

Abstract
The intracellular molecular mechanism that controls the timing of oligodendrocyte differentiation remains unknown. Temple and Raff (1986) previously showed that an oligodendrocyte precursor cell (OPC) can divide a maximum of approximately eight times before its daughter cells simultaneously cease proliferating and differentiate into oligodendrocytes. They postulated that over time the level of an intracellular molecule might synchronously change in each daughter cell, ultimately reaching a level that prohibited additional proliferation. Here, we report the discovery of such a molecule, the cyclin-dependent kinase inhibitor p57Kip2 (Cdkn1c). We show in vitro that all daughters of a clone of OPCs express similar levels of p57Kip2, that p57Kip2 levels increase over time in proliferating OPCs, and that p57Kip2 levels regulate how many times an OPC can divide before differentiating. These findings reveal a novel part of the mechanism by which OPCs measure time and are likely to extend to similar timers in many other precursor cell types.
Keywords: oligodendrocyte, p57Kip2, CDKN1C, myelin, multiple sclerosis, ZFP536, Id4
Introduction
We are interested in understanding what regulates the generation of oligodendrocytes (OLs), the myelin-forming cells of the CNS. OLs are generated from proliferating, immature oligodendrocyte precursor cells (OPCs), which exit the cell cycle and differentiate throughout the CNS at predictable developmental ages (Baumann and Pham-Dinh, 2001). Previous experiments have demonstrated that the precise timing of OL generation is regulated in part by an intracellular molecular clock, which intrinsically determines how many times an OPC can divide before differentiating (Temple and Raff, 1986; Barres et al., 1994a). When stimulated to divide by mitogens, this intracellular OPC clock measures the passage of time independent of additional external cues (Barres et al., 1994a; Gao et al., 1997). Only once sufficient time has passed do dividing OPCs finally respond to environmental cues such as retinoic acid (RA) and thyroid hormone (T3) by dropping out of the cell cycle and differentiating into mature OLs (Barres et al., 1994a; Gao et al., 1997). These external cues are required to trigger the “effector” component of the clock that promotes differentiation, as in the absence of such external cues (e.g., T3) OPCs can proliferate indefinitely in the presence of mitogens (Barres et al., 1994a; Tang et al., 2000). The clock mechanism also relies on mitogen signaling, as in the absence of mitogen stimulation OPCs immediately initiate OL differentiation regardless of the status of the timer or T3 signaling (Raff et al., 1983; Barres et al., 1992, 1993). Interestingly, mitogen withdrawal and the clock mechanism do not induce OL maturation via identical pathways, because mitogen withdrawal and T3 exposure differentially alter gene expression in early differentiating OLs, and genes distinctly required for T3-mediated OL differentiation have been identified (Tokumoto et al., 2001; Billon et al., 2004). The importance of the clock mechanism in regulating the timing of OL generation in vivo is demonstrated by the fact that hypothyroid animals have delayed myelination, whereas hyperthyroid animals have accelerated myelin formation (Walters and Morell, 1981; Dussault and Ruel, 1987). Although the extrinsic cues that trigger the clock effector have been extensively studied, the components of the intracellular timer that intrinsically regulate when an OPC will differentiate remain less well characterized.
Cell cycle arrest is an obligate step in the terminal differentiation of many mammalian cell types, including cells of the nervous system such as OLs (Casaccia-Bonnefil and Liu, 2003). Several groups have investigated the links between cell cycle regulation and OL differentiation. During OL differentiation, activity of the G1-S phase checkpoint complex CyclinE–cyclin-dependent kinase 2 (cdk2) decreases (Ghiani and Gallo, 2001). CyclinE-cdk2 complex formation is inhibited by members of the Cip/Kip family of cyclin-dependent kinase inhibitor proteins (Cunningham and Roussel, 2001), and all three members of this family, p21Cip1, p27Kip1, and p57Kip2, have been implicated in regulating various stages of OL differentiation. A role for p27Kip1 has been demonstrated in both inhibiting OPC proliferation and inducing differentiation in response to T3, whereas p21Cip1 is required for OL differentiation but not cessation of the cell cycle (Casaccia-Bonnefil et al., 1999; Tang et al., 1999; Zezula et al., 2001; Tokumoto et al., 2002). In zebrafish, p57Kip2 has been implicated in the specification of OPCs from uncommitted neural precursor cells (Park et al., 2005).
We report here that p57Kip2 is an important component of the OPC timer as well as the mechanism that couples the regulation of OPC proliferation and differentiation. We show in vitro that all daughters of a clone of OPCs share the same level of p57Kip2 immunoreactivity, that p57Kip2 levels increase over time in proliferating OPCs, and that p57Kip2 levels regulate how many times a given OPC can divide before differentiating. These findings reveal a novel part of the mechanism by which OPCs keep time and are likely to extend to similar timers in many other precursor cell types.
Materials and Methods
Genomic analysis of OL gene expression.
Genomic analyses of gene expression changes induced by OL differentiation were performed as described previously (Dugas et al., 2006). Briefly, postnatal day 7 (P7) OPCs purified by immunopanning were induced to differentiate by mitogen withdrawal and T3 exposure, and RNA samples obtained every 1–2 d after the induction of differentiation were labeled and hybridized to Affymetrix (Santa Clara, CA) U34A-C chips. RNA from acutely purified P12 OLs was also applied to Affymetrix chips. Data from four independent experiments were averaged to obtain the probe set expression values depicted.
Purification and culture of OL-lineage cells.
Purification and culturing of OPCs and OLs were performed as described previously (Dugas et al., 2006). Briefly, OPCs were purified from enzymatically dissociated P7–P8 Sprague Dawley (Charles Rivers, Wilmington, MA) rat brains by immunopanning: after removal of potential contaminating OLs and other cells by Ran-2 and GC antibody binding, OPCs were selected by O4 antibody binding. Nonadherent cells were washed away, and purified OPCs were recovered by trypsinization. Acutely purified OLs (AcOLs) were similarly obtained from enzymatically dissociated P12 rat brains: positive selection by GC antibody binding followed negative selection by Ran-2 and A2B5 antibody binding. Purified OPCs were cultured on poly-d-lysine (pDL)-coated tissue culture plastic or glass coverslips in DMEM-Sato-based medium containing N-acetyl-L-cysteine (5 μg/ml), forskolin (4.2 μg/ml), bovine insulin (5 μg/ml), and ciliary neurotrophic factor (10 ng/ml). OPC proliferation was induced by the addition of mitogenic platelet-derived growth factor-AA (PDGF-AA; 10 ng/ml) and neurotrophin-3 (NT3; 1 ng/ml) (+PDGF) (Barres et al., 1993, 1994b). Differentiation of pure OPCs into OLs was stimulated either by removal of PDGF and NT3 from the medium (-PDGF) or by addition of T3 (+T3) (40 ng/ml) to the culture medium as noted. All cultures were maintained at 37°C in 10% CO2.
Immunostaining.
Immunostaining of OPC and OL cultures for cyclic nucleotide phosphodiesterase 1 (CNP), myelin basic protein (MBP), myelin oligodendrocyte glycoprotein (MOG), and green fluorescent protein (GFP) expression was performed as described previously (Dugas et al., 2006). To stain for p57Kip2 expression, purified cells cultured on glass coverslips were fixed in 4% paraformaldehyde for 10 min at 25°C and then incubated for 1 h in a 50% donkey serum, 1% bovine serum albumin (BSA) and 100 mm l-lysine solution to block nonspecific binding and 0.4% Triton X-100 to permeabilize the cells. Coverslips were then incubated overnight at 4°C in 10% donkey serum, 1% BSA, 100 mm l-lysine solution containing 1/50 goat anti-p57Kip2 (SC-1039; Santa Cruz Biotechnology, Santa Cruz, CA). Finally, coverslips were incubated for 1 h at 25°C in 1% BSA, 100 mm l-lysine solution containing 1/1000 Alexa Fluor-488 donkey anti-goat (A-11055; Invitrogen, Carlsbad, CA) secondary antibody. To stain for p57Kip2 expression in vivo, optic nerves were dissected from rats at various ages and fixed by immersion in 4% paraformaldehyde for 2–3 h at 4°C. Nerves were then equilibrated in 30% sucrose solution overnight at 4°C, mounted in OCT, and 10-μm-thick sections were generated on a cryostat. Sections mounted onto silane-prep slides (S4651; Sigma-Aldrich, St. Louis, MO) were then blocked and stained as described above for p57Kip2 expression. Slides were costained with either 1/100 mouse anti-CC-1 (OP80; Calbiochem, La Jolla, CA) or 1/400 mouse anti-NG2 (MAB5384; Chemicon, Temecula, CA) primary antibody, and 1/1000 Alexa Fluor-594 donkey anti-mouse secondary antibody. All staining was mounted in Vectashield anti-fade medium plus 4′,6′-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA) to allow identification of healthy cell nuclei.
Reverse transcription-PCR.
Total RNA was isolated from cells with the RNeasy mini kit (Qiagen, Valencia, CA), using Qiashredder columns for cell lysis, and inserting Qiagen on-column DNase steps to remove any contaminating genomic DNA. Equivalent amounts of RNA from each sample were reverse transcribed with Superscript III (Invitrogen 18080-044) according to manufacturer protocols. PCRs were then set up with equivalent volumes of completed reverse transcription (RT) reactions, using Platinum TaqDNA polymerase (Invitrogen 10966-026) according to manufacturer protocols. PCR reactions: 94°C, 30 s; 55°C, 30 s; 68°C, 45 s cycles (cycle numbers are as indicated in the figures). Primers for PCRs are as follows: p57Kip2, 5′TGTCCCTCTCCTAACGTGGCTC and 5′CATTGCACAGTTTTCAGATTTCCAC (234 bp product); β-actin, 5′GCATTGTCACCAACTGGGACG and 5′ACCGCTCATTGCCGATAGTG (543 bp product).
OPC transfection and differentiation assay.
OPC transfections were performed as described previously (Dugas et al., 2006). Briefly, purified P8 OPCs were cultured in proliferation-promoting medium for 7 DIV (unless otherwise noted) and then enzymatically lifted by gentle trypsinization. Rinsed 2 × 106 to 3 × 106 aliquots of OPCs were resuspended in 100 μl of Amaxa OPC nucleofection reagent (VPG-100; Amaxa, Gaithersburg, MD) plus 1.5–4.0 μg of pC1-eGFP [CMV-promoter driven enhanced GFP (eGFP) expression; 6084-1, Clontech, Mountain View, CA]; 2.5 μg of pSPORT6-p57Kip2 (CMV-p57Kip2; MMM1013–63856, Open Biosystems, Huntsville, AL); 2.5 μg of pSPORT6-ZFP536 (CMV-ZFP536; MMM1013-7513950, Open Biosystems); 4.0 μg of pSPORT6-Id4 [CMV-Id4; Id4 from Open Biosystems MHS1011-75790 excised with EcoRI (5′) and XhoI (3′) and subcloned into the same sites of the pSPORT6 expression vector, Open Biosystems MMM1013-63856, after excising existing insert]; 100–200 pmol siGenome SMARTpool small interfering RNA (siRNA) targeting rat ZFP536 (M-082235-00; Dharmacon, Lafayette, CO); 100–200 pmol siGenome SMARTpool siRNA targeting rat p27Kip1 (Dharmacon M-090938-00); 100–200 pmol siGenome SMARTpool siRNA targeting rat p57Kip2 (Dharmacon M-098880-00); 100 pmol siGenome individual siRNA duplexes targeting rat p57Kip2 (Dharmacon M-098880-01, -02, -03, -04); 100–200 siControl nontargeting siRNA pool (Dharmacon D-001206-13). Note that, unless otherwise noted, knockdown of p57Kip2 expression was performed with the siRNA pool targeting p57Kip2. OPC-plasmid/siRNA mixes were then electroporated with the Amaxa nucleofection apparatus, O-17 program. For clonal assays, 250–500 cells were added into medium in pDL-coated six-well tissue culture plates or 40 cells were added to pDL-coated glass coverslips. OLs were identified by their characteristic multiple complex, thin, multibranched process-bearing morphology and were easily discerned from the OPCs, which possess very few simple, thick process. Note that, in independent experiments, we found that this characteristic OL morphology was tightly correlated with the expression of the OL maturation marker GalC (data not shown). To assay for differentiation by myelin gene expression, transfected OPCs were plated at either 30,000 cells/pDL-coated glass coverslip in 24-well plates and incubated for 3–4 d in medium lacking PDGF and NT3 or plated at 1000–1500 cells/coverslip and incubated for 7 d in medium containing PDGF and NT3 in the presence or absence of T3. Coverslips were then stained for GFP and either CNP, MBP or MOG expression as described above. Myelin gene expression was scored as the percentage of transfected (GFP+) cells positive for CNP, MBP, or MOG expression. All experiments were scored blind, 100–300 cells were counted per coverslip, and at least three coverslips were scored per data point. To control for variations observed in differentiation of controls between trials (because of slight differences in exact times in cultures, plating densities, staining times, etc.), all experimental conditions were paired with controls performed in parallel experiments derived from common pools of starting cells. All experiments were repeated at least twice with qualitatively similar results obtained.
Results
Regulation of p57Kip2 expression during OL differentiation
To investigate how cell cycle regulation is linked to OL differentiation, we first used gene chip technology to identify the cell cycle control genes that are strongly regulated as OPCs differentiate into OLs in vitro (Dugas et al., 2006). In particular, we observed that the cell cycle inhibitor gene p57Kip2 (or cdkn1c) was strongly upregulated immediately after the initiation of OL differentiation (Fig. 1 A). In fact, p57Kip2 is one of the earliest induced genes in purified OPCs after coupled mitogen withdrawal (removal of PDGF–AA and NT3; -PDGF) and thyroid hormone triiodothyronine exposure (+T3) to induce differentiation. Of the three members of the Cip/Kip family of cell cycle inhibitor genes, only p57Kip2 demonstrated a strong change in expression level (Dugas et al., 2006). The induction of p57Kip2 expression in OLs was confirmed in vivo by comparing p57Kip2 levels in total RNA obtained from acutely purified P7 OPCs and P12 OLs, both on Affymetrix gene chips (Fig. 1 A) and by RT-PCR on independently obtained samples (data not shown).
Figure 1.
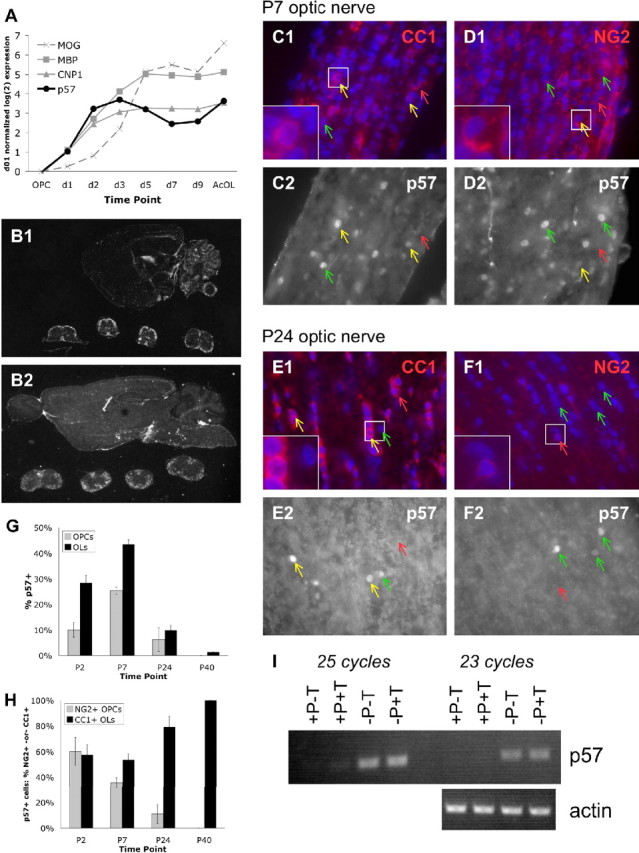
Regulation of p57Kip2 expression. p57Kip2 is expressed in differentiating OLs. A, p57Kip2 is induced in differentiating OLs in vitro. Expression levels of p57Kip2 (black circles, rc_AA998565_at probe set), early induced myelin genes CNP (gray triangles, L16532_at) and MBP (gray squares, average K00512_at, rc_AI044093_at, rc_AI145512_at), and late induced myelin gene MOG (gray dashed X, M99485_at) as assayed on Affymetrix U34A-C gene chips (Dugas et al., 2006). OPC, Purified OPCs; d1–d9, days after induction of OL differentiation in vitro by mitogen withdrawal and T3 exposure; AcOL, acutely purified P12 OLs. All expression values are normalized to OPC expression level and expressed on a log2 scale. B, In situ expression of p57Kip2 in sagittal sections of P7 (B1) and P42 (B2) mouse brain, obtained from St. Jude's BGEM website (www.stjudebgem.org). C–F, Immunostaining of P7 (C, D) and P24 (E, F) optic nerve sections. Sections were costained for CC-1 (red) and p57Kip2 (white) expression (C, E) or NG2 (red) and p57Kip2 (white) expression (D, F). Blue, DAPI nuclear stain. Green arrows indicate cells expressing only p57Kip2; red arrows indicate cells only expressing CC-1 (C, E) or NG2 (D, F); and yellow arrows indicate cells coexpressing p57Kip2 and either CC-1 or NG2. White boxes indicate regions of higher magnification shown in the lower left corners of C1–F1. G, Proportion of NG2+ OPCs (gray bars) and CC-1+ OLs (black bars) that were positive for p57Kip2 expression in the optic nerve at various developmental ages, as assayed by immunostaining. H, Proportion of p57Kip2-positive cells that were NG2+ OPCs (gray bars) or CC-1+ OLs (black bars) in the optic nerve at various developmental ages. At P2, the >100% sum of NG2+ and CC-1+ cells may reflect the presence of a small population of cells that coexpress NG2 and CC-1. In G and H, for each age-staining (p57Kip2 + NG2 and p57Kip2 + CC-1), >100 cells were scored from two distinct optic nerves, except P24-NG2 and P40-NG2 (48 and 23 cells, respectively) because of paucity of NG2+ and p57Kip2+ cells at those ages. All data presented are ± SE of the proportion. I, RT-PCR to assay p57Kip2 (23 and 25 cycles) and β-actin (control, 23 cycles) expression levels in purified P8 OPCs incubated for 6 DIV in medium containing or lacking PDGF/NT3 (± P), containing or lacking T3 (± T).
To more closely examine p57Kip2 expression in vivo, we next analyzed the in situ RNA expression patterns in P7 and adult mice, as determined by the St. Jude Children's Research Hospital Brain Gene Expression Map (BGEM) (Fig. 1 B). At P7, p57Kip2 expression is detected in the white matter areas of the spinal cord and corpus callosum. Interestingly, by adulthood (P42), p57Kip2 expression was still present in white matter regions of the spinal cord but no longer detected in the corpus callosum. To more precisely resolve the in vivo cellular time course of p57Kip2 expression, we next costained rat optic nerves at various developmental ages (P2, P7, P24, P40) for p57Kip2 and markers of immature OPCs (NG2) and mature OLs (CC-1). Expression was detected in the nuclei of both OLs and OPCs at P2 (data not shown), P7 (Fig. 1 C,D), and P24 (Fig. 1 E,F) and almost entirely extinguished by P40 (Fig. 1 G). By determining how many OPCs or OLs were coexpressing p57Kip2 at each age, we found that p57Kip2 expression peaked at P7 (Fig. 1 G), the time at which myelination is initiated in the optic nerve (Barres et al., 1992). When we determined the phenotype of cells expressing p57Kip2 at various ages, we observed that an equal proportion of p57Kip2-expressing cells are OPCs and OLs early in development (P2), and that the majority of p57Kip2-expressing cells are OLs at later time points (Fig. 1 H). These results indicate that p57Kip2 expression peaks during the initiation of myelination in OL-lineage cells and is extinguished in established, stably mature OLs in vivo, at least in the optic nerve and corpus callosum.
OL differentiation can be triggered either by mitogen withdrawal or the intrinsic timer coupled with exposure to T3 (Barres and Raff, 1994). Although either of these conditions will produce mature OLs, the initial genetic programs triggered by these two stimuli are not identical (Tokumoto et al., 2001). To more precisely determine how p57Kip2 expression is induced, we subjected purified P8 OPCs to either mitogen withdrawal, T3 exposure, or both differentiation-promoting stimuli simultaneously (Fig. 1 I). By semiquantitative RT-PCR, we found that mitogen withdrawal strongly induced p57Kip2 expression after 6 d in vitro (DIV) in either the presence or absence of T3; induction of p57Kip2 expression was seen as early as 3 DIV after mitogen withdrawal (data not shown). Conversely, we found that T3 exposure did not induce p57Kip2 expression after 6 DIV. Therefore, it appears that mitogen withdrawal is the stimulus responsible for the rapid induction of p57Kip2 that we observed in our genomics experiments (Fig. 1 A).
p57Kip2 is induced intrinsically in OPCs over time
The previous data have indicated that p57Kip2 expression is robustly upregulated in OPCs induced to differentiate specifically by mitogen withdrawal in vitro, but also that p57Kip2 expression is detected in undifferentiated OPCs in vivo (Fig. 1). Do these NG2+ p57Kip2+ cells in vivo represent OPCs that have been induced recently to differentiate by limiting amounts of mitogen but have not yet extinguished their NG2 expression, or can p57Kip2 expression be induced in OPCs independently of mitogen withdrawal? To investigate the regulation of p57Kip2 expression in undifferentiated OPCs, we cultured purified P8 OPCs for 28 d consistently in proliferation-promoting (+PDGF -T3) medium. To maintain the OPCs at a density that would minimize their spontaneous differentiation into OLs, the OPCs were passaged every 4–7 d by gentle trypsinization, followed by replating at lower densities. Cultures were checked at various times, and at no point were >2–4% of the cells present OLs by morphology (data not shown). By RT-PCR, we found that OPCs increased their p57Kip2 expression over time, such that after 28 DIV in the absence of any differentiation-promoting stimuli, these OPCs expressed p57Kip2 at a level higher than the original population of purified P8 OPCs and at a level similar to OLs generated in vitro by mitogen withdrawal (Fig. 2 A). Interestingly, this intrinsic rise in p57Kip2 expression correlates with a reduction in OPC division rate (Figs. 3 A, 4 A,C) and could potentially account for the slowing in passaged OPC cell division rate reported previously (Tang et al., 2000).
Figure 2.

Regulation of p57Kip2 expression in proliferating OPCs. A, RT-PCR (25 cycles) to detect p57Kip2 or β-actin expression in purified OPCs cultured for 7 DIV (OPC-young) or 28 DIV (OPC-old) in +PDGF -T3 medium or for 4 DIV in -PDGF +T3 medium (OL). B, Quantification of clones uniformly expressing a low level of p57Kip2 (weak), uniformly expressing a high level of p57Kip2 (strong), or showing mixed levels of p57Kip2 expression (mixed) as assayed by immunostaining after 4 DIV in +PDGF -T3 medium (black bars), 7 DIV in +PDGF -T3 medium (gray bars), or 7 DIV in +PDGF +T3 medium (white bars). Clones containing less than two healthy cells were not counted; >20 clones scored/condition. C–H, Generally uniform expression of p57Kip2 in clones of OPCs. OPCs cultured for 7 DIV in +PDGF -T3 medium and then plated at clonal density and cultured for 4 DIV (C–E) or 7 DIV (F–H) in +PDGF -T3 medium were immunostained for p57Kip2 expression (C, D, F–H ) or only stained with secondary antibody (E); pictures of representative cells from within larger clones are shown (8 to >50 cells/clone). C, F, Cells from clones uniformly expressing a high level of p57Kip2. D, G, Cells from clones uniformly expressing a lower level of p57Kip2. H, Cells from the one mixed intensity p57Kip2 clone observed at 7 DIV in +PDGF -T3 (weaker expression bottom left). I, RT-PCR (26 cycles) to detect p57Kip2 or β-actin expression in purified OPCs transfected with a CMV-GFP vector (G) or a CMV-Id4 vector (Id) and then cultured for 3 DIV in -PDGF -T3 medium or 6 DIV in +PDGF ±T3 medium.
Figure 3.

Increasing p57Kip2 slows OPC proliferation and accelerates OPC responsiveness to T3. A, B, Purified OPCs transfected with a CMV-GFP vector alone (black bars) or CMV-GFP plus CMV-p57Kip2 vectors (gray bars) and then plated at clonal density (250 cells/well) and cultured for 4 DIV in PDGF/NT3 containing medium without (A) or with (B) added T3 (+PDGF ±T3). Only clones containing GFP+ cells were scored. Clones containing ≥50% morphologically complex OL-like cells were scored as OL clones, <50% morphologically complex cells were scored as OPC clones, and both OPC and OL clone sizes were binned and plotted as histograms (1, 2, 3–4, 5–8 cells, etc.). In each condition, >70 clones were scored. In both A and B, OPC clone sizes are significantly decreased by p57Kip2 overexpression (p < 0.0001; Student's t test). C–E, In each experiment, the proportions of transfected (GFP+) cells expressing CNP, MBP, or MOG were determined by immunostaining after culturing as described (±SEM; n = 3 each condition). C, Purified OPCs transfected with CMV-GFP (black bars) or CMV-GFP + CMV-p57Kip2 (gray bars) were cultured for 7 DIV in +PDGF +T3 medium before immunostaining (*p < 0.01; **p < 0.001; Student's t test). D, E, Purified OPCs initially cultured for 7 DIV (black bars) or 28 DIV (gray bars) in +PDGF -T3 medium were transfected with CMV-GFP and nontargeting siRNA and cultured for either an additional 7 DIV in +PDGF +T3 medium before immunostaining (p < 0.006; Student's t test) (D) or an additional 3 (CNP, MBP) or 4 (MOG) DIV in -PDGF -T3 medium before immunostaining (*p < 0.001; Student's t test) (E).
Figure 4.

Decreasing p57Kip2 expression accelerates OPC proliferation and decreases OPC responsiveness to T3. A–D, OPCs initially cultured for 7 DIV (A, B) or 28 DIV (C, D) in +PDGF -T3 medium were transfected with a CMV-GFP vector plus nontargeting siRNA (black bars) or siRNA targeting p57Kip2 (gray bars) and then plated at clonal density (A, B, 250 cells/well; C, D, 500 cells/well) and cultured for 4 DIV in PDGF/NT3 containing medium without (A, C) or with (B, D) added T3 (+PDGF ±T3). Only clones containing GFP+ cells were scored. Clones containing ≥50% morphologically complex OL-like cells were scored as OL clones, <50% morphologically complex cells were scored as “OPC clones,” and both OPC and OL clone sizes were binned and plotted as histograms. In each condition, >50 clones were scored. In A–D, OPC clone sizes are significantly increased by p57Kip2 knockdown (A, p < 0.005; B, p < 0.0001; C, p < 0.0001; D, p < 0.05; Student's t test). E, F, Purified OPCs initially cultured for 7 DIV (E) or 28 DIV (F) in +PDGF -T3 medium were transfected as described in A–D and then cultured for an additional 7 d in +PDGF +T3 medium. The proportions of transfected (GFP+) cells expressing CNP, MBP, or MOG were subsequently determined by immunostaining (±SEM; n = 3 each condition; *p < 0.05, **p < 0.01, Student's t test).
When a single OPC is plated at clonal density in the presence of PDGF, all of the cells derived from that single proliferating OPC tend to differentiate synchronously in response to T3 exposure (Temple and Raff, 1986; Barres et al., 1994a). Therefore, any molecule involved in controlling the intracellular timer that regulates the sensitivity of OPCs to T3 would be expected to simultaneously change in all of the cells belonging to a single OPC clone. To determine whether p57Kip2 levels rise synchronously within proliferating OPC siblings, we examined the expression levels of p57Kip2 within individual clones of OPCs. Proliferating OPCs were plated at clonal density in the presence of mitogens with or without T3 for 4 or 7 DIV and then immunostained for p57Kip2 expression. With rare exception, by examining staining intensity, we found that p57Kip2 was expressed uniformly in every cell belonging to a single clone (Fig. 2 B). At 4 d after plating, we found only 1 of 27 clones where individual cells expressed different levels of p57Kip2, and only 3 mixed of 47 total clones at 7 d after plating (Fig. 2 H). Much more common were clones expressing uniformly high (Fig. 2 C,F) or low (Fig. 2 D,G) levels of p57Kip2. Consistent with our observation that p57Kip2 expression increases in cultured, proliferating OPCs over time in the absence of any T3 stimulation (+PDGF -T3 medium), we observed an increase in the number of clones expressing higher levels of p57Kip2 from days 4 to 7: 5 of 27 clones uniformly expressed high levels of p57Kip2 at day 4, which increased to 16 of 24 at day 7. Interestingly, T3 exposure does not increase the rate of clonal p57Kip2 induction (15 of 23 total clones expressed uniformly high levels of p57Kip2 after 7 d in +PDGF +T3 medium), reinforcing the finding that T3 does not regulate p57Kip2 expression. The finding that p57Kip2 expression increases over time in proliferating OPCs but that clones of OPCs rarely contain cells expressing both high and low levels of p57Kip2 implies that induction of p57Kip2 expression occurs approximately simultaneously in all of the cells belonging to a single clone.
p57Kip2 expression is suppressed by Id4 in proliferating OPCs
What factors might be responsible for promoting the gradual rise in p57Kip2 levels observed in continuously proliferating OPCs? Id4 is a known inhibitor of OL differentiation with expression that gradually decreases in older OPCs (Kondo and Raff, 2000a). In theory, high Id4 levels in younger OPCs could inhibit p57Kip2 expression by dimerizing with and inhibiting the activity of the class I basic helix- loop-helix (bHLH) protein E47 (Sun et al., 1991; Riechmann et al., 1994), because E47 has been shown to specifically promote the transcription of p57Kip2 (Rothschild et al., 2006). To test this hypothesis, we transfected purified P7 OPCs that had been cultured initially for 7 d in +PDGF -T3 medium with a plasmid that constitutively expresses full-length Id4, or a control GFP-expressing plasmid. Transfected cells were then cultured for 3 d in the absence of mitogens (-PDGF -T3) or for 6 d in the presence of mitogens with or without T3 (+PDGF ±T3). We found that, in the presence of mitogens, high levels of Id4 could suppress p57Kip2 expression in OPCs regardless of whether T3 was present (Fig. 2 I). Interestingly, we found that Id4 was not similarly able to inhibit the induction of p57Kip2 driven by mitogen withdrawal (-PDGF -T3), indicating that additional factors likely promote p57Kip2 expression when mitogens become limiting.
Increasing p57Kip2 slows OPC proliferation and accelerates the T3-responsive intrinsic differentiation timer
The fact that p57Kip2 expression increases synchronously in clones of proliferating OPCs, independent of T3 stimulation, implies that p57Kip2 could be a component of the intrinsic timer that limits the number of divisions OPCs can undergo in the presence of T3 (Barres et al., 1994a). To investigate this possibility, we began by examining the effects of increasing p57Kip2 levels on OPC proliferation rate and responsiveness to T3. Purified OPCs were cotransfected with plasmids expressing GFP (to identify transfected cells) and p57Kip2 from constitutive CMV promoters, and then plated at clonal density in mitogen-containing media that either contained or lacked T3 for 4 DIV. In pure proliferation-promoting medium (+PDGF -T3), we found that p57Kip2 overexpression significantly reduced OPC proliferation rate when compared with control, GFP-only transfected OPCs (Fig. 3 A, Table 1). In addition, the percentage of clones containing >50% OLs (as assayed by morphological transformation; see Materials and Methods) is slightly increased, potentially corresponding to the low level of spontaneous, T3-independent differentiation previously observed in OPCs after proliferation for an extended period of time (Barres and Raff, 1994). When OPCs were cultured in mitogens in the presence of T3 (Fig. 3 B), we noticed a slight reduction in division rate in response to T3 alone (control transfections) and a stronger reduction in division rate when p57Kip2 overexpression was coupled with T3 exposure (Table 1). p57Kip2 overexpression also greatly increased the number of clones that morphologically differentiated in response to T3, raising the percentage of clones containing a majority of complex, OL-like cells from 11.7 to 41%. p57Kip2-transfected clones apparently also transformed earlier in response to T3, as indicated by the average clone sizes of the OL morphology clones (control 4.3 vs p57Kip2 2.1).
Table 1.
Effects of p57Kip2 induction/repression on OPC cell cycle and morphology
| Average OPC clone size | Hours/division | % OL clones | Average OL clone sizes | |
|---|---|---|---|---|
| Young OPCs, p57 increase | ||||
| -T3 GFP only | 16.7 ± 1.5 | 23.6 ± 2.1 | 1.3 ± 1.3% | 1.0 ± 0.0 |
| -T3 CMV-p57 | 6.0 ± 0.6 | 37.0 ± 3.8 | 14.1 ± 3.9% | 3.1 ± 0.8 |
| +T3 GFP only | 12.6 ± 1.4 | 26.2 ± 3.0 | 11.7 ± 3.7% | 4.3 ± 0.8 |
| +T3 CMV-p57 | 4.9 ± 0.6 | 41.8 ± 5.5 | 41.0 ± 5.6% | 2.1 ± 0.2 |
| Young OPCs, p57 knock-down | ||||
| -T3 siControl | 13.9 ± 1.5 | 25.3 ± 2.7 | 11.1 ± 4.3% | 3.7 ± 1.3 |
| -T3 sip57 | 21.9 ± 2.3 | 21.6 ± 2.2 | 0.0 ± 0.0% | - |
| +T3 siControl | 7.1 ± 0.8 | 33.9 ± 3.7 | 25.5 ± 6.1% | 3.4 ± 1.0 |
| +T3 sip57 | 17.8 ± 1.8 | 23.1 ± 2.3 | 7.4 ± 3.6% | 3.3 ± 0.9 |
| Older OPCs, p57 knock-down | ||||
| -T3 siControl | 2.9 ± 0.2 | 63.2 ± 5.2 | 15.4 ± 5.0% | 1.8 ± 0.4 |
| -T3 sip57 | 6.1 ± 0.6 | 36.9 ± 3.6 | 1.9 ± 1.9% | 1.0 ± 0.0 |
| +T3 siControl | 2.8 ± 0.3 | 64.8 ± 7.3 | 54.7 ± 6.8% | 1.6 ± 0.2 |
| +T3 sip57 | 4.0 ± 0.4 | 48.2 ± 4.6 | 23.1 ± 5.8% | 2.3 ± 0.4 |
Tabulated results of clonal size and morphology assays described in Figure 3, A and B, and Figure 4 A–D. In all conditions, only clones containing transfected (GFP+) cells were counted; 50–80 clones were counted per condition. Young OPCs were transfected after 7 DIV in +PDGF -T3 medium; older OPCs were transfected after 28 DIV in +PDGF -T3 medium and then cultured 4 DIV +PDGF ±T3. ″OPC clones″ contain <50% OLs (by morphology); ″OL clones″ contain ≥50% morphologically complex OL-like cells. Hours per division are calculated as 96 h/log2 (average OPC clone size). All data presented are ± SEM or SE of the proportion in percentage of OL clones.
To further investigate the role of p57Kip2 in regulating the T3-dependent timer that controls OL differentiation, we determined the effect of p57Kip2 overexpression on T3-induced myelin gene expression. Because we had determined previously that OL differentiation occurs in multiple distinct stages (Dugas et al., 2006), we determined the effects of increasing p57Kip2 on markers of both early (CNP, MBP) and late (MOG) stage OL differentiation (Fig. 1 A). Transfected OPCs were cultured for 7 DIV in the presence of both mitogens and T3 and then immunostained for CNP, MBP, or MOG expression (Fig. 3 C). When compared with GFP-only transfected control cells, we found that p57Kip2 overexpression greatly enhanced the percentages of transfected cells expressing early markers of OL differentiation (CNP and MBP) in the presence of T3, resembling the increase in clones containing >50% OL morphology cells we observed previously with increased p57Kip2 expression (Table 1). Therefore, in OPCs, increased p57Kip2 expression appears capable of inducing both complex OL-like morphology and characteristic OL gene expression in the presence of T3. Interestingly, the acceleration of the T3-dependent OPC differentiation timer induced by p57Kip2 expression only augmented the early phase of OL maturation; expression of the late-phase marker MOG was not altered by overexpression of p57Kip2.
Because we had demonstrated that OPCs aged in culture intrinsically increased their p57Kip2 expression levels, we wanted to test whether these older OPCs were also more responsive to T3. Similar to previously published observations (Barres et al., 1994a), when purified P8 OPCs were initially aged in culture for 28 DIV in +PDGF -T3 medium, a much higher percentage of these older OPCs differentiated in response to T3 exposure relative to younger purified OPCs (Fig. 3 D). In contrast to the p57Kip2-overexpressing younger OPCs, these older OPCs generated OLs that robustly expressed not only the early OL differentiation markers CNP and MBP but also the later marker MOG in response to T3. These data indicate that genes in addition to p57Kip2 may be intrinsically induced in older OPCs that promote rapid late-phase differentiation in response to T3.
Is the enhanced differentiation capacity we see in older OPCs specific to the T3-triggered timer, or are older OPCs simply more prone to rapidly mature into OLs? To address this question, we stimulated differentiation in OPCs initially cultured for 7 or 28 DIV in +PDGF -T3 medium by subsequent mitogen withdrawal (Fig. 3 E). Consistent with previous observations (Shi et al., 1998), the data clearly demonstrate that, whereas older OPCs differentiate more rapidly in response to T3 exposure, younger OPCs differentiate more rapidly in response to mitogen withdrawal. These data indicate that in older OPCs, which intrinsically express higher levels of p57Kip2, only differentiation mediated by the T3-dependent clock mechanism is accelerated.
Reducing p57Kip2 accelerates OPC proliferation and retards the T3-responsive intrinsic differentiation timer
Having demonstrated that increased p57Kip2 levels were sufficient to both increase OPC cell cycle time and to accelerate the intrinsic timer that allows T3 to induce OL differentiation, we next wanted to determine whether p57Kip2 was necessary for normal timing of T3-mediated differentiation and cell cycle regulation in OPCs. We first tested whether OPC proliferation rates were normally regulated by p57Kip2 by reducing p57Kip2 levels in both younger, rapidly dividing, and older, more slowly dividing OPCs. After either 7 or 28 d in proliferation-promoting (+PDGF -T3) medium, cultured OPCs were transfected with an siRNA pool targeting p57Kip2 (sip57 pool) to reduce intrinsic p57Kip2 levels, and then plated at clonal density. Knockdown of p57Kip2 expression was confirmed by semiquantitative RT-PCR (Fig. S1A, available at www.jneurosci.org as supplemental material). When compared with controls transfected with a nontargeting siRNA pool, clones of OPCs in which p57Kip2 expression had been knocked down were notably larger when assayed after 4 d in +PDGF -T3 medium (Fig. 4 A,C; Table 1). Significantly, the proliferation rate observed for the older OPCs, in which p57Kip2 levels were initially intrinsically higher, increased from one division per 62.5 h to one division per 36.8 h when p57Kip2 levels were reduced. In addition, the low level of spontaneous morphological transformation indicative of OL differentiation normally seen in +PDGF -T3 medium was almost completely abolished in both younger and older OPCs when p57Kip2 levels were reduced.
To next determine whether p57Kip2 was required for controlling the timing of T3-stimulated OL differentiation, we first cultured the same siRNA-transfected sets of OPCs described above at clonal density in medium containing mitogen and T3 (+PDGF +T3) for 4 DIV (Fig. 4 B,D). With younger OPCs that had only been proliferating for 7 DIV before transfection, the reduction in division rate normally induced by T3 exposure was completely abolished when p57Kip2 levels were reduced (Table 1). There was also a large decrease in the number of clones that morphologically differentiated in response to T3 (from 25.5 to 7.4%). Similarly, when p57Kip2 levels were reduced in older OPCs that had initially been proliferating for 28 DIV before transfection and T3 exposure, a strong reduction in the number of clones that morphologically differentiated in response to T3 was observed (from 54.7 to 23.1%).
To further investigate the extent to which OL differentiation induced by T3 exposure relied on p57Kip2 expression, we purified P8 OPCs transfected with sip57 pool, plated for 7 DIV in mitogens in the presence of T3, and then immunostained for CNP, MBP, and MOG expression. We found that knockdown of p57Kip2 expression in OPCs reduced the numbers of cells that expressed both early and late OL differentiation markers in response to T3 exposure (Fig. 4 E). When this experiment was repeated with OPCs initially proliferated for 28 DIV, in which intrinsic p57Kip2 levels are higher, similar results were obtained (Fig. 4 F). Whereas it is possible that the sip57 pool could have off-target effects by directly silencing genes other than p57Kip2, similar reductions in myelin gene expression were observed with two distinct individual siRNAs targeting nonoverlapping regions of p57Kip2 (Fig. S1B,C, available at www.jneurosci.org as supplemental material). Because it is unlikely that two distinct siRNA sequences would silence the same off-target genes, the effects observed are most likely the direct result of a reduction in p57Kip2 expression. Thus, whereas overexpression of p57Kip2 in the presence of T3 is sufficient only to accelerate the early stage of OL differentiation, p57Kip2 appears to be required in the T3-dependent differentiation timer for the normal promotion of all OL differentiation stages, as assayed both by morphology and myelin gene expression. Cumulatively, these data indicate that p57Kip2 normally plays an important role in regulating the timing of OL differentiation.
Reducing p57Kip2 interferes with the ability of mitogen withdrawal to promote premature OL differentiation
The previous data have indicated that the gradual increase in p57Kip2 expression that we observe in proliferating OPCs intrinsically regulates the normal timing of OL differentiation in response to T3. However, our initial findings were that the withdrawal of mitogens from proliferating OPCs produced a rapid increase in p57Kip2 expression. Could this observed induction of p57Kip2 be contributing to the ability of mitogen withdrawal to short circuit the normal T3-dependent timer of OL differentiation?
To determine whether p57Kip2 is required in the differentiation program triggered immediately in OPCs by mitogen withdrawal, we assayed the effects of p57Kip2 knockdown on the expression of the OL differentiation markers CNP, MBP, and MOG 3–4 d after the removal of mitogens from purified OPCs. We found that purified OPCs transfected with sip57 pool generated fewer mature OLs than control siRNA transfected OPCs in response to mitogen withdrawal (Fig. 5). Specifically, the numbers of MBP+ (Fig. 5 A,B) and MOG+ (Fig. 5 C,D) OLs were significantly reduced by p57Kip2 knockdown (Fig. 5 E). Interestingly, expression of the early differentiation marker CNP was not significantly altered by p57Kip2 reduction during mitogen withdrawal (Fig. 5 E). Together, these results indicate that, in contrast to the T3-dependent timer, the immediate induction of OL differentiation triggered by mitogen withdrawal does not initially require p57Kip2, but that increasing p57Kip2 levels may promote the completion of OL differentiation when mitogens are limiting.
Figure 5.

Reducing p57Kip2 expression retards OPC response to mitogen withdrawal. A–D, Purified OPCs cotransfected with a CMV-GFP vector and either siRNA targeting p57Kip2 (sip57; B, D) or control nontargeting siRNA (siControl; A, C) were plated in -PDGF -T3 medium for 3 DIV and stained for MBP (red) and GFP (white) expression (A, B), or 4 DIV and stained for MOG (red) and GFP (white) expression (C, D). Blue, DAPI nuclear stain. Yellow arrows indicate transfected cells (GFP+) expressing MBP (A, B) or MOG (C, D), green arrows indicate transfected cells not expressing MBP or MOG, and red arrows indicate untransfected cells (GFP-) expressing MBP or MOG. E, Proportion of siControl (green bars), sip57 (red bars), sip27 (blue bars) transfected, or sip57 + sip27 cotransfected (yellow bars) cells expressing CNP, MBP, or MOG after 3 DIV (CNP, MBP) or 4 DIV (MOG) in -PDGF -T3 medium (±SEM; n = 3 each condition; *p < 0.01, Holm–Sidak post hoc test vs control). All sip57, sip27, and sip57+sip27 cotransfections not significantly different from each other except: CNP, sip57 versus sip27 and sip27+sip57; MBP, sip27+sip57 versus sip57 and sip27; MOG, sip27+sip57 versus sip57 and sip27 (all exceptions listed; p < 0.01; Holm–Sidak; all pairwise post hoc test).
What could account for the incomplete suppression of mitogen withdrawal-induced OL differentiation obtained by p57Kip2 knockdown? One explanation could be that siRNA transfection does not completely abolish p57Kip2 expression, resulting in an incomplete phenotype. In addition, other cell cycle arrest genes could partially compensate for the loss of p57Kip2. p27Kip1, a second member of the Cip/Kip family of cell cycle inhibitor genes, has been implicated previously in OL differentiation in vitro (Tang et al., 1999; Miskimins et al., 2002; Tokumoto et al., 2002; Wei et al., 2003, 2004). Additionally, p27Kip1 − / − mice demonstrate a delay, but not a block, in mature OL generation and an increase in OPC proliferation in vivo (Casaccia-Bonnefil et al., 1999). Could both p57Kip2 and p27Kip1 be contributing to the premature OL differentiation triggered by mitogen withdrawal? When p57Kip2 and p27Kip1 are simultaneously knocked down by siRNA transfections, the reductions in myelin gene expression observed after mitogen withdrawal are significantly stronger than those seen in individual knockdowns (Fig. 5 E). In addition, although p57Kip2 − / − knock-out mice die around the time of birth before most myelination has commenced in the CNS (Takahashi et al., 2000), OPC proliferation rates can be assessed in the spinal cord, where OL-lineage cell generation begins at embryonic ages (Hardy, 1997; Casaccia-Bonnefil et al., 1999). p57Kip2 − / − mice did not show significant increases in OPC levels early in the embryonic spinal cord, but p57Kip2 − / − p27Kip1 − / − double mutants had more proliferating OL lineage cells than either single mutant mouse (P. Casaccia-Bonnefil, personal communication), indicating that p27Kip1 may compensate for p57Kip2 loss in regulating the OPC cell cycle in vivo. These results indicate that p57Kip2 and p27Kip1 may synergistically contribute to the immediate cessation of the cell cycle and program of OL differentiation triggered by mitogen withdrawal.
Increasing p57Kip2 promotes spontaneous OL differentiation
If the rapid induction of p57Kip2 expression triggered by mitogen withdrawal contributes to the overriding of the OPC differentiation timer, which results in premature differentiation independent of T3, then acutely increasing p57Kip2 levels in proliferating OPCs should promote OL differentiation in the absence of T3. To test this hypothesis, we overexpressed p57Kip2 in transfected OPCs and then cultured the cells for 7 DIV in +PDGF -T3 medium. When compared with control, GFP-only transfected OPCs, a significant increase was seen in the number of morphologically OL-appearing cells expressing CNP and MBP, early markers of OL differentiation (Fig. 6 A,B,I). A smaller but still significant increase in the number of transfected cells expressing the late-stage differentiation marker MOG was also detected (Fig. 6 E,F,I). However, by comparing these data with Figure 3 C, we observe that p57Kip2 overexpression and T3 exposure act cooperatively to provide a much greater enhancement of OL differentiation. Cumulatively, these data indicate that p57Kip2 is likely more important in the regulation of the intracellular OPC differentiation timer, and that a rapid increase in p57Kip2 may only partially contribute to the ability of mitogen withdrawal to override this intrinsic timer.
Figure 6.
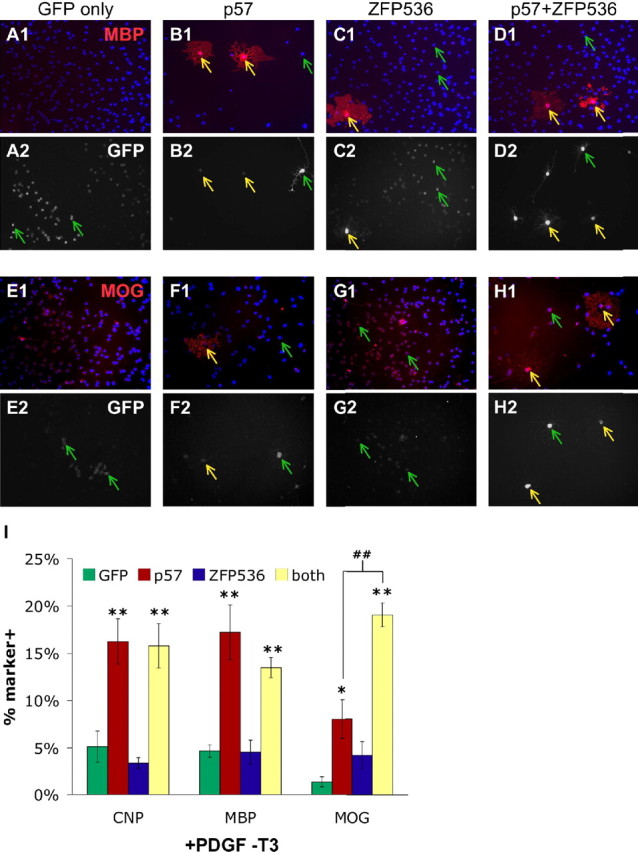
Increasing p57Kip2 and ZFP536 expression promotes OL differentiation. Increasing p57Kip2 expression initiates OL differentiation, and p57Kip2 + ZFP536 together cooperatively enhance late-stage OL differentiation in the absence of differentiation-promoting stimuli. A–H, Purified OPCs transfected with a CMV-GFP vector (GFP only; A, E), or cotransfected with CMV-GFP + CMV-p57Kip2 (p57; B, F), + CMV-ZFP536 (ZFP536; C, G), or both CMV-p57Kip2 + CMV-ZFP536 (p57+ZFP536; D, H) were cultured for 7 DIV in +PDGF -T3 medium. Cells were then costained for MBP (red) and GFP (white) expression (A–D) or MOG (red) and GFP (white) expression (E–H). Blue, DAPI nuclear stain. Yellow arrows indicate transfected cells (GFP+) expressing MBP (A–D) or MOG (E–H), green arrows indicate transfected cells not expressing MBP or MOG. I, Proportion of control (green bars), p57Kip2 (red bars), ZFP536 (blue bars), or p57Kip2 + ZFP536 (yellow bars) transfected cells expressing CNP, MBP, or MOG after 7 DIV in +PDGF -T3 medium (±SEM; n = 6 all conditions except CNP–both condition, n = 3; *p < 0.05, **p < 0.01 vs control Holm–Sidak post hoc test; in all cases, control vs ZFP536, p > 0.05 and p57 vs both, p > 0.05, except ## p < 0.01 Holm–Sidak post hoc test).
p57Kip2 and ZFP536 act cooperatively to promote late-stage oligodendrocyte differentiation
The previous results have consistently demonstrated that p57Kip2 is required, particularly as a component of the T3-dependent timer, for robust induction of both the early and late stages of OL differentiation. However, increasing p57Kip2 appears to be sufficient only to accelerate the early stage of OL differentiation (as measured by increases in CNP and MBP expression, but not MOG expression). Potentially, additional genes not induced by p57Kip2 are required to fully promote the final stage of OL differentiation. A probable candidate gene to fill this role is ZFP536, a recently identified transcription factor that is both induced late in the normal time course of OL differentiation and required specifically to induce the later stage of OL differentiation (Dugas et al., 2006). To investigate whether p57Kip2 and ZFP536 can combine to robustly promote full OL differentiation, we compared the effects of overexpressing p57Kip2 and ZFP536 alone to the effects of coexpressing both p57Kip2 and ZFP536 in OPCs. After culturing transfected OPCs for 7 d in +PDGF -T3 medium, we observed that ZFP536 alone was not able to enhance OL differentiation, as assayed by either change in morphology (data not shown) or by measure of early and late marker gene expression (Fig. 6 C,G,I). In addition, expression of ZFP536 combined with p57Kip2 did not further enhance the ability of p57Kip2 to promote the early phase of OL differentiation (Fig. 6 D,I). However, the combination of ZFP536 and p57Kip2 was able to significantly increase the number of cells expressing the late-phase marker gene MOG over the level of either ZFP536 or p57Kip2 alone (Fig. 6 H,I). In fact, ZFP536 and p57Kip2 appear to act cooperatively, because the increase in MOG+ cells observed with ZFP536-p57Kip2 cotransfections is greater than the sum of the effect seen for ZFP536 and p57Kip2 single-gene transfections. Similar results were observed in the presence of T3 (+PDGF +T3 medium) (data not shown). These results indicate that proteins involved in regulating the intracellular timer of OL differentiation, such as p57Kip2, can be involved in not just initiating but also promoting all stages of OL differentiation. These data should prove useful in both understanding how to promote normal, complete maturation of OLs from undifferentiated OPCs and potentially also in reconstructing the genetic program required to produce a myelinating OL from unspecified, plastic precursor cells.
Discussion
p57Kip2 is an integral component of the timer that normally regulates OL differentiation
The precise regulation of how many times a precursor cell can divide before differentiating is crucial to the normal development of any complex multicellular organism. OPCs present an excellent model system in which to study the normal timing of vertebrate cell differentiation, because OPCs can be highly purified and cultured in defined, serum-free media that either promotes precursor proliferation or OL differentiation (Barres et al., 1994a; Barres and Raff, 1994). Previous studies identified an intrinsic clock that regulates how many times an OPC can divide before it differentiates (Temple and Raff, 1986; Barres et al., 1994a; Gao et al., 1997). This differentiation clock requires the combined activity of both an effector component, which responds to the presentation of external cues such as T3 or RA, and a timer component that intracellularly measures the passage of time as an OPC proliferates, independent of T3 stimulation. Although the external cues that trigger OL differentiation have been extensively studied, to date, the components of the intracellular timer remain less well characterized. Any candidate for the timer component of the clock should possess certain characteristics: its level should change synchronously over time within immature proliferating OPC clones, the change that occurs in older OPCs should be required for T3 to initiate OL differentiation, and the time-monitoring change should be independent of T3 stimulation. Otherwise, early presentation of T3 would accelerate the timer and young proliferating OPCs would rapidly differentiate in response to T3, similar to the immediate differentiation induced by mitogen withdrawal.
Several candidates have been proposed as potential timers of OL differentiation. Protein levels (but not mRNA levels) of the cell cycle inhibitor genes p27Kip1 and p18Ink4c have been observed to increase over time in proliferating OPCs, and increasing levels of these proteins accelerated differentiation in response to T3 (Durand et al., 1997; Tokumoto et al., 2002). Perhaps paradoxically, the same group also demonstrated that p27Kip1 and p18Ink4c protein levels also rise directly in response to T3 stimulation (Tokumoto et al., 2001), raising the possibility that other T3-independent components of the timer may remain to be identified. Here, we propose that the cell cycle inhibitor gene p57Kip2 is an integral component of the timer that normally regulates OL differentiation. Two bHLH proteins that inhibit OL differentiation, Id4 and Hes5, have been characterized as genes with expression that gradually decreases in older OPCs (Kondo and Raff, 2000a,b). Our current data indicate that the ability of Id4 to repress OL differentiation may be mediated in part by the ability of Id4 to suppress p57Kip2 expression.
In vivo, we observed that p57Kip2 expression was transiently upregulated around the age at which myelination is initiated and reduced after the peak of myelination has passed. Early in development, p57Kip2 was detected in both OPCs and OLs, whereas later in development, the majority of p57Kip2-expressing cells were OLs. These data indicate that a rise in p57Kip2 expression is likely a very early event in OL differentiation, potentially marking OPCs that are on the verge of differentiating into OLs. In addition, the extinguishing of p57Kip2 expression indicates that p57Kip2 may be involved in the initiation but not the maintenance of the mature OL phenotype in vivo.
In vitro, we observed that p57Kip2 mRNA and protein expression increases intrinsically in purified OPCs over time in the absence of any extrinsic cues, and that this induction appears to occur synchronously within all the cells of individual OPC clones. With the exception of thyroid hormone receptor (Barres et al., 1994a), this is the first reported observation of a potential intrinsic OPC clock regulator whose level changes synchronously within dividing OPC clones. This could potentially correlate with the synchronous differentiation of OPC clones previously observed in vitro (Temple and Raff, 1986; Barres et al., 1994a). Importantly, exposure to T3 does not alter p57Kip2 expression; thus, p57Kip2 increase could measure time independent of environmental T3 levels. Our data indicate that increasing p57Kip2 accelerates the intracellular timer that regulates T3-mediated OL differentiation, and that reducing p57Kip2 retards this timer. These effects may be mediated, at least in part, by altering cell cycle regulation, because these same manipulations respectively increase and reduce OPC cell cycle time in both the presence and absence of T3. Interestingly, manipulations of p73 activity have similar phenotypic outcomes for OL differentiation (Billon et al., 2004) as we observe here with p57Kip2. This may be a direct consequence of the induction of p57Kip2 expression by p73β (Balint et al., 2002; Vaccarello et al., 2006).
Model for the role of p57Kip2 in OL differentiation
Although the exact mechanism of p57Kip2-mediated promotion of OL differentiation remains to be elucidated, we propose the following model (Fig. 7). In recently generated, rapidly proliferating OPCs (Fig. 7 A), p57Kip2 expression levels are kept low, likely in part by the transcription-suppressing activity of Id4. Low levels of p57Kip2, in combination with low levels of other known cell cycle inhibitors such as p27Kip1 and p18Inc4c (Durand et al., 1997; Tokumoto et al., 2001, 2002) would promote cell cycling and maintenance of the immature OPC phenotype in the presence of mitogens.
Figure 7.
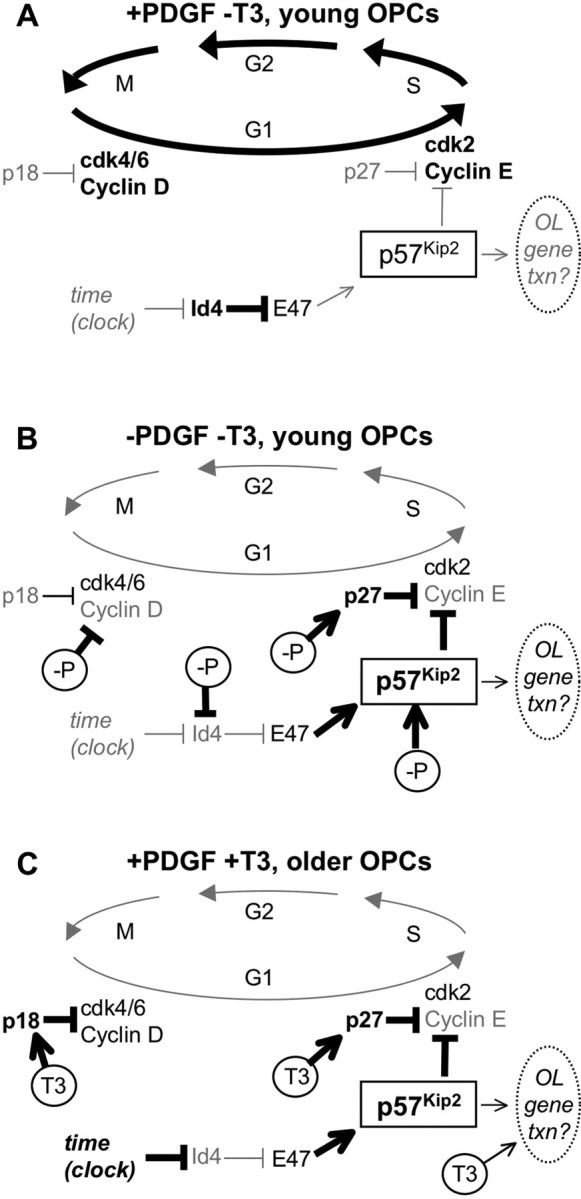
Model for the role of p57Kip2 in OL differentiation. A, In the presence of mitogens and the absence of T3 early in development, p57Kip2 levels are kept low in part by inhibition of E47 by Id4. Low levels of cell cycle inhibitor proteins (e.g., p57, p27, p18) result in disinhibition of CyclinE-cdk2 and CyclinD-cdk4/6 complexes and progression of the cell cycle. B, When mitogens become limiting (e.g., PDGF; -P), p57Kip2 expression is rapidly induced, likely partially (but not wholly) via reduced Id4 levels, resulting in disinhibition of E47 and increase in p57Kip2 production. Mitogen withdrawal also promotes an increase in p27Kip1 levels and a decrease in CyclinD levels, cumulatively leading to the inhibition of CyclinE-cdk2 and CyclinD-cdk4/6 complexes and promotion of cell cycle arrest. C, When exposed to T3 (T3), p18Ink4c and p27Kip1 proteins are induced, leading to a reduction in Cyclin-cdk complex activity. In older OPCs, Id4 levels are reduced via an unknown mechanism independent of T3 activity (time). The resulting activation of E47 leads to an increase in p57Kip2 which, coupled with the actions of T3, promotes cell cycle arrest in older OPCs. Both p57Kip2and T3 may also promote OL differentiation via more direct stimulation of OL gene transcription.
Mitogens are required for the normal operation of the OPC differentiation timer, as when mitogens become limiting OPCs immediately differentiate regardless of age or T3 levels. Mitogen withdrawal may override the timer in part by triggering a rapid increase in p57Kip2 expression (Fig. 7 B). In addition to triggering a rapid decrease in Id4 levels (Kondo and Raff, 2000a), our data indicate that mitogen withdrawal likely promotes p57Kip2 expression through other, Id4-independent pathways. Mitogen withdrawal has also been shown to increase p27Kip1 levels (Tokumoto et al., 2001). Together, higher p27Kip1 and p57Kip2 levels would inhibit CyclinE-cdk2 complex formation. Supporting this model is the fact that inhibition of CyclinE-cdk2 complex formation has been implicated previously in withdrawal of OPCs from the cell cycle (Ghiani and Gallo, 2001), and our demonstration that both p27Kip1 and p57Kip2 contribute to mitogen withdrawal-induced OL differentiation. CyclinD levels also rapidly drop in response to mitogen withdrawal (Tokumoto et al., 2001). Cumulatively, these factors should lead to a rapid cessation of the cell cycle, allowing for the initiation of OL differentiation.
When mitogens and T3 are both present, p57Kip2 intrinsically accumulates more gradually as the timer component of the clock to eventually promote OL differentiation in response to T3 (Fig. 7 C). T3, the effector component of the clock mechanism, may encourage OL differentiation in a number of ways. T3 can slow the cell cycle by increasing several cell cycle inhibitor proteins, such as p18Ink4c and p27Kip1 (Tokumoto et al., 2001), which would serve to decrease CyclinD-cdk4/6 and CyclinE-cdk2 complex formation. However, the reduction in CyclinE and CyclinD stimulated by T3 exposure is not as strong as that observed for mitogen withdrawal (Tokumoto et al., 2001). In young OPCs, high levels of Id4 would serve to inhibit transcription of p57Kip2. The low levels of p57Kip2 coupled with an incomplete suppression of Cyclins likely produces the slowing but not cessation of the cell cycle observed in young OPCs exposed to T3. Over time, p57Kip2 expression increases in concordance with a drop in Id4 levels. Similar increases in p27Kip1 gene expression are likely not observed as a result of falling Id4 levels, because E47 specifically induces p57Kip2 (Kondo and Raff, 2000a; Tokumoto et al., 2002; Rothschild et al., 2006). In the absence of T3 stimulation, the increases in p57Kip2, p27Kip1, and p18Ink4c protein expression observed in older OPCs may only partially inhibit Cyclin-cdk complex formation and incompletely inhibit the cell cycle. Only the cumulative increase in p57Kip2 in older OPCs coupled with the T3-stimulated increases in additional cell cycle inhibitor proteins may be sufficient to completely halt the cell cycle and allow OL differentiation to proceed. Similar cooperation between cell cycle inhibitor proteins in promoting differentiation has been reported previously (Zhang et al., 1998; Zhang et al., 1999; Figliola and Maione, 2004; Myers et al., 2004; Itoh et al., 2007). Finally, once the cell cycle has stopped, both p57Kip2 and T3 may further induce OL differentiation by independent stimulation of other OL genetic programs (Baas et al., 1997).
Although arrest of the cell cycle may be an obligate step in promoting OL differentiation, previous studies have shown it may not be sufficient. Interestingly, although increases in p57Kip2 levels alone were sufficient to initiate OL differentiation (Fig. 6), increases in p27Kip1 levels or other means of arresting the cell cycle were not (Tang et al., 1999; Casaccia-Bonnefil and Liu, 2003). p57Kip2 has been shown previously to influence differentiation independent of its ability to inhibit the cell cycle; p57Kip2 promotes myogenic differentiation by stabilizing the bHLH transcription factor MyoD (Reynaud et al., 2000) and promotes dopamine neuron differentiation via interactions with the orphan nuclear receptor Nurr1 (Joseph et al., 2003). A related Cip/Kip family member, p27Kip1, has been shown to directly influence transcription from the mbp promoter (Miskimins et al., 2002; Wei et al., 2003, 2004). It is possible that p57Kip2 may similarly directly promote myelin gene transcription independent of its interactions with Cyclin-cdk complexes.
Among members of the Cip/Kip family, only deficiencies in p57Kip2 result in developmental lethality (Takahashi et al., 2000). In fact, p57Kip2 has been implicated previously in regulating the differentiation of various tissues, such as the lens, pancreas, blood, chondrocytes, muscle, and dopaminergic neurons (Zhang et al., 1998, 1999; Joseph et al., 2003; MacLean et al., 2004; Scandura et al., 2004; Georgia et al., 2006). The current study adds OLs to the list of cell types with maturation that is regulated by p57Kip2. In future studies, determining whether the unique role of p57Kip2 in regulating the timing of OL differentiation is replicated in these various other cell types may provide valuable insight into how the essential control of vertebrate cell number is developmentally regulated.
Footnotes
This work was supported by the Myelin Repair Foundation, National Multiple Sclerosis Society Grant RG2332, and National Eye Institute Grant R01 EY10257. All protocols are available on request from jcdugas@alum.mit.edu.
References
- Baas D, Bourbeau D, Sarlieve LL, Ittel ME, Dussault JH, Puymirat J. Oligodendrocyte maturation and progenitor cell proliferation are independently regulated by thyroid hormone. Glia. 1997;19:324–332. doi: 10.1002/(sici)1098-1136(199704)19:4<324::aid-glia5>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Balint E, Phillips AC, Kozlov S, Stewart CL, Vousden KH. Induction of p57(KIP2) expression by p73beta. Proc Natl Acad Sci USA. 2002;99:3529–3534. doi: 10.1073/pnas.062491899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA, Raff MC. Control of oligodendrocyte number in the developing rat optic nerve. Neuron. 1994;12:935–942. doi: 10.1016/0896-6273(94)90305-0. [DOI] [PubMed] [Google Scholar]
- Barres BA, Hart IK, Coles HS, Burne JF, Voyvodic JT, Richardson WD, Raff MC. Cell death and control of cell survival in the oligodendrocyte lineage. Cell. 1992;70:31–46. doi: 10.1016/0092-8674(92)90531-g. [DOI] [PubMed] [Google Scholar]
- Barres BA, Schmid R, Sendnter M, Raff MC. Multiple extracellular signals are required for long-term oligodendrocyte survival. Development. 1993;118:283–295. doi: 10.1242/dev.118.1.283. [DOI] [PubMed] [Google Scholar]
- Barres BA, Lazar MA, Raff MC. A novel role for thyroid hormone, glucocorticoids and retinoic acid in timing oligodendrocyte development. Development. 1994a;120:1097–1108. doi: 10.1242/dev.120.5.1097. [DOI] [PubMed] [Google Scholar]
- Barres BA, Raff MC, Gaese F, Bartke I, Dechant G, Barde YA. A crucial role for neurotrophin-3 in oligodendrocyte development. Nature. 1994b;367:371–375. doi: 10.1038/367371a0. [DOI] [PubMed] [Google Scholar]
- Baumann N, Pham-Dinh D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev. 2001;81:871–927. doi: 10.1152/physrev.2001.81.2.871. [DOI] [PubMed] [Google Scholar]
- Billon N, Terrinoni A, Jolicoeur C, McCarthy A, Richardson WD, Melino G, Raff M. Roles for p53 and p73 during oligodendrocyte development. Development. 2004;131:1211–1220. doi: 10.1242/dev.01035. [DOI] [PubMed] [Google Scholar]
- Casaccia-Bonnefil P, Liu A. Relationship between cell cycle molecules and onset of oligodendrocyte differentiation. J Neurosci Res. 2003;72:1–11. doi: 10.1002/jnr.10565. [DOI] [PubMed] [Google Scholar]
- Casaccia-Bonnefil P, Hardy RJ, Teng KK, Levine JM, Koff A, Chao MV. Loss of p27Kip1 function results in increased proliferative capacity of oligodendrocyte progenitors but unaltered timing of differentiation. Development. 1999;126:4027–4037. doi: 10.1242/dev.126.18.4027. [DOI] [PubMed] [Google Scholar]
- Cunningham JJ, Roussel MF. Cyclin-dependent kinase inhibitors in the development of the central nervous system. Cell Growth Differ. 2001;12:387–396. [PubMed] [Google Scholar]
- Dugas JC, Tai Y-C, Speed TP, Ngai J, Barres BA. Functional genomic analysis of oligodendrocyte differentiation. J Neurosci. 2006;26:10967–10983. doi: 10.1523/JNEUROSCI.2572-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand B, Gao FB, Raff M. Accumulation of the cyclin-dependent kinase inhibitor p27/Kip1 and the timing of oligodendrocyte differentiation. EMBO J. 1997;16:306–317. doi: 10.1093/emboj/16.2.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dussault JH, Ruel J. Thyroid hormones and brain development. Annu Rev Physiol. 1987;49:321–334. doi: 10.1146/annurev.ph.49.030187.001541. [DOI] [PubMed] [Google Scholar]
- Figliola R, Maione R. MyoD induces the expression of p57Kip2 in cells lacking p21Cip1/Waf1: overlapping and distinct functions of the two cdk inhibitors. J Cell Physiol. 2004;200:468–475. doi: 10.1002/jcp.20044. [DOI] [PubMed] [Google Scholar]
- Gao FB, Durand B, Raff M. Oligodendrocyte precursor cells count time but not cell divisions before differentiation. Curr Biol. 1997;7:152–155. doi: 10.1016/s0960-9822(06)00060-1. [DOI] [PubMed] [Google Scholar]
- Georgia S, Soliz R, Li M, Zhang P, Bhushan A. p57 and Hes1 coordinate cell cycle exit with self-renewal of pancreatic progenitors. Dev Biol. 2006;298:22–31. doi: 10.1016/j.ydbio.2006.05.036. [DOI] [PubMed] [Google Scholar]
- Ghiani C, Gallo V. Inhibition of cyclin E-cyclin-dependent kinase 2 complex formation and activity is associated with cell cycle arrest and withdrawal in oligodendrocyte progenitor cells. J Neurosci. 2001;21:1274–1282. doi: 10.1523/JNEUROSCI.21-04-01274.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy RJ. Dorsoventral patterning and oligodendroglial specification in the developing central nervous system. J Neurosci Res. 1997;50:139–145. doi: 10.1002/(SICI)1097-4547(19971015)50:2<139::AID-JNR3>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Itoh Y, Masuyama N, Nakayama K, Nakayama KI, Gotoh Y. The cyclin-dependent kinase inhibitors p57 and p27 regulate neuronal migration in the developing mouse neocortex. J Biol Chem. 2007;282:390–396. doi: 10.1074/jbc.M609944200. [DOI] [PubMed] [Google Scholar]
- Joseph B, Wallen-Mackenzie A, Benoit G, Murata T, Joodmardi E, Okret S, Perlmann T. p57(Kip2) cooperates with Nurr1 in developing dopamine cells. Proc Natl Acad Sci USA. 2003;100:15619–15624. doi: 10.1073/pnas.2635658100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo T, Raff M. The Id4 HLH protein and the timing of oligodendrocyte differentiation. EMBO J. 2000a;19:1998–2007. doi: 10.1093/emboj/19.9.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo T, Raff M. Basic helix-loop-helix proteins and the timing of oligodendrocyte differentiation. Development. 2000b;127:2989–2998. doi: 10.1242/dev.127.14.2989. [DOI] [PubMed] [Google Scholar]
- MacLean HE, Guo J, Knight MC, Zhang P, Cobrinik D, Kronenberg HM. The cyclin-dependent kinase inhibitor p57(Kip2) mediates proliferative actions of PTHrP in chondrocytes. J Clin Invest. 2004;113:1334–1343. doi: 10.1172/JCI21252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miskimins R, Srinivasan R, Marin-Husstege M, Miskimins WK, Casaccia-Bonnefil P. p27(Kip1) enhances myelin basic protein gene promoter activity. J Neurosci Res. 2002;67:100–105. doi: 10.1002/jnr.10080. [DOI] [PubMed] [Google Scholar]
- Myers TK, Andreuzza SE, Franklin DS. p18INK4c and p27KIP1 are required for cell cycle arrest of differentiated myotubes. Exp Cell Res. 2004;300:365–378. doi: 10.1016/j.yexcr.2004.07.024. [DOI] [PubMed] [Google Scholar]
- Park HC, Boyce J, Shin J, Appel B. Oligodendrocyte specification in zebrafish requires notch-regulated cyclin-dependent kinase inhibitor function. J Neurosci. 2005;25:6836–6844. doi: 10.1523/JNEUROSCI.0981-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff MC, Miller RH, Noble M. A glial progenitor cell that develops in vitro into an astrocyte or an oligodendrocyte depending on culture medium. Nature. 1983;303:390–396. doi: 10.1038/303390a0. [DOI] [PubMed] [Google Scholar]
- Reynaud EG, Guillier M, Leibovitch MP, Leibovitch SA. Dimerization of the amino terminal domain of p57Kip2 inhibits cyclin D1-cdk4 kinase activity. Oncogene. 2000;19:1147–1152. doi: 10.1038/sj.onc.1203403. [DOI] [PubMed] [Google Scholar]
- Riechmann V, van Cruchten I, Sablitzky F. The expression pattern of Id4, a novel dominant negative helix-loop-helix protein, is distinct from Id1, Id2 and Id3. Nucleic Acids Res. 1994;22:749–755. doi: 10.1093/nar/22.5.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothschild G, Zhao X, Iavarone A, Lasorella A. E Proteins and Id2 converge on p57Kip2 to regulate cell cycle in neural cells. Mol Cell Biol. 2006;26:4351–4361. doi: 10.1128/MCB.01743-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scandura JM, Boccuni P, Massague J, Nimer SD. Transforming growth factor beta-induced cell cycle arrest of human hematopoietic cells requires p57KIP2 up-regulation. Proc Natl Acad Sci USA. 2004;101:15231–15236. doi: 10.1073/pnas.0406771101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Marinovich A, Barres BA. Purification and characterization of adult oligodendrocyte precursor cells from the rat optic nerve. J Neurosci. 1998;18:4627–4636. doi: 10.1523/JNEUROSCI.18-12-04627.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun XH, Copeland NG, Jenkins NA, Baltimore D. Id proteins Id1 and Id2 selectively inhibit DNA binding by one class of helix-loop-helix proteins. Mol Cell Biol. 1991;11:5603–5611. doi: 10.1128/mcb.11.11.5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Nakayama K, Nakayama K. Mice lacking a CDK inhibitor, p57Kip2, exhibit skeletal abnormalities and growth retardation. J Biochem (Tokyo) 2000;127:73–83. doi: 10.1093/oxfordjournals.jbchem.a022586. [DOI] [PubMed] [Google Scholar]
- Tang DG, Tokumoto YM, Raff MC. Long-term culture of purified postnatal oligodendrocyte precursor cells. Evidence for an intrinsic maturation program that plays out over months. J Cell Biol. 2000;148:971–984. doi: 10.1083/jcb.148.5.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang XM, Beesley JS, Grinspan JB, Seth P, Kamholz J, Cambi F. Cell cycle arrest induced by ectopic expression of p27 is not sufficient to promote oligodendrocyte differentiation. J Cell Biochem. 1999;76:270–279. doi: 10.1002/(sici)1097-4644(20000201)76:2<270::aid-jcb10>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Temple S, Raff MC. Clonal analysis of oligodendrocyte development in culture: evidence for a developmental clock that counts cell divisions. Cell. 1986;44:773–779. doi: 10.1016/0092-8674(86)90843-3. [DOI] [PubMed] [Google Scholar]
- Tokumoto YM, Tang DG, Raff MC. Two molecularly distinct intracellular pathways to oligodendrocyte differentiation: role of a p53 family protein. EMBO J. 2001;20:5261–5268. doi: 10.1093/emboj/20.18.5261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokumoto YM, Apperly JA, Gao FB, Raff MC. Posttranscriptional regulation of p18 and p27 Cdk inhibitor proteins and the timing of oligodendrocyte differentiation. Dev Biol. 2002;245:224–234. doi: 10.1006/dbio.2002.0626. [DOI] [PubMed] [Google Scholar]
- Vaccarello G, Figliola R, Cramerotti S, Novelli F, Maione R. p57Kip2 is induced by MyoD through a p73-dependent pathway. J Mol Biol. 2006;356:578–588. doi: 10.1016/j.jmb.2005.12.024. [DOI] [PubMed] [Google Scholar]
- Walters SN, Morell P. Effects of altered thyroid states on myelinogenesis. J Neurochem. 1981;36:1792–1801. doi: 10.1111/j.1471-4159.1981.tb00433.x. [DOI] [PubMed] [Google Scholar]
- Wei Q, Miskimins WK, Miskimins R. The Sp1 family of transcription factors is involved in p27(Kip1)-mediated activation of myelin basic protein gene expression. Mol Cell Biol. 2003;23:4035–4045. doi: 10.1128/MCB.23.12.4035-4045.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Q, Miskimins WK, Miskimins R. Sox10 acts as a tissue-specific transcription factor enhancing activation of the myelin basic protein gene promoter by p27Kip1 and Sp1. J Neurosci Res. 2004;78:796–802. doi: 10.1002/jnr.20342. [DOI] [PubMed] [Google Scholar]
- Zezula J, Casaccia-Bonnefil P, Ezhevsky SA, Osterhout DJ, Levine JM, Dowdy SF, Chao MV, Koff A. p21cip1 is required for the differentiation of oligodendrocytes independently of cell cycle withdrawal. EMBO Rep. 2001;2:27–34. doi: 10.1093/embo-reports/kve008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Wong C, DePinho RA, Harper JW, Elledge SJ. Cooperation between the Cdk inhibitors p27(KIP1) and p57(KIP2) in the control of tissue growth and development. Genes Dev. 1998;12:3162–3167. doi: 10.1101/gad.12.20.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Wong C, Liu D, Finegold M, Harper JW, Elledge SJ. p21(CIP1) and p57(KIP2) control muscle differentiation at the myogenin step. Genes Dev. 1999;13:213–224. doi: 10.1101/gad.13.2.213. [DOI] [PMC free article] [PubMed] [Google Scholar]