D, lanes 4, 7). The steady-state levels of S847-PO4 were relatively unchanged by limited treatment with glutamate, 30 and 150 μm (Fig. 8D, lanes 2, 5). However, in the presence of 2 μm ascomycin (Fig. 6D, lanes 3, 6) or 0.5 μm okadaic acid (Fig. 8D, lanes 4, 7), the levels of S847-PO4 were increased. This is in agreement with our previous finding that PP1 dephos phorylates nNOS at S847 (Rameau et al., 2004). It is also possible that ascomycin or FK506 increases CaMKII phosphorylation and activity, which may contribute to recovery from NMDAR-mediated dephosphorylation of nNOS at this site. Together, these findings suggest that both PP1 and PP2B may act on the same nNOS regulatory pathway, consistent with the ability of PP2B to activate PP1 by dephosphorylating and inactivating the PP1 inhibitor, inhibitor-1 (Cohen, 2002).
Figure 8.

Role of L-type VGCC, AMPAR, and phosphatases in control of glutamate-induced dephosphorylation at S1412 and S847. Western blots were performed on cortical cultured neurons (10–15 DIV), which were treated for 15 min with glutamate alone or glutamate plus nifedipine and CNQX as indicated. A, Lane 1, No glutamate; lane 2, 150 μm glutamate; lane 3, 150 μm glutamate plus 1 μm nifedipine; lane 4, 150 μm glutamate plus 10 μm CNQX; lane 5, 1 μm nifedipine; lane 6, 10 μm CNQX. B, C, Quantification of A. Significance is comparison with treatment with 150 μm glutamate (*p ≤ 0.001, **p ≤ 0.0003). D, Calcineurin and PP1 inhibitors block NMDAR-induced dephosphorylation of S847-PO4 and S1412-PO4. Lane 1, No drug control; lane 2, 30 μm glutamate; lane 3, 30 μm glutamate plus 2 μm ascomycin; lane 4, 30 μm glutamate plus 0.5 μm okadaic acid; lane 5, 150 μm glutamate; lane 6, 150 μm glutamate plus 2 μm ascomycin; lane 7, 150 μm glutamate plus 0.5 μm okadaic acid. E, F, Quantification of D. *p ≤ 0.002; **p ≤ 0.0001.
Phosphorylation of nNOS regulates NO-induced cell death
Exposure of neurons to high levels of glutamate induces excitotoxicity, a form of neuron death that occurs after ischemia and other forms of neurodegenerative insult (Keynes and Garthwaite, 2004). A dependence of this pathology on NO was suggested by the accumulation of nitrotyrosine, a product of protein reaction with peroxynitrite that correlated with cell death (Ischiropoulos, 1998; Eliasson et al., 1999; Estevez et al., 1999; Zanelli et al., 2002; Rameau et al., 2003). We evaluated the contribution to toxicity of an increase in NO production caused by nNOS phosphorylation after stimulation with glutamate. For this purpose, we compared the extent of death of neurons expressing the constitutively active S1412D nNOS mutant to death of neurons expressing wild-type nNOS, using the TUNEL assay to quantify cell death. Surprisingly, in the absence of a glutamate stimulus, the expression of S1412D showed little effect on cell survival relative to control cells expressing wild-type nNOS or GFP (Fig. 9A). This is despite the fact that under the same conditions, the S1412D mutant showed greatly elevated levels of NO production as reflected by increased cGMP levels (Fig. 5F). Strikingly, TUNEL positivity increased up to fourfold relative to the uninfected control (n = 3; p ≤ 0.003) in neurons expressing S1412D (n = 3; p ≤ 0.003), 3 h after a brief (10 min) stimulation with 150 μm glutamate (Fig. 9B), whereas neurons expressing wild-type nNOS appeared initially resistant to the glutamate challenge as shown by decreased twofold (n = 3; p ≤ 0.0001) in TUNEL positivity relative to uninfected control. This suggests that a moderate increase in NO, provided by overexpression of wild-type nNOS could delay or precondition the neurons against glutamate-induced toxicity. However, these results also demonstrate that excess NO production by S1412D nNOS decreases the threshold for glutamate-induced excitotoxicity.
Figure 9.

Constitutive nNOS activation requires a concomitant glutamate stimulus to induce excitotoxicity. A, Comparison of death of neurons expressing GFP, S1412D, or wild-type nNOS using the TUNEL assay. Relative to a GFP control, expression of S1412D or wild-type nNOS did not significantly affect the viability of neurons. B, TUNEL positivity was measured in neurons expressing wild-type or S1412D nNOS after treatment with glutamate, 30 μm (nontoxic) or 150 μm (toxic), or with no treatment, as a control. Expression of S1412D nNOS enhanced the toxicity of a 150 μm glutamate stimulus whereas expression of wild-type nNOS reduced the toxicity, relative to cells without glutamate stimulation. The equation that was used to calculate the effects of the mutants on cell viability is given in Materials and Methods. *p ≤ 0.0001; p ≤ 0.0005.
Discussion
We have demonstrated a regulatory mechanism that couples stimulation of the NMDAR to the control of nNOS in which the influx of Ca2+ after NMDAR activation leads to rapid and transient NO production by nNOS. This regulation depends on distinct, sequential stimulatory and repressive nNOS phosphorylations that directly control enzyme function. The localization of these phosphorylated nNOS species at synapses indicates that the nNOS phosphorylations preferentially take place within synaptic nNOS-PSD-95-NMDAR complexes, and supports their role in mechanisms of synapse modification. The finding that phosphomimetic mutants reproduce the proposed stimulatory and repressive effects of phosphorylation on nNOS activity confirms the regulatory role of the phosphorylations.
Sequential stimulatory and inhibitory phosphorylations
We show that S1412 phosphorylation performed by the kinase Akt is required for NMDAR-induced production of NO. Furthermore, phosphomimetic mutation S1412D activates nNOS in the absence of NMDAR Ca2+ fluxes, which suggests that nNOS activity resulting from S1412 phosphorylation lowers its dependence on Ca2+. CaMKII, which is activated by Ca2+-CaM formed through CaM binding of Ca2+ entering via the NMDAR, carries out the phosphorylation at S847 (Rameau et al., 2004). S847 phosphorylation is thought to block the binding of the activator, Ca2+-CaM (Komeima et al., 2000). This is expected to place the enzyme in a state refractory to direct activation by Ca2+. The sequential course of nNOS phosphorylations indicates that each phosphorylation site provides an independent step in the activation and deactivation of nNOS and that collectively these steps tightly control the production of NO.
Structural mechanism for temporal sequence of phosphorylation
Based on NOS crystallographic structures (Garcin et al., 2004), together with the results presented here, we propose a structural mechanism for the sequential phosphorylations of nNOS (Fig. 10). This builds on our previous swinging domain hypothesis for the rate-limiting delivery of electrons from NOSred to NOSox for NO synthesis (Garcin et al., 2004), as summarized below. In the absence of activating levels of Ca2+-CaM and phosphorylation, the flavin mononucleotide (FMN) domain of NOSred remains in the “locked” state (Craig et al., 2002) observed in the dimeric nNOS reductase crystallographic structure (Garcin et al., 2004). In this locked conformation, the N-terminal FMN domain fits into a “cup” formed by the remaining C-terminal portion of NOSred. This locked position of the FMN domain buries the exposed electron-transferring end of the FMN cofactor against the adjacent flavin adenine dinucleotide (FAD) donor. Thus, FMN can accept incoming electrons, flowing from NADPH through FAD to FMN within NOSred, but cannot deliver outgoing electrons to the NOSox heme.
Figure 10.

Diagram illustrating the structural mechanism for temporal sequence of nNOS phosphorylations. A, In the presence of NADPH (red hexagon) and the absence of phosphorylations and Ca2+-CaM (red circles), the NOSred dimer (for simplicity, only one monomer is shown) remains in the locked state, and is covalently tethered by the extended CaM-binding linker (green dotted line) to the NOSox dimer [twofold symmetric turquoise and green subunits, with arginine substrate channel and opposing heme (labeled Fe) backface oriented forward on the right and left, respectively]. In the locked state, the N-terminal helical portion (magenta coil) of the C-terminal tail (CT) lies across the interface that the FMN domain (yellow) makes with the remainder of NOSred (green). The FMN (pale yellow) and FAD (cyan) flavin cofactors are buried at this interface, and positioned for the FMN to accept electrons (circled e−) from FAD. With activation of NMDAR, the influx of Ca2+ activates Ca2+-CaM to bind NOS, and to turn on a phosphorylation cascade in which PI3 kinase activates Akt to phosphorylate nNOS at CT S1412. B, In the next step, phosphorylation (green star) of S1412 leads to electrostatic repulsion of the CT from NOSred, which together with Ca2+-CaM binding to the NOS linker, frees the FMN domain from its locked conformation. C, Once released, the FMN domain is free to swing into its proposed electron-donating position, allowing electron delivery to the backface of the NOSox heme belonging to the other polypeptide (turquoise) of dimeric NOS. The open conformation of NOSred exposes CT and the AI helix within the autoinhibitory insert (magenta helix and flanking linkers) of the FMN domain. S1412 is rapidly dephosphorylated by the PP1 phosphatase. D, Meanwhile, Ca2+-CaM influx through activated NMDAR activates CaMKII to phosphorylate AI S847. E, With decreased levels of Ca2+-CaM, S847 is dephosphorylated by PPI, and NOSred returns to the locked conformation (A).
The nNOS S1412 phosphorylation site is located in the surface-exposed C-terminal tail, near the end of the ordered α-helix (residues 1397–1413) and the beginning of the flexible C terminus (Garcin et al., 2004). Stimulatory phosphorylation at S1412 by Akt would introduce a bulky negatively charged group into an already electrostatically negative environment and, thus, would be predicted to displace the C-terminal tail by electrostatic repulsion. We propose that phosphorylation of exposed S1412 will release the FMN-domain from its locked conformation, allowing it to become the swinging electron shuttle that delivers electrons from FMN to heme for NO synthesis. Release of the FMN domain would also free the AI helix, exposing previously sequestered S847 for phosphorylation by CaMKII. NMDAR elevates Ca2+, and the formation of Ca2+-CaM binds the covalent linker between the NOSox and NOSred modules to promote NO synthesis by altering the equilibrium between the electron-accepting and electron-delivering positions of the FMN domain. The structural features of the nNOSred module, as well as kinetics of kinase and phosphatase activation, likely influence the temporal sequence of nNOS phosphorylation. Thus, NMDAR-induced program of phosphorylation of nNOS serves as a binary switch for the transient production of NO after NMDAR activation.
Technical approaches and considerations
Conflicting reports in the literature pose ambiguities regarding the protective versus the neurotoxic role of NO in connection with stimulation with glutamate. Exposure to low concentrations of glutamate induces neuroprotective effects in dispersed neuronal cultures (Balazs et al., 1988; Bambrick et al., 1995; Hardingham et al., 2002; Jiang et al., 2005; Soriano et al., 2006). In contrast, high concentrations of glutamate trigger excitotoxicity in cultured neurons (Michaels and Rothman, 1990; Westbrook, 1993; Choi, 1994; Ankarcrona et al., 1995; Raymond et al., 1996). This disparity could be attributable to differences in culture conditions, leading neurons to express different amounts of nNOS and NO because of differences in growth factors, plate coating matrix reagents (Samdani et al., 1997a,b), the developmental age of the culture, and the timing and the intensity of the stimulus (Aizenman et al., 1998). Also, cultured neurons such as those used in this study may differ from tissues in vivo.
NO production has been measured by different methods, including electron paramagnetic resonance (EPR) (Tominaga et al., 1993; Wei et al., 1999), microdialysis (Adachi et al., 2000; Fassbender et al., 2000), and with microsensor probes (Lin et al., 1996; Stingele et al., 1998). Studies with microsensors observed variable concentrations of NO, whereas the readings from microdialysis or EPR studies have been difficult to translate into brain NO concentrations. A sensitive and well characterized method of measuring NO is to use the guanylate–cyclase-coupled NO receptor as a biosensor and to follow the production of cGMP (Garthwaite et al., 1989). We have used this method to assay the activity of nNOS in cultured neurons, but note that it is an indirect measure of NO production.
Synaptic regulation by nNOS–NMDAR coupling
NMDAR activation regulates AMPAR trafficking, potentially leading to LTP and LTD. Both of these processes are regulated by NO (Malenka and Bear, 2004); however, the role of phosphorylation of nNOS has not been examined previously. nNOS mutants that mimicked the active and repressed phosphorylated states were assayed for their effects on trafficking of endogenous GluR1- and GluR2-containing AMPARs. Expression of the S1412D nNOS mutant, which mimics the activated state of nNOS, increased surface expression of the AMPAR subunit, GluR2, relative to expression of either wild-type nNOS or the S847D mutant nNOS, which mimics the repressed state. This is in agreement with previous studies, which suggest that S-nitrosylation of NSF increases surface GluR2 by increasing an NSF interaction with GluR2 that enhances direct insertion of GluR2 at synaptic sites (Huang et al., 2005). Although GluR2S is the predominant GluR2 form in the brain (Kohler et al., 1994), GluR2L subunits make substantial contributions to AMPAR transmission in the second postnatal week (Kolleker et al., 2003). Additional studies are required to assess the role of NO in the differential trafficking of these two GluR2 splice variants. In contrast to GluR2, GluR1 surface levels were elevated by wild-type nNOS and S1412D to the same extent. These results suggest that the GluR1 trafficking machinery may be saturated by NO, even under conditions of expression of wild-type nNOS.
Regulation of phosphorylation of nNOS by VGCC and AMPAR
Glutamate stimulation also activates AMPARs, which depolarize the membrane. Membrane depolarization in turn activates L-type VGCCs (Xia and Storm, 2005). Together, these steps form a coordinated program that can regulate kinases and phosphatases. Because L-type VGCCs activate the Ca2+-CaM-dependent calcineurin (PP2B) pathway (Groth et al., 2003), we examined the possibility that activity of the L-type VGCCs and AMPARs can synergize with NMDAR-mediated Ca2+ fluxes to regulate the phosphatase(s) that dephosphorylate nNOS. NMDAR-induced dephosphorylation at S1412 was blocked by inhibitors of L-type VGCCs and AMPARs. Strikingly, blocking the activity of AMPARs and VGCCs significantly increased basal levels of phosphorylation at S847 and S1412, suggesting that tonic regulation of phosphorylation by phosphatases by VGCCs and AMPAR limits the steady-state levels of nNOS phosphorylation. These results show that Ca2+-entry through neighboring receptors and channels converges to provide regulatory control of phosphorylation of nNOS at S847 and S1412.
Excitotoxicity and failure of the biphasic switch
Excitotoxic death can be blocked by nNOS inhibitors and cerebral ischemia is reduced in nNOS knock-out (nNOS−/−) mice (Keynes and Garthwaite, 2004). The availability of the constitutively active S1412D nNOS mutant, which produces elevated NO in the absence of neuron activity, provides an opportunity to explore the role of NO in excitotoxic death and the NO-mediated cell-death hypothesis. We find that expression of S1412D nNOS by itself does not increase cell death, but that the expression of this nNOS mutant increased neuronal susceptibility to glutamate relative to cells that express either wild-type nNOS or GFP, a control. This suggests that NO overproduction must cooperate with other glutamate-regulated functions to induce excitotoxicity. These glutamate-regulated functions may include mitochondrial activity that produces reactive oxygen species that can combine with high levels of NO to form peroxynitrite. Peroxynitrite may then react with protein to form nitrotyrosine, an established factor in mechanisms of NMDAR-induced excitotoxicity (Huang et al., 1994; Dawson et al., 1996; Ayata et al., 1997; Eliasson et al., 1999; Rameau et al., 2003). These results suggest that glutamate-induced excitotoxicity involves nNOS-dependent pathways as well as ones that are nNOS-independent. Pharmacologic interventions that target both of these pathways may provide a basis for effective neuroprotective therapies that limit the production of toxic metabolites and block excitotoxic death.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant R37AG13620 to E.B.Z. D.S.T. was supported by NIH Training Grant 5T32DC000063 to the New York University Training Program in the Neurosciences and by NIH Fellowship F31MH76617-01. R.F.T. was supported by NIH Training Grant 5T32GM07308. E.D.G. was supported by NIH Grant RO1HL58883 and E.D.G.-H. by the Skaggs Institute for Chemical Biology. We thank Dr. Solomon Snyder for the rat nNOS cDNA and Ling-Yu Chiu for technical assistance. We also thank Dr. Dennis Stuehr for the gift of purified nNOS, the members of the Ziff lab for helpful discussions, and Peter Tukey for a close reading of this manuscript.
References
- Adachi N, Lei B, Soutani M, Arai T. Different roles of neuronal and endothelial nitric oxide synthases on ischemic nitric oxide production in gerbil striatum. Neurosci Lett. 2000;288:151–154. doi: 10.1016/s0304-3940(00)01222-2. [DOI] [PubMed] [Google Scholar]
- Adak S, Santolini J, Tikunova S, Wang Q, Johnson JD, Stuehr DJ. Neuronal nitric-oxide synthase mutant (Ser-1412 → Asp) demonstrates surprising connections between heme reduction, NO complex formation, and catalysis. J Biol Chem. 2001a;276:1244–1252. doi: 10.1074/jbc.M006857200. [DOI] [PubMed] [Google Scholar]
- Adak S, Aulak KS, Stuehr DJ. Chimeras of nitric-oxide synthase types I and III establish fundamental correlates between heme reduction, heme-NO complex formation, and catalytic activity. J Biol Chem. 2001b;276:23246–23252. doi: 10.1074/jbc.M102509200. [DOI] [PubMed] [Google Scholar]
- Aizenman E, Brimecombe JC, Potthoff WK, Rosenberg PA. Why is the role of nitric oxide in NMDA receptor function and dysfunction so controversial? Prog Brain Res. 1998;118:53–71. doi: 10.1016/s0079-6123(08)63200-8. [DOI] [PubMed] [Google Scholar]
- Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- Ayata C, Ayata G, Hara H, Matthews RT, Beal MF, Ferrante RJ, Endres M, Kim A, Christie RH, Waeber C, Huang PL, Hyman BT, Moskowitz MA. Mechanisms of reduced striatal NMDA excitotoxicity in type I nitric oxide synthase knock-out mice. J Neurosci. 1997;17:6908–6917. doi: 10.1523/JNEUROSCI.17-18-06908.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balazs R, Hack N, Jorgensen OS. Stimulation of the N-methyl-D-aspartate receptor has a trophic effect on differentiating cerebellar granule cells. Neurosci Lett. 1988;87:80–86. doi: 10.1016/0304-3940(88)90149-8. [DOI] [PubMed] [Google Scholar]
- Balda MA, Anderson KL, Itzhak Y. Adolescent and adult responsiveness to the incentive value of cocaine reward in mice: role of neuronal nitric oxide synthase (nNOS) gene. Neuropharmacology. 2006;51:341–349. doi: 10.1016/j.neuropharm.2006.03.026. [DOI] [PubMed] [Google Scholar]
- Bambrick LL, Yarowsky PJ, Krueger BK. Glutamate as a hippocampal neuron survival factor: an inherited defect in the trisomy 16 mouse. Proc Natl Acad Sci USA. 1995;92:9692–9696. doi: 10.1073/pnas.92.21.9692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredt DS. Nitric oxide signaling specificity–the heart of the problem. J Cell Sci. 2003;116:9–15. doi: 10.1242/jcs.00183. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Snyder SH. Nitric oxide mediates glutamate-linked enhancement of cGMP levels in the cerebellum. Proc Natl Acad Sci USA. 1989;86:9030–9033. doi: 10.1073/pnas.86.22.9030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW. Glutamate receptors and the induction of excitotoxic neuronal death. Prog Brain Res. 1994;100:47–51. doi: 10.1016/s0079-6123(08)60767-0. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Hillier BJ, Lim WA, Bredt DS. PSD-95 assembles a ternary complex with the N-methyl-d-aspartic acid receptor and a bivalent neuronal NO synthase PDZ domain. J Biol Chem. 1999;274:27467–27473. doi: 10.1074/jbc.274.39.27467. [DOI] [PubMed] [Google Scholar]
- Chung KK, Thomas B, Li X, Pletnikova O, Troncoso JC, Marsh L, Dawson VL, Dawson TM. S-nitrosylation of parkin regulates ubiquitination and compromises parkin's protective function. Science. 2004;304:1328–1331. doi: 10.1126/science.1093891. [DOI] [PubMed] [Google Scholar]
- Cohen PT. Protein phosphatase 1—targeted in many directions. J Cell Sci. 2002;115:241–256. doi: 10.1242/jcs.115.2.241. [DOI] [PubMed] [Google Scholar]
- Dawson VL, Kizushi VM, Huang PL, Snyder SH, Dawson TM. Resistance to neurotoxicity in cortical cultures from neuronal nitric oxide synthase-deficient mice. J Neurosci. 1996;16:2479–2487. doi: 10.1523/JNEUROSCI.16-08-02479.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'Acqua ML, Smith KE, Gorski JA, Horne EA, Gibson ES, Gomez LL. Regulation of neuronal PKA signaling through AKAP targeting dynamics. Eur J Cell Biol. 2006;85:627–633. doi: 10.1016/j.ejcb.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- Eliasson MJ, Huang Z, Ferrante RJ, Sasamata M, Molliver ME, Snyder SH, Moskowitz MA. Neuronal nitric oxide synthase activation and peroxynitrite formation in ischemic stroke linked to neural damage. J Neurosci. 1999;19:5910–5918. doi: 10.1523/JNEUROSCI.19-14-05910.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estevez AG, Crow JP, Sampson JB, Reiter C, Zhuang Y, Richardson GJ, Tarpey MM, Barbeito L, Beckman JS. Induction of nitric oxide-dependent apoptosis in motor neurons by zinc-deficient superoxide dismutase. Science. 1999;286:2498–2500. doi: 10.1126/science.286.5449.2498. [DOI] [PubMed] [Google Scholar]
- Fassbender K, Fatar M, Ragoschke A, Picard M, Bertsch T, Kuehl S, Hennerici M. Subacute but not acute generation of nitric oxide in focal cerebral ischemia. Stroke. 2000;31:2208–2211. doi: 10.1161/01.str.31.9.2208. [DOI] [PubMed] [Google Scholar]
- Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallis B, Corthals GL, Goodlett DR, Ueba H, Kim F, Presnell SR, Figeys D, Harrison DG, Berk BC, Aebersold R, Corson MA. Identification of flow-dependent endothelial nitric-oxide synthase phosphorylation sites by mass spectrometry and regulation of phosphorylation and nitric oxide production by the phosphatidylinositol 3-kinase inhibitor LY294002. J Biol Chem. 1999;274:30101–30108. doi: 10.1074/jbc.274.42.30101. [DOI] [PubMed] [Google Scholar]
- Garcin ED, Bruns CM, Lloyd SJ, Hosfield DJ, Tiso M, Gachhui R, Stuehr DJ, Tainer JA, Getzoff ED. Structural basis for isozyme-specific regulation of electron transfer in nitric-oxide synthase. J Biol Chem. 2004;279:37918–37927. doi: 10.1074/jbc.M406204200. [DOI] [PubMed] [Google Scholar]
- Garthwaite J, Garthwaite G, Palmer RM, Moncada S. NMDA receptor activation induces nitric oxide synthesis from arginine in rat brain slices. Eur J Pharmacol. 1989;172:413–416. doi: 10.1016/0922-4106(89)90023-0. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science. 2000;290:985–989. doi: 10.1126/science.290.5493.985. [DOI] [PubMed] [Google Scholar]
- Groth RD, Dunbar RL, Mermelstein PG. Calcineurin regulation of neuronal plasticity. Biochem Biophys Res Commun. 2003;311:1159–1171. doi: 10.1016/j.bbrc.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. The yin and yang of NMDA receptor signalling. Trends Neurosci. 2003;26:81–89. doi: 10.1016/S0166-2236(02)00040-1. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Nishio M, Naito Y, Yokokura H, Nimura Y, Hidaka H, Watanabe Y. Regulation of neuronal nitric-oxide synthase by calmodulin kinases. J Biol Chem. 1999;274:20597–20602. doi: 10.1074/jbc.274.29.20597. [DOI] [PubMed] [Google Scholar]
- Hisatsune C, Umemori H, Mishina M, Yamamoto T. Phosphorylation-dependent interaction of the N-methyl-d-aspartate receptor epsilon 2 subunit with phosphatidylinositol 3-kinase. Genes Cells. 1999;4:657–666. doi: 10.1046/j.1365-2443.1999.00287.x. [DOI] [PubMed] [Google Scholar]
- Huang Y, Man HY, Sekine-Aizawa Y, Han Y, Juluri K, Luo H, Cheah J, Lowenstein C, Huganir RL, Snyder SH. S-nitrosylation of N-ethylmaleimide sensitive factor mediates surface expression of AMPA receptors. Neuron. 2005;46:533–540. doi: 10.1016/j.neuron.2005.03.028. [DOI] [PubMed] [Google Scholar]
- Huang Z, Huang PL, Panahian N, Dalkara T, Fishman MC, Moskowitz MA. Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science. 1994;265:1883–1885. doi: 10.1126/science.7522345. [DOI] [PubMed] [Google Scholar]
- Ischiropoulos H. Biological tyrosine nitration: a pathophysiological function of nitric oxide and reactive oxygen species. Arch Biochem Biophys. 1998;356:1–11. doi: 10.1006/abbi.1998.0755. [DOI] [PubMed] [Google Scholar]
- Jiang X, Tian F, Mearow K, Okagaki P, Lipsky RH, Marini AM. The excitoprotective effect of N-methyl-d-aspartate receptors is mediated by a brain-derived neurotrophic factor autocrine loop in cultured hippocampal neurons. J Neurochem. 2005;94:713–722. doi: 10.1111/j.1471-4159.2005.03200.x. [DOI] [PubMed] [Google Scholar]
- Keynes RG, Garthwaite J. Nitric oxide and its role in ischaemic brain injury. Curr Mol Med. 2004;4:179–191. doi: 10.2174/1566524043479176. [DOI] [PubMed] [Google Scholar]
- Kohler M, Kornau HC, Seeburg PH. The organization of the gene for the functionally dominant alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor subunit GluR-B. J Biol Chem. 1994;269:17367–17370. [PubMed] [Google Scholar]
- Kolleker A, Zhu JJ, Schupp BJ, Qin Y, Mack V, Borchardt T, Kohr G, Malinow R, Seeburg PH, Osten P. Glutamatergic plasticity by synaptic delivery of GluR-B(long)-containing AMPA receptors. Neuron. 2003;40:1199–1212. doi: 10.1016/s0896-6273(03)00722-0. [DOI] [PubMed] [Google Scholar]
- Komeima K, Hayashi Y, Naito Y, Watanabe Y. Inhibition of neuronal nitric-oxide synthase by calcium/ calmodulin-dependent protein kinase IIalpha through Ser847 phosphorylation in NG108–15 neuronal cells. J Biol Chem. 2000;275:28139–28143. doi: 10.1074/jbc.M003198200. [DOI] [PubMed] [Google Scholar]
- Kornau HC, Schenker LT, Kennedy MB, Seeburg PH. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science. 1995;269:1737–1740. doi: 10.1126/science.7569905. [DOI] [PubMed] [Google Scholar]
- Kornau HC, Seeburg PH, Kennedy MB. Interaction of ion channels and receptors with PDZ domain proteins. Curr Opin Neurobiol. 1997;7:368–373. doi: 10.1016/s0959-4388(97)80064-5. [DOI] [PubMed] [Google Scholar]
- Lin SZ, Chiou AL, Wang Y. Ketamine antagonizes nitric oxide release from cerebral cortex after middle cerebral artery ligation in rats. Stroke. 1996;27:747–752. doi: 10.1161/01.str.27.4.747. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- McBain CJ, Mayer ML. N-methyl-d-aspartic acid receptor structure and function. Physiol Rev. 1994;74:723–760. doi: 10.1152/physrev.1994.74.3.723. [DOI] [PubMed] [Google Scholar]
- Michaels RL, Rothman SM. Glutamate neurotoxicity in vitro: antagonist pharmacology and intracellular calcium concentrations. J Neurosci. 1990;10:283–292. doi: 10.1523/JNEUROSCI.10-01-00283.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michell BJ, Griffiths JE, Mitchelhill KI, Rodriguez-Crespo I, Tiganis T, Bozinovski S, de Montellano PR, Kemp BE, Pearson RB. The Akt kinase signals directly to endothelial nitric oxide synthase. Curr Biol. 1999;9:845–848. doi: 10.1016/s0960-9822(99)80371-6. [DOI] [PubMed] [Google Scholar]
- Rameau GA, Akaneya Y, Chiu L, Ziff EB. Role of NMDA receptor functional domains in excitatory cell death. Neuropharmacology. 2000;39:2255–2266. doi: 10.1016/s0028-3908(00)00066-6. [DOI] [PubMed] [Google Scholar]
- Rameau GA, Chiu LY, Ziff EB. NMDA receptor regulation of nNOS phosphorylation and induction of neuron death. Neurobiol Aging. 2003;24:1123–1133. doi: 10.1016/j.neurobiolaging.2003.07.002. [DOI] [PubMed] [Google Scholar]
- Rameau GA, Chiu LY, Ziff EB. Bidirectional regulation of neuronal nitric-oxide synthase phosphorylation at serine 847 by the N-methyl-d-aspartate receptor. J Biol Chem. 2004;279:14307–14314. doi: 10.1074/jbc.M311103200. [DOI] [PubMed] [Google Scholar]
- Raymond LA, Moshaver A, Tingley WG, Huganir RL. Glutamate receptor ion channel properties predict vulnerability to cytotoxicity in a transfected nonneuronal cell line. Mol Cell Neurosci. 1996;7:102–115. doi: 10.1006/mcne.1996.0008. [DOI] [PubMed] [Google Scholar]
- Robison AJ, Bartlett RK, Bass MA, Colbran RJ. Differential modulation of Ca2+/calmodulin-dependent protein kinase II activity by regulated interactions with N-methyl-d-aspartate receptor NR2B subunits and alpha-actinin. J Biol Chem. 2005;280:39316–39323. doi: 10.1074/jbc.M508189200. [DOI] [PubMed] [Google Scholar]
- Rose SP. God's organism? The chick as a model system for memory studies. Learn Mem. 2000;7:1–17. doi: 10.1101/lm.7.1.1. [DOI] [PubMed] [Google Scholar]
- Samdani AF, Dawson TM, Dawson VL. Nitric oxide synthase in models of focal ischemia. Stroke. 1997a;28:1283–1288. doi: 10.1161/01.str.28.6.1283. [DOI] [PubMed] [Google Scholar]
- Samdani AF, Newcamp C, Resink A, Facchinetti F, Hoffman BE, Dawson VL, Dawson TM. Differential susceptibility to neurotoxicity mediated by neurotrophins and neuronal nitric oxide synthase. J Neurosci. 1997b;17:4633–4641. doi: 10.1523/JNEUROSCI.17-12-04633.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafe GE, Bauer EP, Rosis S, Farb CR, Rodrigues SM, LeDoux JE. Memory consolidation of Pavlovian fear conditioning requires nitric oxide signaling in the lateral amygdala. Eur J Neurosci. 2005;22:201–211. doi: 10.1111/j.1460-9568.2005.04209.x. [DOI] [PubMed] [Google Scholar]
- Sekaran S, Cunningham J, Neal MJ, Hartell NA, Djamgoz MB. Nitric oxide release is induced by dopamine during illumination of the carp retina: serial neurochemical control of light adaptation. Eur J Neurosci. 2005;21:2199–2208. doi: 10.1111/j.1460-9568.2005.04051.x. [DOI] [PubMed] [Google Scholar]
- Soriano FX, Papadia S, Hofmann F, Hardingham NR, Bading H, Hardingham GE. Preconditioning doses of NMDA promote neuroprotection by enhancing neuronal excitability. J Neurosci. 2006;26:4509–4518. doi: 10.1523/JNEUROSCI.0455-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingele R, Wilson DA, Traystman RJ, Hanley DF. Tyrosine confounds oxidative electrochemical detection of nitric oxide. Am J Physiol. 1998;274:H1698–H1704. doi: 10.1152/ajpheart.1998.274.5.H1698. [DOI] [PubMed] [Google Scholar]
- Tominaga T, Sato S, Ohnishi T, Ohnishi ST. Potentiation of nitric oxide formation following bilateral carotid occlusion and focal cerebral ischemia in the rat: in vivo detection of the nitric oxide radical by electron paramagnetic resonance spin trapping. Brain Res. 1993;614:342–346. doi: 10.1016/0006-8993(93)91053-u. [DOI] [PubMed] [Google Scholar]
- Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, Ma Y, Masliah E, Nomura Y, Lipton SA. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. 2006;441:513–517. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- Vanhoutte P, Bading H. Opposing roles of synaptic and extrasynaptic NMDA receptors in neuronal calcium signalling and BDNF gene regulation. Curr Opin Neurobiol. 2003;13:366–371. doi: 10.1016/s0959-4388(03)00073-4. [DOI] [PubMed] [Google Scholar]
- Waxman EA, Lynch DR. N-methyl-d-aspartate receptor subtype mediated bidirectional control of p38 mitogen-activated protein kinase. J Biol Chem. 2005;280:29322–29333. doi: 10.1074/jbc.M502080200. [DOI] [PubMed] [Google Scholar]
- Wei G, Dawson VL, Zweier JL. Role of neuronal and endothelial nitric oxide synthase in nitric oxide generation in the brain following cerebral ischemia. Biochim Biophys Acta. 1999;1455:23–34. doi: 10.1016/s0925-4439(99)00051-4. [DOI] [PubMed] [Google Scholar]
- Westbrook GL. Glutamate receptors and excitotoxicity. Res Publ Assoc Res Nerv Ment Dis. 1993;71:35–50. [PubMed] [Google Scholar]
- Xia Z, Storm DR. The role of calmodulin as a signal integrator for synaptic plasticity. Nat Rev Neurosci. 2005;6:267–276. doi: 10.1038/nrn1647. [DOI] [PubMed] [Google Scholar]
- Zanelli SA, Ashraf QM, Mishra OP. Nitration is a mechanism of regulation of the NMDA receptor function during hypoxia. Neuroscience. 2002;112:869–877. doi: 10.1016/s0306-4522(02)00141-0. [DOI] [PubMed] [Google Scholar]