

Abstract
Purinergic P2Y receptors are one of four types of Gq/11-coupled receptors in rat superior cervical ganglia (SCG) sympathetic neurons. In cultured SCG neurons, purinergic and bradykinin suppression of IM were similar in magnitude and somewhat less than that by muscarinic agonists. The effects of the P2Y receptor agonist UTP on neuronal excitability and discharge properties were studied. Under current clamp, UTP increased action potential (AP) firing in response to depolarizing current steps, depolarized the resting potential, decreased the threshold current required to fire an AP, and decreased spike-frequency adaptation. These effects were very similar to those resulting from bradykinin stimulation and not as profound as from muscarinic stimulation or full M-current blockade. We then examined the P2Y mechanism of action. Like bradykinin, but unlike muscarinic, purinergic stimulation induced rises in intracellular [Ca2+]i. Tests using expression of IP3“sponge” or IP3 phosphatase constructs implicated IP3 accumulation as necessary for purinergic suppression of IM. Overexpression of wild-type or dominant-negative calmodulin (CaM) implicated Ca2+/CaM in the purinergic action. Both sets of results were similar to bradykinin, and opposite to muscarinic, suppression. We also examined modulation of Ca2+ channels. As for bradykinin, purinergic stimulation did not suppress ICa, unless neuronal calcium sensor-1 (NCS-1) activity was blocked by a dominant-negative NCS-1 construct. Our results indicate that P2Y receptors modulate M-type channels in SCG cells via IP3-mediated [Ca2+]i signals in concert with CaM and not by depletion of phosphatidylinositol-4, 5-biphosphate. We group purinergic P2Y and bradykinin B2 receptors together as having a common mode of action.
Keywords: M current, calcium channel, purinergic receptor, PLC signaling, G-protein, patch clamp, sympathetic neuron
Introduction
The M-type K+ current is a prime regulator of neuronal excitability wherever its constituent Kv7 (KCNQ) gene products are expressed (Cooper and Jan, 2003). M-current suppression thus causes increased action potential (AP) firing and bursting, whereas its augmentation inhibits neuronal discharge (Wang et al., 1998; Peretz et al., 2005; Gamper et al., 2006; Yue and Yaari, 2006; Zaika et al., 2006). Although the critical regulatory action of transmitters on M-type channels centers on binding to membrane phosphatidylinositol 4,5-bisphosphate (PIP2), two major modes of inhibition of M-type channels by stimulation of Gq/11-coupled receptors have emerged that use partly distinct, and partly shared, intracellular mechanisms. Both involve activation of phospholipase C (PLC) and hydrolysis of PIP2 but differ in the use of IP3-mediated release of Ca2+ from stores, subsequent [Ca2+]i signals and calmodulin action (Delmas and Brown, 2005). Four Gq/11-coupled receptors have been characterized in superior cervical ganglion (SCG) sympathetic neurons: muscarinic M1, angiotensin AT1, bradykinin (BK) B2, and purinergic P2Y. Muscarinic and angiotensin stimulation do not elicit [Ca2+]i signals and suppress M current by PIP2 depletion (Shapiro et al., 1994; Suh and Hille, 2002; Zhang et al., 2003; Zaika et al., 2006). Conversely, bradykinin suppression involves Ca2+/CaM binding to the channels (Gamper and Shapiro, 2003), which may lower their affinity for PIP2, but such B2 receptor stimulation does not strongly lower global PIP2 abundance (Gamper et al., 2004; Winks et al., 2005; Gamper and Shapiro, 2007; Hughes et al., 2007). High-threshold CaV2 channels are also sensitive to membrane PIP2 abundance and are inhibited during PIP2 depletion (Wu et al., 2002; Gamper et al., 2004). Consistent with specificity among Gq/11-coupled receptors for inducing PIP2 depletion, stimulation of M1 and AT1, but not B2, receptors suppresses endogenous ICa and heterologously expressed Kir3 current in SCG cells (Gamper et al., 2004; Winks et al., 2005).
Purinergic receptors, classified as P2X ionotropic and P2Y metabotropic, are found throughout the nervous system (Burnstock, 2006). Although the P2Y1, P2Y2, and P2Y6 subtypes have been described in mouse and rat sympathetic ganglia (Vartian et al., 2001; Calvert et al., 2004), P2Y6 seems to be primary in rat SCG (Bofill-Cardona et al., 2000), whereas the P2Y1 subtype dominates in mammalian CNS (Moore et al., 2001). In many neuronal types, P2Y receptor stimulation depresses M current via PLC activation, but the intracellular mechanism used from that point on appears intriguingly divergent. In the rat SCG cells also studied here, the action involves intracellular Ca2+ signals (Bofill-Cardona et al., 2000) but does not in frog ganglia (Stemkowski et al., 2002). Moreover, in rat hippocampus, the P2Y1 receptor-mediated suppression of IM is not associated with [Ca2+]i signals and is suggested to be via PIP2 depletion (Filippov et al., 2006). In this work, we systematically investigate the action of P2Y receptor stimulation on neuronal excitability and discharge properties of rat SCG cells and its mode of action on the endogenous IM and ICa in those cells. The purinergic signals are compared with those from muscarinic and bradykinin stimulation and show the purinergic action to increase neuronal excitability via IP3-mediated [Ca2+]i signals.
Materials and Methods
SCG neuron culture and transduction.
Sympathetic neurons were isolated from the SCG of 7- to 14-d-old male rats (Sprague Dawley) and cultured for 2–4 d. Rats were anesthetized with halothane and decapitated. Neurons were dissociated using methods of Bernheim et al. (1991), plated on 4 × 4 mm glass coverslips (coated with poly-l-lysine) and incubated at 37°C (5% CO2). Fresh culture medium containing nerve growth factor (50 ng/ml) was added to the cells 3 h after plating. In some experiments, pertussis toxin (PTX) (100 ng/ml) was added to the culture medium. For exogenous expression of cDNA constructs in SCG neurons, we used the PDS-1000/He biolistic particle delivery system (“gene gun”; Bio-Rad, Hercules, CA), as described recently (Gamper and Shapiro, 2006). Transfection efficiency was assumed to be determined by the random distribution of fired gold particles and was up to 10% of cultured neurons.
Perforated-patch electrophysiology.
Pipettes were pulled from borosilicate glass capillaries (1B150F-4; World Precision Instruments, Sarasota, FL) using a Flaming/Brown micropipette puller P-97 (Sutter Instruments, Novato, CA) and had resistances of 1–4 MΩ when filled with internal solution and measured in standard bath solution. Membrane current was measured with pipette and membrane capacitance cancellation, sampled at 5 ms or 200 μs, and filtered at 1 or 2.5 kHz (for IM and ICa, respectively) by an EPC-9 amplifier and PULSE software (HEKA/InstruTech, Port Washington, NY). In all experiments, the perforated-patch method of recording was used with amphotericin B (600 ng/ml) in the pipette (Rae et al., 1991). Amphotericin was prepared as a stock solution as 60 mg/ml in DMSO. In these experiments, the access resistance was typically 10 MΩ 5–10 min after seal formation. Cells were placed in a 500 μl perfusion chamber through which solution flowed at 1–2 ml/min. Inflow to the chamber was by gravity from several reservoirs, selectable by activation of solenoid valves (Warner Instruments, Hamden, CT). Bath solution exchange was essentially complete by <30 s. Experiments were performed at room temperature.
M currents in SCG cells were studied by holding the membrane potential at −25 mV and applying a 500 ms hyperpolarizing pulse to −60 mV every 3 s. M-current amplitude was measured at −60 mV from the decaying time course of the deactivating current sensitive to the M-channel-specific blocker XE991 [10,10-bis(4-pyridinylmethyl)-9(10H)-anthracenone] (Zaczek et al., 1998). UTP, BK, oxotremorine-M (oxo-M), and XE991 were used at concentrations of 10 μm, 250 nm, 10 μm, and 50 μm, respectively. To evaluate the amplitude of ICa, cells were held at −80 mV, and 15 ms depolarizing steps to 5 mV were applied every 5 s. The amplitude of ICa was usually defined as the inward current sensitive to Cd2+ (100 μm). The external solution used to record M currents contained the following: 150 mm NaCl, 5 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 10 mm glucose, 10 mm HEPES, and 500 nm tetrodotoxin, pH 7.4 with NaOH. For ICa recordings, KCl was reduced to 2.5 mm, and CaCl2 was increased to 5 mm. The pipette solution for voltage-clamp experiments contained the following (in mm): 150 KCl/CsCl, 5 MgCl2, and 10 HEPES to record IM/ICa. For current-clamp experiments, the pipette solution contained the following (in mm): 20 KCl, 100 K-acetate, 5 MgCl2, and 40 HEPES. Data are presented as mean ± SEM. Statistical tests were performed using paired t test or unpaired t test when appropriate.
Imaging.
Fluorescent microscopy was performed with an inverted Nikon (Tokyo, Japan) Eclipse TE300 microscope with an oil-immersion 40×/1.30 numerical aperture objective. A Polychrome IV monochromator (T.I.L.L. Photonics, Martinsreid, Germany) was used as the excitation light source, and a FURA2 71000 filter cube (Chroma Technology, Brattleboro, VT) was used for fura-2 imaging. SCG neurons were bath loaded with fura-2 AM (2 μm) for 30 min at 37°C in the presence of pluronic acid (0.01%). Cells were excited alternatively at 340 and 380 nm (50–200 ms every 2 s), the fluorescence emission was collected by an IMAGO 12-bit cooled CCD camera, and images were stored/analyzed with TILLvisION 4.0 software. These fura-2 signals were not calibrated because of inherent difficulties in calibrating esterified indicator dyes (Zhou and Neher, 1993).
Computer modeling.
Simulations were performed using NEURON version 5.9 (Hines and Carnevale, 2001) on a Macintosh G4 computer (Apple Computers, San Jose, CA). The passive properties of a single compartment model (50 μm diameter) were chosen to reflect the input resistance and charging time constant of SCG neurons to hyperpolarizing current steps (specific membrane resistivity, 50,000 Ωcm2; specific capacitance, 1 μF/cm2). Four voltage-gated and two Ca2+-dependent conductances (available for download through ModelDB at http://senselab.med.yale.edu) were inserted into the model. A fast voltage-gated Na+ conductance (nahh.mod) (Traub et al., 1991) was inserted with a density of 5 mS/cm2. Threshold was adjusted by shifting the voltage dependence of the forward and backward activation rate constants by +20 and +10 mV, respectively. Spike repolarization was achieved with a delayed rectifier K+ conductance (kdr.mod) (Migliore et al., 1995) at a density of 0.3 mS/cm2 (EK of −80 mV). A high-threshold, non-inactivating voltage-gated Ca2+ conductance (cal.mod) (Migliore et al., 1995) at a density of 2 mS/cm2 was inserted with ECa determined by the Goldman–Hodgkin–Katz equation. Intracellular calcium dynamics were modeled including both rapid buffering and Ca2+ pumps (cadifus.mod). Two Ca2+-dependent K+ conductances were included in the model. A BK-type Ca2+-dependent conductance (cagk.mod) at a density of 0.1 mS/cm2 contributed mostly to spike repolarization, whereas an SK-type Ca2+-dependent conductance (gSK of 3.9 · [Ca2+] mS/cm2) contributed mostly to spike-frequency adaptation (SFA). Finally, the M-channel conductance (gM) (im.mod) (Migliore et al., 1995) was added with a density of 0.0175 mS/cm2. It was assumed that the M current was active at rest and therefore contributed to the resting potential of the cell. Therefore, the equilibrium potential of the leak conductance was determined with no M current resulting for an initial resting potential (Vrest) of −50 mV. When gM was active, Vrest in the model became −55 mV.
DNA constructs.
Plasmids containing wild-type (wt) or dominant-negative (DN) vertebrate CaM were given to us by Trisha Davis (University of Washington, Seattle, WA). DN CaM has an alanine substitution in each of the four Ca2+-binding EF hands (D20A, D56A, D93A, and D129A). DN neuronal calcium sensor-1 was given to us by Andreas Jeromin (Mt. Sinai Hospital, Toronto, Ontario, Canada). The IP3 sponges consist of residues 224–604 of the rat type 1 IP3 receptor containing either the R441Q (“high-affinity sponge”) or K508A (“low-affinity sponge”) mutations, made by PCR amplification and given to us by Llewelyn Roderick (Babraham Institute, Cambridge, UK).
Reagents.
The following reagents were used: UTP, TTX, NGF, bradykinin, oxo-M, and collagenase type I (Sigma, St. Louis, MO); fura-2 AM (Invitrogen, Carlsbad, CA); DMEM, fetal bovine serum, and penicillin/streptomycin (Invitrogen); amphotericin B and PTX (Calbiochem, La Jolla, CA); and XE991 (Tocris Bioscience, Ellisville, MO).
Results
We first evaluated the effect of purinergic stimulation on the M current in cultured SCG neurons and compared it with that of muscarinic and BK stimulation. Such a comparison between the effects of stimulating these receptor types was made throughout this study, because the muscarinic and bradykinin actions are the prototypes for the two described mechanisms of Gq/11-mediated suppression of IM, the first being via PIP2 depletion and second via IP3, [Ca2+]i signals, and CaM (Delmas and Brown, 2005). Cells were studied under perforated-patch voltage clamp, and agonists were applied by bath perfusion. Guided by previous work on P2Y receptor-mediated modulation of M current (IM) in these cells (Boehm, 1998; Bofill-Cardona et al., 2000), we chose to use UTP as the purinergic agonist in this study. UTP is not an agonist of the ionotropic P2X receptors in the SCG (Calvert and Evans, 2004). Application of UTP caused a significant suppression of IM in approximately one-half and BK in approximately three-fourths of the neurons examined, which we assume to reflect that proportion of neurons that express UTP-sensitive P2Y or BK receptors, respectively. There were no morphological differences between cells that did or did not respond to UTP. For those neurons that responded to UTP with a suppression of IM >5%, we compared the response with those from bradykinin and the muscarinic agonist oxo-M. Application of UTP, BK, or oxo-M suppressed IM by 54 ± 5% (n = 6), 62 ± 6% (n = 6), and 81 ± 6% (n = 6), respectively (Fig. 1). These values are consistent with those reported previously, although the extent of the purinergic suppression is slightly greater than that seen previously (Bofill-Cardona et al., 2000).
Figure 1.
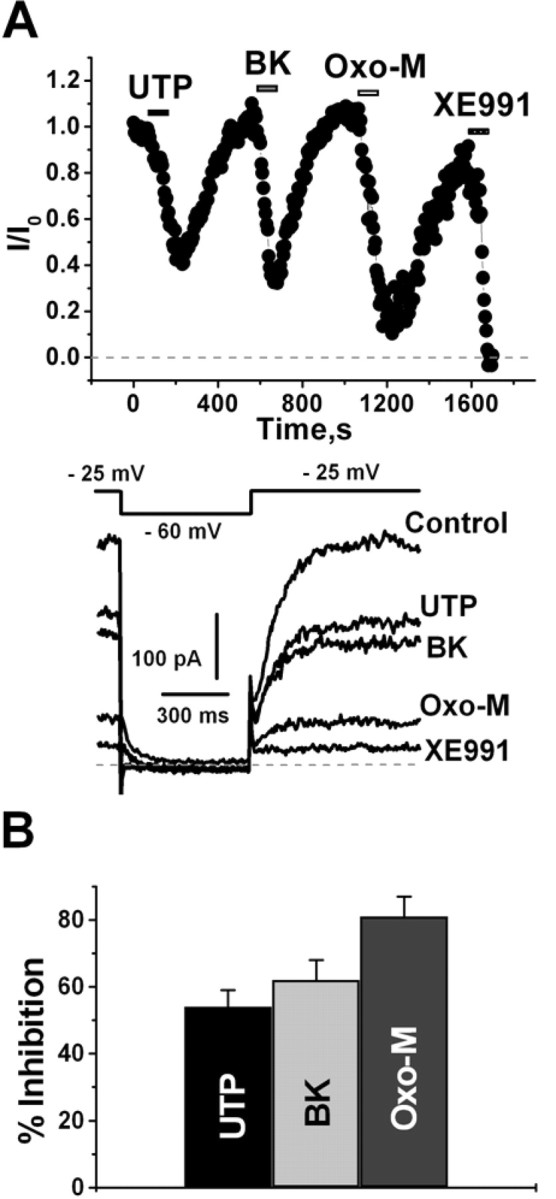
Purinergic stimulation suppresses M current in SCG neurons. Purinergic inhibition of M current recorded from cultured SCG neurons using the pulse protocol depicted in the inset. A, Plotted are normalized M-current amplitudes, quantified as the deactivating time-dependent relaxations at −60 mV from pulses given every 3 s. UTP (10 μm), BK (250 nm), oxo-M (10 μm), or XE991 (50 μm) were bath applied during the periods shown by the bars and representative current traces shown below. B, Bars show mean inhibitions of IM by UTP, BK, or oxo-M.
Purinergic and bradykinin stimulation increases neuronal excitability and decreases spike-frequency adaptation
Given the widely demonstrated control of neuronal excitability by M-current activity (Jones et al., 1995; Gu et al., 2005; Peretz et al., 2005; Peters et al., 2005; Shen et al., 2005; Gamper et al., 2006; Vervaeke et al., 2006; Yue and Yaari, 2006; Zaika et al., 2006), we next assessed the effects of purinergic stimulation on AP firing in our preparation of cultured SCG cells. We also systematically evaluated the effects of BK stimulation, whose actions on neuronal discharge have only been briefly described (Jones et al., 1995). Using perforated-patch current-clamp recording, we evaluated both the active and passive properties of the neurons in response to current steps in the presence or absence of UTP and BK. We compared the effects of UTP and BK with the well described M-current-mediated effects of muscarinic agonists or M-channel blockade. PTX was not used, meaning that Gq/11-mediated actions are not isolated in these experiments. Because our initial voltage-clamp experiments suggested that only approximately half of the neurons express the P2Y receptor, we triaged the cells studied under current clamp so as to characterize only P2Y receptor-expressing neurons. For each neuron studied, the resting potential (Vrest) was monitored for 10 min before application of any agonist. During application of UTP, if Vrest changed outside of the range seen during that 10 min control period, we assumed that neuron to express P2Y receptors, and the cell was studied further. As for the voltage-clamp experiments, approximately half of the neurons satisfied that criterion.
Shown in Figure 2 are representative voltage recordings in response to positive or negative current steps. In control, a 100 pA current injection elicited a short train of APs that terminated well before the end of the current step (Fig. 2A). Application of UTP caused a reversible increase in the number of APs elicited by the 100 pA current step, consistent with suppression of M current (Fig. 2B,C). Subsequent application of BK also caused a reversible increase in AP firing that was slightly greater than that caused by UTP (Fig. 2D,E). Finally, the response to muscarinic stimulation was a spike train that was nearly as intense as that caused by total blockade of M channels by the selective blocker XE991 (Fig. 2F,G). In control, the number of APs elicited by the 2 s, 100 pA depolarizing current was 4.1 ± 1.2 (n = 14). Application of UTP, BK, oxo-M, or XE991 increased the number of such APs to 7.8 ± 0.9 (p < 0.01; n = 9), 8.7 ± 0.8 (p < 0.01; n = 11), 20.2 ± 0.8 (p < 0.001; n = 12), or 22.0 ± 1.8 (p < 0.001; n = 12), respectively. There was not a significant change in the responses to hyperpolarizing current steps, ruling out an effect of the agonists on inward conductances such as Ih.
Figure 2.
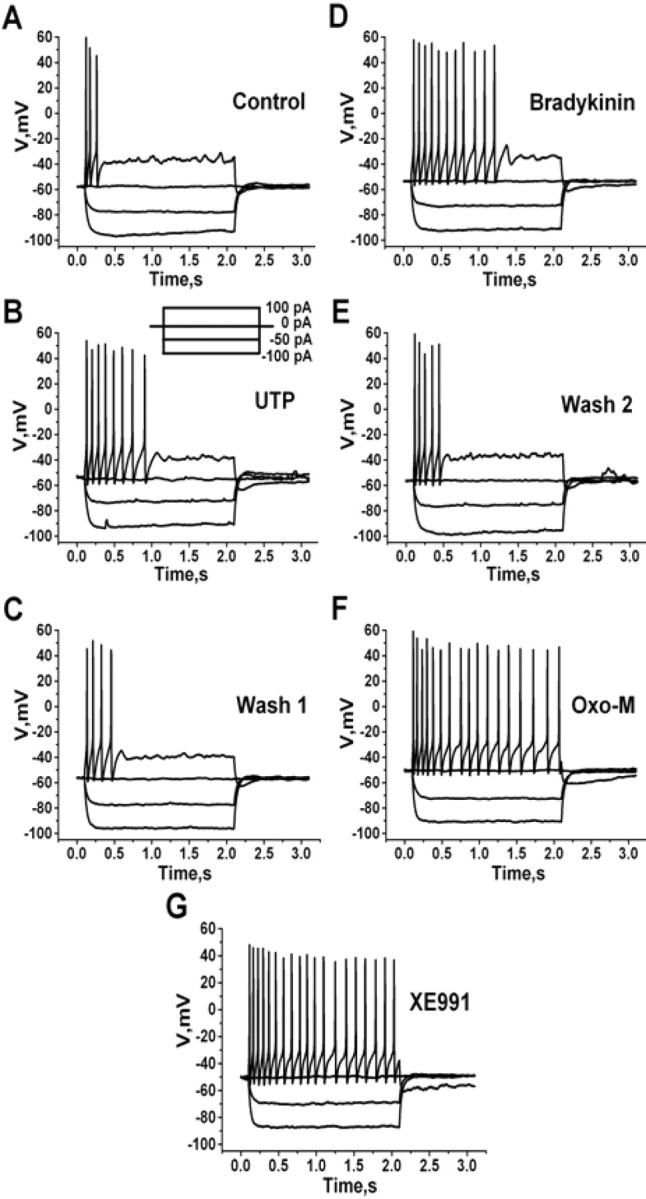
Purinergic stimulation increases action potential firing in SCG neurons. Cultured SCG neurons were studied under perforated-patch current clamp, and voltages were recorded from a family of current pulses (inset). Voltage sweeps are shown in control (A), after UTP (10 μm) (B), first wash (C), after BK (250 nm) (D), second wash (E), after oxo-M (10 μm) (F), or after XE991 (50 μm) (G).
The effect of receptor stimulation on Vrest was quantified as an initial indictor of control over excitability (Fig. 3A). In control, Vrest was −56.2 ± 0.6 mV (n = 14). Application of UTP or BK modestly depolarized Vrest to −53.7 ± 0.4 mV (p < 0.01; n = 9) and −53.1 ± 0.6 mV (p < 0.01; n = 11), respectively. In some experiments, we tested the effect of BK applied first to rule out any residual effect of previous UTP application, and, in those cells, the initial Vrest was −55.7 ± 0.3 mV, which was depolarized by BK to −53.4 ± 0.1 mV (p < 0.01; n = 12), similar to the effect of BK in cells in which UTP was applied first. Application of oxo-M induced a stronger depolarization of Vrest, to −50.9 ± 0.4 mV (p < 0.01; n = 12), nearly as much as that induced by XE991 (−50.0 ± 0.6 mV; p < 0.01; n = 12). We further quantified the effects of stimulation of Gq/11-coupled receptors on excitability by examining the relationship between injected current and AP firing (Fig. 3B,C). In control, the amplitude of the current required to reach threshold for AP generation (APthresh) was 28.8 ± 0.4 pA (n = 20). Application of UTP or BK modestly decreased APthresh to 25.9 ± 1.7 pA (p < 0.05; n = 9) and 24.1 ± 2.1 pA (p < 0.05; n = 11), respectively. Application of oxo-M reduced APthresh more strongly, to 22.1 ± 1.7 pA (p < 0.01; n = 12), nearly as much as from full M-channel blockade by XE991 (21.1 ± 2.3 pA; p < 0.01; n = 12). Evaluated over a range of current amplitudes, UTP and BK modestly, but significantly, increased the number of elicited APs at all amplitudes of injected current (Fig. 3C). The effect of muscarinic stimulation was stronger and was nearly as great as that produced by XE991. In summary, purinergic and BK stimulation both suppressed IM by 50–60%, and both modestly, but significantly, increased somatic excitability to a similar degree.
Figure 3.
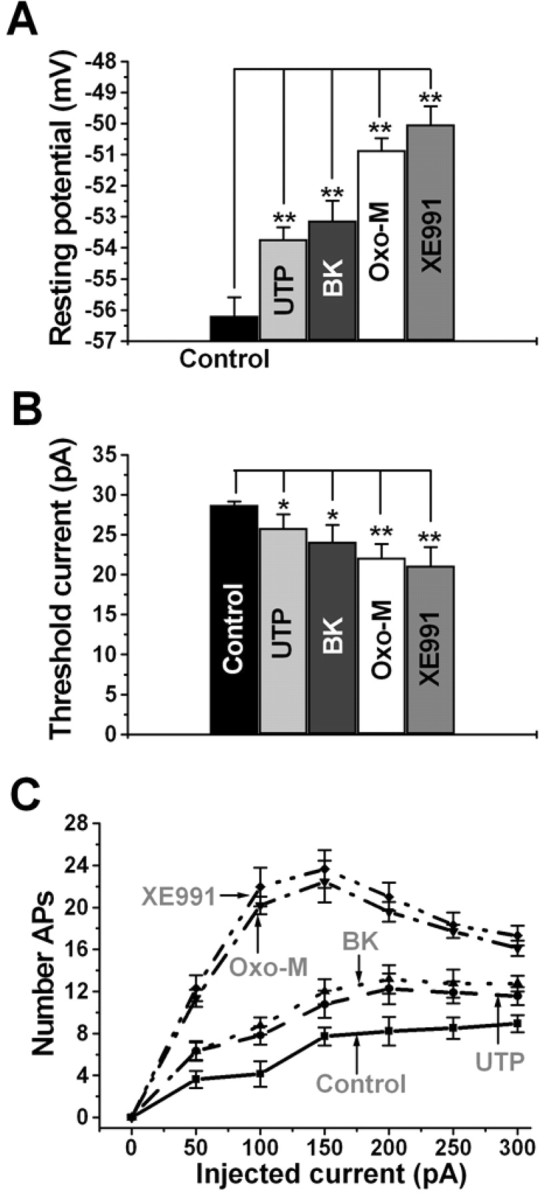
Control of somatic excitability by regulation of M current. Bars show summarized data of the resting potential (A) and the threshold current required for firing of action potentials (B) for SCG neurons in control, after UTP (10 μm), BK (250 nm), oxo-M (10 μm), or XE991 (50 μm). C, Plotted are the number of APs elicited during a 2 s depolarizing current injection over a range of current amplitudes, during the indicated conditions. *p < 0.05; **p < 0.01.
Many neurons, including ganglia cells, exhibit SFA, characterized by the slowing or cessation of AP firing during a maintained stimulatory input. M current has been shown to be a prime contributor to SFA because of its accumulating conductance as the slowly activating M channels turn on during a spike train (Lawrence et al., 2006; Vervaeke et al., 2006; Zaika et al., 2006). We evaluated the effect of P2Y or BK receptor stimulation on SFA during a train of spikes using two related measures: (1) the relationship between interspike interval and spike number, and (2) spike-train duration (Fig. 4A). Spike trains elicited by 100 pA stimulatory currents were analyzed from SCG neurons studied under perforated-patch current clamp. In control, the instantaneous interspike interval rapidly lengthened during the short spike train, which terminated after 0.4 ± 0.1 s (n = 14). In the presence of UTP or BK, the slope of the interspike interval versus spike number relationship was significantly more shallow, and the spike-train duration was lengthened to 1.2 ± 0.1 s (p < 0.01; n = 9) and 1.3 ± 0.1 s (p < 0.01; n = 11), respectively. In accord with its greater effect on IM and on AP firing, muscarinic stimulation more greatly dampened SFA, resulting in a very shallow relationship between interspike interval and spike number, and a spike-train duration that was lengthened to 1.8 ± 0.1 s (p < 0.001; n = 12). Complete blockade of M channels by application of XE991 made that relationship only slightly shallower and resulted in a spike train that lasted throughout the length of the 2 s stimulatory pulse.
Figure 4.
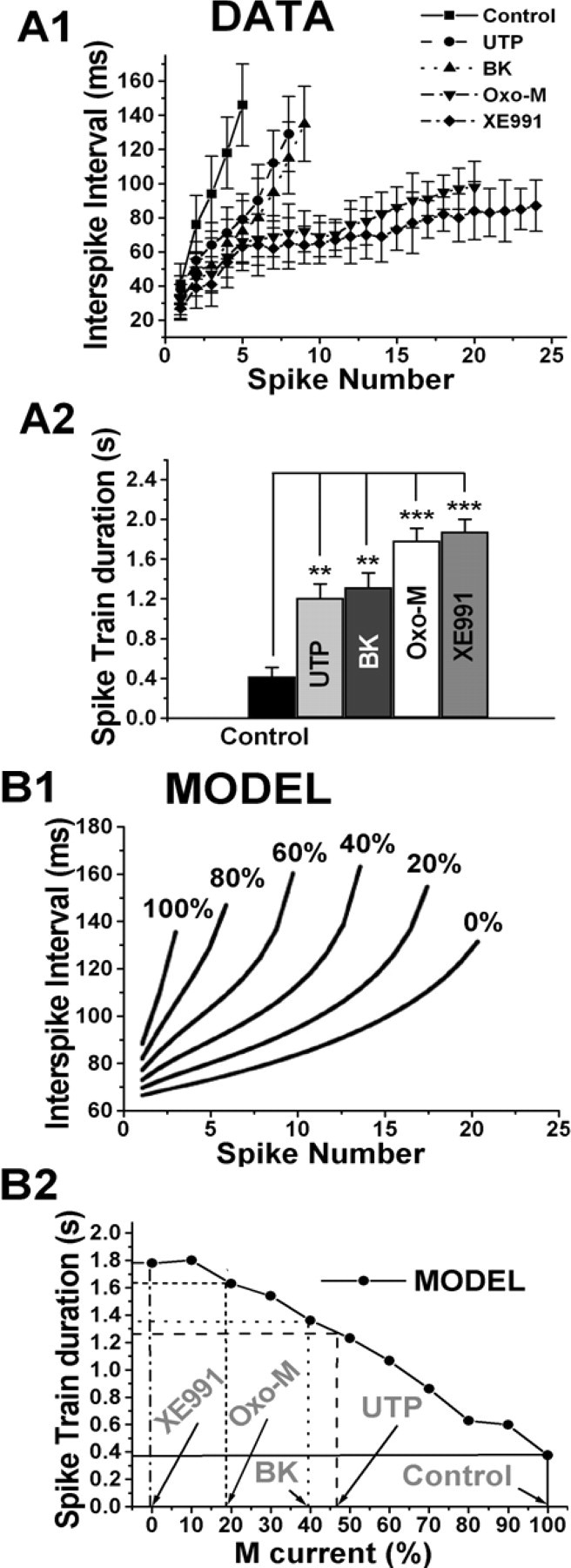
Effects on spike-frequency adaptation and comparison with a computer model of neuronal discharge. Plotted are the instantaneous interspike interval as a function of spike number (A1) and spike-train duration (A2) from spike trains elicited by a 100 pA stimulus applied to SCG neurons under current clamp, in control, after UTP (10 μm), BK (250 nm), oxo-M (10 μm), and XE991 (50 μm). In B1 are shown the simulated curves of interspike interval versus spike number predicted by a neuronal discharge model over the indicated range of fractional M-current activity. In B2 are plotted the simulated dependence of spike-train duration on fractional M-current activity. The observed suppressions of M current by the compounds used in this paper are marked on the abscissa and the predicted spike-train durations on the ordinate for comparison with the data in A2. **p < 0.01, ***p < 0.001.
In a recent paper, we used a multi-conductance neuronal model to successfully predict the effects on AP firing and SFA of suppression of M-channel conductance by angiotensin II (Zaika et al., 2006). The model program, NEURON version 5.7 (Hines and Carnevale, 2001), contains four voltage-gated and two Ca2+-dependent conductances, including gM (Borg-Graham, 1991; Migliore et al., 1995). In the study by Zaika et al. (2006), we included only a BK conductance as a KCa type, but here we also include an SK conductance in the simulation. The values of the six conductances were adjusted to best correlate with the observed behavior of the neurons (for details, see Materials and Methods). In the model, full activity of both SK- and M-type conductances resulted in a response to a depolarizing current of only several APs, but turning off of M current (at first leaving the SK current intact) changed the response to an uninterrupted spike train exhibiting only minimal SFA (data not shown), similar to what we observed (Fig. 2A,G). In Figure 4B are shown the predictions of the model for the interspike interval versus spike number relationship and spike-train duration over a range of fractional gM activation. We examined these model data to see whether the effects on SFA correlate with the degree of suppression of IM for the agonists used in this study. In comparing Figure 4, A1 and B1, one can see that the predicted interspike interval versus spike number relationships at 100, 60, 20, and 0% gM qualitatively match the experimentally observed relationships for control, UTP ≈ BK, oxo-M, and XE991, under which conditions the fractional IM is 100, 38–46, 18, and 0%, respectively. Indicated on the model predictions of spike-train durations are the measured suppressions of IM and the predicted spike-train durations as a result (Fig. 4B2). For control, UTP, BK, oxo-M, and XE991, the predicted spike-train durations are ∼0.4, 1.25, 1.35, 1.65, and 1.8 s, which qualitatively agree with the experimentally observed values (Fig. 4A2).
With the SK conductance included, the model prediction of spike-train duration versus gM includes an inflection in the curve at ∼10% gM, which the model ascribes to an oscillation in the membrane potential from sequential SK-induced hyperpolarization, turning off of M current, depolarization that produces rises in [Ca2+]i via influx through Ca2+ channels, and cycling back to the turning on of SK channels. We observed such membrane oscillations of 2–3 mV in many of our current-clamp recordings, although this phenomenon was not studied further. The reduction of SFA produced by muscarinic stimulation, as measured by the interspike interval versus spike number relationship, was somewhat greater than that predicted by M-current suppression alone, especially at spike numbers >15, in which the model predicts an upward inflection as opposed to the flat relationship actually observed. This is likely attributable to concurrent suppression of ICa by M1 and M4 receptor stimulation and consequential reduction in KCa conductances. Indeed, systematic reduction of ISK in the model reduced SFA for later spikes of a given train. As a result, when gM was 20% of control, reducing ISK or ICa resulted in SFA that approached that of 0% gM, as observed experimentally.
P2Y receptor stimulation induces intracellular Ca2+ signals and requires IP3 for suppression of IM
In SCG neurons, stimulation of bradykinin B2, but not muscarinic M1 nor angiotensin AT1, receptors induce [Ca2+]i signals from IP3-gated stores (Delmas et al., 2005). Although all three receptor types couple to Gq/11 and activate phospholipase C, it is hypothesized that colocalization of B2, but not M1 or AT1, receptors with IP3 receptors in “microdomains” accounts for this selectivity for eliciting [Ca2+]i signals (Delmas and Brown, 2002; Delmas et al., 2002). Previous work has demonstrated P2Y receptor-mediated suppression of IM to be via Gq/11 and to require PLC activity (Bofill-Cardona et al., 2000; Stemkowski et al., 2002). We thus began our investigation of the intracellular mechanism linking P2Y receptor stimulation with suppression of IM by asking whether UTP elicits [Ca2+]i signals in SCG neurons. [Ca2+]i changes were observed using fura-2 loaded into the cells via the bath solution as the AM ester. We tested the ability of UTP, as well as BK and oxo-M, to provoke Ca2+ signals. Only cells in which the resting 340/380 nm ratio was <0.3 were used in these measurements. Although the cells were not voltage clamped, the bath contained TTX to eliminate the possibility of [Ca2+]i rises caused by AP spiking and Ca2+ influx through CaV channels attributable to agonist-induced depolarization of Vrest. Shown in Figure 5A is a representative experiment from two adjoining SCG neurons. The 340/380 nm ratio is shown pseudocolored using the indicated scale. Muscarinic stimulation with oxo-M did not result in a measurable [Ca2+]i signal, but subsequent application of UTP caused an obvious and reversible rise in [Ca2+]i that was not as large as that induced by BK. In such experiments, the resting 340/380 nm ratio was 0.21 ± 0.01 (n = 20), and the rises in the 340/380 nm ratio evoked by application of oxo-M, UTP, and BK were 0.016 ± 0.002 (n = 6), 0.10 ± 0.02 (n = 7), and 0.31 ± 0.03 (n = 15), respectively (Fig. 5B). Our data indicate that, like bradykinin but unlike muscarinic agonists (Cruzblanca et al., 1998), purinergic P2Y receptor agonists induce rises in intracellular Ca2+ in sympathetic ganglia cells.
Figure 5.
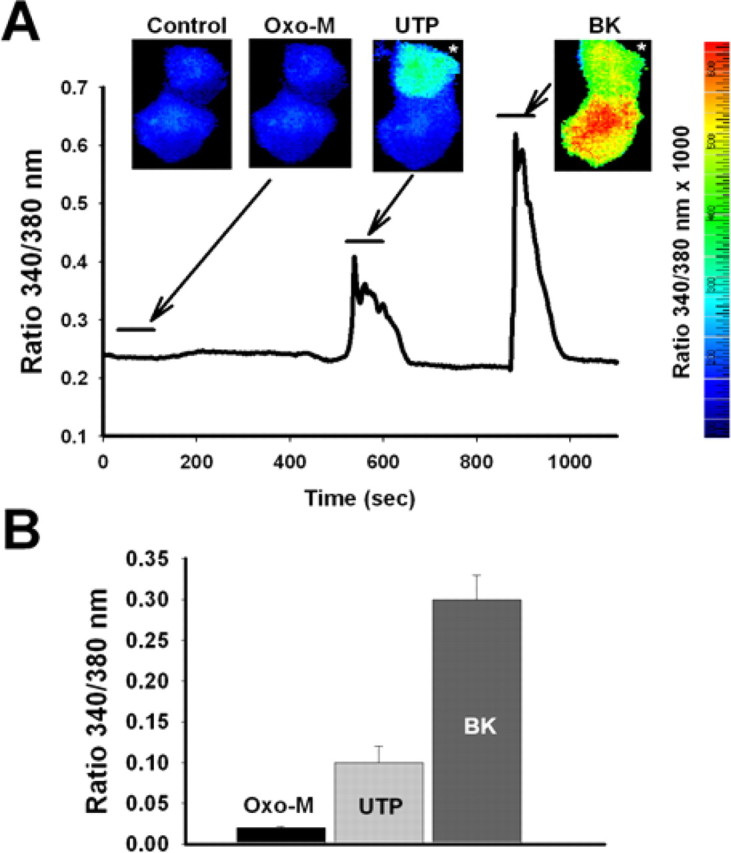
Purinergic stimulation induces intracellular Ca2+ signals. Shown in A is a representative experiment from two SCG neurons loaded with fura-2 dye from the bath as the AM ester. Shown are pseudocolor 340/380 nm ratio images of fura-2 fluorescence from two adjoining neurons in control and during 2 min applications of oxo-M (10 μm), UTP (10 μm), and BK (250 nm). The pseudocolor scale runs from blue (low [Ca2+]) to red (high [Ca2+]). Plotted is the 340/380 nm ratio during the experiment for the topmost neuron (white asterisk). The agonists were applied during the indicated times. Bars in B are the summarized results of these experiments.
P2Y receptor stimulation requires IP3 and functional CaM for suppression of IM
We then probed for the involvement of IP3 in UTP-induced suppression of IM. Our first test used a construct containing a fusion protein of enhanced green fluorescent protein (EGFP) and point mutants of the IP3-binding domain in the N-terminal of the rat type 1 IP3 receptor. A protein consisting only of residues 224–604 has an affinity for IP3 in the range of 92 pm and the R441Q mutant 45 pm, some 1000-fold higher than the native IP3 receptor, whereas the K508A mutant reduces the affinity to only 340 nm. The R441Q, but not the K508A, mutant has been shown to be an effective sponge of IP3 when overexpressed in cells, preventing accumulation of IP3 in the cytoplasm during PLC activation and any downstream [Ca2+]i signal (Uchiyama et al., 2002). Transfection of SCG neurons was performed using a biolistic particle delivery device (gene gun), using recently described procedures (Gamper and Shapiro, 2006), and the neurons were studied the following day under perforated-patch voltage clamp. Figure 6, A and B, shows experiments on neurons transfected with either the K508A low-affinity sponge or the R441Q high-affinity sponge. Whereas the cell overexpressing the low-affinity sponge displayed robust and reversible suppressions of IM by UTP, BK, and oxo-M, the cell overexpressing the high-affinity sponge responded to both UTP and BK with very little suppressions of IM, but the response to oxo-M was unaltered. These data are summarized in Figure 6C (left). In neurons transfected with the low-affinity sponge, the suppressions of IM by UTP, BK, and oxo-M were 54 ± 9% (n = 5), 57 ± 8% (n = 6), and 78 ± 6% (n = 6), respectively. In neurons transfected with the high-affinity sponge, the suppressions of IM by UTP, BK, and oxo-M were 25 ± 5% (p < 0.01; n = 5), 26 ± 6% (p < 0.01; n = 8), and 69 ± 15% (n = 8). There was no effect on tonic IM densities of sponge expression (Fig. 6C, right). In neurons transfected with the K508A sponge or the R441Q sponge, tonic IM densities were 1.0 ± 0.2 and 0.9 ± 0.2 pA/pF (n = 6 and 8), respectively. As another test to determine which receptor types require IP3 accumulation for suppression of IM, we assayed the effects of overexpression of a type 1 IP3-5′phosphatase (IP3-P), whose activity is the first step in IP3 degradation. Overexpression of IP3-P results in rapid dephosphorylation of IP3 molecules as soon as they are made, preventing its accumulation in the cytoplasm (Hirose et al., 1999). As for the IP3 sponge experiments, the constructs were transfected into SCG neurons using the gene gun and studied under perforated-patch voltage clamp. Figure 7, A and B, shows experiments on a control neuron transfected only with EGFP or a neuron cotransfected with EGFP-tagged IP3-P. Whereas the control neuron displayed robust and reversible suppressions of IM by UTP, BK, and oxo-M, the neuron overexpressing IP3-P responded to both UTP and BK with suppressions of IM that were very weak, but the response to oxo-M was still large.
Figure 6.
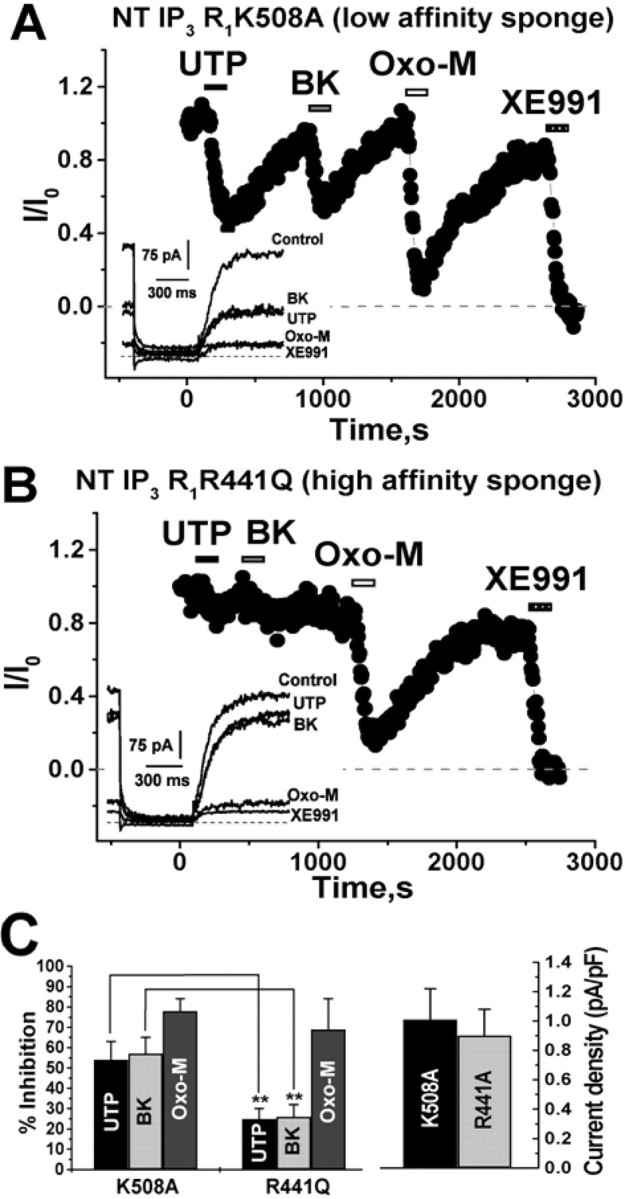
A high-affinity IP3 sponge blocks purinergic inhibition of M current. Plotted are normalized M-current amplitudes, quantified as before, from neurons transfected with the N-terminal IP3 type 1 receptor (NT IP3R1) K508A low-affinity sponge (A) or the N-terminal IP3R1 R441Q high-affinity sponge (B) using the gene gun and studied using perforated-patch recording. UTP (10 μm), BK (250 nm), oxo-M (10 μm), or XE991 (50 μm) were bath applied during the periods shown by the bars. Representative current traces during the experiments are shown in the insets. C, Bars show summarized inhibitions by UTP, BK, or oxo-M (left) or tonic IM densities (right) for cells transfected with the low-affinity (K508A) or the high-affinity (R441Q) sponges. **p < 0.01.
Figure 7.

Purinergic suppression of IM requires cytosolic IP3 and functional CaM. Plotted are normalized M-current amplitudes quantified as before, from SCG cells transfected using the gene gun with EGFP only (A), type 1 IP3-5′phosphatase (B), DN CaM (C), or wt CaM (D). UTP (10 μm), BK (250 nm), oxo-M (10 μm), or XE991 (50 μm) were bath applied during the periods shown by the bars. Representative current traces during the experiments are shown in the insets. Bars in E show summarized data from these experiments for IM suppression by UTP, BK, and oxo-M (left) or tonic IM densities (right). *p < 0.05; **p < 0.01; ***p < 0.001.
Along with the involvement of IP3-mediated [Ca2+]i signals in suppression of IM by bradykinin is the use of CaM as the Ca2+ sensor of M channels. Much like the role of CaM in Ca2+-dependent inactivation of CaV channels (Halling et al., 2006) and activation of SK-type K+ channels (Maylie et al., 2004), CaM binds to M-type channels and inhibits them on [Ca2+]i signals, perhaps by reducing the affinity of PIP2 for the channels (Brown et al., 2007). Given the role of IP3 in the purinergic action shown here, we therefore tested for the involvement of CaM in UTP-mediated suppression of IM. SCG neurons were transfected with either wt CaM or a DN CaM that has an Asp-to-Ala mutation in all four Ca2+-binding sites, rendering it unable to bind Ca2+ (Geiser et al., 1991). If [Ca2+]i signal sensing by CaM mediates purinergic inhibition of M channels, then we predict overexpression of DN CaM to block the suppression because channels will be bound by CaM molecules that cannot sense [Ca2+]i, and we would predict overexpression of wt CaM to amplify tonic Ca2+/CaM modulation because many channels would be bound by Ca2+/CaM at resting [Ca2+]i (Gamper and Shapiro, 2003). Figure 7C shows an experiment on a neuron overexpressing DN CaM. In this cell, the suppression of IM in response to application of UTP or BK was negligible but that from oxo-M application was robust. In Figure 7D, an experiment on a neuron overexpressing wt CaM is shown, in which the responses to UTP and BK were very small and that from oxo-M application was somewhat attenuated.
The suppressions of IM in these four groups of cells are summarized in Figure 7E (left). In neurons transfected only with EGFP, the suppressions of IM by UTP, BK, and oxo-M were 58 ± 8% (n = 7), 61 ± 6% (n = 9), and 80 ± 5% (n = 9), indistinguishable from nontransfected cells. However, in cells transfected with IP3-P, the suppressions of IM by UTP and BK, but not oxo-M, were severely blunted, with responses to UTP, BK, and oxo-M of 22 ± 8% (p < 0.01; n = 5), 23 ± 7% (p < 0.01; n = 6), and 71 ± 9% (n = 6), respectively. Likewise, in cells transfected with DN CaM, only the suppression of IM by oxo-M remained intact. Thus, the responses to UTP, BK, and oxo-M were 18 ± 5% (p < 0.01; n = 5), 20 ± 4% (p < 0.01; n = 5), and 82 ± 7% (n = 5), respectively. The fractional suppressions of IM by UTP and BK were also sharply reduced and that by oxo-M somewhat attenuated in cells overexpressing wt CaM. In those cells, the responses to UTP, BK, and oxo-M were 21 ± 5% (p < 0.01; n = 4), 23 ± 6% (p < 0.01; n = 4), and 50 ± 12% (p < 0.05; n = 4), respectively. The results from the IP3-P and DN CaM experiments are consistent with our scheme of M-channel regulation, in which there is little tonic modulation at rest via IP3 or Ca2+/CaM signals. Thus, there should be little effect on tonic IM densities of lowering resting [IP3] by transfection of the IP3-P or occlusion of tonic CaM action by DN CaM overexpression. Conversely, overexpression of wt CaM might greatly amplify tonic Ca2+/CaM inhibition because the IC50 of the sensitivity of KCNQ2/3 channels for [Ca2+]i is ∼70 nm, in the range of resting [Ca2+]i, and many more channels will now be tonically bound by Ca2+/CaM (Gamper and Shapiro, 2003). Indeed, this is what we observed (Fig. 7E, right). In neurons transfected with EGFP only, IP3-P, or DN CaM, tonic IM densities were very similar: 1.2 ± 0.3, 1.1 ± 0.2, and 1.2 ± 0.2 pA/pF (n = 9, 6, and 5), respectively. However, in cells overexpressing wt CaM, the tonic IM density was reduced by more than half, to 0.5 ± 0.2 pA/pF (p < 0.01; n = 4). Moreover, the reduction of tonic IM currents in those cells is similar to that produced by BK or UTP, as if Ca2+/CaM-mediated suppression is now tonically strong, consistent with our hypothesis.
The attenuation of muscarinic suppression of IM in cells overexpressing wt CaM was surprising and somewhat different from the unaffected action of oxo-M in such cells reported previously (Gamper and Shapiro, 2003). One possible reason for the effect involves myrisotylated alanine-rich C kinase substrate (MARCKS) protein that binds to both PIP2 and Ca2+/CaM in a manner in which binding of the latter is thought to favor unbinding of the former (McLaughlin and Murray, 2005). At low [Ca2+]i, MARCKS can sequester much PIP2 and then release it on [Ca2+]i rises. Thus, overexpression of wt CaM may greatly increase the fraction of MARCKS proteins bound by Ca2+/CaM, resulting in increased tonic [PIP2]. Similar to the effect of increasing tonic [PIP2] by overexpression of phosphatidyl-4-phosphate 5-kinase [PI(4)5-kinase], muscarinic suppression of IM would then be reduced (Winks et al., 2005). It has been suggested that Ca2+/CaM inhibits M channels by reducing their affinity for PIP2 (Brown et al., 2007; Gamper and Shapiro, 2007). A second, related, explanation is that overexpression of wt CaM may result in many channels unbinding PIP2, again resulting in increased tonic [PIP2] in the vicinity of the channels.
In summary, the IP3-P and IP3 sponge data indicate that P2Y receptor-mediated suppression of IM requires cytosolic IP3 accumulation. They also provide more evidence for previous conclusions suggesting M-current suppression by BK, but not oxo-M, to also use an IP3-mediated signal (Cruzblanca et al., 1998). The blockade of purinergic and BK inhibitions in cells overexpressing DN CaM is consistent with those agonists acting via [Ca2+]i signals in concert with CaM, whose actions are now occluded by the overexpressed DN CaM bound to the channels. Overexpression of wt CaM likely results in most channels already bound by Ca2+/CaM and tonic currents already suppressed before any receptor stimulation, with little additional IP3/[Ca2+]i-dependent suppression observed. Our data suggest that P2Y receptors in SCG neurons, like B2 and unlike M1 receptors, modulate M current through CaM action.
P2Y receptor stimulation does not suppress ICa unless neuronal Ca2+ sensor-1 activity is blocked
Two groups have shown CaV2 channels to be regulated by plasma-membrane PIP2 and its depletion to underlie M1 receptor-mediated inhibition (Wu et al., 2002; Gamper et al., 2004). Although both muscarinic and bradykinin stimulation provoke robust PLC-mediated PIP2 hydrolysis in SCG neurons, the former, but not the latter, inhibits PIP2-sensitive voltage-gated Ca2+ channels in those cells (Gamper et al., 2004) (but see Lechner et al., 2005). We suggested this differential action on the Ca2+ current (ICa) to be attributable to receptor-specific depletion of PIP2, arising from concurrent stimulation of PIP2 synthesis by bradykinin, but not muscarinic, agonists. In this model, bradykinin, but not muscarinic, stimulation produces IP3-mediated [Ca2+]i signals, resulting in acceleration of phosphatidylinositol-4-kinase (PI4)-kinase activity by neuronal Ca2+ sensor-1 (NCS-1) (Delmas et al., 2005). Given the similar mode of action of P2Y and B2 receptors on M current seen in this study, we therefore tested the action of purinergic stimulation on ICa in SCG cells. As for bradykinin, we found purinergic stimulation to cause little suppression of ICa. Figure 8A shows an example of a neuron studied under perforated-patch voltage clamp under conditions that isolate ICa. Because stimulation of P2Y receptors has been reported to also activate Go/i-mediated signaling pathways (Filippov et al., 2003), SCG cultures were treated overnight with pertussis toxin to isolate Gq/11-coupled receptor signaling (Beech et al., 1992). Application of neither UTP nor BK caused an appreciable suppression of ICa, whereas oxo-M produced a robust response in the same cell. In response to application of UTP, BK, or oxo-M, ICa was suppressed by 4.5 ± 1.0% (n = 7), 6.2 ± 0.8% (n = 7), and 52.1 ± 4.2% (n = 7), respectively (Fig. 8D, control). This result suggests that, as for B2 receptor stimulation, P2Y receptor stimulation does not appreciably lower PIP2 abundance. We then tested for the involvement of NCS-1-mediated stimulation of PIP2 synthesis by transfecting a DN mutant that cannot bind Ca2+ (Koizumi et al., 2002). In that scenario, cells overexpressing DN NCS-1 should have [Ca2+]i-dependent stimulation of PIP2 synthesis aborted, allowing PIP2 abundance to fall during P2Y or BK stimulation and ICa to be suppressed. Neurons were transfected either with enhanced yellow fluorescent protein (EYFP) only or with EYFP-tagged DN NCS-1. Figure 8, B and C, show examples from both groups of neurons. In the cell transfected only with EYFP, UTP and BK caused very little suppression of ICa, but oxo-M induced robust suppression, as in Figure 8A. However, in the cell transfected with DN NCS-1, UTP and BK now induced a significant suppression of ICa, as did oxo-M. These data are summarized in Figure 8D. In neurons transfected with EYFP only, application of UTP, BK, and oxo-M suppressed ICa by 5.8 ± 3.6% (n = 5), 8.2 ± 3.2% (n = 5), and 53.2 ± 5.7% (n = 5). In neurons transfected with DN NCS-1, the suppressions were 31.4 ± 4.7% (p < 0.01; n = 4), 36.6 ± 6.7% (p < 0.01; n = 4), and 59.7 ± 8.3% (n = 4), respectively. There was no significant effect on tonic ICa current amplitudes of NCS-1 overexpression. For nontransfected neurons or those transfected with EYFP only or with NCS-1, ICa densities were 44.4 ± 4.1, 50.4 ± 6.1, and 52.9 ± 3.2 pA/pF (n = 7,5,4), respectively. These results are in accord with our model of selective depletion of PIP2 by muscarinic, but not purinergic or bradykinin, stimulation. Thus, we suggest that stimulation of both P2Y and B2 receptors concurrently stimulate PIP2 synthesis, at least in part, via NCS-1 action, compensating for PIP2 depletion by PLC activity. With NCS-1 action blocked by overexpression of a DN form, [PIP2] is allowed to fall during stimulation of either receptor, and stimulation of either now induces significant suppression of ICa.
Figure 8.
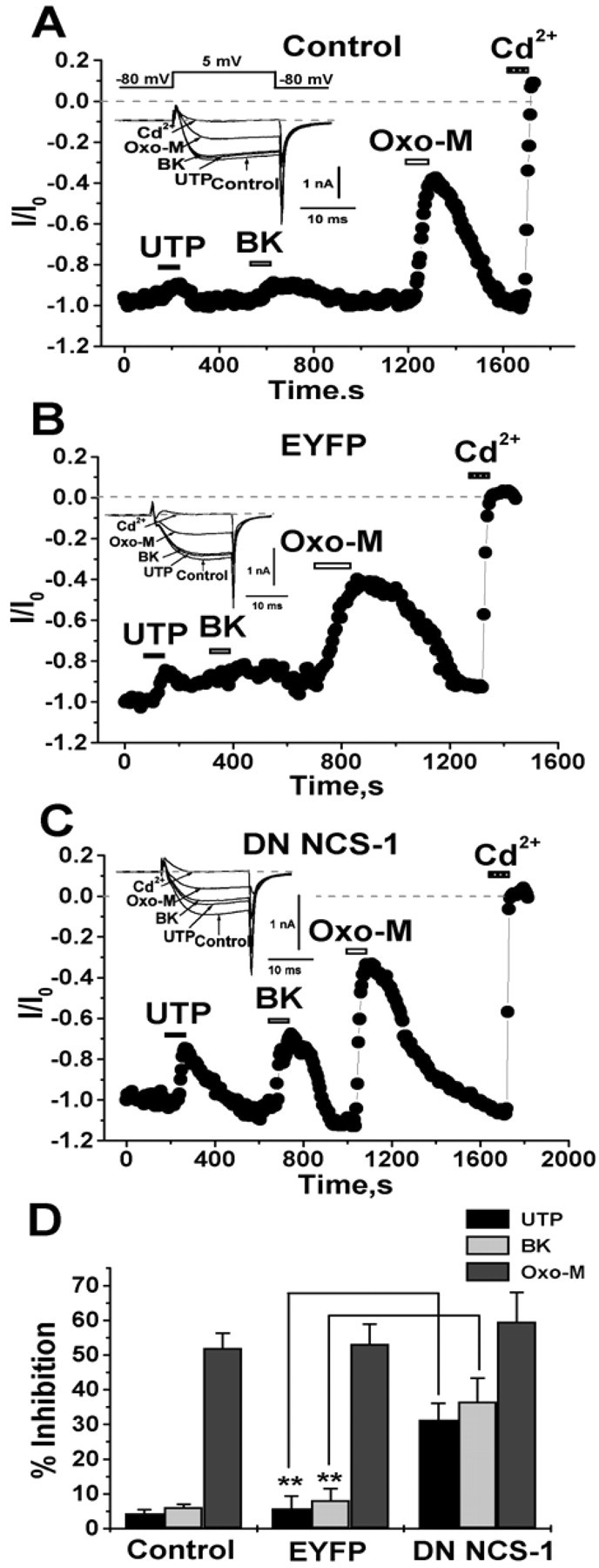
Purinergic and bradykinin stimulation do not suppress ICa unless NCS-1 activity is blocked. Plotted are normalized ICa amplitudes from nontransfected SCG neurons (A) or those transfected using the gene gun with EYFP only (B), or EYFP-tagged DN NCS-1 (C) and studied using perforated-patch recordings. SCG cultures were treated overnight with PTX (100 ng/ml) to block Go/i-mediated actions. UTP (10 μm), BK (250 nm), oxo-M (10 μm), or CdCl2 (100 μm) were bath applied during the periods shown by the bars. Representative current traces are shown in the insets. D, Bars show summarized data for suppressions of ICa by UTP, BK, and oxo-M for the three groups of cells. **p < 0.01.
Discussion
We suggest that the action of Gq/11-coupled receptors can be divided into two fundamental modes that have in common PLC activation and hydrolysis of PIP2. The first mode is based on the widening discovery that many ion channels are regulated by a membrane PIP2 abundance that can be strongly lowered by PLC (Suh and Hille, 2005). This mechanism seems particularly established for M1 receptor suppression of M current in sympathetic ganglia and likely for AT1 receptors as well (Shapiro et al., 1994; Zaika et al., 2006; Suh and Hille, 2007). Both receptor types share the lack of provoked IP3-mediated [Ca2+]i signals and dual inhibition of both M-type K+ and Ca2+ channels. The second mode is used by Gq/11-coupled receptors that do provoke such [Ca2+]i signals, which then activate multiple Ca2+-binding signaling proteins. The bradykinin B2 receptor of ganglia cells is the best described in this category. Because of concurrent stimulation of PIP2 synthesis by bradykinin (Xu et al., 2003) via [Ca2+]i-dependent activation of NCS-1 and acceleration of PI4-kinase activity, PIP2 levels do not appreciably fall, M channels are inhibited instead by Ca2+/CaM action, and Ca2+ channels are little affected (Gamper et al., 2004; Winks et al., 2005; Brown et al., 2007). However, is this action in SCG cells unique to bradykinin? Our work here suggests not and places the purinergic P2Y receptors of these cells in the category using the second mode along with the B2 receptor. For both, the action of IP3 receptors is required, CaM functions as the Ca2+-sensor mediating suppression of M current, and Ca2+ channels are not modulated. Figure 9 summarizes the bipartite division of Gq/11-coupled receptor mechanisms hypothesized in sympathetic ganglia cells.
Figure 9.

Schematic representation of the two modes of Gq/11-coupled receptor mechanisms in SCG neurons. Both pathways involve activation of Gq/11 G-proteins, PLC, and PIP2 hydrolysis. The “mode 1” pathway suppresses M current (IM) and Ca2+ current (ICa) attributable to depletion of PIP2. Mode 1 does not elicit [Ca2+]i signals. The “mode 2” pathway does elicit [Ca2+]i signals and concurrently stimulates PIP2 synthesis via NCS-1. Released Ca2+ binds to NCS-1 and to CaM. Ca2+/CaM binds to M channels, resulting in their inhibition.
Along with muscarine and several peptides, purinergic agonists were soon identified as regulators of neuronal excitability (Siggins et al., 1977) via depression of M current (Adams et al., 1982; Lopez and Adams, 1989; Tokimasa and Akasu, 1990) in frog sympathetic neurons. The mechanism of action of neuronal P2Y receptors on IM has been controversial and may depend on precise neuronal type or receptor isoform. Previous work in the rat SCG cells studied here, in which the dominant subtype is likely P2Y6, suggested the requirement for [Ca2+]i signals (Bofill-Cardona et al., 2000), consistent with our results, but, in frog sympathetic neurons in which the subtype is not clear, the PIP2-depletion mode is suggested (Stemkowski et al., 2002). Recently, P2Y1 receptor-mediated suppression of M current in hippocampus was studied, and the lack of [Ca2+]i signals seen during purinergic stimulation in those cells points toward a PIP2-depletion mechanism there as well. It seems that the basic division between (Filippov et al., 2006) modes of action hinges on the ability of an agonist to provoke [Ca2+]i signals during receptor stimulation, and the critical factor for induction of such [Ca2+]i release has been suggested to be colocalization of certain plasma-membrane receptors with endoplasmic reticulum-membrane IP3 receptors (Delmas et al., 2004). Thus, if IP3 is produced in the microdomain of the IP3 receptor, [Ca2+]i is the relevant signal, and downstream Ca2+-dependent actions of CaM and NCS-1 are initiated; if not, PIP2 depletion is the mode of action. Perhaps the divergent results in different cells among different P2Y receptor subtypes predict differential localization of receptor isoforms in microdomains containing IP3 receptors. Alternatively, different neuronal types may use different membrane protein organizations. Although we do not observe suppression of ICa by bradykinin unless the NCS-1 pathway is blocked (Gamper et al., 2004; this study), Lechner et al. (2005) observe such an inhibition that they ascribe to depletion of PIP2. The most likely reason for the discrepancy is differential tonic PIP2 levels, perhaps caused by the different medium/growth conditions used by the two laboratories (Chuang et al., 2001).
Filippov and colleagues, using SCG cells as a vehicle for heterologous expression of four different P2Y receptor subtypes, observed an primarily voltage-dependent and PTX-sensitive suppression of ICa and a wholly PTX-insensitive suppression of IM by UTP (Filippov et al., 1998, 1999, 2000, 2003). The depression of ICa seen in those studies had all the hallmarks of the fast “willing-reluctant” type of modulation mediated by Go/i-coupled receptors in many neuronal types (Hille, 1994) and none of the characteristics of the slower Gq/11-mediated pathway typified by M1 receptors that we ascribe to depletion of PIP2 (Gamper et al., 2004). We here found purinergic stimulation of PTX-treated neurons to likewise normally have little effect on ICa. The suppression of IM in the papers cited above seems similar to that observed by stimulation of native P2Y receptors (Bofill-Cardona et al., 2000; this study), although additional mechanistic information from the heterologous-expression studies is not available. As in the case here using native receptors, stimulation of expressed P2Y2 receptors increased AP firing, consistent with closure of M channels (Filippov et al., 1998). Interestingly, Filippov and colleagues observed little modulation by UTP of either type of channel in nontransfected neurons (which would use native receptors), whereas we observe suppression of IM in approximately half the SCG cells studied. Perhaps our use of younger rats (1–2 weeks), and slightly longer culture (2–3 d), provides the explanation. Because colocalization of P2Y6 receptors with IP3 receptors may underlie the mechanistic specificity described in this study, we appreciate the opportunity to study the actions of the native receptors.
We suggest that stimulation of either B2 or P2Y receptors does not appreciably deplete membrane PIP2 attributable, at least in part, to NCS-1-mediated stimulation of PIP2 synthesis. Such agonist stimulation of PIP2 synthesis is by now widely documented and has been suggested to be often obligatory to account for the mass of IP3 produced during PLC activation (Loew, 2007). Independent evidence for receptor-specific depletion of PIP2 comes from the tubby probe that binds PIP2, but not IP3 (Santagata et al., 2001). Translocation of tubby was minor during bradykinin stimulation in the same cells in which muscarinic translocation was large, but, during acute blockade of PI4-kinase, the translocations were equal (Hughes et al., 2007). In our scheme, blockade of NCS-1 or PI4-kinase activity allows [PIP2] to fall and ICa to be suppressed. This scenario presents us with a conundrum, however, because the prevention of IP3 accumulation by overexpression of the IP3 sponge or the IP3-P should prevent the [Ca2+]i signals that not only activate CaM but also NCS-1, the molecule that we implicate in stimulation of PIP2 synthesis. With both CaM and NCS-1 activity prevented, why should stimulation of B2 or P2Y receptors not deplete PIP2, as by M1 receptor stimulation? The Hille group has shown some [Ca2+]i signal (either released from stores or via Ca2+ influx) to be necessary for full PLC activity in tsA-201 tissue-culture cells in which overexpression of IP3-P slowed and weakened diacylglycerol production and KCNQ2/3 channel inhibition by expressed M1 receptors (Horowitz et al., 2005; Suh et al., 2006). However in the SCG neurons in which muscarinic suppression of M current is strongest, M1 receptor stimulation does not produce detectable [Ca2+]i rises (Beech et al., 1991) [unlike stimulation of expressed M1 receptors in tsA-201 cells (Shapiro et al., 2000)], and we did not observe any effect on muscarinic suppression of IM of the IP3 sponge or IP3-P. Two possible explanations emerge. The first is receptor-specific acceleration of PI(4)5-kinase activity as well, which “virtual cell” modeling suggests is required for compensation of PIP2 levels in the face of PLC activity (Xu et al., 2003; Loew, 2007). Thus, whereas BK and P2Y receptor triggering may depress M current primarily through IP3-mediated [Ca2+]i signals, leading to blockade of IM suppression when IP3 accumulation is prevented, the receptor-stimulated acceleration of PIP2 synthesis may partly be via [Ca2+]i signals and partly through receptor-specific stimulation of PI(4)5-kinase via an, as yet, unknown pathway. The second possibility is receptor-specific turn on of store-operated Ca2+ entry that might require coupling of G-protein-coupled receptors and IP3 receptor but not IP3 (Kiselyov et al., 2005). Other aspects of this issue have been discussed recently (Gamper and Shapiro, 2007).
In our comparison among agonists, the effects on excitability generally correlated with degree of M-channel inhibition, although the increase in firing by UTP and BK seemed qualitatively weaker than that by oxo-M. Our finding that neither purinergic nor bradykinin simulation normally suppress ICa give a hypothesis why this may be so. Simultaneous inhibition of M channels from Gq/11-coupled M1 receptor stimulation and of Ca2+ channels from combined M1 and Go/i-coupled M4 receptor stimulation (Hille, 1994) predict a synergistic decrease in K+ current braking action by the reduced IM combined with the reduced KCa conductances from suppression of Ca2+ influx, especially likely given the strong role ascribed to the SK channels in controlling the medium afterhyperpolarization (Kawai and Watanabe, 1986; Davies et al., 1996). It is likely that additional mechanisms make a contribution to SFA in these neurons as well. We look forward to a more complete model of simultaneous modulation of multiple ion channels by the same receptor and activation by ionotropic and metabotropic receptors by the same agonist (e.g., acetylcholine, ATP, and glutamate) to understand the multifaceted potential of neurotransmitter-mediated regulation of neuronal function.
Footnotes
This work was supported by National Institutes of Health–National Institute of Neurological Disorders and Stroke Grant RO1 NS43394 (M.S.S.). We thank Pamela Martin for expert technical assistance and Nikita Gamper for comments on this manuscript.
References
- Adams PR, Brown DA, Constanti A. Pharmacological inhibition of the M-current. J Physiol (Lond) 1982;332:223–262. doi: 10.1113/jphysiol.1982.sp014411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beech DJ, Bernheim L, Mathie A, Hille B. Intracellular Ca2+ buffers disrupt muscarinic suppression of Ca2+ current and M current in rat sympathetic neurons. Proc Natl Acad Sci USA. 1991;88:652–656. doi: 10.1073/pnas.88.2.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beech DJ, Bernheim L, Hille B. Pertussis toxin and voltage dependence distinguish multiple pathways modulating calcium channels of rat sympathetic neurons. Neuron. 1992;8:97–106. doi: 10.1016/0896-6273(92)90111-p. [DOI] [PubMed] [Google Scholar]
- Bernheim L, Beech DJ, Hille B. A diffusible second messenger mediates one of the pathways coupling receptors to calcium channels in rat sympathetic neurons. Neuron. 1991;6:859–867. doi: 10.1016/0896-6273(91)90226-p. [DOI] [PubMed] [Google Scholar]
- Boehm S. Selective inhibition of M-type potassium channels in rat sympathetic neurons by uridine nucleotide preferring receptors. Br J Pharmacol. 1998;124:1261–1269. doi: 10.1038/sj.bjp.0701956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bofill-Cardona E, Vartian N, Nanoff C, Freissmuth M, Boehm S. Two different signaling mechanisms involved in the excitation of rat sympathetic neurons by uridine nucleotides. Mol Pharmacol. 2000;57:1165–1172. [PubMed] [Google Scholar]
- Borg-Graham L. Modelling the non-linear conductances of excitable membranes. In: Chad J, Wheal H, editors. Cellular neurobiology: a practical approach. Oxford, UK: IRL/Oxford UP; 1991. [Google Scholar]
- Brown DA, Hughes SA, Marsh SJ, Tinker A. Regulation of M(Kv7.2/7.3) channels in neurons by PIP2 and products of PIP2 hydrolysis: significance for receptor-mediated inhibition. J Physiol (Lond) 2007;582:917–925. doi: 10.1113/jphysiol.2007.132498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnstock G. Purinergic signaling—an overview. Novartis Found Symp. 2006;276:26–48. discussion 48–57:275–281. [PubMed] [Google Scholar]
- Calvert JA, Evans RJ. Heterogeneity of P2X receptors in sympathetic neurons: contribution of neuronal P2X1 receptors revealed using knockout mice. Mol Pharmacol. 2004;65:139–148. doi: 10.1124/mol.65.1.139. [DOI] [PubMed] [Google Scholar]
- Calvert JA, Atterbury-Thomas AE, Leon C, Forsythe ID, Gachet C, Evans RJ. Evidence for P2Y1, P2Y2, P2Y6 and atypical UTP-sensitive receptors coupled to rises in intracellular calcium in mouse cultured superior cervical ganglion neurons and glia. Br J Pharmacol. 2004;143:525–532. doi: 10.1038/sj.bjp.0705959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang HH, Prescott ED, Kong H, Shields S, Jordt SE, Basbaum AI, Chao MV, Julius D. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature. 2001;411:957–962. doi: 10.1038/35082088. [DOI] [PubMed] [Google Scholar]
- Cooper EC, Jan LY. M-channels: neurological diseases, neuromodulation, and drug development. Arch Neurol. 2003;60:496–500. doi: 10.1001/archneur.60.4.496. [DOI] [PubMed] [Google Scholar]
- Cruzblanca H, Koh DS, Hille B. Bradykinin inhibits M current via phospholipase C and Ca2+ release from IP3-sensitive Ca2+ stores in rat sympathetic neurons. Proc Natl Acad Sci USA. 1998;95:7151–7156. doi: 10.1073/pnas.95.12.7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies MP, An RH, Doevendans P, Kubalak S, Chien KR, Kass RS. Developmental changes in ionic channel activity in the embryonic murine heart. Circ Res. 1996;78:15–25. doi: 10.1161/01.res.78.1.15. [DOI] [PubMed] [Google Scholar]
- Delmas P, Brown DA. Junctional signaling microdomains: bridging the gap between the neuronal cell surface and Ca2+ stores. Neuron. 2002;36:787–790. doi: 10.1016/s0896-6273(02)01097-8. [DOI] [PubMed] [Google Scholar]
- Delmas P, Brown DA. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci. 2005;6:850–862. doi: 10.1038/nrn1785. [DOI] [PubMed] [Google Scholar]
- Delmas P, Wanaverbecq N, Abogadie FC, Mistry M, Brown DA. Signaling microdomains define the specificity of receptor-mediated InsP3 pathways in neurons. Neuron. 2002;34:209–220. doi: 10.1016/s0896-6273(02)00641-4. [DOI] [PubMed] [Google Scholar]
- Delmas P, Crest M, Brown DA. Functional organization of PLC signaling microdomains in neurons. Trends Neurosci. 2004;27:41–47. doi: 10.1016/j.tins.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Delmas P, Coste B, Gamper N, Shapiro MS. Phosphoinositide lipid second messengers: new paradigms for calcium channel modulation. Neuron. 2005;47:179–182. doi: 10.1016/j.neuron.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Filippov AK, Webb TE, Barnard EA, Brown DA. P2Y2 nucleotide receptors expressed heterologously in sympathetic neurons inhibit both N-type Ca2+ and M-type K+ currents. J Neurosci. 1998;18:5170–5179. doi: 10.1523/JNEUROSCI.18-14-05170.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippov AK, Webb TE, Barnard EA, Brown DA. Dual coupling of heterologously-expressed rat P2Y6 nucleotide receptors to N-type Ca2+ and M-type K+ currents in rat sympathetic neurones. Br J Pharmacol. 1999;126:1009–1017. doi: 10.1038/sj.bjp.0702356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippov AK, Brown DA, Barnard EA. The P2Y(1) receptor closes the N-type Ca2+ channel in neurones, with both adenosine triphosphates and diphosphates as potent agonists. Br J Pharmacol. 2000;129:1063–1066. doi: 10.1038/sj.bjp.0703185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippov AK, Simon J, Barnard EA, Brown DA. Coupling of the nucleotide P2Y4 receptor to neuronal ion channels. Br J Pharmacol. 2003;138:400–406. doi: 10.1038/sj.bjp.0705043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippov AK, Choi RC, Simon J, Barnard EA, Brown DA. Activation of P2Y1 nucleotide receptors induces inhibition of the M-type K+ current in rat hippocampal pyramidal neurons. J Neurosci. 2006;26:9340–9348. doi: 10.1523/JNEUROSCI.2635-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper N, Shapiro MS. Calmodulin mediates Ca2+-dependent modulation of M-type K+ channels. J Gen Physiol. 2003;122:17–31. doi: 10.1085/jgp.200208783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper N, Shapiro MS. Exogenous expression of proteins in neurons using the biolistic particle delivery system. Methods Mol Biol. 2006;337:27–38. doi: 10.1385/1-59745-095-2:27. [DOI] [PubMed] [Google Scholar]
- Gamper N, Shapiro MS. Target-specific PIP2 signalling: how might it work? J Physiol (Lond) 2007;582:967–975. doi: 10.1113/jphysiol.2007.132787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper N, Reznikov V, Yamada Y, Yang J, Shapiro MS. Phosphatidylinositol 4,5-bisphosphate signals underlie receptor-specific Gq/11-mediated modulation of N-type Ca2+ channels. J Neurosci. 2004;24:10980–10992. doi: 10.1523/JNEUROSCI.3869-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper N, Zaika O, Li Y, Martin P, Hernandez CC, Perez MR, Wang AY, Jaffe DB, Shapiro MS. Oxidative modification of M-type K+ channels as a mechanism of cytoprotective neuronal silencing. EMBO J. 2006;25:4996–5004. doi: 10.1038/sj.emboj.7601374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiser JR, van Tuinen D, Brockerhoff SE, Neff MM, Davis TN. Can calmodulin function without binding calcium? Cell. 1991;65:949–959. doi: 10.1016/0092-8674(91)90547-c. [DOI] [PubMed] [Google Scholar]
- Gu N, Vervaeke K, Hu H, Storm JF. Kv7/KCNQ/M and HCN/h, but not KCa2/SK channels, contribute to the somatic medium after-hyperpolarization and excitability control in CA1 hippocampal pyramidal cells. J Physiol (Lond) 2005;566:689–715. doi: 10.1113/jphysiol.2005.086835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halling DB, Aracena-Parks P, Hamilton SL. Regulation of voltage-gated Ca2+ channels by calmodulin. Sci STKE. 2006;2006:er1. doi: 10.1126/stke.3182006er1. [DOI] [PubMed] [Google Scholar]
- Hille B. Modulation of ion-channel function by G-protein-coupled receptors. Trends Neurosci. 1994;17:531–536. doi: 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- Hines ML, Carnevale NT. NEURON: a tool for neuroscientists. The Neuroscientist. 2001;7:123–135. doi: 10.1177/107385840100700207. [DOI] [PubMed] [Google Scholar]
- Hirose K, Kadowaki S, Tanabe M, Takeshima H, Iino M. Spatiotemporal dynamics of inositol 1,4,5-trisphosphate that underlies complex Ca2+ mobilization patterns. Science. 1999;284:1527–1530. doi: 10.1126/science.284.5419.1527. [DOI] [PubMed] [Google Scholar]
- Horowitz LF, Hirdes W, Suh BC, Hilgemann DW, Mackie K, Hille B. Phospholipase C in living cells: activation, inhibition, Ca2+ requirement, and regulation of M current. J Gen Physiol. 2005;126:243–262. doi: 10.1085/jgp.200509309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes S, Marsh S, Tinker A, Brown D. PIP2-dependent inhibition of M-type (Kv7.2/7.3) potassium channels: direct on-line assessment of PIP2 depletion by Gq-coupled receptors in single living neurons. Pflügers Arch. 2007 doi: 10.1007/s00424-007-0259-6. in press. [DOI] [PubMed] [Google Scholar]
- Jones S, Brown DA, Milligan G, Willer E, Buckley NJ, Caulfield MP. Bradykinin excites rat sympathetic neurons by inhibition of M current through a mechanism involving B2 receptors and Gaq/11. Neuron. 1995;14:399–405. doi: 10.1016/0896-6273(95)90295-3. [DOI] [PubMed] [Google Scholar]
- Kawai T, Watanabe M. Blockade of Ca-activated K conductance by apamin in rat sympathetic neurones. Br J Pharmacol. 1986;87:225–232. doi: 10.1111/j.1476-5381.1986.tb10175.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiselyov K, Kim JY, Zeng W, Muallem S. Protein-protein interaction and function of TRPC channels. Pflügers Arch. 2005;451:116–124. doi: 10.1007/s00424-005-1442-2. [DOI] [PubMed] [Google Scholar]
- Koizumi S, Rosa P, Willars GB, Challiss RA, Taverna E, Francolini M, Bootman MD, Lipp P, Inoue K, Roder J, Jeromin A. Mechanisms underlying the neuronal calcium sensor-1-evoked enhancement of exocytosis in PC12 cells. J Biol Chem. 2002;277:30315–30324. doi: 10.1074/jbc.M201132200. [DOI] [PubMed] [Google Scholar]
- Lawrence JJ, Saraga F, Churchill JF, Statland JM, Travis KE, Skinner FK, McBain CJ. Somatodendritic Kv7/KCNQ/M channels control interspike interval in hippocampal interneurons. J Neurosci. 2006;26:12325–12338. doi: 10.1523/JNEUROSCI.3521-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner SG, Hussl S, Schicker KW, Drobny H, Boehm S. Presynaptic inhibition via a phospholipase C- and phosphatidylinositol bisphosphate-dependent regulation of neuronal Ca2+ channels. Mol Pharmacol. 2005;68:1387–1396. doi: 10.1124/mol.105.014886. [DOI] [PubMed] [Google Scholar]
- Loew LM. Where does all the PIP2 come from? J Physiol (Lond) 2007;582:945–951. doi: 10.1113/jphysiol.2007.132860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez HS, Adams PR. A G protein mediates the inhibition of the voltage-dependent potassium M current by muscarine, LHRH, substance P and UTP in bullfrog sympathetic neurons. Eur J Neurosci. 1989;1:529–542. doi: 10.1111/j.1460-9568.1989.tb00360.x. [DOI] [PubMed] [Google Scholar]
- Maylie J, Bond CT, Herson PS, Lee WS, Adelman JP. Small conductance Ca2+-activated K+ channels and calmodulin. J Physiol (Lond) 2004;554:255–261. doi: 10.1113/jphysiol.2003.049072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin S, Murray D. Plasma membrane phosphoinositide organization by protein electrostatics. Nature. 2005;438:605–611. doi: 10.1038/nature04398. [DOI] [PubMed] [Google Scholar]
- Migliore M, Cook EP, Jaffe DB, Turner DA, Johnston D. Computer simulations of morphologically reconstructed CA3 hippocampal neurons. J Neurophysiol. 1995;73:1157–1168. doi: 10.1152/jn.1995.73.3.1157. [DOI] [PubMed] [Google Scholar]
- Moore DJ, Chambers JK, Wahlin JP, Tan KB, Moore GB, Jenkins O, Emson PC, Murdock PR. Expression pattern of human P2Y receptor subtypes: a quantitative reverse transcription-polymerase chain reaction study. Biochim Biophys Acta. 2001;1521:107–119. doi: 10.1016/s0167-4781(01)00291-3. [DOI] [PubMed] [Google Scholar]
- Peretz A, Degani N, Nachman R, Uziyel Y, Gibor G, Shabat D, Attali B. Meclofenamic acid and diclofenac, novel templates of KCNQ2/Q3 potassium channel openers, depress cortical neuron activity and exhibit anticonvulsant properties. Mol Pharmacol. 2005;67:1053–1066. doi: 10.1124/mol.104.007112. [DOI] [PubMed] [Google Scholar]
- Peters HC, Hu H, Pongs O, Storm JF, Isbrandt D. Conditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance and behavior. Nat Neurosci. 2005;8:51–60. doi: 10.1038/nn1375. [DOI] [PubMed] [Google Scholar]
- Rae J, Cooper K, Gates P, Watsky M. Low access resistance perforated patch recordings using amphotericin B. J Neurosci Methods. 1991;37:15–26. doi: 10.1016/0165-0270(91)90017-t. [DOI] [PubMed] [Google Scholar]
- Santagata S, Boggon TJ, Baird CL, Gomez CA, Zhao J, Shan WS, Myszka DG, Shapiro L. G-protein signaling through tubby proteins. Science. 2001;292:2041–2050. doi: 10.1126/science.1061233. [DOI] [PubMed] [Google Scholar]
- Shapiro MS, Wollmuth LP, Hille B. Angiotensin II inhibits calcium and M current channels in rat sympathetic neurons via G proteins. Neuron. 1994;12:1319–1329. doi: 10.1016/0896-6273(94)90447-2. [DOI] [PubMed] [Google Scholar]
- Shapiro MS, Roche JP, Kaftan EJ, Cruzblanca H, Mackie K, Hille B. Reconstitution of muscarinic modulation of the KCNQ2/KCNQ3 K+ channels that underlie the neuronal M current. J Neurosci. 2000;20:1710–1721. doi: 10.1523/JNEUROSCI.20-05-01710.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Hamilton SE, Nathanson NM, Surmeier DJ. Cholinergic suppression of KCNQ channel currents enhances excitability of striatal medium spiny neurons. J Neurosci. 2005;25:7449–7458. doi: 10.1523/JNEUROSCI.1381-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siggins GR, Gruol DL, Padjen AL, Formans DS. Purine and pyrimidine mononucleotides depolarise neurones of explanted amphibian sympathetic ganglia. Nature. 1977;270:263–265. doi: 10.1038/270263a0. [DOI] [PubMed] [Google Scholar]
- Stemkowski PL, Tse FW, Peuckmann V, Ford CP, Colmers WF, Smith PA. ATP-inhibition of M current in frog sympathetic neurons involves phospholipase C but not InsP3, Ca2+, PKC, or Ras. J Neurophysiol. 2002;88:277–288. doi: 10.1152/jn.2002.88.1.277. [DOI] [PubMed] [Google Scholar]
- Suh B, Hille B. Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron. 2002;35:507–520. doi: 10.1016/s0896-6273(02)00790-0. [DOI] [PubMed] [Google Scholar]
- Suh BC, Hille B. Regulation of ion channels by phosphatidylinositol 4,5-bisphosphate. Curr Opin Neurobiol. 2005;15:370–378. doi: 10.1016/j.conb.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Suh BC, Hille B. Regulation of KCNQ channels by manipulation of phosphoinositides. J Physiol (Lond) 2007;582:911–916. doi: 10.1113/jphysiol.2007.132647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh BC, Inoue T, Meyer T, Hille B. Rapid chemically induced changes of PtdIns(4,5)P2 gate KCNQ ion channels. Science. 2006;314:1454–1457. doi: 10.1126/science.1131163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokimasa T, Akasu T. ATP regulates muscarine-sensitive potassium current in dissociated bull-frog primary afferent neurones. J Physiol (Lond) 1990;426:241–264. doi: 10.1113/jphysiol.1990.sp018136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub RD, Wong RK, Miles R, Michelson H. A model of a CA3 hippocampal pyramidal neuron incorporating voltage-clamp data on intrinsic conductances. J Neurophysiol. 1991;66:635–650. doi: 10.1152/jn.1991.66.2.635. [DOI] [PubMed] [Google Scholar]
- Uchiyama T, Yoshikawa F, Hishida A, Furuichi T, Mikoshiba K. A novel recombinant hyperaffinity inositol 1,4,5-trisphosphate (IP3) absorbent traps IP3, resulting in specific inhibition of IP3-mediated calcium signaling. J Biol Chem. 2002;277:8106–8113. doi: 10.1074/jbc.M108337200. [DOI] [PubMed] [Google Scholar]
- Vartian N, Moskvina E, Scholze T, Unterberger U, Allgaier C, Boehm S. UTP evokes noradrenaline release from rat sympathetic neurons by activation of protein kinase C. J Neurochem. 2001;77:876–885. doi: 10.1046/j.1471-4159.2001.00290.x. [DOI] [PubMed] [Google Scholar]
- Vervaeke K, Gu N, Agdestein C, Hu H, Storm JF. Kv7/KCNQ/M channels in glutamatergic hippocampal axons and their role in regulation of excitability and transmitter release. J Physiol (Lond) 2006;576:235–256. doi: 10.1113/jphysiol.2006.111336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- Winks JS, Hughes S, Filippov AK, Tatulian L, Abogadie FC, Brown DA, Marsh SJ. Relationship between membrane phosphatidylinositol-4,5-bisphosphate and receptor-mediated inhibition of native neuronal M channels. J Neurosci. 2005;25:3400–3413. doi: 10.1523/JNEUROSCI.3231-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Bauer CS, Zhen XG, Xie C, Yang J. Dual regulation of voltage-gated calcium channels by PtdIns(4,5)P2. Nature. 2002;419:947–952. doi: 10.1038/nature01118. [DOI] [PubMed] [Google Scholar]
- Xu C, Watras J, Loew LM. Kinetic analysis of receptor-activated phosphoinositide turnover. J Cell Biol. 2003;161:779–791. doi: 10.1083/jcb.200301070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue C, Yaari Y. Axo-somatic and apical dendritic Kv7/M channels differentially regulate the intrinsic excitability of adult rat CA1 pyramidal cells. J Neurophysiol. 2006;95:3480–3495. doi: 10.1152/jn.01333.2005. [DOI] [PubMed] [Google Scholar]
- Zaczek R, Chorvat RJ, Saye JA, Pierdomenico ME, Maciag CM, Logue AR, Fisher BN, Rominger DH, Earl RA. Two new potent neurotransmitter release enhancers, 10,10-bis(4-pyridinylmethyl)-9(10H)-anthracenone and 10,10-bis(2-fluoro-4-pyridinylmethyl)-9(10H)-anthracenone: comparison to linopirdine. J Pharmacol Exp Ther. 1998;285:724–730. [PubMed] [Google Scholar]
- Zaika O, Lara LS, Gamper N, Hilgemann DW, Jaffe DB, Shapiro MS. Angiotensin II regulates neuronal excitability via phosphatidylinositol 4,5-bisphosphate-dependent modulation of Kv7 (M-type) K+ channels. J Physiol (Lond) 2006;575:49–67. doi: 10.1113/jphysiol.2006.114074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Craciun LC, Mirshahi T, Rohacs T, Lopes CM, Jin T, Logothetis DE. PIP2 activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron. 2003;37:963–975. doi: 10.1016/s0896-6273(03)00125-9. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Nehere E. Mobile and immobile calcium buffers in bovine adrenal chromaffin cells. J Physiol. 1993;469:245–273. doi: 10.1113/jphysiol.1993.sp019813. [DOI] [PMC free article] [PubMed] [Google Scholar]