

Abstract
Coordinated proteolysis of synaptic proteins is required for synaptic plasticity, but a mechanism for recruiting the ubiquitin-proteasome system (UPS) into dendritic spines is not known. NAC1 is a cocaine-regulated transcriptional protein that was found to complex with proteins in the UPS, including cullins and Mov34. NAC1 and the proteasome were cotranslocated from the nucleus into dendritic spines in cortical neurons in response to proteasome inhibition or disinhibiting synaptic activity with bicuculline. Bicuculline also produced a progressive accumulation of the proteasome and NAC1 in the postsynaptic density. Recruitment of the proteasome into dendrites and postsynaptic density by bicuculline was prevented in neurons from mice harboring an NAC1 gene deletion or in neurons transfected with mutated NAC1 lacking the proteasome binding domain. These experiments show that NAC1 modulates the translocation of the UPS from the nucleus into dendritic spines, thereby suggesting a potential missing link in the recruitment of necessary proteolysis machinery for synaptic remodeling.
Keywords: NAC1, proteasome, cullin, translocate, dendritic spine, POZ/BTB
Introduction
Precisely regulated proteolysis of synaptic proteins is now recognized as a critical component of synaptic plasticity (DiAntonio and Hicke, 2004; Bingol and Schuman, 2005; Yi and Ehlers, 2005). Most synaptic proteins are degraded by the ubiquitin-proteasome system (UPS), whereby covalent attachment of polyubiquitin to lysine residues targets proteins for degradation by the proteasome (Glickman and Ciechanover, 2002). Although the importance of the UPS in remodeling the postsynaptic density (PSD) has been clearly demonstrated using proteasome inhibitors (Colledge et al., 2003; Ehlers, 2003; Karpova et al., 2006), and increased synaptic activity was recently shown to induce proteasome accumulation in dendritic spines (Bingol and Schuman, 2006), the mechanisms underlying the recruitment of the proteasome and the enzymes coordinating the polyubiquitin reaction into dendritic spines are unknown (Glickman and Raveh, 2005).
Contrasting a relatively poor understanding of how the UPS is translocated to cellular compartments of high protein turnover, significant advances have been made toward understanding how specific proteins are targeted to the UPS for degradation. One such targeting mechanism involves the binding of cullin-based ubiquitin ligases to proteins containing a Pox virus and Zinc finger/Bric-a-brac/Tramtrack/Broad complex (POZ/BTB) N-terminal binding motif (Pintard et al., 2004; Allen et al., 2005; Kwon et al., 2006; Schaefer and Rongo, 2006). The POZ/BTB proteins act as substrate acceptors by containing additional protein–protein interaction motifs that bind with and facilitate ubiquitination of specific protein substrates (Pintard et al., 2004). Most proteins in the POZ/BTB family are transcription factors containing a zinc finger DNA-binding domain, or are actin-binding proteins possessing a Kelch motif. A previously described cocaine-regulated protein, NAC1, is a member of the POZ/BTB family of proteins, but the lack of zinc finger and Kelch domains indicates that NAC1 could have unique properties (Cha et al., 1997). Supporting this possibility, NAC1 translocates from the nucleus to the cytoplasm of cultured cells in an activity-dependent manner (Korutla et al., 2005).
NAC1 was originally described as an immediate early gene product that is upregulated by psychostimulant drug administration (Cha et al., 1997), and using genetic strategies to regulate NAC1 levels in vivo, it was found that NAC1 modulates the development of psychostimulant-induced behavioral plasticity (Kalivas et al., 1999; Mackler et al., 2000; Wang et al., 2003). Psychostimulant-induced plasticity and addiction have been strongly linked to enduring changes in excitatory transmission in the prefrontal cortex and nucleus accumbens (Wolf, 1998; Kalivas et al., 2005), posing synaptic translocation of NAC1 and proteins complexing with NAC1 via its POZ/BTB interaction domain as a mechanism for regulating excitatory synaptic plasticity. Given the aforementioned role of POZ/BTB-containing proteins as ubiquitination substrate acceptors (Pintard et al., 2004), we hypothesized that NAC1 contributes to the recruitment of components of the proteasome complex from the nucleus into dendritic spines. By confirming this hypothesis, the present study identifies a new cellular mechanism for trafficking the UPS to sites of high protein turnover, such as into dendritic spines during synaptic activity.
Materials and Methods
Yeast two-hybrid screening.
Constructs of two splice variants of NAC1 [short NAC1 (sNAC1) and long NAC1 (lNAC1)] were described previously (Korutla et al., 2002). sNAC1, lNAC1, or POZ/BTB were subcloned in pGBKT7 vector (Clontech, Mountain View, CA) and fused to galactosidase 4 (Gal4)-binding domain (BD) as bait. After the transformation into AH109 yeast and self-activation test, mouse adult cDNA library fused to Gal4-activation domain (AD) was transformed into the AH109 together with bait plasmids. The mating mixtures were plated on synthetic medium lacking tryptophan, leucine, adenine, and histidine, but containing 5 mm 3-aminotriazole (Sigma, St Louis, MO). One week after transformation, 3 × 106 colonies were screened. The positive clone plasmids were rescued from yeast cells and a cotransformation test was performed to verify the interactions between the baits and their preys. The positive prey plasmids were sequenced to identify the encoding protein.
GST fusion protein pulldowns.
To obtain glutathione S-transferase (GST) fusion proteins, plasmids expressing lNAC1, sNAC1, dNAC1 (NAC1 lacking the POZ/BTB sequence), and POZ/BTB were subcloned into the GST fusion vector pGEX-2T (Pfizer-Pharmacia LKB Biotechnology, Piscataway, NJ). Additionally, lNAC1, sNAC1, Mov34, and cullin 3 (Cul3) were subcloned into the T7 plink vector at appropriate restriction enzyme sites for in vitro translation and 35S labeling. GST proteins were bound to glutathione Sepharose 4B beads (GE Healthcare, Piscataway, NJ) in glutathione buffer [containing (in mm) 20 HEPES, pH 7.5, 200 NaCl, 1 MgCl2, 1 DTT, 0.2 EDTA, 0.5% NP-40, and 0.1 PMSF] on ice for 1 h. After washing, 35S-labeled Mov34, Cul3, and lNAC1 were incubated with glutathione Sepharose beads bound with a GST fusion protein at room temperature (RT) for 30 min or 1 h. After washing with glutathione buffer several times, beads were boiled in sample buffer (2% SDS, 10% glycerol, 62 mm Tris, pH 6.8) and loaded onto SDS-PAGE gel, followed by autoradiography.
Generation of recombinant adeno-associated virus.
The rat lNAC1 coding sequence was amplified using whole-brain cDNA and the PCR product expressed as a N-terminal and C-terminal fusion protein with the hemagglutinin (HA) tag and the hexahistidine tag, respectively, in a recombinant adeno-associated virus (rAAV) backbone containing the 1.1 kb cytomegalovirus enhancer/chicken β-actin (CBA) promoter, the woodchuck post-transcriptional regulatory element (WPRE), and the bovine growth hormone (bGH) polyA flanked by inverted terminal repeats. The same rAAV-CBA-WPRE-bGH backbone encoding the humanized renilla green fluorescent protein (GFP) was used as a control. AAV pseudotyped vectors (virions containing a 1:1 ratio of AAV1 and AAV2 capsid proteins with AAV2 inverted terminal repeats) were generated as described previously (Klugmann et al., 2005). Briefly, human embryonic kidney 293 cells were transfected with the AAV cis-plasmid, the AAV1 (pH21) and AAV2 (pRV1) packaging plasmids and the adenovirus helper plasmid (pFΔ6) by standard calcium phosphate transfection methods. Forty-eight hours after transfection, cells were harvested and the vector purified using heparin affinity columns as described previously (During et al., 2003). Genomic titers were determined using the Prism 7700 sequence detector system (Applied Biosystems, Foster City, CA) with primers designed to WPRE as described previously (Clark et al., 1999).
Generation of NAC1 KO mice.
All experiments were approved by the Institutional Animal Care and Use Committee of the Medical University of South Carolina and conducted in accordance with the National Institutes of Health (NIH) Principles of Laboratory Animal Care (1985). The mouse NAC1 gene was cloned from a 129SvJ BAC (bacterial artificial chromosome) library (Research Genetics, Huntsville, AL) as described previously (Mackler et al., 2003). The targeting vector containing the Escherichia coli lacZ gene encoding β-galactosidase and the neomycin-resistance gene driven by the mouse phosphoglycerate kinase promoter (pHM4) (Kaestner et al., 1994) was inserted into exon 1. Exon 1 was chosen because it encodes for the POZ/BTB functional domain of the NAC1 protein. Thus, deletion of exon 1 of the NAC1 gene produces a functional null allele. The construct was electroporated into mouse 129SvTac embryonic stem (ES) cells (a gift from Dr. P. Labosky, University of Pennsylvania, Philadelphia, PA) and neomycin resistant colonies were selected for Southern blot analysis. One hundred and ninety-eight resistant clones were analyzed by PCR using a common 5′ primer (5′-GGCCGCTAGTAGCTCTTACTT-3′) and specific 3′ primers derived from exon 1 (5′-GTCGCAACTTGGAGAGCTAAC-3′) for the wild-type (WT) allele, resulting in an amplified fragment of 243 bp and the lacZ gene (5′-CAAAGCGCCATTCGCCATTCA-3′) for the mutant allele resulting in an amplified fragment of 340 bp. Targeted ES cells were injected into C57BL/6 blastocysts to obtain eight germ line chimeras. Heterozygote mice were mated to give rise to mice of all three genotypes.
GFP-NAC1 fusion protein construction.
cDNAs encoding full-length lNAC1, sNAC1, or dNAC1 were amplified by PCR and subcloned into PCR 2.1 TOPO TA cloning vector to get TOPO-lNAC1, -sNAC1, or -dNAC1. These TOPO constructs were digested with EcoRI and BamHI to get the two full-length isoforms or truncated form and subcloned into the multiple cloning sites of pEGFP-N1 (Clontech) cut with the same restriction enzymes.
Antibodies.
Antibodies used in these experiments include mouse monoclonal antibodies against the α7 20S proteasome subunit (Biomol, Plymouth Meeting, PA), HA (Cell Signaling, Danvers, MA), B23 (Invitrogen, Carlsbad, CA), Cul3 (BD Biosciences, San Diego, CA), α-tubulin (Santa Cruz Biotechnology, Santa Cruz, CA), PSD-95 (Sigma), and GAD67 (Millipore, Temecula, CA), rabbit polyclonal antibodies against HA (Bethyl, Montgomery, TX), 20S proteasome (subunit α5; BostonBiochem, Cambridge, MA; or subunits α/β; Biomol) and Cul3 (Lab Vision, Fremont, CA), and goat polyclonal antibodies against HA (Bethyl).
Protein subfraction, immunoblotting, and immunoprecipitation.
Cortical neurons were scratched from culture dishes and collected in ice-cold PBS buffer. PSD fractions from cortical culture were purified essentially as described previously (Toda et al., 2003). Briefly, the synaptosomal membrane fraction was resuspended in a buffer A (1% Triton X-100, 20 mm HEPES-NaOH, and 100 mm NaCl, pH 7.2) and incubated at 4°C for 30 min with gentle rotation. After a 20 min centrifugation (10,000 × g, 4°C), the pellet was resuspended in buffer B (15 mm HEPES-NaOH, 0.15 mm NaCl, 1% SDS, 10 mm EDTA, and 1 mm DTT, pH 7.5). After 1 h of incubation at 4°C and an additional 20 min centrifugation (10,000 × g, 4°C), the supernatant was recovered as the PSD fraction. For immunoblotting, 5–7.5 μg of protein were loaded in SDS-PAGE and transferred to a polyvinylidene fluoride membrane. For immunoprecipitation experiments, AAV-infected neurons were resuspended in NP-40 lysis buffer (150 mm NaCl, 50 mm Tris, 1% NP-40, pH 8.0), homogenized, and centrifuged to remove cellular debris. After preclearing with protein-A Sepharose (GE Healthcare) and incubation with anti-20S, anti-Cul3, or anti-α-tubulin for 90 min at 4°C, complexes were precipitated with protein-A Sepharose and detected by immunoblotting with anti-HA. Signals were detected by ECL Plus (GE Healthcare). Quantification of band density was performed by NIH ImageJ and normalized to α-tubulin.
Neuron cultures, transfection, infection, and drug treatment.
Primary cortical neurons were obtained from day 18 rat or mouse embryos, and dissociated using trypsin and trituration through a Pasteur pipette. Then cells were plated at a density of 2 × 105 cells on 35 mm poly-l-lysine-coated plastic dishes (Nunc, Naperville, IL) in Neurobasal media (Invitrogen) supplemented with 2% B-27 (Invitrogen), l-glutamine (0.5 mm). Cells were grown in a humidified incubator with 5% CO2 at 37°C. At 3 d in vitro (DIV), 5 μm β-cytosine arabinoside (Sigma) was added to the medium. At 7 DIV, and subsequently once a week, one-half of the medium was changed with fresh medium. Approximately 10% of the infected neurons could be immunostained by anti-GAD67 (supplemental Fig. 3, available at www.jneurosci.org as supplemental material), indicating that the cortical culture contained GABAergic neurons. At 14 DIV, cortical cultures were transfected with the mixture of 1.0 μg of plasmid DNA and 2.5 μl of Lipofectamine 2000 (Invitrogen), or were infected by 1 μl of AAV-NAC1 or AAV-hrGFP stock (titer, 5 × 1011 viral genomes/ml). After 48 h of plasmid transfection or 7 d of infection by AAV, these dishes were used for treatment, immunoblotting, or immunocytochemistry. For analysis of neuronal survival in parallel to immunocytochemistry, the fluorescent nuclear dye 4,6-diamidino-2-pheylindole (DAPI) was used. Nuclear chromatin morphology was analyzed with a 63× objective. Nonviable neurons were recognized by nuclear condensation and/or fragmented chromatin. No effect on neuron viability was observed by NAC1 infection or in NAC1 knock-out (KO) neurons. Filamentous (F)-actin staining with the fluorescent dye phallotoxin showed that the morphology of dendrites and cell bodies were also not affected by these treatments.
Drugs were dissolved in DMSO or water at 1000× of treatment concentration. Neurons were treated with 0.1% DMSO, 20S proteasome inhibitor MG132 (10 μm for 6 h; Biomol), bicuculline (40 μm for 12 h; Sigma), protein kinase C (PKC) activator phorbol 12-myristate 13-acetate (PMA, 10 μm for 1 h; Promega, Madison, WI), and/or PKC inhibitor bisindolylmaleimide VIII (BIM, 1 μm for 1, 6, or 12 h; Sigma). Then cells were washed with PBS, and collected in PBS for immunoblotting or fixed for immunocytochemistry.
Immunocytochemistry and confocal imaging.
Neurons were fixed in 4% paraformaldehyde/4% sucrose/PBS for 10 min at RT, followed by permeabilization in 0.1% Triton X-100/PBS for 10 min. Cultures were then incubated in PBS with 3% bovine serum albumin (BSA) for 30 min at RT to block nonspecific staining, followed by incubation with primary antibodies in 3% BSA/PBS for 12 h at 4C. Then cultures were rinsed three times with PBS and incubated with Alexa Fluor 488-, 594- and/or 647-conjugated secondary antibodies (Invitrogen) in 3% BSA/PBS. To counterstain the nucleus, cultures were incubated with Sytox green or DAPI nucleic acid stain (Invitrogen) in PBS for 5 min. F-actin was labeled by fluorescent phallotoxin (Invitrogen). Confocal images of neurons were acquired with a Zeiss (Thornwood, NY) LSM 510 confocal microscope using a 63× oil-immersion objective. Fluorochromes were excited using the argon laser at 488 nm for Alexa Fluor 488 and Sytox green, the helium/neon laser at 543 or 633 nm for Alexa Fluor 594 or 647, and the titanium–sapphire laser at 720 nm for DAPI. Fluorescent intensity was measured along a line crossing the nucleus and cytoplasm with NIH ImageJ. Quantification of the subcellular distribution was determined by the obtaining the ratio of fluorescence intensity (nucleus vs cytoplasm) from six to eight neurons in each treatment group within a randomly placed 300 × 300 μm grid. Alternatively, neurons were rated by an individual unaware of the treatment for the presence or absence of fluorescence in the nucleus versus cytoplasm (see Fig. 3d). All quantifications were made between sister cultures by individuals unaware of the treatment groups.
Figure 3.

Translocation of NAC1 and 20S from the nucleus to the cytoplasm and dendritic shaft. a, NAC1 and the 20S were translocated into the cytoplasm by MG132-induced inhibition of the proteasome (10 μm, 6 h), PMA-induced activation of PKC (10 μm, 1 h), or bicuculline disinhibition (40 μm, 2 h). DMSO was the control incubation (0.1%, 12 h). Neurons were either infected with AAV-NAC1 or not infected (No AAV). Arrows show cells with complete translocation of 20S and NAC1 from the nucleus to cytoplasm by MG132. Scale bars, 20 μm. b, Individual intensity plot of 20S cellular fluorescence corresponding to cells labeled 1 and 2 in a. c, Quantification of relative fluorescence of 20S in the nucleus versus cytoplasm after all treatments shown in a using relative intensity plots. Data were collected by an individual unaware of the treatment group and are shown as the mean proportion of fluorescence for n = 6–8 per treatment group. d, Quantification of the relative distribution of NAC1 labeling in the nucleus, nucleus and cytoplasm, and cytoplasm only (in the case of MG132, some cells had no measurable labeling in the nucleus). Quantification was made by an individual unaware of the treatment groups classify each cell in a culture dish (n = 6 dishes per treatment). For c and d, the data are presented as the mean proportion of fluorescence for n = 6–8 in each treatment group. *p < 0.05, compared with 0.1% DMSO using a Kruskal–Wallis test; +p < 0.05, comparing between treatments with or without AAV infection (c) or with or without BIM (d)
Statistics.
The ratio of subcellular distribution was compared between different treatments using a Kruskal–Wallis test. Protein levels were compared across different times using a one-way ANOVA. Least significant difference tests were applied for multiple comparisons and p < 0.05 was considered statistically significant. Statistical analyses were conducted using SPSS 11 (SPSS, Chicago, IL).
Results
NAC1 associates with cullins and Mov34, a proteasome subunit
This study was initially begun to determine whether, akin to other POZ/BTB proteins, NAC1 bound to cullin proteins, and therefore functioned as a substrate acceptor for ubiquitination (Pintard et al., 2004). A yeast 2-hybrid (Y2H) assay was used to screen a GAL4-AD/cDNA library constructed from adult mouse brain using NAC1-GAL4-BD fusion protein as bait. Of the 3 × 106 library clones screened, positive clones were found to encode Cul3, a cullin protein that is a critical subunit in cullin-based E3 ubiquitin ligases (Glickman and Ciechanover, 2002), and Mov34, a 19S ATPase regulatory subunit in the 26S proteasome (Mason et al., 1998; Ambroggio et al., 2004; Burger-Kentischer et al., 2005). To verify the interaction between NAC1 with Cul3 and Mov34, isolated prey plasmids were retransformed into AH109 and only Cul3 or Mov34/Gal4BD × lNac1/Gal4AD cotransformed yeast cells proliferated on the selecting medium.
The interactions identified in the Y2H assay were verified by GST pulldown assays in vitro. To confirm that the POZ/BTB domain of NAC1 interacted with cullins and Mov34, either the long (lNAC1) or short (sNAC1) isoforms of NAC1 (Korutla et al., 2002), an NAC1 mutant lacking the POZ/BTB domain (dNAC1), or POZ/BTB alone (Fig. 1a) were fused to GST, and Cul3, Cul4, Mov34, and NAC1 were labeled with 35S-methionine by in vitro translation. GST-lNAC1, GST-sNAC1, and GST-POZ/BTB specifically pulled down 35S-labeled Cul3 (Fig. 1b), Cul4 (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), and Mov34 (Fig. 1c), whereas dNAC1 and GST alone were ineffective. Although the cullins contain a complementary POZ/BTB binding domain, Mov34 does not, indicating that interaction with the POZ/BTB domain of NAC1 results from the tertiary conformation of Mov34.
Figure 1.

NAC1 forms a complex with cullin-based ubiquitin ligase and Mov34 in the 26S proteasome. a, Diagram of NAC1 constructs used in GST-pulldown experiment and transfection in cultured cells. b, GST-pulldown showing complex formation between Cul3 and NAC1, and dependence on the POZ/BTB binding domain. c, GST-pulldown showing POZ/BTB-dependent complex between Mov34 and NAC1. d, Immunoprecipitation with 20S proteasome subunit or Cul3 reveals interaction with HA-tagged NAC1 immunoblot (IB) in AAV-NAC1-infected primary cortical cultures. No coimmunoprecipitation of HA-tagged NAC1 was found using α-tubulin to immunoprecipitate. However, HA-tagged NAC1 could be seen in whole-cell lysates. e, Coimmunoprecipitation of endogenous NAC1 (IB) using striatal homogenates from WT and NAC1-KO mice. Immunoprecipitation by 19S antibody revealed an interaction with endogenous NAC1, whereas immunoprecipitation with anti-α-tubulin did not. X, Lane not loaded with protein. Arrows indicate presumed NAC1 immunoreactivity. IP, Immunoprecipitation.
Immunoprecipitation in primary cortical neurons infected with HA-tagged NAC1 packaged in an adeno-associated viral construct (AAV-NAC1) also revealed an interaction between NAC1 and Cul3 (Fig. 1d). Because an antibody was not available for Mov34 and Mov34 is a regulatory subunit of the 26S proteasome complex (Tsurumi et al., 1995; Burger-Kentischer et al., 2005), we used antibodies directed against the 19S or 20S proteasome (core particles in the proteolytic domain of the 26S proteasome) (Hegde, 2004) to immunoprecipitate NAC1 in AAV-NAC1-infected cortical neurons (Fig. 1d) or in tissue dissected from adult rat striatum (Fig. 1e). In infected cortical cells, NAC1 was precipitated by either anti-20S or anti-Cul3, but not by anti-α-tubulin (Fig. 1d). In adult striatal tissue, endogenous NAC1 was precipitated by anti-19S, but not anti-α-tubulin (Fig. 1e). Note that the antibody directed against endogenous NAC1 recognized multiple bands in whole-cell lysates from mice harboring an NAC1 gene deletion (KO) as well as from WT mice. However, after immunoprecipitation with 19S antibody, the NAC1 antibody recognized only a single band in WT tissue that was absent in KO tissue.
Additional evaluation of the interaction between NAC1 and the UPS was conducted using confocal microscopy to identify colocalization of NAC1 with Cul3 or 20S. Primary cells from rat prefrontal cortex were infected with AAV-NAC1 (>85% efficiency), and HA-tagged NAC1 was localized predominantly to the nucleus (Fig. 2a). Counting infected neurons in four dishes revealed that only 17 of 158 HA-labeled neurons contained NAC1 in both the nucleus and cytoplasm (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). NAC1 was generally distributed unevenly throughout the nucleus in speckles, and sometimes as a large spherical aggregate that did not colocalize with the nucleolus (see the lack of coregistration with B23 nucleolus labeling in Fig. 2a). Cells infected with a control AAV expressing green fluorescent protein (AAV-GFP) were immunostained for 20S or Cul3 (Fig. 2b). Cul3 showed substantial labeling in the nucleus and soma. Consistent with previous reports examining 20S distribution in neurons (Patrick et al., 2003; Wojcik and DeMartino, 2003), 20S staining was higher in the nucleus than in the somatic cytoplasm, and under the staining conditions used, relatively little 20S was identified in the dendrites. When AAV-NAC1-infected cells were double labeled for 20S or Cul3, both proteins were colocalized with NAC1 in the nucleosome (Fig. 2 c–e). Although some colocalization was apparent in the nuclear speckles, there was extremely high colocalization in the cytoplasm, especially with the 20S proteasome, which often appeared almost entirely colocalized with NAC1 in the cytoplasmic compartment (Fig. 2d).
Figure 2.
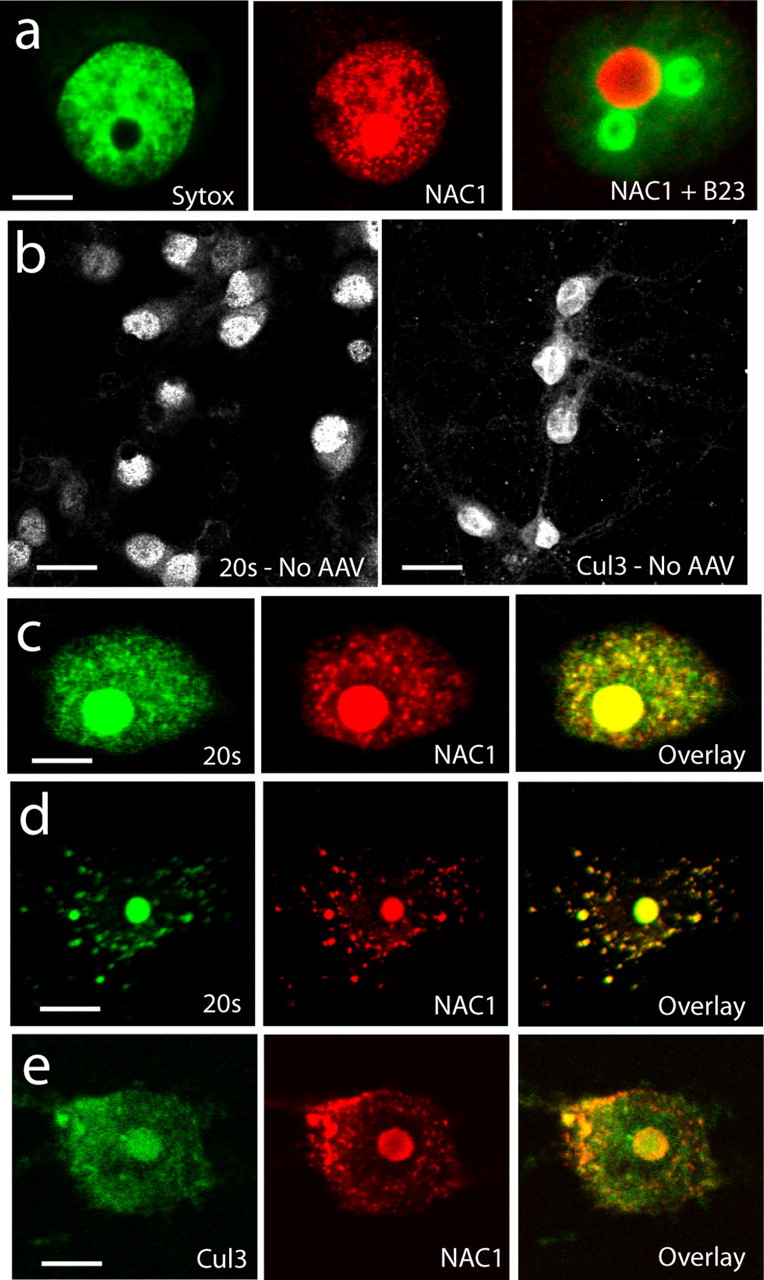
Colocalization of NAC1 with Cul3 and 20S proteasome in primary cortical neurons. a, Neurons infected with AAV-NAC1 show NAC1 labeling in the nucleus (compare with nuclear labeling with Sytox green) and sometimes contained a large nucleosome that is not the nucleolus (compare with B23 labeling of nucleolus). b, Labeling for 20S and Cul3 in AAV-GFP-infected neurons shows 20S and Cul3 to be localized mainly to the nucleus and somatic cytoplasm. c, Colocalization (yellow color in overlay) of NAC1 and 20S in neuron with primarily nuclear NAC1. d, Colocalization of NAC1 and 20S in neuron showing cytoplasmic NAC1. e, Colocalization of Cul3 with NAC1. Scale bars: a, c–e, 5 μm; b, 20 μm.
Cytoplasmic translocation of NAC1 associated 20S and Cul3
We found previously that NAC1 translocates from the nucleus to the cytoplasm in PC-12 cells by depolarizing stimuli or activation of PKC (Korutla et al., 2005). Given the protein interactions and colocalization between NAC1 and 19S, 20S, or Cul3 (Figs. 1, 2), we examined whether stimulating the translocation of NAC1 in primary cortical cells caused 20S and Cul3 to cotranslocate from the nucleus to the cytoplasm. When cells were not infected or infected with control AAV-GFP and treated with 0.1% DMSO control vehicle for 12 h, the 20S subunit was predominantly in the nucleus, and infection with AAV-NAC1 concentrated 20S into the NAC1-containing nuclear speckles and nucleosome (Fig. 3a). Stimulation of the cortical neurons with the PKC activator PMA (10 μm/1 h) produced an increase in the relative amount of 20S in the cytoplasm of uninfected or AAV-GFP-infected cells (Fig. 3a). Surprisingly, the relative increase in cytoplasmic fluorescence by PMA was not enhanced by AAV-NAC1 infection; although, in NAC1-infected neurons, the 20S labeling became more punctate and generally colocalized with NAC1 (Fig. 3a). To quantify the effect by AAV-NAC1 infection on the relative amounts of nuclear and non-nuclear 20S, cells were double labeled with the nuclear stain DAPI to demarcate the nucleus, and relative fluorescent intensity was quantified (Fig. 3b,c). Also, cells were counted and rated as labeled only in the nucleus or labeled in both the nucleus and cytoplasm (Fig. 3d). Quantification revealed no difference in the ability of PMA to translocate 20S or NAC1 from the nucleus to the cytoplasm after AAV-NAC1 infection.
It was reasoned that inhibiting the proteasome would be an impetus for compensatory translocation of nuclear proteasome into the cytoplasm (Hegde, 2004). After inhibiting the proteasome with MG132 (10 μm/6 h), 20S (Fig. 3a) or 19S (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) were translocated into the cytoplasm in both AAV-NAC1-infected and control neurons. However, in contrast with PMA, proteasome translocation was augmented by AAV-NAC1 infection. Quantification revealed that, although control cells showed significant increases in cytoplasmic versus nuclear fluorescence in response to MG132, the increase was significantly greater after AAV-NAC1 infection (Fig. 3c). Remarkably, in a portion of AAV-NAC1-infected neurons, MG132 induced the translocation of virtually all detectable 20S and NAC1 out of the nucleus (Fig. 3a, arrows). Similar translocation of 19S occurred when cells were treated with another proteasome inhibitor, lactacystin (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Labeling cortical cultures for GAD, a marker of GABAergic neurons, revealed that ∼10% of the cells were GABAergic (supplemental Fig. 3, available at www.jneurosci.org as supplemental material), and increasing overall cell excitability by adding the GABAA receptor antagonist bicuculline to hippocampal cultures has been shown to induce changes in PSD protein content (Ehlers, 2003). The addition of bicuculline (40 μm/12 h) did not affect the relative amount of 20S in the nucleus versus cytoplasm of control cells, but significant translocation was observed in AAV-NAC-infected neurons (Fig. 3). As in Figure 2, in AAV-NAC1-infected neurons, the 20S always showed substantial colocalization with NAC1, regardless of the treatment. Similar bicuculline-induced cotranslocation of NAC1 and 19S was observed in AAV-NAC1-infected, but not AAV-GFP-infected cortical cells (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Cul3 demonstrated both cytoplasmic and nuclear labeling (Fig. 4). Regardless of whether or not the neurons were infected by AAV-NAC1, ∼80% of the labeling intensity was in the nucleus relative to the cytoplasm (AAV-NAC1, nucleus, 79.4 ± 4.6%, cytoplasm, 20.6 ± 1.7%, n = 6; AAV-GFP, nucleus, 78.2 ± 2.3%, cytoplasm, 21.8 ± 0.9%, n = 6). Although the overall labeling distribution was not quantitatively altered by bicuculline, the amount of puncate Cul3 labeling in the dendritic shaft appeared to be increased and was in partial coregistration with NAC1 (Fig. 4b,c). Interestingly, the greatest amount of colocalization in dendrites was organized in punctate structures along the dendritic shaft (Fig. 4c). This pattern of colocalization raised the possibility that NAC1 was translocating Cul3 and 20S into dendritic spines (see below).
Figure 4.
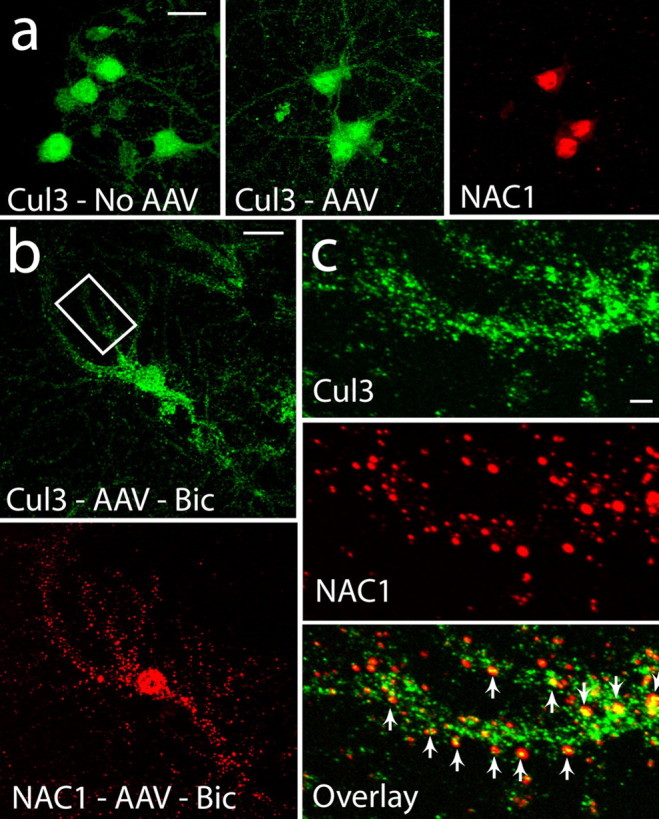
Translocation of Cul3 and NAC1 by bicuculline into dendrites. a, Distribution of Cul3 in uninfected cells is nuclear, cytoplasmic, and dendritic and after infection, NAC1 is highly localized to the nucleus. b, After bicuculline (40 μm, 12 h), significant amounts of NAC1 were translocated into the dendritic tree. c, NAC1 is colocalized with Cul3 in puncta in the dendritic shaft. Arrows indicate colocalized puncta. Scale bars: a, b, 20 μm; c, 5 μm.
NAC1 gene deletion prevents bicuculline- and MG132-induced translocation of the 20S proteasome
The fact that PKC activation produced equivalent increases in cytoplasmic 20S in control and AAV-NAC1-infected neurons argues that PKC-dependent translocation did not require NAC1. In contrast, because bicuculline and MG132 translocated 20S only in infected cells or to a greater extent in infected cells, respectively (Fig. 3c), a requirement for NAC1 was indicated. To evaluate these possibilities, primary cortical neuron cultures were made from embryonic mice lacking the NAC1 gene (NAC1 KO) (Mackler et al., 2007). The primary nuclear distribution of 20S in NAC1 KO neurons was identical to that in WT neurons (Fig. 5b). Consistent with PKC-mediated translocation not requiring NAC1, substantial 20S translocation to the cytoplasm was produced by incubation with PMA in uninfected KO neurons (Fig. 5a,c). Conversely, translocation of 20S was not induced by either MG132 or bicuculline in uninfected KO cells (Fig. 5a,d,e). Importantly, the lack of effect by MG132 in uninfected KO cells is in contrast to neurons containing endogenous NAC1 where MG132 produced a significant increase in translocation to the cytoplasm, albeit less so than after AAV-NAC1 infection (compare Figs. 3c, 5a). Interestingly, in uninfected KO neurons, MG132 moved 20S to the nuclear envelope, but not beyond (Fig. 5, compare b, d). The deficit in cytoplasmic translocation induced by bicuculline or MG132 after NAC1 gene deletion was rescued by infecting KO neurons with AAV-NAC1 (Fig. 5a,d,e), confirming that NAC1 is necessary and sufficient for this response.
Figure 5.

Deletion of the NAC1 gene or elimination of POZ/BTB domain prevents NAC1 and 20S translocation by MG132 and bicuculline, but not by PMA. a, Relative fluorescence of 20S in nuclear versus cytoplasmic compartments in each treatment condition. Data are shown as the mean proportion of fluorescence in the nuclear versus cytoplasmic compartments. b, Predominately nuclear localization of 20S in NAC1 KO neurons. c, PMA translocates 20S in both KO and KO cells infected with AAV-NAC1. d, MG132 translocates 20S only in rescued KO cells. e, Bicuculline translocates 20S only in rescued KO cells. The box refers to high-power micrographs shown in Figure 6e. f, Neurons were transfected with either full-length NAC1 linked to GFP or truncated NAC1 linked to GFP that lacked the N-terminal POZ/BTB domain. Neither dNAC1 nor 20S were translocated by bicuculline. Scale bars: b–e, 20 μm; f, 5 μm. *p < 0.05, compared with 1% DMSO using a Kruskal–Wallis test; +p < 0.05, comparing between treatments with or without AAV infection.
The data in Figures 3 and 5 indicate that translocation of the 20S proteasome by PKC activation proceeds by an NAC1-independent mechanism, possibly via direct phosphorylation of the proteasome (Glickman and Raveh, 2005), which is distinct from the NAC1-dependent mechanism underlying translocation of 20S by bicuculline or MG132. This distinction was further investigated by showing that although incubation of cortical neurons with the PKC inhibitor BIM abolished PMA-induced cotranslocation of NAC1 and 20S from the nucleus to the cytoplasm, it did not affect cotranslocation induced by bicuculline or MG132 (supplemental Fig. 4, available at www.jneurosci.org as supplemental material).
POZ/BTB domain is necessary for translocation on NAC1 and the 20S proteasome
Based on the GST pulldown experiments (Fig. 1b,c), the POZ/BTB binding domain in NAC1 is necessary for complexing with the cullins and Mov34. To determine whether the POZ/BTB domain was also necessary for cotranslocating NAC1 and 20S, cDNAs encoding full-length NAC1 or NAC1 lacking the POZ/BTB binding domain (dNAC1) (Fig. 1a) were transfected into primary cortical neurons. Neurons transfected with full-length NAC1 showed cotranslocation of NAC1 and 20S in response to bicuculline, whereas both NAC1 and 20S were retained in the nucleus of neurons transfected with dNAC1 after exposure to bicuculline (Fig. 5f). Similar to bicuculline treatment, neurons transfected with dNAC1 did not show translocation of dNAC1 after incubation with MG132 (data not shown).
NAC1 translocates the 20S proteasome into dendritic spines and the PSD
In most instances, the bicuculline-induced translocation of NAC1, 20S, and Cul3, from the nucleus increased cytoplasmic labeling not only in the soma, but also in the dendritic tree (Figs. 3–5). This posed the possibility that NAC1 translocates the UPS into dendritic spines, thereby contributing to the previously reported bicuculline-induced postsynaptic remodeling (Ehlers, 2003). To verify translocation into dendritic spines, AAV-NAC1-transfected cortical neurons were treated with bicuculline (40 μm/12 h) and double labeled for the presynaptic marker protein synaptophysin and NAC1, or were triple labeled for NAC1, 20S, and F-actin (a dendritic spine cytoskeletal protein critical for synaptic remodeling) (Kasai et al., 2003; Carlisle and Kennedy, 2005). Double labeling for NAC1 and synaptophysin in the absence of bicuculline treatment showed the lack of translocation of NAC1 into dendrites and into apposition with synaptophysin at presumed points of synaptic contact (Fig. 6a). In contrast, treatment with bicuculline caused NAC1 to translocate into the dendrites and come into apposition with synaptophysin-labeled presynaptic terminals (Fig. 6a). Triple labeling showed that cotranslocation of NAC1 and 20S by bicuculline resulted in significant colocalization of both proteins in F-actin-labeled clusters (Fig. 6c). A similar marked colocalization of NAC1, 20S, and F-actin was induced by bicuculline in dendrites of NAC1 KO neurons that had been rescued by AAV-NAC1 infection (Fig. 6d,e). To further determine whether bicuculline-induced translocation was moving NAC1 into the PSD, neurons were double labeled for PSD-95 (scaffolding protein in high concentration in the PSD) (Schnell et al., 2000) and NAC1 (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Colocalization between PSD-95 and NAC1 was frequent along the dendritic shaft.
Figure 6.

Bicuculline translocates NAC1 and 20S into putative dendritic spines labeled for F-actin. a, Neurons double labeled for NAC1 (green) and the presynaptic marker protein synaptophysin (Syn; red). The panels compare AAV-NAC1-infected neurons with and without bicuculline (Bic) treatment (40 μm, 12 h). Arrows in the bottom indicate close apposition of NAC1 and synaptophysin staining. b, A neuron triple labeled for F-actin (green), NAC1 (white), and 20S (red). c, High-power micrograph of neuron in b showing colocalization of F-actin, NAC1, and 20S (yellow-green labeling, indicated by arrows). d, Neurons from NAC1-KO culture that were rescued by infection with AAV-NAC1 show colocalization (arrows) of F-actin, NAC1, and 20S. e, Another rescued KO cell showing colocalization (arrows; for whole-cell labeling, see box in low-resolution micrograph in Fig. 5E). Scale bars: a–e, 20 μm for low-resolution panels; 5 μm for high-resolution panels.
Although the confocal evidence implicates cotranslocation of NAC1 and the proteasome into dendritic spines and the PSD, immunoblotting of the PSD subcellular fraction was also conducted for NAC1, 20S, 19S, Cul3, and PSD-95 in cortical neurons after bicuculline. To more clearly associate the cotranslocation of these proteins with previous immunocytochemistry, a time course of bicuculline-induced translocation of NAC1 was compared between immunocytochemistry and immunobloting (Fig. 7 b–d). Immunocytochemistry revealed translocation of NAC1 by 6 h after bicuculline, increasing to an apparent maximum by 24 and 48 h (Fig. 7b). More quantitative immunoblotting revealed a similar pattern except that by 2 h after bicuculline, NAC1 was significantly increased in the PSD subfraction of AAV-NAC1-infected cells (Fig. 7c,d). By 12 h after bicuculline, all four cotranslocated proteins were increased, and 20S, Cul3, and AAV-NAC1 remained elevated at 48 h. In contrast, PSD-95 was not significantly elevated in AAV-NAC1-infected cells at any bicuculline incubation time. Interestingly, when control AAV-GFP-infected neurons were assayed, even though confocal immunocytochemistry did not reveal significant translocation of 19S or 20S by bicuculline, the level of both subunits was elevated at 12 h, presumably because of the presence of endogenous NAC1. This possibility was confirmed by showing that, in cultured neurons from NAC1-KO mice, bicuculline did not alter PSD content of 19S or 20S. In GFP transfected cells, PSD-95 was significantly elevated at 12 and 48 h after bicuculline, as was reported previously in cultured hippocampal neurons (Ehlers, 2003). Importantly, bicuculline treatment did not alter whole-cell content of 19S, 20S, or Cul3 at 12 h after bicuculline exposure demonstrating, that the increase in the PSD subfraction was not the result of an overall cellular increase in protein content (Table 1).
Figure 7.

Bicuculline translocates NAC1, 20S, and Cul3 into PSD. a, Time course of bicuculline-induced translocation of NAC1 from the nucleus to the cytoplasm and dendritic shaft. b, Representative immunoblots of the time course of bicuculline-induced translocation of NAC1, 20S, and Cul3 into the PSD subfraction obtained from cultured cortical neurons infected with AAV-NAC1 or AAV-NAC1. c, Summary of immunobloting for proteins in the PSD subfraction. PSD levels of protein were measured in cultures prepared from cortical cells transfected with AAV-NAC1 or AAV-GFP, or from untransfected NAC1-KO cells. n = 6 for each group, and the data are shown as the mean ± SEM percentage change in normalized band density from time 0. Scale bar: a, low magnification, 20 μm; high magnification, 5 μm. *p < 0.05, compared with time 0 h using one-way ANOVA followed by a least significant difference test.
Table 1.
Bicuculline (40 μm, 12 h) produced no change in NAC1, 20S, 19S, Cul3, or PSD95 in whole-cell lysates of cortical neurons infected with AAV-NAC1 or AAV-GFP
| Treatment | PSD Proteins |
||||
|---|---|---|---|---|---|
| 19S | 20S | Cul3 | PSD-95 | HA-NAC1 | |
| AAV-GFP | |||||
| Control | 100 ± 5.6 | 100 ± 3.7 | 100 ± 13.3 | 100 ± 7.7 | NA |
| Bicuculline | 107.7 ± 4.7 | 106.7 ± 6.0 | 110.1 ± 11.2 | 98.0 ± 15.4 | NA |
| AAV-NAC1 | |||||
| Control | 100 ± 16.4 | 100 ± 8.2 | 100 ± 8.1 | 100 ± 4.0 | 100 ± 13.7 |
| Bicuculline | 108.1 ± 13.2 | 86.1 ± 5.0 | 100.7 ± 5.1 | 81.2 ± 13.6 | 110.2 ± 11.1 |
Cells were incubated in bicuculline for 12 h before harvesting. n = 5 for all groups, data are shown as the mean ± SEM percentage change from GFP- or NAC1-infected cells incubated with control buffer. NA, Not applicable because cells were not infected with HA-tagged NAC1.
Discussion
Targeted ubiquitination and proteolytic degradation are critical mechanisms controlling protein stability and localization in synapses (Patrick, 2006). Thus, increasing excitatory neurotransmission involves increased activity of the UPS in synapses and dendritic spines (Yi and Ehlers, 2005; Bingol and Schuman, 2006). However, the mechanisms for recruiting the UPS into dendritic spines are unclear. The present study demonstrates that NAC1 is required for translocating the UPS from a region of high concentration in the nucleus to dendritic spines in response to bicuculline-induced decrease in synaptic inhibition, or into the cytoplasm in response to proteasome dysfunction. The translocation involved NAC1 complexing with cullin proteins in cullin-based ubiquitin ligases or Mov34, a subunit of the 26S proteasome (Mason et al., 1998). Both the protein–protein associations and the translocation of NAC1 depend on an intact POZ/BTB domain. Translocation was stimulated by incubating primary cortical neuron cultures with the GABAA receptor antagonist bicuculline, which is consistent with many studies showing that bicuculline stimulates synaptic protein ubiquitination and proteasomal degradation, as well as proteasome-dependent membrane trafficking of glutamate receptors (Colledge et al., 2003; Ehlers, 2003; Patrick et al., 2003).
The abundance and activity of the UPS are responsive to cell needs for protein degradation, the recruitment to synapses in response to bicuculline being a prime example (Glickman and Raveh, 2005; Yi and Ehlers, 2005; Patrick, 2006). Under unstimulated conditions, the majority of proteasome resides in the nucleus and rough endoplasmic reticulum (Brooks et al., 2000; Wojcik and DeMartino, 2003). One immunocytochemical study estimated that only 60% of synapses have detectable proteasome (Patrick et al., 2003). Although the nuclear UPS serves an important role in gene transcription (Muratani and Tansey, 2003), dramatic proteasome translocation between the nucleus and cytoplasm is apparent during the mitotic cycle (Kawahara and Yokosawa, 1992; Lafarga et al., 2002). Examples of mitotic translocation are consistent with nuclear UPS being a reservoir of UPS available for recruitment to the cytoplasm or synapses in response to proteasome inhibition or bicuculline, respectively (Yi and Ehlers, 2005; Patrick, 2006). To explain the recruitment of the UPS to cellular compartments with high proteolytic demand, research has focused on chaperone proteins that affect the assembly of the proteasome and the efficiency of protein ubiquitination or that target specific substrate proteins to the UPS (Hegde, 2004; Glickman and Raveh, 2005). In contrast, chaperone proteins regulating the movement of the UPS between compartments have been hypothesized but not identified (Glickman and Raveh, 2005). Recently, the existence of such a trafficking mechanism was indicated by research showing that depolarization of hippocampal cells caused proteasome sequestration in dendritic spines, but the mechanism for transporting the proteasome was not identified (Bingol and Schuman, 2006). The current findings demonstrate that NAC1 serves a role in translocating the UPS from the nucleus into the cytoplasm and dendritic spines, thereby providing a mechanism for spine sequestration. Thus, in neurons lacking NAC1, or transfected with NAC1 lacking the critical POZ/BTB protein–protein interaction domain, stimulating synaptic activity with bicuculline did not significantly move the 20S proteasome subunit into dendrites.
In addition to being necessary for UPS translocation, having an N-terminus POZ/BTB domain places NAC1 into the family of proteins with the potential to function as substrate acceptors for ubiquitination (Pintard et al., 2004). However, to function as a substrate acceptor, the NAC1 primary structure should contain additional protein–protein interaction domains, such as Kelch or MATH domains, which are known to target ubiquitin-dependent degradation of nuclear factor erythroid 2-related factor 2 and meiotic-1, respectively (Pintard et al., 2004; Zhang et al., 2005). NAC1 lacks a protein–protein interaction domain known to serve this function, arguing that its primary role may be to localize the UPS to sites of increased cellular activity rather than target substrates for ubiquitination. Although the POZ/BTB domain is well established as a protein–protein interaction domain, and the current study shows that it is required for binding and translocating the cullins and proteasome, it is unclear how it mediates translocation of NAC1 from the nucleus to cytoplasm and ultimately targets NAC1 to dendritic spines. Also, it is unclear how NAC1, with a single POZ/BTB domain translocates both cullins and the proteasome. However, because Cul3 is distributed throughout the nucleus, soma and dendrites under basal conditions (Figs. 2, 4), translocation of the 19S and 20S subunits from the nucleus by NAC1 appeared to be the primary effect of bicuculline stimulation. Although NAC1 was necessary to translocate the UPS after bicuculline or proteasome inhibition, it is important to note that NAC1 is not the only mechanism for UPS cytoplasmic translocation, because PKC activation moved the 20S proteasome into the cytoplasm in the absence of NAC1, possibly via direct phosphorylation of a proteasome subunit (Hegde, 2004; Glickman and Raveh, 2005).
NAC1 synthesis is regulated in an immediate early gene manner by increased synaptic activity (e.g., psychostimulant drug administration) (Cha et al., 1997; Mackler et al., 2003). Consistent with a role for NAC1 in mediating activity-dependent UPS translocation and a role for activity regulated UPS in synaptic plasticity, upregulating NAC1 in vivo prevented the development of behavioral sensitization to repeated cocaine administration, whereas downregulating NAC1 produced a presensitized phenotype (Kalivas et al., 1999; Mackler et al., 2000). Behavioral sensitization to cocaine has been linked in many studies to short and long-term plasticity in excitatory synapses (Bonci and Malenka, 1999; Thomas et al., 2001; Saal et al., 2003; Boudreau and Wolf, 2005; Brebner et al., 2005), posing NAC1 recruitment of the UPS into synapses as a compensatory response to minimize the neuropathological consequences of cocaine abuse (Kalivas et al., 2005). A compensatory role for UPS activation in synapses in mitigating addiction neuropathology is consistent with the emerging indications that UPS dysfunctions contribute to neurodegenerative disorders, such as Alzheimer's, Huntington's, and Parkinson's diseases (Hegde, 2004). Thus, in all of these neurodegenerative diseases, proteins with putative etiologic involvement have been identified that affect UPS activity (e.g., Ab peptide, polyglutamine-containing proteins, and parkin). The fact that NAC1 was required for mobilizing the UPS into the cytoplasm in response to pharmacologically inhibiting the proteasome indicates that NAC1 could be an important cellular response to overproduction of these proteins associated with classic neurodegenerative diseases. Thus, the discovery of NAC1 as a protein regulating translocation of the UPS from the nucleus into the cytoplasm opens new possibilities for understanding not only physiological cellular functions, such as UPS regulation of memory acquisition and consolidation (Wood et al., 2005; Yeh et al., 2006), but also pathological processes such as addiction and neurodegenerative diseases.
Footnotes
This work was supported, in part, by National Institutes of Health Grants DA-03906, DA-11809, and RR-015455, Veterans Administration Grant DVA-MR-393, the University of Pennsylvania Research Foundation, and the Joseph Alexander Foundation.
References
- Allen E, Ding J, Wang W, Pramanik S, Chou J, Yau V, Yang Y. Gigaxonin-controlled degradation of MAP1B light chain is critical to neuronal survival. Nature. 2005;438:224–228. doi: 10.1038/nature04256. [DOI] [PubMed] [Google Scholar]
- Ambroggio XI, Rees DC, Deshaies RJ. JAMM: a metalloprotease-like zinc site in the proteasome and signalosome. PLoS Biol. 2004;2:E2. doi: 10.1371/journal.pbio.0020002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingol B, Schuman EM. Synaptic protein degradation by the ubiquitin proteasome system. Curr Opin Neurobiol. 2005;15:536–541. doi: 10.1016/j.conb.2005.08.016. [DOI] [PubMed] [Google Scholar]
- Bingol B, Schuman EM. Activity-dependent dynamics and sequestration of proteasomes in dendritic spines. Nature. 2006;441:144–1148. doi: 10.1038/nature04769. [DOI] [PubMed] [Google Scholar]
- Bonci A, Malenka R. Properties and plasticity of excitatory synapses on dopaminergic and GABAergic cells in the ventral tegmental area. J Neurosci. 1999;19:3723–3730. doi: 10.1523/JNEUROSCI.19-10-03723.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau AC, Wolf ME. Behavioral sensitization to cocaine is associated with increased AMPA receptor surface expression in the nucleus accumbens. J Neurosci. 2005;25:9144–9151. doi: 10.1523/JNEUROSCI.2252-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brebner K, Wong TP, Liu L, Liu Y, Campsall P, Gray S, Phelps L, Phillips AG, Wang YT. Nucleus accumbens long-term depression and the expression of behavioral sensitization. Science. 2005;310:1340–1343. doi: 10.1126/science.1116894. [DOI] [PubMed] [Google Scholar]
- Brooks P, Fuertes G, Murray R, Bose S, Knecht E, Rechsteiner M, Hendil K, Tanaka K, Dyson J, Rivett J. Subcellular localization of proteasomes and their regulatory complexes in mammalian cells. Biochem J. 2000;346:155–161. [PMC free article] [PubMed] [Google Scholar]
- Burger-Kentischer A, Finkelmeier D, Thiele M, Schmucker J, Geiger G, Tovar GE, Bernhagen J. Binding of JAB1/CSN5 to MIF is mediated by the MPN domain but is independent of the JAMM motif. FEBS Lett. 2005;579:1693–1701. doi: 10.1016/j.febslet.2005.01.080. [DOI] [PubMed] [Google Scholar]
- Carlisle HJ, Kennedy MB. Spine architecture and synaptic plasticity. Trends Neurosci. 2005;28:182–187. doi: 10.1016/j.tins.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Cha XY, Pierce RC, Kalivas PW, Mackler SA. NAC-1, a rat brain mRNA, is increased in the nucleus accumbens three weeks after chronic cocaine self-administration. J Neurosci. 1997;17:6864–6871. doi: 10.1523/JNEUROSCI.17-18-06864.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark KR, Liu X, McGrath JP, Johnson PR. Highly purified recombinant adeno-associated virus vectors are biologically active and free of detectable helper and wild-type viruses. Hum Gene Ther. 1999;10:1031–1039. doi: 10.1089/10430349950018427. [DOI] [PubMed] [Google Scholar]
- Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK, Lu H, Bear MF, Scott JD. Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron. 2003;40:595–607. doi: 10.1016/s0896-6273(03)00687-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiAntonio A, Hicke L. Ubiquitin-dependent regulation of the synapse. Annu Rev Neurosci. 2004;27:223–246. doi: 10.1146/annurev.neuro.27.070203.144317. [DOI] [PubMed] [Google Scholar]
- During MJ, Young D, Baer K, Lawlor P, Klugmann M. Development and optimization of adeno-associated virus vector transfer into the central nervous system. Methods Mol Med. 2003;76:221–236. doi: 10.1385/1-59259-304-6:221. [DOI] [PubMed] [Google Scholar]
- Ehlers MD. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci. 2003;6:231–242. doi: 10.1038/nn1013. [DOI] [PubMed] [Google Scholar]
- Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- Glickman MH, Raveh D. Proteasome plasticity. FEBS Lett. 2005;579:3214–3223. doi: 10.1016/j.febslet.2005.04.048. [DOI] [PubMed] [Google Scholar]
- Hegde AN. Ubiquitin-proteasome-mediated local protein degradation and synaptic plasticity. Prog Neurobiol. 2004;73:311–357. doi: 10.1016/j.pneurobio.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Kaestner K, Montoliu L, Kern H, Thulke M, Schutz G. Universal β-galactosidase vectors for promoter analysis and gene targeting. Gene. 1994;148:67–70. doi: 10.1016/0378-1119(94)90234-8. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Duffy P, Mackler SA. Interrupted expression of NAC-1 augments the behavioral responses to cocaine. Synapse. 1999;33:153–159. doi: 10.1002/(SICI)1098-2396(199908)33:2<153::AID-SYN5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Volkow N, Seamans J. Unmanageable motivation in addiction: a pathology in prefrontal-accumbens glutamate transmission. Neuron. 2005;45:647–650. doi: 10.1016/j.neuron.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Karpova A, Mikhaylova M, Thomas U, Knopfel T, Behnisch T. Involvement of protein synthesis and degradation in long-term potentiation of Schaffer collateral CA1 synapses. J Neurosci. 2006;26:4949–4955. doi: 10.1523/JNEUROSCI.4573-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai H, Matsuzaki M, Noguchi J, Yasumatsu N, Nakahara H. Structure-stability-function relationships of dendritic spines. Trends Neurosci. 2003;26:360–368. doi: 10.1016/S0166-2236(03)00162-0. [DOI] [PubMed] [Google Scholar]
- Kawahara H, Yokosawa H. Cell cycle-dependent change of proteasome distribution during embryonic development of the ascidian Halocynthia roretzi. Dev Biol. 1992;151:27–33. doi: 10.1016/0012-1606(92)90210-8. [DOI] [PubMed] [Google Scholar]
- Klugmann M, Wymond Symes C, Leichtlein CB, Klaussner BK, Dunning J, Fong D, Young D, During MJ. AAV-mediated hippocampal expression of short and long Homer 1 proteins differentially affect cognition and seizure activity in adult rats. Mol Cell Neurosci. 2005;28:347–360. doi: 10.1016/j.mcn.2004.10.002. [DOI] [PubMed] [Google Scholar]
- Korutla L, Wang PJ, Lewis DM, Neustadter JH, Stromberg MF, Mackler SA. Differences in expression, actions and cocaine regulation of two isoforms for the brain transcriptional regulator NAC1. Neuroscience. 2002;110:421–429. doi: 10.1016/s0306-4522(01)00518-8. [DOI] [PubMed] [Google Scholar]
- Korutla L, Champtiaux N, Shen HW, Klugman M, Kalivas PW, Mackler SA. Activity-dependent subcellular localization of NAC1. Eur J Neurosci. 2005;22:397–403. doi: 10.1111/j.1460-9568.2005.04208.x. [DOI] [PubMed] [Google Scholar]
- Kwon JE, La M, Oh KH, Oh YM, Kim GR, Seol JH, Baek SH, Chiba T, Tanaka K, Bang OS, Joe CO, Chung CH. BTB domain-containing speckle-type POZ protein (SPOP) serves as an adaptor of Daxx for ubiquitination by Cul3-based ubiquitin ligase. J Biol Chem. 2006;281:12664–12672. doi: 10.1074/jbc.M600204200. [DOI] [PubMed] [Google Scholar]
- Lafarga M, Fernandez R, Mayo I, Berciano MT, Castano JG. Proteasome dynamics during cell cycle in rat Schwann cells. Glia. 2002;38:313–328. doi: 10.1002/glia.10075. [DOI] [PubMed] [Google Scholar]
- Mackler S, Pacchio A, Degnan R, Homan YX, Conti AC, Kalivas P, Blendy JA. NAC1 regulates acute responses to psychomotor stimulants. J Neurosci. 2007 in press. [Google Scholar]
- Mackler SA, Korutla L, Cha XY, Koebbe MJ, Fournier KM, Bowers MS, Kalivas PW. NAC-1 is a brain POZ/BTB protein that can prevent cocaine-induced sensitization in the rat. J Neurosci. 2000;20:6210–6217. doi: 10.1523/JNEUROSCI.20-16-06210.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackler SA, Homan YX, Korutla L, Conti AC, Blendy JA. The mouse nac1 gene, encoding a cocaine-regulated Bric-a-brac tramtrac broad complex/pox virus and zinc finger protein, is regulated by AP1. Neuroscience. 2003;121:355–361. doi: 10.1016/s0306-4522(03)00376-2. [DOI] [PubMed] [Google Scholar]
- Mason GG, Murray RZ, Pappin D, Rivett AJ. Phosphorylation of ATPase subunits of the 26S proteasome. FEBS Lett. 1998;430:269–274. doi: 10.1016/s0014-5793(98)00676-0. [DOI] [PubMed] [Google Scholar]
- Muratani M, Tansey W. How ubiquitin-proteasome system controls transcription. Mol Cell Biol. 2003;4:1–10. doi: 10.1038/nrm1049. [DOI] [PubMed] [Google Scholar]
- Patrick GN. Synapse formation and plasticity: recent insights from the perspective of the ubiquitin proteasome system. Curr Opin Neurobiol. 2006;16:90–94. doi: 10.1016/j.conb.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Patrick GN, Bingol B, Weld HA, Schuman EM. Ubiquitin-mediated proteasome activity is required for agonist-induced endocytosis of GluRs. Curr Biol. 2003;13:2073–2081. doi: 10.1016/j.cub.2003.10.028. [DOI] [PubMed] [Google Scholar]
- Pintard L, Willems A, Peter M. Cullin-based ubiquitin ligases: Cul3-BTB complexes join the family. EMBO J. 2004;23:1681–1687. doi: 10.1038/sj.emboj.7600186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron. 2003;37:577–582. doi: 10.1016/s0896-6273(03)00021-7. [DOI] [PubMed] [Google Scholar]
- Schaefer H, Rongo C. KEL-8 is a substrate receptor for CUL3-dependent ubiquitin ligase that regulates synaptic glutamate receptor turnover. Mol Biol Cell. 2006;17:1250–1260. doi: 10.1091/mbc.E05-08-0794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell E, Chetkovich D, Nicoll R, Bredt D. PSD-95 involvement in maturation of excitatory synapses. Science. 2000;290:1364–1368. [PubMed] [Google Scholar]
- Thomas MJ, Beurrier C, Bonci A, Malenka RC. Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat Neurosci. 2001;4:1217–1223. doi: 10.1038/nn757. [DOI] [PubMed] [Google Scholar]
- Toda S, Alguacil LF, Kalivas PW. Repeated cocaine administration changes the function and subcellular distribution of adenosine A1 receptor in the rat nucleus accumbens. J Neurochem. 2003;87:1478–1484. doi: 10.1046/j.1471-4159.2003.02121.x. [DOI] [PubMed] [Google Scholar]
- Tsurumi C, DeMartino GN, Slaughter CA, Shimbara N, Tanaka K. cDNA cloning of p40, a regulatory subunit of the human 26S proteasome, and a homolog of the Mov-34 gene product. Biochem Biophys Res Commun. 1995;210:600–608. doi: 10.1006/bbrc.1995.1701. [DOI] [PubMed] [Google Scholar]
- Wang PJ, Stromberg M, Replenski S, Snyder-Mackler A, Mackler SA. The relationship between cocaine-induced increases in NAC1 and behavioral sensitization. Pharmacol Biochem Behav. 2003;75:49–54. doi: 10.1016/s0091-3057(03)00040-6. [DOI] [PubMed] [Google Scholar]
- Wojcik C, DeMartino G. Intracellular localization of proteasomes. Int J Biochem Cell Biol. 2003;35:579–589. doi: 10.1016/s1357-2725(02)00380-1. [DOI] [PubMed] [Google Scholar]
- Wolf ME. The role of excitatory amino acids in behavioral sensitization to psychomotor stimulants. Prog Neurobiol. 1998;54:679–720. doi: 10.1016/s0301-0082(97)00090-7. [DOI] [PubMed] [Google Scholar]
- Wood MA, Kaplan MP, Brensinger CM, Guo W, Abel T. Ubiquitin C-terminal hydrolase L3 (Uchl3) is involved in working memory. Hippocampus. 2005;15:610–621. doi: 10.1002/hipo.20082. [DOI] [PubMed] [Google Scholar]
- Yeh SH, Mao SC, Lin HC, Gean PW. Synaptic expression of glutamate receptor after encoding of fear memory in the rat amygdala. Mol Pharmacol. 2006;69:299–308. doi: 10.1124/mol.105.017194. [DOI] [PubMed] [Google Scholar]
- Yi JJ, Ehlers MD. Ubiquitin and protein turnover in synapse function. Neuron. 2005;47:629–632. doi: 10.1016/j.neuron.2005.07.008. [DOI] [PubMed] [Google Scholar]
- Zhang DD, Lo SC, Sun Z, Habib GM, Lieberman MW, Hannink M. Ubiquitination of Keap1, a BTB-Kelch substrate adaptor protein for Cul3, targets Keap1 for degradation by a proteasome-independent pathway. J Biol Chem. 2005;280:30091–30099. doi: 10.1074/jbc.M501279200. [DOI] [PubMed] [Google Scholar]