

Abstract
Plasticity of synaptic transmission between parallel fiber (PF) and Purkinje neurons (PNs) is widely accepted as a cellular model for certain forms of cerebellar learning. Whereas the signaling cascades involved in postsynaptically expressed bidirectional long-term changes at PF–PN synapses are well investigated, data on presynaptically expressed long-term potentiation (LTP) are incomplete and controversial. Here we used transgenic mice that express a fluorescent protein Ca2+ sensor in PFs to demonstrate LTP of PF presynaptic Ca2+ transients after PF stimulation with 120 pulses at 4 Hz. Potentiation of the presynaptic Ca2+ transients correlated with the expression of simultaneously recorded LTP of PF–PN synaptic transmission and was suppressed by a protein kinase A inhibitor. Moreover, this presynaptically expressed form of LTP clearly required activation of an NMDA receptor/nitric oxide pathway, in contrast with the majority of previous reports. Blockade of NMDA receptors did not affect the PF Ca2+ transients induced during 4 Hz stimulation, indicating that the NMDA receptors required for the induction of presynaptic PF LTP are not localized in PFs.
Keywords: calcium (Ca), cerebellum, nitric oxide, NMDA, presynaptic mechanisms, synaptic plasticity
Introduction
Most synapses in the brain are endowed with mechanisms that enable persistent modifications of their efficacy. Synaptic plasticity is important for the formation and stability of neuronal circuits and provides substrates for memory that is likely to be used for learning. A plethora of molecular mechanisms exist that allow the induction and maintenance (expression) of synaptic plasticity and, moreover, a given type of synapse can express several distinct mechanisms for both presynaptic and postsynaptic long-term potentiation (LTP) and long-term depression (LTD) (Sanes and Lichtman, 1999; Malenka and Bear, 2004). The different mechanisms underlying synaptic plasticity most likely correspond to different learning rules at the circuit level.
In the cerebellum, LTP at parallel fiber (PF) to Purkinje neuron (PN) synapses was first postulated by Marr (1969) in his perceptron model of the cerebellar cortex and then modified by Albus (1971) by proposing LTD at this synapse. The first form of PF–PN long-term plasticity described on the basis of experimental data was LTD (Ito and Kano, 1982) and is often referred to as “cerebellar LTD.” LTP was demonstrated only much later at this synapse (Salin et al., 1996). The only form of PF–PN LTD described to date is expressed postsynaptically, whereas the first reports on PF–PN LTP implied a presynaptic expression mechanism (Salin et al., 1996; Ito, 2001). More recently, a postsynaptic form of PF–PN LTP has been characterized (Lev-Ram et al., 2002, 2003; Coesmans et al., 2004). Postsynaptically expressed LTD requires PF activity coupled with high levels of intracellular Ca2+ concentration in PNs and is mediated by metabotropic glutamate (mGlu) receptor and NMDA/nitric oxide (NO) signaling pathways that are likely to act synergistically (Lev-Ram et al., 1995, 1997; Coesmans et al., 2004; Shin and Linden, 2005). Postsynaptic PF–PN LTP is induced by synaptic activity combined with lower postsynaptic calcium levels and complements postsynaptic LTD to bidirectional plasticity (Lev-Ram et al., 2002, 2003; Coesmans et al., 2004).
There is compelling evidence that expression of presynaptic PF–PN LTP is dependent on a cAMP/protein kinase A (PKA) pathway (Salin et al., 1996; Storm et al., 1998; Lonart et al., 2003), and it has been reported that this form of LTP is NMDA receptor and NO independent (Salin et al., 1996; Lev-Ram et al., 2002). However, there is one report that suggested that an NMDA/NO cascade is involved in presynaptically expressed PF–PN LTP (Jacoby et al., 2001). Any stringent evaluation of presynaptic PF–PN LTP has been hindered by the fact that the locus of presynaptic expression could only be indirectly inferred from changes in paired-pulse facilitation (Salin et al., 1996; Storm et al., 1998; Lonart et al., 2003; van Beugen et al., 2006) and an increase in the frequency of miniature EPSCs after forskolin application (Chen and Regehr, 1997; Jacoby et al., 2001).
Here we used transgenic mice that express a fluorescent protein calcium sensor in PFs to directly evaluate a presynaptic expression mechanism for PF–PN LTP. Pharmacological manipulation, imaging of presynaptic PF calcium transients, and extracellular and intracellular recordings of PF-to-PN synaptic transmission directly demonstrated that presynaptically expressed PF–PN LTP correlates with an increase in PF Ca2+ transients and appears to involve an NMDA/NO cascade.
Materials and Methods
Slice preparation.
All experimental procedures were conducted in line with RIKEN guidelines and were approved by the RIKEN Animal Experiments Committee and the RIKEN Safety Center. Cerebellar slices were prepared from adult (4–6 weeks old) transgenic mice expressing the fluorescent protein calcium sensor GCaMP2 (Diez-Garcia et al., 2005, 2006) or from standard laboratory ICR mice. Animals were deeply anesthetized with halothane and decapitated. The cerebellum was dissected and placed in ice-cold artificial CSF (ACSF) containing the following (in mm): 125 NaCl, 3 KCl, 1 MgSO4, 2 CaCl2, 1 NaH2PO4, 25 NaHCO3, and 10 d-glucose bubbled with 95% O2/5% CO2 (pH 7.4, 295–305 mOsm). The cerebellar cortex was glued onto a block of 3% agar. The agar block with the cerebellar cortex was trimmed and glued to the stage of a Vibratome (VT1000s; Leica, Nussloch, Germany) so that the cerebellar cortex was cut in the frontal plane (coronal slices, 300 μm thick). The slices were incubated for ≥1 h in a chamber filled with 95%O2/5% CO2-equilibrated ACSF at room temperature (24–25°C) before recording.
Imaging and electrophysiology.
For calcium imaging, slices were submerged in a perfused recording chamber mounted below epifluorescence optics consisting of a 10× water-immersion objective (Nikon Microscope, Tokyo, Japan) and a 1× projection lens (Leica) or a standard fixed-stage microscope (Nikon Microscope) using a 40× water-immersion objective (Nikon Microscope). The fluorescence excitation was delivered using a halogen lamp (Moritex, Tokyo, Japan) and a 470–490 nm excitation filter. Fluorescence light was filtered through a 505 nm dichroic mirror and a 520–540 nm bandpass filter. Images were acquired by a cooled charge-coupled device system (Sensicam; PCO Computer Optics, Kelheim, Germany) at a frame rate of 40 or 100 Hz for sweeps of 2 s. Data were recorded using a 4 × 4 binning with macros under Image Pro (Media Cybernetics, Silver Spring, MD) and with synchronized illumination control and stimulation via a Master 8 controller (A.M.P.I., Jerusalem, Israel). Color-coded maps of ΔF/F were obtained using custom-made macros under Image Pro (Media Cybernetics). The time course of fluorescence bleaching was estimated using fluorescence measurements from nonresponsive areas or sweeps without stimulation. Fluorescence traces were divided by the normalized bleaching curve. Background fluorescence (e.g., fluorescence from PFs below the stimulated PFs) was not subtracted. For electrical stimulation, current pulses (200 μs, 10–100 μA) at 0.04 Hz (Figs. 1–4), 0.033 Hz (Fig. 5), or 0.05 Hz (Fig. 6) were delivered through a glass electrode mounted on remote-controlled manipulators (Luigis and Neumann, Ratingen, Germany). The stimulating electrode containing ACSF (0.1–0.5 MΩ) was placed in the molecular layer of the cerebellar cortex for PF stimulation. Extracellular recordings were obtained with glass pipettes filled with ACSF (2–4 MΩ) and positioned at the cell body of PNs. The stimulus intensity and electrode locations were adjusted to induce one or two PN spikes after PF stimulation. Spike events were detected off-line using pClamp 9 software (Molecular Devices, Foster City, CA). We took whole-cell patch-clamp recordings from PN somata in coronal slices visualized using a 60× water-immersion lens. Patch electrodes (3–4 MΩ) contained a solution of the following composition (in mm): 120 potassium gluconate, 10 HEPES, 10 EGTA, 5 KCl, 3.5 MgCl, 4 NaCl, 8 sucrose, 4 Na2ATP, and 0.2 Na2GTP, pH 7.3 with KOH (osmolarity adjusted to 300 mOsm). Cells were held in voltage-clamp mode at −70 mV. Series resistance was monitored by applying voltage pulses (10 ms, 5 mV), and only cells with stable series resistance were include in the analysis. The LTP induction protocol was applied at least 30 min after establishing the whole-cell configuration to allow sufficient time for the diffusion of EGTA into the dendritic tree. The ACSF included picrotoxin (50 μm) during all recordings to prevent inhibitory synaptic transmission. Calcium-free ACSF was prepared by replacing Ca2+ with equimolar Mg2+. Values are expressed as mean ± SEM; nonparametric statistics (Mann–Whitney U test) determined the level of statistical significance between groups of data using SPSS (Chicago, IL) software.
Figure 1.
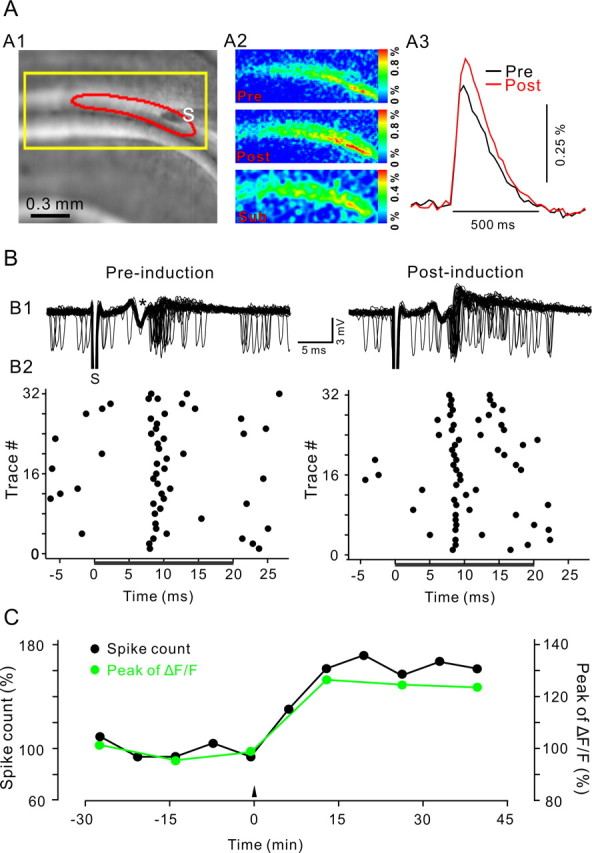
LTP of PF presynaptic Ca2+ transients and PF-to-PN synaptic transmission induced by 4 Hz PF stimulation. A, LTP induction induced a long-term augmentation of PF Ca2+ transients. A1, Fluorescent image of a coronal cerebellar slice from a GCaMP2 transgenic mouse with a stimulation electrode (S) placed in the molecular layer. The yellow rectangle indicates the cropped region used in A2. A2, Color-coded map of peak Ca2+ transients in response to a single PF stimulation pulse, before (Pre) and after (Post) LTP induction. The bottom map (Sub) shows the difference between the preinduction and postinduction maps of the Ca2+ transient. A3, Time course of ΔF/F values obtained from the responsive area (boxed area in A1). B, Extracellularly recorded spiking activity of a PN recorded simultaneously with the Ca2+ imaging shown in A. B1, Superposition of 32 representative recording epochs of spiking activity before (left) and 45 min after (right) 4 Hz stimulation. The time of a single PF stimulus is indicated by stimulus artifact (S); PF action potential volley is indicated by an asterisk. B2, Raster plot of spike events derived from traces shown in B1. Spikes were counted in the interval between 0 and 20 ms after PF stimulation (black bar) to derive the spike count measure plotted in C. C, Time course of the normalized spike count during the first 20 ms after PF stimulation (black) and peak of Ca2+ transients (green). Individual Ca2+ transients and spike count measurements are averages of 32 sweeps recorded at 0.04 Hz. All data are from the same experiment.
Figure 2.
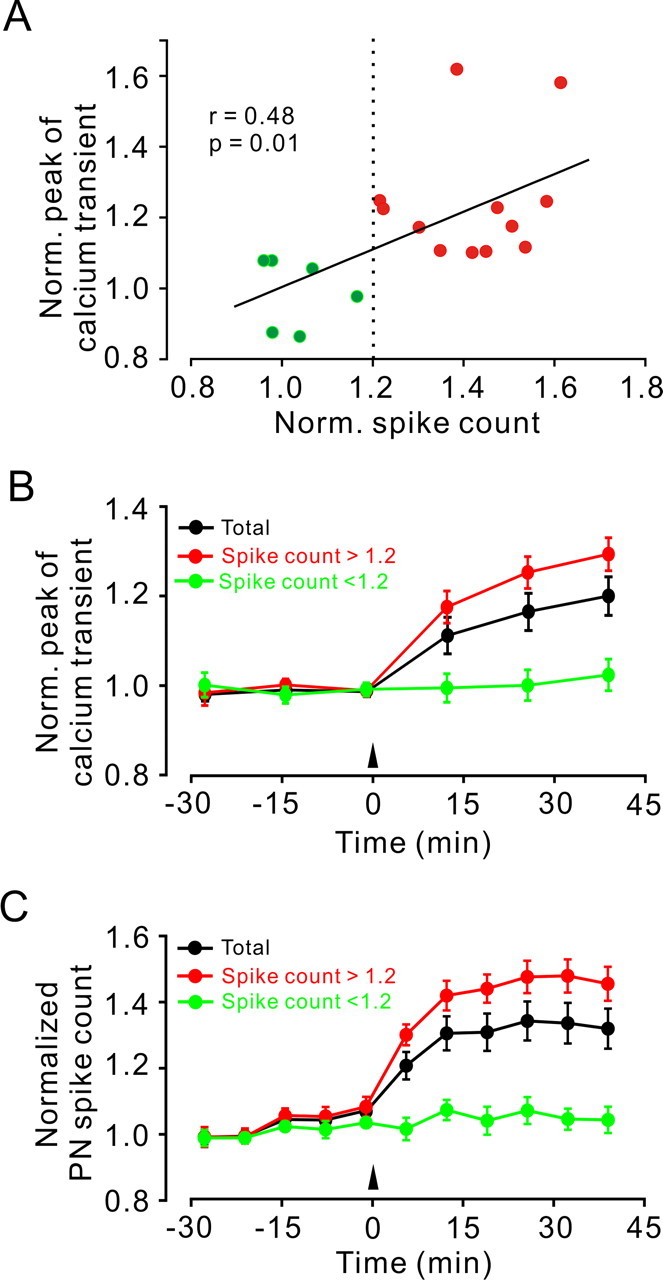
Analysis of 18 experiments conducted as illustrated in Figure 1. A, A plot of the normalized change in peak amplitude of PF Ca2+ transient versus normalized change in PN spike count measured after 4 Hz PF stimulation. Green and red colors along with the dashed line indicate experiments in which the change in spike count was below or above 1.2-fold, respectively. The solid line indicates linear regression (r = 0.48). B, Time course of the change in peak amplitude of PF Ca2+ transient from all experiments (black; n = 18), from experiments in which the spike count change exceeded 1.2-fold (red; n = 12) or was <1.2 (green; n = 6). C, Time course of the change in spike count from the same experiments as in B. Data points in B and C are mean ± SEM. Norm., Normalized.
Figure 3.

PF presynaptic LTP requires glutamate receptor but not AMPA, mGlu1, or CB1 receptor activation. A, LTP of the PF Ca2+ transient was prevented by blockers of ionotropic and mGlu receptors (50 μm d-APV, 20 μm NBQX, and 1 μm JNJ16259685, the mGlu1 antagonist; n = 5 experiments). B–D, Blockade of AMPA receptors (20 μm NBQX; n = 7 experiments), mGlu1 receptors (1 μm JNJ16259685; n = 6 experiments), and CB1 cannabinoid receptors (2 μm AM251; n = 7 experiments) did not prevent LTP of PF Ca2+ transients. Controls in A–C are the same as shown in Figure 2B (n = 18; average over all experiments). Data points are mean ± SEM. Norm., Normalized; JNJ, JNJ16259685.
Figure 4.

4 Hz PF–PN LTP required activation of NMDA receptors and NO. A, B, LTP of PF Ca2+ transients (A) and PN spike count (B) was prevented by d-APV (50 μm; n = 9 experiments). C, Accumulative Ca2+ signals during 4 Hz stimulation under control conditions and in the presence of d-APV (grand average of signals obtained in n = 10 control and n = 9 experiments with d-APV in the bath; p = 0.458). D, An inhibitor of NOS (l-NNA, 10 μm) prevented LTP induction. Controls in A–C are the same as shown in Figure 2B (n = 18; average over all experiments). Data points are mean ± SEM. Norm., Normalized.
Figure 5.

LTP of PF calcium transients is induced by 4 and 8 Hz but not by 1 and 2 Hz PF stimulation. Time courses of PF Ca2+ transient peak amplitude before and after 120 PF pulses at 1 Hz (•; n = 6), 2 Hz (○; n = 6), 4 Hz (▴; n = 7), and 8 Hz (▾; n = 7) are shown. Note that LTP was induced with 4–8 Hz PF stimulation but not with 1–2 Hz stimulation. Data points are mean ± SEM. Norm., Normalized.
Figure 6.

Intracellularly recorded 4 Hz PF–PN LTP also required activation of NMDA receptors and NO. A1, Representative examples of EPSCs elicited by paired-pulse stimulation in control conditions and in the presence of 50 μm d-APV before (solid trace) and after (dashed trace) 4 Hz stimulation. Each trace is an average of six pairs of EPSCs to two PF stimuli delivered 50 ms apart. A2, Summary of the experiments in which the 4 Hz stimulation induction protocol was delivered under control conditions (•; n = 15) and in the presence of 50 μm d-APV (○; n = 7). A3, Time course of the mean PPR (second EPSC/first EPSC, normalized to baseline levels). B1–B3, Same as A1–A3, except that control conditions (•; n = 15) were compared with recordings in the presence of 10 μm l-NNA (○; n = 7). Data points are mean ± SEM
Chemicals.
Reagents included picrotoxin, 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX), d-aminophos-phonovaleric acid (d-APV), 3,4-dihydro-2H-pyrano[2,3] β-quinolin-7-yl)(cis-4-methoxycyclohexyl) methanone (JNJ16259685), NG-nitro-l-arginine (l-NNA), N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinoline sulfonamide (H-89), and N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-di-chlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (AM251). All drugs were purchased from Tocris Cookson (Bristol, UK), except picrotoxin (Sigma-Aldrich, St. Louis, MO) and H-89 (LC Laboratories, Woburn, MA).
Results
LTP of PF calcium transients and PF–PN synaptic transmission
Previous studies indicated that stimulation of PF with 120 pulses at 4 or 8 Hz can induce LTP at PF–PN synapses. Expression of this form of LTP was associated with a decrease in paired-pulse facilitation; therefore, this LTP might be associated with an increased probability of transmitter release (Salin et al., 1996; Storm et al., 1998; Lev-Ram et al., 2002, 2003; Lonart et al., 2003). Hereafter, we will refer to this form of plasticity as (presynaptic) 4 Hz PF LTP. A decrease in paired-pulse facilitation correlates with an increase in synaptic strength and the amplitude of the PF presynaptic calcium transient (Mintz et al., 1995). To directly test whether 4 Hz PF LTP is expressed presynaptically, we monitored PF presynaptic calcium transients before and after 4 Hz stimulation in coronal slices prepared from transgenic mice that express a fluorescent protein Ca2+ sensor exclusively in granule cell axons (PFs) within the molecular layer of the cerebellar cortex (Diez-Garcia et al., 2005).
Single test pulses were delivered to the molecular layer at a frequency of 0.04 Hz inducing Ca2+ transients within a bundle (beam) of PFs (Fig. 1A). These Ca2+ transients were abolished by removing extracellular Ca2+ (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), but not when applying blockers of glutamatergic synaptic transmission (Diez-Garcia et al., 2005, 2006), reconfirming that they represent PF Ca2+ transients. Spontaneous and evoked action potential firings were recorded from a single PN located ∼1.5 mm lateral to the stimulation electrode simultaneously with the calcium imaging (Fig. 1B). Calcium transients and the number of spikes measured during the 20 ms period after PF stimulation (hereafter referred to as spike count) remained stable throughout the 30 min period of baseline recording (Fig. 1B,C). After the baseline recording, PFs were stimulated with 120 pulses delivered at 4 Hz. Presynaptic and postsynaptic responses were monitored for an additional 40 min period. In the experiment illustrated in Figure 1, the 4 Hz PF stimulation induced LTP of both the PF Ca2+ transient and the spike count. Similar results were obtained in 12 of 18 experiments. In the remaining six experiments, 4 Hz stimulation did not affect the presynaptic or postsynaptic responses. To investigate the relationship between LTP of PF Ca2+ transients and LTP of the spike count, we performed a correlation analysis of these two measures. The change in presynaptic calcium transient correlated with the change in spike count (Fig. 2A) (r = 0.48; p = 0.01), strongly suggesting that LTP of PF Ca2+ transients and LTP of the spike count are, under our conditions, corresponding measures for the same form of presynaptically expressed PF LTP.
Averaged over all 18 experiments, the PF transient increased by 22.6 ± 4.2% (at 40 min after 4 Hz stimulation; p < 0.001) (Fig. 2B), and the spike count increased by 30.9 ± 6.1% (at 40 min after 4 Hz stimulation; p < 0.001) (Fig. 2C). In the 12 experiments in which the spike count increase exceeded 20%, the PF calcium transient increased by 29.5 ± 3.6% (at 40 min after 4 Hz stimulation; p < 0.001) (Fig. 2B), and the spike count increased by 45.1 ± 5.2% (at 40 min after 4 Hz stimulation; p < 0.001) (Fig. 2C). In the remaining six experiments (spike count increase <20%), PF calcium transients and mean spike count did not change significantly from baseline values (p = 0.89) (Fig. 2B,C).
4 Hz PF–PN LTP required activation of NMDA receptors and NO
To test for the involvement of any known glutamate receptor(s) that may mediate PN postsynaptic responses, we delivered the 4 Hz stimulation protocol in the presence of a mixture containing 20 μm NBQX, 50 μm d-APV, and 1 μm JNJ16259685 to block AMPA, NMDA, and mGlu1 receptors, respectively. We could not detect LTP of PF Ca2+ transients (at 40 min after 4 Hz stimulation, 99.8 ± 2.4% of baseline, not significantly different from baseline; p = 0.84; n = 5) (Fig. 3A). To test whether postsynaptic spiking activity of PN is required for induction of PF–PN LTP, AMPA receptors were blocked by 20 μm NBQX, a condition known to abolish the postsynaptic response of PN to single PF stimuli (Knöpfel et al., 1990; Konnerth et al., 1990). In the presence of NBQX, test and 4 Hz stimulation-induced changes in PN spiking activity were not seen (data not shown), but we could still observe LTP of PF Ca2+ transients (at 40 min after 4 Hz stimulation, 117 ± 3.5% of baseline, not significantly different from control LTP; p = 0.65; n = 7) (Fig. 3B). Also, blockade of mGlu1 receptors with 1 μm JNJ16259685 alone did not affect LTP of PF Ca2+ transients (at 40 min after 4 Hz stimulation, 122.1 ± 5.3% of baseline, p = 0.002; not significantly different from control LTP, p = 0.589; n = 6) (Fig. 3C). Recently, it was shown that presynaptic PF LTP is controlled through endocannabinoid signaling triggered by climbing fiber activity (van Beugen et al., 2006). To test for an involvement of endocannabinoid signaling (in the absence of climbing fiber stimulation), we applied the CB1 receptor antagonist AM251 (2 μm), but this did not prevent LTP of PF Ca2+ transients (at 40 min after 4 Hz stimulation, 112.7 ± 3.8% of baseline, p = 0.026; not significantly different from control LTP, p = 0.297; n = 7) (Fig. 3D). These results indicate, by exclusion, that NMDA receptors might be involved in 4 Hz PF LTP.
To directly test the requirement for NMDA receptor activation in presynaptic PF LTP, we used d-APV alone. In the presence of d-APV, PF calcium transients and PN postsynaptic responses to PF stimulation were no different from control conditions (data not shown). However, LTP of PF Ca2+ transients and LTP of PN spike count did not occur in the presence of d-APV (at 40 min after 4 Hz stimulation, 103.3 ± 2.5% and 107.5 ± 4.3% of baseline, p = 0.136 and p = 0.19, respectively; n = 9) (Fig. 4A,B). This raises the question of the locus of the NMDA receptor dependence of 4 Hz PF LTP. Immunhistochemical studies have indicated a weak expression of NMDA receptors in PFs (Petralia et al., 1994), and evidence of a functional role for NMDA receptors at PFs has been reported (Casado et al., 2002). Other studies aimed at testing the functional involvement of NMDA receptors in the generation of paired-pulse-induced presynaptic Ca2+ transients failed to provide positive evidence for a function of PF NMDA receptors (Diez-Garcia et al., 2005; Shin and Linden, 2005). Consistent with the results of these reports, we did not detect any change in the accumulation of presynaptic Ca2+ during 4 Hz stimulation between control stimulations and stimulations in the presence of d-APV (Fig. 4C). The mean of the calcium transient remained unchanged over the first 3.75 s during the LTP induction protocol (control, 120.6 ± 13.9%; in the presence of d-APV, 137.2 ± 17.2%; p = 0.549) (Fig. 4C). NMDA receptors are present on cerebellar molecular layer interneurons and can be readily activated by glutamate released from PFs, likely followed by the activation of NO synthase (NOS) (Akazawa et al., 1994; Carter and Regehr, 2000). NO, in turn, has been implicated in various forms of presynaptically expressed LTP (Arancio et al., 1996; Hardingham and Fox, 2006). To test for the possible involvement of an NMDA/NO cascade, we used an inhibitor of NOS, L-NNA (10 μm) and, as shown in Figure 4D, this efficiently prevented the induction of 4 Hz PF LTP (100.2 ± 2.4% of baseline at 40 min after 4 Hz stimulation, not significantly different from baseline; p = 0.885; n = 18).
LTP of PF Ca2+ transients is induced by PF stimulation at 4 and 8 Hz but not at 1 and 2 Hz
To examine which stimulation frequency is required for induction of LTP of PF Ca2+ transients, we applied 200 stimulation pulses at 1, 2, 4, and 8 Hz (Fig. 5). LTP was induced by 4 Hz PF stimulation (121.9 ± 6.4% of baseline at 40 min after stimulation; p = 0.002; n = 6) and also by 8 Hz PF stimulation (121.5 ± 5.8% of baseline at 40 min after stimulation, not significantly different from 4 Hz LTP; p = 0.673; n = 7). However, LTP of PF Ca2+ transients was not observed after 1 Hz (101.36 ± 5.4% of baseline at 40 min after stimulation; p = 0.686; n = 6) or 2 Hz PF stimulation (103.8 ± 6.2% of baseline at 40 min after stimulation; p = 0.628; n = 6).
Intracellular recordings confirm an involvement of an NMDA/NO cascade in presynaptically expressed 4 Hz PF LTP
Our finding that activity of NMDA receptors and NO was required for 4 Hz PF LTP differs from several previous studies (Salin et al., 1996; Lev-Ram et al., 2002, 2003) (but see Jacoby et al., 2001). We therefore confirmed our findings using standard intracellular recording techniques in PNs of standard laboratory mice. Under control conditions, 4 Hz stimulation induced LTP of PF EPSCs (144.2 ± 9.0% of baseline at 30 min after 4 Hz stimulation; p < 0.001; n = 15) (Fig. 6A1,A2) and a decrease in paired-pulse ratio (PPR) (91.4 ± 1.9% of baseline at 30 min after 4 Hz stimulation; p < 0.001 vs baseline; n = 15) (Fig. 6A1,A3). Potentiation of EPSCs and a decrease in the PPR did not occur in the presence of 50 μm d-APV [EPSC: 96.6 ± 7.7% of baseline at 30 min after 4 Hz stimulation, unchanged from baseline, p = 0.8, n = 7 (Fig. 6A2); PPR: 100.2 ± 2.4% of baseline at 30 min after 4 Hz stimulation, unchanged from baseline, p = 0.8, n = 7 (Fig. 6A3)]. Similarly, application 10 μm l-NNA, a NOS inhibitor, prevented the expression of 4 Hz PF–PN LTP; the mean amplitudes of EPSCs measured 30 min after 4 Hz stimulation did not change from baseline (95.8 ± 6.2%; p = 0.209; n = 7) (Fig. 6B1,B2), and the PPR did not change (100.2 ± 2.0% of baseline; p = 0.783; n = 7) (Fig. 6B3).
LTP of PF Ca2+ transients is suppressed by the PKA inhibitor H-89
It has been shown that forskolin application can replicate presynaptic PF LTP but also can affect the excitability of PFs (Salin et al., 1996; Chen and Regehr, 1997; van Beugen et al., 2006). Comparing the effects of forskolin on PF excitability and PF Ca2+ transients, Chen and Regehr (1997) concluded that the elevation of cAMP levels by forskolin increases release at PF–PC synapses via a presynaptic mechanism that does not alter calcium influx. To determine whether the effect of elevated cAMP levels is downstream to calcium influx in synaptically induced presynaptic PF LTP, we examined the effect of the PKA inhibitor H-89 on LTP of simultaneously recorded Ca2+ transients and PF EPSCs. Consistent with previous reports (Salin et al., 1996; Jacoby et al., 2001), 10 μm H-89 prevented the induction of presynaptic PF LTP [control EPSCs: 135.4 ± 7.8% of baseline at 30 min after 4 Hz stimulation, p = 0.002, n = 7 (Fig. 7A1,A2); control PPR: 91.4 ± 1.9% of baseline at 30 min after 4 Hz stimulation, p = 0.002, n = 7 (Fig. 7A2); EPSCs in the presence of H-89: 93.6 ± 3.5% of baseline at 30 min after 4 Hz stimulation, p = 0.017, n = 7 (Fig. 7A1,A2); PPR in the presence of H-89: 98.4 ± 3.6% of baseline at 30 min after 4 Hz stimulation, p = 0.841, n = 7 (Fig. 7A2)]. Importantly, H-89 also prevented the potentiation of the PF Ca2+ transient (Fig. 7B1,B2). Instead of the potentiation seen under control conditions (112.8 ± 3.6% of baseline at 30 min; p = 0.008; n = 9) (Fig. 7B1,B2), 4 Hz stimulation in the presence of H-89 caused a small but significant decrease in the Ca2+ transients (93.2 ± 1.9% of baseline at 30 min after 4 Hz stimulation; p = 0.001; n = 13) (Fig. 7B2). We did not study this decrease further, but it is possible that inhibition of LTP of PF Ca2+ transients by H-89 uncovered a novel form of presynaptic LTD.
Figure 7.

LTP of PF Ca2+ transients and PF-to-PN synaptic transmission was depressed by the PKA blocker H-89. EPSCs (A1, A2) and presynaptic calcium transients (B1, B2) were elicited by paired-pulse stimulation. A1, Representative examples of EPSCs in control conditions and in the presence of 10 μm H-89, before (solid trace) and after (dashed trace) application of the 4 Hz stimulation induction protocol. Each trace is an average of eight sweeps. A2, Time course of mean changes in the amplitude of the first EPSC (top) and the PPR (bottom) in control conditions (•; n = 7) and in the presence of 10 μm H-89 (○; n = 7). B1, PF Ca2+ transients in control conditions (left) and in the presence of 10 μm H-89 (right) before (solid traces) and 20–40 min after (dashed traces) 4 Hz stimulation. Traces are grand averages over 8 (control) and 13 (H-89) experiments. B1, Time course of mean changes in the amplitude of the PF Ca2+ transients in control conditions (•; n = 8) and in the presence of 10 μm H-89 (○; n = 13). In the experiments involving H-89, the drug was present throughout baseline and postinduction recording periods. Error bars indicate ±SEM.
Discussion
The main finding of this study is that LTP at the PF–PN synapse induced by 120 stimulation pulses applied at 4 Hz occurs alongside a clear potentiation of presynaptic Ca2+ transients in the PFs that requires the activation of PKA, NMDA receptors, and NO. A presynaptic expression mechanism for 4 Hz PF LTP was previously proposed on the basis of paired-pulse facilitation measurements, but here we demonstrate this directly. Our demonstration of LTP of presynaptic calcium transients does not exclude additional modifications of the coupling between presynaptic Ca2+ and the release machinery as proposed by Chen and Regehr (1997). However, LTP of the Ca2+ transients sufficiently explains presynaptic expression of 4 Hz PF LTP, including the requirement for PKA activation. Measuring Ca2+ transients was facilitated by the use of transgenic mice that express a fluorescent protein calcium sensor within the cerebellar cortex exclusively in PFs (Diez-Garcia et al., 2005). In this way, we avoided any uncertainty as to the source of the measured Ca2+ signals.
The involvement of NO has previously been used as a hallmark to differentiate 4 Hz presynaptic LTP from 1 Hz postsynaptic LTP, with the 4 Hz LTP being NO independent (Lev-Ram et al., 2002). Our data contradict the conclusion of that study but do overlap with a previous report by Hartell and coworkers (Jacoby et al., 2001). Tsien and coworkers (Lev-Ram et al., 2002) suggested that a more detailed experimental analysis would be desirable to resolve the contradiction between their results and that of Hartell and coworkers (Jacoby et al., 2001). Although we have no explanation for their contradictory results (Lev-Ram et al., 2002), our study provides fresh, independent, and direct evidence supporting the conclusions of Hartell and coworkers (Jacoby et al., 2001) as to the requirement of NO for presynaptically expressed PF LTP.
Importantly, the involvement of an NMDA/NO cascade is not specific to this transgenic mouse strain because we confirmed these main conclusions in a standard laboratory mouse strain (Fig. 6). NO contributes to the mechanisms underlying presynaptic plasticity at other synaptic pathways (Arancio et al., 1996; Hardingham and Fox, 2006). It has been proposed that NO is produced by PFs and that this production increases after tetanic PF stimulation (Kimura et al., 1998). Therefore, the expression of LTP may be caused by an increased tonic effect of NO or a stimulation-induced potentiation of NO production (Kimura et al., 1998). However, stimulation-induced changes in NO are not NMDA receptor dependent (Kimura et al., 1998), and blocking neuronal NOS after LTP induction did not reverse NO-dependent presynaptic PF LTP (Jacoby et al., 2001).
It has been reported that 1 Hz PF stimulation-induced, postsynaptically expressed LTP (Lev-Ram et al., 2002) as well as 1 Hz PF stimulation paired with climbing fiber stimulation or postsynaptic depolarization-induced, postsynaptically expressed LTD are NO dependent (Lev-Ram et al., 1997; Shin and Linden, 2005). Tsien and coworkers (Lev-Ram et al., 2002) concluded that postsynaptic LTP is mediated by NO without postsynaptic Ca2+ elevation. More recently, compelling evidence provided by Hansel and coworkers (Coesmans et al., 2004) demonstrated that postsynaptically expressed PF–PN plasticity is bidirectional where the size of the postsynaptic calcium load determines the polarity of synaptic gain change. In this model, small Ca2+ loads trigger LTP, “intermediate” Ca2+ loads have no effect on postsynaptic responsivity, and large Ca2+ loads trigger LTD (Coesmans et al., 2004). Reports on the role of NO in postsynaptic PF–PN plasticity (Lev-Ram et al., 1997; Shin and Linden, 2005) can be reconciled with the Ca2+ limb of the signaling cascade by assuming that NO either acts synergistically or in parallel with postsynaptic calcium signals to induce bidirectional postsynaptic PF–PN plasticity. If so, 4 Hz PF stimulation may result in intermediate postsynaptic NO/Ca2+ signals that lead to no significant net change in the postsynaptic responsivity. Consistent with this model, our results show little residual change in PF EPSCs after 4 Hz stimulation in the presence of H-89 (to block presynaptically expressed LTP but not postsynaptically expressed PF–PN synaptic plasticity). Notwithstanding any small 4 Hz PF stimulation-induced effect on postsynaptic responsivity at the PF–PN synapses (either postsynaptic LTP or postsynaptic LTD), our measurements of presynaptic calcium transients (supported by PPF measurements) unequivocally characterize the presynaptic component of PF–PN plasticity independent from any possible additional postsynaptic changes.
In contrast to 4/8 Hz stimulation, PF stimulation at 1 and 2 Hz did not induce LTP of PF Ca2+ transients. This observation is in full agreement with the conclusion that 1 Hz PF LTP is expressed entirely postsynaptically (Lev-Ram et al., 2002, 2003) and with the previously established frequency dependency of presynaptic PF LTP (Salin et al., 1996; Lev-Ram et al., 2002).
Our results are consistent with other studies to show that the influx of calcium into PFs, cAMP production, and subsequent activation of PKA, is necessary for induction of presynaptic PF LTP (Salin et al., 1996; Storm et al., 1998) but not consistent with the concept that elevation of cAMP and activation of PKA is exclusively downstream to calcium influx in the signaling cascade underlying presynaptic PF LTP (Chen and Regehr, 1997). Indeed, the depression of LTP of PF calcium transients by H-89 indicated that the PKA signaling cascade feeds back to alter Ca2+ influx during expression of presynaptic PF LTP. This along with the requirement of NO for the LTP of PF Ca2+ transients points toward a difference to the scheme proposed by Hartell and coworkers (Jacoby et al., 2001), who suggested that the action of NO is “downstream” to that of cAMP (which in turn was considered downstream to Ca2+ influx). However, this portion of the conclusion of the study by Hartell and coworkers (Jacoby et al., 2001) was based on chemical LTP induction (forskolin application, NO donor application), procedures that may well have different effects to the more physiologically relevant synaptic stimulation protocol used in this study.
It may also be that some of the discrepancies in the existing literature arise from different populations of synapses studied (Sims and Hartell, 2006). Here, we stimulated a bundle of PFs in coronal cerebellar slices, a condition under which the possible recruitment of synapses formed by the ascending branch of the granule cell axon is minimized.
The presence of (functional) NMDA receptors on PFs has been controversial. Our data are in line with previous conclusions from experiments designed to test whether NMDA receptors contribute to the presynaptic Ca2+ signals induced by paired-pulse stimulation of PFs (Diez-Garcia et al., 2005; Shin and Linden, 2005). In extension to these previous studies, we also tested whether the action of NMDA receptors could contribute to PF Ca2+ accumulation during 4 Hz stimulation and found that this was also not the case (Fig. 4C). Available evidence therefore suggests that the NMDA receptors required for induction of 4 Hz LTP are not localized on PFs but rather on molecular layer interneurons (Akazawa et al., 1994; Carter and Regehr, 2000). A similar conclusion was recently reached with respect to the localization of NMDA receptors (and the source of NO) involved in postsynaptic PF–PN LTD (Shin and Linden, 2005). If the activity of interneurons regulates both presynaptic and postsynaptic PF–PN plasticity, we propose that the previously assumed learning rules for PF–PN synapses need to be revised. Moreover, although it is now clear that cerebellar long-term synaptic plasticity is not limited to one type of synapse nor at a given synapse to one mechanistically defined form, concerted work of many laboratories is still required to understand how the various forms of cerebellar synaptic plasticity engage in cerebellar function.
Footnotes
This work was supported by the RIKEN Brain Science Institute. We thank the members of the Knöpfel laboratory and Dr. Ruth Empson for helpful discussions.
References
- Akazawa C, Shigemoto R, Bessho Y, Nakanishi S, Mizuno N. Differential expression of five N-methyl-D-aspartate receptor subunit mRNAs in the cerebellum of developing and adult rats. J Comp Neurol. 1994;347:150–160. doi: 10.1002/cne.903470112. [DOI] [PubMed] [Google Scholar]
- Albus JS. A theory of cerebellar function. Math Biosci. 1971;10:25–61. [Google Scholar]
- Arancio O, Kiebler M, Lee CJ, Lev-Ram V, Tsien RY, Kandel ER, Hawkins RD. Nitric oxide acts directly in the presynaptic neuron to produce long-term potentiation in cultured hippocampal neurons. Cell. 1996;87:1025–1035. doi: 10.1016/s0092-8674(00)81797-3. [DOI] [PubMed] [Google Scholar]
- Carter AG, Regehr WG. Prolonged synaptic currents and glutamate spillover at the parallel fiber to stellate cell synapse. J Neurosci. 2000;20:4423–4434. doi: 10.1523/JNEUROSCI.20-12-04423.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casado M, Isope P, Ascher P. Involvement of presynaptic N-methyl-D-aspartate receptors in cerebellar long-term depression. Neuron. 2002;33:123–130. doi: 10.1016/s0896-6273(01)00568-2. [DOI] [PubMed] [Google Scholar]
- Chen C, Regehr WG. The mechanism of cAMP-mediated enhancement at a cerebellar synapse. J Neurosci. 1997;17:8687–8694. doi: 10.1523/JNEUROSCI.17-22-08687.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coesmans M, Weber JT, De Zeeuw CI, Hansel C. Bidirectional parallel fiber plasticity in the cerebellum under climbing fiber control. Neuron. 2004;44:691–700. doi: 10.1016/j.neuron.2004.10.031. [DOI] [PubMed] [Google Scholar]
- Diez-Garcia J, Matsushita S, Mutoh H, Nakai J, Ohkura M, Yokoyama J, Dimitrov D, Knöpfel T. Activation of cerebellar parallel fibers monitored in transgenic mice expressing a fluorescent Ca2+ indicator protein. Eur J Neurosci. 2005;22:627–635. doi: 10.1111/j.1460-9568.2005.04250.x. [DOI] [PubMed] [Google Scholar]
- Diez-Garcia J, Akemann W, Knöpfel T. In vivo calcium imaging from genetically specified target cells in mouse cerebellum. NeuroImage. 2006;34:859–869. doi: 10.1016/j.neuroimage.2006.10.021. [DOI] [PubMed] [Google Scholar]
- Hardingham N, Fox K. The role of nitric oxide and GluR1 in presynaptic and postsynaptic components of neocortical potentiation. J Neurosci. 2006;26:7395–7404. doi: 10.1523/JNEUROSCI.0652-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M. Cerebellar long-term depression: characterization, signal transduction, and functional roles. Physiol Rev. 2001;81:1143–1195. doi: 10.1152/physrev.2001.81.3.1143. [DOI] [PubMed] [Google Scholar]
- Ito M, Kano M. Long-lasting depression of parallel fiber-Purkinje cell transmission induced by conjunctive stimulation of parallel fibers and climbing fibers in the cerebellar cortex. Neurosci Lett. 1982;33:253–258. doi: 10.1016/0304-3940(82)90380-9. [DOI] [PubMed] [Google Scholar]
- Jacoby S, Sims RE, Hartell NA. Nitric oxide is required for the induction and heterosynaptic spread of long-term potentiation in rat cerebellar slices. J Physiol (Lond) 2001;535:825–839. doi: 10.1111/j.1469-7793.2001.t01-1-00825.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S, Uchiyama S, Takahashi HE, Shibuki K. cAMP-dependent long-term potentiation of nitric oxide release from cerebellar parallel fibers in rats. J Neurosci. 1998;18:8551–8558. doi: 10.1523/JNEUROSCI.18-21-08551.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knöpfel T, Audinat E, Gahwiler BH. Climbing fibre responses in olivo-cerebellar slice cultures. I. Microelectrode recordings from Purkinje cells. Eur J Neurosci. 1990;2:726–732. doi: 10.1111/j.1460-9568.1990.tb00463.x. [DOI] [PubMed] [Google Scholar]
- Konnerth A, Llano I, Armstrong CM. Synaptic currents in cerebellar Purkinje cells. Proc Natl Acad Sci USA. 1990;87:2662–2665. doi: 10.1073/pnas.87.7.2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev-Ram V, Makings LR, Keitz PF, Kao JP, Tsien RY. Long-term depression in cerebellar Purkinje neurons results from coincidence of nitric oxide and depolarization-induced Ca2+ transients. Neuron. 1995;15:407–415. doi: 10.1016/0896-6273(95)90044-6. [DOI] [PubMed] [Google Scholar]
- Lev-Ram V, Jiang T, Wood J, Lawrence DS, Tsien RY. Synergies and coincidence requirements between NO, cGMP, and Ca2+ in the induction of cerebellar long-term depression. Neuron. 1997;18:1025–1038. doi: 10.1016/s0896-6273(00)80340-2. [DOI] [PubMed] [Google Scholar]
- Lev-Ram V, Wong ST, Storm DR, Tsien RY. A new form of cerebellar long-term potentiation is postsynaptic and depends on nitric oxide but not cAMP. Proc Natl Acad Sci USA. 2002;99:8389–8393. doi: 10.1073/pnas.122206399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev-Ram V, Mehta SB, Kleinfeld D, Tsien RY. Reversing cerebellar long-term depression. Proc Natl Acad Sci USA. 2003;100:15989–15993. doi: 10.1073/pnas.2636935100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonart G, Schoch S, Kaeser PS, Larkin CJ, Sudhof TC, Linden DJ. Phosphorylation of RIM1alpha by PKA triggers presynaptic long-term potentiation at cerebellar parallel fiber synapses. Cell. 2003;115:49–60. doi: 10.1016/s0092-8674(03)00727-x. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Marr D. A theory of cerebellar cortex. J Physiol (Lond) 1969;202:437–470. doi: 10.1113/jphysiol.1969.sp008820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintz IM, Sabatini BL, Regehr WG. Calcium control of transmitter release at a cerebellar synapse. Neuron. 1995;15:675–688. doi: 10.1016/0896-6273(95)90155-8. [DOI] [PubMed] [Google Scholar]
- Petralia RS, Wang YX, Wenthold RJ. The NMDA receptor subunits NR2A and NR2B show histological and ultrastructural localization patterns similar to those of NR1. J Neurosci. 1994;14:6102–6120. doi: 10.1523/JNEUROSCI.14-10-06102.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salin PA, Malenka RC, Nicoll RA. Cyclic AMP mediates a presynaptic form of LTP at cerebellar parallel fiber synapses. Neuron. 1996;16:797–803. doi: 10.1016/s0896-6273(00)80099-9. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW. Can molecules explain long-term potentiation? Nat Neurosci. 1999;2:597–604. doi: 10.1038/10154. [DOI] [PubMed] [Google Scholar]
- Shin JH, Linden DJ. An NMDA receptor/nitric oxide cascade is involved in cerebellar LTD but is not localized to the parallel fiber terminal. J Neurophysiol. 2005;94:4281–4289. doi: 10.1152/jn.00661.2005. [DOI] [PubMed] [Google Scholar]
- Sims RE, Hartell NA. Differential susceptibility to synaptic plasticity reveals a functional specialization of ascending axon and parallel fiber synapses to cerebellar Purkinje cells. J Neurosci. 2006;26:5153–5159. doi: 10.1523/JNEUROSCI.4121-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm DR, Hansel C, Hacker B, Parent A, Linden DJ. Impaired cerebellar long-term potentiation in type I adenylyl cyclase mutant mice. Neuron. 1998;20:1199–1210. doi: 10.1016/s0896-6273(00)80500-0. [DOI] [PubMed] [Google Scholar]
- van Beugen BJ, Nagaraja RY, Hansel C. Climbing fiber-evoked endocannabinoid signaling heterosynaptically suppresses presynaptic cerebellar long-term potentiation. J Neurosci. 2006;26:8289–8294. doi: 10.1523/JNEUROSCI.0805-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]