

Abstract
The glutamate–glutamine cycle is thought to be integral in continuously replenishing the neurotransmitter pool of glutamate. Inhibiting glial transfer of glutamine to neurons leads to rapid impairment in physiological and behavioral function; however, the degree to which excitatory synaptic transmission relies on the normal operation of this cycle is unknown. In slices and cultured neurons from rat hippocampus, we enhanced the transfer of glutamine to neurons, a fundamental step in this cycle, by adding exogenous glutamine. Although raising glutamine augments synaptic transmission by increasing vesicular glutamate, access to this synthetic pathway by exogenously applied glutamine to neurons is delayed and slow, challenging mechanisms linking the rapid effects of pharmacological inhibitors to decreased vesicular glutamate. We find that pharmacological inhibitors of glutamine synthetase or system A transporters cause an acute depression of basal synaptic transmission that is rapidly reversible, which is unlikely to be attributable to the rapid loss of vesicular glutamate. Furthermore, release of vesicular glutamate remains robust even during the prolonged removal of glutamine from pure neuronal cultures. We conclude that neurons have the capacity to store or produce glutamate for long periods of time, independently of glia and the glutamate–glutamine cycle.
Keywords: glutamate–glutamine cycle, hippocampus, CA1, synaptic transmission, system A, glutamine synthetase
Introduction
Excitatory neurons use the amino acid glutamate as a neurotransmitter in the mammalian CNS. To sustain chemical transmission, these neurons accumulate high concentrations of glutamate in synaptic vesicles. All cells synthesize glutamate from intermediates in the tricarboxylic acid (TCA) cycle (Stryer, 1995). However, glutamate biosynthesis through the TCA cycle is thought to be insufficient to support the neurotransmitter pool, because exclusive supply of glutamate through this pathway would deplete the TCA cycle of key intermediates (Hertz and Zielke, 2004). Neurons lack enzymes (e.g., pyruvate carboxylase) present in other cells that serve to replenish the TCA cycle (Shank et al., 1985). Neurons, then, apparently cannot maintain both energy production and a neurotransmitter pool independently. Instead, neuronal glutamate, or its synthetic precursor, may originate from an exogenous source.
A cycle in which glutamate and glutamine are exchanged between neurons and glia arose from studies of neuronal metabolism (van den Berg and Garfinkel, 1971; Benjamin and Quastel, 1975). In the glutamate–glutamine cycle, glutamate released during synaptic transmission is transported into glia by the EAAT2 (excitatory amino acid transporter 2) family of plasma membrane glutamate transporters (Danbolt, 2001). Glutamine synthetase, an enzyme found in astrocytes and other glia, converts glutamate into glutamine (Martinez-Hernandez et al., 1977). Glutamine is then transported by system N transporters from glia into the extracellular space where it is retrieved by neuronal system A neutral amino acid transporters (Chaudhry et al., 2002). In neurons, phosphate-activated glutaminase (PAG) deaminates glutamine, producing glutamate (Kvamme et al., 2000). Finally, glutamate is transported into synaptic vesicles by vesicular glutamate transporters, thus completing the cycle (Fremeau et al., 2004).
Although many of the molecular components of the cycle have been identified, the physiological role of the glutamate–glutamine cycle in synaptic transmission is poorly understood (Rothman et al., 2003). Models based on estimates of metabolic rates in bulk tissue suggest that astrocytes contribute significantly to neuronal metabolism and support astrocytes as the site of glutamate to glutamine conversion (Gruetter et al., 2001). Inhibition of glutamine synthetase in hippocampal slice cultures decreases immunogold labeling of astrocytic glutamine and neuronal glutamate, while increasing labeling of astrocytic glutamate (Laake et al., 1999). This redistribution further supports an essential role for astrocytic glutamine synthetase in the generation of nerve terminal glutamate.
In fact, it has been suggested that uninterrupted transfer of glutamine from astrocytes to neurons is necessary for synaptic function. Inhibition of glutamine synthetase causes an acute depression in retinal function (Winkler et al., 1999; Barnett et al., 2000) as well as an acute impairment in performance on a learning task in chicks (Hertz et al., 1996). Given these dramatic effects, we sought to assay synaptic transmission directly in rat hippocampal slices and dissociated neuronal cultures under conditions in which the glutamate–glutamine cycle was perturbed. We show that pharmacological inhibition of the glutamate–glutamine cycle fails to abolish synaptic transmission, even during intense synaptic activation. Our findings indicate that even in the absence of glial support, neurons can maintain a releasable pool of vesicular glutamate for long periods of time.
Materials and Methods
Primary hippocampal culture.
Hippocampi from postnatal day 0 (P0) rats were dissociated in HBSS with papain/EDTA (Worthington Biochemical, Lakewood, NJ) for 15 min at 37°C, treated with trypsin inhibitor for 5 min, washed in Neurobasal A (Invitrogen, Eugene, OR) containing 5% FBS (Invitrogen), and triturated. After trituration, the cells were plated on poly-d-lysine-coated coverslips at a density of 100,000 per 12 mm coverslip. After 1 d, the medium was replaced with Neurobasal A containing 5% FBS, 2% B27 supplement (Invitrogen), 1% penicillin-streptomycin, and 1 mm l-glutamine (Invitrogen). Ten micromolars fluoro-deoxy uridine in 1/5 volume of fresh medium were added on day 2–3 to inhibit glial growth, and one-half volume fresh medium was exchanged on day 9.
Banker culture.
The cortex from postnatal day 0 (P0) to P2 was dissociated in HBSS with papain/EDTA for 10 min at 37°C, treated with trypsin inhibitor, triturated, and resuspended in MEM with Earle's containing 10% FBS. After resuspension, cells were plated in 75 mm flasks. One day and 4 d after plating, fresh medium was added. After 7 d, the flask was shaken vigorously overnight to remove contaminating glia, fibroblasts, and neurons. After agitation, HBSS with papain/EDTA was added and cells were plated in MEM with Earle's containing 10% FBS at a density of 25,000 per well in a six-well plate. Two to 5 d after plating, serum medium was replaced with Neurobasal containing 10% FBS, 4% B27 supplement, 1% penicillin-streptomycin, and 1 mm glutamine.
Dissociated hippocampal neurons derived from embryonic day 16 (E16) to E18 rats using the above procedure were plated on poly-d-lysine-coated coverslips with paraffin dots at a density of 100,000 per 12 mm coverslip. Cells were left to settle on the coverslips overnight before the coverslips were flipped over into the wells containing the glial feeder layer. Two to 5 d after plating, 10 μm fluoro-deoxy uridine was added in 1/5 volume fresh medium.
Electrophysiology.
Transverse hippocampal slices (300–400 μm) were prepared from Sprague Dawley rats (12–24 d old) as described previously. The slices were maintained in a submerged chamber for both recovery (at least 1 h) and recording at room temperature (24–28°C). Perfusion medium contained (in mm) 119 NaCl, 2.5 KCl, 2.5 CaCl2, 1.4 MgSO4, 1 NaH2PO4, 26.2 NaHCO3, and 11 glucose, saturated with 95% O2-5% CO2. The perfusion rate was 2 ml/min.
Field EPSPs (fEPSPs) were evoked in the CA1 stratum radiatum by stimulation of Schaffer collaterals with glass pipettes filled with either 1 m NaCl or extracellular recording solution, and were recorded with 3–5 MΩ glass pipettes filled with either 1 m NaCl or extracellular recording solution using the Axopatch-1D amplifier (Molecular Devices, Union City, CA). fEPSPs were filtered at 2 kHz, digitized at 10 kHz, and stored on computers using IgorPro (Wavemetrics, Lake Oswego, OR). Test stimuli consisted of 100 μs pulses of constant voltage delivered by stimulus isolation units (A.M.P.I., Jerusalem, Israel). The baseline stimulation rate was 0.1 Hz for all experiments. For input–output experiments, slices from control and glutamine incubated groups were interleaved. In hyperkalemic conditions, slices were perfused with solution containing (in mm) 29 NaCl, 90 KCl, 26 NaHCO3, 10 glucose, 1 NaH2PO4, 1.3 MgSO4, and 2.5 CaCl2, saturated with 95% O2-5% CO2. Data points obtained during hyperkalemic stimulation and stimulus artifacts were blanked for clarity. Control conditions were made iso-osmotic with the experimental condition using sucrose to avoid osmolarity artifacts. Picrotoxin was included in all solutions to isolate glutamatergic transmission. d-(−)-2-Amino-5-phosphonopentanoic acid (d-APV) was included except when indicated to prevent NMDA-dependent plasticity.
Somatic whole-cell voltage-clamp recordings in acute slices were obtained from visually identified CA1 pyramidal cells using 2.5–5 MΩ glass electrodes filled with (in mm) 107.5 Cs-gluconate, 20 HEPES, 0.2 EGTA, 8 Na-gluconate, 8 tetraethylammonium-Cl, 4 Mg-ATP, 0.3 Na-GTP, and 5 QX-314, pH 7.27, 280 mOsm. Cells in which the series or input resistance varied by 20% during the experiment were discarded. Stimulus artifacts were blanked. mEPSCs were measured in the presence of 0.5 μm TTX and were recorded and analyzed off line with customized software, using an amplitude threshold of 6 pA. Entries in cumulative distribution histograms ranged from 90 to 330 events.
For cultured neurons, the extracellular recording solution contained (in mm) 140 NaCl, 2.4 KCl, 4 CaCl2, 4 MgSO4, 10 HEPES, and 10 glucose. The pH was adjusted with NaOH to 7.27. In Na+-free incubation solution, the NaCl was replaced with choline chloride and the pH was adjusted with KOH. For high-K+ stimulation, the neurons were placed in a solution containing (in mm) 52.4 NaCl, 90 KCl, 10 HEPES, 10 glucose, 4 CaCl2, and 4 MgSO4.
Averaged data represent the mean ± SEM. Statistical significance was determined using the Student's t test at a significance level of p < 0.05 unless otherwise indicated.
d-APV, γ-d-glutamylglycine (γ-DGG), dl-threo-β-benzyloxyaspartic acid (dl-TBOA), and tetrodotoxin were obtained from Tocris (Ellisville, MO). All other compounds were obtained from Sigma (St. Louis, MO).
Results
Glutamine synthetase inhibition depresses synaptic transmission acutely, but not through a glutamate–glutamine cycle block
Glial synthesis of glutamine from synaptically released glutamate initiates the return of this neurotransmitter back to presynaptic terminals. l-Methionine sulfoximine (MSO) inhibits this conversion by blocking glutamine synthetase (Lamar and Sellinger, 1965; Manning et al., 1969). Application of MSO (10 mm) in rat hippocampal slices acutely decreased the fEPSP recorded in stratum radiatum area CA1 to 73.91 ± 2.36% of control; however, exogenous glutamine (4 mm) did not offset the inhibitory effect of MSO, which reduced the fEPSP to 79.41 ± 1.66% of control in the presence of glutamine (n = 9 slices; p > 0.05, compared with MSO alone) (Fig. 1A). Therefore, the acute depression cannot be related to decreased glutamine synthesis, rather it may be attributable to a nonspecific MSO effect. In some preparations, MSO requires hours of incubation to inhibit glutamine synthetase completely. Incubating slices in MSO (40 mm) for 1–6 h and then returning the slices to control solutions did not specifically affect basal synaptic transmission (Fig. 1B), suggesting that glutamine synthetase is not necessary for maintaining the vesicular pool of glutamate at Schaffer collateral synapses.
Figure 1.

MSO causes an acute decrease in synaptic transmission that appears to be independent of glutamate–glutamine cycle inhibition. A, fEPSPs recorded in stratum radiatum of CA1 in rat hippocampal slices (n = 9) are inhibited by MSO (10 mm); however, glutamine (Gln; 4 mm) does not prevent the effect. B, Input–output relationships in slices incubated in MSO (40 mm; n = 11) or control solution (n = 9) for >4 h are not significantly different (p > 0.05 for all fiber volley amplitudes). Representative traces at three fiber volley amplitudes are shown to the right for a control slice (top) and a MSO incubated slice (bottom). FV, Fiber volley. Calibration: 0.5 mV, 10 ms.
Inhibition of glutamine uptake produces a rapidly reversible, acute depression
Although glutamine synthetase may not generate the glutamine that serves as the biosynthetic precursor of vesicular glutamate, neurons may still import glutamine from other sources through neutral amino acid transporters. Glutamine entry into neurons through system A transporters can be prevented by α-(methylamino)isobutyric acid (MeAIB), a competitive substrate used to define this family of transporters (Reimer et al., 2000; Sugawara et al., 2000; Varoqui et al., 2000; Yao et al., 2000). Acute application of MeAIB (25 mm) produced a small decrease in the fEPSP slope that was rapidly reversible, suggesting a role for extracellular glutamine in maintaining excitatory synaptic transmission (Fig. 2A). To determine whether long-term restriction of glutamine entry produces a persistent depression in synaptic transmission, input–output curves were collected in slices incubated in MeAIB (25 mm) for 1–6 h. We hypothesized that, with spontaneous glutamate release ongoing while the slices are in the incubating chamber, blocking the glutamate–glutamine cycle for an extended period of time would eventually deplete vesicular glutamate; however, basal synaptic transmission was unaffected by the prolonged incubation, similar to MSO-incubated slices (Fig. 2B).
Figure 2.

MeAIB produces a rapidly reversible acute depression of synaptic transmission. A, Depression of fEPSPs recorded in stratum radiatum of CA1 is observed after MeAIB (25 mm) application (n = 7). B, Incubation in MeAIB (25 mm) for more than 4 h does not change the input–output curve (control, n = 16; MeAIB, n = 14; p > 0.05 for all fiber volley amplitudes). Representative traces in control slices (top) and MeAIB-incubated slices (bottom) are shown to the right. FV, Fiber volley. Calibration: 0.5 mV, 10 ms.
Enhancing extracellular glutamine increases vesicular glutamate release
To avoid nonspecific effects, slices incubated in MSO and MeAIB were recorded in a solution lacking these inhibitors. Our data suggest that relieving inhibition of glutamine synthetase or system A transport may allow rapid replenishment of glutamine and vesicular glutamate. If this were the case, glia should be able to restore glutamine to neurons very quickly and neurons should be able to convert that glutamine to vesicular glutamate in minutes. Thus, enhancing the transfer of glutamine to neurons should increase vesicular glutamate with the same efficiency. To test this hypothesis, we exposed neurons to increased extracellular glutamine concentrations. Extracellular glutamine is estimated to be between 200 and 900 μm in vivo (Gjessing et al., 1972; Lerma et al., 1986), but may be absent in tissue slices (Kapetanovic et al., 1993). Hippocampal slices incubated in physiological concentrations of glutamine do not show any difference in quantal amplitude (control, −9.95 ± 0.50 pA; 900 μm glutamine, −10.70 ± 0.55 pA; n = 9 for each; p > 0.05) or frequency (control, 0.69 ± 0.21 Hz; 900 μm glutamine, 0.58 ± 0.21 Hz; n = 9 for each; p > 0.05) compared with slices incubated in our standard extracellular solution. Incubating hippocampal slices in 4 mm glutamine resulted in an increase in the input–output relationship of the fEPSP mediated by AMPA receptors when compared with control slices (Fig. 3A). fEPSPs mediated by NMDA receptors in glutamine-incubated slices were similarly augmented (Fig. 3B). Increases in the ratio of both AMPA and NMDA postsynaptic potentials to the presynaptic fiber volley in glutamine-treated slices indicate a presynaptic change in release.
Figure 3.
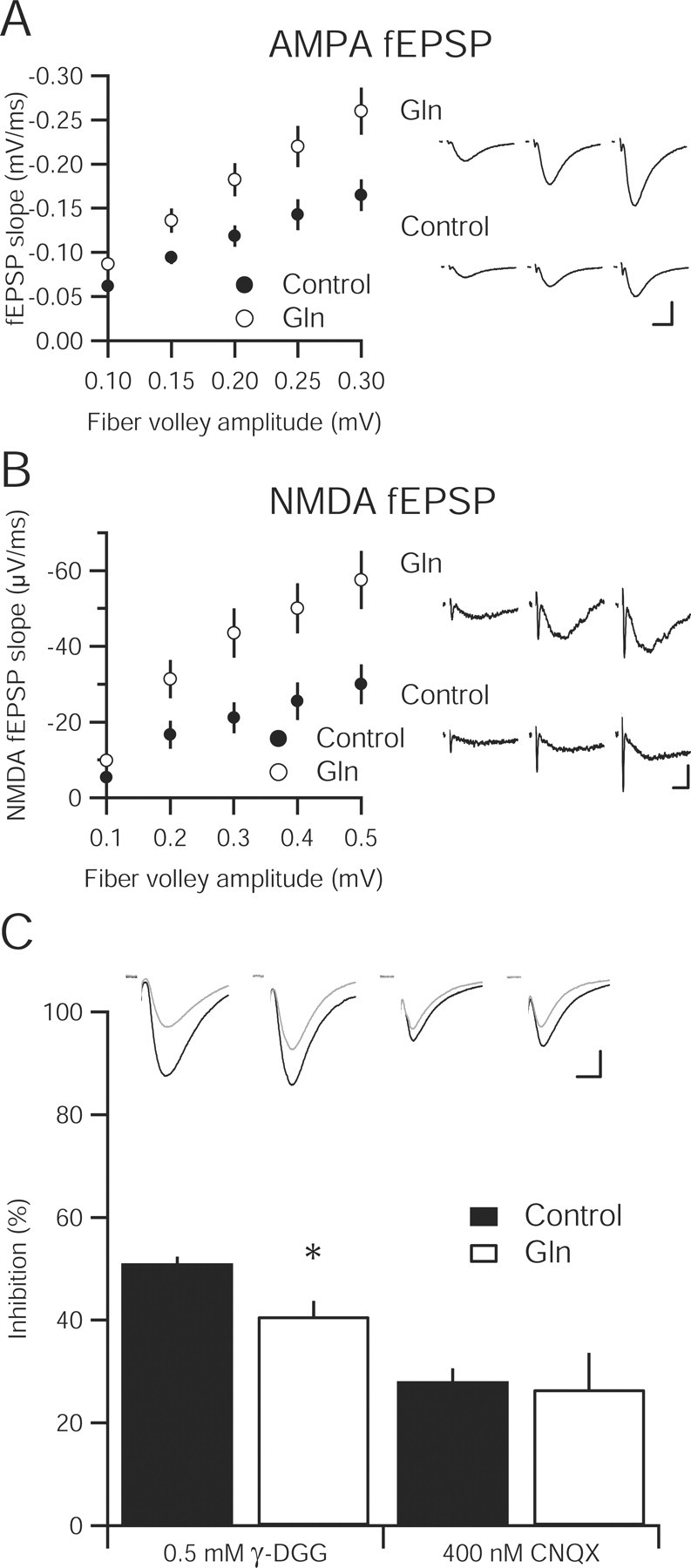
Increasing extracellular glutamine enhances synaptic transmission and vesicular glutamate content in hippocampal slice. A, Input–output relationship of the AMPA-mediated fEPSPs in hippocampal slices incubated in 4 mm glutamine (n = 16) compared with a 4 mm sucrose osmotic control (n = 15) are statistically greater at all fiber volley amplitudes (p < 0.05). Sample fEPSPs at different fiber volley amplitudes from each group are shown to the right. Calibration: 0.5 mV, 10 ms. B, Input–output curve of NMDA-mediated fEPSP in 4 mm glutamine-incubated slices (n = 13) is significantly enhanced over control slices (n = 12). Representative traces from three fiber volley amplitudes are shown to the right for glutamine-incubated (top) and control (bottom) slices. Calibration: 0.1 mV, 10 ms. C, Inhibition of evoked EPSCs by the low-affinity AMPA receptor antagonist γ-DGG (0.5 mm) is reduced in 4 mm glutamine-incubated slices (n = 12) relative to control (n = 10; * p < 0.05). However, the high-affinity antagonist CNQX (400 nm) produces similar inhibition in both control (n = 7) and glutamine-incubated slices (n = 6; p > 0.05). Inset, Sample traces before (black traces) and after (gray traces) γ-DGG and CNQX application. Calibration: 25 pA, 10 ms.
To determine whether the presynaptic change is caused by an increase in probability of release or an increase in the vesicular concentration of glutamate, we measured quantal size in glutamine-treated slices. Surprisingly, miniature EPSC (mEPSC) amplitude (control, −11.22 ± 0.70 pA, n = 9; 4 mm glutamine, −10.89 ± 0.32 pA, n = 10; p > 0.05) and frequency (control, 0.84 ± 0.35 Hz, n = 9; 4 mm glutamine, 0.48 ± 0.12 Hz, n = 10; p > 0.05) were unaffected in slices incubated in glutamine for up to 4 h (Fig. 4A). When slices were exposed to the same concentration of glutamine for longer than 4 h, an increase in quantal amplitude (control, −10.73 ± 0.54 pA, n = 7; 4 mm glutamine, −14.29 ± 1.12 pA, n = 8; p < 0.05) was observed with no change in frequency (control, 0.45 ± 0.16 Hz, n = 7; 4 mm glutamine, 0.47 ± 0.18 Hz, n = 8; p > 0.05) (Fig. 4B). This increased vesicular glutamate originated from extracellular glutamine that had entered through system A transporters because incubation of slices in both glutamine and MeAIB prevented the augmentation in mEPSC amplitude (4 mm glutamine, −13.16 ± 0.67 pA, n = 18; 4 mm glutamine plus 25 mm MeAIB, −11.48 ± 0.55 pA, n = 18; p > 0.05) with no change in frequency (4 mm glutamine, 0.27 ± 0.03 Hz, n = 18; 4 mm glutamine plus 25 mm MeAIB, 0.39 ± 0.07 Hz, n = 18; p > 0.05) (Fig. 4C).
Figure 4.
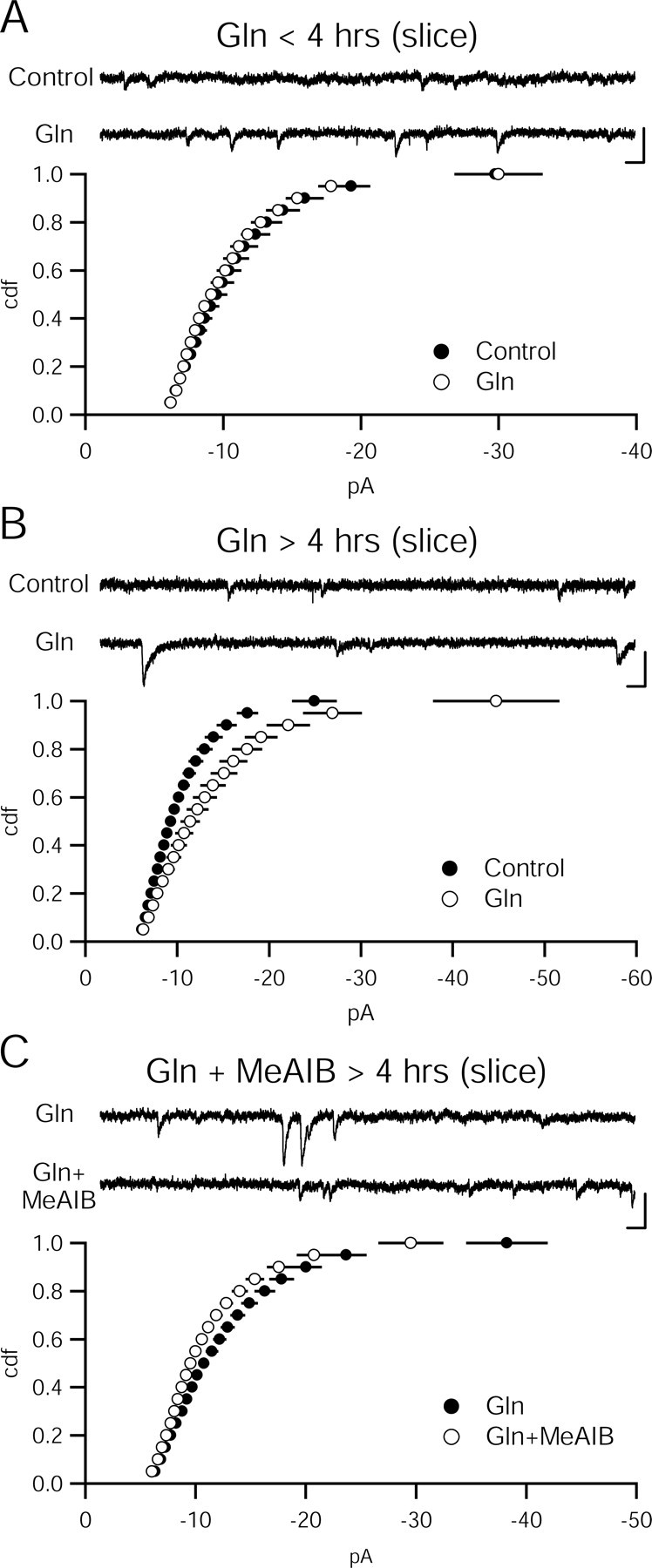
Prolonged incubation in glutamine increases quantal amplitude through a system A-dependent mechanism. A, Quantal amplitude does not change in slices incubated in glutamine (Gln; 4 mm) for <4 h. Cumulative distribution function of amplitudes are not different between glutamine (n = 7) and control slices (n = 9; p > 0.05, Kolmogorov–Smirnov test). Sample recordings from control (top) and glutamine-incubated (bottom) cells. Calibration: 10 pA, 100 ms. B, After more than 4 h of glutamine incubation, quantal amplitude is increased. Cumulative distribution function of amplitudes are significantly increased in glutamine (n = 8) slices over control slices (n = 7; p < 0.05, Kolmogorov–Smirnov test). Sample recordings from control (top) and glutamine-incubated (bottom) cells are shown. Calibration: 10 pA, 100 ms. C, Glutamine enhancement relies on system A transporters. Cumulative distribution function of amplitudes is reduced in slices incubated in glutamine and MeAIB (n = 18) relative to slices incubated in glutamine alone (n = 18; p < 0.05, Kolmogorov–Smirnov test). Sample recordings from glutamine-incubated (top) and glutamine-incubated (bottom) cells. Calibration: 10 pA, 100 ms.
The glutamate–glutamine cycle has also been proposed to support inhibitory transmission mediated by GABA. In this model, GABAergic neurons use glutamic acid decarboxylase (GAD) to convert glutamate generated from exogenous glutamine into GABA (Bak et al., 2006). The amplitude (control, 10.71 ± 0.51 pA, n = 10; 4 mm glutamine, 11.08 ± 0.32 pA, n = 17; p > 0.05) and frequency (control, 0.69 ± 0.10 Hz, n = 10; 4 mm glutamine, 0.71 ± 0.08 Hz, n = 17; p > 0.05) of spontaneous miniature IPSCs mediated by GABA, however, were unaffected by incubation of slices in 4 mm glutamine for more than 4 h. These results are consistent with GABA synthesis relying, instead, on direct uptake of extracellular glutamate (Mathews and Diamond, 2003).
To determine whether the amount of vesicular glutamate is increased in glutamine incubated slices, we used the low-affinity AMPA receptor antagonist, γ-DGG, to assay the concentration of synaptic glutamate (Watkins et al., 1990; Liu et al., 1999; Wadiche and Jahr, 2001; Christie and Jahr, 2006). In control slices, γ-DGG (0.5 mm) decreased the evoked AMPA EPSC recorded in whole-cell mode by 50%. After glutamine incubation, γ-DGG inhibition was reduced, indicating an increase in synaptically released glutamate (Fig. 3C). In contrast, inhibition by the high-affinity antagonist 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) was not significantly different between control slices and slices incubated in 4 mm glutamine for more than 4 h (Fig. 3C).
The delay in glutamine-induced augmentation of synaptic transmission might be attributable to an inability of glutamine to access transporters in the slice preparation. Dissociated hippocampal neurons are grown in a monolayer and are readily accessible to exogenously applied substances. Incubating cultured hippocampal neurons in high glutamine concentrations for up to 4 h at 34°C also did not produce an increase in quantal amplitude (control, −14.23 ± 1.09 pA, n = 16; 4 mm glutamine, −14.55 ± 0.64 pA, n = 19; p > 0.05) or frequency (control, 1.67 ± 0.60 Hz, n = 16; 4 mm glutamine, 1.95 ± 0.54 Hz, n = 19; p > 0.05) (Fig. 5A). To increase our ability to detect changes in vesicular glutamate release, we locally perfused 500 mm sucrose to engage the readily releasable pool of vesicles (Rosenmund and Stevens, 1996). Sucrose-evoked currents in cultures exposed to high glutamine for less than 4 h did not differ from control cultures (Fig. 5C). If cultures were exposed to glutamine for longer than 4 h, mEPSC amplitude did increase (control, −13.42 ± 0.82 pA, n = 16; 4 mm glutamine, −18.53 ± 1.60 pA, n = 16; p < 0.05), although the frequency was not significantly different (control, 2.95 ± 1.36 Hz, n = 16; 4 mm glutamine, 3.30 ± 0.73 Hz, n = 16; p > 0.05) (Fig. 5B). The sucrose-evoked current increased as well, although not significantly (Fig. 5C). Thus, the delay in the enhancement must arise either from a delay in glutamine uptake or conversion.
Figure 5.
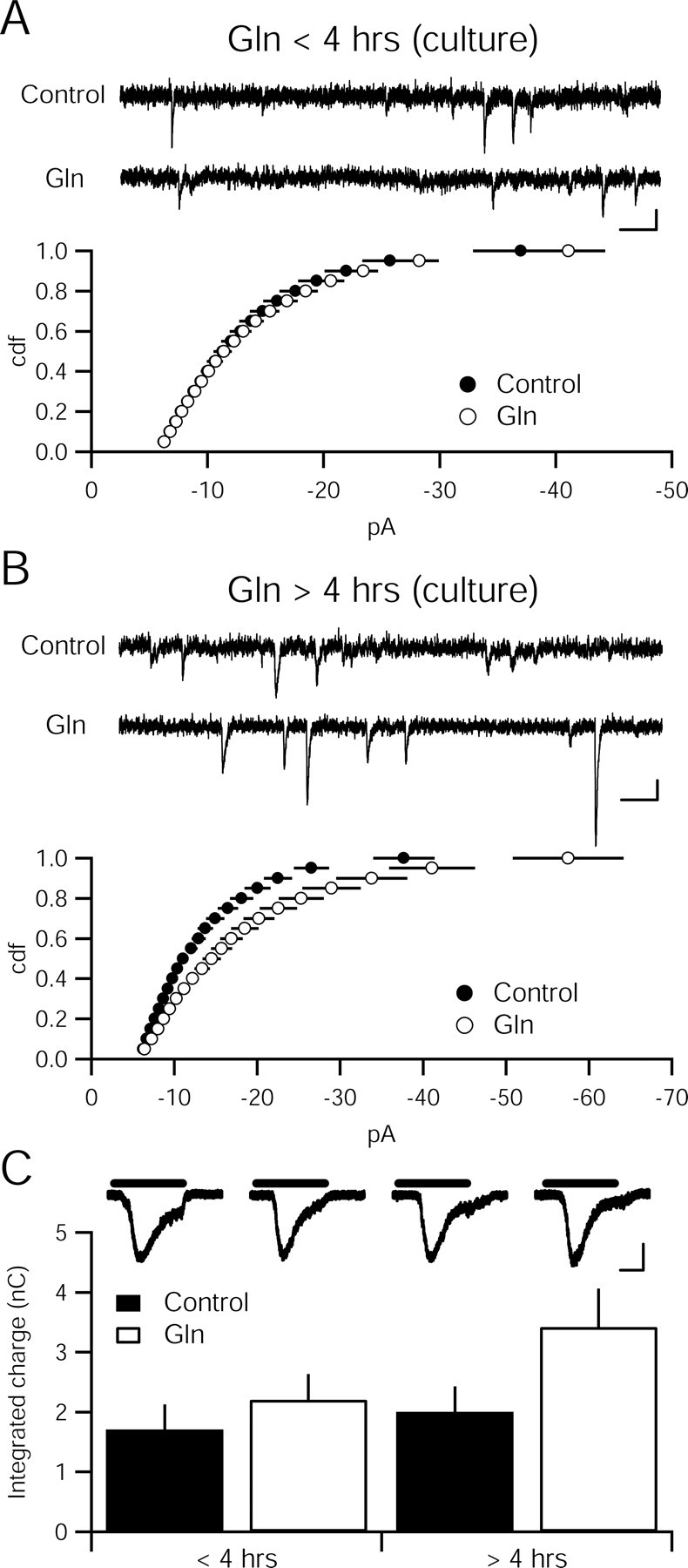
Glutamine enhances quantal amplitude in dissociated hippocampal culture with the same time course as in hippocampal slices. A, Cumulative distribution function of amplitudes is not different between 4 mm glutamine (Gln)-incubated (n = 18) and control cultures (n = 20; p > 0.05, Kolmogorov–Smirnov test). Sample recordings from control (top) and glutamine-incubated (bottom) cells. Calibration: 10 pA, 100 ms. B, Cumulative distribution function of amplitudes is different between 4 mm glutamine-incubated (n = 15) and control cultures (n = 14; p < 0.05, Kolmogorov–Smirnov test). Sample recordings from control (top) and glutamine-incubated (bottom) cells. Calibration: 10 pA, 100 ms. C, Current evoked by sucrose (500 mm) does not differ between cultures incubated in 4 mm glutamine (<4 h, n = 19; >4 h, n = 16) relative to control coverslips (<4 h, n = 16; >4 h, n = 16; p > 0.05). Inset, Representative traces show sucrose-evoked currents. Black bars indicate application of 500 mm sucrose. Calibration: 400 pA, 1 s.
The time course of the glutamine-induced increase in vesicular glutamate was monitored directly by stimulating the Schaffer collateral pathway in the presence of high glutamine. The fEPSP slope was unchanged for ∼1 h. The slope then gradually increased over the next 3 h (Fig. 6A). Whereas the augmentation appeared sooner than in unstimulated slices, a 1 h delay remained. Given the slow effects of exogenously applied glutamine, rapid recovery after either glutamine synthetase or system A inhibition (Figs. 1, 2) seems unlikely.
Figure 6.
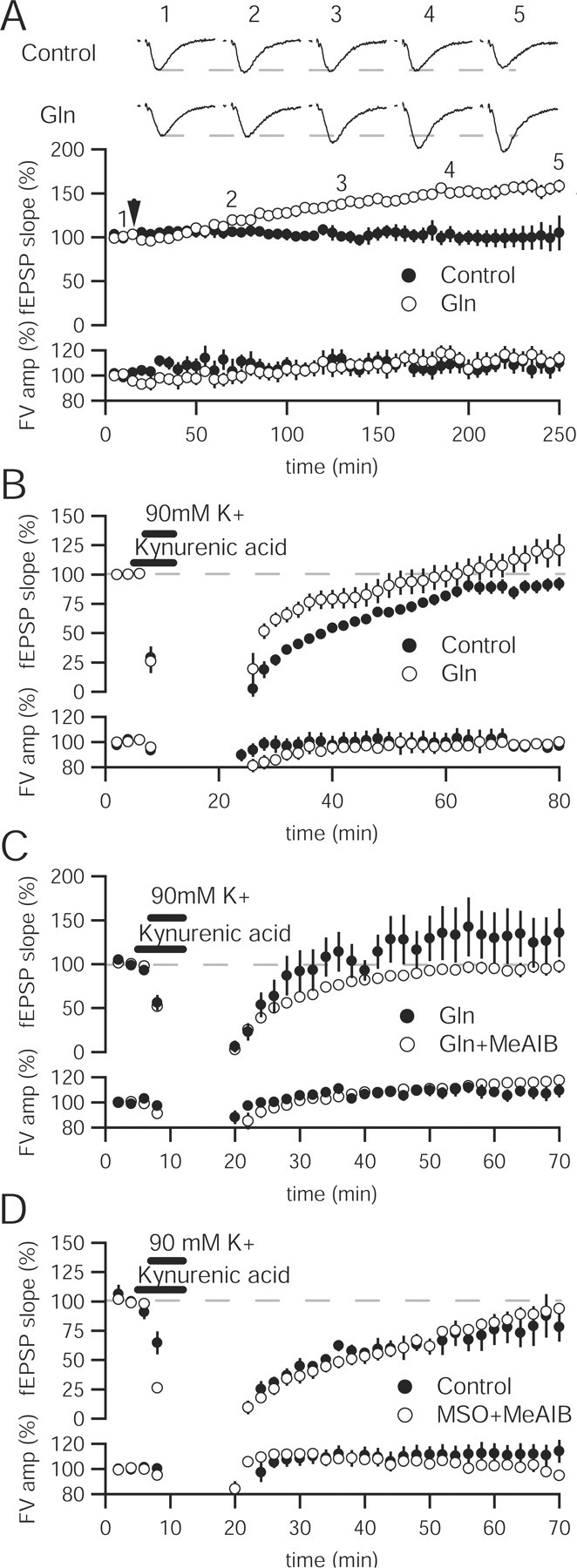
Glutamine enhancement of synaptic transmission is activity-dependent. A, Time course of glutamine enhancement shows that population activity does not increase appreciably until 1 h after application. Sample traces in control (top) and 4 mm glutamine (Gln)-incubated (bottom) slices at baseline, 1, 2, 3, and 4 h after glutamine application. Calibration: 0.5 mV, 10 ms. B, Application of hyperkalemic solution results in an immediate increase in fEPSP slope in the presence of 4 mm glutamine (n = 11) relative to control (n = 6) and produces an enhancement over baseline after 60 min. C, Immediate enhancement by 4 mm glutamine (n = 7) after hyperkalemic stimulation is lessened by addition of 25 mm MeAIB (n = 8), whereas the persistent enhancement after 60 min is abolished. D, Recovery of synaptic transmission after intense hyperkalemic stimulation does not differ in the presence of 40 mm MSO and 25 mm MeAIB (n = 7) relative to control (n = 5) slices.
The difference in time before glutamine augmentation between spontaneously active slices and stimulated slices could reflect resolution of the assay (quantal event vs population response) or a role for activity in regulating the access of extracellular glutamine to vesicular glutamate. To distinguish between these two possibilities, we monitored the recovery of fEPSPs after hyperkalemic stimulation. Potassium depolarization has been shown to rapidly turn over a large percentage of the recycling pool of vesicles (Ryan et al., 1993; Klingauf et al., 1998; Kuromi and Kidokoro, 1999). In control slices, application of 90 mm K+ for 5 min results in an acute depression followed by recovery of the fEPSP slope (Fig. 6B). Kynurenic acid, a low-affinity antagonist of ionotropic glutamate receptors, was present during the high K+ to protect against excitotoxicity in the slice. When glutamine was added after stimulation, the fEPSP slope increased immediately, remained higher through the recovery, and overshot baseline responses (Fig. 6B). The enhancement depends on glutamine entry through system A transporters because adding glutamine and MeAIB together after hyperkalemic stimulation mitigates the enhanced recovery and abolishes the long-lasting enhancement (Fig. 6C).
If high activity improves utilization of the glutamate–glutamine cycle, adding inhibitors may produce a more persistent hyperkalemic decrease in glutamatergic transmission. MSO and MeAIB were added after 5 min high K+ application. The rate and level of recovery in the presence of glutamate–glutamine cycle blockers was not statistically different from control slices after hyperkalemic stimulation (Fig. 6D). Thus, tissue slices can sustain synaptic transmission in the absence of glutamate–glutamine cycling, even after intense stimulation.
Synaptic transmission remains robust during metabolic restriction
The inability to affect synaptic transmission in hippocampal slices after interfering with the glutamate–glutamine cycle was surprising given the reported profound effects of MSO alone on glutamate release, visual function, and behavior. One explanation is that apposition of glia and neurons in tissue slices may be too intimate to interrupt the cycle pharmacologically. Banker cultures (Goslin et al., 1998), which suspend neurons above a confluent layer of glia, physically separate neurons from glia and allow for experimental intervention in the cycle. After allowing neurons to grow in the presence of a glial monolayer for 8–14 d in medium containing 1 mm glutamine, pure neuronal cultures (with no glia) were placed in a different well in the presence or absence of 1 mm glutamine. Removal of glutamine from pure neuronal cultures did not affect quantal amplitude (control, −13.66 ± 1.06 pA, n = 33; without glutamine, −12.43 ± 0.92 pA, n = 28; p > 0.05), quantal frequency (control, 2.57 ± 0.47 Hz, n = 33; without glutamine, 3.19 ± 0.85 Hz, n = 28; p > 0.05), or the sucrose-evoked current (control, 0.79 ± 0.13 nC, n = 20; without glutamine, 1.06 ± 0.29 nC, n = 18; p > 0.05) even after more than 4 h without glutamine. The absence not only of glutamine, but also glia, suggests that neurons can function for a considerable time independently of glia and the glutamate–glutamine cycle.
Having stripped neurons of all exogenous sources of glutamate postulated by the glutamate–glutamine cycle, we tested the hypothesis that glutamate could be taken up directly through plasma membrane glutamate transporters as is the case at inhibitory nerve terminals (Mathews and Diamond, 2003). Pure neuronal cultures were exposed to 5 min of hyperkalemic stimulation followed by incubation in a glutamine-free medium containing 100 μm dl-TBOA, a glutamate transporter inhibitor. These neurons still displayed robust sucrose evoked responses (control, 1.92 ± 0.44 nC, n = 11; 100 μm dl-TBOA, 1.53 ± 0.24 nC, n = 13; p > 0.05), as well as similar quantal size (control, −14.99 ± 2.49 pA, n = 13; 100 μm dl-TBOA, −14.20 ± 1.72 pA, n = 14; p > 0.05) and frequency (control, 2.64 ± 1.16 Hz, n = 13; 100 μm dl-TBOA, 2.53 ± 0.62 Hz, n = 14; p > 0.05) that did not differ from dl-TBOA-free cultures.
Discussion
It is generally accepted that the neurotransmitter pool of glutamate is maintained by the glutamate–glutamine cycle (Kandel et al., 2000). Although the time scale at which this cycle operates is unclear, it has been proposed to replenish activity-dependent loss of glutamate rapidly. Our results indicate that even when the nerve terminal is supplied by exogenous glutamine, it takes many hours to access the glutamate–glutamine cycle. Furthermore, attempts to acutely deplete the neurotransmitter pool of glutamate failed to demonstrate necessity of the glutamate–glutamine cycle for many hours.
Pharmacology of glutamate–glutamine inhibition
The acute, rapidly reversible effects of MSO and MeAIB on excitatory synaptic transmission are inconsistent with inhibition of the glutamate–glutamine cycle. Specific glutamine synthetase inhibition can only be ascertained if exogenous glutamine is able to mitigate the effects of MSO, which was not the case in our preparation. MSO has been shown to have nonspecific effects at much lower concentrations than those used here and in previous papers (Zielinska et al., 2004). At millimolar concentrations, MSO induces glutamate release and excitotoxicity independent of glutamine synthetase inhibition in cortical tissue slices (Shaw et al., 1999). At higher millimolar concentrations, MSO actually increases extracellular glutamine content by blocking glutamine uptake into glia and neurons (Albrecht and Norenberg, 1990).
Involvement of system A transporters in supporting excitatory synaptic transmission has been offered from hippocampal neuron–glia cocultures. A rapid depression of mEPSC amplitude was observed minutes after perfusion of 25–40 mm MeAIB in cocultures, but not in pure neuronal cultures (Armano et al., 2002). This mild decrease in quantal amplitude was accompanied by a paradoxical increase in quantal frequency, which was attributed to increased symport of Na+ through system A producing an increase in intracellular Ca2+ through Na+/Ca2+ exchange (Armano et al., 2002). Alternatively, application of MeAIB, a competitive, nonmetabolizable substrate for system A transporters, at high concentrations could result in transport and membrane depolarization through the electrogenic system A transporter (Chaudhry et al., 2002). The acute effect seen here on fEPSPs might be attributed to membrane depolarization rather than acute disruption of glutamine entry into neurons. Supporting the nonspecific effect of both drugs was the lack of a persistent decrease after many hours of incubation in contrast to the prolonged potentiation of synaptic transmission in high extracellular glutamine.
Re-evaluating the glutamate–glutamine cycle
Several previous studies also suggest that the glutamate–glutamine cycle may not play a central role in directly maintaining the vesicular glutamate pool. In the retina, direct uptake of released glutamate accounts for a significant contribution to vesicular glutamate at photoreceptors (Winkler et al., 1999; Hasegawa et al., 2006). Plasma membrane glutamate transporters are also present at bipolar cell terminals (Palmer et al., 2003). Preventing the conversion of glutamine to glutamate by genetically deleting PAG has little effect on quantal size (Masson et al., 2006). In these mice, synaptic transmission is only impaired under intense stimulation and, even then, is not completely abrogated; although, these effects do lead to deficits in basic behavior. However, in a separate study using a spatial learning paradigm, glutamine synthetase inhibition by MSO did not decrease performance relative to control mice (Blin et al., 2002).
Cellular and subcellular distribution of glutamate–glutamine cycle components also argues against vesicular glutamate being responsive to interruptions in glutamine influx. Immunolocalization of system A transporters in neurons finds only rare puncta at axon terminals. Instead, the majority of staining is found in the somatodendritic compartment, consistent with slow access of transported glutamine to vesicles at presynaptic terminals (Conti and Melone, 2006).
Regional differences in glutamate–glutamine cycle utilization
Additional analysis shows substantial heterogeneity in the cellular distribution of PAG. Labeling in pontine neurons that project mossy fibers onto granule cells in the cerebellum is much denser than that of granule cells and their parallel fiber terminals, as well as climbing fiber terminals (Laake et al., 1999).
The heterogeneity identified in the cerebellum indicates that neuronal reliance on the glutamate–glutamine cycle may depend on regional differences in the functional properties of synaptic transmission. Effects of glutamate–glutamine cycle inhibition have been demonstrated most convincingly in the retina (Pow and Robinson, 1994; Pow and Crook, 1996; Barnett et al., 2000). The peculiar metabolic demands of the photoreceptor-bipolar cell synapse may require a more substantial astrocytic role. Photoreceptors release glutamate tonically in a graded manner in response to a reduction in light levels (Sterling, 1998). This tonic release may require higher levels of glutamate than can be produced de novo by the cell from glucose.
Differences in the anatomical relationship between neurons and glia may be related to the degree to which neurons rely on the glutamate–glutamine cycle. The photoreceptor–bipolar cell synapse is ensheathed by Müller cells, large glial cells spanning the inner retina. In contrast, only 40% of synapses in area CA1 of hippocampus are closely apposed to astroglia (Ventura and Harris, 1999). The low association between glia and neurons may reflect the reduced transmission needs of CA3 pyramidal cells, which fire phasically at a maximum of 10 Hz. Whether metabolic demands, anatomical relationships, or some other factor determine the degree to which particular neurons depend on the glutamate–glutamine cycle remains unknown.
Alternative sources of vesicular glutamate
What, then, allows neurons, in which glutamate–glutamine cycling is not necessary, to maintain a neurotransmitter pool of glutamate? Cytoplasmic glutamate has been estimated at 1–10 mm (Carlson et al., 1989; McMahon and Nicholls, 1991) whereas mitochondrial glutamate has also been estimated at 5–10 mm (Roberg et al., 1999). Glutamate concentrations in vesicles, however, are estimated to be in the hundreds of millimolars range (Maycox et al., 1988; Carlson et al., 1989). If we estimate that each vesicle contains ∼2,300 molecules of glutamate based on a 40 nm diameter vesicle (Takamori et al., 2006) and concentration of 150 mm (Burger et al., 1989), and the total vesicle pool contains 200 vesicles (Sudhof, 2000), then the number of glutamate molecules stored in vesicles is ∼5 × 105. A lower bound estimate of the cytoplasmic pool of glutamate, assuming a cytoplasmic volume of 1.5 pl (Peet et al., 2004) and a glutamate concentration of 1 mm (Maycox et al., 1988; Carlson et al., 1989), yields 9 × 108 molecules of glutamate. The total cytoplasmic pool of glutamate, if able to contribute solely to the neurotransmitter pool of glutamate, could act as a significant reservoir for synaptic transmission. Whether hyperkalemic stimulation accesses the reserve pool of vesicles is unclear, leaving another possible store of glutamate. Either reserve could temporarily buffer synaptic transmission against acute insult or deprivation.
In addition to a possible store of glutamate, neurons might also be able to use glutamate that has been synthesized de novo, while still maintaining TCA cycle intermediates. Pyruvate carboxylation, which had been thought to be restricted to astrocytes, has been observed previously in neurons, although the significance of this observed flux is in dispute (Hassel and Brathe, 2000; Waagepetersen et al., 2001). Glutamine entry through system A transporters could enter a distinct metabolic pool (Rae et al., 2003; Shokati et al., 2005). Exogenous glutamine could serve an anaplerotic function, replenishing TCA cycle intermediates as opposed to the neurotransmitter pool of glutamate. An increase in glutamate synthesized de novo from glucose could result from this increased anaplerotic glutamine. The shunt of glutamine away from glutaminase and into the TCA cycle could account for the delay in augmentation of synaptic transmission and vesicular glutamate. Finally, a ready supply of extracellular glucose could be sufficient to maintain the basic energetic and metabolic needs of neurons, and also satisfy the additional demands for generating a neurotransmitter pool of glutamate. We are currently unable to distinguish between a reduction in vesicular glutamate and an inability to generate ATP, which prevents filling of vesicles with glutamate, exocytosis or endocytosis of synaptic vesicles, or other ATP-dependent processes in synaptic transmission. We speculate that the ability of a neuron to maintain synaptic transmission may be intimately tied to its energy use.
Based on our results, the neuronal TCA cycle seems capable of maintaining the neurotransmitter pool of glutamate, as well as meeting basic energetic demands for several hours in the absence of known exogenous sources. Rather than an exclusive flow of carbons from vesicle to astrocyte and back, we support a model in which neurons are also capable of de novo synthesis of glutamate from TCA cycle intermediates, which can then be replenished from glutamine, glucose, or any other source of carbons that can be transported into the cell. Multiple biosynthetic pathways ensure a robust supply of vesicular glutamate to support synaptic transmission in neurons.
Footnotes
This work was supported by a National Science Foundation Graduate Research Fellowship (K.K.) and the National Institutes of Health (R.N.). We thank Keith Brown, Pierre Apostolides, Dr. Zhaolin Hua, and Lars Funke for preparing cultures and Dr. Anastassios Tzingounis, Dr. Robert Edwards, Dr. David Copenhagen, and members of the Nicoll and Edwards laboratories for helpful discussion and preparation of this manuscript.
References
- Albrecht J, Norenberg MD. L-methionine-DL-sulfoximine induces massive efflux of glutamine from cortical astrocytes in primary culture. Eur J Pharmacol. 1990;182:587–589. doi: 10.1016/0014-2999(90)90061-a. [DOI] [PubMed] [Google Scholar]
- Armano S, Coco S, Bacci A, Pravettoni E, Schenk U, Verderio C, Varoqui H, Erickson JD, Matteoli M. Localization and functional relevance of system a neutral amino acid transporters in cultured hippocampal neurons. J Biol Chem. 2002;277:10467–10473. doi: 10.1074/jbc.M110942200. [DOI] [PubMed] [Google Scholar]
- Bak LK, Schousboe A, Waagepetersen HS. The glutamate/GABA-glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J Neurochem. 2006;98:641–653. doi: 10.1111/j.1471-4159.2006.03913.x. [DOI] [PubMed] [Google Scholar]
- Barnett NL, Pow DV, Robinson SR. Inhibition of Muller cell glutamine synthetase rapidly impairs the retinal response to light. Glia. 2000;30:64–73. doi: 10.1002/(sici)1098-1136(200003)30:1<64::aid-glia7>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- Benjamin AM, Quastel JH. Metabolism of amino acids and ammonia in rat brain cortex slices in vitro: a possible role of ammonia in brain function. J Neurochem. 1975;25:197–206. doi: 10.1111/j.1471-4159.1975.tb06953.x. [DOI] [PubMed] [Google Scholar]
- Blin M, Crusio WE, Hevor T, Cloix JF. Chronic inhibition of glutamine synthetase is not associated with impairment of learning and memory in mice. Brain Res Bull. 2002;57:11–15. doi: 10.1016/s0361-9230(01)00631-1. [DOI] [PubMed] [Google Scholar]
- Burger PM, Mehl E, Cameron PL, Maycox PR, Baumert M, Lottspeich F, De Camilli P, Jahn R. Synaptic vesicles immunoisolated from rat cerebral cortex contain high levels of glutamate. Neuron. 1989;3:715–720. doi: 10.1016/0896-6273(89)90240-7. [DOI] [PubMed] [Google Scholar]
- Carlson MD, Kish PE, Ueda T. Characterization of the solubilized and reconstituted ATP-dependent vesicular glutamate uptake system. J Biol Chem. 1989;264:7369–7376. [PubMed] [Google Scholar]
- Chaudhry FA, Reimer RJ, Edwards RH. The glutamine commute: take the N line and transfer to the A. J Cell Biol. 2002;157:349–355. doi: 10.1083/jcb.200201070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie JM, Jahr CE. Multivesicular release at Schaffer collateral-CA1 hippocampal synapses. J Neurosci. 2006;26:210–216. doi: 10.1523/JNEUROSCI.4307-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti F, Melone M. The glutamine commute: lost in the tube? Neurochem Int. 2006;48:459–464. doi: 10.1016/j.neuint.2005.11.016. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Fremeau RT, Jr, Voglmaier S, Seal RP, Edwards RH. VGLUTs define subsets of excitatory neurons and suggest novel roles for glutamate. Trends Neurosci. 2004;27:98–103. doi: 10.1016/j.tins.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Gjessing LR, Gjesdahl P, Sjaastad O. The free amino acids in human cerebrospinal fluid. J Neurochem. 1972;19:1807–1808. doi: 10.1111/j.1471-4159.1972.tb06226.x. [DOI] [PubMed] [Google Scholar]
- Goslin K, Asmussen H, Banker G. Rat hippocampal neurons in low-density culture. In: Banker G, Goslin K, editors. In culturing nerve cells. Cambridge, MA: MIT; 1998. pp. 339–371. [Google Scholar]
- Gruetter R, Seaquist ER, Ugurbil K. A mathematical model of compartmentalized neurotransmitter metabolism in the human brain. Am J Physiol Endocrinol Metab. 2001;281:E100–E112. doi: 10.1152/ajpendo.2001.281.1.E100. [DOI] [PubMed] [Google Scholar]
- Hasegawa J, Obara T, Tanaka K, Tachibana M. High-density presynaptic transporters are required for glutamate removal from the first visual synapse. Neuron. 2006;50:63–74. doi: 10.1016/j.neuron.2006.02.022. [DOI] [PubMed] [Google Scholar]
- Hassel B, Brathe A. Neuronal pyruvate carboxylation supports formation of transmitter glutamate. J Neurosci. 2000;20:1342–1347. doi: 10.1523/JNEUROSCI.20-04-01342.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz L, Zielke HR. Astrocytic control of glutamatergic activity: astrocytes as stars of the show. Trends Neurosci. 2004;27:735–743. doi: 10.1016/j.tins.2004.10.008. [DOI] [PubMed] [Google Scholar]
- Hertz L, Gibbs ME, O'Dowd BS, Sedman GL, Robinson SR, Sykova E, Hajek I, Hertz E, Peng L, Huang R, Ng KT. Astrocyte-neuron interaction during one-trial aversive learning in the neonate chick. Neurosci Biobehav Rev. 1996;20:537–551. doi: 10.1016/0149-7634(95)00020-8. [DOI] [PubMed] [Google Scholar]
- Kandel E, Schwartz J, Jessell T. Principles of neural science. Ed 4. New York: McGraw-Hill; 2000. [Google Scholar]
- Kapetanovic IM, Yonekawa WD, Kupferberg HJ. Time-related loss of glutamine from hippocampal slices and concomitant changes in neurotransmitter amino acids. J Neurochem. 1993;61:865–872. doi: 10.1111/j.1471-4159.1993.tb03597.x. [DOI] [PubMed] [Google Scholar]
- Klingauf J, Kavalali ET, Tsien RW. Kinetics and regulation of fast endocytosis at hippocampal synapses. Nature. 1998;394:581–585. doi: 10.1038/29079. [DOI] [PubMed] [Google Scholar]
- Kuromi H, Kidokoro Y. The optically determined size of exo/endo cycling vesicle pool correlates with the quantal content at the neuromuscular junction of Drosophila larvae. J Neurosci. 1999;19:1557–1565. doi: 10.1523/JNEUROSCI.19-05-01557.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvamme E, Roberg B, Torgner IA. Phosphate-activated glutaminase and mitochondrial glutamine transport in the brain. Neurochem Res. 2000;25:1407–1419. doi: 10.1023/a:1007668801570. [DOI] [PubMed] [Google Scholar]
- Laake JH, Takumi Y, Eidet J, Torgner IA, Roberg B, Kvamme E, Ottersen OP. Postembedding immunogold labelling reveals subcellular localization and pathway-specific enrichment of phosphate activated glutaminase in rat cerebellum. Neuroscience. 1999;88:1137–1151. doi: 10.1016/s0306-4522(98)00298-x. [DOI] [PubMed] [Google Scholar]
- Lamar C, Jr, Sellinger OZ. The inhibition in vivo of cerebral glutamine synthetase and glutamine transferase by the convulsant methionine sulfoximine. Biochem Pharmacol. 1965;14:489–506. doi: 10.1016/0006-2952(65)90222-4. [DOI] [PubMed] [Google Scholar]
- Lerma J, Herranz AS, Herreras O, Abraira V, Martin del Rio R. In vivo determination of extracellular concentration of amino acids in the rat hippocampus. A method based on brain dialysis and computerized analysis. Brain Res. 1986;384:145–155. doi: 10.1016/0006-8993(86)91230-8. [DOI] [PubMed] [Google Scholar]
- Liu G, Choi S, Tsien RW. Variability of neurotransmitter concentration and nonsaturation of postsynaptic AMPA receptors at synapses in hippocampal cultures and slices. Neuron. 1999;22:395–409. doi: 10.1016/s0896-6273(00)81099-5. [DOI] [PubMed] [Google Scholar]
- Manning JM, Moore S, Rowe WB, Meister A. Identification of L-methionine S-sulfoximine as the diastereoisomer of L-methionine SR-sulfoximine that inhibits glutamine synthetase. Biochemistry. 1969;8:2681–2685. doi: 10.1021/bi00834a066. [DOI] [PubMed] [Google Scholar]
- Martinez-Hernandez A, Bell KP, Norenberg MD. Glutamine synthetase: glial localization in brain. Science. 1977;195:1356–1358. doi: 10.1126/science.14400. [DOI] [PubMed] [Google Scholar]
- Masson J, Darmon M, Conjard A, Chuhma N, Ropert N, Thoby-Brisson M, Foutz AS, Parrot S, Miller GM, Jorisch R, Polan J, Hamon M, Hen R, Rayport S. Mice lacking brain/kidney phosphate-activated glutaminase have impaired glutamatergic synaptic transmission, altered breathing, disorganized goal-directed behavior and die shortly after birth. J Neurosci. 2006;26:4660–4671. doi: 10.1523/JNEUROSCI.4241-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews GC, Diamond JS. Neuronal glutamate uptake contributes to GABA synthesis and inhibitory synaptic strength. J Neurosci. 2003;23:2040–2048. doi: 10.1523/JNEUROSCI.23-06-02040.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maycox PR, Deckwerth T, Hell JW, Jahn R. Glutamate uptake by brain synaptic vesicles. Energy dependence of transport and functional reconstitution in proteoliposomes. J Biol Chem. 1988;263:15423–15428. [PubMed] [Google Scholar]
- McMahon HT, Nicholls DG. Transmitter glutamate release from isolated nerve terminals: evidence for biphasic release and triggering by localized Ca2+ J Neurochem. 1991;56:86–94. doi: 10.1111/j.1471-4159.1991.tb02566.x. [DOI] [PubMed] [Google Scholar]
- Palmer MJ, Taschenberger H, Hull C, Tremere L, von Gersdorff H. Synaptic activation of presynaptic glutamate transporter currents in nerve terminals. J Neurosci. 2003;23:4831–4841. doi: 10.1523/JNEUROSCI.23-12-04831.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peet JA, Bragin A, Calvert PD, Nikonov SS, Mani S, Zhao X, Besharse JC, Pierce EA, Knox BE, Pugh EN., Jr Quantification of the cytoplasmic spaces of living cells with EGFP reveals arrestin-EGFP to be in disequilibrium in dark adapted rod photoreceptors. J Cell Sci. 2004;117:3049–3059. doi: 10.1242/jcs.01167. [DOI] [PubMed] [Google Scholar]
- Pow DV, Crook DK. Direct immunocytochemical evidence for the transfer of glutamine from glial cells to neurons: use of specific antibodies directed against the d-stereoisomers of glutamate and glutamine. Neuroscience. 1996;70:295–302. doi: 10.1016/0306-4522(95)00363-n. [DOI] [PubMed] [Google Scholar]
- Pow DV, Robinson SR. Glutamate in some retinal neurons is derived solely from glia. Neuroscience. 1994;60:355–366. doi: 10.1016/0306-4522(94)90249-6. [DOI] [PubMed] [Google Scholar]
- Rae C, Hare N, Bubb WA, McEwan SR, Broer A, McQuillan JA, Balcar VJ, Conigrave AD, Broer S. Inhibition of glutamine transport depletes glutamate and GABA neurotransmitter pools: further evidence for metabolic compartmentation. J Neurochem. 2003;85:503–514. doi: 10.1046/j.1471-4159.2003.01713.x. [DOI] [PubMed] [Google Scholar]
- Reimer RJ, Chaudhry FA, Gray AT, Edwards RH. Amino acid transport system A resembles system N in sequence but differs in mechanism. Proc Natl Acad Sci USA. 2000;97:7715–7720. doi: 10.1073/pnas.140152797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberg B, Torgner IA, Kvamme E. Inhibition of glutamine transport in rat brain mitochondria by some amino acids and tricarboxylic acid cycle intermediates. Neurochem Res. 1999;24:809–814. doi: 10.1023/a:1020941510764. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Rothman DL, Behar KL, Hyder F, Shulman RG. In vivo NMR studies of the glutamate neurotransmitter flux and neuroenergetics: implications for brain function. Annu Rev Physiol. 2003;65:401–427. doi: 10.1146/annurev.physiol.65.092101.142131. [DOI] [PubMed] [Google Scholar]
- Ryan TA, Reuter H, Wendland B, Schweizer FE, Tsien RW, Smith SJ. The kinetics of synaptic vesicle recycling measured at single presynaptic boutons. Neuron. 1993;11:713–724. doi: 10.1016/0896-6273(93)90081-2. [DOI] [PubMed] [Google Scholar]
- Shank RP, Bennett GS, Freytag SO, Campbell GL. Pyruvate carboxylase: an astrocyte-specific enzyme implicated in the replenishment of amino acid neurotransmitter pools. Brain Res. 1985;329:364–367. doi: 10.1016/0006-8993(85)90552-9. [DOI] [PubMed] [Google Scholar]
- Shaw CA, Bains JS, Pasqualotto BA, Curry K. Methionine sulfoximine shows excitotoxic actions in rat cortical slices. Can J Physiol Pharmacol. 1999;77:871–877. [PubMed] [Google Scholar]
- Shokati T, Zwingmann C, Leibfritz D. Contribution of extracellular glutamine as an anaplerotic substrate to neuronal metabolism: a re-evaluation by multinuclear NMR spectroscopy in primary cultured neurons. Neurochem Res. 2005;30:1269–1281. doi: 10.1007/s11064-005-8798-8. [DOI] [PubMed] [Google Scholar]
- Sterling P. Retina. In: Shepherd GM, editor. The synaptic organization of the brain. New York: Oxford UP; 1998. pp. 205–255. [Google Scholar]
- Stryer L. Biochemistry. Ed 4. New York: W. H. Freeman; 1995. [Google Scholar]
- Sudhof TC. The synaptic vesicle cycle revisited. Neuron. 2000;28:317–320. doi: 10.1016/s0896-6273(00)00109-4. [DOI] [PubMed] [Google Scholar]
- Sugawara M, Nakanishi T, Fei YJ, Huang W, Ganapathy ME, Leibach FH, Ganapathy V. Cloning of an amino acid transporter with functional characteristics and tissue expression pattern identical to that of system A. J Biol Chem. 2000;275:16473–16477. doi: 10.1074/jbc.C000205200. [DOI] [PubMed] [Google Scholar]
- Takamori S, Holt M, Stenius K, Lemke EA, Gronborg M, Riedel D, Urlaub H, Schenck S, Brugger B, Ringler P, Muller SA, Rammner B, Grater F, Hob JS, De Groot BW, Mieskes G, Moriyama Y, Klingauf J, Grubmuller H, Heuser J. Molecular anatomy of a trafficking organelle. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- van den Berg CJ, Garfinkel D. A stimulation study of brain compartments. Metabolism of glutamate and related substances in mouse brain. Biochem J. 1971;123:211–218. doi: 10.1042/bj1230211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varoqui H, Zhu H, Yao D, Ming H, Erickson JD. Cloning and functional identification of a neuronal glutamine transporter. J Biol Chem. 2000;275:4049–4054. doi: 10.1074/jbc.275.6.4049. [DOI] [PubMed] [Google Scholar]
- Ventura R, Harris KM. Three-dimensional relationships between hippocampal synapses and astrocytes. J Neurosci. 1999;19:6897–6906. doi: 10.1523/JNEUROSCI.19-16-06897.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waagepetersen HS, Qu H, Schousboe A, Sonnewald U. Elucidation of the quantitative significance of pyruvate carboxylation in cultured cerebellar neurons and astrocytes. J Neurosci Res. 2001;66:763–770. doi: 10.1002/jnr.10061. [DOI] [PubMed] [Google Scholar]
- Wadiche JI, Jahr CE. Multivesicular release at climbing fiber-Purkinje cell synapses. Neuron. 2001;32:301–313. doi: 10.1016/s0896-6273(01)00488-3. [DOI] [PubMed] [Google Scholar]
- Watkins JC, Pook PC, Sunter DC, Davies J, Honore T. Experiments with kainate and quisqualate agonists and antagonists in relation to the sub-classification of “non-NMDA” receptors. Adv Exp Med Biol. 1990;268:49–55. doi: 10.1007/978-1-4684-5769-8_6. [DOI] [PubMed] [Google Scholar]
- Winkler BS, Kapousta-Bruneau N, Arnold MJ, Green DG. Effects of inhibiting glutamine synthetase and blocking glutamate uptake on b-wave generation in the isolated rat retina. Vis Neurosci. 1999;16:345–353. doi: 10.1017/s095252389916214x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao D, Mackenzie B, Ming H, Varoqui H, Zhu H, Hediger MA, Erickson JD. A novel system A isoform mediating Na+/neutral amino acid cotransport. J Biol Chem. 2000;275:22790–22797. doi: 10.1074/jbc.M002965200. [DOI] [PubMed] [Google Scholar]
- Zielinska M, Stafiej A, Law RO, Albrecht J. Effects of methionine sulfoximine on the glutamine and glutamate content and cell volume in rat cerebral cortical slices: involvement of mechanisms not related to inhibition of glutamine synthesis. Neurotoxicology. 2004;25:443–449. doi: 10.1016/j.neuro.2003.10.003. [DOI] [PubMed] [Google Scholar]