

Abstract
Repeated stress enhances vulnerability to neural dysfunction that is cumulative over the course of the lifespan. This dysfunction contributes to cognitive deficits observed during aging. In addition, aging is associated with dysregulation of the limbic–hypothalamic–pituitary–adrenal (LHPA) axis, leading to a delayed termination of the stress response. This delay, in turn, increases exposure to glucocorticoids and exacerbates the likelihood of neural damage. Here we asked whether similar effects could emerge at an early age as a result of genetic variations in the level or function of the brain glucocorticoid receptor (GR). We investigated the effect of forebrain-specific overexpression of GR on LHPA axis activity. Transgenic mice with GR overexpression in forebrain (GRov) display normal basal circulating adrenocorticotropic hormone and corticosterone levels. However, young GRov mice exhibit a number of LHPA alterations, including a blunted initial response to acute restraint stress followed by a delayed turn-off of the stress response. This deficit in negative feedback is paradoxical in the face of elevated GR levels, resembles the stress response in aged animals, and continues to worsen as GRov mice age. The neuroendocrine dysregulation in young GRov mice is coupled with a mild cognitive deficit, also consistent with the accelerated aging hypothesis. The molecular basis of this phenotype was examined using microarray analysis of the hippocampus, which revealed a broad downregulation of glutamate receptor signaling in GRov mice. Thus, even in the absence of chronic stress, elevation of GR gene expression can lead to an increased allostatic load and result in an “aging-like” phenotype in young animals.
Keywords: glucocorticoid receptor, neuroendocrine, cognitive, aging, stress, hippocampus
Introduction
Glucocorticoids modulate a broad range of neural functions, including neuronal excitability and plasticity, neurogenesis, neuronal death, stress reactivity, and emotional responses (Akil and Morano, 1995; McEwen and Sapolsky, 1995; De Kloet et al., 2005). Their actions are mediated by two ligand-dependent transcription factors, the glucocorticoid receptor (GR) and the mineralocorticoid receptor. In response to a stressful event, GR appears to function in part as a sensor of salient and/or threatening stimuli, and its activation results in increased anxiety behavior and alterations in learning and memory. On the other hand, GR activation also serves to limit further activation of the stress system via negative feedback mechanisms (Akil, 2005). This dual role is encoded anatomically, with structures such as the amygdala being implicated in the activation of the stress response and others such as the hippocampus (HC) and the prefrontal cortex being largely inhibitory of LHPA axis activity (Lopez et al., 1999; Herman et al., 2003).
Stress-related neuropsychiatric diseases are accompanied by alterations in glucocorticoids (McEwen, 1998), and the normalization of the LHPA system is thought to be a prerequisite to clinical improvement (Ising et al., 2005). The specific role of brain GR in stress and mood disorders has been investigated in a number of animal models (Pepin et al., 1992; Reichardt et al., 1998; Tronche et al., 1999; Wei et al., 2004; Boyle et al., 2005). We overexpressed GR specifically in forebrain and demonstrated a very distinctive phenotype consisting of increased reactivity to the environment along with emotional lability. Transgenic mice with GR overexpression in forebrain (GRov) display a significant increase in anxiety-like and depressant-like behaviors, yet they are also supersensitive to antidepressants and show enhanced sensitization to cocaine (Wei et al., 2004).
Glucocorticoids also modulate learning and memory (Diamond et al., 1992; Oitzl and De Kloet, 1992; Conrad et al., 1999). Chronic elevation of glucocorticoids is thought to cause damage to the HC, a brain region important for both information processing and termination of the stress response. Previous findings have shown that aged animals exhibit prolonged secretion of glucocorticoids after stress, and this is associated with impairment of cognitive function (Landfield et al., 1978; Sapolsky et al., 1983; McEwen and Sapolsky, 1995). This led to the view that common mechanisms may impair hippocampal control of the LHPA axis and impact spatial learning and memory. Indeed, prolonged exposure to high levels of glucocorticoids accelerates senescent hippocampal degeneration (Sapolsky, 1992). This body of work points to the importance of cumulative exposure to stress and glucocorticoids in the course of an individual's lifetime and shows that excessive and repeated stress can result in an “allostatic load” that has profound affective and cognitive consequences (McEwen, 1998).
It has become clear that vulnerability to psychiatric and neurological diseases is highly modified by reactivity to environmental stressors (Caspi and Moffitt, 2006). To investigate the possible functional role of genetic differences in stress responsivity, we used the GRov mouse model to ascertain how innate differences in the neural expression of a stress-related gene can modify stress reactivity and cognitive function both in the presence and absence of environmental stressors.
Materials and Methods
Animals
The generation of transgenic mice overexpressing GR specifically in forebrain was described previously (Wei et al., 2004). GRov mice were established by breeding founders and their progeny to C57BL/6J mice. Mice were maintained on a 14/10 light/dark schedule with lights on between 5:00 A.M. and 7:00 P.M. All transgenic mice are maintained as hemizygotes. All experiments were performed with male GRov mice and their wild-type (WT) littermates. Mice were between 2 and 6 months of age at the time of testing, except for the aging study, in which mice were between 18 and 22 months of age. Mice were housed individually for 1 week before each study. All experiments were conducted in accordance with the principles and procedures outlined in the National Institutes of Health guidelines for the care and use of animals.
Neuroendocrinological experiments
Blood samples were collected by decapitation <20 s after opening the cage. Blood was placed immediately in iced Eppendorf tubes containing EDTA.
Restraint stress.
The restraint device consists of a 9 × 12 cm piece of flexible Teflon attached to a 9 × 3 cm platform with Plexiglas ends containing a tail slot and air holes. The Teflon is wrapped snugly around the mouse and fastened with Velcro straps. Plasma was collected at the onset of restraint stress (t = 0), during stress (t = 15 and 30 min), and after the end of exposure to stress (t = 60, 90, and 150 min; i.e., 30, 60, and 120 min after the termination of 30 min of restraint stress). In the aging study, plasma was collected at the onset of restraint stress (t = 0), during stress (t = 15 and 30 min), and after stress (t = 60 and 90 min). The test was performed between 7:00 and 10:00 A.M.
Chronic stress protocol.
Mice from each genotype were randomly assigned to one of the following experimental groups: (1) undisturbed (control); (2) twice daily exposure to intraperitoneal injections of 0.9% saline (injection stress; 10 ml/kg); or (3) twice daily exposure to chronic unpredictable stress (CUS). CUS consisted of forced swimming, tail suspension, open field, light–dark box, elevated plus maze (EPM), cold, crowding, wet bedding, loud noise, intraperitoneal injection of saline, and reversed light cycle paradigms. Stress exposure took place once in the morning and once in the afternoon for 14 consecutive days. Plasma was collected at 2 h after lights on in the morning, which was 16 h after the last stress session on the last day.
Dexamethasone suppression test.
Mice were injected intraperitoneally with different doses of dexamethasone (Dex; 10, 20, 40, and 100 μg/kg; American Pharmaceutical Partners, Schaumburg, IL) or 0.9% saline at 11:00 A.M. Plasma was collected 6 h later.
Corticotropin-releasing hormone challenge test.
Mice were injected intraperitoneally with 40 μg/kg corticotropin-releasing hormone (CRH; American Peptide, Sunnyvale, CA) dissolved in 0.9% saline containing 0.05% ascorbic acid at 7:00 A.M. Plasma was collected at various time points (t = 0, 15, 30, 60, and 120 min).
Dex/CRH test.
Six hours after application of 40 μg/kg Dex given at 11:00 A.M., mice were injected intraperitoneally with 40 μg/kg CRH and killed 60 min later.
Adrenocorticotropic hormone stimulation test.
Cortrosyn (Amphastar Pharmaceuticals, Rancho Cucamonga, CA) was reconstituted in 0.4 ml of normal saline. The Cortrosyn solution was then diluted in PBS. Mice were injected intraperitoneally with 40 μg/kg Cortrosyn or vehicle saline at 7:00 A.M. Plasma was collected 30 min later.
For the restraint stress studies in naive GRov and WT mice and the CRH challenge study, plasma adrenocorticotropic hormone (ACTH) was determined using an ACTH kit (Quest Diagnostics, Lyndhurst, NJ). Plasma corticosterone (CORT) was assayed using a highly specific antibody developed in our laboratory. Cross-reactivities to related compounds (e.g., cortisol) were <3%. Intra-assay and interassay variations were <10% (data not shown).
For the remaining studies involving chronic stress, aging, dexamethasone suppression, and Dex/CRH test, we assayed plasma ACTH using the kit provided by MP Biomedicals (Orangeburg, NY) because the ACTH kit from Quest Diagnostics was discontinued during the period of these studies. For these studies and the ACTH stimulation test, plasma CORT concentrations were also analyzed using the kit provided by MP Biomedicals. It should be noted, however, that each study included its own control WT mice, and all comparisons were made within each study.
Morris water maze test
The Morris water maze (MWM) was used to test for spatial learning and memory. The protocol was described previously (Hebda-Bauer et al., 2005). Briefly, mice were given 4 trials/d with a 3–8 min intertrial interval. A regular trial began by placing a mouse in the water at one of four randomly assigned starting positions. If a mouse had not located the platform within 60 s, it was removed from the water and placed on the platform for 30 s. Mice were tested for 6 d to assess initial learning. On day 7, the mice were given a 60 s probe trial in which the platform was removed. The three subsequent trials on day 7 had the submerged platform present in its original location. On days 8–10, the submerged platform was located in the opposite quadrant of the pool to assess reversal learning. A second probe trial was performed on day 11 with the platform removed. A video tracking system (Ethovision; Noldus Technology, Sterling, VA) was used to measure distance traveled and number of platform crossings in the goal quadrant and comparable platform locations in the other three quadrants.
Microarray analysis and quantitative real-time PCR
The microarray and the quantitative real-time PCR (qRT-PCR) experiments were performed and analyzed as described previously (Stead et al., 2006). Briefly, mice were killed by decapitation in the morning. The hippocampus was rapidly dissected and stored at −80°C before processing. Total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA), followed by an additional clean-up step using RNeasy RNA purification (Qiagen, Valencia, CA). RNA quality and concentration were both determined by absorbance at 260 and 280 nm and by analysis using an Agilent (Palo Alto, CA) bioanalyzer. First- and second-strand cDNA syntheses were performed as detailed in the Affymetrix (Santa Clara, CA) Expression Analysis Technical Manual, using 10 μg of total RNA. In vitro transcription (IVT) of cDNA to biotinylated cRNA was performed with the MEGAscript T7 High Yield Transcription kit (Ambion, Austin, TX). Products of second-strand cDNA and IVT were purified with the Affymetrix GeneChip Sample Cleanup Module. IVT products were prepared for hybridization to Affymetrix Mouse Genome 430_2.0 GeneChips per instructions. Arrays were hybridized for 18 h at 45°C, washed, stained on an Affymetrix fluidics station using the standard EukGEWS2v4_450 protocol, and scanned.
GeneChip signal intensity data were interpreted by use of a custom chip description file (filename MM430_MM_REFSEQ_6). Design of the custom chip description file was described in detail previously (Dai et al., 2005). This file is available at http://brainarray.mbni.med.umich.edu/Brainarray/Database/CustomCDF/CDF_download_v6.asp. Cell intensity files from the Affymetrix GeneChip arrays were normalized by the robust multiarray average algorithm available at www.bioconductor.org.
For qRT-PCR, the template cDNA was synthesized from 0.4 μg of total RNA using iScript cDNA synthesis kit (Bio-Rad, Hercules, CA) and quantified using Quant-It PicoGreen dsDNA kit (Invitrogen). Primers were designed to generate amplicons of 70–150 bp in length at the 3′ of the target mRNA (supplemental Table S1, available at www.jneurosci.org as supplemental material). Most primers were designed to flank intronic sequences. Each primer pair was tested using a fivefold dilution series of pooled cDNA template to ensure that the efficiency of amplifications was linear and that the primers specifically produced a single amplified product. qRT-PCR was performed using the iQ SYBR Green PCR kit (Bio-Rad). PCR products were amplified using an iCycler RT-PCR system (Bio-Rad) for 40 cycles using parameters of 95°C for 15 s, 60°C for 15 s, and 72°C for 15 s. The cycle threshold was calculated with iCycler software.
Statistical analysis
Statistical comparisons were performed by unpaired two-tailed t tests and ANOVAs followed by post hoc tests as necessary. In the MWM test, two-way ANOVAs with repeated measures using the Mixed Model procedure in SAS were used to analyze regular trial performance across days, and ANOVAs were used to analyze probe trial performance. Results were expressed as mean ± SEM.
Results
Altered endocrine response to restraint stress in young GRov mice
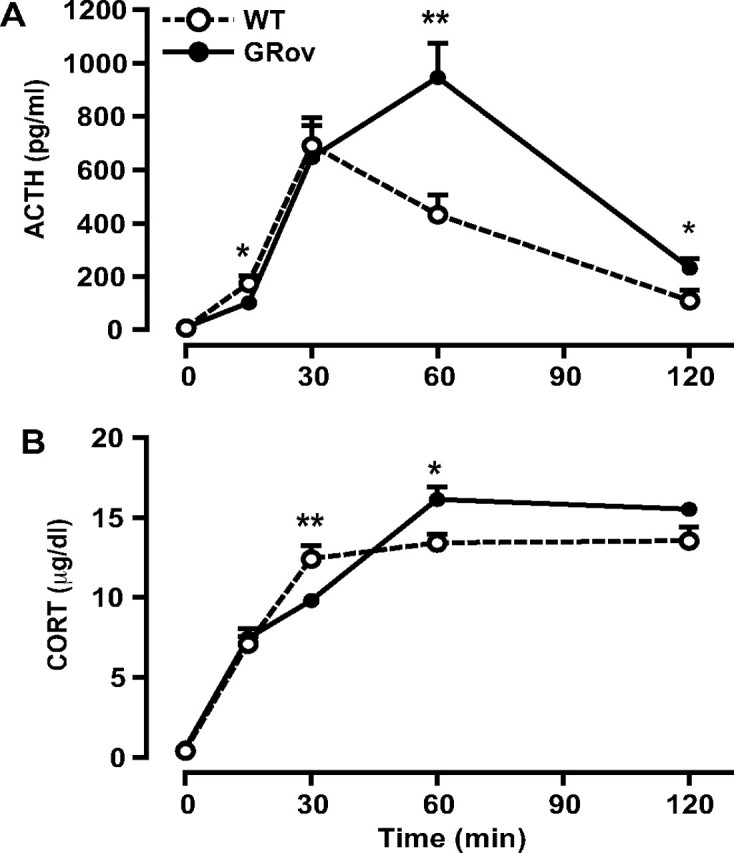
We have previously demonstrated that GRov mice express the GR transgene primarily in the forebrain, with transgene-specific GR mRNA detected in cerebral cortex, olfactory nuclei, tenia tecta, piriform cortex, olfactory tubercle, caudate–putamen, nucleus accumbens, septum, bed nucleus of the stria terminalis (BNST), median preoptic nucleus, thalamus, paraventricular hypothalamic nucleus (PVN), arcuate nucleus, ventromedial and dorsomedial hypothalamic nuclei, posterior hypothalamic area, hippocampus (HC-CA1, -CA2, -CA3, and dentate gyrus), central, basolateral, and basomedial amygdaloid nuclei, and mammillary nuclei [supporting information in Wei et al. (2004)]. The most intense signals for transgene-specific GR mRNA were observed in the HC. Similarly, in situ hybridization experiments using a probe for GR mRNA showed significantly higher levels of total GR mRNA in forebrain regions of GRov mice, with a particularly significant increase in GR mRNA levels in regions that are typically thought of as modulating emotional reactivity and stress responsiveness, including prefrontal cortex, nucleus accumbens, BNST, central nucleus of the amygdala, PVN, and the HC [supporting information in Wei et al. (2004)]. Previous studies showed that GRov mice exhibited normal HPA axis activity basally and in response to a mild stressor, 5 min free exploration in the EPM; this brief exposure to the EPM led only to a mild activation of the HPA axis in WT controls, with ACTH and CORT levels peaking at 137 pg/ml and 6.7 μg/dl, respectively (Wei et al., 2004). In contrast, the 30 min acute restraint stress paradigm of the current study led to a strong activation of the HPA axis in WT controls, with ACTH and CORT levels peaking at ∼904 pg/ml and 16 μg/dl, respectively, which would be expected to engage GR in forebrain and trigger negative feedback. This strong stressor produced a distinct pattern of ACTH response in GRov mice, with a lower peak but an extended duration (Fig. 1A). At the onset of restraint stress (t = 0), GRov and WT mice did not differ in basal ACTH levels. However, the stress response of GRov mice seemed initially attenuated, because their ACTH levels did not reach the same magnitude at the 30 min time point as that of WT mice (Fig. 1A) (p < 0.05). Surprisingly, the stress response of GRov mice was also more prolonged. Although ACTH levels decreased dramatically at the 60 min time point for both groups, GRov mice had significantly higher levels than those of WT controls at this time point (GRov, 78.03 ± 16.63 pg/ml; WT, 38.66 ± 7.11 pg/ml; p < 0.05).
Figure 1.

Alteration in the dynamics of the endocrine response to acute restraint stress in GRov mice. ACTH (A) and CORT (B) responses to restraint stress are shown at various time points. GRov mice exhibited a blunted initial response to acute restraint stress and a prolonged termination of the stress response. ANOVA revealed a significant genotype × time point interaction in CORT response to the stress (p < 0.001). *p < 0.05 and ***p < 0.001 versus WT group at the same time point. n = 4–10 mice per time point per genotype.
GR overexpression also produced an altered CORT profile during and after restraint stress (Fig. 1B). GRov and WT mice did not differ in basal CORT levels at the beginning of the restraint stress. However, CORT levels were slower to rise in GRov mice at the 15 min time point than in WT mice (p < 0.05) but reached the same magnitude at the 30 min time point (p = 0.18). Interestingly, at the 60 min time point, CORT levels in GRov mice remained as high as at the 30 min time point, unlike the lower levels of WT mice (p < 0.001). Even at the 90 min time point, CORT levels in GRov mice were still significantly higher than those of WT controls (p < 0.05). These results suggest that, under conditions that strongly engage the HPA axis, overexpression of GR in forebrain results in a blunted initial response to restraint stress coupled with a prolonged duration of the neuroendocrine response. This pattern is paradoxical, because one would expect increased GR in the HC to produce a more efficient negative feedback. Instead, it resembles the termination pattern of the stress response seen in aged individuals and led to the series of studies below.
Normal HPA axis regulation in the dexamethasone suppression test in young GRov mice
To further characterize the LHPA response in GRov mice, we used the dexamethasone suppression test (DST). ACTH levels were effectively suppressed by Dex at the 100 μg/kg dose (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material). CORT levels were significantly suppressed with 20, 40, and 100 μg/kg Dex (supplemental Fig. S1B, available at www.jneurosci.org as supplemental material). There were no genotype differences in ACTH and CORT levels in the DST. These results suggest that GR overexpression in forebrain does not alter the feedback regulation of LHPA axis in the DST. This is consistent with the notion that Dex exerts its inhibitory effects on the LHPA axis primarily by acting at the level of the pituitary (Cole et al., 2000), and we have previously reported normal levels of GR mRNA in the anterior pituitary of GRov mice (Wei et al., 2004).
Abnormal HPA axis activity in response to CRH challenge and Dex/CRH test in young GRov mice
To assess the impact of GR overexpression on responsiveness to a neuroendocrine challenge, we measured CORT and ACTH levels after intraperitoneal administration of 40 μg/kg CRH. The ACTH response to CRH challenge showed a significant interaction between genotype and time (Fig. 2A) (p < 0.001). ACTH levels were slower to rise in GRov mice at the 15 min time point than in WT mice (p < 0.05). There was no genotype difference in ACTH at the 30 min time point, when WT mice reached their maximum ACTH response to CRH stimulation. However, GRov mice showed a greater ACTH response to CRH at the 60 min time point (p < 0.01), and their levels remained significantly elevated relative to WT at 120 min (p < 0.05). These results suggest that GRov mice show a prolonged ACTH response to CRH challenge, although they exhibit an attenuated response at the 15 min time point, as indicated by a significant difference between genotypes across time points (p < 0.05).
Figure 2.
Abnormal endocrine response to CRH challenge in GRov mice. A, GRov mice exhibited an exaggerated ACTH response to CRH (p < 0.05). ANOVA yielded a significant genotype × time point interaction in ACTH response to CRH challenge (p < 0.001). B, The CORT response to CRH administration also yielded a significant genotype × time point interaction (p < 0.001). *p < 0.05 and **p < 0.01 versus WT group at the same time point. n = 5–7 mice per time point per genotype.
Similarly, we observed a significant genotype by time interaction in the CORT response to CRH challenge (Fig. 2B) (p < 0.001). When compared with WT controls, GRov mice showed an attenuated CORT response to CRH at the 30 min time point (p < 0.01) and an increased CORT response at the 60 min time point (p < 0.05). GRov mice still exhibited a trend toward increased CORT secretion to CRH challenge at the 120 min time point (p = 0.06).
We then examined the CORT and ACTH responses to the combined Dex/CRH test. Six hours after intraperitoneal injection with 40 μg/kg Dex, mice were injected with 40 μg/kg CRH (i.p.) and killed 60 min later. GRov mice showed higher ACTH levels (GRov, 484.89 ± 64.56 pg/ml; WT, 272.44 ± 23.87 pg/ml; n = 8 per genotype; p < 0.01) and CORT levels (GRov, 9.79 ± 0.65 μg/dl; WT, 7.74 ± 0.44 μg/dl; n = 8 per genotype; p < 0.05) than WT controls. These findings underscore the delayed turn-off of the stress response in GRov mice.
We also performed an ACTH stimulation test in GRov mice. There was no difference in ACTH-induced secretion of CORT between GRov and WT mice (saline injection, GRov, 3.06 ± 1.29; WT, 3.36 ± 0.62 μg/dl; ACTH injection, GRov, 13.96 ± 1.45; WT, 13.40 ± 1.20 μg/dl; n = 6 per group per genotype), suggesting equivalent adrenal sensitivity to ACTH.
Increased impact of repeated chronic stress on resting stress hormone levels in young GRov mice
Although the above studies demonstrated altered responsiveness to an acute stressor or neuroendocrine challenge, we sought to determine whether there were sustained differences after chronic stress between the transgenic and WT mice. We asked whether, after chronic stress followed by a period of rest, stress hormones would return to normal or remain elevated. This addresses whether the impact of environmental stimuli may differ as a function of genotype. Mice were exposed to 14 d of either CUS or repeated intraperitoneal saline injection stress.
There was no difference in ACTH levels between genotypes in the control groups. Mice exposed to CUS, regardless of genotype, did not differ in circulating ACTH levels compared with their respective controls (Fig. 3A). Both GRov and WT mice showed an increase in CORT levels after CUS compared with their respective controls (Fig. 3B), but there was no genotype difference after CUS. It is likely that, by the time we obtained our measures, the impact of CUS on ACTH levels had elapsed or was actively suppressed by the enhanced resting levels of CORT in both groups of animals. This suggests that CUS produces a lasting effect on CORT levels but does not discriminate between genotypes.
Figure 3.

Increased morning basal HPA axis activity to repeated chronic stress in GRov mice. A, B, GRov mice exposed to repeated intraperitoneal saline injection stress exhibited significantly higher basal ACTH (A) and CORT (B) levels in the morning than undisturbed GRov controls. GRov mice had significantly higher CORT levels than WT mice after repeated injection stress. **p < 0.01 versus WT group under the same treatment. #p < 0.05 and ##p < 0.01 versus respective control groups. n = 7–8 mice per group per genotype.
We observed a different pattern with the repeated injection stress. This stressor did not result in a sustained effect on ACTH or CORT levels in WT mice. In contrast, GRov mice exhibited significantly elevated resting ACTH levels after injection stress when compared with undisturbed GRov controls (Fig. 3A) (p < 0.05), and a tendency for higher ACTH levels than WT mice after injection stress (p = 0.09). Although injection stress did not induce increased CORT in WT mice, CORT levels were significantly higher in GRov mice exposed to injection stress than in undisturbed GRov controls (Fig. 3B) (p < 0.01) and their WT counterparts (p < 0.01).
This combination of studies reveals that, in WT mice, CUS is more potent than injection stress in producing lasting changes in resting levels of glucocorticoids, possibly because CUS does not produce as much habituation as repetition of the same stressor. However, GRov mice, while exhibiting the same response to the CUS, do show a differential and sustained response to injection stress, again supporting the notion of prolonged responsiveness to certain (less potent) stressors. These results suggest that GRov mice are unable to habituate to the mild repeated saline injection stressor as readily as their WT littermates and that this lack of habituation causes their prolonged stress response.
Altered restraint stress response in aged GRov mice
To assess whether overexpression of GR in the forebrain alters the basal CORT levels with aging, plasma was collected in young and aged mice at 2 h after lights on. There was no age effect on morning basal CORT levels in WT or in GRov mice (WT, young, 0.40 ± 0.14; old, 0.35 ± 0.09 μg/dl; GRov, young, 0.56 ± 0.19; old, 0.59 ± 0.23 μg/dl; n ≥ 5 per group per genotype).
Because the termination pattern of the endocrine response to restraint stress in young GRov mice was reminiscent of older animals, we studied this response in aged GRov and WT mice. There were no genotype differences in morning basal CORT and ACTH levels in aged mice. The restraint stress paradigm resulted in an exaggerated ACTH response in old GRov mice (Fig. 4A), as indicated by an overall genotype effect across time points (p < 0.002). At the end of restraint stress (i.e., t = 30 min), ACTH levels of GRov mice were significantly higher than those of WT littermates (p < 0.01) and remained elevated at the 60 min time point (p = 0.06). Aged GRov mice also showed an exaggerated CORT response to restraint stress in comparison with their WT littermates (Fig. 4B), as indicated by an overall genotype effect across time points (p < 0.05). These results suggest that long-term overexpression of GR in forebrain leads to exacerbated ACTH and CORT responses to acute restraint stress not only in young adult animals but also during aging.
Figure 4.

Exaggerated response to acute restraint stress in aged GRov mice. ACTH (A) and CORT (B) responses to restraint stress are shown at various time points. There were significant differences between aged GRov mice and WT littermates in ACTH (p < 0.01) and CORT (p < 0.05) responses across time points. **p < 0.01 versus WT group at the same time point. n = 5–10 mice per time point per genotype.
We compared the rate of recovery from restraint stress in the aged GRov and WT mice to that of young mice, because young and aged mice were tested in different sessions. CORT values at the termination of restraint stress (i.e., 30 min time point) were normalized to 100%. This point was chosen because the magnitude of the stress response is commonly reported as showing no alteration with aging (Sapolsky et al., 1983; Morano et al., 1994), whereas the recovery from stress shows age-related changes. In comparison with young WT animals, old WT mice showed a delayed CORT recovery to restraint stress (Fig. 5A) (age effect, p < 0.05). Post hoc tests demonstrated a significant difference between young and old WT mice 30 min after the termination of the stressor (i.e., t = 60 min; p < 0.05), supporting the view that aging results in decreased HPA feedback in WT mice. In contrast, aging did not alter the termination pattern of the stress response in GRov mice (Fig. 5B). Interestingly, young GRov mice exhibited a very similar CORT recovery pattern after acute restraint stress to that found in aged WT mice (Fig. 5C). Thus, CORT levels of young GRov mice after stress showed the delayed return to baseline typically induced by aging. These analyses suggest that overactivation of GR signaling in forebrain accelerates age-related dysfunction of LHPA axis activity.
Figure 5.

Termination of CORT response to acute restraint stress in young GRov mice resembles pattern observed in aged WT mice. A, Aged WT mice display a delayed CORT recovery after restraint stress in comparison with young WT mice. B, Young and old GRov mice show a similar termination pattern of the stress response. C, Young GRov exhibit a delayed CORT recovery pattern to restraint stress like that found in aged WT mice. *p < 0.05 versus group at the same time point. #p < 0.05 and ##p < 0.01 versus respective 30 min restraint stress groups. n = 5–10 mice per time point per genotype.
Mild cognitive impairment in young GRov mice
This series of endocrine studies led us to hypothesize that young GRov mice may have altered hippocampal function. GRov mice were tested in the MWM to evaluate their spatial learning and memory ability. Statistical analysis of learning curves during the initial learning phase of the MWM reveals a significant effect of time but not genotype (time, p < 0.001; genotype, p > 0.05). Both GRov and WT mice learned equally well over time, as shown by decreasing distance traveled to the goal across the 6 test days (Fig. 6A). Mice of both genotypes also showed a preference for the goal quadrant in the probe trial on day 7, as demonstrated by significantly more crossings over the platform location in the goal quadrant versus comparable locations in the other quadrants (data not shown) (WT, p < 0.01; GRov, p < 0.001). Thus, initial spatial learning and memory of GRov mice appear to be intact.
Figure 6.

Mild cognitive impairment of young GRov mice in the MWM. A, Distance traveled during the initial learning phase. GRov and WT mice learned equally well, as shown by a similar decrease in distance traveled across test days. B, Distance traveled during reversal learning. GRov mice did not learn to the same extent as WT mice, as exhibited by a flatter learning curve than WT mice (p = 0.08). C, Number of platform crossings during the reversal learning probe trial. GRov mice did not show a preference for the goal quadrant, unlike the preference shown by WT mice (p < 0.05). *p < 0.05. n = 13 mice per genotype. Opp, Opposite; AdjL, adjacent left; AdjR, adjacent right.
During reversal learning, both GRov and WT mice again showed significant learning over time, as demonstrated by decreasing distance traveled to the goal across test days (Fig. 6B) (p < 0.001). However, GRov mice exhibited a flatter learning curve than WT mice across the 3 reversal test days (p = 0.08). GRov mice, unlike their WT counterparts, also did not show a preference for the goal quadrant during the probe trial on day 11, as shown by the number of platform crossings measured (Fig. 6C) (WT, p < 0.05; GRov, p > 0.05). Thus, GRov mice learned during reversal learning, but not to the same extent as WT mice, and their long-term memory in this task is impaired. These results demonstrate that initial spatial learning and memory is preserved in GRov mice, but their ability to adjust or adapt to changes in a learning environment is attenuated.
Altered glutamatergic system in the HC of young GRov mice
The HC plays a critical role in feedback inhibition of the stress response and in spatial learning and memory. Moreover, the HC is the forebrain region that showed the highest level of transgene-specific GR mRNA in GRov mice (Wei et al., 2004). We therefore focused on the HC to identify possible molecular mechanisms whereby GR overexpression leads to the aging-like neuroendocrine pattern coupled with the mild cognitive deficit that we observed.
We performed an expression profiling study using microarray technology to evaluate the global change in basal gene expression in the HC of GRov mice. Systematic analysis of the arrays revealed several hundred transcripts that were differentially expressed in the HC between GRov and WT mice (supplemental Table S2, available at www.jneurosci.org as supplemental material), including genes implicated in growth, synaptogenesis, and memory impairment associated with aging. The most striking result is dysregulation of the glutamate receptor signaling system in GRov mice (Table 1). In contrast, the GABA receptor signaling system is not significantly altered (data not shown).
Table 1.
Dysregulation of glutamate receptor signaling in HC
| Gene | RefSeq ID | Microarray |
qRT-PCR |
||
|---|---|---|---|---|---|
| FC | p | FC | p | ||
| GRIA2 | NM_013540 | −1.21 | 0.057 | −15.70 | 0.003 |
| GRIA3 | NM_016886 | −1.20 | 0.040 | −4.89 | 0.017 |
| GRIA4 | NM_019691 | −1.11 | 0.018 | −3.19 | 0.133 |
| GRIN2C | NM_010350 | −1.12 | 0.061 | −3.04 | 0.092 |
| GRM1 | NM_016976 | −1.25 | 0.034 | −9.50 | 0.040 |
| GRM3 | NM_181850 | −1.17 | 0.083 | −2.28 | 0.095 |
| GRM5 | XM_149971 | −1.21 | 0.049 | NC | NC |
| GRM7 | NM_177328 | −1.13 | 0.093 | ND | ND |
| SLC1A6 | NM_009200 | −1.21 | 0.028 | −2.70 | 0.137 |
| SLC1A7 | NM_146255 | −1.12 | 0.055 | ND | ND |
| SLC38A1 | NM_134086 | −1.18 | 0.064 | −6.69 | 0.022 |
Fold changes (FC) of gene expression represent GRov mice compared with WT. GRov, n = 5; WT, n = 6. NC, No change; ND, not done.
Ingenuity pathway analysis identified the glutamate receptor signaling canonical pathway as the greatest significant alteration in the HC between GRov and WT mice (p < 0.0001). An independent algorithm based on gene set enrichment analysis (Mootha et al., 2003) was also used to analyze the microarray data and showed significant differences in glutamate receptor activity (p < 0.01). Genes within this pathway that were found differentially expressed were all decreased in the HC of GRov animals. Glutamate is the major mediator of excitatory neurotransmission in the CNS, acting via activation of the several classes of glutamate receptors that are widely expressed throughout the brain, and localized presynaptically, postsynaptically, or extrasynaptically on neurons or on glial cells. In the young GRov mice, we observed decreased expression of several subunits of the ionotropic ligand-gated ion-channel receptors, including GRIA2, GRIA3, and GRIA4 (AMPA receptor subunits) and GRIN2C (NMDA receptor subunit), and of the metabotropic G-protein-coupled receptors (mGluRs), including GRM1, GRM3, GRM5, and GRM7. We also observed a significant downregulation of mRNA levels of the glutamate transporters, SLC1A6, SLC1A7, and SLC38A1. These 11 genes involved in glutamate receptor signaling and downregulated by microarray analysis were further tested for differential expression using qRT-PCR. Of the 11 genes assayed, six were confirmed as downregulated, two showed a trend toward downregulation, two failed analysis for technical reasons related to inefficient primers, and only one did not show differential expression by qRT-PCR analysis.
Discussion
In this study, we demonstrate that overexpression of GR in the forebrain leads to an altered pattern of LHPA responsiveness, generally characterized by a delayed termination of the stress response. The response pattern resembles a neuroendocrine phenotype seen in aging and associated with decreased functionality of the hippocampus. In addition, GRov mice express mild cognitive impairment in the MWM, again suggestive of hippocampal dysfunction. This neuroendocrine and neurocognitive phenotype is accompanied by a significant alteration in the expression of a number of genes in the HC, with a remarkably consistent decrease in mRNA levels of glutamate signaling genes. Together, these findings demonstrate that increased neural activity of a stress-sensing gene, even in the absence of substantial environment triggers, can accelerate hippocampal dysregulation and result in endocrine and cognitive sequelae that mimic some aspects of aging.
GRov mice show a normal basal pattern of ACTH and CORT secretion, accompanied by normal basal expression levels of several genes critical to the control of the stress response, including the mineralocorticoid receptor in HC, CRH in PVN, and proopiomelanocortin in anterior pituitary (Wei et al., 2004). Thus, the fundamental elements of the basal control of the stress system are not perturbed by GR overexpression in forebrain, and the GRov mouse is not chronically exposed to abnormal levels of stress hormones. GRov and WT mice also show similar ACTH and CORT responses to a relatively mild stressor such as the EPM (Wei et al., 2004).
Restraint stress, however, resulted in an unexpected response pattern. Given the elevated levels of GR in the HC of GRov mice and the importance of the HC in inhibiting the stress response (Herman et al., 2003), we expected particularly effective negative feedback in GRov mice. There is an inverse relationship between levels of hippocampal GR and levels of stress responses, with lower levels of hippocampal GR associated with impaired negative feedback inhibition (Weidenfeld and Feldman, 1993; Boyle et al., 2005, 2006; Ridder et al., 2005) and higher levels of hippocampal GR associated with greater negative feedback inhibition (Liu et al., 1997; Ridder et al., 2005). In this study, GRov mice responded with lower levels of stress hormones during the activation phase of the stress response. However, they showed significantly elevated CORT levels during the stress termination period, a pattern indicative of impaired negative feedback inhibition of the LHPA axis and suggesting a defect in hippocampal function. Moreover, the prolonged stress response is also evident in aged GRov mice relative to their age-matched controls.
A possible endocrine interpretation of the restraint stress results is that the slow rate of rise of CORT observed during the first 15 min after restraint in the young animals leads to a decrease in rate-sensitive fast feedback and/or intermediate level-sensitive feedback, and thus delayed termination of the stress response (Dallman and Yates, 1969; Jones et al., 1972; Young et al., 1991; Young and Vazquez, 1996). However, in the CRH challenge study, GRov and WT mice showed a comparable rate of rise, yet GRov mice went on to secrete significantly higher levels of stress hormones at 60 min. Similarly, the deficient termination of the stress response in the aged GRov mice relative to aged WT cannot be interpreted in terms of differences caused by the initial rate of rise.
An alternate interpretation for the restraint stress results is that GRov mice may have increased overall reactivity to stressful stimuli, consonant with their increased anxiety-like behavior in tests such as the EPM (Wei et al., 2004). However, we would have predicted that such an increase in emotionality would have impacted the activation phase of the stress response, leading to increased initial levels of stress hormones rather than the unaltered response seen in the EPM or the blunted initial response to restraint stress. It remained conceivable, however, that GRov mice were unable to adjust to the psychological stressor as readily as their WT littermates, resulting in a prolonged stress response.
Therefore, the most parsimonious explanation is that GRov mice show defective negative feedback. This feedback depends on neural circuitry, especially the HC (Herman and Cullinan, 1997), which is typically altered during aging (Sapolsky et al., 1986; Jacobson and Sapolsky, 1991; Young and Vazquez, 1996), and may be altered by the transgene at an earlier age.
The extended stress response in GRov mice was also observed after exposure to chronic stress for 14 d. In response to a mild and habituating stressor (repeated saline injection), GRov mice exhibited increased resting levels of glucocorticoids. Thus, although their basal circulating levels are normal under unstressed conditions, GRov mice show longer and more profound repercussions of mild repeated saline injection stress that may well lead to neural sequelae.
These neuroendocrine studies led us to hypothesize that GRov mice may have altered hippocampal function. The HC participates in episodic and declarative memory and is especially important for accurate and reliable contextual memory (McEwen, 1998). The cognitive deficit in GRov mice, although relatively mild, is consistent with the hypothesis that hippocampal function is disrupted. Because analysis of hippocampal morphology did not reveal major differences between GRov and WT mice (A. Pletsch, unpublished work), we focused on the possibility that there are functional changes in the HC. Gene expression profiling revealed a significant downregulation of several glutamate receptor subunits and glutamate transporters in the HC of GRov mice. Although the impact of this decrease of gene expression on functional circuitry and glutamatergic signaling of the HC remains to be elucidated, the findings strongly suggest that GR overexpression is functionally damaging to hippocampal cells and point to molecular mechanisms that would explain both the neuroendocrine and the cognitive features of the GRov mouse.
The hippocampal glutamatergic system is implicated in the control of negative feedback on the LHPA axis. Our laboratory has delineated a bisynaptic inhibitory circuit arising from the hippocampus and terminating near the CRH neurons of the hypothalamus, where it restrains the activity of these neurons. The initial component of this pathway arises from the subiculum region of the HC and is glutamatergic. These neurons project to the BNST, synapsing on GABAergic neurons. These inhibitory BNST neurons project to the hypothalamus and relay information to the PVN to inhibit secretion of CRH (Herman et al., 1989, 2003; Cullinan et al., 1993; Herman and Cullinan, 1997). Thus, the decrease in negative feedback in GRov mice could result from the dysregulation of the glutamatergic output, which may fail to fully activate the inhibitory circuit that terminates the stress response.
Glutamate signaling is also essential in controlling the structural and functional plasticity of the synapse and plays a critical role in learning and memory mechanisms within the HC (Kim and Diamond, 2002). A large body of work demonstrates the key role of altered glutamatergic mechanisms in glucocorticoid-induced cognitive deficits (Sapolsky, 1992) and in deficits associated with normal aging. Several age-related changes in glutamatergic function have been described, including a reduced capacity for glutamate uptake and a loss in the number of high-affinity transporters in the terminals of aged animals (Segovia et al., 2001). Numerous reports have focused on age-associated decreases in the hippocampal density of glutamatergic receptors (Segovia et al., 2001). The loss of GRIA2 may be particularly critical in aging, because higher expression of GRIA2 relative to other subunits results in low calcium permeability of AMPA receptors in pyramidal neurons (Geiger et al., 1995). This protects against neuronal cell death, as excessive calcium influx through glutamate receptors is known to lead to neuronal degeneration. It is remarkable that all of these age-related changes in the glutamatergic system are apparent in GRov mice.
In summary, we have shown that elevated levels of GR, even in the absence of a stressful environment, can be damaging both at the molecular and functional levels in the HC. The process may be self-amplifying in that alterations in glutamatergic function lead to a delay in negative steroid feedback. The prolonged elevation in glucocorticoids levels after stress coupled with higher levels of GR accelerates the stress-induced process of neuronal damage, leading to the GRov phenotype. Hence, our findings show that variations in GR levels are likely to contribute to differential vulnerability to stress and to the associated acceleration of neural damage. Our results corroborate the hypothesis that normal brain function requires optimal levels of the glucocorticoid signaling and that deviation from this optimal level in either direction is highly deleterious (De Kloet et al., 1998).
We have previously reported that forebrain overexpression of GR led to increased environmental reactivity and emotional lability (Wei et al., 2004), mimicking some aspects of bipolar illness. Interestingly, bipolar subjects also exhibit abnormalities in negative steroid feedback (Watson et al., 2004, 2007), along with deficits in cognitive function that appear to be GR mediated (Watson et al., 2006) and are ameliorated after treatment with a GR antagonist (Young et al., 2004). Thus, the GRov mouse represents a model of increased vulnerability to the impact of the environment on neural function, a feature likely to play a role in many brain-related diseases, including mood and aging-related disorders.
Footnotes
This work was supported by National Institutes of Health Grants 5 P01 MH42251 (S.J.W., H.A.) and R01 DA13386 (H.A.), the Nancy Pritzker Network for Research on Depression, and the Bristol Myers Squibb Unrestricted Research Award (H.A.).
References
- Akil H. Stressed and depressed. Nat Med. 2005;11:116–118. doi: 10.1038/nm0205-116. [DOI] [PubMed] [Google Scholar]
- Akil H, Morano M. Stress. In: Bloom F, Kupfer D, editors. Psychopharmacology: the fourth generation of progress. New York: Raven; 1995. pp. 773–785. [Google Scholar]
- Boyle MP, Brewer JA, Funatsu M, Wozniak DF, Tsien JZ, Izumi Y, Muglia LJ. Acquired deficit of forebrain glucocorticoid receptor produces depression-like changes in adrenal axis regulation and behavior. Proc Natl Acad Sci USA. 2005;102:473–478. doi: 10.1073/pnas.0406458102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle MP, Kolber BJ, Vogt SK, Wozniak DF, Muglia LJ. Forebrain glucocorticoid receptors modulate anxiety-associated locomotor activation and adrenal responsiveness. J Neurosci. 2006;26:1971–1978. doi: 10.1523/JNEUROSCI.2173-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi A, Moffitt TE. Gene-environment interactions in psychiatry: joining forces with neuroscience. Nat Rev Neurosci. 2006;7:583–590. doi: 10.1038/nrn1925. [DOI] [PubMed] [Google Scholar]
- Cole MA, Kim PJ, Kalman BA, Spencer RL. Dexamethasone sup-pression of corticosteroid secretion: evaluation of the site of action by receptor measures and functional studies. Psychoneuroendocrinology. 2000;25:151–167. doi: 10.1016/s0306-4530(99)00045-1. [DOI] [PubMed] [Google Scholar]
- Conrad CD, Lupien SJ, McEwen BS. Support for a bimodal role for type II adrenal steroid receptors in spatial memory. Neurobiol Learn Mem. 1999;72:39–46. doi: 10.1006/nlme.1998.3898. [DOI] [PubMed] [Google Scholar]
- Cullinan WE, Herman JP, Watson SJ. Ventral subicular interaction with the hypothalamic paraventricular nucleus: evidence for a relay in the bed nucleus of the stria terminalis. J Comp Neurol. 1993;332:1–20. doi: 10.1002/cne.903320102. [DOI] [PubMed] [Google Scholar]
- Dai M, Wang P, Boyd AD, Kostov G, Athey B, Jones EG, Bunney WE, Myers RM, Speed TP, Akil H, Watson SJ, Meng F. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005;33:e175. doi: 10.1093/nar/gni179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallman MF, Yates FE. Dynamic asymmetries in the corticosteroid feedback path and distribution-metabolism-binding elements of the adrenocortical system. Ann NY Acad Sci. 1969;156:696–721. doi: 10.1111/j.1749-6632.1969.tb14008.x. [DOI] [PubMed] [Google Scholar]
- De Kloet ER, Vreugdenhil E, Oitzl MS, Joels M. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19:269–301. doi: 10.1210/edrv.19.3.0331. [DOI] [PubMed] [Google Scholar]
- De Kloet ER, Joels M, Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci. 2005;6:463–475. doi: 10.1038/nrn1683. [DOI] [PubMed] [Google Scholar]
- Diamond DM, Bennett MC, Fleshner M, Rose GM. Inverted-U relationship between the level of peripheral corticosterone and the magnitude of hippocampal primed burst potentiation. Hippocampus. 1992;2:421–430. doi: 10.1002/hipo.450020409. [DOI] [PubMed] [Google Scholar]
- Geiger JR, Melcher T, Koh DS, Sakmann B, Seeburg PH, Jonas P, Monyer H. Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron. 1995;15:193–204. doi: 10.1016/0896-6273(95)90076-4. [DOI] [PubMed] [Google Scholar]
- Hebda-Bauer EK, Watson SJ, Akil H. Cognitive performance is highly sensitive to prior experience in mice with a learning and memory deficit: failure leads to more failure. Learn Mem. 2005;12:461–471. doi: 10.1101/lm.94105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman JP, Cullinan WE. Neurocircuitry of stress: central control of the hypothalamo-pituitary-adrenocortical axis. Trends Neurosci. 1997;20:78–84. doi: 10.1016/s0166-2236(96)10069-2. [DOI] [PubMed] [Google Scholar]
- Herman JP, Schafer MK, Young EA, Thompson R, Douglass J, Akil H, Watson SJ. Evidence for hippocampal regulation of neuroendocrine neurons of the hypothalamo-pituitary-adrenocortical axis. J Neurosci. 1989;9:3072–3082. doi: 10.1523/JNEUROSCI.09-09-03072.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman JP, Figueiredo H, Mueller NK, Ulrich-Lai Y, Ostrander MM, Choi DC, Cullinan WE. Central mechanisms of stress integration: hierarchical circuitry controlling hypothalamo-pituitary-adrenocortical responsiveness. Front Neuroendocrinol. 2003;24:151–180. doi: 10.1016/j.yfrne.2003.07.001. [DOI] [PubMed] [Google Scholar]
- Ising M, Kunzel HE, Binder EB, Nickel T, Modell S, Holsboer F. The combined dexamethasone/CRH test as a potential surrogate marker in depression. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:1085–1093. doi: 10.1016/j.pnpbp.2005.03.014. [DOI] [PubMed] [Google Scholar]
- Jacobson L, Sapolsky R. The role of the hippocampus in feedback regulation of the hypothalamic-pituitary-adrenocortical axis. Endocr Rev. 1991;12:118–134. doi: 10.1210/edrv-12-2-118. [DOI] [PubMed] [Google Scholar]
- Jones MT, Brush FR, Neame RL. Characteristics of fast feedback control of corticotrophin release by corticosteroids. J Endocrinol. 1972;55:489–497. doi: 10.1677/joe.0.0550489. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Diamond DM. The stressed hippocampus, synaptic plasticity and lost memories. Nat Rev Neurosci. 2002;3:453–462. doi: 10.1038/nrn849. [DOI] [PubMed] [Google Scholar]
- Landfield PW, Waymire JC, Lynch G. Hippocampal aging and adrenocorticoids: quantitative correlations. Science. 1978;202:1098–1102. doi: 10.1126/science.715460. [DOI] [PubMed] [Google Scholar]
- Liu D, Diorio J, Tannenbaum B, Caldji C, Francis D, Freedman A, Sharma S, Pearson D, Plotsky PM, Meaney MJ. Maternal care, hippocampal glucocorticoid receptors, and hypothalamic-pituitary-adrenal responses to stress. Science. 1997;277:1659–1662. doi: 10.1126/science.277.5332.1659. [DOI] [PubMed] [Google Scholar]
- Lopez JF, Akil H, Watson SJ. Neural circuits mediating stress. Biol Psychiatry. 1999;46:1461–1471. doi: 10.1016/s0006-3223(99)00266-8. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Stress, adaptation, and disease: allostasis and allostatic load. Ann NY Acad Sci. 1998;840:33–44. doi: 10.1111/j.1749-6632.1998.tb09546.x. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Sapolsky RM. Stress and cognitive function. Curr Opin Neurobiol. 1995;5:205–216. doi: 10.1016/0959-4388(95)80028-x. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Morano MI, Vazquez DM, Akil H. The role of the hippocampal mineralocorticoid and glucocorticoid receptors in the hypothalamo-pituitary-adrenal axis of the aged Fisher rat. Mol Cell Neurosci. 1994;5:400–412. doi: 10.1006/mcne.1994.1050. [DOI] [PubMed] [Google Scholar]
- Oitzl MS, De Kloet ER. Selective corticosteroid antagonists modulate specific aspects of spatial orientation learning. Behav Neurosci. 1992;106:62–71. doi: 10.1037//0735-7044.106.1.62. [DOI] [PubMed] [Google Scholar]
- Pepin MC, Pothier F, Barden N. Impaired type II glucocorticoid-receptor function in mice bearing antisense RNA transgene. Nature. 1992;355:725–728. doi: 10.1038/355725a0. [DOI] [PubMed] [Google Scholar]
- Reichardt HM, Kaestner KH, Tuckermann J, Kretz O, Wessely O, Bock R, Gass P, Schmid W, Herrlich P, Angel P, Schutz G. DNA binding of the glucocorticoid receptor is not essential for survival. Cell. 1998;93:531–541. doi: 10.1016/s0092-8674(00)81183-6. [DOI] [PubMed] [Google Scholar]
- Ridder S, Chourbaji S, Hellweg R, Urani A, Zacher C, Schmid W, Zink M, Hortnagl H, Flor H, Henn FA, Schutz G, Gass P. Mice with genetically altered glucocorticoid receptor expression show altered sensitivity for stress-induced depressive reactions. J Neurosci. 2005;25:6243–6250. doi: 10.1523/JNEUROSCI.0736-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapolsky RM. Cambridge, MA: MIT; 1992. Stress, the aging brain and the mechanisms of neuron death. [Google Scholar]
- Sapolsky RM, Krey LC, McEwen BS. The adrenocortical stress-response in the aged male rat: impairment of recovery from stress. Exp Gerontol. 1983;18:55–64. doi: 10.1016/0531-5565(83)90051-7. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM, Krey LC, McEwen BS. The adrenocortical axis in the aged rat: impaired sensitivity to both fast and delayed feedback inhibition. Neurobiol Aging. 1986;7:331–335. doi: 10.1016/0197-4580(86)90159-4. [DOI] [PubMed] [Google Scholar]
- Segovia G, Porras A, Del Arco A, Mora F. Glutamatergic neurotransmission in aging: a critical perspective. Mech Ageing Dev. 2001;122:1–29. doi: 10.1016/s0047-6374(00)00225-6. [DOI] [PubMed] [Google Scholar]
- Stead JD, Neal C, Meng F, Wang Y, Evans S, Vazquez DM, Watson SJ, Akil H. Transcriptional profiling of the developing rat brain reveals that the most dramatic regional differentiation in gene expression occurs postpartum. J Neurosci. 2006;26:345–353. doi: 10.1523/JNEUROSCI.2755-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schutz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- Watson S, Gallagher P, Ritchie JC, Ferrier IN, Young AH. Hypothalamic-pituitary-adrenal axis function in patients with bipolar disorder. Br J Psychiatry. 2004;184:496–502. doi: 10.1192/bjp.184.6.496. [DOI] [PubMed] [Google Scholar]
- Watson S, Thompson JM, Ritchie JC, Nicol Ferrier I, Young AH. Neuropsychological impairment in bipolar disorder: the relationship with glucocorticoid receptor function. Bipolar Disord. 2006;8:85–90. doi: 10.1111/j.1399-5618.2006.00280.x. [DOI] [PubMed] [Google Scholar]
- Watson S, Gallagher P, Smith MS, Nicol Ferrier I, Young AH. The dex/CRH test and the DST in patients with bipolar disorder and major depressive disorder. Psychoneuroendocrinology. 2007;32:92–94. [Google Scholar]
- Wei Q, Lu XY, Liu L, Schafer G, Shieh KR, Burke S, Robinson TE, Watson SJ, Seasholtz AF, Akil H. Glucocorticoid receptor overexpression in forebrain: a mouse model of increased emotional lability. Proc Natl Acad Sci USA. 2004;101:11851–11856. doi: 10.1073/pnas.0402208101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidenfeld J, Feldman S. Glucocorticoid feedback regulation of adrenocortical responses to neural stimuli: role of CRF-41 and corticosteroid type I and type II receptors. Neuroendocrinology. 1993;58:49–56. doi: 10.1159/000126511. [DOI] [PubMed] [Google Scholar]
- Young AH, Gallagher P, Watson S, Del-Estal D, Owen BM, Ferrier IN. Improvements in neurocognitive function and mood following adjunctive treatment with mifepristone (RU-486) in bipolar disorder. Neuropsychopharmacology. 2004;29:1538–1545. doi: 10.1038/sj.npp.1300471. [DOI] [PubMed] [Google Scholar]
- Young EA, Vazquez D. Hypercortisolemia, hippocampal glucocorticoid receptors, and fast feedback. Mol Psychiatry. 1996;1:149–159. [PubMed] [Google Scholar]
- Young EA, Haskett RF, Murphy-Weinberg V, Watson SJ, Akil H. Loss of glucocorticoid fast feedback in depression. Arch Gen Psychiatry. 1991;48:693–699. doi: 10.1001/archpsyc.1991.01810320017003. [DOI] [PubMed] [Google Scholar]