

Abstract
Immunotherapies for various neurodegenerative diseases have recently emerged as a promising approach for clearing pathological protein conformers in these disorders. This type of treatment has not been assessed in models that develop neuronal tau aggregates as observed in frontotemporal dementia and Alzheimer's disease. Here, we present that active immunization with a phosphorylated tau epitope, in P301L tangle model mice, reduces aggregated tau in the brain and slows progression of the tangle-related behavioral phenotype. Females had more tau pathology than males but were also more receptive to the immunotherapy. The tau antibodies generated in these animals recognized pathological tau on brain sections. Performance on behavioral assays that require extensive motor coordination correlated with tau pathology in corresponding brain areas, and antibody levels against the immunogen correlated inversely with tau pathology. Interestingly, age-dependent autoantibodies that recognized recombinant tau protein but not the immunogen were detected in the P301L mice. To confirm that anti-tau antibodies could enter the brain and bind to pathological tau, FITC-tagged antibodies purified from a P301L mouse, with a high antibody titer against the immunogen, were injected into the carotid artery of P301L mice. These antibodies were subsequently detected within the brain and colocalized with PHF1 and MC1 antibodies that recognize pathological tau. Currently, no treatment is available for clearing tau aggregates. Our present findings may lead to a novel therapy targeting one of the major hallmarks of Alzheimer's disease and frontotemporal dementia.
Keywords: tau, P301L, tangles, transgenic mice, immunization, behavior
Introduction
An emerging treatment for Alzheimer's disease (AD) is immunotherapy to clear amyloid-β (Aβ). Another important target in AD and frontotemporal dementia is the neurofibrillary tangles and/or their pathological tau protein conformers, whose presence correlates well with the degree of dementia (Terry, 1994; Goedert, 2004). The objective of immunotherapy for tau pathology is that anti-tau antibodies can clear tau aggregates that may affect neuronal viability. Tau is a soluble protein that promotes tubulin assembly, microtubule stability, and cytoskeletal integrity. Although tau pathology is likely to occur after Aβ aggregation based on Down syndrome studies, analyses of AD brains and mouse models indicate that these pathologies are likely to be synergistic (Sigurdsson et al., 1996, 1997; Gotz et al., 2001b; Lewis et al., 2001; Delacourte et al., 2002; Oddo et al., 2004; Ribe et al., 2005). Hence, targeting both pathologies may substantially increase treatment efficacy. To date, no tau mutations have been observed in AD; however, in frontotemporal dementia, mutations in the tau protein on chromosome 17 (FTDP-17) are a causative factor in the disease, which further supports tau-based therapeutic approaches (Poorkaj et al., 1998; Spillantini et al., 1998). Transgenic (Tg) mice expressing these mutations have modeled many aspects of the disease and are valuable tools to study the pathogenesis of tangle-related neurodegeneration and to assess potential therapies. One of these models, P301L mice (Lewis et al., 2000), recapitulates many of the features of frontotemporal dementia, although the CNS distribution of the tau aggregates results primarily in sensorimotor abnormalities, which complicates cognitive assessment. Homozygous lines of these mice have an early onset of CNS pathology and associated functional impairments, which renders this model ideal for the initial assessment of the feasibility of immunotherapy targeting pathological tau conformers.
Other tau-related therapeutic approaches include the following: (1) drugs that inhibit the kinases or activate the phosphatases that affect the state of tau phosphorylation (Iqbal and Grundke-Iqbal, 2004; Noble et al., 2005), (2) microtubule stabilizing drugs (Michaelis et al., 2005; Zhang et al., 2005), (3) compounds that interfere with tau aggregation (Pickhardt et al., 2005), and (4) drugs that promote heat shock protein-mediated clearance of tau (Dickey et al., 2005). Although all these approaches are certainly worth pursuing, target specificity and toxicity are of a concern, which emphasizes the importance of developing concurrently other types of tau-targeting treatments, such as immunotherapy.
In the present study, we sought to determine the effectiveness of active immunization directed against phosphorylated tau conformers in the CNS. Clearance of extracellular tangles may reduce associated pathology, and numerous reports of neuronal uptake of antibodies suggest that intracellular tangles and pretangles may also be affected (Fabian and Ritchie, 1986; Fabian and Petroff, 1987; Liu et al., 1989; Dietzschold et al., 1992; Aihara et al., 1994; Mohamed et al., 2002). Toward this end, homozygous P301L mice were immunized with a phosphorylated tau epitope with subsequent analysis of tau pathology and associated functional impairments. While these studies were underway, the feasibility of this approach was strengthened by findings indicating that vaccination with recombinant α-synuclein in transgenic mice reduces intraneuronal α-synuclein aggregates (Masliah et al., 2005b).
Materials and Methods
Peptides
The peptide immunogen Tau379–408[P-Ser396,404] was synthesized at the Keck facility (Yale University, New Haven, CT), by the solid-phase technique on a p-methyl-benzhydrylamine resin, using a Biosearch SAM 2 synthesizer (Biosearch Technologies, Novato, CA). The peptide was cleaved from the resin with hydrofluoric acid and then extracted with ether and acetic acid before lyophilization. Subsequently, it was purified by HPLC with the use of a reverse-phase support medium (Delta-Bondapak; Waters, Milford, MA) on a 0.78 × 30 cm column with a 0–66% linear gradient of acetonitrile in 0.1% trifluoroacetic acid.
Antibody purification and FITC labeling
A polystyrene mini column (Pierce, Rockford, IL) was loaded with 1 ml of GammaBind plus Sepharose matrix (GE Healthcare, Little Chalfont, Buckinghamshire, UK) and allowed to settle. The column was washed extensively (at least 5 bed volumes) with binding buffer (0.01 m sodium phosphate, 0.15 m NaCl, and 0.01 m EDTA, pH 7.0) to return the pH to the neutral range. The column was then incubated with 1 ml of PBS-diluted mouse plasma for 5 min at room temperature. This was then reapplied once more over the column to increase the yield of bound antibodies. Unbound protein was washed from the column with the binding buffer. Sequentially, 250 μl of elution buffer (0.5 m acetic acid, pH 3.0) was applied to the column and aliquots of elution fractions were collected. The samples were quantified using the BCA protein assay kit (Pierce) and were also resolved on a 10% SDS-PAGE gel. The appropriate fractions containing the cleanest separation of IgG from other serum proteins (typically fractions 5–8) were pooled and dialyzed over night at 4°C in 0.1 m Tris buffer, pH 7.4, to quickly neutralize the elution reagent's pH, before using the samples for additional experiments.
For the labeling studies, FITC (Sigma, St. Louis, MO) was covalently conjugated to the IgG via primary amines. Briefly, 1 mg of IgG was dissolved in 100 mm sodium carbonate-bicarbonate buffer, pH 9.0, to a final reaction volume of 0.25 ml, and reacted with 5× molar ratio of FITC by adding 50 μl of FITC (1 mg/ml) very slowly in 5 μl aliquots. The FITC was solubilized in anhydrous dimethyl sulfoxide (DMSO). The reaction vial was then covered with aluminum foil and incubated for 8 h at 4°C with gentle stirring. Ammonium chloride was then added to 5 mm and the reaction was incubated at 4°C for 2 h. Lastly, xylene cyanol was added to 0.1%, and to separate unbound FITC, the mixture was applied to a PD-10 (Sephadex G-25M; GE Healthcare) column gel bed that had been equilibrated with PBS solution. Subsequently, the flow-through was collected, and the column was eluted with 2.5 ml of PBS, in 0.25 ml fractions. Two bands were visible during elution, and the conjugate was present in the first band (fractions 3–5), as determined by measuring the absorbance of each fraction at 280 nm and then at 495 nm (The and Feltkamp, 1970). The IgG was purified from a mouse with a high titer against the immunogen (Phos-tau) and purified mouse IgG (Sigma) from pooled mouse plasma was used in controls.
Fibrillogenicity
Aliquots of the tau peptide immunogen prepared in 0.1 m Tris, pH 7.4, were incubated for different times, and their fibril formation compared with that of Aβ1–42. In vitro fibrillogenesis was evaluated by an assay based on the fluorescence emission by thioflavin T (ThT), as we described previously (Sigurdsson et al., 2001, 2004). ThT binds specifically to β-sheet structure and this binding produces a shift in its emission spectrum and a fluorescent enhancement proportional to the amount of amyloid formed. After the incubation period, 50 mm glycine, pH 9.2, and 2 μm ThT were added to a final volume of 200 μl containing 3 μg of the immunogen or the P301L tau peptide (with or without equimolar concentration of antibodies purified from the immunized mice). Fluorescence was measured at excitation of 435 nm and emission of 485 nm on a SpectraMax M2 multidetection plate reader (Molecular Devices, Sunnyvale, CA).
Neurotoxicity
Potential neurotoxicity of the tau derivative (10 μm) was evaluated at 6 d in a human neuroblastoma cell line (SK-N-SH) using the standard MTT assay as described by the manufacturer (Roche, Indianapolis, IN). Aβ1–40 and Aβ1–42 were used as control peptides.
Animal studies
Mice
The studies were performed in the Tg P301L mouse model that develops neurofibrillary tangles in several brain regions and spinal cord [Taconic, Germantown, NY (Lewis et al., 2000)]. Although this model is not ideal for AD, it is an excellent model to study the consequences of tangle development and for screening therapy that may prevent the generation of these aggregates. Another advantage of these animals is the relatively early onset of pathology. In the homozygous line that we maintain, minor sensorimotor abnormalities associated with tau pathology can be observed at least as early as 3 months. These changes are progressive but the animals remain relatively healthy at least until 8 months of age. In other words, at that time point the animals ambulate and feed themselves, and they can perform the behavioral tasks sufficiently well to allow us to monitor treatment effect. At ∼12 months of age, these mice become severely impaired with hindleg paralysis, and cannot ambulate or feed themselves properly, and have to be killed.
Vaccine administration
Phos-tau peptide was mixed with Adju-Phos adjuvant (Brenntag Biosector, Frederikssund, Denmark) at a concentration of 1 mg/ml and the solution was rotated overnight at 4°C before administration to allow the peptide to adsorb onto the aluminum phosphate particles. The mice received a subcutaneous injection of 100 μl followed by a second injection 2 weeks later and then monthly thereafter. In the first study, vaccination started at 2 months of age and continued until the animals were 5 months of age at which time the animals were perfused and their organs were collected for analysis. In the latter study, the mice were immunized starting at 2 months of age and continued until the animals were 8 months. The mice went through a battery of sensorimotor tests at 5 months and again at 8 months of age before being killed. Control mice received the adjuvant alone. For gender split, see Tables 1–3.
Table 1.
| Dentate gyrus (MC1) |
Dentate gyrus (PHF1) |
Motor cortex (MC1) |
Brainstem (MC1) |
|||||
|---|---|---|---|---|---|---|---|---|
| M | F | M | F | M | F | M | F | |
| 2–5 months | ||||||||
| Tg control | 8.03 ± 2.44 | 37.05 ± 6.37+++ | 8.91 ± 3.62 | 23.96 ± 1.19++ | 0.89 ± 0.33 | 2.74 ± 0.48++ | 0.38 ± 0.12 | 4.72 ± 1.89++ |
| Tg Phos-tau | 6.94 ± 3.03 | 4.13 ± 1.26**** | 13.72 ± 6.89 | 2.78 ± 2.15*** | 0.09 ± 0.07* | 0.06 ± 0.03*** | 0.12 ± 0.04* | 0.19 ± 0.16** |
| 2–8 months | ||||||||
| Tg control | 7.39 ± 3.40 | 30.21 ± 2.17+++ | 9.46 ± 6.78 | 24.54 ± 5.19 | 2.09 ± 1.60 | 2.83 ± 1.54 | 1.88 ± 1.37 | 5.12 ± 1.29 |
| Tg Phos-tau | 3.00 ± 1.89 | 17.18 ± 4.19**/++ | 7.32 ± 5.67 | 15.08 ± 6.38* | 0.08 ± 0.07** | 0.36 ± 0.12* | 0.23 ± 0.18* | 1.39 ± 0.31**/++ |
aTreatment effect within gender: Significantly more effect of the immunization on reducing pathological tau was observed in females (eight of eight groups) than males (four of eight groups) compared with controls. *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001, significantly different from control mice within the same gender.
bGender differences: Significant gender differences were observed between some of the groups. In the 2−5 month study, male (n = 8) and female (n = 6) controls differed in all the areas analyzed but not Phos-tau-immunized males (n = 5) and females (n = 7). In the 2−8 month study (six males and six females per group), this difference was not as group specific with differences observed in the dentate gyrus (MC1) within both treatment groups and in the brainstem in the Phos-tau-immunized mice. +p ≤ 0.05, ++p ≤ 0.01, and +++p ≤ 0.001, respectively, significantly different from males within the same treatment group.
Table 2.
Western blot analysis: treatment effect within gendera and genderb differences(percentage density relative to total tau values)
| Insoluble tau: PHF1 |
Soluble tau: PHF1 |
Insoluble tau: MC1 |
Soluble tau: MC1 |
|||||
|---|---|---|---|---|---|---|---|---|
| M | F | M | F | M | F | M | F | |
| 2–5 months | ||||||||
| Tg control | 184 ± 35 | 96 ± 33 | 122 ± 15 | 169 ± 57 | 12 ± 3 | 17 ± 3 | 7 ± 1 | 3 ± 1+ |
| Tg Phos-tau | 116 ± 12 | 95 ± 13 | 282 ± 46** | 218 ± 52 | 18 ± 6 | 27 ± 8 | 20 ± 9 | 7 ± 2* |
| 2–8 months | ||||||||
| Tg control | 39 ± 11 | 96 ± 24 | 111 ± 15 | 55 ± 6++ | 11 ± 3 | 15 ± 6 | 54 ± 14 | 12 ± 4++ |
| Tg Phos-tau | 38 ± 13 | 68 ± 14 | 77 ± 14 | 62 ± 5 | 11 ± 2 | 32 ± 21 | 56 ± 13 | 9 ± 3++ |
aTreatment effect within gender: In the 2−5 month study, significantly more effect of the immunization on pathological tau was observed between males in levels of soluble PHF1 tau and in females in levels of soluble MC1. *p ≤ 0.05, **p ≤ 0.01, significantly different from control mice within the same gender.
bGender differences: Significant gender differences were observed between some of the groups, but these differences were not as prevalent as in the immunohistochemical analysis. In the 2−5 month study, male (n = 8) and female (n = 6) controls differed in soluble tau levels (MC1) but not Phos-tau-immunized males (n = 5) and females (n = 7). A similar pattern was observed in the immunohistochemical analysis with differences only observed in control groups. In the 2−8 month study (six males and six females per group), this difference was not as group specific with differences observed in soluble tau (MC1, PHF1) within controls. In the Phos-tau-immunized mice, gender differences were detected in soluble tau (MC1). +p ≤ 0.05, ++p ≤ 0.01, significantly different from males within the same treatment group.
Table 3.
Western blot analysis: treatment effect within gendera and genderb differences[ratio of values in Table 2 (soluble/insoluble)]
| Ratio (soluble/insoluble) |
||||
|---|---|---|---|---|
| PHF1 |
MC1 |
|||
| M | F | M | F | |
| 2–5 months | ||||
| Tg control | 0.83 ± 0.14 | 2.04 ± 0.41++ | 0.76 ± 0.15 | 0.16 ± 0.04++ |
| Tg Phos-tau | 2.72 ± 0.62** | 2.36 ± 0.50 | 1.55 ± 0.90 | 0.36 ± 0.11 |
| 2–8 months | ||||
| Tg control | 4.26 ± 1.19 | 1.00 ± 0.38+ | 5.73 ± 1.64 | 2.07 ± 0.88 |
| Tg Phos-tau | 2.98 ± 0.61 | 1.31 ± 0.45 | 5.20 ± 1.39 | 0.83 ± 0.34++ |
aTreatment effect within gender: In the 2−5 month study, the ratio of soluble/insoluble tau differed between treated and control mice in PHF1 blots for males. **p ≤ 0.01, significantly different from control mice within the same gender.
bGender differences: In the 2−5 month study, male (n = 8) and female (n = 6) controls differed in tau ratio (soluble/insoluble; PHF1 and MC1) but not Phos-tau-immunized males (n = 5) and females (n = 7). A similar pattern was observed in the immunohistochemical analysis with differences only observed in control groups. In the 2−8 month study (six males and six females per group), this difference was not as group specific with differences observed in tau ratio (soluble/insoluble; PHF1) within controls and in the Phos-tau-immunized mice (MC1). +p ≤ 0.05, ++p ≤ 0.01, significantly different from males within the same treatment group.
Intracarotid injection of FITC-labeled antibodies
Mice were anesthetized with 2% isoflurane and maintained with 1.5% isoflurane in 70% N2O and 30% O2. After exposing the carotid sheath, left common carotid artery (CCA), external carotid artery (ECA), and internal carotid artery (ICA) were exposed via a midline incision. A silk suture was tied to the distal end of the ECA, and the left CCA, ICA, and pterygopalatine artery were temporarily tied. A 30 gauge needle connected to PE-10 tubing (Becton Dickinson, San Diego, CA) was attached to a 1 ml syringe, mounted on a pump, and the FITC–IgG conjugate was then administered over a period of 10–15 min through the needle punctured upward into the common carotid. Subsequently, the needle was slowly withdrawn and glue was applied to the site of injection to prevent postoperative bleeding. One hour later, the mice were perfused transaortically with PBS and periodate–lysine–paraformaldehyde (PLP) and postfixed in PLP overnight. The brains were then placed overnight in a phosphate buffer solution containing 20% glycerol and 2% DMSO, and subsequently serial coronal 40 μm sections of the brain were prepared and subject to fluorescent microscopy. Our previous experience with developing peptidic ligands for magnetic resonance imaging of amyloid plaques indicates that identical method of intracarotid injection, without the use of an osmotic agent, does not disrupt the blood–brain barrier (Wadghiri et al., 2003; Sigurdsson et al., 2007). Work by others supports this observation (for review, see Rapoport, 2000). We decided on the 1 h interval based on previous studies of uptake of proteins into brain neurons after intracarotid injection (Carro et al., 2000).
Antibody response
The mice were bled before the commencement of the study and a week after each injection. The antibody response to the vaccine was determined by serial dilution of plasma using an ELISA as we described previously (Sigurdsson et al., 2001, 2004), in which 0.5 μg per well of the peptide immunogen was coated onto microtiter wells (Immulon 2HB; Thermo Electron Corporation, Milford, MA). The anti-tau autoantibodies in the plasma of Tg and control animals were also detected by ELISA on plates coated with 0.5 μg per well of recombinant full-length wild-type (wt) or P301L human tau protein. For detection, goat anti-mouse IgG linked to a horseradish peroxidase (GE Healthcare) was used at 1:3000 dilution. Tetramethyl benzidine (Pierce) was the substrate.
Expression and purification of recombinant tau protein
pcDNA3.1(+) mutant tau P301L construct or its wild-type equivalent, expressing tau which has two N-terminal exons (58 residues) and contains all the microtubule binding repeats in the longest human tau isoform, was generously provided by Dr. M. Hutton (Mayo Clinic, Jacksonville, FL). The cDNAs were subcloned in pET30a vector using standard methods. The plasmid DNA was prepared using the Qiagen protocol (Qiagen, Chatsworth, CA), and used to transform Escherichia coli BL21-DE3 competent cells. The clones were sequenced after transformation to verify the presence of the desired mutation and to confirm that the rest of the tau sequence was identical with the published human cDNA sequence.
Tau purification was performed as described previously (Connell et al., 2001). Transformed bacteria were grown in LB broth containing kanamycin. The expression plasmid within the transformed cells was then induced to express the tau protein of interest by adding IPTG (isopropyl β-d-thiogalactoside). The bacterial suspension was then centrifuged and the pellets were resuspended in 50 mm MES buffer, pH 6.5, with protease inhibitors (Complete; Roche), sonicated, and centrifuged at 15,000 × g at 4°C for 25 min. The supernatant was added to equal volume of 0.5 m NaCl to separate out tau in its soluble form. This solution was boiled until a white precipitate formed, and was subsequently centrifuged at 4°C for 2 h at 100,000 × g, and then tau was precipitated out of the supernatant with ammonium sulfate. After centrifugation at 4°C for 30 min at 15,000 × g, the supernatant was removed and the tau pellets were resuspended in 50 mm MES, pH 6.5, containing 1 mm DTT and 50 mm NaCl. After dialysis, the tau was purified with FPLC.
Histology
Mice were anesthetized with sodium pentobarbital (120 mg/kg, i.p.) and perfused transaortically with PBS, and the brains were processed as described previously (Sigurdsson et al., 2001, 2004; Sigurdsson, 2005). Briefly, the right hemisphere was immersion fixed overnight in PLP, whereas the left hemisphere was snap-frozen for tau protein analysis (see below, Western blotting). After fixation, the brain was moved to a phosphate buffer solution containing 20% glycerol and 2% DMSO and stored at 4°C until sectioned. Serial coronal brain sections (40 μm) were cut on a freezing microtome and every 10th section processed as free-floating sections and stained with two tau antibodies that recognize abnormal tau protein (PHF1, MC1). The remaining series were placed in ethylene glycol cryoprotectant and stored at −20°C until used.
Tau antibodies.
Staining was performed as described previously (Sigurdsson et al., 2001, 2004). Briefly, sections were incubated in 0.3% H2O2 for 15 min to block endogenous peroxidase activity, and then incubated in mouse-on-mouse (MOM) blocking reagent from the MOM immunodetection kit (Vector Laboratories, Burlingame, CA) to block nonspecific binding for 1 h at room temperature. The sections were incubated in the tau primary antibodies at a 1:50 to 1:100 dilution (MC1, PHF1; generously provided by Dr. Peter Davies, Albert Einstein College of Medicine, Bronx, NY). The anti-mouse IgG secondary antibody from the MOM kit was used at a 1:2000 dilution.
Epitope unmasking.
For heat-induced epitope retrieval, brain sections were mounted on microscopic slides and allowed to dry. Subsequently, the racked slides were placed in sodium citrate buffer (10 mm sodium citrate, 0.05% Tween 20, pH 6.0) that had been preheated in a microwave at 700 W. A circle of 1 min heating and 1 min cooling was performed for 10–20 min. After the heat processing, the slides were allowed to cool in buffer at room temperature for at least 20 min. The sections were rinsed in PBS at least three times for 1 min each time before proceeding with standard histology procedure with the MC1 and PHF1 antibodies.
Mice that received intracarotid injection of FITC-labeled purified mouse IgG were perfused 1 h after the injection with PBS until the perfusate was clear and subsequently with PLP for 10 min followed by further PLP fixation overnight. The brain was then placed overnight in the glycerol/DMSO phosphate buffer, and then immediately sectioned, mounted, and coverslipped using fluorescence compatible mounting media (DakoCytomation, High Wycombe, UK). Adjacent sections were stained free-floating with PHF1 or MC1 anti-tau antibodies followed by reaction with Texas Red-labeled anti-mouse IgG. Sections were counterstained with 4′-6-diamidino-2-phenylindole (DAPI), which labels nuclei. In contrast to the regular immunohistochemistry described above, blocking solution was not used before applying the primary antibody.
Image analysis
Analysis of tissue sections was quantified with a Bioquant (Nashville, TN) image analysis system. The software uses hue, saturation, and intensity to segment objects in the image field. Thresholds were established with accurately identified objects on a standard set of slides and these segmentation thresholds remained constant throughout the analysis session. After establishing the threshold parameter, the image field was digitized with a frame grabber. The Bioquant software corrects for heterogeneity in background illumination (blank field correction) and calculates the measurement parameter for the entire field. For quantitative image analysis of immunohistochemistry, we initially selected the granular layer of the dentate gyrus, which consistently contained intraneuronal tau aggregates (pretangles and tangles). This observation concurs with the original characterization of this model (Lewis et al., 2000). We analyzed the motor cortex and brainstem in these animals as well because tau pathology in those regions may relate to the motor abnormalities observed in this model. An individual blind to the experimental conditions of the study performed all procedures. Sample numbers were randomized before the start of the tissue processing, and the code was broken only after the analysis was complete. For each antibody stain (PHF1, MC1), we sampled every 10th section from the mouse brain. In the dentate gyrus, the measurement was the percentage of area in the measurement field at 200× magnification occupied by reaction product with the tip of the dentate gyrus at the left edge of the field. Four to five sections were analyzed per animal. In the motor cortex, the measurement was the percentage of neuronal staining in the field at 100× magnification with the thickest region of the cingulum positioned at the lower left edge of the field. In the brainstem, the measurement was the percentage of neuronal staining in the field at 100× magnification with the center of the top edge of the field positioned below the Aqueduct of Sylvius. Five sections were analyzed per animal in those two brain regions.
Western blotting
Brains were homogenized in Tris-buffered saline (TBS) (10 mm Tris/150 mm NaCl, pH 7.4) containing protease inhibitors (1 tablet in 10 ml of TBS; Complete mini protease inhibitor mixture tablet; Roche) and phosphatase inhibitors (1 mm NaF, 1 mm PMSF, 0.4 μm Na3VO4, and 0.5 μm okadaic acid). After initial preclearance centrifugation at 20,800 × g for 5 min, the postnuclear supernatants were centrifuged at 100,000 × g for 60 min at 4°C in Beckman TL-100 ultracentrifuge (Beckman, Palo Alto, CA), and the high-speed supernatants were collected. The resulting pellets were resuspended with an equal volume of homogenization buffer containing 0.1% SDS, pH 8.0, to generate high-speed soluble samples.
Equal amount of protein (BCA assay) was loaded and the samples were electrophoresed on 10% SDS-PAGE gels and transferred to nitrocellulose membranes. All blots were blocked (5% nonfat milk and 0.1% Tween 20 in TBS), and then incubated with various primary antibodies overnight. Subsequently, the blots were washed and incubated for 1 h at room temperature with peroxidase-conjugated, goat anti-rabbit or anti-mouse IgG (1:2000; GE Healthcare), followed with washing and detection of bound antibodies (ECL; Pierce). Immunoreactivity of tau proteins was analyzed from scanned films using UN-SCAN-IT software. To compare the relative amount of tau protein, the densities of the immunoreactive bands corresponding to phospho-tau were normalized and reported, relative to the amounts of total tau protein instead of actin levels, because some studies reported that changes in pathophysiological conditions and interactions with extracellular matrix components can alter actin protein synthesis, rendering actin unsuitable as an internal standard (Schmittgen and Zakrajsek, 2000; Fulga et al., 2007).
Antibodies for tau histology and blots
PHF1 mAb (kindly provided by Dr. Peter Davies) was used at 1:500 for immunoblots and 1:50 for immunohistochemistry. PHF1 mAb recognizes phosphorylated serines 396 and 404 located outside the microtubule-binding repeat on the C terminal of PHF tau protein (Otvos et al., 1994). MC1 tau antibody from Dr. Peter Davies was used as well (Jicha et al., 1999). Both these antibodies recognize the neurofibrillary tangles in the P301L mouse model (Lewis et al., 2000). MC1 is a conformation dependent IgG1 antibody that is similar to Alz-50 (IgM). Its reactivity depends on both the N terminus (amino acids 7–9), and an amino acid sequence of tau (amino acids 313–322) in the third microtubule binding domain that is both necessary and sufficient for in vitro formation of filamentous aggregates of tau similar to those seen in AD (Jicha et al., 1999). The pathological conformation of tau recognized by MC1 may precede the aggregation of tau into filaments and the resultant neurofibrillary degeneration seen in AD (Jicha et al., 1999). 3G6 monoclonal antibody (generously provided by Prof. Brian Anderton, Kings College London, London, UK), which recognizes total tau and anti-actin polyclonal antibody (1:2000; Sigma), were used as controls on Western blots.
Behavioral studies
Several tests were performed in the mice to determine (1) whether the tau-targeting therapy prevented or reversed the age-related sensorimotor abnormalities observed in the P301L mice, and (2) whether the immunization had any behavioral effects per se. Cognitive analysis of these homozygous animals has not been previously reported and we determined that their sensorimotor defects would not enable them to properly navigate the radial arm maze, which we have used extensively to assess the cognitive status of Tg2576 mice that deposit Aβ within the brain (Sigurdsson et al., 2004; Asuni et al., 2006). Hence, we used an object recognition test that requires less navigation. The tests were as follows: (1) locomotor activity; (2) motor and reflex behaviors: (a) traverse beam test and (b) accelerating rotarod; and (3) memory test: object recognition.
Sensorimotor tests
Before testing, the mice were adapted to the room with lights on for 15 min.
Locomotor activity.
Exploratory locomotor activity was recorded in a circular open field activity chamber (70 cm in diameter). A video camera mounted above the chamber automatically recorded horizontal movements in the open field in each dimension (i.e., x, y, and two z planes). Total distance was measured in centimeters traveled and is defined as sequential movement interruptions of the animal (white) measured relative to the background (black). The duration of the behavior was timed for 15 min. Results are reported based on distance traveled (in centimeters), mean resting time, and velocity (mean and maximum) of the animal.
Rotarod.
The animals were placed onto the rod (diameter, 3.6 cm) apparatus to assess differences in motor coordination and balance by measuring forelimb and hindlimb motor coordination and balance (Rotarod 7650 accelerating model; Ugo Basile, Biological Research Apparatus, Varese, Italy). This procedure was designed to assess motor behavior without a practice confound. The animals were habituated to the apparatus by receiving training sessions of two trials, sufficient to reach a baseline level of performance. Then, the mice were tested an additional three times, with increasing speed. During habituation, the rotarod was set at 1.0 rpm, which was gradually raised every 30 s, and was also wiped clean with 30% ethanol solution after each session. A soft foam cushion was placed beneath the apparatus to prevent potential injury from falling. Each animal was tested for three sessions, with each session separated by 15 min, and measures were taken for latency to fall or invert (by clinging) from the top of the rotating barrel.
Traverse beam.
This task tests balance and general motor coordination and function integration. Mice were assessed by measuring their ability to traverse a graded narrow wooden beam to reach a goal box [modification of Torres et al. (1994) specifically examining hindlimb function]. The mice were placed on a 1.1-cm-wide beam that is 50.8 cm long and suspended 30 cm above a padded surface by two identical columns. Attached at each end of the beam is a shaded goal box. Mice were placed on the beam in a perpendicular orientation to habituate, and were then monitored for a maximum of 60 s. The number of foot slips each mouse had before falling or reaching the goal box were recorded for each of four successive trials. Errors are defined as foot slips and were recorded numerically. To prevent injury from falling, a soft foam cushion was always kept underneath the beam. Animals that fell off were placed back in their position before the fall.
Cognitive test: object recognition
The spontaneous object recognition test that was used measures deficits in short-term memory and was conducted in a square-shaped open-field box (48 cm square, with 18-cm-high walls constructed from black Plexiglas), raised 50 cm from the floor. The light intensity was set to 30 lx. On the day before the tests, mice were individually habituated in a session in which they were allowed to explore the empty box for 15 min. During training sessions, two novel objects were placed at diagonal corners in the open field and the animal was allowed to explore for 15 min. For any given trial, the objects in a pair were 10 cm high, and composed of the same material so that they could not readily be distinguished by olfactory cues. The time spent exploring each object was recorded by a tracking system (San Diego Instruments, San Diego, CA), and at the end of the training phase, the mouse was removed from the box for the duration of the retention delay (retention delay, 3 h). Normal mice remember a specific object after a delay of up to 1 h and spend the majority of their time investigating the novel object during the retention trial. During retention tests, the animals were placed back into the same box, in which one of the previous familiar objects used during training was replaced by a second novel object, and allowed to explore freely for 6 min. A different object pair was used for each trial for a given animal, and the order of exposure to object pairs as well as the designated sample and novel objects for each pair were counterbalanced within and across groups. The time spent exploring the novel and familiar objects was recorded for the 6 min. The percentage short-term memory score is the time spent exploring any one of the two objects (training session) compared with the novel one (retention session).
The objective of these experiments was to evaluate the effects of the vaccination on selected sensorimotor and cognitive behaviors. The homozygous P301L mice have tangle pathology as early as 3 months of age and those animals were tested at 5 and 8 months of age.
Data analysis
All the data were analyzed with GraphPad (San Diego, CA) Prism 4.3. The amount of tau aggregates on Western blots, the immunoreactivity on brain sections within the dentate gyrus, motor cortex, and brainstem, the locomotor activity measurements (distance, Vmax, Vmean, rest time), and the object recognition test were analyzed by an unpaired t test. When the data failed at least two of three normality tests (Kolmogorov–Smirnov, D'Agostino and Pearson omnibus, and Shapiro–Wilk normality tests), nonparametric Mann–Whitney test was used. The test was two-tailed when gender differences were analyzed but otherwise one-tailed, because we predicted that the immunotherapy would clear tau pathology that would slow the progression of behavioral impairments. Neurotoxicity was analyzed by one-way ANOVA and Newman–Keuls post hoc test. The data from the traverse beam and rotarod were analyzed by two-way ANOVA repeated measures and a Bonferroni post hoc test. Correlation between behavioral outcome and tau pathology as assessed by immunohistochemistry or Western blotting was analyzed by Pearson r correlation or Spearman rank correlation if the data failed normality test.
Results
In vitro assays
To determine the therapeutic potential of tau-based immunotherapy, we designed a phosphorylated immunogen, Tau379–408[P-Ser396,404], that was likely to elicit generation of antibodies that would selectively detect highly phosphorylated tau protein as found in AD and tangle mouse models. The tau protein like the Aβ peptide has the propensity to form β-sheets, which has been associated with toxicity. To confirm that our immunogen was not likely to have toxic effects in vivo, we determined with thioflavin T assay that it was not fibrillogenic with a fluorescence value of 0.9 ± 0.9 (SEM) at 10 d, whereas Aβ1–42 at the same time point had a value of 29.7 ± 2.2. Similar values were observed at 7 and 14 d. Furthermore, the tau-derived immunogen was not neurotoxic to SK-N-SH neurons compared with neurons treated with Aβ1–42 for 6 d (10 μm; 20% reduction in neuronal viability; p < 0.01) as assessed by the MTT assay (data not shown).
Animal studies
Antibody response and autoantibodies
We first immunized homozygous Tg P301L mice from 2 to 5 months of age with the Phos-tau peptide in aluminum adjuvant (n = 12; 5 males and 7 females). Control Tg P301L animals received adjuvant alone (n = 14; 8 males, 6 females). The mice elicited a robust antibody response against the immunogen, whereas minimal reactivity was observed in control mice (Fig. 1A). Surprisingly, autoantibodies that recognized recombinant tau (P301L and wild type) were observed in both control (alum-treated) and immunized (Phos-tau) Tg mice (Fig. 1B), but there were no autoantibodies in untreated control wild-type mice (data not shown). The level of autoantibodies observed may be indicative of a blood–brain barrier (BBB) dysfunction, and tau from damaged neurons may enter the systemic circulation and stimulate an antibody response. The autoantibody levels in the control mice are not significantly different from the immunized mice (Fig. 1B). Although the mean values in the controls are about double those observed in the immunized mice, this difference was mainly because of an outlier and hence the substantial variance in the control values.
Figure 1.
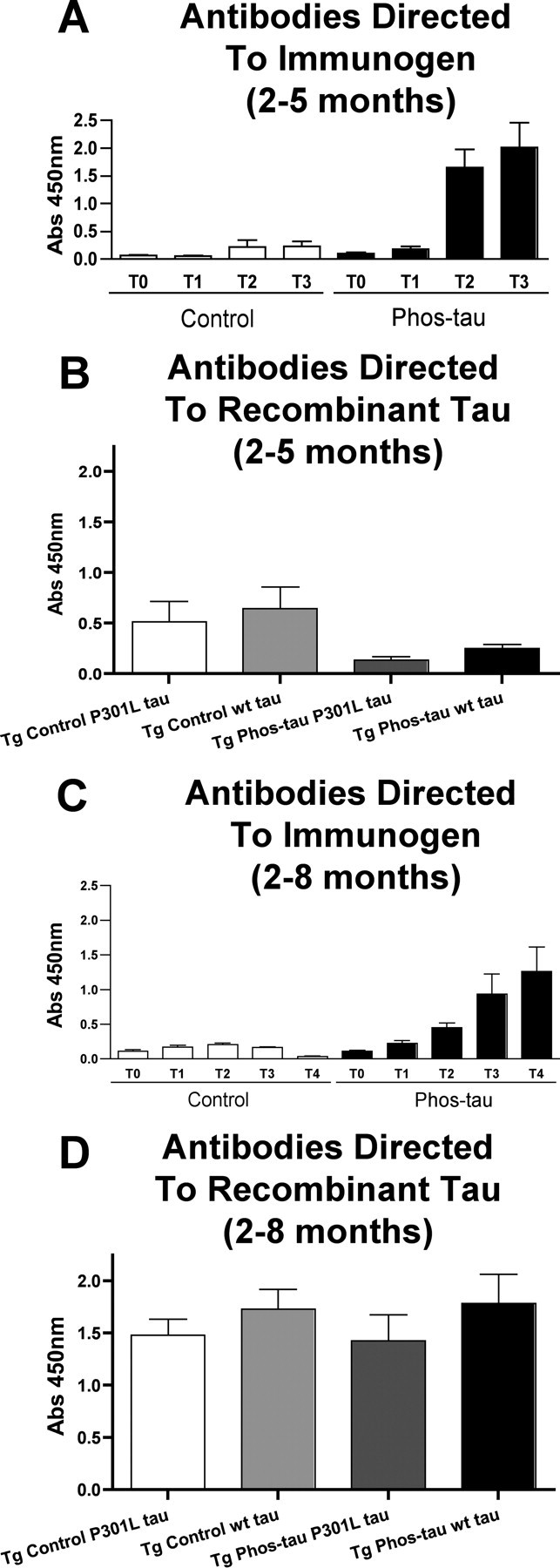
Phospho-tau derivative peptide is highly immunogenic in P301L mice, but autoantibodies against tau are detected. Homozygous Tg P301L mice were immunized from 2 months of age with a phosphorylated tau peptide (Phos-tau, Tau379–408[P-Ser396,404]; n = 24). Control Tg P301L animals received aluminum adjuvant alone (n = 25). Plasma samples from the animals were analyzed by ELISA, and the brains were analyzed biochemically and immunohistochemically at 5 months of age (Phos-tau, n = 14; controls, n = 12) and at 8 months of age (Phos-tau, n = 12; controls, n = 12). A, C, Shown is the generation of IgG antibodies (1:200 plasma dilution) against the immunogen at various time points as determined by Phos-tau peptide ELISA assay [A, 2–5 months: T0, T1, T2, T3 = 0, 7, 11, and 14 weeks; C, 2–8 months: T0, T1, T2, T3, T4 = 0, 7, 11, 14, and 26 weeks]. Control mice had low levels of autoantibodies that recognized the immunogen. B, D, Autoantibodies that recognized both wild-type and P301L human tau were observed in controls and immunized mice within both groups at the end of each study (B, 2–5 months; D, 2–8 months), but the levels did not differ significantly between the groups. Similar reactivity was observed toward wild-type and P301L human tau. Error bars indicate SEM.
A similar immune response was observed in another group of homozygous P301L mice that were immunized from 2 to 8 months of age (n = 12 per group; 6 males and 6 females per group). Again, a robust antibody response was elicited against the Phos-tau derivative, and some, although much less, reactivity was observed in control mice (Fig. 1C). Again, the plasma obtained at the end of the study in both the controls and immunized mice contained autoantibodies that recognized recombinant tau (P301L and wild type) (Fig. 1D). In these 8-month-old animals, the levels of these autoantibodies were substantially higher than in the 5-month-old mice in the previous study (Fig. 1B). It is well known that autoantibodies increase with age, and we have observed a similar phenomenon in the Tg2576 mouse model of cerebral Aβ amyloidosis although those animals are usually well into their second year when appreciable levels of autoantibodies are detected (Sigurdsson et al., 2004; Asuni et al., 2006).
Cross-reactivity between antibodies recognizing the immunogen and recombinant tau (wt and P301L) was minimal, and the autoantibodies had a similar affinity toward wt and P301L tau. These results imply that the autoantibodies are primarily recognizing normal tau as opposed to pathogenic tau.
Tau pathology
In the original description of a hemizygous line of this model, the most extensive pathology was observed in the brainstem and spinal cord (Lewis et al., 2000). The homozygous line that we used also has extensive pathology in these regions but appears to have more pathology in the hippocampal and cortical regions than hemizygous animals, which is as expected because of their homozygosity.
At 5 months of age, quantitative analysis of neuronal immunoreactivity in the dentate gyrus with two antibodies that selectively stain Alzheimer's tau protein revealed 74% (p < 0.01) (Figs. 2A, 3A,B) and 52% (p < 0.05) (Figs. 2B, 3C,D) reduction in MC1 and PHF1 immunoreactivity, respectively, in immunized mice compared with control mice that received adjuvant only. Antibodies generated against the immunogen, Tau379–408[P-Ser396,404], could potentially mask binding of the PHF1 antibody on brain sections because the PHF1 epitope contains the phosphorylated serine residues 396 and 404. It is theoretically unlikely that the conformational MC1 epitopes would be unmasked because those do not overlap with the immunogen. To clarify this issue, we performed standard unmasking procedure (see Materials and Methods) without any obvious improvement in PHF1 or MC1 staining relative to sections from control Tg mice (data not shown).
Figure 2.
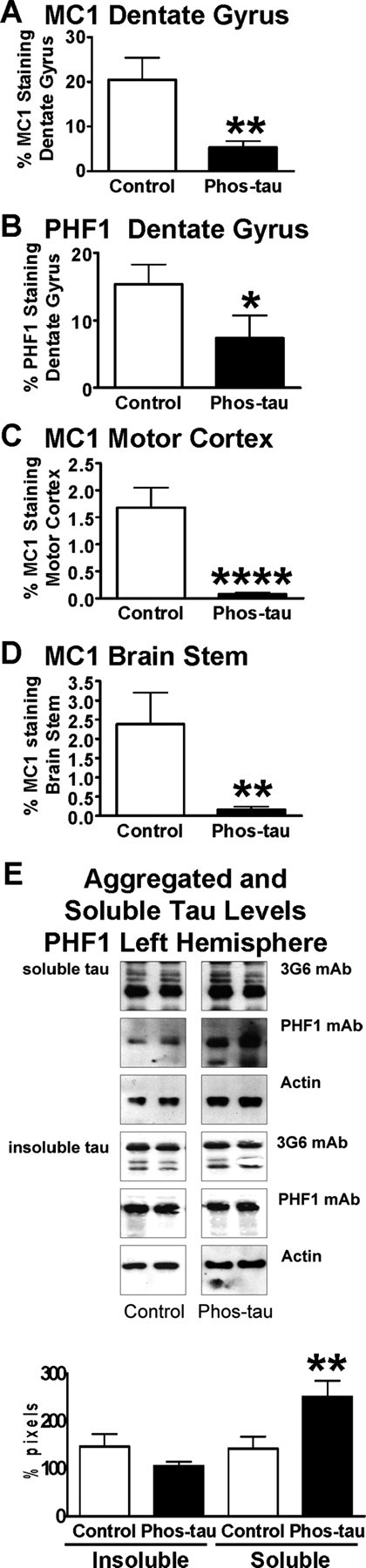
The vaccine reduces tau aggregates in the brains of P301L tangle mice at 5 months of age. A, Quantitative analysis of MC1 immunoreactivity within the granular layer of the dentate gyrus revealed a 74% reduction (**p < 0.01) in immunized mice compared with control Tg mice that received adjuvant alone. B, Likewise, PHF1 immunoreactivity within the granular layer of the dentate gyrus was reduced by 52% (*p < 0.05) in immunized mice compared with controls. C, Additional confirmation of a therapeutic effect was obtained by analysis of the motor cortex, in which MC1 neuronal staining was reduced by 96% (****p < 0.0001) compared with control Tg mice. D, Likewise, in the brainstem, MC1 neuronal staining was reduced by 93% (**p = 0.01) compared with control Tg mice. E, Densitometric analysis of PHF1 blots revealed a strong trend for reduction in insoluble tau (28% reduction; p = 0.09) and a significant increase in soluble tau (77% increase; **p = 0.01) in the immunized mice compared with control Tg mice, relative to total tau levels. Additional analysis of the ratio of soluble tau to insoluble tau indicated a significant increase in the immunized group on PHF1 blots (89% increase; p = 0.01), suggesting a mobilization of tau from its insoluble form to soluble form in these treated animals. The panel shows representative blots from control and Phos-tau-immunized mice. The PHF1 antibody recognizes phosphorylated serines 396 and 404 located outside the microtubule-binding repeat on the C terminus of PHF tau protein. Antibodies against total tau (3G6) and actin were used as controls. The same amount of protein was loaded in each line. Mean values are presented with SEM.
Figure 3.
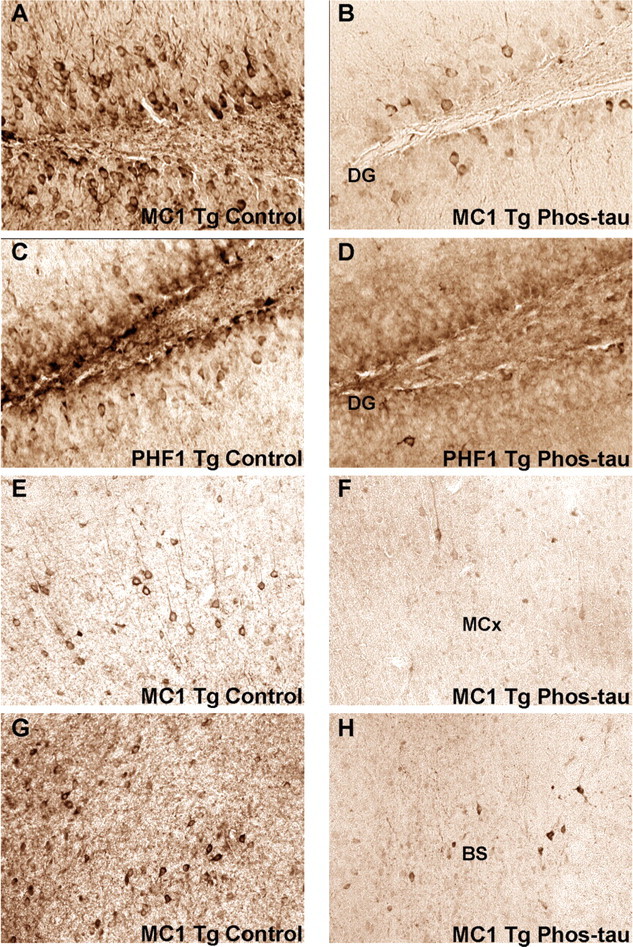
The immunotherapy reduces pathological tau in neurons. Representative examples of the histological regions that were analyzed in MC1- and PHF1-stained brain sections of mice at 5 months in the 2–5 month study group. Neuronal tau aggregates were cleared in the dentate gyrus (DG), the motor cortex (MCx), and the brainstem (BS) in immunized mice compared with control mice. The dentate gyrus develops extensive tau pathology at an early age in the homozygous P301L mice and tau pathology in the motor cortex and brainstem may relate to the motor deficits in this model. A, B, Coronal section through the dentate gyrus of MC1-stained section in control (A) versus immunized (B) Tg mouse (original magnification, 200×). C, D, Coronal section through the dentate gyrus of PHF1-stained section in a control (C) versus immunized (D) Tg mouse (original magnification, 200×). E, F, Coronal section through the motor cortex of MC1-stained section in a control (E) versus immunized (F) Tg mouse (original magnification, 100×). G, H, Coronal section through the brainstem below the aqueduct of Sylvius of MC1-stained section in a control (G) versus immunized (H) Tg mouse (original magnification, 100×). The following magnifications were used for the quantitative analysis: dentate gyrus, 200×; motor cortex, 100×; brainstem, 100×.
MC1 recognizes earlier stage of tau pathology than PHF1 (Jicha et al., 1999), and these intermediates should be more easily cleared than higher order aggregates, which may explain the greater reduction in MC1 compared with PHF1 immunoreactivity. Alternatively, the mechanism responsible for clearing pathology selects for an abnormal conformation of tau, and MC1-reactive tau corresponds to those species.
To confirm this finding in other brain regions that may better relate to the motor abnormalities in the model, neuronal MC1 staining was quantified in the motor cortex and brainstem, and immunoreactivity was reduced by 96% (p < 0.0001) (Figs. 2C, 3E,F) and 93% (p = 0.01) (Figs. 2D, 3G,H), respectively, compared with control Tg mice. No significant correlation was observed between antibody levels against the immunogen, recombinant wild-type tau, or P301L tau versus the immunohistochemical analysis. Western blot analysis did not show as robust treatment effect as observed with immunohistochemistry (Fig. 2E). Densitometric analysis of the PHF1 blots revealed a strong trend for reduction in insoluble tau (28% reduction; p = 0.09) and a significant increase in soluble tau (77% increase; p = 0.01) in the immunized mice compared with control Tg mice. No significant changes were observed on MC1 blots, although there was a strong trend for an increase in soluble tau (167% increase; p = 0.10) (data not shown). The Western blot data did not correlate exactly with the immunohistochemical findings, although a similar gender differences were detected, and a more robust treatment effect was observed with both types of analysis in the 5-month-old group compared with the 8-month-old animals. Any discrepancy may at least in part be related to the artificial antibody epitopes that are generated in the blots. It is well established that both the MC1 and PHF1 antibodies show greater specificity toward pathological tau on histological sections than in Western blots (Greenberg et al., 1992; Rye et al., 1993; Jicha et al., 1997; Weaver et al., 2000). For example, the MC1 antibody binds to recombinant tau on Western blots (Jicha et al., 1997), indicating its lack of specificity under those conditions as opposed to on fixed brain sections, on which it specifically binds to abnormal tau conformation (Weaver et al., 2000). Furthermore, the conformational MC1 epitope is likely to be in large part denatured in the SDS-PAGE as evident by its relatively limited reactivity on the blots (data not shown).
Additional analysis of the ratio of soluble tau to insoluble tau indicated a significant increase in the immunized group on PHF1 blots (89% increase; p = 0.01), suggesting a mobilization of tau from its insoluble form to soluble form in these treated animals. Furthermore, a positive correlation was observed between antibody levels against the immunogen versus band density in PHF1 blots (insoluble tau, r = −0.60, p < 0.01; ratio of soluble to insoluble tau, r = 0.46, p < 0.03).
At 8 months of age, quantitative analysis of tau immunoreactivity in the dentate gyrus revealed a 47% reduction (p < 0.05) (Fig. 4A) in MC1 staining and a strong trend (40% reduction; p = 0.12) (Fig. 4B) for reduced PHF1 immunoreactivity. As at 5 months of age, MC1 neuronal staining was more robustly reduced in the motor cortex (76%; p = 0.02) (Fig. 4C) and brainstem (78%; p = 0.005) (Fig. 4D) than in the dentate gyrus. Biochemical analysis of the whole left hemisphere of the brain did not reveal significant differences in pelletable tau or soluble tau on PHF1 or MC1 blots (data not shown). Hence as expected, no correlation was observed between antibody levels against the immunogen versus band density on PHF1 or MC1 blots of brain homogenate.
Figure 4.
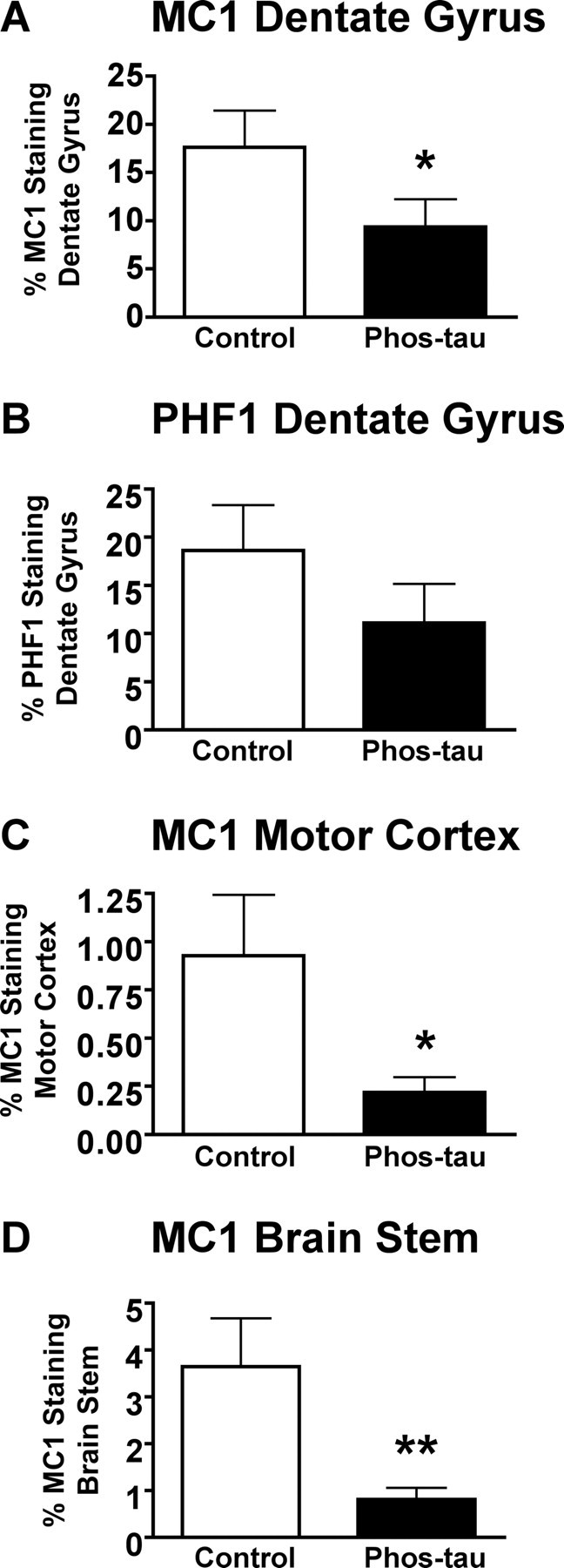
Immunotherapy from 2 to 8 months reduces brain tau pathology. A, Quantitative analysis of MC1 immunoreactivity within the granular layer of the dentate gyrus revealed a 47% reduction (*p < 0.05) in immunized mice compared with control Tg mice that received adjuvant alone. B, There was a strong trend for a diminished PHF1 immunoreactivity within the granular layer of the dentate gyrus (40% reduction; p < 0.12) in immunized mice compared with controls. C, As at 5 months of age (Fig. 2C), MC1 neuronal staining of the motor cortex revealed a more pronounced therapeutic effect (76%; *p = 0.02) than in the dentate gyrus. D, Likewise, neuronal MC1 staining was substantially reduced in the brainstem of immunized mice compared with controls (78%; **p = 0.005). Mean values are presented with SEM.
Regarding gender differences in tau pathology; more extensive tau pathology in females than males has been described in a hemizygous line of the P301L model (Lewis et al., 2000, 2001; Sahara et al., 2002). We took this into account in group assignments of the homozygous line of the P301L mice that we used. In the 5-month-old mice, when we compared gender differences in tau pathology as assessed by immunohistochemistry, the female controls had more tau pathology in all areas analyzed than male controls but no gender differences were observed between immunized mice (Table 1). When we reanalyzed the immunohistochemical data based on gender, greater treatment effect was observed among the females than males (Table 1). A similar gender-related pattern was observed on Western blots (Table 2), particularly in the ratio of soluble to insoluble tau (Table 3), but gender-specific treatment effect was not pronounced on the blots.
As in the treatment study that lasted from 2 to 5 months, significant gender differences in immunohistochemical tau pathology were also observed in the 2–8 month study group. Again, females had more pathology than males although only in one brain region in controls (MC1: dentate gyrus) and in two brain regions in immunized mice (MC1: dentate gyrus and brainstem) (Table 1). In contrast, in the 2–5 month study group, the males and females in the control group differed in all the brain regions analyzed but no gender differences were observed in the immunized mice (Table 1). As in the 2–5 month group, greater treatment effect was observed among the females than males when we reanalyzed the immunohistochemical data based on gender (Table 1). A similar gender-related pattern was observed on Western blots with the females having more insoluble tau and less soluble tau than their male counterparts (Tables 2, 3). However, gender-specific treatment effect was not observed at this time point.
Antibody characterization
The high level of autoantibodies observed in the animals complicates characterization of the antibodies that were generated toward the immunogen. Purified antibodies from several of the immunized mice stained specifically tau aggregates/tangles in neuronal cell bodies of P301L mice as the PHF antibody and did not react with tau in wild-type mice (Fig. 5). However, the PHF1 antibody stained more neuronal processes than the antibodies from the immunized mice and their pattern of neuronal perikarya staining often differed. Some staining of pretangles/tangles was occasionally observed with antibodies purified from control animals with high levels of tau autoantibodies, although most of them resulted in minimal or no staining in P301L or wild-type mice (Fig. 5). These findings indicate that the immunized mice generate antibodies that specifically recognize pathological tau aggregates in the P301L mouse, but some animals may contain autoantibodies with these properties. Likewise, the antibodies from the immunized mice stained tangle pathology in Alzheimer's brain, but as in the mice, neuronal processes were not prominently stained, compared with the PHF1 antibody (Fig. 6).
Figure 5.
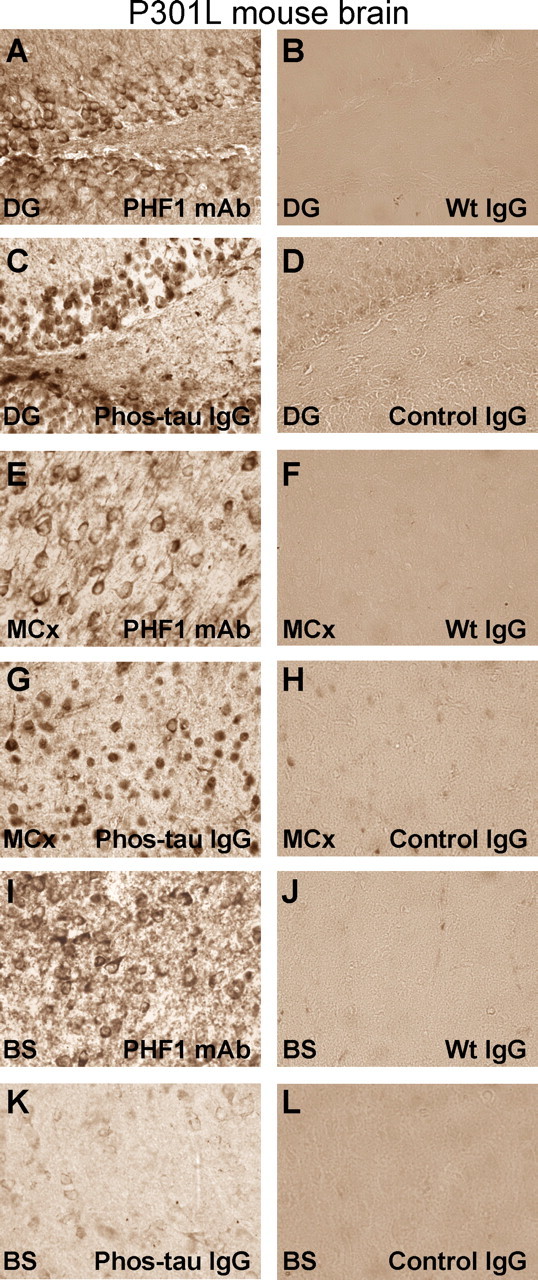
Purified antibodies from immunized mice stain tau aggregates/tangles in neuronal cell bodies in P301L mice. A–L, Adjacent coronal brain sections through the dentate gyrus (A–D), motor cortex (E–H), and brainstem (I–L; immediately below the Aqueduct of Sylvius) in a P301L transgenic mouse with tau pathology. A, The PHF1 antibody reveals the typical staining of tau aggregates/tangles in neuronal cell bodies as previously reported in this model (Lewis et al., 2000). Most of the staining is associated with the neuronal plasma membrane. B, D, Pooled mouse IgG from wild-type mice [Wt IgG (Sigma)] (B) or antibodies from mice immunized with the adjuvant only (Control IgG) (D) do not decorate neurons in the dentate gyrus. C, Antibodies from mice immunized with the Phos-tau peptide, which contains the PHF1 epitope, lack extensive dendritic staining, but stain primarily neuronal cell bodies within the dentate gyrus, but the pattern is not identical to the PHF1 staining. E, G, I, K, Similar staining pattern as in A and C is obsered in the motor cortex (E, G) and brainstem (I, K) after immunoreactivity with the PHF1 antibody and the polyclonal antibody from an immunized mouse, respectively. However, this particular polyclonal antibody stained neurons in the brainstem less intensely than in the dentate gyrus and motor cortex. F, H, J, L, These images depict adjacent coronal sections to those shown in E, G, I, and K, which were stained with purified antibodies from Tg control mice that received adjuvant alone (Control IgG) or pooled mouse IgG (Wt IgG). Staining with those antibodies resulted in minimal or no staining. No immunostaining was observed in wild-type mice with the antibodies purified from immunized mice (data not shown). These findings indicate that the immunized mice generate antibodies that specifically recognize pathological tau aggregates in the P301L mouse. Staining was performed as detailed in Materials and Methods with PHF1 and purified IgG used at a 1:250 and 10 μg/ml dilutions, respectively. Original magnification, 400×.
Figure 6.
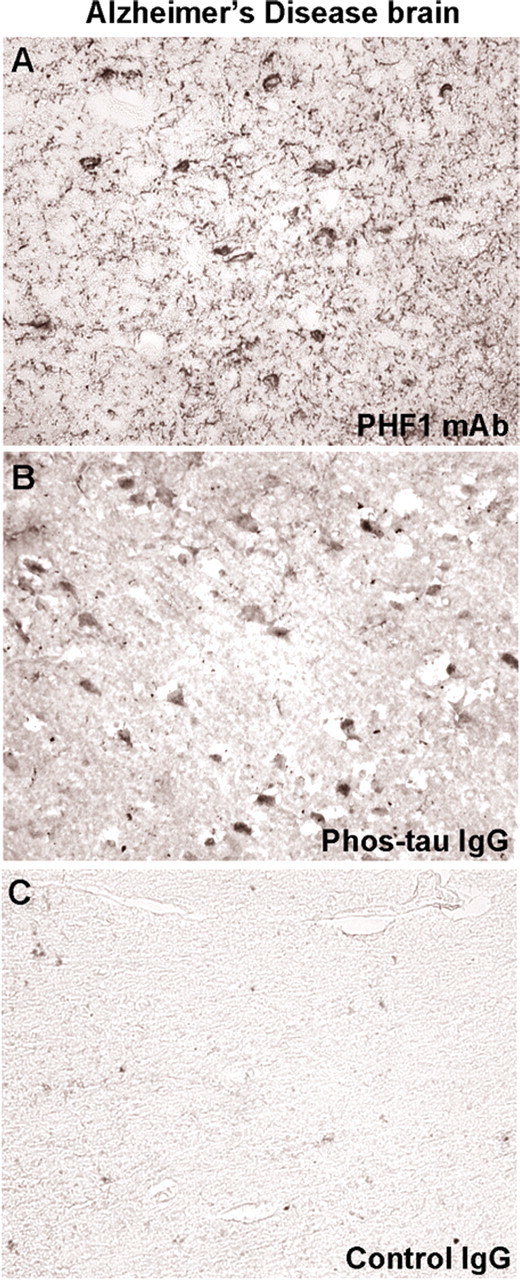
Purified antibodies from immunized mice stain tau aggregates/tangles in neuronal cell bodies in Alzheimer's disease similar to the PHF1 antibody. A, PHF1 staining of the entorhinal cortex from an Alzheimer's brain reveals the typical staining of cell bodies and dystrophic neurites as previously described for this antibody. B, The polyclonal IgG antibody derived from immunized mouse stains neuronal cell bodies, although this was dissimilar to PHF1 monoclonal antibody and dystrophic neurites are not prominent. C, Antibodies purified from a control mouse that received adjuvant alone do not result in appreciable staining. Overall, the staining pattern with these different antibodies is comparable with that observed in the P301L mouse (Fig. 5).
By staining selected mouse brain sections with secondary antibody against mouse IgG, intracerebral antibodies that label neurons were detected in the P301L mice (Fig. 7). To confirm that anti-tau antibodies would gain access into the brain, we FITC-tagged purified IgG antibodies from an immunized mouse that had generated very high levels of antibodies against the immunogen. The FITC-labeled IgG was subsequently injected into the carotid artery of 8-month-old P301L mice and their brains were harvested 1 h later for analysis of antibody uptake. Several neurons in various brain regions showed typical green FITC fluorescence (Fig. 8), and when the sections were incubated with PHF1 or MC1, these antibodies colocalized with the FITC-labeled IgG (Fig. 9). Figures 7–9 demonstrate perinuclear staining that is highly localized, with the distribution of neuronal staining clearly demarcating the neurons and accumulated predominantly in the apical part of cell soma and probably also partially in the plasma membrane. This staining pattern is similar to that described by others of the endosomal–lyzosomal compartments of neurons (Cataldo et al., 1997). Carotid injection in another set of 8-month-old P301L mice of FITC-labeled purified IgG from a control mouse that received only the alum adjuvant, resulted in some FITC fluorescence within the brain, although it did not appear to be neuronal and it did not colocalize with PHF1 or MC1 staining (data not shown). Identical approach in wild-type mice with the same antibodies did not lead to FITC fluorescence within the brain (data not shown). These findings suggest that leakage of the BBB in the P301L model may perhaps explain treatment efficacy. The observation of fluorescence in the brain after such an injection is also likely to be dependent on the presence of tau aggregates. Our previous studies are in agreement with this view, showing lack of appreciable uptake/binding of anti-tau antibodies (3G6 and PHF1) after 24 h incubation with cultured neurons (neuroblastoma cells and primary cultures) that do not have tau pathology (Asuni et al., 2004).
Figure 7.
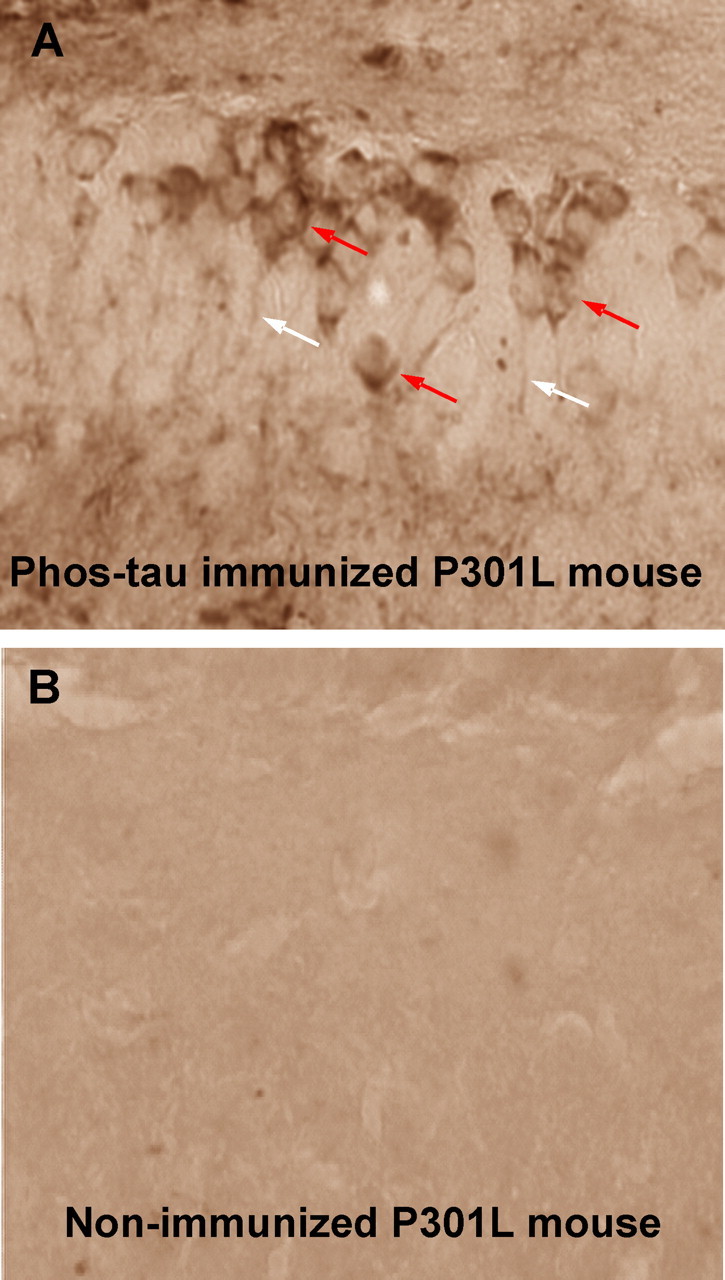
Intracerebral antibodies that label neurons are detected in the immunized P301L mice. A, A coronal brain section, representative of a high-titer P301L mouse, stained with an anti-IgG secondary antibody (1:50; Vectastain Elite kit) through the dentate gyrus of the hippocampus. Note the staining of neuronal cell bodies (red arrows) and processes (white arrows) indicating the presence of IgG. No immunostaining is observed in a representative nonimmunized P301L mouse of a similar age (B) or in a wild-type mouse (data not shown) under these staining conditions. Original magnification, 400×.
Figure 8.

FITC-tagged IgG antibodies from an immunized mouse label neurons in the brain after intracarotid injection into an 8-month-old P301L mouse. A coronal brain section through the brachium of the inferior colliculus revealing FITC-labeled neurons (arrows). Counterstain with DAPI (blue) shows nuclei of the neurons. Some FITC labeling was observed in control P301L mice of the same age that were injected with tagged IgG antibodies from pooled mouse plasma (Sigma), but neurons were not detected (data not shown). No appreciable FITC fluorescence was observed in wild-type mice of the same age that received intracarotid injection of IgG antibodies from an immunized mouse or control IgG (data not shown).
Figure 9.
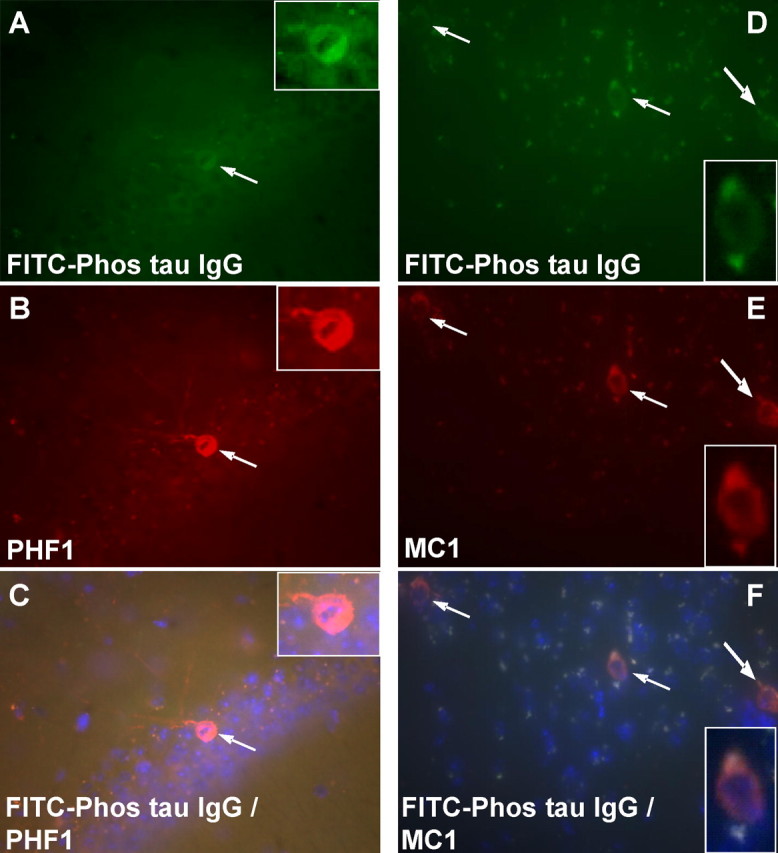
Neurons that label with the injected FITC-tagged antibody from an immunized mouse stain with MC1 and PHF1 antibodies. A–C, A coronal brain section through the pyramidal layer of the hippocampus. Note the FITC-labeled neurons in A that stain with PHF1 antibody that was applied to the section (B; Texas Red-tagged secondary antibody). The section was counterstained with DAPI that stains nuclei in blue and double-labeled neurons are orange (C). Original magnification, 200×. D–F, A coronal brain section through the nucleus of the brachium inferior colliculus. Note the FITC-labeled neurons in D that stain with MC1 antibody that was applied to the section (E; Texas Red-tagged secondary antibody). The section was counterstained with DAPI, which stains nuclei in blue, and double-labeled neurons are orange (F). Original magnification, 200×.
Behavior
At 5 months of age, the animals in the 2–8 month treatment group underwent their first behavioral testing to determine whether the therapy was associated with functional improvements. The immunization increased the time the animals were able to stay on the accelerating rotarod (p < 0.02) (Fig. 10A) and reduced the number of foot slips in the traverse beam task (p < 0.001) (Fig. 10B). Also, the vaccinated mice attained higher maximum velocity (Vmax, p = 0.004) (Fig. 10C) in the locomotor activity test but were not significantly different from Tg controls in the distance traveled, average speed (Vmean) or the resting time (data not shown). These animals continued to receive monthly immunizations and were retested on the same behavioral tests at 8 months of age. The mice were subsequently killed for biochemical and histological analysis of tau pathology. At 8 months of age, the treated animals continued to perform better than Tg controls on the rotarod and traverse beam, although the differences between the groups were not as substantial as during the previous comparison (rotarod, p < 0.05) (Fig. 10A) (traverse beam, p = 0.05) (Fig. 10B). The locomotor activity of the groups was similar at this time point (Fig. 10C, Vmax) (other data not shown). Overall, the groups performed worse at 8 months compared with 5 months. Because of the relatively poor mobility of the animals at this age, cognitive assessment was only performed with an object recognition test. This test indicated that both the immunized mice and their controls were cognitively normal, with the animals spending 60–70% of their time exploring the novel object (Fig. 10D), which is similar to wild-type mouse performance (our unpublished observations).
Figure 10.
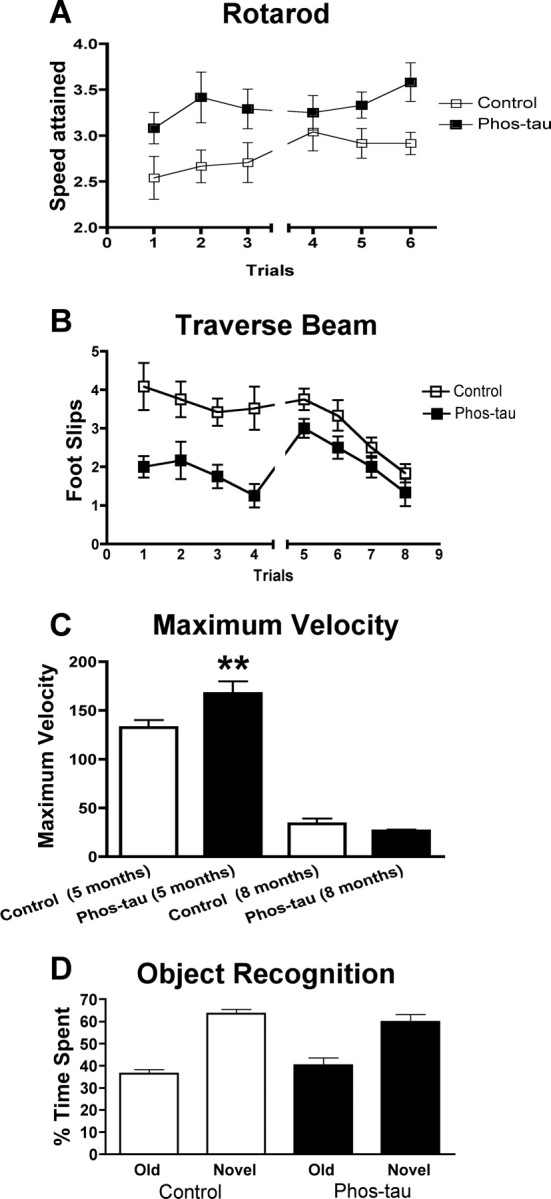
Immunotherapy from 2 to 8 months of age slows the progression of behavioral abnormalities in P301L mice. A shows the performance of Tg P301L-immunized and Tg control mice, trained to remain on a rotarod, and the speed attained during the task. The immunization increased the time the animals were able to stay on the rotarod both at 5 months (trials 1–3, p < 0.02) and 8 months (trials 4–6, p < 0.05). B shows the number of foot slips the animals had during the traverse beam task. The immunization greatly reduced number of foot slips during the performance of the task at 5 months (p < 0.001) and at 8 months (p = 0.05). C, There was no significant difference in the distance traveled, average speed (Vmean), or the resting time at 5 and 8 months (data not shown), but there was an increase in the maximum velocity (Vmax) attained by the phospho-tau-immunized Tg animals (**p = 0.004) at 5 months, compared with Tg controls. Vmax did not differ between the groups at 8 months. D, No difference was observed between the groups in the object recognition task that measures short-term memory. Both the immunized P301L mice and their transgenic controls spent a comparable time exploring the novel object that differed substantially from the time they spent with the old object. This finding indicates that both groups had normal short-term memory at 8 months of age. Error bars indicate SEM.
Interestingly, performance at 8 months on behavioral assays that require extensive motor coordination (traverse beam and rotarod) correlated with tau pathology (MC1) in corresponding brain areas (motor cortex vs traverse beam: r = 0.47, p = 0.01; motor cortex vs rotarod: r = −0.37, p = 0.04; brainstem vs traverse beam: r = 0.47, p = 0.01; brainstem vs rotarod: r = −0.45, p = 0.02). In addition, antibody levels against the immunogen but not the recombinant tau proteins (wild type and P301L) at the time of killing (T4) correlated with tau pathology in the brainstem (MC1, r = −0.48, p < 0.01) and dentate gyrus (MC1, r = −0.37, p = 0.04; PHF1, r = −0.44, p = 0.02), and there was a trend for a similar correlation in the motor cortex (MC1, r = −0.23, p = 0.14).
Regarding gender differences in behavior, it is of note that although tau pathology clearly varied depending on gender, this pattern was not seen in the behavioral analysis. The only significant difference between males and females within the same group was observed in the rotarod at 5 months (p < 0.05), with the females performing better (3.6 ± 0.2 rpm) than the males (2.8 ± 0.1 rpm).
Discussion
These results demonstrate that (1) vaccination of P301L mice with a phospho-tau epitope leads to the generation of antibodies that enter the brain; (2) these antibodies bind to abnormal tau like a monoclonal antibody (PHF1) against a similar epitope, although their staining pattern is not identical; (3) this type of immunotherapy reduces the extent of aggregated tau in the brain and slows the progression of the behavioral phenotype of these animals; and (4) as expected, the therapeutic effect decreases as the functional impairments advance in these animals.
Regarding the mechanism of tau-based immunotherapy, it is well established that ∼0.1% of circulating IgG is found within the CNS (Nerenberg and Prasad, 1975), and it may enter through regions that are deficient in BBB (Broadwell and Sofroniew, 1993), as has been shown for an antibody targeting Aβ (Banks et al., 2002). Also, IgG can be transported slowly across the BBB via adsorptive-mediated transcytosis (Zlokovic et al., 1990). In various neurological disorders such as AD, the BBB is thought to be compromised, suggesting that a substantially greater percentage of circulating IgG can be found within the CNS. Increased permeability of the BBB has been observed in plaque-depositing AD model mice (Poduslo et al., 2001; LaRue et al., 2004) but has not been assessed in tangle AD model mice, although our observations indicate that the BBB may be impaired in the P301L model. In addition to antibody uptake into the brain, it is also well established that antibody-secreting cells from the periphery can enter the brain and secrete the antibodies locally (Knopf et al., 1998). In the mouse immunotherapy studies targeting Aβ, IgG has been routinely found within the brain associated with extracellular Aβ deposits and phagocytic microglia (Schenk, 2002), and in the present study we detected neuronal antibodies within the brain by immunohistochemistry. In addition, we detected FITC-labeled IgG in brains of P301L mice but not in wild-type mice after intracarotid injection, which indicates that antibodies can enter the brain from the periphery in the P301L model. The absence of labeling in the wild-type mice can be explained by their intact BBB and/or their lack of tau aggregates. It is interesting to note that the levels of autoantibodies against tau increased with age, suggesting that age-related impairments of the BBB associated with the progression of brain pathology in these animals may expose the tau proteins as an antigen to the immune system. Transport of antibodies within the CNS has not been thoroughly investigated but IgG transport within and across cells in the periphery is essential for effective humoral immunity. Also, several studies have shown that antibodies can be found within neurons, which supports the feasibility of our approach (Fabian and Ritchie, 1986; Fabian and Petroff, 1987; Liu et al., 1989; Dietzschold et al., 1992; Aihara et al., 1994; Mohamed et al., 2002). The antibodies may enter the cells via pinocytosis (e.g., receptor-mediated endocytosis or fluid-phase endocytosis) (Lobo et al., 2004).
In cells that do not have surface antigens recognized by the antibody as is likely the case for tau antibodies, receptor-mediated uptake may occur via Fcγ or FcRn receptors (Lobo et al., 2004), and Fcγ receptors have been located on neurons (Mohamed et al., 2002; Andoh and Kuraishi, 2004). Indeed, it has been demonstrated that central neurons that project to the periphery such as motor neurons take up IgG at their synapses by retrograde axonal transport through receptors for the Fc portion of IgG (Fabian and Petroff, 1987; Mohamed et al., 2002). Additional receptors found on neurons that have been shown to mediate antibody uptake include the Thy1.1 receptor (Fabian, 1990) and the lipoprotein receptor-related protein (LRP) (Herz et al., 1990; Kounnas et al., 1995). Furthermore, LRP-mediated endocytosis has been shown to result in lysosomal degradation (Gonias et al., 2004). It can be postulated that, as a consequence of P301L tau-mediated neurodegeneration, the endosomal–lysosomal pathway is activated, which may facilitate uptake of antibodies into the neuronal cytoplasm. Abnormally folded tau may be taken up in the lysosomes by the protein quality control mechanism of the neurons, and this may be the site of its interaction with the antibodies, because those should be in the endosomal compartment subsequent to their receptor-mediated uptake. Once the endosomes fuse with the lysosomes, the binding of the pathological tau to the antibodies may then trigger degradation by the lysosomes. In addition to the intracellular interaction, clearance of extracellular tangles may also reduce associated pathology. While these studies were underway, recent findings from a related field support the validity of our approach and subsequent observations. Like tau in AD, α-synuclein aggregates intracellularly within the brain in Parkinson's disease, and immunization with α-synuclein in mice with this pathology clears these aggregates, which occurred most likely through the endosomal–lysosomal pathway (Masliah et al., 2005b). Another more recent report shows that antibodies against Aβ can be internalized in AD neuronal culture models of Aβ accumulation and clear intraneuronal Aβ aggregates via the endosomal–lysosomal pathway, which protects against synaptic alternations (Tampellini et al., 2007), further supporting our findings and interpretations.
Less effort has been spent on developing therapy targeting pathological tau conformers than their Aβ counterparts. With regard to immunotherapy, Aβ plaque clearance in the AN 1792 trial did not appear to affect tangle pathology (Nicoll et al., 2003; Ferrer et al., 2004; Masliah et al., 2005a), but in a mouse model Aβ immunotherapy cleared early tau pathology although hyperphosphorylated tau aggregates were not affected (Oddo et al., 2004), as in the human studies. These findings emphasize the need for therapy targeting pathological tau conformers. As we and others have observed in Aβ plaque mouse models, it may not be necessary to clear plaques to observe a cognitive benefit (Sigurdsson, 2006). This concept may also apply to tau pathology, because suppression of transgenic tau in a different P301L mouse model has been shown to improve memory although neurofibrillary tangles remained (Santacruz et al., 2005). In that model as well as in the htau model, region-specific dissociation of neuronal loss and neurofibrillary pathology has been observed (Andorfer et al., 2005; Spires et al., 2006), which suggests toxic effects of early stage pathological tau conformers that cannot easily be detected, analogous to Aβ oligomers. Other transgenic mice expressing human tau, including the model that we immunized, have shown tau pathology and neuronal loss (Paris et al., 1999; Lewis et al., 2000; Gotz et al., 2001a), suggesting a causal relationship. Any differences between these models may be attributable to a variety of factors such as tau sequence, expression levels, promoter, and mouse strain.
With this issue in mind, it is interesting that, although the model we used has age-related progression in tau pathology and functional impairments, immunohistochemical analysis revealed a similar degree of tau pathology at 5 and 8 months and the animals performed at a comparable level on the rotarod and the traverse beam at these different ages. However, their locomotor activity was substantially less at 8 months compared with 5 months. It is also of note that a significant correlation was observed between performance on the rotarod and the traverse beam and tau pathology in two of the three brain areas analyzed, namely the motor cortex and the brainstem, both of which have a prominent role in motor coordination, which indicates a direct relationship between the main pathological feature of this model and associated functional impairments.
Our immunotherapy approach was substantially more effective in the early stages of functional impairments in the animals (at 5 months) than at a later time point (at 8 months). These findings indicate that clearance of early-stage pathological tau conformers may be of a therapeutic benefit. These smaller aggregates should also be easier to clear than late-stage neurofibrillary tangles. In other words, the antibodies may be delaying the time course of the tau pathology, but with the age-related increase in the rate of tau aggregation the clearance becomes less efficient. As previously reported in the original heterozygous line of this model (Lewis et al., 2000), the homozygous females had more extensive neurofibrillary pathology than the males as observed by immunohistochemistry (Table 1). It was, therefore, surprising that the immunotherapy was more effective in the females. We are not aware of similar gender differences in therapeutic outcome in transgenic models of tau pathology. This phenomenon cannot be explained by gender differences in antibody response because males and females had an equivalent mean level of antibody response (for 1:200 dilution Abs at 450 nm, ∼2). A possible explanation is that more of the aggregated tau in the females is available for antibody-mediated disassembly because of its higher levels within the neurons. In the male animals, the lower amount of pathological tau may be more easily sequestered and thereby less accessible for antibody binding. A related possibility is that, because of its higher levels and presumably more rapid assembly, the tau aggregates in the females may be more soluble and hence more easily removed.
The Western blot data did not correlate exactly with the immunohistochemical findings although a similar gender differences were detected, and a more robust treatment effect was observed with both types of analysis in the 5-month-old group compared with the 8-month-old animals. Any discrepancy may at least in part be related to the artificial antibody epitopes that are generated in the blots. It is well established that both the MC1 and PHF1 antibodies show greater specificity toward pathological tau on histological sections than in Western blots (Greenberg et al., 1992; Rye et al., 1993; Jicha et al., 1997; Weaver et al., 2000).
It should be emphasized that these tau immunotherapy studies were performed in transgenic mice that are homozygous for the tau mutation P301L. These animals have a very aggressive phenotype with tau pathology detected within a few months of age, with related severe functional impairments observed a few months later. Our approach should be more effective in transgenic animals with a phenotype progression that more resembles the human condition, and eventually in humans with frontotemporal dementia and/or AD in which pathology develops over years or decades instead of months.
Overall, our present findings support the feasibility of immunotherapy targeting pathological tau conformers that may benefit AD patients and individuals with frontotemporal dementia caused by tau mutations.
Footnotes
This work was supported by National Institutes of Health–National Institute on Aging Grants AG20197 and AG5891 and by the Alzheimer's Association. We thank Dr. Peter Davies (Albert Einstein College of Medicine, Bronx, NY) for the PHF1 and MC1 antibodies, Dr. Brian Anderton (King's College London, London, UK) for the 3G6 antibody, Dr. Mike Hutton (Mayo Clinic Jacksonville, Jacksonville, FL) for the recombinant tau cDNA, Dr. Yong-sheng Li (New York University School of Medicine) for his help with the intracarotid injections of the FITC-labeled antibodies, and Dr. Blas Frangione for his support and encourangaement over the years.
References
- Aihara N, Tanno H, Hall JJ, Pitts LH, Noble LJ. Immunocytochemical localization of immunoglobulins in the rat brain: relationship to the blood-brain barrier. J Comp Neurol. 1994;342:481–496. doi: 10.1002/cne.903420402. [DOI] [PubMed] [Google Scholar]
- Andoh T, Kuraishi Y. Direct action of immunoglobulin G on primary sensory neurons through Fc gamma receptor I. FASEB J. 2004;18:182–184. doi: 10.1096/fj.02-1169fje. [DOI] [PubMed] [Google Scholar]
- Andorfer C, Acker CM, Kress Y, Hof PR, Duff K, Davies P. Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J Neurosci. 2005;25:5446–5454. doi: 10.1523/JNEUROSCI.4637-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asuni AA, Knudsen E, Frangione B, Wisniewski T, Sigurdsson EM. Antibody mediated modulation of Aβ induced neurotoxicity in cell culture. Neurobiol Aging. 2004;25:S581–S582. [Google Scholar]
- Asuni AA, Boutajangout A, Scholtzova H, Knudsen E, Li YS, Quartermain D, Frangione B, Wisniewski T, Sigurdsson EM. Vaccination of Alzheimer's model mice with Aβ derivative in alum adjuvant reduces Aβ burden without microhemorrhages. Eur J Neurosci. 2006;24:2530–2542. doi: 10.1111/j.1460-9568.2006.05149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks WA, Terrell B, Farr SA, Robinson SM, Nonaka N, Morley JE. Passage of amyloid-β protein antibody across the blood-brain barrier in a mouse model of Alzheimer's disease. Peptides. 2002;23:2223–2226. doi: 10.1016/s0196-9781(02)00261-9. [DOI] [PubMed] [Google Scholar]
- Broadwell RD, Sofroniew MV. Serum proteins bypass the blood-brain fluid barriers for extracellular entry to the central nervous system. Exp Neurol. 1993;120:245–263. doi: 10.1006/exnr.1993.1059. [DOI] [PubMed] [Google Scholar]
- Carro E, Nunez A, Busiguina S, Torres-Aleman I. Circulating insulin-like growth factor I mediates effects of exercise on the brain. J Neurosci. 2000;20:2926–2933. doi: 10.1523/JNEUROSCI.20-08-02926.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Barnett JL, Pieroni C, Nixon RA. Increased neuronal endocytosis and protease delivery to early endosomes in sporadic Alzheimer's disease: neuropathologic evidence for a mechanism of increased β-amyloidogenesis. J Neurosci. 1997;17:6142–6151. doi: 10.1523/JNEUROSCI.17-16-06142.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell JW, Gibb GM, Betts JC, Blackstock WP, Gallo J, Lovestone S, Hutton M, Anderton BH. Effects of FTDP-17 mutations on the in vitro phosphorylation of tau by glycogen synthase kinase 3β identified by mass spectrometry demonstrate certain mutations exert long-range conformational changes. FEBS Lett. 2001;493:40–44. doi: 10.1016/s0014-5793(01)02267-0. [DOI] [PubMed] [Google Scholar]
- Delacourte A, Sergeant N, Champain D, Wattez A, Maurage CA, Lebert F, Pasquier F, David JP. Nonoverlapping but synergetic tau and APP pathologies in sporadic Alzheimer's disease. Neurology. 2002;59:398–407. doi: 10.1212/wnl.59.3.398. [DOI] [PubMed] [Google Scholar]
- Dickey CA, Eriksen J, Kamal A, Burrows F, Kasibhatla S, Eckman CB, Hutton M, Petrucelli L. Development of a high throughput drug screening assay for the detection of changes in tau levels—proof of concept with HSP90 inhibitors. Curr Alzheimer Res. 2005;2:231–238. doi: 10.2174/1567205053585927. [DOI] [PubMed] [Google Scholar]
- Dietzschold B, Kao M, Zheng YM, Chen ZY, Maul G, Fu ZF, Rupprecht CE, Koprowski H. Delineation of putative mechanisms involved in antibody-mediated clearance of rabies virus from the central nervous system. Proc Natl Acad Sci USA. 1992;89:7252–7256. doi: 10.1073/pnas.89.15.7252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian RH. Uptake of antineuronal IgM by CNS neurons: comparison with antineuronal IgG. Neurology. 1990;40:419–422. doi: 10.1212/wnl.40.3_part_1.419. [DOI] [PubMed] [Google Scholar]
- Fabian RH, Petroff G. Intraneuronal IgG in the central nervous system: uptake by retrograde axonal transport. Neurology. 1987;37:1780–1784. doi: 10.1212/wnl.37.11.1780. [DOI] [PubMed] [Google Scholar]
- Fabian RH, Ritchie TC. Intraneuronal IgG in the central nervous system. J Neurol Sci. 1986;73:257–267. doi: 10.1016/0022-510x(86)90150-4. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Boada RM, Sanchez Guerra ML, Rey MJ, Costa-Jussa F. Neuropathology and pathogenesis of encephalitis following amyloid-β immunization in Alzheimer's disease. Brain Pathol. 2004;14:11–20. doi: 10.1111/j.1750-3639.2004.tb00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulga TA, Elson-Schwab I, Khurana V, Steinhilb ML, Spires TL, Hyman BT, Feany MB. Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat Cell Biol. 2007;9:139–148. doi: 10.1038/ncb1528. [DOI] [PubMed] [Google Scholar]
- Goedert M. Tau protein and neurodegeneration. Semin Cell Dev Biol. 2004;15:45–49. doi: 10.1016/j.semcdb.2003.12.015. [DOI] [PubMed] [Google Scholar]
- Gonias SL, Wu LH, Salicioni AM. Low density lipoprotein receptor-related protein: regulation of the plasma membrane proteome. Thromb Haemost. 2004;91:1056–1064. doi: 10.1160/TH04-01-0023. [DOI] [PubMed] [Google Scholar]
- Gotz J, Chen F, Barmettler R, Nitsch RM. Tau filament formation in transgenic mice expressing P301L tau. J Biol Chem. 2001a;276:529–534. doi: 10.1074/jbc.M006531200. [DOI] [PubMed] [Google Scholar]
- Gotz J, Chen F, Van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301L tau transgenic mice induced by Aβ42 fibrils. Science. 2001b;293:1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- Greenberg SG, Davies P, Schein JD, Binder LI. Hydrofluoric acid-treated tau PHF proteins display the same biochemical properties as normal tau. J Biol Chem. 1992;267:564–569. [PubMed] [Google Scholar]
- Herz J, Kowal RC, Ho YK, Brown MS, Goldstein JL. Low-density-lipoprotein receptor-related protein mediates endocytosis of monoclonal-antibodies in cultured-cells and rabbit liver. J Biol Chem. 1990;265:21355–21362. [PubMed] [Google Scholar]
- Iqbal K, Grundke-Iqbal I. Inhibition of neurofibrillary degeneration: a promising approach to Alzheimer's disease and other tauopathies. Curr Drug Targets. 2004;5:495–502. doi: 10.2174/1389450043345254. [DOI] [PubMed] [Google Scholar]
- Jicha GA, Bowser R, Kazam IG, Davies P. Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res. 1997;48:128–132. doi: 10.1002/(sici)1097-4547(19970415)48:2<128::aid-jnr5>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Jicha GA, Berenfeld B, Davies P. Sequence requirements for formation of conformational variants of tau similar to those found in Alzheimer's disease. J Neurosci Res. 1999;55:713–723. doi: 10.1002/(SICI)1097-4547(19990315)55:6<713::AID-JNR6>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Knopf PM, Harling-Berg CJ, Cserr HF, Basu D, Sirulnick EJ, Nolan SC, Park JT, Keir G, Thompson EJ, Hickey WF. Antigen-dependent intrathecal antibody synthesis in the normal rat brain: tissue entry and local retention of antigen-specific B cells. J Immunol. 1998;161:692–701. [PubMed] [Google Scholar]
- Kounnas MZ, Moir RD, Rebeck GW, Bush AI, Argraves WS, Tanzi RE, Hyman BT, Strickland DK. LDL receptor-related protein, a multifunctional ApoE receptor, binds secreted β-amyloid precursor protein and mediates its degradation. Cell. 1995;82:331–340. doi: 10.1016/0092-8674(95)90320-8. [DOI] [PubMed] [Google Scholar]
- LaRue B, Hogg E, Sagare A, Jovanovic S, Maness L, Maurer C, Deane R, Zlokovic BV. Method for measurement of the blood-brain barrier permeability in the perfused mouse brain: application to amyloid-β peptide in wild type and Alzheimer's Tg2576 mice. J Neurosci Methods. 2004;138:233–242. doi: 10.1016/j.jneumeth.2004.04.026. [DOI] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul MM, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- Liu HM, Atack JR, Rapoport SI. Immunohistochemical localization of intracellular plasma proteins in the human central nervous system. Acta Neuropathol (Berl) 1989;78:16–21. doi: 10.1007/BF00687397. [DOI] [PubMed] [Google Scholar]
- Lobo ED, Hansen RJ, Balthasar JP. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci. 2004;93:2645–2668. doi: 10.1002/jps.20178. [DOI] [PubMed] [Google Scholar]
- Masliah E, Hansen L, Adame A, Crews L, Bard F, Lee C, Seubert P, Games D, Kirby L, Schenk D. Aβ vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease. Neurology. 2005a;64:129–131. doi: 10.1212/01.WNL.0000148590.39911.DF. [DOI] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Adame A, Alford M, Crews L, Hashimoto M, Seubert P, Lee M, Goldstein J, Chilcote T, Games D, Schenk D. Effects of α-synuclein immunization in a mouse model of Parkinson's disease. Neuron. 2005b;46:857–868. doi: 10.1016/j.neuron.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Michaelis ML, Ansar S, Chen Y, Reiff ER, Seyb KI, Himes RH, Audus KL, Georg GI. β-Amyloid-induced neurodegeneration and protection by structurally diverse microtubule-stabilizing agents. J Pharmacol Exp Ther. 2005;312:659–668. doi: 10.1124/jpet.104.074450. [DOI] [PubMed] [Google Scholar]
- Mohamed HA, Mosier DR, Zou LL, Siklos L, Alexianu ME, Engelhardt JI, Beers DR, Le WD, Appel SH. Immunoglobulin Fc gamma receptor promotes immunoglobulin uptake, immunoglobulin-mediated calcium increase, and neurotransmitter release in motor neurons. J Neurosci Res. 2002;69:110–116. doi: 10.1002/jnr.10271. [DOI] [PubMed] [Google Scholar]
- Nerenberg ST, Prasad R. Radioimmunoassays for Ig classes G, A, M, D, and E in spinal fluids: normal values of different age groups. J Lab Clin Med. 1975;86:887–898. [PubMed] [Google Scholar]
- Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-β peptide: a case report. Nat Med. 2003;9:448–452. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F, Gaynor K, Wang L, LaFrancois J, Feinstein B, Burns M, Krishnamurthy P, Wen Y, Bhat R, Lewis J, Dickson D, Duff K. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci USA. 2005;102:6990–6995. doi: 10.1073/pnas.0500466102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Aβ immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43:321–332. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Otvos L, Jr, Feiner L, Lang E, Szendrei GI, Goedert M, Lee VM. Monoclonal antibody PHF-1 recognizes tau protein phosphorylated at serine residues 396 and 404. J Neurosci Res. 1994;39:669–673. doi: 10.1002/jnr.490390607. [DOI] [PubMed] [Google Scholar]
- Paris D, Town T, Parker TA, Tan J, Humphrey J, Crawford F, Mullan M. Inhibition of Alzheimer's β-amyloid induced vasoactivity and proinflammatory response in microglia by a cGMP-dependent mechanism. Exp Neurol. 1999;157:211–221. doi: 10.1006/exnr.1999.7055. [DOI] [PubMed] [Google Scholar]
- Pickhardt M, Gazova Z, von Bergen M, Khlistunova I, Wang Y, Hascher A, Mandelkow EM, Biernat J, Mandelkow E. Anthraquinones inhibit tau aggregation and dissolve Alzheimer's paired helical filaments in vitro and in cells. J Biol Chem. 2005;280:3628–3635. doi: 10.1074/jbc.M410984200. [DOI] [PubMed] [Google Scholar]
- Poduslo JF, Curran GL, Wengenack TM, Malester B, Duff K. Permeability of proteins at the blood-brain barrier in the normal adult mouse and double transgenic mouse model of Alzheimer's disease. Neurobiol Dis. 2001;8:555–567. doi: 10.1006/nbdi.2001.0402. [DOI] [PubMed] [Google Scholar]
- Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M, Schellenberg GD. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43:815–825. doi: 10.1002/ana.410430617. [DOI] [PubMed] [Google Scholar]
- Rapoport SI. Osmotic opening of the blood-brain barrier: principles, mechanism, and therapeutic applications. Cell Mol Neurobiol. 2000;20:217–230. doi: 10.1023/A:1007049806660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribe EM, Perez M, Puig B, Gich I, Lim F, Cuadrado M, Sesma T, Catena S, Sanchez B, Nieto M, Gomez-Ramos P, Moran MA, Cabodevilla F, Samaranch L, Ortiz L, Perez A, Ferrer I, Avila J, Gomez-Isla T. Accelerated amyloid deposition, neurofibrillary degeneration and neuronal loss in double mutant APP/tau transgenic mice. Neurobiol Dis. 2005;20:814–822. doi: 10.1016/j.nbd.2005.05.027. [DOI] [PubMed] [Google Scholar]
- Rye DB, Leverenz J, Greenberg SG, Davies P, Saper CB. The distribution of Alz-50 immunoreactivity in the normal human brain. Neuroscience. 1993;56:109–127. doi: 10.1016/0306-4522(93)90567-y. [DOI] [PubMed] [Google Scholar]
- Sahara N, Lewis J, McGowan E, Dickson DW, Hutton M, Yen SHC, Lee G. Temporal expression and phosphorylation of tau in P301L mutant tau transgenic mice. Neurobiol Aging. 2002;23:S552–S553. [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk D. Amyloid-β immunotherapy for Alzheimer's disease: the end of the beginning. Nat Rev Neurosci. 2002;3:824–828. doi: 10.1038/nrn938. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Zakrajsek BA. Effect of experimental treatment on housekeeping gene expression: validation by real-time, quantitative RT-PCR. J Biochem Biophys Methods. 2000;46:69–81. doi: 10.1016/s0165-022x(00)00129-9. [DOI] [PubMed] [Google Scholar]
- Sigurdsson EM. Histological staining of amyloid-β in mouse brains. Methods Mol Biol. 2005;299:299–308. doi: 10.1385/1-59259-874-9:299. [DOI] [PubMed] [Google Scholar]
- Sigurdsson EM. Immunotherapy for conformational diseases. Curr Pharm Des. 2006;12:2569–2585. doi: 10.2174/138161206777698837. [DOI] [PubMed] [Google Scholar]
- Sigurdsson EM, Lorens SA, Hejna MJ, Dong XW, Lee JM. Local and distant histopathological effects of unilateral amyloid-β 25–35 injections into the amygdala of young F344 rats. Neurobiol Aging. 1996;17:893–901. doi: 10.1016/s0197-4580(96)00169-8. [DOI] [PubMed] [Google Scholar]
- Sigurdsson EM, Lee JM, Dong XW, Hejna MJ, Lorens SA. Bilateral injections of amyloid-β 25–35 into the amygdala of young Fischer rats: behavioral, neurochemical, and time dependent histopathological effects. Neurobiol Aging. 1997;18:591–608. doi: 10.1016/s0197-4580(97)00154-1. [DOI] [PubMed] [Google Scholar]
- Sigurdsson EM, Scholtzova H, Mehta PD, Frangione B, Wisniewski T. Immunization with a non-toxic/non-fibrillar amyloid-β homologous peptide reduces Alzheimer's disease associated pathology in transgenic mice. Am J Pathol. 2001;159:439–447. doi: 10.1016/s0002-9440(10)61715-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson EM, Knudsen E, Asuni A, Fitzer-Attas C, Sage D, Quartermain D, Goni F, Frangione B, Wisniewski T. An attenuated immune response is sufficient to enhance cognition in an Alzheimer's disease mouse model immunized with amyloid-β derivatives. J Neurosci. 2004;24:6277–6282. doi: 10.1523/JNEUROSCI.1344-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson EM, Wadghiri YZ, Mosconi L, Blind JA, Knudsen E, Asuni A, Scholtzova H, Tsui WH, Li Y, Sadowski M, Turnbull DH, deLeon M, Wisniewski T. A non-toxic ligand for voxel-based MRI analysis of plaques in AD transgenic mice. Neurobiol Aging. 2007 doi: 10.1016/j.neurobiolaging.2006.12.018. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Bird TD, Ghetti B. Frontotemporal dementia and Parkinsonism linked to chromosome 17: a new group of tauopathies. Brain Pathol. 1998;8:387–402. doi: 10.1111/j.1750-3639.1998.tb00162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires TL, Orne JD, Santacruz K, Pitstick R, Carlson GA, Ashe KH, Hyman BT. Region-specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. Am J Pathol. 2006;168:1598–1607. doi: 10.2353/ajpath.2006.050840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tampellini D, Magrané J, Takahashi RH, Li F, Lin MT, Almeida CG, Gouras GK. Internalized antibodies to the Aβ domain of APP reduce neuronal Aβ and protect against synaptic alterations. J Biol Chem. 2007;282:18895–18906. doi: 10.1074/jbc.M700373200. [DOI] [PubMed] [Google Scholar]
- Terry RD. Neuropathological changes in Alzheimer disease. Prog Brain Res. 1994;101:383–390. doi: 10.1016/s0079-6123(08)61964-0. [DOI] [PubMed] [Google Scholar]
- The TH, Feltkamp TE. Conjugation of fluorescein isothiocyanate to antibodies II. A reproducible method. Immunology. 1970;18:875–881. [PMC free article] [PubMed] [Google Scholar]
- Torres EM, Perry TA, Blockland A, Wilkinson LS, Wiley RG, Lappi DA, Dunnet SB. Behavioural, histochemical and biochemical consequences of selective immunolesions in discrete regions of the basal forebrain cholinergic system. Neuroscience. 1994;63:95–122. doi: 10.1016/0306-4522(94)90010-8. [DOI] [PubMed] [Google Scholar]
- Wadghiri YZ, Sigurdsson EM, Sadowski M, Elliott JI, Li Y, Scholtzova H, Tang CY, Aguinaldo G, Pappolla M, Duff K, Wisniewski T, Turnbull DH. Detection of Alzheimer's amyloid in transgenic mice using magnetic resonance microimaging. Magn Reson Med. 2003;50:293–302. doi: 10.1002/mrm.10529. [DOI] [PubMed] [Google Scholar]
- Weaver CL, Espinoza M, Kress Y, Davies P. Conformational change as one of the earliest alterations of tau in Alzheimer's disease. Neurobiol Aging. 2000;21:719–727. doi: 10.1016/s0197-4580(00)00157-3. [DOI] [PubMed] [Google Scholar]
- Zhang B, Maiti A, Shively S, Lakhani F, McDonald-Jones G, Bruce J, Lee EB, Xie SX, Joyce S, Li C, Toleikis PM, Lee VM, Trojanowski JQ. Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc Natl Acad Sci USA. 2005;102:227–231. doi: 10.1073/pnas.0406361102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV, Skundric DS, Segal MB, Lipovac MN, Mackic JB, Davson H. A saturable mechanism for transport of immunoglobulin G across the blood-brain barrier of the guinea pig. Exp Neurol. 1990;107:263–270. doi: 10.1016/0014-4886(90)90144-h. [DOI] [PubMed] [Google Scholar]