

Abstract
The blood–brain barrier (BBB) restricts molecular and cellular trafficking between the blood and the CNS. Although astrocytes are known to control BBB permeability, the molecular determinants of this effect remain unknown. We show that angiotensinogen (AGT) produced and secreted by astrocytes is cleaved into angiotensin II (AngII) and acts on type 1 angiotensin receptors (AT1) expressed by BBB endothelial cells (ECs). Activation of AT1 restricts the passage of molecular tracers across human BBB-derived ECs through threonine-phosphorylation of the tight junction protein occludin and its mobilization to lipid raft membrane microdomains. We also show that AGT knock-out animals have disorganized occludin strands at the level of the BBB and a diffuse accumulation of the endogenous serum protein plasminogen in the CNS, compared with wild-type animals. Finally, we demonstrate a reduction in the number of AGT-immunopositive perivascular astrocytes in multiple sclerosis (MS) lesions, which correlates with a reduced expression of occludin similarly seen in the CNS of AGT knock-out animals. Such a reduction in astrocyte-expressed AGT and AngII is dependent, in vitro, on the proinflammatory cytokines tumor necrosis factor-α and interferon-γ. Our study defines a novel physiological role for AngII in the CNS and suggests that inflammation-induced downregulation of AngII production by astrocytes is involved in BBB dysfunction in MS lesions.
Keywords: astrocytes, tight junctions, renin-angiotensin system, CNS, neuroinflammation, lipid rafts, human, mouse, lymphocytes, immune privilege
Introduction
The blood–brain barrier (BBB) regulates CNS homeostasis by controlling the passage of blood-borne molecules and cells from the periphery to the CNS. Compromise of BBB integrity is a hallmark of several neurological disorders, including brain and spinal cord trauma, brain neoplasms, and multiple sclerosis (MS). In MS, BBB disruption contributes to the multifocal infiltration of autoreactive inflammatory immune cells, leading to tissue destruction and neurological impairment.
The BBB consists of specialized endothelial cells (ECs) strongly anastomosed to one another through multiprotein tight junction (TJ) complexes. This cerebral microvasculature is in close apposition to a dense network of astrocytic endfeet, which promote the formation and maintenance of the BBB through contact-dependent and soluble factor-induced mechanisms (Abbott, 2002; Nedergaard et al., 2003; Abbott et al., 2006). Tao-Cheng and Brightman (1988) first demonstrated that soluble astrocytic factors reinduced a BBB phenotype in rodent brain-derived ECs grown in vitro, a phenomenon confirmed in other systems (Neuhaus et al., 1991; Stanimirovic et al., 1995; Prat et al., 2001). The identity of the astrocytic molecule(s) responsible for this effect remains unclear.
The blood-borne octapeptide angiotensin II (AngII) is generated from angiotensinogen (AGT) via cleavage by renin and angiotensin converting enzyme-1 (ACE1), an enzymatic cascade defined as the renin-angiotensin system (RAS). Although the peripheral blood endocrine RAS regulates blood volume, arterial pressure, and kidney filtration rate and is a critical systemic player in fluid homeostasis (de Gasparo et al., 2000), the biological functions of the CNS RAS range from control of cerebral blood flow to memory retention and neuronal regeneration (Morimoto and Sigmund, 2002; Saavedra, 2005). Furthermore, in a seminal observation, Kakinuma suggested that in rodents, AngII and one of its metabolites in the brain, AngIV, are also involved in CNS vascular repair after injury (Kakinuma et al., 1998; Yanai et al., 2000). In the rodent CNS, AGT-immunopositive astroglial cells are found in the hippocampus, thalamus, hypothalamus, anterior and posterior cingulate cortex, basal and lateral amygdala, caudate nucleus, globus pallidus as well as in the brainstem. In the cortex, AGT expression seems restricted to neurons (Stornetta et al., 1988; Intebi et al., 1990; Mungall et al., 1995).
In this study, we hypothesized that AGT metabolites are secreted by human astrocytes and act on angiotensin receptors (AT) on brain ECs to promote BBB maintenance through posttranslational TJ protein modifications.
Materials and Methods
Primary cell isolation and culture.
BBB-ECs were isolated from CNS tissue specimens from temporal lobe resections from young adults undergoing surgery for the treatment of intractable epilepsy, as described previously (Stanimirovic et al., 1997; Prat et al., 2000). Informed consent and ethic approval was given before surgery (ethics approval number HD04.046). Although cultures express endothelial markers factor VIII, Ulex Agglutenens Europaensis-1 binding sites, and antigen HT-7 until passages (P) 7–8, only early passages (P1–P3) were used in this study to prevent variability in the expression of occludin at later passages. No immunoreactivity for β-tubulin, α-myosin, or glial fibrillary acidic protein (GFAP) could be detected, confirming the absence of contaminating smooth muscle cells or astrocytes. Occasional CD68-positive cells (microglia) were seen (<1% at P2), and their number decreased along passages because of their strong adherence to plastic and their nonproliferative nature.
Astrocyte-conditioned media (ACM) was harvested once a week from confluent flasks of human fetal astrocyte cultures. To obtain astrocyte cultures, fetal CNS tissue (cerebral hemispheres) was obtained at 17–23 weeks of gestation following Canadian Institute of Health Research-approved guidelines. Astrocyte isolation has been described previously (D'Souza et al., 1995; Wosik et al., 2001; Jack et al., 2005). Human fetal astrocytes were used between P2 and P4, and cultures were determined to be >90% pure, as determined by GFAP immunostaining.
Tissue and immunohistochemistry.
Formaldehyde-fixed and paraffin-embedded archival CNS tissue from control and MS patients was obtained from the Neuropathology Department at Notre Dame Hospital. Six brain specimens from MS patients were examined, three male (51, 42, and 42 years of age) and three female (49, 58, and 68 years of age); evolution of disease was between 2 and 20 years of age. Four brain specimens from Parkinson's patients (all male, 65–72 years of age) and two brains from control patients (traumatic death, 51 years of age and unknown age, male) were investigated. In all control cases, cause of death was non-neurological, and autopsies were performed within 6–24 h from time of death. The generation of AGT deficient mice has been described previously (Methot et al., 2001; Lochard et al., 2003). Paraffin-embedded whole mouse brains were used from wild-type (wt) and AGT−/− null animals.
For immunohistochemistry and fluorescence staining, 3-μm-thick tissue sections were deparaffinized in three successive changes of toluene and rehydrated in 100% and 95% ethanol, water, and PBS. For luxol fast blue (LFB) and hematoxylin and eosin (H&E), slides were incubated at 60°C overnight in LFB solution (0.95% Solvent Blue 38, 90% ethanol, 0.5% acetic acid) and rinsed with 95% ethanol and water. Differentiation was done with repeated 15 s washes in 0.05% lithium carbonate, 20 s in 70% ethanol, and water. Slides were subsequently incubated in hematoxylin solution for 8 min, rinsed in water, and differentiated in 1% lithium carbonate (15 s). A 1 min incubation in eosin solution (20% eosin stock solution, 10% acetic acid, 50% ethanol in water) preceded dehydration with two washes of ethanol (95% and 100%, 1 min) and a toluene wash (2 min). Slides were mounted with Permount. All reagents are from Sigma (Oakville, Ontario, Canada).
For AGT and S100 stains, enzymatic antigen retrieval was performed with trypsin (0.1% trypsin, 0.1% CaCl2 in 20 mm Tris, pH 7.6) for 10 min at 37°C. For occludin and zonula occludens-1 (ZO-1), enzymatic antigen retrieval with proteinase K was done (20 μg/ml in Tris EDTA buffer, pH 8) for 15 min at 37°C. For GFAP and junctional adhesion molecule-1 (JAM-1), sodium citrate (0.3% sodium citrate in 4 mm HCl solution) heat antigen retrieval was required, whereas an EDTA (1 mm) heat antigen retrieval was necessary for the claudin-5 stain. For albumin, AT1, AT2, caveolin-1 (cav-1), and plasminogen, no antigen retrieval was necessary. GFAP and AGT immunostainings could not be performed concurrently, because they require different antigen retrieval protocols. Sections were subsequently permeabilized with 1% Triton X-100 [5 min, room temperature (RT)] and blocked by incubation in HHG (1 mm HEPES, 2% horse serum, 10% goat serum in HBSS; Sigma) plus 0.5% Triton X-100 for 1 h at RT. Primary antibodies against AT1, AT2, cav-1, plasminogen (all 1:50; Santa Cruz Biotechnology, Santa Cruz, CA), S100 (1:100), albumin (1:1000; Abcam, Cambridge, MA), occludin (1:50), ZO-1 (1:50), Claudin-5 (1:100) (all from Zymed, South San Francisco, CA), GFAP (1:500; DakoCytomation, Glostrup, Denmark), and JAM-1 (1:50; BD Biosciences, Franklin Lakes, NJ) were incubated overnight at 4°C. Anti-AGT (1:200; US Biologicals, Swampscott, MA) was incubated for 1 h at RT. After washing, AT1, AT2, AGT, Claudin-5, GFAP, JAM-1, plasminogen, occludin, and ZO-1 stains were amplified using biotin-conjugated anti-rabbit (1:400) and anti-mouse (1:200) antibodies followed by streptavidin-fluorescein isothiocyanate (FITC; 1:1000; DakoCytomation) and streptavidin-Cy3 (1:400; Jackson ImmunoResearch, West Grove, PA). Albumin, Cav-1, and S100 stainings were revealed with Cy3-conjugated anti-rabbit (1:400) and anti-mouse antibody (1:400; Jackson ImmunoResearch). Nuclei were stained with Hoechst 33258 dye (1:2000; 10 min, RT; Invitrogen, Carlsbad, CA) or with TO-PRO3 (1:300 in PBS, 15 min, pretreatment with RNase A 100 μg/ml, 30 min; Invitrogen, Eugene, OR). For immunohistochemistry, the AGT signal was visualized using anti-rabbit biotin followed by horseradish peroxidase-conjugated avidin complex and revealed using chromogen diaminobenzidine (Vector Laboratories, Burlingame, CA) enhanced with nickel ammonium sulfate. In all cases, control stainings were performed omitting the primary antibody, and no immunopositive cells could be detected. Stainings were visualized using either a Leica DM6000 microscope with OpenLab software or the Leica SP5 confocal microscope and analyzed using the Leica LAS AF software.
Semiquantitative analysis of occludin signal intensity and strand thickness were performed as follows: 30 x-y planar images (0.1 μm thickness) of occludin immunostainings were recorded sequentially and reconstructed (collapsed) as a 3 μm x-y-z image. Occludin strand immunostaining intensity and thickness were calculated using the Leica LAS AF software. All microscopy data were recovered by two investigators blinded to the disease/treatment group (n > 40 for each CNS condition studied).
Flow cytometry.
Human astrocytes were permeabilized with 4% paraformaldehyde and 0.1% Saponin (Sigma), incubated with recombinant mouse anti-GFAP conjugated to Alexa 488 (1:500; BD Biosciences) and rabbit anti-AGT (1:200) for 1 h at 4°C, washed three times with 1% FCS/PBS, incubated for 30 min with mouse anti-rabbit phycoerythrin secondary antibody (1:500; BD Biosciences), and then visualized on a BD LSRII flow cytometer. Analysis was performed using the BD-FACSDiva software.
Permeability assays.
BBB-ECs grown in primary cultures were used to generate an in vitro model of the human BBB, as reported previously (Prat et al., 2002; Alter et al., 2003; Biernacki et al., 2004). On day 0, BBB-ECs were plated on gelatin-coated 3 μm pore size Boyden chambers (BD Biosciences) at a density of 2.104 cells per well in EC culture media and were supplemented, where applicable, with 40% (v/v) ACM or ACM plus ACE1 inhibitor [ACM treated with an ACE1 inhibitor, Captopril (10−7 m; Sigma) every 24 h for 5 d] and were allowed to grow for 72 h to reach confluency. During this period, cells were treated every 24 h with AngII (10−8 m; Sigma), Losartan (10−7 m; a generous gift from Dr. J. Chan, CHUM Hôtel-Dieu, University of Montreal, Montréal, Quebec, Canada), PD123,319 (10−7 m; Sigma), CGP-42112A (10−8 m; Sigma), or L-162,313 (10−8 m; Sigma). On day 3, 1 h after the last treatment, FITC-labeled bovine serum albumin (FITC-BSA; Invitrogen) (50 μg/ml) or 14C-sucrose (250 nCi/ml; MP Biomedicals, Aurora, OH) were added to the upper chambers, and their diffusion across the EC monolayer was assessed by sampling upper and lower chambers at specific time points. Fluorescence intensity and radioactivity in the media were monitored using a FL600 microplate fluorescent reader (Biotek, Winooski, VT) and liquid scintillation analyzer (A2100; Packard Instruments, Downers Grove, IL). Molecular tracer diffusion representing BBB permeability is calculated using the formula (BSA lower chamber) × 100/(BSA upper chamber), as reported previously (Ifergan et al., 2006). When applicable, confluent Boyden chambers were treated for 1 h with raft disrupting agents MβCD (5 mm), filipin (2 μg/ml), or nystatin (50 μg/ml) (all from Sigma) and washed before the addition of FITC-BSA.
In the coculture experiments, BBB-ECs (2.104 cells) and astrocytes (2.104 cells) were plated on the luminal and abluminal sides of the transwell, respectively, allowing the astrocytes to directly contact the BBB-ECs through the pores of the transwell. Permeability experiments were performed using FITC-labeled BSA as described above.
Lipid raft isolation and analysis.
For the isolation of lipid rafts, six T75 flasks of confluent BBB-ECs per condition were washed with ice-cold PBS and scraped on ice. After centrifugation, the pellet was resuspended in 1 ml of 1% Brij58 (Sigma) in separation buffer (150 mm NaCl, 25 mm Tris HCl, pH 7.4) with protease inhibitors (BD Biosciences), and cells were solubilized for 30 min on ice. The cell lysate was transferred to a glass homogenizer, dounced with 10 strokes of a loose fitting pestle, and mixed with 1 ml of 85% sucrose (Sigma) in separation buffer (w/v). The resulting 42.5% sucrose cell lysate was overlaid with 6 ml of 35% sucrose and 4 ml of 5% sucrose (w/v in separation buffer) and centrifuged for 24 h at 39,000 rpm at 4°C in a Beckman SW41 rotor. Twelve 1 ml fractions were collected at the meniscus, top to bottom. Cholesterol, phospholipid, and protein concentrations in each fraction were assayed using commercially available kits: Amplex red cholesterol assay kit (Invitrogen), Phospholipids B Enzymatic colorimetric method kit (Wako, Richmond, VA), and BCA Protein assay kit (Pierce, Rockford, IL).
Reverse transcription-PCR, ELISA.
All reagents used were from Invitrogen. RNA was isolated using the TRIzol reagent. Three micrograms of RNA per reaction were used in a reverse transcription-PCR with 3.3 μm random hexamer primers, 3 mm dNTPs, RNase-OUT recombinant ribonuclease inhibitor, 3 mm DTT, and 400 U Maloney murine leukemia virus-reverse transcription for 1 h at 42°C and 10 min at 75°C to produce cDNA. Two microliters of cDNA were then amplified using 5 U TaqDNA polymerase, 1.5 mm MgCl2, 0.2 mm dNTPs, and 50 pmol forward (F) and reverse (R) primers. AGT primers have been published previously (Juillerat-Jeanneret et al., 2004). Primers used were: AGT F, 5′-tcc acc tcg tca tcc aca-3′; R, 5′-ggc tcc cag ata gag aga-3′; ACE1 F, 5′-acc tca acc tgc atg cct ac-3′; R, 5′-cag cat cga ctt gtt cca ga-3′; GAPDH F, 5′-caa agt tgt cat gga tga cc-3′; R, 5′-cca tgg aga agg ctg ggg-3′. The reaction mixture was placed in an Eppendorf “mastercycler” thermal cycler for 10 min at 94°C followed by 30 cycles of 1 min at 94°C, 45 s at 60°C, and 1 min at 72°C. After amplification, 20 μl of each sample was resolved on a 1.5% agarose gel with ethidium bromide.
AngII enzyme immunoassays were performed on undiluted culture supernatants according to the manufacturer instructions (SPI Bio, Massy, France). When applicable, cells were treated for 24 h with human recombinant interferon-γ (IFNγ) and/or tumor necrosis factor-α (TNFα) (R&D Systems, Minneapolis, MN) before harvesting of media.
Immunoprecipitation and Western blot analysis.
For AT1 and AT2 Western blot analysis, BBB-ECs were lysed in NP-40 buffer (10 mm Tris-HCl, 10 mm NaCl, 3 mm MgCl2, and 0.5% NP-40) supplemented with protease inhibitors. Thirty micrograms of total protein were electrophoresed on a 12% SDS-PAGE under reducing conditions. Proteins were transferred to a polyvinylidene difluoride membrane (Bio-Rad, Hercules, CA) and blocked for 1 h at RT in 5% milk. Membranes were incubated overnight with rabbit anti-human AT1 or AT2 antibody (1:250 in milk; Santa Cruz Biotechnology) followed by a 1 h incubation with horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG antibody (Dako Cytomation). Specific binding was visualized using the ECL system (GE Healthcare, Piscataway, NJ).
To assess for GM1 localization in sucrose gradient fractions, 5 μl of each fraction was run on a 12% gel, transferred and blocked as before, and incubated with HRP-cholera toxin B subunit (1:10,000 in milk; 1 h at RT; Invitrogen). Other Western blots on sucrose fractions were performed by running 10–35 μl of each fraction on a 6–12% gel under reducing conditions, transfer and blocking as before, and overnight incubation with the primary antibody: rabbit anti-occludin (1:100), mouse anti-JAM-1 (1:500), goat anti-CD59 (1:200; R&D Systems), mouse anti-transferrin receptor (1:1000; BD Biosciences). HRP-conjugated secondary antibody was incubated for 1 h at RT. All signals were revealed using the ECL reagents.
For immunoprecipitations (IPs), whole-cell lysates or sucrose fractions were incubated overnight at 4°C with agarose bead-conjugated monoclonal anti-P-Thr or anti-P-Tyr antibodies on a rotary shaker (8 μl of compact beads per reaction; Santa Cruz Biotechnology). Agarose beads were spun down, washed three times in PBS, resuspended in loading buffer, and run on a 12% SDS-PAGE under reducing conditions. Western blots for occludin were performed as above.
Densitometric analyses were performed on a Bio-Rad Gel Doc System using the Quantity One software and are expressed as percentage of fraction 12 or as percentage of untreated, when applicable.
Statistical analysis.
Permeability curves after different treatments were evaluated using repeated-measures two-way ANOVA followed by a Bonferroni posttest comparing all pairs of data points. All data points are expressed as mean ± SEM from at least n = 3 experiments, each conducted in duplicate. Permeability 72 h after raft-disrupting drug treatment was evaluated by one-way ANOVA. Results of AGT+/S100+ cell counts were evaluated using one way-ANOVA, followed by Bonferroni's multiple comparison test; scatter plots show individual data points and mean. Differences of occludin peak strand fluorescence intensity and strand thickness were evaluated using an unpaired Student's t test.
Results
Human astrocytes produce AngII; brain endothelium expresses cognate receptors
Reverse transcription-PCR was performed using primers specific for human AGT (Juillerat-Jeanneret et al., 2004) and ACE1. As seen in Figure 1a, primary cultures of human astrocytes express AGT as well as ACE1 mRNA. Supernatants were also collected after 1 and 3 d in culture, and levels of AngII were assessed by ELISA: the active AngII metabolite is secreted by human astrocytes (12.9 ± 1.6 pg/ml at 24 h) and although human BBB-derived ECs do not produce AngII, they do produce ACE1 (data not shown). GFAP-expressing astrocytes grown in primary culture in vitro were also subjected to intracellular immunostainings for AGT and analyzed by flow cytometry (Fig. 1b). As shown, the majority (80.6%) of GFAP+ astrocytes were also positive for AGT, confirming that the source of AGT in these cultures was astrocytes and not contaminating neurons.
Figure 1.
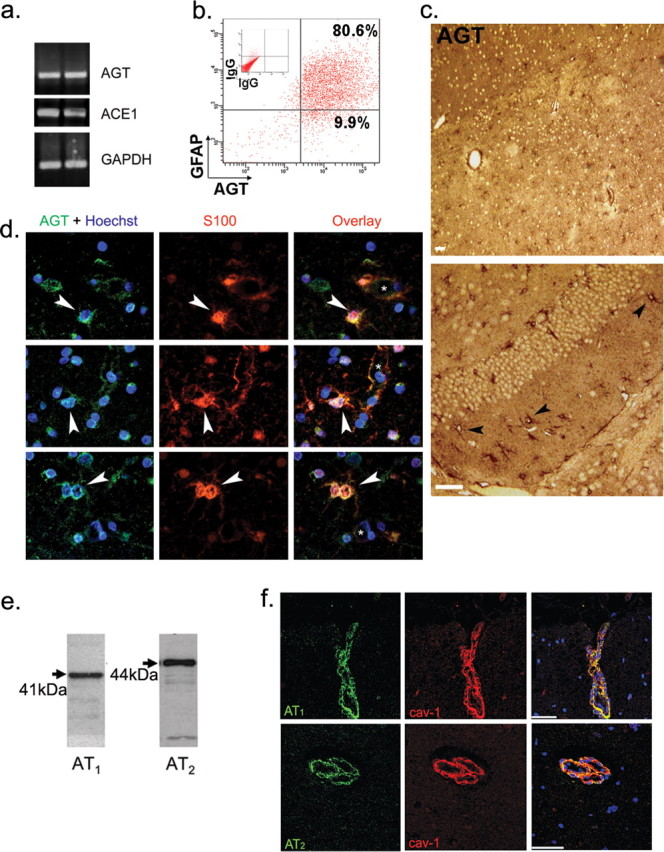
Angiotensinogen, Angiotensin II, and angiotensin receptor expression in human brain. a, PCR of human astrocytes in culture for AGT and ACE1, two astrocyte preparations are shown. b, Flow cytometry analysis of human fetal astrocytes in culture immunostained for GFAP and AGT (n = 3). c, Immunohistochemistry on normal human CNS sections (hemispheric and cerebellar) reveals the presence of numerous AGT-positive cells (n = 8 sections; 2 CNS samples). Scale bars, 20 μm. d, Immunohistofluorescence on normal human subcortical white matter shows S100-positive astrocytes (red) are immunopositive for AGT (green), as evidenced by the yellow color in the overlay pictures. Vessel lumen is shown with an asterisk. Nuclei were stained with Hoechst (blue). e, Western blots for angiotensin receptors AT1 and AT2 on human BBB-ECs. f, Immunohistofluorescence of normal human subcortical white matter shows brain capillaries are immunopositive for both cav-1 (red) as well as for AT1 and AT2 (green); overlays are shown on the right; TO-PRO3-stained nuclei are blue. Scale bars, 50 μm.
Immunostainings for AGT [low-magnification histochemistry (Fig. 1c) and higher magnification histofluorescence (Fig. 1d)] were further performed on normal human brain sections using antibodies specific for AGT and S100, a marker of perivascular astrocyte (Ghandour et al., 1981; Kacem et al., 1998). Low-magnification immunochemistry staining for AGT revealed an abundance of astrocyte-like cells in the hemispheric and cerebellar white matter of human CNS material (Fig. 1c) and confirmed high numbers of AGT+ astrocytes in hypothalamus, thalamus, hippocampus, corpus callosum, and in several brainstem nuclei, including solitary tract nuclei (data not shown). Additional AGT+ cells, likely pyramidal neurons (by morphology), were also seen in the hemispheric cortical gray matter and in cerebellar neurons (data not shown). S100 (red)-positive parenchymal and perivascular astrocytes express AGT (green), as seen in the overlay pictures (Fig. 1d, yellow, arrows; vessel lumen indicated with an asterisk), confirming that in the white matter, perivascular astrocytes express AGT. From these results, we conclude that human astrocytes thus express AGT in vitro and in situ in the brain, which can subsequently be cleaved into AngII through the action of ACE1 expressed by astrocytes and brain ECs.
To ascertain that human brain ECs have the capacity to respond to astrocyte-derived AGT products, AT1 and AT2 receptor expression on BBB-ECs was assessed by Western blot. BBB-ECs express both AT1 and AT2 when grown in primary cultures (Fig. 1e) and in situ, as shown by immunohistofluorescence staining on human brain capillaries, discernible by cav-1 coexpression (Fig. 1f).
Astrocyte-secreted AngII decreases EC permeability via the AT1 receptor
Confluent BBB-EC monolayers in vitro take on a cobblestone appearance and form a semipermeable barrier that restricts diffusion of soluble tracers. We and others have demonstrated previously that astrocyte-derived soluble factors in ACM decrease BBB-EC permeability to soluble tracers (Stanimirovic et al., 1995; Prat et al., 2001). In a modified Boyden chamber permeability assay using human BBB-ECs, 40% ACM (v/v) induced an important decrease in permeability to the large molecular weight tracer FITC-BSA (Fig. 2a). This effect was long lasting, as evidenced by the important difference in tracer diffusion up to 72 h (n = 3 independent experiments, in duplicate; p < 0.0001 at 72 h). Addition of AngII (10−8 m; 72 h) to BBB-ECs paralleled the effect of ACM (Fig. 2b). Similar results were obtained using the small molecular weight tracer 14C-sucrose, a marker of paracellular transport. The permeability of untreated BBB-EC monolayers to 14C-sucrose was 21.5% at 3 h, ACM and AngII reduced the permeability of ECs to 13.1 and 8.7%, respectively (p < 0.05; n = 2 in duplicate). At 6 h, 14C-sucrose permeability was further reduced from 41.6% (untreated) to 21.2% (AngII) and 24.4% (ACM; p < 0.01) (data not shown).
Figure 2.

Astrocyte-secreted angiotensin II decreases endothelial cell permeability via the AT1 receptor. a, Permeability of BBB-EC monolayers grown in the modified Boyden chamber assay to FITC-BSA is substantially decreased when cells are grown in the presence of astrocyte-conditioned media (filled square; ACM 40% v/v) when compared with cells grown under basal conditions (open circle; untreated) (n = 3, in duplicate; *p < 0.005, p < 0.0001, Bonferroni posttest at 48 and 72 h, respectively). b, A similar decrease in permeability is seen when BBB-ECs are grown in the presence of AngII (filled circle;10−8 m, AngII) or the angiotensin receptor AT1 agonist L-162,313 (filled triangle; 10−8 m; n = 3, in duplicate; *p < 0.005, compared with nontreated cells). AT2 agonist CGP-42112A (filled square; 10−8 m) did not affect permeability. c, d, BBB-EC monolayers were grown in the presence of ACM with or without the AT1 receptor antagonist Losartan (c) (filled square; 10−7 m) or the AT2 receptor antagonist PD123,319 (d) (filled diamond; 10−7 m). The AT1 receptor antagonist blocks the permeability decreasing the effect of ACM; the AT2 antagonist does not (n = 3; in duplicate, *p < 0.005 between indicated curves). e, Astrocyte cultures were treated with ACE1 inhibitor (filled diamond; Captopril, 5 d at 10−7 m) to reduce AngII levels in ACM. Captopril-treated ACM did not promote BBB integrity, compared with untreated ACM (n = 3, in duplicate; *p < 0.005 between indicated curves). f, BBB-ECs were cocultured with astrocytes (filled triangle), with ACM (filled square), or alone (open circle) for 72 h to demonstrate that astrocyte-EC proximity is important for barrier properties (n = 2, in duplicate; *p < 0.01). All data are expressed as mean ± SEM.
To assess whether astrocyte-produced AngII was responsible for the BBB-EC barrier promoting effects of ACM, BBB-ECs were cultured in ACM, in conjunction with AT1 or AT2 nonpeptide receptor antagonists Losartan or PD123,319 (de Gasparo et al., 2000), respectively (10−7 m). AT1 antagonist Losartan reversed the barrier promoting effects of ACM, increasing the permeability of BBB-ECs to levels of untreated ECs (Fig. 2c) (n = 3; in duplicate; p < 0.005). In contrast, AT2 antagonist PD123,319 did not abrogate the effect of ACM on permeability (Fig. 2d). Antagonists alone had no effect (data not shown). In addition, media from astrocytes treated with Captopril (10−7 m), an ACE1 inhibitor, was unable to reproduce the effects of ACM (Fig. 2e) (n = 3; in duplicate; p < 0.005). In separate experiments, astrocytes were grown on the reverse side of the transwell and allowed to contact BBB-ECs in a coculture assay. Using this system, we could demonstrate an even greater effect of astrocytes on the permeability of BBB-ECs (p < 0.01; compared with ACM) (Fig. 2f), supporting the notion that astrocyte-EC proximity is important in the regulation of BBB permeability.
To confirm that AngII decreases BBB permeability through the AT1 receptor, the effects of the selective synthetic AT1 and AT2 receptor nonpeptide agonists L-162,313 and CGP-42112A, respectively (Vianello et al., 1998), were assessed. Treatment of BBB-EC monolayers with L-162,313 (10−8 m) but not with CGP-42112A (10−8 m) resulted in a significant reduction of permeability, similar to both ACM and AngII (Fig. 2b) (n = 3; in duplicate; p < 0.005, compared with nontreated cells). Our data demonstrate that astrocyte-produced AngII promotes barrier properties in BBB-ECs through ligation of the AT1 receptor.
Astrocytic factors and AngII promote occludin concentration into lipid rafts
Nusrat et al. (2000) have shown that in the T84 gut epithelial cell line, hyperphosphorylated forms of the TJ protein occludin localized to detergent insoluble lipid rafts. We tested whether endothelial TJs of the BBB were concentrated in lipid rafts and whether this concentration is necessary for barrier function. Lipid rafts from BBB-ECs were isolated by sucrose flotation after solubilization with Brij58. After centrifugation, 12 fractions were collected from top (fraction 1) to bottom (fraction 12). High phospholipid and cholesterol concentration, as well as enrichment of the lipid raft markers GM1-ganglioside and CD59, demonstrate the presence of lipid rafts in fractions 4 and 5 (Fig. 3a). To determine whether BBB TJs localized to lipid rafts, rafts were isolated from BBB-ECs and blotted for TJ proteins JAM-1 and occludin. JAM-1 was strongly concentrated in lipid rafts of BBB-ECs grown under basal conditions (Fig. 3b). In contrast, occludin expression was low in BBB-EC lipid rafts when cells were not supplemented. After treatment of BBB-ECs with ACM or AngII (10−8 m) (Fig. 3b), expression of occludin shifted to lipid raft fractions: densitometry analysis of Western blots revealed that 23% (with ACM) and 28% (with AngII) of total occludin were found in lipid rafts, compared with 11% in the untreated BBB-ECs (n = 4; p < 0.05). In contrast, no change in JAM-1 localization was seen, because 20, 23, and 21% of total JAM-1 were found in the rafts of untreated, ACM, and AngII treated BBB-ECs, respectively (n = 4). Protein determination in gradient fractions (Fig. 3a, white squares) showed that protein levels are below the detection threshold in lipid rafts, confirming that occludin expression relative to total protein content was clearly enriched in BBB-EC rafts and suggesting convergence of TJ proteins to rafts.
Figure 3.

Lipid raft profiles of blood–brain barrier endothelial cells. a, Characterization of BBB-EC sucrose density fractions with regards to cholesterol concentration (filled triangle; left axis; in μg/ml), phospholipid content (filled diamond; right axis; in values of absorbance), and protein concentration (open square; far right axis; in mg/ml). Lipid rafts are concentrated in fractions 4 and 5 (rectangular outline) where peak values of cholesterol and phospholipids are seen. Fractions 4 and 5 also show a high concentration of lipid raft markers GM1 ganglioside and CD59 but no evidence of transferrin receptor (TfR), a marker of nonlipid raft membrane. b, Western blots for the TJ protein JAM-1 in sucrose density fractions show a similar concentration of JAM-1 in lipid raft fractions under all conditions tested (untreated, 20%; ACM, 23%; AngII, 21% of total occludin). The TJ molecule occludin is weakly expressed in lipid rafts when cells are grown under basal conditions (untreated) but is enriched in fractions 4 and 5 when BBB-ECs are grown in the presence of ACM or AngII (10 nm) (untreated, 12%; ACM, 23%; AngII, 28% of total occludin). c, When BBB-ECs grown in the presence of ACM are treated for 1 h before the isolation of lipid rafts with the raft disrupting agent MβCD, lipid rafts are dissolved as evidenced by the sharp decrease in cholesterol concentration in fractions 4 and 5 as well as the absence of occludin. Similar data were obtained with filipin and nystatin (data not shown). d, Permeability of BBB-EC monolayers grown in ACM, 72 h after a 1 h treatment with the raft disrupting drug MβCD (17.79 ± 5.31%), filipin (20.05 ± 5.2%), or nystatin (17.03 ± 4.9%) is strongly increased when compared with cells grown in ACM alone (−11.99 ± 2.91%). Percentage values are expressed compared with permeability of cells grown under basal conditions (*p < 0.05; n = 3, in duplicate). e, Immunoprecipitation experiments using antibodies specific to phosphorylated forms of threonine (P-Thr) or tyrosine (P-Tyr): whole-cell lysates of BBB-ECs grown in the presence of ACM or AngII show an upregulation of P-Thr occludin (169 and 228% of untreated, respectively) and a downregulation of P-Tyr occludin (37 and 11% of untreated, respectively) compared with cells grown under basal conditions. f, When raft (R, fractions 4 and 5) and soluble (S, fractions 11 and 12) fractions are immunoprecipitated separately, only the raft fractions display a decrease in P-Tyr occludin (6 and 7% of occludin, respectively), whereas P-Thr occludin upregulation (254 and 351% of untreated, respectively) is evident in both raft and soluble fractions, particularly after AngII treatment.
To assess the functional significance of TJ protein assembly in lipid rafts, BBB-ECs were grown in Boyden chambers in the presence of ACM and treated for 1 h with lipid raft disrupting agents methyl-β-cyclodextrin (MβCD; 5 mm), filipin (2 μg/ml), or nystatin (50 μg/ml) (Pike, 2004) before addition of FITC-BSA. A marked decrease in cholesterol and the absence of occludin was evident in fractions 4 and 5 of MβCD, filipin, or nystatin-treated BBB-ECs (Fig. 3c, MβCD), confirming lipid raft disruption. Seventy-two hours after treatment, MβCD, filipin, or nystatin in conjunction with ACM induced a marked increase in permeability of BBB-ECs to FITC-BSA (Fig. 3d), compared with cells grown in ACM alone. When compared with nontreated cells (showing ∼50% permeability at 72 h, as before), BBB-ECs grown in ACM plus lipid raft disrupting agents showed a 17.8 ± 5.3% increase in permeability, compared with a decrease of 12 ± 2.9% with ACM alone. Our data suggest that AngII secreted by astrocytes induces, at the level of the human BBB, the functional rearrangement of specific TJ proteins to lipid rafts, a phenomenon necessary to promote BBB integrity.
Astrocytic factors and AngII influence occludin phosphorylation in lipid rafts
Occludin phosphorylation is an essential determinant of its localization and function. To assess the effects of ACM and AngII on occludin phosphorylation, cell lysates from BBB-ECs were immunoprecipitated using anti-phosphorylated-tyrosine (anti-P-Tyr) or anti-phosphorylated-threonine (anti-P-Thr) antibodies and blotted for occludin (Fig. 3e). ACM and AngII induced a downregulation of P-Tyr occludin (37 and 11% of untreated, respectively) and an upregulation of P-Thr occludin (162 and 228% of untreated, respectively) when compared with untreated BBB-ECs. No change in total occludin could be detected. To determine whether these changes occurred at the level of membrane microdomain-associated occludin, lipid rafts from BBB-ECs were isolated as before, and anti-P-Tyr or anti-P-Thr immunoprecipitations were performed on lipid raft (fractions 4–5) and soluble fractions (fractions 11–12). Immunoprecipitated lipid raft-derived proteins enriched for phosphorylated-Tyr and -Thr were subsequently immunoblotted, as shown in Figure 3f. BBB-EC treatment with ACM or AngII induced a downregulation of P-Tyr occludin in lipid rafts only (R lanes, 6 and 7% of untreated, respectively) and an upregulation of P-Thr occludin in lipid rafts (254 and 351% of untreated, respectively) and to a lesser extent in the soluble occludin pool (S lanes) (Fig. 3f). In addition, a second occludin band of higher molecular weight appears in AngII-treated cells, likely representing the hyperphosphorylated form of the protein, as described previously (Nusrat et al., 2000).
Pro-inflammatory cytokines disrupt AngII production by human astrocytes in vitro
Because neuroinflammatory conditions are associated with a BBB breakdown, we further evaluated the effects of the proinflammatory cytokines IFNγ and TNFα on the production of AGT/AngII by astrocytes. Figure 4a shows a substantial decrease of AGT mRNA in astrocytes treated with IFNγ, TNFα (100 U/ml; 24 h), or both. This downregulation was further confirmed by ELISA, which shows an almost complete absence of AngII peptide in the media of astrocytes after 24 h treatment with either inflammatory cytokine (Fig. 4b). Moreover, a decrease in ACE1 mRNA was seen after treatment with IFNγ (100 U/ml; 24 h) as well as with the combined treatment IFNγ and TNFα (100 U/ml; 24 h) (Fig. 4a), suggesting a reduction in the cleavage of AGT to AngII.
Figure 4.

Proinflammatory cytokines disrupt angiotensin production by astrocytes in vitro. a, PCR analysis of AGT expression by human astrocytes grown in vitro shows a marked decrease in AGT expression when astrocytes are cultured in the presence of proinflammatory cytokines IFN-γ and/or TNF-α (100 U/ml; 24 h). Astrocytes also show a decrease expression in ACE1 when treated with IFNγ or both IFNγ/TNFα (100 U/ml; 24 h). b, Twenty-four hour supernatants of human astrocytes grown in the presence of IFNγ and/or TNFα show decreased levels of angiotensin II as assessed by ELISA (n = 4 cytokines alone; n = 8 untreated and both cytokines; *p < 0.01, IFNγ, TNFα, IFNγ plus TNFα when compared with untreated).
Decreased number of AGT immunopositive astrocytes in MS lesions
Infiltration of inflammatory cells into the brain parenchyma is a hallmark of MS. To test whether such inflammation can affect AGT expression in situ, we assessed AGT expression by perivascular S100-positive astrocytes in six MS brains. Areas counted were categorized in three groups with respect to demyelination and cellular infiltration as assessed by LFB (myelin) and H&E staining: active plaques (white matter showing demyelination and cellular infiltration; n = 14 sections from six MS patients) (Fig. 5g–l), inactive plaques (demyelination without cellular infiltration; n = 10 sections from six MS patients) (Fig. 5d–f), and normal-appearing white matter (NAWM; no demyelination nor infiltration; n = 11 sections from six MS patients) (Fig. 5a–c). LFB/H&E as well as AGT (green) and S100 (red) stains are shown for each category of MS lesions in Figure 5a–l and demonstrate an important reduction in perivascular astrocyte expression of AGT (green) in active lesions (i and l) and in chronic lesions (f), compared with NAWM (c) or control human CNS (data not shown). The corner insets of Fig 5, b and c, display a greater magnification of a S100-positive astrocyte immunopositive for AGT (arrowhead). In parallel, the corner insets of Fig. 5, k and l, display S100-positive astrocytes negative for AGT (arrowhead).
Figure 5.
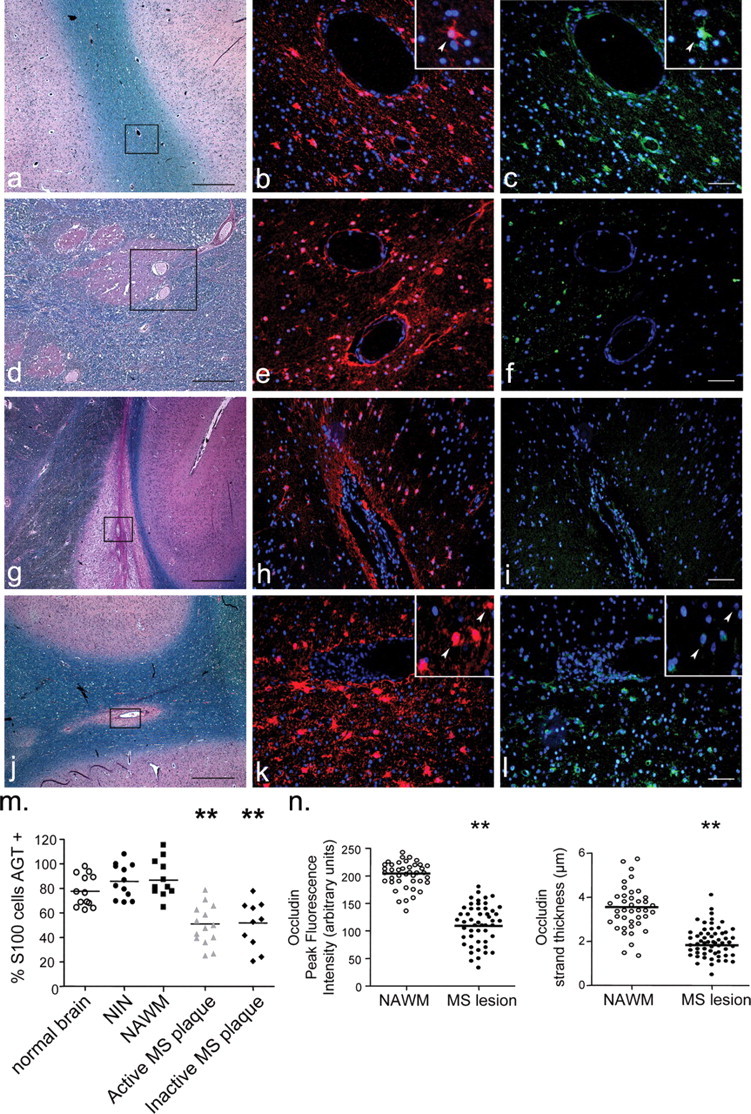
Staining of multiple sclerosis brain for angiotensinogen and S100. LFB and H&E stains, AGT (green), and S100 (red) immunohistofluorescence of brain specimens from MS patients. a–c, Normal-appearing white matter of MS brain; d–f, inactive plaque; g–i, j–l, active plaque. GFAP and AGT immunostainings could not be performed simultaneously, because they require different antigen retrieval protocols, and therefore S100 was used as the perivascular astrocyte marker. Rectangular outlines in LFB/H&E show the area analyzed with regards to AGT/S100 expression. Greater magnification of S100 astrocyte immunopositive for AGT is shown in NAWM (corner insets in b and c; arrowheads) as well as S100 astrocytes negative for AGT in active lesions (corner insets in k and l; arrowheads). Scale bars: black, 250 μm; white, 50 μm. m, Percentage of S100 astrocytes immunopositive for AGT in human was 77.78 ± 3.58% in normal brain (n = 13; 2 patients), 87.64 ± 5.32% in NAWM (n = 11 sections) of six MS brain, and 85.85 ± 4.01% in brain samples from four NIN patients (n = 14 sections from 4 patients). Active (50.96 ± 4.76%; n = 14 sections) and inactive (51.79 ± 6.89%; n = 10 sections, both from 6 MS patients) MS lesions show a significant decrease in the number of AGT-expressing astrocytes (**p < 0.01 for active and inactive plaques compared with normal brain; p < 0.001 compared with NAWM or NIN; all numbers expressed as mean ± SEM). n, Quantification of occludin strand peak fluorescence intensity and thickness in NAWM and active MS lesions reveals a marked decrease of occludin immunosignal in active MS lesions (**p < 0.01; mean ± SEM; from >50 vessels per group).
The percentage of S100 astrocytes immunopositive for AGT was 77.8 ± 3.6% in white matter in normal brain, 87.6 ± 5.3% in NAWM in MS, and 85.8 ± 4.01% in noninflammatory neurological disease (NIN) brain (Fig. 5m). In contrast, in both active (50 ± 4.8%) and inactive (51.8 ± 6.9%) MS lesions, a marked decrease in the number of AGT-positive astrocytes was evident (p < 0.01 for active and inactive plaques compared with normal brain; p < 0.001 for active and inactive plaques compared with NAWM or NIN) (Fig. 5m). This effect was not caused by an increase in the absolute number of astrocytes in MS plaques (as a result of reactive astrogliosis), because the percentage of nuclei that are S100 positive was similar between groups, with the exception of active plaques, showing a decrease in the percentage of S100-positive nuclei, most likely because of the presence of infiltrating immune cells. Immunostainings for AGT and S100 in control normal brain (two patients; n = 13 sections) and tissue from NIN controls (four samples from Parkinson's disease; n = 12 sections) were similar to those performed on NAWM (data not shown).
Decreased occludin expression in MS lesions
In MS patients, brain capillaries found in active lesions also showed a marked reduction in peak occludin strand fluorescence intensity (111 ± 21; n = 55 strands in 12 capillaries) compared with brain capillaries from NAWM (205 ± 16; n = 50 strands in 15 capillaries; p < 0.0001) (Fig. 5n). MS lesions additionally presented a significant reduction in occludin strand thickness around brain capillaries (1.87 ± 0.48 μm; n = 55 strands; 12 capillaries; p < 0.01) compared with NAWM tissue (3.62 ± 0.87 μm; n = 50 strands; 15 capillaries).
Decreased occludin expression in brain capillaries of AGT-deficient mice
Brain sections from wt and AGT knock-out (AGT−/−) mice were stained for the TJ protein occludin. Distinct strands of occludin were visible in brain capillaries. Stacks of 70–100 images were acquired per vessel using the Leica SP5 confocal microscope and collapsed onto a one-dimensional x-y-z projection. Figure 6a shows brain capillaries from wt (top panels) and AGT−/− brains (bottom panels). The peak fluorescence intensity (in arbitrary units) of each strand as well as its thickness (in micrometers) was assessed using the LAS AF Leica software. Occludin strands from AGT−/− brains displayed a significantly reduced peak strand fluorescence intensity (122.4 ± 5.3; n = 54 strands in eight capillaries) compared with brain capillaries from wt mice (215.6 ± 2.8; n = 41 strands in seven capillaries; p < 0.0001) (Fig. 6b). AGT−/− brains also presented a significant reduction in occludin strand thickness (0.99 ± 0.03 μm; n = 59 strands; eight capillaries; p < 0.0001) compared with wt mice (1.78 ± 0.07 μm; n = 40 strands; seven capillaries). Morphologically, occludin-positive strands in AGT−/− animals were also more frequently discontinued and displayed a beaded appearance, suggestive of TJ strand disorganization. Expression of additional TJ proteins, such as claudin-5, ZO-1, and JAM-1 were not affected in the CNS of AGT−/− animals, when compared with wt mice (Fig. 6c). Together, our results demonstrate that in AGT-deficient mice, as well as in MS lesions, occludin expression is diminished at TJ strands.
Figure 6.
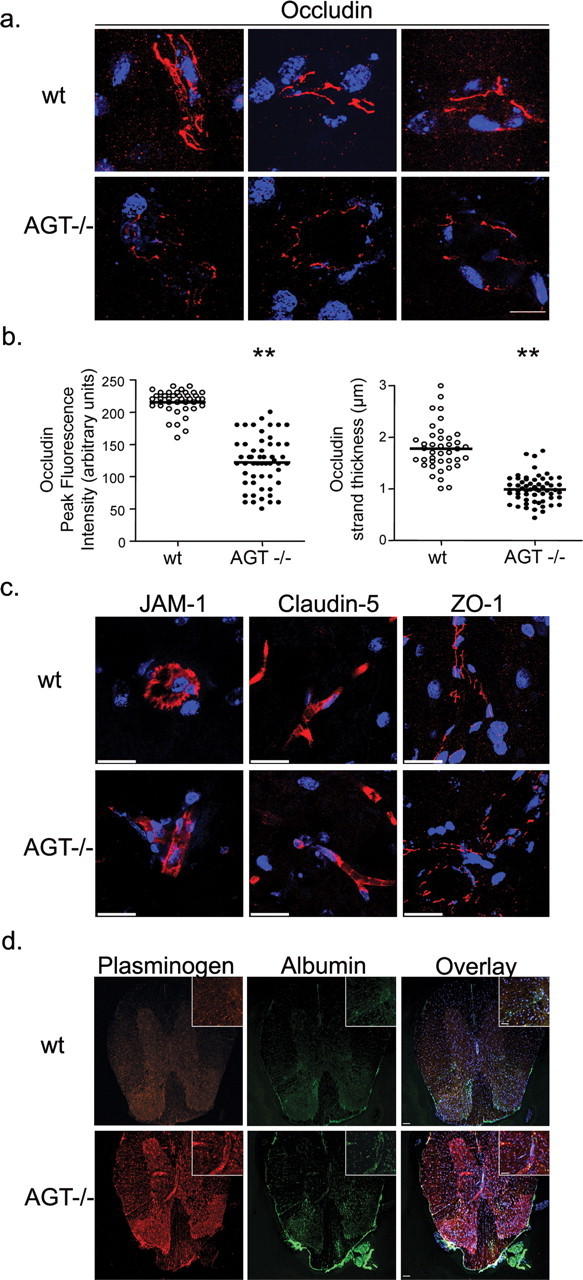
Tight junction protein expression and BBB dysfunction in the CNS of angiotensinogen null animals. a, x-y-z projection of occludin staining in the brain capillary of wt and AGT−/− animals; occludin staining (red) of wt mouse brain (top panels; 3 different microvessels shown) is more intense and continuous than occludin staining of AGT−/− (bottom 3 panels). Scale bar, 20 μm. b, Quantification of occludin strand peak fluorescence intensity and thickness in wt and AGT−/− brain reveals a marked decrease in AGT−/− animals (**p < 0.0001; mean ± SEM). c, x-y-z projection of JAM-1, claudin-5, and ZO-1 staining (all in red) in brain capillaries of wt and AGT−/− animals. x-y-z projections were reconstructed from 70–100 individual images acquired by confocal microscopy. Scale bar, 20 μm. d, x-y planar images of plasminogen and albumin immunostainings in the CNS of wt and AGT−/− animals. Scale bars: 50 μm; inset, 25 μm. TO-PRO3-stained nuclei appear in blue.
Leakage of serum proteins into the CNS of AGT−/− animals
To confirm the functional in vivo significance of AGT disruption in the CNS, adult mice CNS sections from AGT−/− and wt animals were immunostained for the serum proteins plasminogen and albumin, proteins normally not found in the CNS. Plasminogen immunostainings in AGT−/− animals revealed a discrete and diffuse perivascular and parenchymal signal, confirming chronic accumulation of high molecular weight peripheral blood proteins in the CNS (Figs. 6d, 7). The distribution of serum protein leakage in the CNS of AGT−/− animals correlated with previously reported sites of AGT expression in the CNS of rodents (Mungall et al., 1995). The percentage of vessels that showed perivascular accumulation of plasminogen was assessed for each CNS region and revealed important accumulation in the hippocampus (48%), thalamus (43%), and motor and sensory cortex (27%). Interestingly, the highest proportion of vessels in which plasminogen accumulation was detected in AGT−/− animals was found in the corpus callosum (50%), a region universally affected in MS. Such staining was not observed in wt animals and could not be found in AGT−/− mice when the anti-plasminogen Ab was preabsorbed with fresh serum. Accumulation of the endogenous serum protein albumin was also detected in the CNS parenchyma of AGT−/− animals, although albumin immunostaining was less intense than that of plasminogen and seemed to be concentrated around large meningeal vessels (Fig. 6d).
Figure 7.

Plasminogen accumulation in various CNS regions of AGT null animals. Sagittal sections of the CNS of AGT−/− animals were immunostained for plasminogen and caveolin-1. x-y planar images were acquired by confocal microscopy, and the proportion of vessels around which plasminogen accumulation was detected was compared with the total number of caveolin-1+ vessels per field (percentage of plasminogen+ vessels). Representative sagittal section of AGT−/− CNS stained with hematoxylin and eosin and counterstained with Luxol fast blue is shown (n = 2 animals; three fields per region).
CNS sections from AGT−/− and wt animals were also immunostained for the presence of immune cells, to evaluate whether AGT-deficient animals had greater immune surveillance of the CNS. Anti-CD3 immunostainings revealed the presence of occasional but rare lymphocytes in the CNS of either wt or AGT−/− animals (less than one immunopositive cell per field; n > 30 fields studied), suggesting that AGT metabolites are not involved in the control of immune cell migration to the CNS (data not shown).
Discussion
In this study, we demonstrate that astrocyte-derived AngII acts via the AT1 receptor on human brain ECs to decrease the permeability of the BBB. This effect is driven by the mobilization of the TJ protein occludin into brain EC lipid rafts and the change in its phosphorylation status within these membrane microdomains. We further demonstrate that in MS plaques, the number of perivascular AGT-expressing astrocytes, as well as occludin immunoreactivity on microvessels, is decreased compared with noninfiltrated white matter from MS patients. Such a decrease in AGT production could be reproduced in vitro when human astrocytes were cultured in the presence of TNFα and IFNγ. Finally, we show that in AGT-deficient mice, occludin-positive immunofluorescence is diminished and disorganized at TJ strands, whereas claudin-5, JAM-1, and ZO-1 remain unaffected.
As shown in experimental BBB models, astrocyte-derived soluble factors are essential for the development and maintenance of an impermeable BBB. Immediately ex vivo brain microvascular ECs lose their transendothelial electrical resistance (TEER) (Rubin et al., 1991) and become permeable to soluble tracers (Prat et al., 2001). Strong TEER and better barrier properties can be reinduced when cells are grown in the presence of ACM (Arthur et al., 1987; Neuhaus et al., 1991; Abbott, 2002). Our data support previous reports showing AGT expression by rodent astrocytes (Clemens et al., 1986; Deschepper et al., 1986; Lynch et al., 1987; Stornetta et al., 1988) and demonstrate AT1 and AT2 receptor expression by human microvascular brain ECs in situ. Our study provides evidence that astrocyte-produced AngII, acting on human BBB-ECs via the AT1 receptor, is an important effector of astrocytic reinduction of BBB integrity in vitro, a situation analogous to vascular repair of the BBB after injury. Kakinuma et al. (1998) and Yanai et al. (2000) found that in AGT-deficient mice, BBB reconstruction was incomplete after cold injury. We demonstrate that astrocyte-derived AngII drives the mobilization of the TJ protein occludin into specialized membrane microdomains in BBB-ECs, leading to the induction of a better barrier. In fact, AGT-deficient mice exhibit a significantly lower expression of occludin at BBB-TJ strands in vivo, confirming the validity of the in vitro findings. This observation is associated with an increased permeability to the endogenous serum proteins plasminogen and albumin but not with an increased immune surveillance by lymphocytes, suggesting that although astrocyte-secreted AngII influence the ability of BBB-ECs to restrict the passage of soluble molecules from the periphery to the CNS, additional signals are required to induce immune privilege. Whether other TJ proteins, such as claudins or JAMs, are involved in the ability of the BBB to protect the CNS against immune cell invasion remains to be established.
The phosphorylation status of occludin also dictates protein targeting to the membrane and is crucial for TJ formation (Farshori and Kachar, 1999; Hirase et al., 2001; Huber et al., 2001; Petty and Lo, 2002). In vitro, Tyr phosphorylation of occludin increases barrier permeability in peripheral and umbilical vein ECs (Lampugnani et al., 1997; Wachtel et al., 1999). In vivo, BBB disruption secondary to cerebral ischemia correlates with enhanced Tyr phosphorylation of occludin on brain capillaries (Kago et al., 2006). Our data using human BBB-ECs further demonstrates that astrocyte-secreted AGT metabolites are required to induce Tyr dephosphorylation and Thr phosphorylation of occludin, a phenomenon associated with occludin concentration within cholesterol-enriched membrane microdomains (rafts) and needed to promote a more competent BBB.
MS is the prototypical neuroinflammatory disease in which BBB breach is observed, leading to infiltration of blood-borne molecules and cells into the brain. We show that human astrocytes exposed to IFNγ/TNFα downregulate not only AGT/AngII expression but also ACE1, thereby interfering with occludin function and BBB impermeability to soluble molecules. This is paralleled in situ by the decrease in AGT-expressing astrocytes around blood vessels and occludin-expressing ECs in MS plaques when compared with normal or noninfiltrated and nondemyelinated white matter of MS patients. Because AGT−/− animals do not display increased numbers of immune cells patrolling the CNS and because astrocytes (in vitro) downregulate AGT production when treated with proinflammatory cytokines, we must conclude that AGT and occludin disruption in MS lesions are likely to be the reflection of local inflammatory mediators and thus are a consequence, not a cause, of inflammation. However, the fact that both active (infiltrated) and chronic (demyelinated but without immune cells) MS plaques display similar levels of AGT-expressing astrocytes tends to signify that reactive astrocytes fail to reinitiate AGT expression, once the infiltrating immune cells leave the CNS. In this specific case, we postulate that either astrocytes acquire compensatory molecular signals, which can functionally replace AngII, or that BBB injury and leakage occur in chronic inactive lesions but are below the threshold of detection by standard magnetic resonance imaging with gadolinium injection.
Our data could provide a biological explanation for the higher prevalence of CNS and vascular malformations seen in newborns of women treated with inhibitors of the renin-angiotensin system (Cooper et al., 2006). One could speculate that AGT metabolites are critical for BBB and peripheral vascular formation during the prenatal period, a phenomenon that closely resembles vascular repair. Many studies have described a detrimental role of the RAS with regards to brain edema in experimental animal models of stroke (Blezer et al., 2001; Ito et al., 2001; Kucuk et al., 2002; Lou et al., 2004), observations seemingly divergent from ours. Such differences can be explained by the fact that most studies were conducted in hypertensive animals, a model in which subclinical BBB dysfunction is certainly present, and during which CNS compensatory mechanisms probably occur. One could also argue that in humans, diuretics and calcium channel blockers that stimulate the RAS and increase AngII production are just as effective as ACE inhibitors in decreasing cerebral edema during stroke, suggesting that the exact role of the RAS during ischemia is still not resolved (Hansson, 2000; Nissen et al., 2004; Ovbiagele et al., 2005).
Our study defines a new physiological role for AngII at the level of the human BBB and provides a novel molecular mechanism to explain BBB disruption in MS lesions. We can postulate that RAS-related agents may be seen as novel candidate molecules to either allow greater access of molecules across the BBB, as would be beneficial for the treatment of CNS tumors and neurodegenerative conditions, or conversely to restrict the movement of soluble inflammatory molecules from the blood to the CNS, as would be beneficial in MS.
Footnotes
This work was supported by grants from the Canadian Institute of Health Research (CIHR) (INO-Neuroscience Mental Health and Addiction and MOP81088), the Multiple Sclerosis Society of Canada (MSSC), and the Canadian Foundation for Innovation. A.P. holds the Donald Paty Career Development Award of the MSSC and is a Research Scholar (Junior 1) from the Fonds de Recherche en Santé du Québec. K.W. holds a fellowship from the MSSC. A.D.-D. and R.C. hold a CIHR-sponsored neuroinflammation training grant studentship. We thank Stephanie Jack for her work in setting up the MS brain bank, Dr. T. Vincent for his work with BBB-ECs/astrocyte cocultures, and Dr. Chan for the generous gift of the AT1 antagonist as well as Dr. Nathalie Arbour, Hania Kébir, and Igal Ifergan for critical reading of this manuscript.
References
- Abbott NJ. Astrocyte-endothelial interactions and blood–brain barrier permeability. J Anat. 2002;200:629–638. doi: 10.1046/j.1469-7580.2002.00064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood–brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- Alter A, Duddy M, Hebert S, Biernacki K, Prat A, Antel JP, Yong VW, Nuttall RK, Pennington CJ, Edwards DR, Bar-Or A. Determinants of human B cell migration across brain endothelial cells. J Immunol. 2003;170:4497–4505. doi: 10.4049/jimmunol.170.9.4497. [DOI] [PubMed] [Google Scholar]
- Arthur FE, Shivers RR, Bowman PD. Astrocyte-mediated induction of tight junctions in brain capillary endothelium: an efficient in vitro model. Brain Res. 1987;433:155–159. doi: 10.1016/0165-3806(87)90075-7. [DOI] [PubMed] [Google Scholar]
- Biernacki K, Prat A, Blain M, Antel JP. Regulation of cellular and molecular trafficking across human brain endothelial cells by Th1- and Th2-polarized lymphocytes. J Neuropathol Exp Neurol. 2004;63:223–232. doi: 10.1093/jnen/63.3.223. [DOI] [PubMed] [Google Scholar]
- Blezer EL, Nicolay K, Koomans HA, Joles JA. Losartan versus enalapril on cerebral edema and proteinuria in stroke-prone hypertensive rats. Am J Hypertens. 2001;14:54–61. doi: 10.1016/s0895-7061(00)01231-0. [DOI] [PubMed] [Google Scholar]
- Clemens DL, Clauser E, Celio MR, Inagami T. Generation of angiotensinogen by cultured neuroblastoma and glioma cells. Brain Res. 1986;364:205–211. doi: 10.1016/0006-8993(86)90832-2. [DOI] [PubMed] [Google Scholar]
- Cooper WO, Hernandez-Diaz S, Arbogast PG, Dudley JA, Dyer S, Gideon PS, Hall K, Ray WA. Major congenital malformations after first-trimester exposure to ACE inhibitors. N Engl J Med. 2006;354:2443–2451. doi: 10.1056/NEJMoa055202. [DOI] [PubMed] [Google Scholar]
- de Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev. 2000;52:415–472. [PubMed] [Google Scholar]
- Deschepper CF, Bouhnik J, Ganong WF. Colocalization of angiotensinogen and glial fibrillary acidic protein in astrocytes in rat brain. Brain Res. 1986;374:195–198. doi: 10.1016/0006-8993(86)90411-7. [DOI] [PubMed] [Google Scholar]
- D'Souza S, Alinauskas K, McCrea E, Goodyer C, Antel JP. Differential susceptibility of human CNS-derived cell populations to TNF-dependent and independent immune-mediated injury. J Neurosci. 1995;15:7293–7300. doi: 10.1523/JNEUROSCI.15-11-07293.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farshori P, Kachar B. Redistribution and phosphorylation of occludin during opening and resealing of tight junctions in cultured epithelial cells. J Membr Biol. 1999;170:147–156. doi: 10.1007/s002329900544. [DOI] [PubMed] [Google Scholar]
- Ghandour MS, Langley OK, Labourdette G, Vincendon G, Gombos G. Specific and artefactual cellular localizations of S 100 protein: an astrocyte marker in rat cerebellum. Dev Neurosci. 1981;4:66–78. doi: 10.1159/000112742. [DOI] [PubMed] [Google Scholar]
- Hansson L. Results of the STOP-Hypertension-2 trial. Blood Press Suppl. 2000;2:17–20. [PubMed] [Google Scholar]
- Hirase T, Kawashima S, Wong EY, Ueyama T, Rikitake Y, Tsukita S, Yokoyama M, Staddon JM. Regulation of tight junction permeability and occludin phosphorylation by Rhoa-p160ROCK-dependent and -independent mechanisms. J Biol Chem. 2001;276:10423–10431. doi: 10.1074/jbc.M007136200. [DOI] [PubMed] [Google Scholar]
- Huber JD, Egleton RD, Davis TP. Molecular physiology and pathophysiology of tight junctions in the blood–brain barrier. Trends Neurosci. 2001;24:719–725. doi: 10.1016/s0166-2236(00)02004-x. [DOI] [PubMed] [Google Scholar]
- Ifergan I, Wosik K, Cayrol R, Kebir H, Auger C, Bernard M, Bouthillier A, Moumdjian R, Duquette P, Prat A. Statins reduce human blood–brain barrier permeability and restrict leukocyte migration: relevance to multiple sclerosis. Ann Neurol. 2006;60:45–55. doi: 10.1002/ana.20875. [DOI] [PubMed] [Google Scholar]
- Intebi AD, Flaxman MS, Ganong WF, Deschepper CF. Angiotensinogen production by rat astroglial cells in vitro and in vivo. Neuroscience. 1990;34:545–554. doi: 10.1016/0306-4522(90)90163-x. [DOI] [PubMed] [Google Scholar]
- Ito H, Takemori K, Suzuki T. Role of angiotensin II type 1 receptor in the leucocytes and endothelial cells of brain microvessels in the pathogenesis of hypertensive cerebral injury. J Hypertens. 2001;19:591–597. doi: 10.1097/00004872-200103001-00011. [DOI] [PubMed] [Google Scholar]
- Jack CS, Arbour N, Manusow J, Montgrain V, Blain M, McCrea E, Shapiro A, Antel JP. TLR signaling tailors innate immune responses in human microglia and astrocytes. J Immunol. 2005;175:4320–4330. doi: 10.4049/jimmunol.175.7.4320. [DOI] [PubMed] [Google Scholar]
- Juillerat-Jeanneret L, Celerier J, Chapuis BC, Nguyen G, Wostl W, Maerki HP, Janzer RC, Corvol P, Gasc JM. Renin and angiotensinogen expression and functions in growth and apoptosis of human glioblastoma. Br J Cancer. 2004;90:1059–1068. doi: 10.1038/sj.bjc.6601646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kacem K, Lacombe P, Seylaz J, Bonvento G. Structural organization of the perivascular astrocyte endfeet and their relationship with the endothelial glucose transporter: a confocal microscopy study. Glia. 1998;23:1–10. [PubMed] [Google Scholar]
- Kago T, Takagi N, Date I, Takenaga Y, Takagi K, Takeo S. Cerebral ischemia enhances tyrosine phosphorylation of occludin in brain capillaries. Biochem Biophys Res Commun. 2006;339:1197–1203. doi: 10.1016/j.bbrc.2005.11.133. [DOI] [PubMed] [Google Scholar]
- Kakinuma Y, Hama H, Sugiyama F, Yagami K, Goto K, Murakami K, Fukamizu A. Impaired blood–brain barrier function in angiotensinogen-deficient mice. Nat Med. 1998;4:1078–1080. doi: 10.1038/2070. [DOI] [PubMed] [Google Scholar]
- Kucuk M, Kaya M, Kalayci R, Cimen V, Kudat H, Arican N, Elmas I, Korkut F. Effects of losartan on the blood–brain barrier permeability in long-term nitric oxide blockade-induced hypertensive rats. Life Sci. 2002;71:937–946. doi: 10.1016/s0024-3205(02)01772-1. [DOI] [PubMed] [Google Scholar]
- Lampugnani MG, Corada M, Andriopoulou P, Esser S, Risau W, Dejana E. Cell confluence regulates tyrosine phosphorylation of adherens junction components in endothelial cells. J Cell Sci. 1997;110:2065–2077. doi: 10.1242/jcs.110.17.2065. [DOI] [PubMed] [Google Scholar]
- Lochard N, Silversides DW, van Kats JP, Mercure C, Reudelhuber TL. Brain-specific restoration of angiotensin II corrects renal defects seen in angiotensinogen-deficient mice. J Biol Chem. 2003;278:2184–2189. doi: 10.1074/jbc.M209933200. [DOI] [PubMed] [Google Scholar]
- Lou M, Blume A, Zhao Y, Gohlke P, Deuschl G, Herdegen T, Culman J. Sustained blockade of brain AT1 receptors before and after focal cerebral ischemia alleviates neurologic deficits and reduces neuronal injury, apoptosis, and inflammatory responses in the rat. J Cereb Blood Flow Metab. 2004;24:536–547. doi: 10.1097/00004647-200405000-00008. [DOI] [PubMed] [Google Scholar]
- Lynch KR, Hawelu-Johnson CL, Guyenet PG. Localization of brain angiotensinogen mRNA by hybridization histochemistry. Brain Res. 1987;388:149–158. doi: 10.1016/s0006-8993(87)80008-2. [DOI] [PubMed] [Google Scholar]
- Methot D, vanKats JP, Lochard N, Tremblay F, Silversides DW, Reudelhuber TL. Development and application of a biological peptide pump for the study of the in vivo actions of angiotensin peptides. Am J Hypertens. 2001;14:38S–43S. doi: 10.1016/s0895-7061(01)02068-4. [DOI] [PubMed] [Google Scholar]
- Morimoto S, Sigmund CD. Angiotensin mutant mice: a focus on the brain renin-angiotensin system. Neuropeptides. 2002;36:194–200. doi: 10.1054/npep.2002.0894. [DOI] [PubMed] [Google Scholar]
- Mungall BA, Shinkel TA, Sernia C. Immunocytochemical localization of angiotensinogen in the fetal and neonatal rat brain. Neuroscience. 1995;67:505–524. doi: 10.1016/0306-4522(95)00044-j. [DOI] [PubMed] [Google Scholar]
- Nedergaard M, Ransom B, Goldman SA. New roles for astrocytes: redefining the functional architecture of the brain. Trends Neurosci. 2003;26:523–530. doi: 10.1016/j.tins.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Neuhaus J, Risau W, Wolburg H. Induction of blood–brain barrier characteristics in bovine brain endothelial cells by rat astroglial cells in transfilter coculture. Ann NY Acad Sci. 1991;633:578–580. doi: 10.1111/j.1749-6632.1991.tb15667.x. [DOI] [PubMed] [Google Scholar]
- Nissen SE, Tuzcu EM, Libby P, Thompson PD, Ghali M, Garza D, Berman L, Shi H, Buebendorf E, Topol EJ. Effect of antihypertensive agents on cardiovascular events in patients with coronary disease and normal blood pressure: the CAMELOT study: a randomized controlled trial. JAMA. 2004;292:2217–2225. doi: 10.1001/jama.292.18.2217. [DOI] [PubMed] [Google Scholar]
- Nusrat A, Parkos CA, Verkade P, Foley CS, Liang TW, Innis-Whitehouse W, Eastburn KK, Madara JL. Tight junctions are membrane microdomains. J Cell Sci. 2000;113:1771–1781. doi: 10.1242/jcs.113.10.1771. [DOI] [PubMed] [Google Scholar]
- Ovbiagele B, Kidwell CS, Starkman S, Selco SL, Rajajee V, Razinia T, Saver JL. Angiotensin 2 type 2 receptor activity and ischemic stroke severity. Neurology. 2005;65:851–854. doi: 10.1212/01.wnl.0000175984.29283.6d. [DOI] [PubMed] [Google Scholar]
- Petty MA, Lo EH. Junctional complexes of the blood–brain barrier: permeability changes in neuroinflammation. Prog Neurobiol. 2002;68:311–323. doi: 10.1016/s0301-0082(02)00128-4. [DOI] [PubMed] [Google Scholar]
- Pike LJ. Lipid rafts: heterogeneity on the high seas. Biochem J. 2004;378:281–292. doi: 10.1042/BJ20031672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prat A, Biernacki K, Becher B, Antel JP. B7 expression and antigen presentation by human brain endothelial cells: requirement for proinflammatory cytokines. J Neuropathol Exp Neurol. 2000;59:129–136. doi: 10.1093/jnen/59.2.129. [DOI] [PubMed] [Google Scholar]
- Prat A, Biernacki K, Wosik K, Antel JP. Glial cell influence on the human blood–brain barrier. Glia. 2001;36:145–155. doi: 10.1002/glia.1104. [DOI] [PubMed] [Google Scholar]
- Prat A, Biernacki K, Lavoie JF, Poirier J, Duquette P, Antel JP. Migration of multiple sclerosis lymphocytes through brain endothelium. Arch Neurol. 2002;59:391–397. doi: 10.1001/archneur.59.3.391. [DOI] [PubMed] [Google Scholar]
- Rubin LL, Barbu K, Bard F, Cannon C, Hall DE, Horner H, Janatpour M, Liaw C, Manning K, Morales J, Porter S, Tanner L, Tomasellli K, Yednock T. Differentiation of brain endothelial cells in cell culture. Ann NY Acad Sci. 1991;633:420–425. doi: 10.1111/j.1749-6632.1991.tb15631.x. [DOI] [PubMed] [Google Scholar]
- Saavedra JM. Brain angiotensin II: new developments, unanswered questions and therapeutic opportunities. Cell Mol Neurobiol. 2005;25:485–512. doi: 10.1007/s10571-005-4011-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanimirovic D, Shapiro A, Wong J, Hutchison J, Durkin J. The induction of ICAM-1 in human cerebromicrovascular endothelial cells (HCEC) by ischemia-like conditions promotes enhanced neutrophil/HCEC adhesion. J Neuroimmunol. 1997;76:193–205. doi: 10.1016/s0165-5728(97)00057-x. [DOI] [PubMed] [Google Scholar]
- Stanimirovic DB, Ball R, Durkin JP. Evidence for the role of protein kinase C in astrocyte-induced proliferation of rat cerebromicrovascular endothelial cells. Neurosci Lett. 1995;197:219–222. doi: 10.1016/0304-3940(95)11941-o. [DOI] [PubMed] [Google Scholar]
- Stornetta RL, Hawelu-Johnson CL, Guyenet PG, Lynch KR. Astrocytes synthesize angiotensinogen in brain. Science. 1988;242:1444–1446. doi: 10.1126/science.3201232. [DOI] [PubMed] [Google Scholar]
- Tao-Cheng JH, Brightman MW. Development of membrane interactions between brain endothelial cells and astrocytes in vitro. Int J Dev Neurosci. 1988;6:25–37. doi: 10.1016/0736-5748(88)90026-3. [DOI] [PubMed] [Google Scholar]
- Vianello B, Clauser E, Corvol P, Monnot C. Functional interactions of L-162,313 with angiotensin II receptor subtypes and mutants. Eur J Pharmacol. 1998;347:113–118. doi: 10.1016/s0014-2999(98)00081-8. [DOI] [PubMed] [Google Scholar]
- Wachtel M, Frei K, Ehler E, Fontana A, Winterhalter K, Gloor SM. Occludin proteolysis and increased permeability in endothelial cells through tyrosine phosphatase inhibition. J Cell Sci. 1999;112:4347–4356. doi: 10.1242/jcs.112.23.4347. [DOI] [PubMed] [Google Scholar]
- Wosik K, Becher B, Ezman A, Nalbantoglu J, Antel JP. Caspase 8 expression and signaling in Fas injury-resistant human fetal astrocytes. Glia. 2001;33:217–224. doi: 10.1002/1098-1136(200103)33:3<217::aid-glia1020>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Yanai K, Saito T, Kakinuma Y, Kon Y, Hirota K, Taniguchi-Yanai K, Nishijo N, Shigematsu Y, Horiguchi H, Kasuya Y, Sugiyama F, Yagami K, Murakami K, Fukamizu A. Renin-dependent cardiovascular functions and renin-independent blood–brain barrier functions revealed by renin-deficient mice. J Biol Chem. 2000;275:5–8. doi: 10.1074/jbc.275.1.5. [DOI] [PubMed] [Google Scholar]