

Abstract
Several murine genetic models of Huntington's disease (HD) have been developed. Murine genetic models are crucial for identifying mechanisms of neurodegeneration in HD and for preclinical evaluation of possible therapies for HD. Longitudinal analysis of mutant phenotypes is necessary to validate models and to identify appropriate periods for analysis of early events in the pathogenesis of neurodegeneration. Here we report longitudinal characterization of the murine Hdh (CAG)150 knock-in model of HD. A series of behavioral tests at five different time points (20, 40, 50, 70, and 100 weeks) demonstrates an age-dependent, late-onset behavioral phenotype with significant motor abnormalities at 70 and 100 weeks of age. Pathological analysis demonstrated loss of striatal dopamine D1 and D2 receptor binding sites at 70 and 100 weeks of age, and stereological analysis showed significant loss of striatal neuron number at 100 weeks. Late-onset behavioral abnormalities, decrease in striatal dopamine receptors, and diminished striatal neuron number observed in this mouse model recapitulate key features of HD. The Hdh (CAG)150 knock-in mouse is a valid model to evaluate early events in the pathogenesis of neurodegeneration in HD.
Keywords: Huntington disease, striatum, neurodegeneration, polyglutamine, trinucleotide repeat, mouse
Introduction
Huntington's disease (HD) is a dominantly inherited human neurodegenerative disorder characterized by progressive motor impairment, cognitive decline, and psychiatric problems. Early degeneration of striatal medium spiny neurons (MSNs) and striatal atrophy are hallmarks of HD, although cortical degeneration may also occur before the onset of clinical features of HD (Vonsattel and DiFiglia, 1998; Rosas et al., 2002, 2004, 2005; Aylward et al., 2004). With disease progression, neuronal loss becomes more global, affecting numerous brain areas. HD is caused by a CAG repeat expansion in exon 1 of the huntingtin gene, which encodes a widely expressed protein [huntingtin (htt)] (Huntington's Disease Collaborative Research Group, 1993). HD belongs to a family of inherited trinucleotide repeat disorders including spinobulbar muscular atrophy (Kennedy's disease), dentatorubro-pallidal Luysian atrophy, and spinocerebellar ataxias 1, 2, 3, 6, 7, and 17 (La Spada et al., 1991; Cummings, 2000). These polyglutamine diseases share similar features, including expanded CAG repeats within translated exons, anticipation, and regionally specific CNS pathology. These diseases may share common mechanisms of pathogenesis (Orr, 2001; Hardy and Orr, 2006). Since the discovery of the huntingtin gene in 1993, a variety of murine genetic models have been generated (Mangiarini et al., 1996; Reddy et al., 1998; Hodgson et al., 1999; Schilling et al., 1999; Shelbourne et al., 1999; Wheeler et al., 2000; Lin et al., 2001), essentially replacing older excitotoxic models. Murine models are potentially valuable as tools in exploring the pathogenesis of HD, enabling the search for potential therapeutic drug targets, and in preclinical evaluation of potential therapies. The available murine models can be separated into two categories: transgenic models (ectopic expression of the huntingtin mutation) and knock-in models (expression of the mutant huntingtin gene under control of the normal regulatory elements). These models vary in the length of repeat, the level of expression of the mutant htt protein, and whether the mutant allele is truncated, full length, human, or murine. Validation of models requires careful characterization of their phenotypic features. An accurate model should recapitulate key behavioral and pathological features of HD in an age-dependent manner. Longitudinal characterization of murine genetic models is crucial for defining periods when early events are likely to be occurring in the pathogenesis of neurodegeneration and for identifying behavioral and/or pathological milestones for use in trials of proposed interventions.
We report the results of longitudinal behavioral and pathological evaluation of the Hdh (CAG)150 knock-in model. Using a series of behavioral tests at five different time points (20, 40, 50, 70, and 100 weeks), we demonstrate an age-dependent, late-onset behavioral phenotype with significant motor abnormalities beginning at 70 weeks of age. Pathological analysis demonstrated loss of striatal dopamine D1 and D2 receptor binding sites at 70 and 100 weeks of age, and stereological analysis showed significant loss of striatal neuron number at 100 weeks.
Materials and Methods
Hdh (CAG)150 mouse model
All experiments were performed with the Hdh (CAG)150 mice murine model of HD maintained on a mixed genetic background, 129/Ola and C57BL/J6 (Lin, 2001) with the current Hdh (CAG)150 line expressing 75–90% C57BL/6 (P. J. Detloff, unpublished data). For all experiments, female and male Hdh (CAG)150 mice with the same genetic background were mated to produce F1 homozygous (HOM), heterozygous (HET), and wild-type (WT) Hdh (CAG)150 mice. Both male and female Hdh (CAG)150 mice and their WT littermates were used. All animals were housed in cages grouped by gender and were provided with food and water ad libitum. Animals were housed in a specific pathogenic-free condition with a 12 h light/dark cycle maintained at 23°C. All procedures were conducted in strict compliance with the Guide for the Care and Use of Laboratory Animals as adopted by the National Institutes of Health and approved by the Committee on Use and Care of Animals (UCUCA), University of Michigan, and the Veterinary Medical Unit (VMU) at the Ann Arbor Veterans Affairs Medical Center.
Genotyping
All mice studied were genotyped as follows; the genotyping protocol was adapted from one used previously (Lin et al., 2001). DNA was extracted from a 1 cm tail snip using a DNeasy Tissue kit (Qiagen, Valencia, CA). PCR amplification was performed using a Taq polymerase kit (Invitrogen, Carlsbad, CA) and primers (Invitrogen) designed to flank the 150 CAG repeat in the Hdh locus (forward primer, CCCATTCATTGCCTTGCTGCTAGG; reverse primer, CCTCTGGACAGGGAACAGTGTTGG). PCR conditions were as follows: 94°C for 5 min, followed by 29 cycles at 94°C for 30 s, 63°C for 30 s, 72°C for 3 min, and a final extension at 72°C for 5 min. DNA products were separated by standard 1.5% agarose gel electrophoresis and were visualized with ethidium bromide using a Gel Doc system (Bio-Rad, Hercules, CA). Expected bands were as follows: 379 bp for WT band and 829 bp for mutant band. PCR products were originally sequenced to ensure flanking primers contained the Hdh gene product. To confirm reliability of genotyping over time, periodic CAG sizing was performed outside our laboratory (Laragen, Los Angeles, CA) to confirm consistency of the CAG repeat number, which ranged from 145 to 150 CAG repeats.
Euthanasia
All animals were killed according to national guidelines. Euthanasia was performed by decapitation. This method was approved by the UCUCA and the VMU.
Behavioral examination
Three cohorts of mice (HOM, HET, and WT littermate controls) were used for each time point (20, 40, 50, 70, and 100 weeks). Time points are selected based on previously published neuronal intranuclear inclusion (NII) data (Tallaksen-Greene et al., 2005). In that study, diffuse nuclear htt immunoreactivity was apparent within MSNs ∼20 weeks of age, and some htt-immunoreactive NIIs were apparent by 40 weeks. By 70 weeks, many striatal neurons contained NIIs, and by 100 weeks, > 90% of striatal neurons contained NIIs. All behavioral evaluations were videotaped and performed with the examiner blind to genotype. Behavioral evaluations commenced on Wednesdays and ended on Fridays of the following week, a total of 8 d of behavioral trials with the first 3 d for acclimation and a break on Saturdays and Sundays. All behavioral trials were performed in the mornings between the hours of 9:00 A.M. and noon. Weights, footprint analysis, and activity cage measurements were taken only on the last day of trials. Brains were collected on the last day of testing.
The six behavioral assays used are described in detail below. They were (1) the accelerating rotarod, (2) the hanging wire test, (3) the tail suspension test, (4) activity monitoring, (5) footprint analysis, and (6) balance beam tests.
Hanging wire
A standard wire cage lid is used with masking tape placed around the perimeter of the lid to prevent animals from escaping. After an animal is placed on the lid, the lid is shaken gently to induce a firm grip, and the lid is inverted. The lid is held horizontally 20 cm above the cage litter. Latency to fall is recorded for up to 2 min. As described by Crawley (2000), normal mice are able to hang upside down for several minutes.
Tail suspension
Animals are suspended by their tail for a total of 1 min, and the presence of hindlimb clasping or hindlimb folding into the abdomen (Lin et al., 2001) >2 s is scored as “clasping.” Seizures were also noted if any occurred. An “escape” was scored during a trial if a mouse crawled up onto the tester's hand within the 60 s trial. To escape, an animal must grab its tail while suspended upside down and then crawl up and onto the examiner's hand.
Accelerating rotarod and balance beam
Rotarod.
The accelerating rotarod model number 7650 from Ugo Basile (Varese, Italy) was used. Mice are habituated for three consecutive daily trials, followed by 2 d of rest and tested once per day for five consecutive daily trials on an accelerating protocol (4–40 rpm over 5 min). The latency to fall is measured in each trial up to 5 min.
Balance beam.
Motor coordination, balance, and hindlimb placement were evaluated by assessing the ability of mice to traverse two types of balance beams to reach an enclosed safety platform (Perry et al., 1995). Each mouse was tested for its ability to traverse two different styles of 41-cm-long scored Plexiglas beams. One was cylindrical 11 mm in diameter, and the other was square and 5 mm wide. Beams were placed horizontally 50 cm above a table. A bright light illuminated the start platform, and a darkened enclosed 8000 cm3 escape box (20 × 20 × 20 cm) was situated at the end of the beam (PlasticTech, Ann Arbor, MI). Time to traverse each beam was recorded for each trial with a 20 s maximum cutoff, and falls were scored as 20 s. The number of foot slips and whether a mouse dragged itself across the beam on its abdomen (“hindlimb drag”) was also recorded.
Analyses of rotarod and balance beam results.
The first three trials were used to assess learning, and the last five trials were used to measure motor performance. In normal animals, the largest improvement in rotarod latency to fall times and time to cross the balance beam are observed in the first three trials with a ceiling by the fourth trial. Trials 4–8 are analyzed separately for motor performance after the animals have been trained in the first three trials.
Footprint analysis
Forepaws and hindpaws of the animals were painted with different colors of nontoxic paint, and the animals are allowed to walk across a 83-cm-long unroofed corridor (7 cm wide × 11 cm tall) lined with paper into an enclosed escape area (36 cm long × 7 cm wide × 11 cm tall). Measurements of stride length (the distance between one stride to the next for the same paw), base length (the distance between the two forepaws or hindpaws), and overlap (the distance between forepaw and hindpaw placement) were determined. Measurements were made for seven continuous strides and averaged per animal.
Automated activity cages
Activity cages with photo beam sensors (Advanced Concepts, Ann Arbor, MI) were used to measure general motor function, spontaneous activity, and movement over a 2 h test session. Four photo beam sensors are located at each end of the activity cage, 2 cm apart, 2.5 cm high, and 4 cm away from the edge. The pairs of beams are situated 15 cm apart, and a normal housing cage is placed into the activity apparatus to contain the mouse for the testing session. Beam breaks are automatically scored when any of the four beams are broken. Animals are tested over a 2 h period.
Brain harvesting and storage
Brains were extracted immediately after decapitation and divided in the sagittal plane. One hemisphere was coated with M-1 embedding matrix (Shandon, Pittsburgh, PA), frozen in crushed dry ice, wrapped in aluminum foil, and stored at −80°C. The other hemisphere was immersion fixed in 4% paraformaldehyde for 24 h, cryoprotected in 20% sucrose in 0.1 m phosphate buffer for an additional 24 h at 4°C, and frozen by placing the brains on boats made from aluminum foil that were floating on isopentane (cooled on dry ice at least 30 min). This hemisphere was also stored at −80°C until time of sectioning. Fixed hemispheres were used for stereological analysis, whereas the corresponding fresh frozen hemispheres were used for autoradiographic receptor binding analyses.
Receptor autoradiography
All studies were performed on coded samples with genotype information removed. Autoradiographic studies were performed as described previously (Tallaksen-Greene et al., 2003). Fresh frozen brains were serially cut on a cryostat in 15 μm sections at −12°C and thaw mounted onto gelatin-coated slides and stored at −80°C until use. Assays for D1 and D2 dopamine receptors were performed as follows: slides with sections were incubated in 25 mm Tris, 100 mm NaCl, 1 mm MgCl2, 0.001% ascorbic acid, and 1 μm pargyline, pH 7.2, with radioactive ligand 0.55 nm [3H]SCH23390 (specific activity, 89 Ci/mmol) for D1 and 0.75 nm [3H]spiperone (specific activity, 96 Ci/mmol) with 100 nm mianserin for D2 for 2.5 h at room temperature. Nonspecific binding was determined in the presence of 1 μm cis-flupenthixol for D1 and 50 μm dopamine for D2, respectively. After incubation, slides were subjected to two cold washes at 4°C and then one 10 min room-temperature wash with incubation buffer lacking radioactive ligand, followed by a quick rinse in double-distilled water before drying under a stream of warm air. For dopamine transporters (DATs), slides were prewashed in a 4°C solution containing 50 mm Tris-HCl, 5 mm KCl, and 300 mm NaCl, pH 7.9, for 5 min, followed by a 1 h incubation at 4°C in the same buffer with the addition of 10 nm [3H]mazindol (specific activity, 23.1 Ci/mmol) and 300 nm desipramine. Blank conditions contained 100 μm nomifensine. Slides were subjected to a final wash under the same conditions as the prewash for 3 min. For GABAA/benzodiazepine receptors, slides were prewashed in 50 mm Tris-citrate buffer, pH 7.2, at 4°C for 30 min, air dried, and incubated in 50 mm Tris-citrate buffer, pH 7.2, with 5 nm [3H]flunitrazepam (specific activity, 85 Ci/mmol) for 1 h at 4°C. Nonspecific binding was determined in ligand buffer with 2 μm clonazepam. Slides were subjected to one rapid rinse and two 5 min washes in 4°C 50 mm Tris-citrate buffer, pH 7.2, before drying under warm air.
D1 and D2 3H-ligands were purchased from Amersham Biosciences (Cardiff, UK). [3H]Mazindol and [3H]flunitrazepam were purchased from DuPont Biotechnology Systems (Wilmington, DE).
For receptor autoradiography, three slides per subject were used for each radioligand assay: two slides for total binding and the third to define nonspecific labeling with a minimum of two consecutive sections taken at fixed intervals (15 μm) mounted per slide. Slides and 14C standards are arrayed in a standard x-ray cassette, and an autoradiogram is generated by direct apposition of the tissue to the emulsion side of the tritium imaging plate (BAS-TR2025; Fuji Photo Film, Tokyo, Japan) for a period of 19–22 h.
Autoradiograms were analyzed by quantitative densitometry using an MCID-M2 image analysis system (Interfocus, Cambridge, UK). Anatomic locations of selected regions of interest were determined using an approach designed to ensure accuracy and consistency of the structures identified (Morrow et al., 1998). Briefly, for each brain section, we overlaid a matching transparent stereotaxic template, adapted from the mouse brain atlas (Paxinos and Franklin, 2001), on the digitized brain images displayed on the video monitor and aligned the images using prominent anatomical landmarks. Optical density (OD) measurements were taken bilaterally in a minimum of six brain sections, and measurements were made in each section in which the structure was visible, sampling as large an area as possible. Tissue 14C concentrations were determined by comparison of ODs with a calibration curve obtained from coexpressed standards. Specific binding was determined by subtracting nonspecific binding from the total binding.
Immunohistochemistry
Hemispheres previously fixed in 4% paraformaldehyde as described above were serially sectioned at 40 μm sagittally throughout the entire hemisphere. Every fourth section was used for stereological analysis. Free-floating sections were then stained with a primary specific neuronal antibody, NeuN (Chemicon, Temecula, CA) at a dilution of 1:1000 for 1 h. Sections were processed without primary NeuN antibody to assess background staining. No staining was visualized in these control sections. Detection of immunoreactivity was performed using the Vectastain Elite kit (Vector Laboratories, Burlingame, CA), and diaminobenzidine substrate was used as the chromogen according to the manufacturer's protocol. Sections were then mounted on gelatin-coated slides and air dried after dehydration with graded alcohols and xylene. Coverslips were affixed with Permount.
Stereology
Unbiased stereological counts of striatal neurons were obtained from the striatum of animals at 70 and 100 weeks of age using the StereoInvestigator software (MicroBrightField, Colchester, VT). The optical fractionator method was used to generate an estimate of neuronal number with only darkly stained NeuN-immunoreactive cells counted in an unbiased selection of serial sections in a defined volume of the striatum. Striatal borders were delineated on a NeuN-stained section by reference to a mouse brain atlas (Paxinos and Franklin, 2001). The striatum was defined to encompass both the dorsal and ventral striatum. Striatal volume was reconstructed by the StereoInvestigator software using the Cavalieri principle. Serially cut sagittal tissue sections (every fourth section) were analyzed for one entire hemisphere of animals in each genotype cohort (n = 4 per group).
Statistical analyses
All studies were performed blind to genotype. Comparisons of different groups (between genotypes) was performed on receptor binding, stereology, and weight data using a one-way ANOVA with post hoc comparisons made using the Tukey honestly significant difference test when p < 0.05. Repeated-measures ANOVA was performed for each behavioral test to determine whether there was an effect from genotypes or from repeated training. If there was no change after repeated training, the trials were collapsed, and a one-factor ANOVA was performed. A critical p < 0.05 was used for statistical significance in all analyses. Huynh-Feldt adjustment was used in correcting for violation of sphericity when necessary to adjust nonuniform variance across days or groups. The Kruskal–Wallis test was used for analysis of activity cage data because of the unequal variances of the groups. The SPSS (Chicago, IL) statistical software package was used.
Results
Survival and general observations
All mice survived to 100 weeks; however, by 100 weeks, HOM Hdh (CAG)150 mice exhibited noticeable resting tremor, unsteady movements, and staggering gait. No seizures were noted in any animal. We looked for sexual dichotomy in the analyses of all our behavioral evaluations and found no differences between genders.
HOM Hdh (CAG)150 mice exhibit weight loss over time
Hdh (CAG)150 homozygotes exhibited progressive weight loss with age (Fig. 1 a). Significant differences in average body weight between HOM mutants and wild type were observed at 70 and 100 weeks [70 weeks: WT, 33.6 ± 1.8 g (mean ± SEM); HOM, 26.9 ± 0.9 g; p < 0.05; 100 weeks: WT, 28.9 ± 1.7 g; HOM, 18.75 ± 0.76 g; p < 0.05]. Similar patterns in weight loss were observed when analyzed separately by gender (data not shown). This weight loss was not apparent at earlier ages, although some nonsignificant differences in weight were observed ∼50 weeks of age.
Figure 1.

Hdh (CAG)150 mice exhibit decreased locomotor activity and progressive weight loss. a, Homozygotes show weight loss over time with significant weight loss at 70 and 100 weeks compared with heterozygotes and wild types. Weights were taken from three cohorts of mice on the last day of behavioral testing (WT, HET, and HOM) at 20, 40, 50, 70, and 100 weeks. Weights are expressed as mean weight ± SEM. **p < 0.05, a significant difference between wild types and heterozygotes. b, Hdh (CAG)150 HET and HOM mice exhibit a trend toward decreasing spontaneous activity beginning at 40 weeks. At 100 weeks, differences are significant compared with WT mice (*Kruskal–Wallis χ2 < 0.05). Data are expressed as mean ± SEM; n = 7–15 per group.
Hdh (CAG)150 mice show decreased motor activity
Mice were assessed for general locomotion and normal exploratory behavior by 2 h trials in automated activity cages. At 100 weeks, HOM mice exhibited significantly reduced activity [371.8 ± 60.2 (mean ± SEM) beam breaks/2 h; Kruskal–Wallis χ2 < 0.05] compared with wild type (804.2 ± 138.6 beam breaks/2 h) (Fig. 1 b), suggesting that HOM mice have abnormal motor behavior. This reduction is also observed at 70 weeks, although not as robust (HOM, 388.7 ± 43.8 beam breaks/2 h; Kruskal–Wallis χ2 > 0.05; WT, 540 ± 135.8 beam breaks/2 h) (Fig. 1 b). The general activity of HET mice was similar to control mice up to 100 weeks (613.9 ± 115.3 beam breaks/2 h; Kruskal–Wallis χ2 > 0.05).
Hdh (CAG)150 mice show motor impairment on the accelerated rotarod
At 100 weeks, HOM Hdh (CAG)150 mice fell significantly earlier during the last five trials [120.34 ± 19.42 s (mean ± SEM); p < 0.05] compared with WT (212.38 ± 27.1 s) (Fig. 2 d) and HET (195.28 ± 22.2 s) Hdh (CAG)150 mice. Motor learning as assessed by improvement over the course of the first three trials was not impaired in Hdh (CAG)150 homozygotes at 100 weeks of age (F (2,24) = 5.70; p < 0.05 effect of training). Rotarod performance impairment was dependent on gene dosage, because 100-week-old HET Hdh (CAG)150 mice were able to maintain equal levels of motor performance as WT mice (F (2,22) = 3.95; p < 0.05). Furthermore, the impairment of motor performance in 100-week-old homozygotes was not found in younger mice, indicating a late-onset deficit. No differences existed between the three cohorts of mice in motor learning or performance at 50 and 70 weeks of age (F (2,11) = 0.56 at 50 weeks and F (2,19) = 0.14 at 70 weeks for motor learning, effect of training, p > 0.05; F (2,10) = 1.14 at 50 weeks and F (2,19) = 0.18 at 70 weeks for motor performance, effect of genotype, p > 0.05) (Fig. 2c). At 20 weeks, the opposite of impairment was seen for Hdh HET and HOM mice, who performed better than WT controls in both motor learning and motor performance (F (2,23) = 21.2 for effect of trials and F (2,20) = 7.2 for effect of genotype) (Fig. 2 a). This effect was maintained for heterozygotes at 40 weeks but only significant for performance compared with homozygotes (F (2,20) = 4.27 for genotype effect, p < 0.05) (Fig. 2 b).
Figure 2.

Hdh (CAG)150 mice exhibit decreased motor performance compared with WT mice on the accelerating rotarod. Animals were placed on a rotarod accelerating from 4 to 40 rpm within 5 min. Data from 20 weeks (a), 40 weeks (b), 70 weeks (c), and 100 weeks (d) are shown. To determine the rate of task learning, the maximum speed at the time of falling off the rotarod was plotted against the trial number over the first 3 d. Performance over days 4–8 measures motor performance. Note that HET and HOM mice initially learn the rotarod task more rapidly in the first three trials than WT controls at 20 weeks (F (2,23) = 21.2 for effect of trials and F (2,20) = 7.2 for effect of genotype; WT, n = 6; HET, n = 13; HOM, n = 7; p < 0.05). Hdh (CAG)150 mutant mice show no impairment in rotarod performance earlier than 70 weeks. All groups exhibited significant improvement in performance as training progressed; there were no differences between Hdh (CAG)150 mutant mice compared with WT at 40 and 70 weeks (WT, n = 3; HET, n = 9; HOM, n = 10; F (2,19) = 0.175 for effect of genotype; p > 0.05). When mice were examined at 100 weeks, homozygotes exhibited a significant decrease in latency to fall (120.34 ± 19.2 s, p < 0.05 compared with WT, 212.38 ± 27.1 s). Heterozygotes fell after 195.28 ± 22.2 s, which was not significantly different from WT controls (p > 0.05; WT, n = 9; HET, n = 8; HOM, n = 8; F (2,22) = 3.98 for effect of genotype). *p < 0.05. Values are expressed as mean ± SEM seconds.
Hdh (CAG)150 mice show abnormal gait
Footprint analysis was a sensitive indicator of gait abnormalities in Hdh (CAG)150 mice (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). One hundred-week-old Hdh (CAG)150 HOM and HET mice exhibit statistically significant differences from WT controls in several measures of gait. Significant differences were found in stride lengths and base lengths [hindpaw stride length: WT, 6.86 ± 0.22 cm (mean ± SEM); HET, 5.8 ± 0.25 cm; HOM, 5.8 ± 0.22; p < 0.05 vs WT; forepaw stride length: WT, 6.8 ± 0.14 cm; HET, 5.8 ± 0.30 cm; HOM, 5.5 ± 0.32 cm; p < 0.05 vs WT; front base length: WT, 4.13 ± 0.2 cm; HET, 3.47 ± 0.10 cm; HOM, 3.40 ± 0.11 cm; p < 0.05 vs WT]. Gait abnormalities were not as profound in younger mutant mice. For example, 70 week Hdh (CAG)150 HOM and HET mutants did not show statistically significant alterations in these same measures [WT, 4.03 ± 2.0 cm (mean ± SEM); HET, 3.86 ± 0.17 cm; p = 0.092 vs WT; HOM, 3.47 ± 0.09 cm; p = 0.056 vs WT]. Hdh (CAG)150 homozygotes also had more difficulty traversing the corridor, often leaning on one wall for support, which can be seen in the bias of gait prints to one side of the paper and the complete loss of gait pattern (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). WT mice at 100 weeks showed no loss of gait pattern over the mice tested at earlier ages, maintaining a consistent base length and very little deviance in overlap between hindpaw and frontpaw. However, WT mice show increased stride length when comparing 20 weeks of age with 100 weeks of age [right and left stride lengths: 20 weeks, 6.25 ± 0.23, 3.47 ± 0.10, and 6.33 ± 0.2 cm (mean ± SEM), respectively, compared with 100 weeks, 6.84 ± 0.1 and 6.86 ± 0.2 cm, respectively; p < 0.05]. Measurement of gait features, however, added little to inspection of footprint records. When measurement of gait features showed significant differences, gait was already qualitatively abnormal, and measurements failed to capture qualitative differences between mutant and WT gaits. Measurements, for example, did not show the tendency of 100-week-old HOM mutants to lean against one wall of the apparatus, a useful qualitative indicator.
Hdh (CAG)150 mutants require more time to traverse balance beam
Limb coordination and balance was measured with the balance beam task. At 70 weeks, Hdh (CAG)150 homozygotes and heterozygotes exhibited a trend toward greater time to traverse the 11 mm round and 5 mm square beams than WT controls (11 mm round beam, F (2,19) = 1.38; 5 mm square beam, F (2,18) = 2.4; effect for genotype, p > 0.05) (Fig. 3 a,c, respectively). Motor abnormalities became significant at 100 weeks; both HET and HOM mutants took longer to traverse the beams compared with age-matched WT littermates [11 mm round beam: WT, 6.09 ± 0.78 s (mean ± SEM); HET, 9.08 ± 1.69 s; HOM, 13.92 ± 1.51 s; p < 0.05; 5 mm square beam: WT, 10.11 ± 1.37 s; HET, 16.77 ± 1.78 s; HOM, 18.75 ± 0.76 s; p < 0.05] (Fig. 3 b,d, respectively). When motor learning was assessed for the 70- and 100-week-old animals only, the 100-week-old Hdh (CAG)150 homozygotes showed no improvement during the first three trials. One hundred-week-old heterozygotes did, however, decrease their time to traverse the 11 mm round beam in the first three trials, demonstrating motor learning was preserved. Deficits in motor performance were not seen in younger mutants. At 20, 40, and 50 weeks, there were no differences between genotypes in latency to cross the balance beams (11 mm round, F (2,10) = 0.013; 5 mm square, F (2,10) = 0.179; effect for genotype, p > 0.05) (supplemental Fig. 2a,b, respectively, available at www.jneurosci.org as supplemental material). The penetrance of abnormalities increased with increased gene dosage. For example, at 100 weeks of age, 80% of Hdh (CAG)150 HOM mice failed to traverse the 5 mm beam, whereas 50% of Hdh (CAG)150 HET mice failed compared with 0% of the wild types. An interesting and obvious difference between Hdh (CAG)150 mutant mice and WT littermates was the manner in which they traversed the balance beams. As early as 40 weeks, Hdh (CAG)150 HOM mice exhibited a “hindlimb drag” behavior in which the thorax and abdomen were pressed against the surface of the beam and hindlimbs were laterally wrapped around the beams. The forelimbs were used to pull the mouse across, resulting in a dragging motion across the beams (Fig. 4 a). WT mice displayed an upright posture with exact forelimb and hindlimb placement on the beams. The percentage of mice that displayed hindlimb drag increased with age and with the narrower 5 mm square beam (Fig. 4 b). By 100 weeks, Hdh (CAG)150 mutant mice displayed significant differences of hindlimb drag (heterozygotes, 63%; homozygotes, 90%) compared with WT controls (11%) (Pearson χ2 analysis, p < 0.05). This behavior probably contributed to the increased latency of mutant mice to traverse the beams. The Hdh (CAG)150 mutant mice display the same type of hindlimb drag that has been previously described in R6/2 mice (Carter et al., 1999). Hindlimb slips were also scored, however, because Hdh (CAG)150 mutant mice primarily displayed hindlimb drag instead of showing clear foot slips; this confounded the scoring of hind-leg slips, and these results were not useful.
Figure 3.

Hdh (CAG)150 mice require more time to transverse the balance beam. Mice were placed on a lit platform and traversed beams of different diameters (5 mm square and 11 mm round) to reach a safe chamber on the far side. Mice were tested at 20, 40, 50, 70, and 100 week time points. a, Eleven millimeter round beam (70 weeks); b, 11 mm round beam (100 weeks); c, 5 mm square beam (70 weeks); d, 5 mm square beam (100 weeks). Hdh (CAG)150 mutant mice show no motor abnormalities up to and including 50 weeks of age (see supplemental Fig. 2, available at www.jneurosci.org as supplemental material). a, c, Although nonsignificant, at 70 weeks both HET and HOM mice required more time to traverse the 11 and 5 mm beams compared with WT mice (11 mm beam, F (2,19) = 1.38; 5 mm beam, F (2,18) = 2.4; effect for genotype, p > 0.05). b, d, At 100 weeks, both HET and HOM mice required significantly more time to traverse the balance beams compared with WT mice (11 mm beam: WT, 6.09 ± 0.78; HET, 9.08 ± 1.69; HOM, 13.92 ± 1.51 s; p < 0.05; 5 mm beam: WT, 10.11 ± 1.37; HET, 16.77 ± 1.78; HOM, 18.75 ± 0.76 s; p < 0.05). *p < 0.05, significance compared with WT; **p < 0.05, significance compared with HET. Data are expressed as mean ± SEM in seconds.
Figure 4.

Hdh (CAG)150 mice displayed hindlimb drag. a, Hdh (CAG)150 mice displayed hindlimb drag as early as 40 weeks, where the abdomen is pressed against the surface of the beam and hindlimbs are dragged across the beams. b, The percentage of Hdh (CAG)150 mice presenting hindlimb drag progressively increased with age. By 100 weeks, 63% of HET and 90% of HOM displayed hindlimb drag compared with only 11% for WT controls (Pearson χ2 analysis, *p < 0.05). Generally, WT mice displayed an upright posture with exact forelimb and hindlimb placement on the beams.
No loss of muscle power in aged Hdh (CAG)150 mice
To test whether the motor impairments are related to loss of muscle power, the hanging wire test was used. At 100 weeks, we found no significant difference in the latency to fall between Hdh (CAG)150 homozygotes (92.0 ± 8.5 s; mean ± SEM) and heterozygotes (104.9 ± 6.0 s) compared with WT controls (89.8 ± 11.9 s). Similarly, no differences existed at any of the earlier time points. Because there was no difference in the repeated-measures ANOVA, the last five trials were collapsed and averaged (p > 0.05).
Hdh (CAG)150 mice exhibit clasping
We performed a tail suspension test that has been used to detect general neurological abnormalities and has been shown to induce seizures in abnormal mice (Lin, 2001). Animals were suspended by their tail for a total of 1 min, and animals were scored as clasping if the paws were held for >2 s. Only HOM mice exhibited clasping behavior compared with HET and WT mice at all time points (Fig. 5 a). A small percentage of HOM mice exhibited clasping at 20 weeks compared with WT littermates; however, the clasping phenomenon did not occur at 40 or 50 weeks but was common at 70 and 100 weeks. We did not observe any seizures during this test, nor were any seizures reported during routine handling. Additionally, we also looked at the percentage of mice that could escape as described by Lin et al. (2001), a general measure of overall fitness and health. Animals were scored as escape if they could crawl up and onto the tester's hand within the 60 s clasping trial. We found that the percentage of HOM mice able to escape was actually higher than HET mice compared with WT controls at 20 and 40 weeks (Fig. 5 b). This effect could be attributable to the size of the mouse because HOM mice are generally smaller in size, which may allow them to climb up more easily. Escape was not observed at time points beyond 40 weeks in all groups.
Figure 5.

Hdh (CAG)150 HOM mice display clasping and escape behaviors. a, HOM mice displayed clasping behavior as early as 20 weeks, and the number of mice presenting this behavior increased at 70 and 100 weeks. Clasping was not observed at the 40 or 50 week time points. b, Before motor deficits, Hdh (CAG)150 mice exhibited escape behavior at 20 and 40 weeks. A larger percentage of Hdh (CAG)150 mice were able to escape compared with WT controls at both time points.
Neuropathology
Receptor binding
Autoradiographic receptor analyses provided evidence of significant progressive, mutant allele-dependent morphological alterations in the Hdh (CAG)150 mouse. Binding sites were selected based on their regional distribution in the brain. GABAA/benzodiazepine receptor sites were chosen to survey neuronal integrity in many brain regions. These binding sites are highly expressed in most brain regions. An important exception is the striatum, where flunitrazepam binding is relatively low compared with other brain regions. Dopamine D1 and D2 receptor binding sites were chosen for their high expression by MSN dendrites and have been shown to be abnormal in HD patients as well as transgenic and other knock-in mouse models of HD (Weeks et al., 1996; Cha et al., 1998; Kennedy et al., 2005; van Oostrom et al., 2005). Additionally, we chose to look at DATs, a good presynaptic marker for nigrostriatal afferent projections.
Quantitative receptor autoradiographic analyses revealed no differences in GABAA/benzodiazepine binding sites between genotypes in any brain region (Fig. 6). We found significant alterations in striatal D1 and D2 receptor binding and in DAT binding at 100 weeks (Fig. 6). DAT binding was significantly decreased in the striatum of HOM mutants to 49% of WT levels. No DAT reduction was seen in HET mice [WT, 81.5 ± 4.9 pCi/g (mean ± SEM); HOM, 41.7 ± 8.2 pCi/g; p < 0.05]. Striatal D1 receptor binding sites decreased by 50% in heterozygotes and 86% in the homozygotes in the ventral striatum compared with WT controls and 42 and 79% in the dorsal striatum, respectively (Fig. 6) [ventral striatum: WT, 36.7 ± 1.7 pCi/g (mean ± SEM); HET, 18.4 ± 4.8 pCi/g; HOM, 5.3 ± 0.6 pCi/g; dorsal striatum: WT, 49.2 ± 5.3 pCi/g; HET, 28.5 ± 6.2 pCi/g; HOM, 10.5 ± 1.5 pCi/g; p < 0.05]. Striatal D2 receptor binding sites exhibited a 17% decrease in heterozygotes and 32% decrease in homozygotes in the ventral striatum and 17 and 42% compared with wild type in the dorsal striatum, respectively (Fig. 6) [ventral striatum: WT, 101.5 ± 3.6 pCi/g (mean ± SEM); HET, 83.8 ± 4.3 pCi/g; HOM, 69.3 ± 0.5; dorsal striatum: WT, 95.2 ± 2.2 pCi/g; HET, 78.6 ± 3.7 pCi/g; HOM, 55.6 ± 2.6 pCi/g; p < 0.05]. These reductions are late onset. Striatal D1 and D2 receptor binding at earlier time points showed smaller reductions in homozygotes compared with wild type at 70 weeks (Fig. 7) [ventral striatum: WT, 73.9 ± 6.5 pCi/g (mean ± SEM); HET, 57.7 ± 6.2 pCi/g; HOM, 36.4 ± 6.9 pCi/g; dorsal striatum: WT, 63.2 ± 4.5 pCi/g; HET, 49.4 ± 7.1 pCi/g; HOM, 31.8 ± 9.0 pCi/g; p < 0.05] and no reduction at 40 weeks (data not shown). Heterozygotes showed a lesser effect than homozygotes at 70 weeks with D2 receptor binding showing a greater effect than D1 receptor binding (Fig. 7) [ventral striatum: WT, 151.6 ± 4.7 pCi/g (mean ± SEM); HET, 140.6 ± 12.6 pCi/g; HOM, 104.2 ± 4.7 pCi/g; p < 0.05 vs WT and HOM and HET vs HOM; dorsal striatum: WT, 146.1 ± 11.7 pCi/g; HET, 108.3 ± 8.0 pCi/g; HOM, 90.9 ± 8.6 pCi/g; p < 0.05 vs WT), indicating a gene dose effect.
Figure 6.
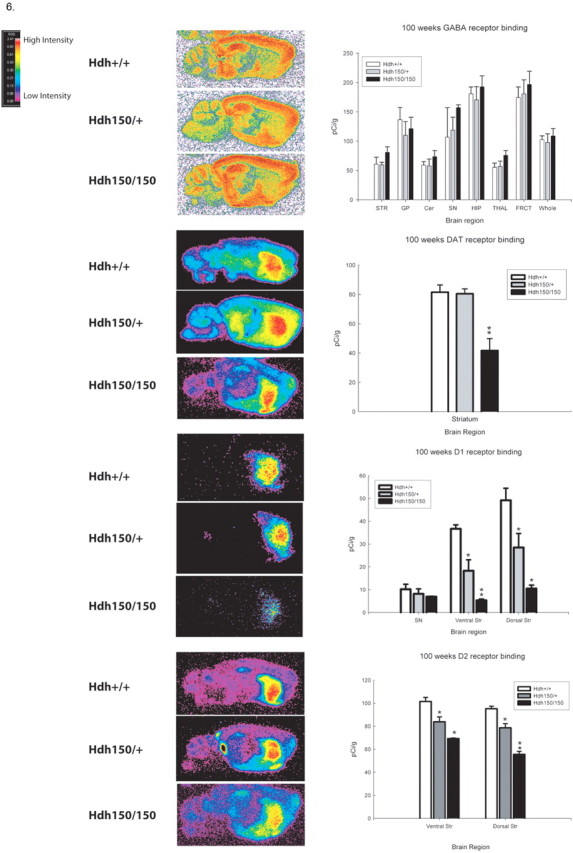
Hdh (CAG)150 mice exhibit alterations in striatal DAT, D1, and D2 receptor binding at 100 weeks. Pseudocolor images of receptor binding are shown. Autoradiographs were analyzed by quantitative densitometry using an MCID-M2 image analysis system. Histograms show the results of densitometric analysis of film images converted into picocuries of 3H-ligand bound per gram of protein. Regions analyzed are the cerebellum (Cer), substantia nigra (SN), globus pallidus (GP), ventral striatum and dorsal striatum (STR), hippocampus (HIP), thalamus (THAL), frontal cortex (FRCT), and whole brain (Whole). *p < 0.05, significant differences from WT; **p < 0.05, significant differences between HET and WT. All values are expressed as mean pCi/g ± SEM.
Figure 7.

Hdh (CAG)150 mice show alterations in D1 and D2 receptor binding as early as 70 weeks. HOM and HET Hdh (CAG)150 mice exhibit significant reductions of striatal D1 (a) and D2 (b) receptor binding compared with WT at 70 weeks. *p < 0.05, significant differences from WT; **p < 0.05, significant differences between HET and WT. All values are expressed as mean pCi/g ± SEM. Ventral Str, Ventral striatum; Dorsal Str, dorsal striatum; SN, substantia nigra.
Stereology
Consistent with the receptor binding results, stereological analyses of Hdh (CAG)150 brains revealed a significant reduction in estimated striatal neuron number and striatal volume at 100 weeks of age compared with WT controls (p < 0.05) (Fig. 8) [NeuN: WT, 1,255,311.0 ± 105,371.1 neurons (mean ± SEM); HET, 529,703.7 ± 51,551.75 neurons; HOM, 718,355.1 ± 51,257.16 neurons; p < 0.05 vs WT]. This reflects a mean 42.8% reduction in striatal neuron number in homozygotes and a mean 57.8% reduction in heterozygotes compared with WT controls. Homozygote striatal neurons were also morphologically distinct. They appeared atrophic and were shaped irregularly. Additionally, at 100 weeks of age, average striatal volume was significantly reduced for HOM compared with HET and WT littermates (Fig. 8 c) [WT, 4.43 × 109 ± 2.45 × 108 μm3 (mean ± SEM); HOM, 2.64 × 109 ± 1.7 × 108 μm3; p < 0.05 vs WT]. Homozygotes exhibited a mean 40.4% reductions in volume compared with WT controls. Striatal neuron number and volume were also analyzed at 70 weeks but showed no significant differences in striatal neuron count or volume between groups (data not shown). In summary, Hdh (CAG)150 mutant mice showed an ∼50% loss in striatal perikarya, concomitant with a 40% reduction in striatal volume in homozygotes, and this loss occurred between 70 and 100 weeks of age.
Figure 8.
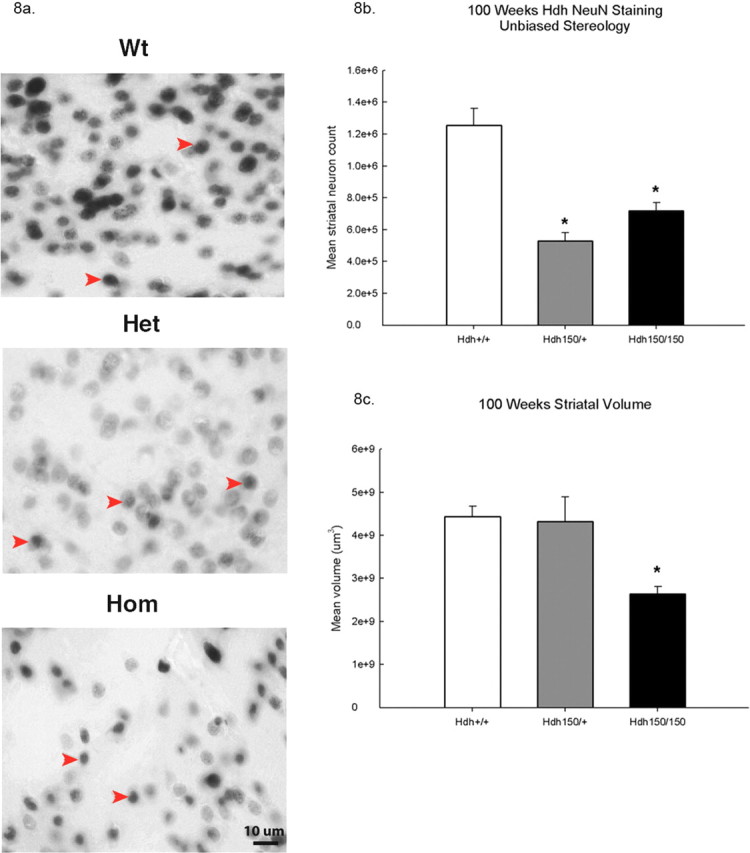
Hdh (CAG)150 mice show reductions in striatal NeuN-positive cells and striatal volume. a, Representative images at 100× of NeuN-immunoreactive striatal neurons at 100 weeks. Only darkly stained NeuN-immunoreactive cells were counted. Arrows indicate typical darkly stained striatal perikarya. Scale bar, 10 μm. b, At 100 weeks, HOM mice show a 42.8% reduction in striatal neuron number, and HET mice show a 57.8% reduction compared with WT. c, Striatal volume was also decreased in Hdh (CAG)150 mice. HOM mice exhibited a 40% reduction in striatal volume at 100 weeks compared with wild type (n = 4 per group; *p < 0.05).
Discussion
We provide a longitudinal characterization of the murine Hdh (CAG)150 knock-in model of HD. We found that Hdh (CAG)150 mice exhibit age-dependent weight loss, motor deficits, and striatal pathology that recapitulate important features of HD. Hdh (CAG)150 mice developed motor deficits by 70 weeks. Older Hdh (CAG)150 mice performed as well as wild type on the rotarod and balance beam during the 3 d of acclimation, suggesting that motor learning was preserved, and performed normally on the hanging wire test, suggesting absence of muscle weakness or nonspecific debilitation. Behavioral deficits observed in Hdh (CAG)150 mice parallel decreases in striatal D1 and D2 receptor binding sites at 70 weeks, which precede significant loss of striatal neuron number and volume observed at 100 weeks. The receptor binding data are consistent with imaging studies in human HD patients, as well as other mouse models of HD, which show decreases in D1 and D2 binding (Weeks et al., 1996; Ginovart et al., 1997; Cha et al., 1998; Kennedy et al., 2005; van Oostrom et al., 2005). Hdh (CAG)150 mice have no overt changes in other brain regions, as shown by GABAA/benzodiazepine binding. These results are consistent with our previous description of almost uniquely high expression of striatal NIIs in this line (Tallaksen-Greene et al., 2005).
We used six different methods to evaluate behavior. Accelerating rotarod, balance beam, and footprint analysis showed evidence of progressive declines in motor function. The balance beam test was quantitative and sensitive, revealing subtle motor deficits and demonstrating differences between WT, HET, and HOM mice at 100 weeks. Observation of balance beam performance provided complementary qualitative information because mutant mice were not only slower but used a different strategy to cross beams. The rotarod was less predictive of genotype, with similar results for WT and HET mutant mice. Footprint analysis discriminated WT and mutants well, but quantification of this method is less useful. Measurement of gait features showed significant differences, but these measurements added little to qualitative inspection of footprint patterns. In preliminary experiments, we evaluated younger (<50 weeks) Hdh (CAG)150 mice with a battery of more “cognitive” tests. No deficiencies were found with the Morris water maze, fear conditioning, or open-field tests (B. C. McKinney, G. G. Murphy, M. Y. Heng, and R. L. Albin, unpublished data).
The original publication of the Hdh (CAG)150 mice reported seizures, as well as some behavioral deficits that occurred earlier than reported here (Lin et al., 2001). This discrepancy may reflect that our current line has more C57BL/6 background (75–90% C57BL/6; Detloff, unpublished data) than the Hdh (CAG)150 mice used in the initial study (50–75% C57BL/6). The C57BL/6 background causes reduction in phenotype severity in one other mouse model of HD (Van Raamsdonk et al., 2007). We never witnessed seizures in our mice. Absence of epilepsy is congruent with HD and eliminates the possibility that seizures could be the cause of pathological changes.
Our pathological analyses suggest that striatal neurons pass through a period of neuronal dysfunction before actual neuronal degeneration. At 70 weeks, we found significant declines in both striatal D1 and D2 receptor binding sites in HOM mutants and of striatal D2 binding sites in HET mutants, but no change in striatal neuron number or volume. Changes in striatal D1 and D2 receptor binding sites at 70 weeks likely reflect neuronal dysfunction (Li et al., 2003; Yu et al., 2003) rather than neuronal degeneration. At 100 weeks, D1 and D2 receptor binding sites were diminished significantly in both HOM and HET mutants, and there was similar loss of neuronal number in both HOM and HET mutants. Striatal volume measurements, however, were abnormal in 100-week-old HOM mutants only. Because we identified neurons with NeuN immunoreactivity, it is plausible that some striatal neuron perikarya are intact in HET mutants but lost the capacity to express NeuN protein. To maintain consistency across genotypes, only darkly stained striatal neuron perikarya were counted. Given the normal striatal volume in 100-week-old heterozygotes, this raises the possibility that many striatal neurons in 100-week-old heterozygotes are dysfunctional rather than deceased. In HOM mutants, the concurrent loss of striatal neuron number and volume is strong evidence of striatal neuron degeneration. This result is consistent also with the loss of striatal DAT binding sites in 100-week-old HOM mutants. Positron emission tomography imaging studies indicate that more advanced HD is accompanied by loss of nigrostriatal dopaminergic innervation (Backman et al., 1997; Bohnen et al., 2000). The more severe pathological findings in HOM mutants is consistent with data indicating that HD homozygotes and SCA3 homozygotes have more aggressive disease than HET patients (Lang et al., 1994; Sobue et al., 1996; Squitieri et al., 2003).
Because the great majority of striatal neurons are MSNs, the substantial changes we report in D1/D2 receptor binding and striatal neuron number in 100-week-old homozygotes imply dysfunction/degeneration of MSNs. The loss of both D1 and D2 binding sites is consistent with dysfunction/degeneration of both major pools of MSNs. The selective loss of striatal D2 binding sites in HET 70-week-old mutants is intriguing in view of the probable early loss of MSNs projecting to the lateral globus pallidus in HD (Reiner et al., 1988).
Since the identification of huntingtin, multiple mouse models have been generated: transgenic chimeric models that express truncated forms of the human mutant HD allele, transgenic chimeric models that express full-length human mutant HD alleles, and knock-in models that express varying length CAG repeat expansions within the murine huntingtin homolog. The R6/2 transgenic line, expressing an exon 1 fragment of htt with a range of 148–153 repeats, is widely used because of its aggressive phenotype and provides clear experimental end points (Mangiarini et al., 1996; Hickey et al., 2005; Morton et al., 2005; Stack et al., 2005). The aggressive phenotype makes R6/2 very useful for preclinical pharmacology, but it is not an exact genetic or neuropathological analog of HD. R6/2 mice have widespread NII expression, a high incidence of epilepsy, diabetes, cardiac dysfunction, and neuromuscular junction abnormalities (Hurlbert et al., 1999; Meade et al., 2002; Ribchester et al., 2004; Mihm et al., 2007). The R6/2 line is a plausible model of juvenile-onset HD, in which the effects of the expanded polyglutamine repeat occurs in the context of a developing brain.
The YAC128 expresses the full-length mutant human HD gene (Slow et al., 2003) and shows behavioral deficits detectable at 6 months with striatal neuronal loss at 9 and 12 months. YAC128 mice show a 9% decrease in striatal neuron number at 9 months and a 15% decrease by 12 months. Studies of other striatal neuron markers, such as striatal neuron neurotransmitter receptor binding sites, have not been reported. Results with YAC128 mice may be different from HD, where there is substantial striatal neuron, and probably cortical neuron loss, before onset of the overt phenotype (Aylward et al., 2004; Rosas et al., 2004).
Unlike transgenic models of HD, knock-in models provide expression of mutant huntingtin in an appropriate genomic and protein context. The Hdh (CAG)150 knock-in model is unique from other knock-in models in that it is nonchimeric, expressing only a polyglutamine expansion in the murine Hdh gene, and this change is not accompanied by foreign DNA sequences or selectable markers. Behavioral data and pathological data have been published on several knock-in models, but none thus far have provided extensive longitudinal behavioral examination and correlative neuropathological data. A striking feature of the Hdh (CAG)150 knock-in model is that the high-frequency expression of NIIs is almost unique to the striatum and that neuropil aggregates are expressed probably in striatal projection neuron terminals, even in mice of advanced age (Tallaksen-Greene et al., 2005). These findings are consistent with the recent reports of the chimeric exon 1 140 CAG knock-in mouse model of HD exhibiting the late and progressive behavioral and neuropathological phenotype (Menalled et al., 2003). Other previous knock-in models have reported mild motor behavioral deficits and neurodegeneration.
The relatively specific expression of NIIs within striatal neurons, selective decrease in striatal neurotransmitter receptor binding sites, diminished striatal neuron number, and late onset of behavioral abnormalities observed in Hdh (CAG)150 mice recapitulate key features of HD. A recent striatal gene expression study also indicates significant similarity of Hdh (CAG)150 mice to HD (Kuhn et al., 2007). Because of its milder phenotype and late onset of behavioral abnormalities, the Hdh (CAG)150 knock-in line has not widely been used as a model for preclinical pharmacological evaluation. The expedience of an aggressive phenotype must be balanced against the possibility that the phenotype of other mouse models may be driven by molecular processes that do not mimic HD. The Hdh (CAG)150 model will be most useful for investigating the pathogenesis of neurodegeneration in HD. We have shown a prolonged period of neuronal dysfunction before neuronal death in this line, which will allow examination of the early events in the pathogenesis of HD.
Footnotes
This work was supported by a Veterans Administration Merit Review Grant and by grants from the Huntington Disease Society of America and the High Q Foundation. We acknowledge Lauren Cline, Kevin Duong, Chad Green, Erin Katz, Alex Martusiewicz, Adam Myrold, and Rebecca York for their technical assistance. We thank Dr. Tom Morrow for use of his MCID system and Dr. Kirk Frey for use of his stereology apparatus.
References
- Aylward EH, Sparks BF, Field KM, Yallapragada V, Shpritz BD, Rosenblatt A, Brandt J, Gourley LM, Liang K, Zhou H, Margolis MD, Ross CA. Onset and rate of striatal atrophy in preclinical Huntington disease. Neurology. 2004;63:66–72. doi: 10.1212/01.wnl.0000132965.14653.d1. [DOI] [PubMed] [Google Scholar]
- Backman L, Robins-Wahlin TB, Lundin A, Ginovart N, Farde L. Cognitive deficits in Huntington's disease are predicted by dopaminergic PET markers and brain volumes. Brain. 1997;120(12):2207–2217. doi: 10.1093/brain/120.12.2207. [DOI] [PubMed] [Google Scholar]
- Bohnen NI, Koeppe RA, Meyer P, Ficaro E, Wernette K, Kilbourne MR, Kuhl DE, Frey KA, Albin RL. Decreased striatal monoaminergic terminals in Huntington disease. Neurology. 2000;54:1753–1759. doi: 10.1212/wnl.54.9.1753. [DOI] [PubMed] [Google Scholar]
- Carter RJ, Humby T, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ. Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation. J Neurosci. 1999;19:3248–3257. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha JH, Kerner JA, Alsdorf SA, Mangiarini L, Davies SW, Penney JB, Bates GP, Young AB. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human Huntington disease gene. Proc Natl Acad Sci USA. 1998;95:6480–6485. doi: 10.1073/pnas.95.11.6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawley JN. What's wrong with my mouse? Behavioral phenotyping of transgenic and knockout mice. New York: Wiley; 2000. [Google Scholar]
- Cummings HY. Fourteen and counting; unraveling trinucleotide repeat diseases. Hum Mol Genet. 2000;9:909–916. doi: 10.1093/hmg/9.6.909. [DOI] [PubMed] [Google Scholar]
- Ginovart N, Lundin A, Farde L, Halldin C, Backman L, Swahn CG, Pauli S, Sedvall G. PET study of the pre- and post-synaptic dopaminergic markers for the neurodegenerative process in Huntington's disease. Brain. 1997;120:503–514. doi: 10.1093/brain/120.3.503. [DOI] [PubMed] [Google Scholar]
- Hardy J, Orr H. The genetics of neurodegenerative diseases. J Neurochem. 2006;97:1690–1699. doi: 10.1111/j.1471-4159.2006.03979.x. [DOI] [PubMed] [Google Scholar]
- Hickey MA, Gross GG, Levine MS, Chesselet MF. Early behavioral deficits in R6/2 mice suitable for use in preclinical drug testing. Neurobiol Dis. 2005;20:1–11. doi: 10.1016/j.nbd.2005.01.024. [DOI] [PubMed] [Google Scholar]
- Hodgson JG, Gutekunst CA, Leavitt BR, LePiane F, Singaraja R, Smith DJ, Bissada N, McCutcheon K, Nasir J, Jamot L, Li XJ, Stevens ME, Rosemond E, Roder JC, Phillips AG, Rubin EM, Hersch SM, Hayden M. A YAC mouse model for Huntington's disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 1999;23:181–182. doi: 10.1016/s0896-6273(00)80764-3. [DOI] [PubMed] [Google Scholar]
- Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Hurlbert MS, Wasmeier C, Kaddis FG, Hutton JC, Freed CR. Mice transgenic for an expanded CAG repeat in the Huntington's disease gene develop diabetes. Diabetes. 1999;48:649–651. doi: 10.2337/diabetes.48.3.649. [DOI] [PubMed] [Google Scholar]
- Kennedy L, Shelbourne PF, Dewar D. Alterations in dopamine and benzodiazepine receptor binding precede overt neuronal pathology in mice modeling early Huntington disease pathogenesis. Brain Res. 2005;1039:14–21. doi: 10.1016/j.brainres.2005.01.029. [DOI] [PubMed] [Google Scholar]
- Kuhn A, Hodges A, Strand AD, Sengstag T, Kooperberg C, Becanovic K, Pouladi MA, Sathasivam K, Cha JH, Hannan AJ, Hayden MR, Leavitt BR, Dunnett SB, Ferrante RJ, Albin RL, Shelbourne P, Delorenzi M, Augood SJ, Faull RL, Olson JM, et al. Mutant huntingtin's effect on striatal gene expression in mice recapitulate changes observed in human Huntington's disease brain and do not differ with mutant huntingtin length or wild-type huntingtin dosage. Hum Mol Genet. 2007;16:1845–1861. doi: 10.1093/hmg/ddm133. [DOI] [PubMed] [Google Scholar]
- Lang AE, Tsuda T, Hutterer J, St. George-Hyslop P. Homozygous inheritance of the Machado-Joseph disease gene. Ann Neurol. 1994;36:443–447. doi: 10.1002/ana.410360318. [DOI] [PubMed] [Google Scholar]
- La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fishchbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352:77–79. doi: 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- Li JY, Plomann M, Brundin P. Huntington's disease: a synaptopathy? Trends Mol Med. 2003;9:414–420. doi: 10.1016/j.molmed.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Lin CH, Tallaksen-Greene S, Chien WM, Clearly JA, Jackson WS, Crouse AB, Ren S, Li XJ, Albin RL, Detloff PJ. Neurological abnormalities in a knock-in mouse model of Hungtington's disease. Hum Mol Genet. 2001;10:137–144. doi: 10.1093/hmg/10.2.137. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- Meade CA, Fusco FR, Del Mar N, Hersch S, Goldowitz D, Reiner A. Cellular localization and development of neuronal intranuclear inclusions in striatal and cortical neurons in R6/2 transgenic mice. J Comp Neurol. 2002;449:241–269. doi: 10.1002/cne.10295. [DOI] [PubMed] [Google Scholar]
- Menalled LB, Sison JD, Dragatsis I, Zeitlin S, Chesselet MF. Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington's disease with 140 CAG repeats. J Comp Neurol. 2003;465:11–26. doi: 10.1002/cne.10776. [DOI] [PubMed] [Google Scholar]
- Mihm MJ, Schanbacher BL, Altschuld RA, Bauer JA, Hoyt KR. Cardiac dysfunction in the R6/2 mouse model of Hungtington's disease. Neurobiol Dis. 2007;25:297–308. doi: 10.1016/j.nbd.2006.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow TJ, Damneman PJ, Casey KL. Regional changes in forebrain activation during the early and late phase of formalin nociception: analysis using cerebral blood flow in the rat. Pain. 1998;75:355–365. doi: 10.1016/s0304-3959(98)00016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton AJ, Hunt MJ, Hodges AK, Lewis PD, Redfern AJ, Dunnett SB, Jones L. A combination drug therapy improves cognition and reverses gene expression changes in a mouse model of Huntington's disease. Eur J Neurosci. 2005;21:855–870. doi: 10.1111/j.1460-9568.2005.03895.x. [DOI] [PubMed] [Google Scholar]
- Orr HT. Beyond the Qs in the polyglutamine diseases. Genes Dev. 2001;15:925–932. doi: 10.1101/gad.888401. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. Ed 2. San Diego: Academic; 2001. [Google Scholar]
- Perry TA, Torres EM, Czech C, Beyreuther K, Richards S, Dunnett SB. Cognitive and motor function in transgenic mice carrying excess copies of the 695 and 751 amino acid isoforms of the amyloid precursor protein gene. Alzheimer's Res. 1995;1:5–14. [Google Scholar]
- Reddy PH, Charles V, Garret L, Pike-Buchanan L, Whetsell WO, Jr, Miller G, Tagle DA. Behavioral abnormalities and selective neuronal loss in HD transgenic mice expressing mutated full-length HD cDNA. Nat Genet. 1998;20:198–202. doi: 10.1038/2510. [DOI] [PubMed] [Google Scholar]
- Reiner A, Albin RL, Anderson KD, D'Amato CJ, Penney JB, Young AB. Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci USA. 1988;85:5733–5737. doi: 10.1073/pnas.85.15.5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribchester RR, Wood NI, Hinks T, Gillingwater TH, Wishart TM, Court FA, Morton AJ. Progressive abnormalities in skeletal muscle and neuromuscular junctions of transgenic mice expressing the Huntington's disease mutation. Eur J Neurosci. 2004;20:3092–3114. doi: 10.1111/j.1460-9568.2004.03783.x. [DOI] [PubMed] [Google Scholar]
- Rosas HD, Hersch S, Glessner M, Ferrante RJ, Salat DH, van der Kouwe A, Jenkins BG, Dale AM, Fischl B. Regional and progressive thinning of the cortical ribbon in Huntington's disease. Neurology. 2002;58:695–701. doi: 10.1212/wnl.58.5.695. [DOI] [PubMed] [Google Scholar]
- Rosas HD, Koroshetz WJ, Chen YI, Skeuse C, Vangel M, Cudkowicz ME, Caplan K, Marek K, Seidman LJ, Makris N, Jenkins BG, Goldstein JM. Evidence for more widespread cerebral pathology in early HD: an MRI-based morphometric analysis. Neurology. 2004;60:1615–1620. doi: 10.1212/01.wnl.0000065888.88988.6e. [DOI] [PubMed] [Google Scholar]
- Rosas HD, Zaleta AK, Greve DN, Salat DH, Fischl B. Regional cortical thinning in preclinical Huntington disease and its relationship to cognition. Neurology. 2005;65:745–747. doi: 10.1212/01.wnl.0000174432.87383.87. [DOI] [PubMed] [Google Scholar]
- Schilling G, Sharp AH, Jinnah HA, Duan K, Kotzuk JA, Slunt HH, Ratovitski T, Cooper JK, Jenkins NA, Copeland NG, Price DL, Ross CA, Borchelt DR. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum Mol Genet. 1999;8:397–407. doi: 10.1093/hmg/8.3.397. [DOI] [PubMed] [Google Scholar]
- Shelbourne PF, Hevner RF, Johnston HM, Tecott L, Lewandoski M, Ennis M, Ramirez L, Li Z, Iannicola C, Littman DR, Myers RM. A Huntington's disease CAG expansion at the murine Hdh locus is unstable and associated with behavioral abnormalities in mice. Hum Mol Genet. 1999;8:763–774. doi: 10.1093/hmg/8.5.763. [DOI] [PubMed] [Google Scholar]
- Slow EJ, Rogers D, Coleman SH, Graham RK, Deng Y, Oh R, Bissada N, Hossain SM, Yang YZ, Li XJ, Simpson EM, Gutekunst CA, Leavitt BR, Hayden MR. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12:1555–1567. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- Sobue G, Nakao N, Shimada N, Mitsuma T, Maruyama H, Kawakami H, Nakamura S. Homozygosity for Machado-Joseph disease gene enhances phenotypic severity. J Neurol Neurosurg Psychiatry. 1996;60:354–356. doi: 10.1136/jnnp.60.3.354-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squitieri F, Gellera C, Cannella M, Mariotti C, Cislaghi G, Rubinsztein DC, Almqvist EW, Turner D, Bachoud-Levi AC, Simpson SA, Delatycki M, Maglione V, Hayden MR, Donato SD. Homozygosity for CAG mutation in Huntington disease is associated with a more severe clinical course. Brain. 2003;126:946–955. doi: 10.1093/brain/awg077. [DOI] [PubMed] [Google Scholar]
- Stack EC, Smith K, Cormier K, Del Signore SJ, Guelin E, Ryu H, Hersch SM, Ferrante RJ. Chronology of behavioral symptoms and neuropathological sequela in R6/2 Huntington's disease transgenic mice. J Comp Neurol. 2005;490:354–370. doi: 10.1002/cne.20680. [DOI] [PubMed] [Google Scholar]
- Tallaksen-Greene SJ, Ordway JM, Crouse AB, Jackson WS, Detloff PJ, Albin RL. Hprt(CAG)146 mice: age of onset of behavioral abnormalities, time course of neuronal intranuclear inclusion accumulation, neurotransmitter marker alterations, mitochondrial function markers, and susceptibility to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. J Comp Neurol. 2003;465:205–219. doi: 10.1002/cne.10855. [DOI] [PubMed] [Google Scholar]
- Tallaksen-Greene SJ, Hunter JM, Detloff PJ, Albin RL. Neuronal intranuclear inclusions and neuropil aggregates in HdhCAG(150) knockin mice. Neuroscience. 2005;131:843–852. doi: 10.1016/j.neuroscience.2004.10.037. [DOI] [PubMed] [Google Scholar]
- van Oostrom JC, Maguire RP, Verschuuren-Bemelmans CC, Veenma-van der Duin L, Pruim J, Roos RA, Leenders KL. Striatal dopamine D2 receptors, metabolism, and volume in preclinical Huntington disease. Neurology. 2005;65:941–943. doi: 10.1212/01.wnl.0000176071.08694.cc. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Metzler M, Slow E, Pearson J, Schwab C, Carroll J, Graham RK, Leavitt BR, Hayden MR. Phenotypic abnormalities in the YAC128 mouse model of Huntington disease are penetrant on multiple genetic backgrounds and modulated by strain. Neurobiol Dis 26. 2007;1:189–200. doi: 10.1016/j.nbd.2006.12.010. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- Weeks RA, Harding AE, Brooks DJ. Striatal D1 and D2 dopamine receptor loss in asymptomatic mutation carriers of Huntington's disease. Ann Neurol. 1996;40:49–54. doi: 10.1002/ana.410400110. [DOI] [PubMed] [Google Scholar]
- Wheeler VC, Gutekunst CA, Vrbanac V, Weaver M, Li XJ, Li SH, Yi H, Vonsattel JP, Gusella JF, Hersch S, Auerbach W, Joyner AL, MacDonald ME. Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in HdhQ92 and HdhQ111 knock-in mice. Hum Mol Genet. 2000;9:503–513. doi: 10.1093/hmg/9.4.503. [DOI] [PubMed] [Google Scholar]
- Yu ZX, Li SH, Evans J, Pillarisetti A, Li H, Li XJ. Mutant huntingtin causes context-dependent neurodegeneration in mice with Huntington's disease. J Neurosci. 2003;23:2193–2202. doi: 10.1523/JNEUROSCI.23-06-02193.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]