

Abstract
To elucidate the molecular mechanisms underlying the light-sensitive retinal degeneration caused by the rhodopsin mutation P23H, which causes retinitis pigmentosa (RP) in humans, we expressed Xenopus laevis, bovine, human, and murine forms of P23H rhodopsin in transgenic X. laevis rod photoreceptors. All P23H rhodopsins caused aggressive retinal degeneration associated with low expression levels and retention of P23H rhodopsin in the endoplasmic reticulum (ER), suggesting involvement of protein misfolding and ER stress. However, light sensitivity varied dramatically between these RP models, with complete or partial rescue by dark rearing in the case of bovine and human P23H rhodopsin, and no rescue for X. laevis P23H rhodopsin. Rescue by dark rearing required an intact 11-cis-retinal chromophore binding site within the mutant protein and was associated with truncation of the P23H rhodopsin N terminus. This yielded an abundant nontoxic ∼27 kDa form that escaped the ER and was transported to the rod outer segment. The truncated protein was produced in the greatest quantities in dark-reared retinas expressing bovine P23H rhodopsin and was not observed with X. laevis P23H rhodopsin. These results are consistent with a mechanism involving enhanced protein folding in the presence of 11-cis-retinal chromophore, with ER exit assisted by proteolytic truncation of the N terminus. This study provides a molecular mechanism for light sensitivity observed in other transgenic models of RP and for phenotypic variation among RP patients.
Keywords: rhodopsin, ER stress, signal transduction, protein misfolding, photoreceptor, neuronal death
Introduction
The turnover of proteins, including degradation of damaged or folding-defective proteins, is a normal part of cellular metabolism. However, large or prolonged accumulations of misfolded proteins can induce endoplasmic reticulum (ER) stress responses, overwhelm quality control pathways, and cause cell death. Many human disorders such as Alzheimer's disease, Huntington's disease, and cystic fibrosis are associated with misfolding and aggregation of integral membrane and secreted proteins (Rao and Bredesen, 2004). Some forms of retinitis pigmentosa (RP) may also belong to this class of diseases.
RP is a group of disorders characterized by retinal degeneration (RD), which involves death of rod photoreceptors, gradual loss of cone photoreceptors, and progressive loss of vision. Defects in the rhodopsin gene are the most common cause of autosomal dominant RP, with >100 missense mutations thus far identified (http://www.sph.uth.tmc.edu/Retnet/). Rhodopsin is a heptahelical G-protein-coupled receptor synthesized at high levels in the cell body or rod inner segment (RIS) and subsequently transported to a specialized light-sensitive organelle called the rod outer segment (ROS), which consists of a large stack of flattened membranous disks ensheathed within a plasma membrane. The majority of RP-causing rhodopsin mutations, including the most prevalent mutation P23H (Dryja et al., 1990), cause misfolding and retention in the ER of cultured cells (Kaushal and Khorana, 1994). Cell culture studies also suggest that the mechanism of RD involves cellular stress responses (Illing et al., 2002). Our previous study of transgenic Xenopus laevis expressing frog P23H rhodopsin established that the mutant protein was predominantly confined to the ER, with no effect of dark rearing or disruption of visual transduction on the rate of RD (Tam et al., 2006). However, in other animal models, P23H rhodopsin traffics normally to the ROS, and RD is exacerbated by light (Naash et al., 1996; Organisciak et al., 2003; Galy et al., 2005). In humans, light may also affect disease progression caused by the rhodopsin mutations T17M and P23H, where the lower retina, which is exposed to brighter illumination from above, is preferentially affected (Li et al., 1994; Cideciyan et al., 1998)
Therapy for P23H rhodopsin-induced RP poses a challenge because of its autosomal dominant nature. Currently, gene replacement combined with knockdown of the mutant protein is the only preventative therapy that can be envisioned because of poor understanding of the disease mechanisms. Greater understanding of the molecular mechanisms underlying this form of RP would potentially allow a wider range of novel therapeutic interventions to be developed. Therefore, it is crucial to reconcile the controversies in previous studies (i.e., whether P23H rhodopsins significantly misfold in vivo and whether light exposure plays a role in causing/exacerbating the disease process). In this study, we generated and examined transgenic X. laevis models of RP expressing P23H rhodopsins from species examined in other transgenic animal models (i.e. X. laevis, murine, bovine, and human). We established their expression levels, subcellular localization, and ability to induce RD. We further determined whether they exhibited differences in light sensitivity and examined the underlying mechanisms.
Materials and Methods
Molecular biology.
Transgene expression constructs were based on XOP0.8-eGFP-N1 (Tam et al., 2006). The green fluorescent protein (GFP) cDNA was excised with EcoRI and NotI and replaced with the various rhodopsin cDNAs. The epitope-tagged X. laevis wild-type (WT) and P23H rhodopsin cDNAs were identical to those described previously (Tam and Moritz, 2006). The bovine rhodopsin cDNA was obtained from D. Oprian (Brandeis University, Boston, MA), and mouse and human rhodopsin cDNAs were obtained by RT-PCR of mRNA. P23H mutations and the rhoΔ2–23 mutation were introduced by methods described previously (Nelson and Long, 1989), and completed transgene cDNAs were verified by DNA sequencing. For transgenesis, expression vectors were linearized by digestion with FseI (New England Biolabs, Beverly MA) and purified using the QIAquick Gel Extraction kit (Qiagen, Valencia, CA). For expression in cultured cells, the mutant rhodopsin cDNAs were subcloned into the EcoRI/NotI sites of pMT3 (Franke et al., 1988).
Generation and rearing of transgenic X. laevis.
Transgenic X. laevis tadpoles were either generated by the methods described previously (Kroll and Amaya, 1996; Tam and Moritz, 2006; Tam et al., 2006) or by mating mature primary transgenic frogs with nontransgenic animals. Embryos were housed in 4 L tanks in an 18°C incubator on a 12 h light/dark cycle. Twenty-four hours after fertilization, G418 selection was performed (Moritz et al., 2002; Tam et al., 2006). Dark-reared animals were housed in the same incubator as cyclic-reared animals, in tanks wrapped in several layers of aluminum foil. At 14 d postfertilization (dpf) (corresponding to developmental stage 48), normally developed X. laevis were killed, and one eye was fixed in 4% paraformaldehyde, whereas the contralateral eye was solubilized as described previously (Tam et al., 2006) for use in dot blot or Western blot analyses.
Dot blots.
Dot blots and data analysis were performed as described previously, using X. laevis eye extracts, monoclonal antibody (mAb) B630N (Adamus et al., 1991) and mAb 1D4 (MacKenzie et al., 1984) (Chemicon, Temecula, CA) primary antibodies, IR-dye800-conjugated secondary antibodies, and a LICOR Odyssey imaging system (Tam et al., 2006). Each blot included standards containing 100% X. laevis rhodopsin (from wild-type retinas) and 100% transgenic rhodopsin (from cell culture extracts), allowing the percentage of transgenic rhodopsin expression levels to be determined from the ratio of mAb B630N (recognizes total rhodopsin) to mAb 1D4 label (recognizes only transgenic rhodopsins). Standard curves derived from serial dilutions of control samples were used to determine the linear range of both antibodies. Nonparametric statistical methods requiring only ordinal data were used for data analysis because of nonnormal (i.e., skewed and bimodal) data distributions.
Cell culture, transfection, and Western blot analysis.
Human embryonic kidney 293S (HEK293S) cells were cultured in DMEM/F-12 medium and transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the recommendations of the manufacturer. After 12 h, medium was replaced, and the cells were cultured in the dark in the presence of 50 μm 11-cis-retinal. After 36 h, cells were solubilized in 1% N-dodecyl maltoside (Anatrace, Maumee, OH) with 0.1 mg/ml PMSF in PBS for 1 h on ice, followed by centrifugation at 36,000 × g for 30 min. Western blot analysis was performed as described previously (Tam and Moritz, 2006), using mAb B630N or mAb A5-3 (Adamus et al., 1991) at 1:10 and mAb 1D4 at 1:750.
Immunohistochemistry and confocal microscopy.
Fixed eyes were embedded and cryosectioned as described previously (Tam et al., 2006). Frozen sections were labeled with mAb 2B2 (Hicks and Molday, 1986) (cell culture supernatant) at 1:10 dilution, or mAb 1D4 at 1:1250, followed by 1:750 dilution of Cy3-conjugated secondary antibody (Jackson ImmunoResearch, West Grove, PA) and counterstained with Alexa 488-conjugated wheat germ agglutinin (Invitrogen, Eugene, OR) and Hoechst 33342 (Sigma-Aldrich, St. Louis, MO) as described previously (Moritz et al., 1999). Sections were imaged using a Zeiss 510 laser-scanning confocal microscope at 10× or 40× (water immersion) objectives.
Results
P23H, but not wild-type forms of X. laevis, bovine, human, and murine rhodopsins, causes retinal degeneration in X. laevis
Transgenic X. laevis expressing X. laevis P23H rhodopsin undergo RD, but the rod death is not light sensitive. Moreover, the vast majority of mutant rhodopsin colocalizes with calnexin, indicating ER retention caused by misfolding (Tam and Moritz, 2006). However, in other animal models expressing murine and human P23H, rod death is exacerbated by light exposure, and the mutant protein is found in the ROS (Naash et al., 1996; Organisciak et al., 2003). Because differences in the primary sequence of rhodopsins of different species could lead to subtle variations in folding and protein stability, we compared the characteristics of wild-type and P23H X. laevis, murine, bovine, and human rhodopsins in transgenic X. laevis retinas. We generated groups of primary transgenic X. laevis expressing wild-type and P23H rhodopsins under control of the X. laevis rod opsin promoter and performed dot blots on retinal extracts from each animal. Each blot was performed in duplicate (results represent the average) and was probed with mAbs recognizing total rhodopsin (B6–30N) or transgenic rhodopsins only (1D4).
Transgenic WT X. laevis (an epitope-tagged version referred to as c-rho), murine, bovine, and human rhodopsins all expressed at a similar range of levels (Fig. 1C). As expected (because each data point represents an independently derived primary transgenic animal), expression levels varied significantly from retina to retina. Of importance is the fact that a relatively high expression level (>10%) was reached in a subset of animals expressing each type of rhodopsin. Moreover, expression of these WT rhodopsins did not cause rod cell death, because decreased total rhodopsin content was not observed (Fig. 1A,E). Histology of frozen sections showed densely packed rods with long ROS, which confirmed the lack of RD (Fig. 1G, top panels). Therefore, X. laevis rods are capable of efficiently expressing rhodopsins derived from many species, with no apparent adverse effects.
Figure 1.
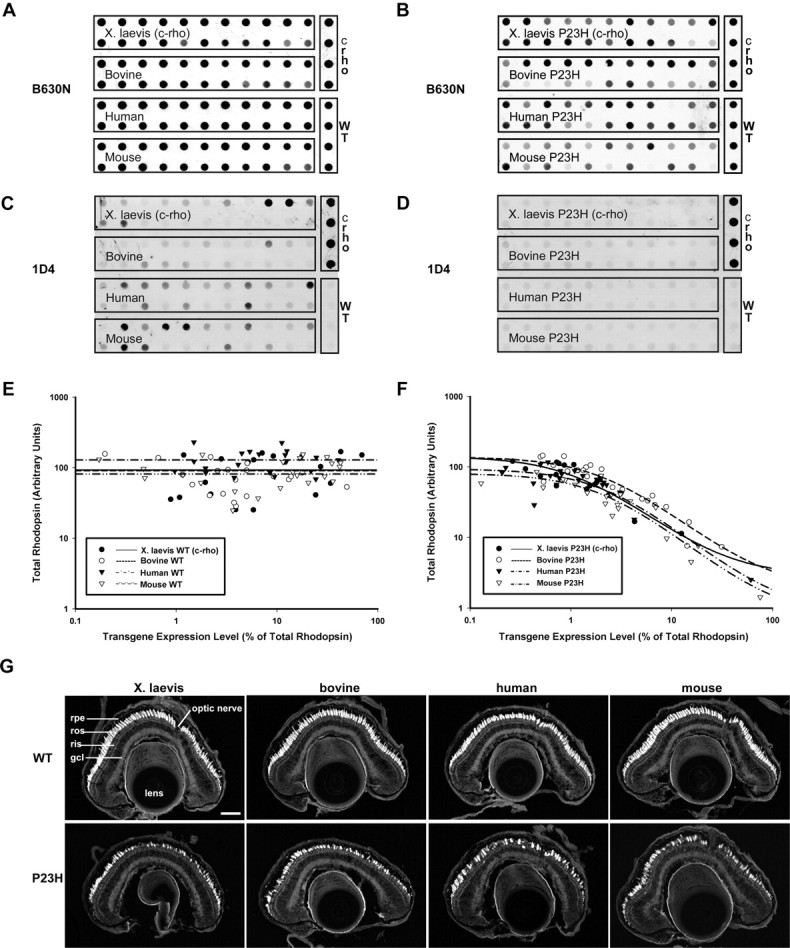
Quantitative and qualitative analysis of X. laevis retinas expressing WT or P23H rhodopsins from different species by dot blot and confocal microscopy. A–D, Samples of eye extracts from primary transgenic X. laevis expressing X. laevis, bovine, human, or mouse wild-type rhodopsins (A, C) or P23H rhodopsins (B, D) were dotted on membranes and probed with mAb B630N (recognizing total rhodopsin) or mAb 1D4 (recognizing transgenic rhodopsin). Samples containing 100% transgenic rhodopsin from transfected HEK293S cells (c-rho) and 100% endogenous wild-type rhodopsin from nontransgenic retinas (WT) were included as standards. Transgenic wild-type rhodopsins were expressed at a wide range of levels, whereas P23H rhodopsins were uniformly expressed at low levels. E, F, Fluorescent signals from dot blots were quantified and used to generate plots of transgenic rhodopsin expression level versus total rhodopsin. Regardless of species, expression of P23H rhodopsins resulted in diminution of endogenous rhodopsin levels indicative of RD, whereas expression of transgenic WT rhodopsins did not correlate with lower endogenous rhodopsin levels. G, Confocal micrographs of cryosections from transgenic retinas expressing WT (top row) or P23H rhodopsins (bottom row) stained with wheat germ agglutinin. The P23H retinas shown have intermediate levels of RD as indicated by both loss of rods and shortened ROS. gcl, Ganglion cell layer; rpe, retinal pigment epithelium. Scale bar, 100 μm.
In contrast, all four forms of P23H rhodopsin expressed at very low levels as seen by the relatively low signal intensity on dot blots (Fig. 1D). Expression levels <1% were tolerated with little or no loss of total rhodopsin, but increasing expression was correlated with decreasing total rhodopsin, indicating RD (Fig. 1B,F). Of the four P23H rhodopsins investigated, the bovine form tended to express at the highest levels (i.e., curve shifted to the right). In Figure 1, E and F, transgenic rhodopsin is presented as a percentage of total rhodopsin to normalize for rod loss because of RD. Thus, even low intensity signals can represent high expression levels in retinas where few rods/ROS remain. Compared with retinas expressing their WT counterparts, the P23H rhodopsins clearly induce RD (loss of rods as seen by gaps between adjacent cells in frozen sections) (Fig. 1G, bottom panels). Retinas exhibiting moderate levels of RD are shown in Figure 1G. These results indicate that all P23H rhodopsins examined cause RD, most likely because of a common attribute of P23H rhodopsins.
RD induced by bovine P23H rhodopsin is rescued by dark rearing
We previously determined that RD in transgenic retinas expressing X. laevis P23H rhodopsin was not light sensitive (Tam and Moritz, 2006). Because bovine P23H rhodopsin displayed greater efficiency of biosynthesis/trafficking, we assessed the light sensitivity of primary transgenic X. laevis expressing bovine P23H rhodopsin by comparing a group of dark-reared animals to a group reared in cyclic light (Fig. 2A–C). Dark rearing completely rescued RD in these animals at 14 dpf as indicated by normal total rhodopsin levels regardless of mutant rhodopsin expression levels (Fig. 2C). In contrast, many animals reared in cyclic light manifested reduced total rhodopsin levels, indicating the presence of RD. Again, the dot blot results were confirmed by histology. Rod loss and shortened ROS were seen in cyclic light but not in constant dark (Fig. 2J). Dark rearing also greatly increased the total amounts of bovine P23H rhodopsin protein detected by mAb 1D4 (Fig. 2B). In some retinas, expression levels of rhodopsin exceeded 10% of total rhodopsin without inducing RD (i.e., comparable with retinas expressing transgenic wild-type rhodopsins).
Figure 2.
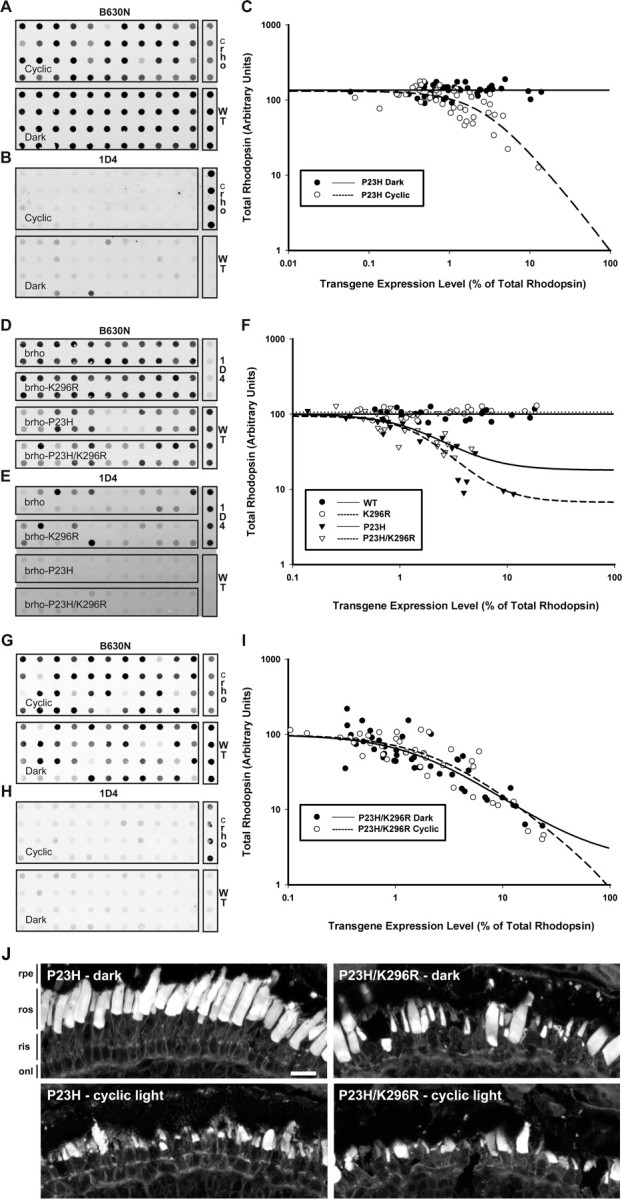
Dark rearing, but not abolishing P23H rhodopsin signal transduction, rescues P23H-induced RD. A, B, Samples of eye extracts from transgenic animals expressing bovine P23H rhodopsin raised in either cyclic light or constant dark were dotted on membranes and probed with mAb B630N (total rhodopsin; A) or mAb 1D4 (transgenic rhodopsin; B). C, Control samples were included as in Figure 1, and used to derive plots of expression level versus total rhodopsin. D–F, Similar dot blots (D, E) and plot (F) were generated using samples of eye extracts obtained from transgenic animals expressing bovine wild-type, K296R, P23H, or P23H/K296R rhodopsin raised in cyclic light. Dark rearing completely rescued P23H-induced degeneration (i.e., higher mutant rhodopsin levels did not correlate with reduced total rhodopsin content), whereas K296R only had a marginal effect. G–I, Dot blots and plot derived from animals expressing bovine P23H/K296R reared in cyclic light or constant darkness. The K296R mutation abolishes the rescuing effect of dark rearing. J, Confocal micrographs of cryosections from transgenic retinas stained with wheat germ agglutinin. Photoreceptors of animals reared in cyclic light expressing bovine P23H and P23H/K296R rhodopsins, or reared in darkness and expressing P23H/K296R rhodopsin, exhibited varying degrees of RD. Photoreceptors of dark-reared bovine P23H retinas did not degenerate, in agreement with the dot blot results. onl, Outer nuclear layer; rpe, retinal pigment epithelium. Scale bar, 20 μm.
Bovine P23H rhodopsin does not cause RD through transducin activation
To determine whether the toxic effects of light exposure in these animals were mediated by P23H rhodopsin signal transduction, we examined a P23H/K296R rhodopsin double mutant. K296R rhodopsin is unable to bind chromophore and does not activate transducin (Cohen et al., 1993), and therefore the double mutant should also be incapable of initiating the signal transduction cascade. We generated four groups of animals expressing bovine wild-type rhodopsin, bovine P23H rhodopsin, bovine K296R rhodopsin, or a bovine P23H/K296R double mutant. Wild-type and K296R rhodopsins were indistinguishable; both expressed at similar levels and neither caused significant degeneration in agreement with our previous observations (Tam and Moritz, 2006; Tam et al., 2006). As before, P23H rhodopsin induced RD. The P23H/K296R double mutant did not significantly rescue RD and therefore was not equivalent to dark rearing (Fig. 2D–F,J). However, comparison of total rhodopsin levels did suggest a subtle rescuing effect of limited statistical significance (Kruskal–Wallis test followed by multiple comparisons test; p = 0.113). We therefore performed a second completely independent experiment, comparing only P23H and P23H/K296R groups (data not shown). We observed a similar trend, again of limited statistical significance (Mann–Whitney test; p = 0.093).
Altogether, these results indicate that light exposure is required for bovine P23H rhodopsin-induced RD, but that light-associated toxicity is not primarily mediated by visual transduction through P23H rhodopsin. Our data suggest, at most, a subtle modulating effect caused by rhodopsin P23H signal transduction.
Rescue by dark rearing requires an intact 11-cis-retinal chromophore binding site
Binding of 11-cis-retinal promotes folding of rhodopsin in cultured cells (Noorwez et al., 2004). Therefore, to determine whether the rescuing effect of dark rearing was mediated by binding of 11-cis-retinal to the mutant protein, we examined the effect of dark versus cyclic light rearing on primary transgenic animals (n = 44 per group) expressing the bovine P23H/K296R rhodopsin double mutant, in which the chromophore binding site is eliminated. Like the single P23H mutant, P23H/K296R caused aggressive retinal degeneration when raised in cyclic light. However, RD was not ameliorated by dark rearing (Fig. 2G–I,J). Furthermore, expression levels of the mutant protein remained low in dark rearing conditions, similar to those observed in animals expressing bovine P23H rhodopsin in cyclic light. Thus, rescue of rod survival in the dark was associated with the capacity of the mutant protein to bind chromophore.
N-terminal- and C-terminal-specific anti-rhodopsin antibodies exhibit different labeling patterns for P23H rhodopsin
We examined the intracellular localization of the four wild-type and four P23H rhodopsins in frozen retinal sections immunolabeled with anti-rhodopsin N-terminal monoclonal antibody mAb 2B2 (Hicks and Molday, 1986) and anti-rhodopsin C-terminal mAb 1D4 (MacKenzie et al., 1984) (Fig. 3). Neither mAb 2B2 nor mAb 1D4 significantly cross-react with the endogenous X. laevis rhodopsin (Tam and Moritz, 2006; Tam et al., 2006).
Figure 3.
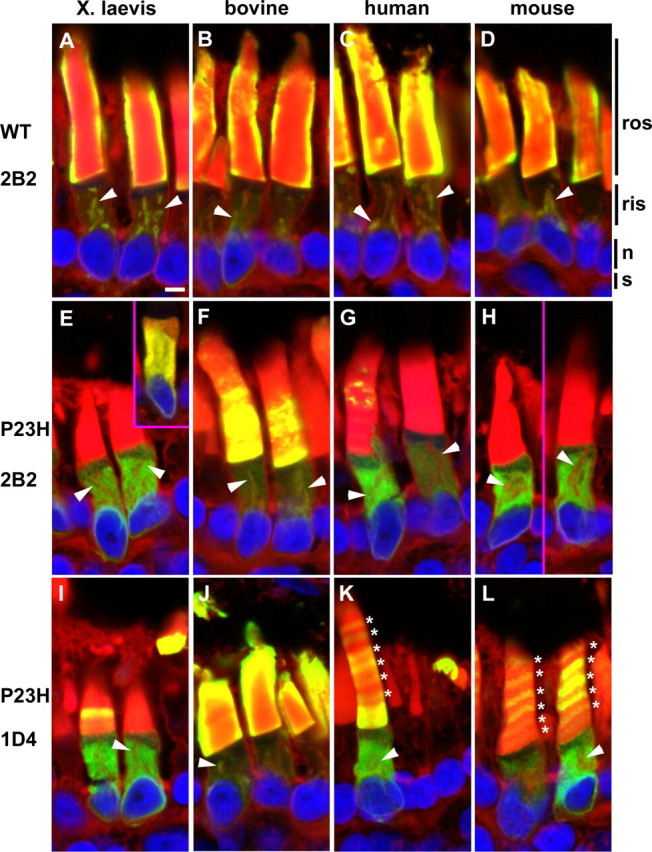
P23H rhodopsins exhibit different cellular localizations depending on the antibody epitope. A–L, Confocal micrographs of cyrosections of retinas expressing transgenic wild-type rhodopsins (A–D), or P23H rhodopsins (E–L). Cryosections were labeled with either mAb 2B2, which recognizes the N terminus of transgenic rhodopsin (A–H), or mAb 1D4 (I–L), which recognizes the C terminus (green), and counterstained with WGA (red) and Hoechst nuclear stain (blue). Overlapping of red and green signals is represented by yellow. Wild-type rhodopsins from all species localized primarily to the ROS as well as Golgi membranes (arrowheads) in the RIS as is normal for rhodopsin (A–D). X. laevis (E), human (G), and mouse (H) P23H rhodopsins localized primarily to the RIS in a diffuse pattern indicative of ER distribution when labeled with mAb 2B2 and colocalized with the ER marker calnexin (E inset; anti-calnexin shown in red). Only occasional mAb 2B2 label was observed in the ROS (G). Bovine P23H rhodopsin (F) also localized to the RIS but significantly more was also in the ROS compared with the other species. mAb 1D4 labeling of P23H rhodopsins (I–L) indicated greater ROS localization than was evident in mAb 2B2 labeling. For bovine P23H rhodopsin (J), very high ROS concentrations of protein were detected as seen by the “halo” of labeling. For human and mouse P23H rhodopsins (K, L), regular spaced bands in the ROS (*) were often observed. n, Nucleus; s, synapse. Scale bar, 5 μm.
Regardless of species, wild-type rhodopsins detected with mAb 2B2 were primarily found in the ROS, with small quantities detectable in the RIS (Fig. 3A–D). RIS labeling was primarily confined to internal membranes consistent with Golgi or rhodopsin transport carriers [as indicated by colocalization with wheat germ agglutinin (Fig. 3, arrowheads)]. ROS labeling typically showed a “haloing” artifact, in which the periphery of the ROS is intensely labeled, but not the interior. This effect has been described previously (Luo et al., 2004; Tam et al., 2006) and is likely caused by extremely dense labeling at the ROS periphery (because of high rhodopsin concentrations combined with dense membrane packing) that physically blocks access of the primary antibody to the central ROS. The wild-type rhodopsin labeling pattern observed with mAb 1D4 (C-terminal epitope) was virtually identical to mAb 2B2 (data not shown). These results indicate that X. laevis rods are capable of proper biosynthesis of wild-type rhodopsins and their normal transport to the ROS.
In contrast, the mAb 2B2 immunolabeling pattern for X. laevis, human, and mouse P23H rhodopsins was primarily confined to the ER and absent from the Golgi, with only very small quantities of antibody label observed in the ROS (Fig. 3E,G,H). ER localization was confirmed by double-label with anti-calnexin (Fig. 3E, inset). In rods expressing bovine P23H rhodopsin, mAb 2B2 labeling was visible in the ER, but there was also a greater proportion of labeling present in the ROS and Golgi (Fig. 3F). However, although this ROS mAb 2B2 labeling is clearly visible, it does not form the “halo” around the periphery of the ROS (as seen with the wild-type rhodopsins). This indicates that the bovine P23H rhodopsin was present in the ROS, but at relatively low levels. This result is in agreement with the quantitative dot blot results.
Unlike the wild-type rhodopsins, the P23H rhodopsins generated a mAb 1D4 immunolabeling pattern that was distinct from that of mAb 2B2. In all cases, after examination of serial sections, mAb 1D4 consistently labeled ROS more extensively (Fig. 3I–L) than mAb 2B2 (Fig. 3E–H). This difference was most pronounced for bovine P23H rhodopsin (Fig. 2F,J) and least apparent for X. laevis P23H rhodopsin (Fig. 3E,I). The effect was intermediate for human (Fig. 3G,K) and mouse (Figure 3H,L) P23H rhodopsin. Interestingly, the mAb 1D4 labeling pattern for human and mouse P23H rhodopsins formed a regular banded pattern suggesting temporal regulation (see below).
Altogether, these results indicate that the majority of the P23H rhodopsin present in the ROS is a truncated form that lacks the N-terminal mAb 2B2 epitope, whereas the protein present in the RIS is full length, because it is recognized by both mAb 2B2 and mAb 1D4. Moreover, the extent of this phenomenon is dependent on the overall amino acid sequence context surrounding the P23H mutation.
The N terminus of bovine P23H rhodopsin is truncated in rods
To confirm whether the N terminus of P23H rhodopsin is truncated, as suggested by our immunohistochemical results, we examined samples of extracts from primary transgenic eyes expressing bovine P23H rhodopsin by Western blot. Duplicate blots (Fig. 4A) were probed with either mAb 1D4 (recognizing the rhodopsin C terminus) (lanes a–d) or mAb A5-3 (Adamus et al., 1991) (recognizing the rhodopsin N terminus) (lanes e–h). Interestingly, very little full-length P23H rhodopsin was present; instead, the predominant species was a truncated rhodopsin that migrated with a relative molecular mass of 26,300 (lanes c and d). This lower molecular mass protein was only detectable using mAb 1D4 (lanes c and d), and not mAb A5-3 (lanes g and h), confirming truncation of the N terminus. The Mr obtained for full-length rhodopsin was 32,700 (lanes b and f), and the Mr for de-glycosylated rhodopsin (PNGaseF treated) was 28,600 (lanes a and e). The ratio of the Mr values for truncated and full-length deglycosylated rhodopsin (0.93) indicates a deletion of 23 amino acid residues (full-length rhodopsin contains 348 residues). In a duplicate analysis, we obtained a value of 30 amino acid residues.
Figure 4.
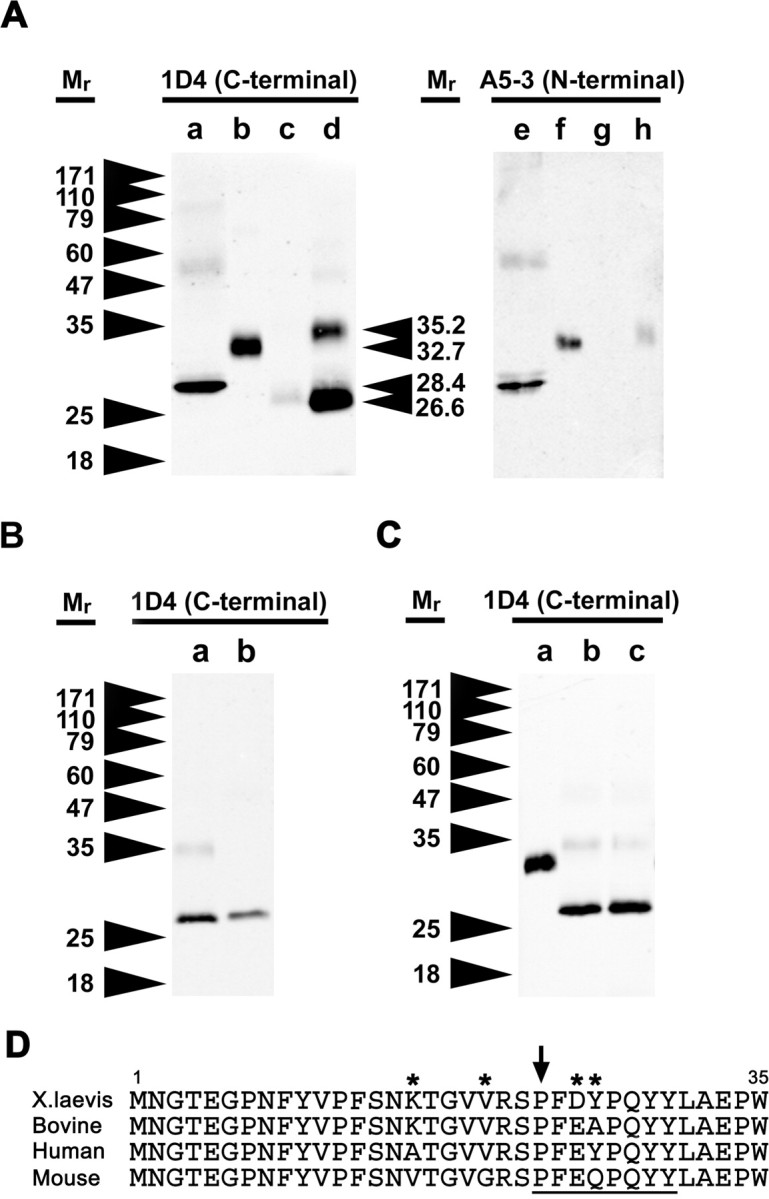
Western blot analysis of bovine wild-type rhodopsin and bovine P23H rhodopsin from transgenic X. laevis eyes indicate that bovine P23H rhodopsin exists primarily in an N-terminally truncated form and is more abundant in dark-reared retinas. A, Western blot analysis was performed using samples of bovine rhodopsin deglycosylated with PNGaseF (a, e) and extracts from transgenic X. laevis eyes expressing bovine wild-type rhodopsin in cyclic light (b, f), bovine P23H rhodopsin in cyclic light (c, g), and bovine P23H rhodopsin in constant dark (d, h). Western blots were probed with mAb 1D4 (recognizing the C terminus; a–d) or mAb A5-3 (recognizing the N terminus; e–h). Retinas expressing bovine P23H rhodopsin contained large quantities of a truncated rhodopsin that was nonreactive with mAb A5-3. B, Western blot of extracts of retinas expressing bovine P23H rhodopsin (a) and N-terminal truncated rhodopsin (ρΔ2–23) (b) demonstrating that the truncated form of bovine P23H rhodopsin is missing at least 23 amino acids from the N terminus. C, Western blot of extracts of retinas expressing bovine wild-type reared in cyclic light (a) and P23H/K296R rhodopsin reared in darkness (b) or cyclic light (c). P23H/K296R rhodopsin is truncated in both dark and cyclic light conditions. D, Sequence alignment of the N-terminal 35 amino acid residues of X. laevis, bovine, human, and mouse rhodopsin transgenes. Amino acid positions that vary among species are indicated by asterisks, and the region encompassing the putative cleavage site is underlined. The P23 residue is indicated by an arrow.
We also expressed a truncated bovine rhodopsin lacking residues 2–23 (rhoΔ2–23) in X. laevis rods (Fig. 4B, lane b). RhoΔ2–23 retains the methionine start codon but lacks both glycosylation sites. The relative mobility of rhoΔ2–23 was very similar to (but slightly larger than) truncated P23H rhodopsin (Fig. 4B, lane a), confirming a deletion of approximately but not <23 residues in this molecule. The sequence of the extracellular N termini of the rhodopsins from the four species is shown in Figure 4D, including the putative cleavage region.
A less abundant higher molecular weight species (Mr 35,200), which was A5-3 reactive and therefore possessed an intact N terminus, was detected in dark-reared retinal samples (Fig. 4A, lane h). This band had a slightly higher Mr than full-length rhodopsin, but not sufficiently high to represent a dimeric form of the truncated protein. The size discrepancy may be attributable to an altered glycosylation pattern of the full-length P23H rhodopsin. EndoH digestion confirmed that the Mr 35,200 band was glycosylated, whereas the lower cleavage product was not glycosylated (data not shown). Therefore, this band likely represents the 2B2 reactive material seen in ROS and RIS by confocal microscopy (Fig. 3F).
N-terminal truncation of P23H rhodopsin does not require chromophore binding
To determine whether truncation of the N terminus of P23H rhodopsin was a direct consequence of a conformational state promoted by binding of the 11-cis-retinal chromophore, we examined retinal extracts derived from animals expressing the bovine P23H/K296R double mutant by Western blot (Fig. 4C). Truncated P23H rhodopsin was the principle form observed regardless of whether the animals were dark (lane b) or cyclic light reared (lane c), indicating that although an intact chromophore binding site was essential for rescue of RD by dark rearing, it was not essential for truncation of the N terminus.
Dark rearing results in significantly increased truncated bovine P23H rhodopsin levels
To quantitatively investigate the effects of light exposure on bovine P23H rhodopsin biosynthesis, we repeated the comparisons between rearing in cyclic light and constant darkness on F1 tadpoles (siblings with identical transgene copy number and integration site). Solubilized eye extracts were quantified by dot blot and Western blot analysis. Total rhodopsin levels in dark-reared bovine P23H animals were the same as their nontransgenic siblings (data not shown) but were significantly higher than in light-reared animals (Fig. 5A) (p = 0.00013 by Mann–Whitney U test), confirming the finding in primary transgenic animals that dark rearing rescues RD.
Figure 5.
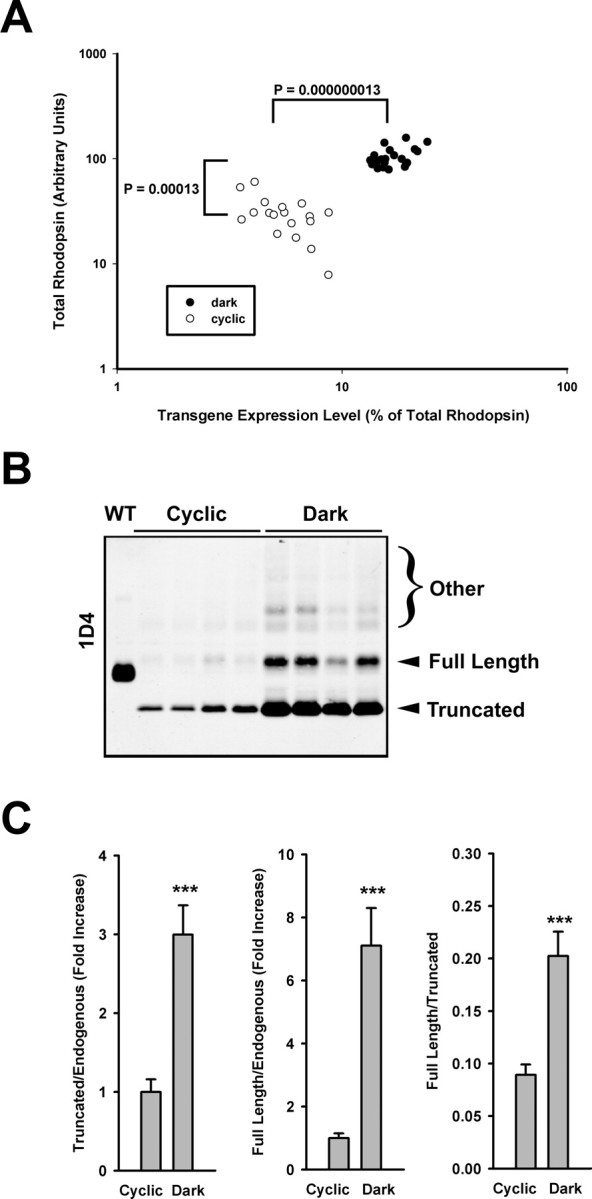
Dark rearing rescues RD in F1 transgenic animals expressing bovine P23H rhodopsin and is associated with an increase in P23H rhodopsin expression levels. A, Retinal extracts of G418-selected F1 offspring were subjected to dot-blot analysis using mAbs 1D4 and B6–30N (as described for Figs. 1 and 3), and total and percentage transgenic rhodopsin levels were similarly determined. Dark rearing rescued total rhodopsin levels in F1 offspring and also resulted in a significant increase in expression levels of the transgenic P23H rhodopsin. B, mAb 1D4 Western blot of samples from the same experiment demonstrating the dramatic increase in total bovine P23H rhodopsin under dark-rearing conditions, consisting primarily of the lower Mr cleaved form. Extracts of four representative eyes from each group are shown, as well as a control expressing wild-type bovine rhodopsin (wt). C, Bar graphs illustrate the differences in relative abundances of cleaved and full-length material (relative to endogenous mAb B630N reactive material and relative to each other) obtained from similar blots with additional samples (n = 14 for each group). All illustrated differences were statistically significant with p < 0.0001 (t test). Error bars are ±SEM.
Bovine P23H rhodopsin increased both in absolute amount and as a relative proportion of total rhodopsin. Absolute transgenic bovine P23H rhodopsin levels increased ∼11 times when animals were reared in the dark (Table 1). Correcting for retinal degeneration [i.e., percentage ratio of transgenic P23H rhodopsin (full length plus truncated) to endogenous rhodopsin], median expression level increased from 5% of total rhodopsin in cyclic light-reared retinas to 16% of total rhodopsin in constant dark-reared retinas (p = 0.000000013; Mann–Whitney U test) (Fig. 5A). Thus, the increase in mutant rhodopsin was not solely attributable to the arrest of cell death (i.e., more rods/ROS therefore more rhodopsin) but was also caused by greater abundance per cell.
Table 1.
Estimated percentage contribution of various rhodopsin species to total rhodopsin in bovine P23H F1 retinas
| Endogenous | Cleaved P23H | Full-length P23H | Other | Total | |
|---|---|---|---|---|---|
| Dark | 86% | 11% | 2.2% | 1.0% | 100% |
| Cyclic (corrected for RD) | 96% | 3.6% | 0.3% | 0.3% | 100% |
| Cyclic (no correction) | 29% | 1.1% | 0.1% | 0.1% | 30% |
To determine the extent to which the full-length and truncated forms of bovine P23H rhodopsin contributed to the total increase observed after dark rearing, we determined the ratios of the Mr 35,200 band (i.e., full length) to the Mr 26,300 band (i.e., truncated) on a Western blot probed with mAb 1D4 (Fig. 5B). Both forms of the mutant rhodopsin increased in the dark relative to endogenous rhodopsin (Fig. 5C, left and middle panels), but the truncated P23H rhodopsin was always far more abundant: 10 times the level of full-length P23H rhodopsin in cyclic light-reared retinas and five times in dark-reared retinas (Fig. 5C, right panel). Based on the results presented in Figure 5, we estimated the relative contributions of various rhodopsin species to total rhodopsin in these F1 animals under conditions of cyclic light and dark rearing (Table 1).
Human P23H rhodopsin behaves similarly to bovine P23H rhodopsin: N-terminal truncation and rescue of RD by dark rearing
We examined the influence of dark rearing on transgenic retinas expressing human P23H rhodopsin to determine whether a similar rescue would occur. We compared total rhodopsin levels from primary transgenic animals (two groups; n = 33), raised either in constant dark or cyclic light. Dark rearing resulted in significant rescue of RD (Mann–Whitney test; p = 0.043) (Fig. 6A). However, the extent of rescue was less complete than that observed in eyes expressing bovine P23H rhodopsin.
Figure 6.
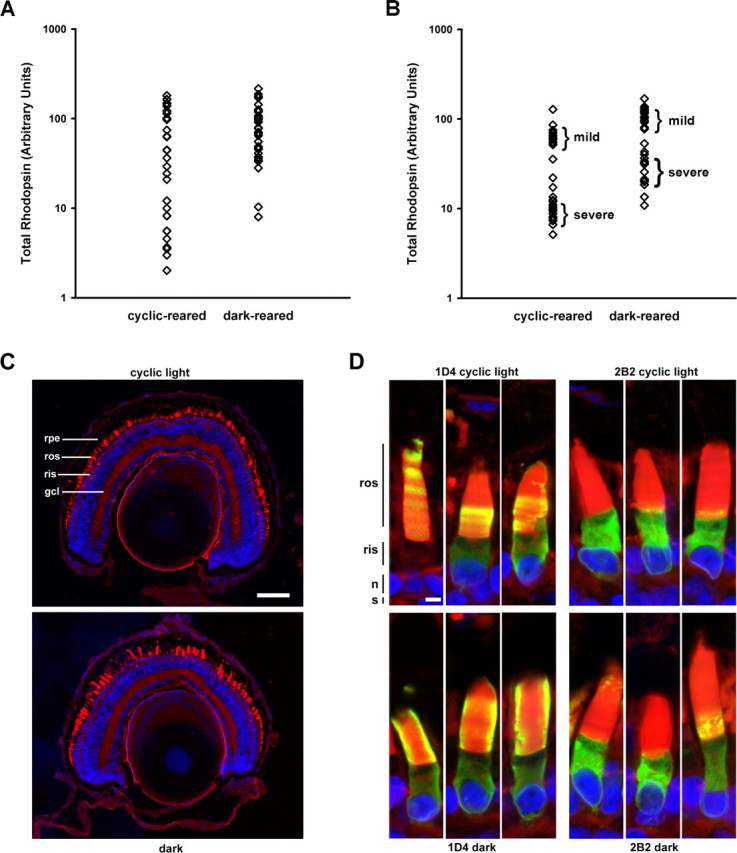
Dark rearing rescues retinas expressing human P23H rhodopsin and is associated with a truncated mutant protein. A, B, Total rhodopsin levels from cyclic light or constant dark-reared primary transgenic animals (A) or F1 transgenic offspring (B) expressing human P23H rhodopsin. Dark-reared retinas on average contained more rhodopsin than their cyclic light-reared counterparts. F1 offspring segregated into two groups, which exhibited either a mild or severe RD phenotype. C, Confocal micrographs of retinal cryosections from F1 offspring from the severe phenotype group. The cyclic light-reared retinas showed greater loss of rods than did the dark-reared retinas. D, Immunolabeling of cryosections with mAbs 2B2 and 1D4. Regardless of light-rearing conditions, mAb 2B2 primarily labeled RIS, whereas mAb 1D4 labeling was significantly present in both RIS and ROS. In cyclic light-reared retinas, the labeling assumed a banded pattern, whereas in dark-reared retinas, continuous labeling along the length of the ROS was observed. WGA (red), Hoechst nuclear stain (blue), mAb 2B2, or 1D4 (green) are shown. gcl, Ganglion cell layer; n, nucleus; s, synapse; rpe, retinal pigment epithelium. Scale bars: C, 100 μm D, 5 μm.
We subsequently verified this effect in dark- and cyclic-reared F1 transgenic offspring (two groups; n = 33) (Fig. 6B). A bimodal distribution of phenotypes was observed in animals that survived G418 selection (severe and mild RD), likely associated with two independent transgene integration sites. Both phenotypes were mitigated by dark rearing, and the results were statistically significant (Mann–Whitney test; p = 0.001). Confocal microscopy of contralateral eyes from F1 transgenic offspring confirmed more extensive rod death in cyclic light-reared retinas than dark-reared retinas (Fig. 6C). We also immunolabeled cryosections of transgenic F1 retinas expressing human P23H rhodopsin with mAb 1D4 and mAb 2B2 (Fig. 6D). In both cyclic light- and dark-reared retinas, mAb 2B2 labeling was very similar (i.e., primarily in the RIS in an ER-like distribution) (Fig. 6D, right panels). In contrast, as well as labeling the ER, mAb 1D4 significantly labeled the ROS (Fig. 6D, left panels). This was even more pronounced in constant dark-reared retinas (Fig. 6D, bottom left panels) compared with cyclic light-reared retinas (Fig. 6D, top left panels). mAb 1D4 intensely labeled the periphery of the ROS in dark-reared retinas indicating dense packing of the transgenic mutant rhodopsin, whereas in cyclic light-reared retinas labeling extended throughout the interior of the ROS indicating low transgenic mutant rhodopsin density. This suggests that although misfolding of human P23H rhodopsin occurred in both light and dark, more truncated P23H rhodopsin exited the ER and was transported to the ROS under dark conditions. The retinas shown in Figure 6 represent the severe phenotype.
In many mAb 1D4 labeled cyclic light-reared retinas expressing mouse (Fig. 2L) or human P23H rhodopsin (Figs. 2K, 6D), we observed a regular pattern of banding in the ROS. The bands occurred with a periodicity of ∼2 μm intervals, which is consistent with the 12 h light/dark cycle and the daily rate of new disc incorporation at the base of X. laevis ROS (Besharse et al., 1977). Regular banding was not observed with dark-reared retinas labeled with mAb 1D4 or cyclic light- or dark-reared retinas labeled with mAb 2B2. Together, these results suggest that the periodic variation in efficiency of transport of P23H rhodopsin to the ROS was coupled to increased abundance of the N-terminally truncated protein in the dark. The regular banding was distinct from the random banding we reported previously, which may be caused by random transgene silencing or “position effect variegation” (Moritz et al., 2001).
Western blot analysis confirmed that human P23H rhodopsin was truncated in transgenic F1 animals (Fig. 7A, lanes g, i) but was present at relatively low levels compared with bovine P23H rhodopsin (Fig. 7A, lane d). Again, there was an increase in the amount of truncated human P23H rhodopsin produced in dark-reared retinas over cyclic light-reared retinas (Fig. 7A, compare lanes g with f and i with h).
Figure 7.

Effects of rhodopsin sequence, expression system, and 11-cis-retinal on proteolytic cleavage of P23H rhodopsin. A, Western blot probed with mAb 1D4 showing samples of nontransgenic X. laevis eye extract (a) and extracts from eyes expressing wild-type bovine rhodopsin (b), bovine P23H rhodopsin (cyclic reared) (c), bovine P23H rhodopsin (dark reared) (d), human wild-type rhodopsin (e), human P23H rhodopsin (F1, mild phenotype, cyclic reared) (f), human P23H rhodopsin (F1, mild phenotype, dark reared) (g), human P23H rhodopsin (F1, severe phenotype, cyclic reared) (h), human P23H rhodopsin (F1, severe degeneration, dark reared) (i), extracts of HEK293S cells expressing bovine P23H rhodopsin (j), or bovine wild-type rhodopsin (k). Lanes a and f–i contain four times as much sample as lanes b–e, and lanes f–k were scanned at a more sensitive setting. HEK293S cells were cultured in the presence of 50 μm 11-cis-retinal. B, Western blot probed with 1D4 showing extracts of HEK293S cells expressing wild-type or P23H bovine rhodopsins cultured in the presence or absence of 50 μm 11-cis-retinal. Arrowheads indicate heterogeneously glycosylated (H), mature glycosylated (M), and truncated (T) rhodopsins. C, Graphical representation of the effects of 11-cis-retinal on the ratio of truncated to mature glycosylated rhodopsins (n = 3 for each condition). Although the overall levels of P23H rhodopsin increased in the presence of 11-cis-retinal, the relative abundance of the truncated product decreased by approximately twofold.
P23H rhodopsin is also truncated in cultured human epithelial cells
To determine whether the proteolytic activity responsible for truncation is unique to rods or present in other cell types, we expressed bovine P23H rhodopsin in HEK293S cells. On a Western blot, a band of the same relative mobility as the truncated bovine P23H rhodopsin from transgenic retinas was detected (Fig. 7A, lane j, band T). This truncated product is similar to the “band D” cleavage product reported previously (Noorwez et al., 2004). Wild-type bovine rhodopsin expressed in HEK293S cells (Fig. 7A, lane j) was cleaved very inefficiently compared with P23H rhodopsin (Fig. 7A, lane k). The predominant species are full-length mature glycosylated (Fig. 7A, band M) and heterogeneously glycosylated (band H). A comparison of bovine and human P23H rhodopsins expressed in X. laevis rods and bovine P23H rhodopsin expressed in HEK293S cells indicated that the cleavage product was identical in size in all cases (Fig. 7A, band T). Thus, cells of both human and X. laevis origin, and both photoreceptors and epithelial cells are capable of truncating P23H rhodopsin.
We examined the effects of the photosensitive rhodopsin chromophore 11-cis-retinal on the expression and relative abundance of the cleaved and full-length forms of P23H rhodopsin produced by HEK293S cells. We transfected HEK293S cells with bovine P23H and wild-type rhodopsins and cultured them in the presence and absence of 11-cis-retinal in the dark. Solubilized cell extracts were analyzed by Western blot (Fig. 7B). 11-cis-retinal increased the yield of mature glycosylated rhodopsin for both P23H and wild-type rhodopsins (band M) and decreased the relative abundance of cleaved P23H rhodopsin (band T) (Fig. 7B,C). Thus, in cultured kidney cells, the availability of 11-cis-retinal during rhodopsin biosynthesis aided full-length P23H rhodopsin in adopting a conformational state capable of exiting the ER.
Discussion
Light-exacerbated RD caused by mutations in the N terminus of rhodopsin occurs in mouse, dog, Drosophila, and rat animal models of RP, as well as human RP patients (Li et al., 1994; Naash et al., 1996; Cideciyan et al., 1998, 2005; Organisciak et al., 2003; Galy et al., 2005; Paskowitz et al., 2006). However, no molecular mechanisms have yet been identified that explain the toxic effects of light. This study provides insights into the molecular events that occur in rods expressing P23H rhodopsin. We demonstrate the following: (1) dark rearing increased ER exit and trafficking of P23H rhodopsins to the ROS and rescued RD; (2) the ability to bind 11-cis-retinal chromophore was essential for this rescue effect; (3) the N terminus of P23H rhodopsin underwent a specific proteolytic cleavage, which eliminated both N-linked glycosylation sites and the H23 residue; (4) increased levels of full-length and truncated P23H rhodopsin were present in dark-reared retinas; and (5) greater rescue correlated with increased abundance of cleaved P23H rhodopsin.
Although P23H rhodopsins from all species examined caused RD, the extent of rescue by dark rearing depended on the primary sequences. All full-length P23H rhodopsins were present at very low levels but were detectable by immunofluorescence using an N terminus-specific antibody (mAb 2B2). Relative to WT rhodopsins, the intracellular distribution of full-length P23H rhodopsins exhibited increased ER label, supporting the hypothesis that P23H rhodopsin misfolds and is retained in the ER where it causes ER stress and cell death. In animals raised in the dark, there was a striking correlation between rescue of RD and the accumulation of an N-terminally truncated form of P23H rhodopsin. In animals expressing X. laevis P23H rhodopsin, neither production of truncated rhodopsin nor rescue was observed. In contrast, in animals expressing bovine P23H rhodopsin, there was abundant truncated rhodopsin and virtually complete rescue of RD. These species-specific differences suggest that rescue was not caused by a general effect of reduced cellular stress in the dark.
Superimposing the K296R mutation on P23H in bovine rhodopsin completely eliminated the rescuing effect of dark rearing. This indicates that interaction between the P23H opsin and the 11-cis-retinal chromophore is required for the enhanced ER exit of P23H rhodopsin observed in darkness. The photoinstability of the chromophore explains the light-dependent nature of P23H rhodopsin-induced RD. In the dark, increased availability of chromophore could stabilize or promote folding of P23H rhodopsin through hydrophobic interactions with amino acid residues that comprise the 11-cis-retinal binding pocket. This theory is supported by in vitro data from this and other studies (Noorwez et al., 2004) in which HEK293 cells produced more full-length P23H rhodopsin when cultured in the presence of 11-cis-retinal. Moreover, administration of vitamin A, an 11-cis-retinal precursor, is beneficial to RP patients (Berson et al., 1993a,b; Berson, 2000). However, light-influenced changes in the expression of chaperones (Li and Guy, 2001; Levitan et al., 2005) or components of the ER-associated degradation (ERAD) pathway may also influence the equilibrium between folding, cleavage and degradation. Interestingly, this result also suggests that 11-cis-retinal is available to opsin in the RIS during biosynthesis and not only in the ROS.
In contrast, light-activated P23H rhodopsin signal transduction did not contribute significantly to the progression of RD. Large amounts of truncated rhodopsin present in ROS were well tolerated by rods; therefore, signaling contributed by truncated P23H rhodopsin was not deleterious to the cell. Moreover, elimination of chromophore binding and transducin activation (P23H/K296R double mutant) resulted in a slight but reproducible rescuing trend, but this effect was not statistically significant. In transducin knock-out mice expressing a murine P23H rhodopsin, RD was ameliorated but not abolished (Samardzija et al., 2006). These results further support P23H rhodopsin misfolding, rather than signal transduction, as the major cause of RD.
Regular banding of mAb 1D4 label (which primarily detects truncated transgenic rhodopsin) was consistently observed in the ROS of cyclic reared rods expressing human P23H rhodopsins, but not with mAb 2B2 (which detects full-length transgenic rhodopsin). Thus, trafficking of truncated human P23H rhodopsin to the ROS cycled up and down over a 24 h period, whereas trafficking of full-length P23H rhodopsin did not. This is consistent with a model in which during periods of darkness, ER exit of truncated rhodopsin increases, whereas the full-length protein may accumulate but does not efficiently leave the ER. The increase in full-length P23H rhodopsin observed in dark-reared retinas may indicate that truncation of the mutant protein is rate limiting for ER exit.
It is possible that truncated rhodopsin represents a stable intermediate in the normal degradative pathway of misfolded rhodopsin. However, this pathway would be unusual in that degradation is initiated from within the secretory pathway, whereas conventional views of ERAD hold that misfolded proteins are retrotranslocated into the cytoplasm before degradation. In this case, 11-cis-retinal may inhibit the initial truncation event, as well as further retrotranslocation and degradation of truncated rhodopsin, resulting in accumulation of both full-length and truncated forms. Alternatively, N-terminal truncation may represent an event that is not required for degradation of misfolded rhodopsin. In this case, 11-cis-retinal may promote production of the full-length abnormally glycosylated intermediate, which may also be targeted for ERAD or inefficiently transported, but which is the substrate of the truncation reaction. After truncation, the mutant rhodopsin is no longer subject to quality control mechanisms and is efficiently transported. Importantly, the truncated product itself does not promote survival, although its production circumvents accumulation of misfolded protein and subsequent ER stress. However, the precise mechanisms underlying this proteolytic pathway remain to be elucidated.
N-terminal truncation of P23H rhodopsin may enhance exit of the mutant protein from the ER and contribute to rescue of RD, although we cannot exclude the possibility that it is a coincident but unrelated event. Several lines of evidence support an active role of truncation in rescue. First, quantification of full-length and truncated bovine P23H rhodopsin show that under all light-rearing conditions, the full-length form remained a minor fraction of the total mutant rhodopsin (present at <20% of truncated levels). Second, there was a strong correlation between the abundance of truncated P23H and extent of rescue for the different P23H rhodopsin species examined (i.e., bovine > human > X. laevis). Third, transport of truncated rhodopsin to the ROS increased in the dark, whereas there was no evidence for increased transport of full-length P23H rhodopsin in the dark. Finally, truncation removes both glycosylation sites and the mutant residue. Thus, it is reasonable to hypothesize that removal of the N terminus of P23H rhodopsin aids in circumventing ER quality control mechanisms associated with glycosylation, or directly contributes to improved folding by removal of H23. The abundance of truncated bovine P23H rhodopsin and the position of the cleavage site argue against this being a superfluous event. However, N-terminal truncation occurred in both cyclic light and dark-reared retinas and in the absence of chromophore binding and thus does not appear to contribute to the photosensitivity of RD. Nevertheless, truncation could act in concert with chromophore binding to enhance rescue by dark rearing.
It will be crucial to identify the protease responsible for truncation of the P23H rhodopsin N terminus. Truncations of P23H rhodopsin have been reported (in abstract form) in transgenic mice as well as in HEK293 cells (Noorwez et al., 2004) (Fig. 7), suggesting that this proteolytic event is relevant in other species and cell types (potentially including human photoreceptors) and not an artifact of X. laevis photoreceptors. Given the relative paucity of mAb 2B2 labeling in the Golgi and ROS, truncation of the N terminus probably occurs in the ER. It is unlikely that the proteosome is responsible for cleavage, because this would first require retrotranslocation of the protein into the cytoplasm. However, limited proteolysis may occur within the ER as has been described for some proteins (Wu et al., 1997; Loo and Clarke, 1998; Bass et al., 2000).
The value of protecting RP patients from light has been debated (Berson, 1971). However, our results suggest a subpopulation of patients, who could be identified by screening for mutations in the N terminus of rhodopsin, may have light-sensitive RD. Protecting these individuals from light, or promoting folding and/or truncation through other means, are potential therapies suggested by our study. Our future research efforts will establish whether these mechanisms influence the progress of RD in human patients with P23H rhodopsin, and RD caused by other rhodopsin mutations.
Footnotes
This work was supported by the Canadian Institutes of Health Research. O.L.M. is a Michael Smith Scholar (Michael Smith Foundation for Health Research) and a W. K. Stell scholar (Foundation Fighting Blindness, Canada). We thank Dr. R. S. Molday for providing mAb 2B2, Dr. P. Hargrave for providing mAbs B630N and A5-3, Dr. D. Deretic for providing mAb 11D5, and Dr. D. Oprian for providing 11-cis retinal and the bovine rhodopsin cDNA.
References
- Adamus G, Zam ZS, Arendt A, Palczewski K, McDowell JH, Hargrave PA. Anti-rhodopsin monoclonal antibodies of defined specificity: characterization and application. Vision Res. 1991;31:17–31. doi: 10.1016/0042-6989(91)90069-h. [DOI] [PubMed] [Google Scholar]
- Bass J, Turck C, Rouard M, Steiner DF. Furin-mediated processing in the early secretory pathway: sequential cleavage and degradation of misfolded insulin receptors. Proc Natl Acad Sci USA. 2000;97:11905–11909. doi: 10.1073/pnas.97.22.11905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berson EL. Light deprivation for early retinitis pigmentosa. A hypothesis. Arch Ophthalmol. 1971;85:521–529. doi: 10.1001/archopht.1971.00990050523001. [DOI] [PubMed] [Google Scholar]
- Berson EL. Nutrition and retinal degenerations. Int Ophthalmol Clin. 2000;40:93–111. doi: 10.1097/00004397-200010000-00008. [DOI] [PubMed] [Google Scholar]
- Berson EL, Rosner B, Sandberg MA, Hayes KC, Nicholson BW, Weigel-DiFranco C, Willett W. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol. 1993a;111:761–772. doi: 10.1001/archopht.1993.01090060049022. [DOI] [PubMed] [Google Scholar]
- Berson EL, Rosner B, Sandberg MA, Hayes KC, Nicholson BW, Weigel-DiFrano C, Willett W. Vitamin A supplementation for retinitis pigmentosa. Arch Ophthalmol. 1993b;111:1456–1459. doi: 10.1001/archopht.1993.01090110014001. [DOI] [PubMed] [Google Scholar]
- Besharse JC, Hollyfield JG, Rayborn ME. Turnover of rod photoreceptor outer segments. II. Membrane addition and loss in relationship to light. J Cell Biol. 1977;75:507–527. doi: 10.1083/jcb.75.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cideciyan AV, Hood DC, Huang Y, Banin E, Li ZY, Stone EM, Milam AH, Jacobson SG. Disease sequence from mutant rhodopsin allele to rod and cone photoreceptor degeneration in man. Proc Natl Acad Sci USA. 1998;95:7103–7108. doi: 10.1073/pnas.95.12.7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cideciyan AV, Jacobson SG, Aleman TS, Gu D, Pearce-Kelling SE, Sumaroka A, Acland GM, Aguirre GD. In vivo dynamics of retinal injury and repair in the rhodopsin mutant dog model of human retinitis pigmentosa. Proc Natl Acad Sci USA. 2005;102:5233–5238. doi: 10.1073/pnas.0408892102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen GB, Yang T, Robinson PR, Oprian DD. Constitutive activation of opsin: influence of charge at position 134 and size at position 296. Biochemistry. 1993;32:6111–6115. doi: 10.1021/bi00074a024. [DOI] [PubMed] [Google Scholar]
- Dryja TP, McGee TL, Reichel E, Hahn LB, Cowley GS, Yandell DW, Sandberg MA, Berson EL. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature. 1990;343:364–366. doi: 10.1038/343364a0. [DOI] [PubMed] [Google Scholar]
- Franke RR, Sakmar TP, Oprian DD, Khorana HG. A single amino acid substitution in rhodopsin (lysine 248-leucine) prevents activation of transducin. J Biol Chem. 1988;263:2119–2122. [PubMed] [Google Scholar]
- Galy A, Roux MJ, Sahel JA, Leveillard T, Giangrande A. Rhodopsin maturation defects induce photoreceptor death by apoptosis: a fly model for RhodopsinPro23His human retinitis pigmentosa. Hum Mol Genet. 2005;14:2547–2557. doi: 10.1093/hmg/ddi258. [DOI] [PubMed] [Google Scholar]
- Hicks D, Molday RS. Differential immunogold-dextran labeling of bovine and frog rod and cone cells using monoclonal antibodies against bovine rhodopsin. Exp Eye Res. 1986;42:55–71. doi: 10.1016/0014-4835(86)90017-5. [DOI] [PubMed] [Google Scholar]
- Illing ME, Rajan RS, Bence NF, Kopito RR. A rhodopsin mutant linked to autosomal dominant retinitis pigmentosa is prone to aggregate and interacts with the ubiquitin proteasome system. J Biol Chem. 2002;28:28. doi: 10.1074/jbc.M204955200. [DOI] [PubMed] [Google Scholar]
- Kaushal S, Khorana HG. Structure and function in rhodopsin. 7. Point mutations associated with autosomal dominant retinitis pigmentosa. Biochemistry. 1994;33:6121–6128. doi: 10.1021/bi00186a011. [DOI] [PubMed] [Google Scholar]
- Kroll KL, Amaya E. Transgenic Xenopus embryos from sperm nuclear transplantations reveal FGF signaling requirements during gastrulation. Development. 1996;122:3173–3183. doi: 10.1242/dev.122.10.3173. [DOI] [PubMed] [Google Scholar]
- Levitan A, Trebitsh T, Kiss V, Pereg Y, Dangoor I, Danon A. Dual targeting of the protein disulfide isomerase RB60 to the chloroplast and the endoplasmic reticulum. Proc Natl Acad Sci USA. 2005;102:6225–6230. doi: 10.1073/pnas.0500676102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QB, Guy CL. Evidence for non-circadian light/dark-regulated expression of Hsp70s in spinach leaves. Plant Physiol. 2001;125:1633–1642. doi: 10.1104/pp.125.4.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ZY, Jacobson SG, Milam AH. Autosomal dominant retinitis pigmentosa caused by the threonine-17-methionine rhodopsin mutation: retinal histopathology and immunocytochemistry. Exp Eye Res. 1994;58:397–408. doi: 10.1006/exer.1994.1032. [DOI] [PubMed] [Google Scholar]
- Loo TW, Clarke DM. Quality control by proteases in the endoplasmic reticulum. Removal of a protease-sensitive site enhances expression of human P-glycoprotein. J Biol Chem. 1998;273:32373–32376. doi: 10.1074/jbc.273.49.32373. [DOI] [PubMed] [Google Scholar]
- Luo W, Marsh-Armstrong N, Rattner A, Nathans J. An outer segment localization signal at the C terminus of the photoreceptor-specific retinol dehydrogenase. J Neurosci. 2004;24:2623–2632. doi: 10.1523/JNEUROSCI.5302-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie D, Arendt A, Hargrave P, McDowell JH, Molday RS. Localization of binding sites for carboxyl terminal specific anti-rhodopsin monoclonal antibodies using synthetic peptides. Biochemistry. 1984;23:6544–6549. doi: 10.1021/bi00321a041. [DOI] [PubMed] [Google Scholar]
- Moritz OL, Tam BM, Knox BE, Papermaster DS. Fluorescent photoreceptors of transgenic Xenopus laevis imaged in vivo by two microscopy techniques. Invest Ophthalmol Vis Sci. 1999;40:3276–3280. [PubMed] [Google Scholar]
- Moritz OL, Tam BM, Papermaster DS, Nakayama T. A functional rhodopsin-green fluorescent protein fusion protein localizes correctly in transgenic Xenopus laevis retinal rods and is expressed in a time-dependent pattern. J Biol Chem. 2001;276:28242–28251. doi: 10.1074/jbc.M101476200. [DOI] [PubMed] [Google Scholar]
- Moritz OL, Biddle KE, Tam BM. Selection of transgenic Xenopus laevis using antibiotic resistance. Transgenic Res. 2002;11:315–319. doi: 10.1023/a:1015612022976. [DOI] [PubMed] [Google Scholar]
- Naash ML, Peachey NS, Li ZY, Gryczan CC, Goto Y, Blanks J, Milam AH, Ripps H. Light-induced acceleration of photoreceptor degeneration in transgenic mice expressing mutant rhodopsin. Invest Ophthalmol Vis Sci. 1996;37:775–782. [PubMed] [Google Scholar]
- Nelson RM, Long GL. A general method of site-specific mutagenesis using a modification of the Thermus aquaticus polymerase chain reaction. Anal Biochem. 1989;180:147–151. doi: 10.1016/0003-2697(89)90103-6. [DOI] [PubMed] [Google Scholar]
- Noorwez SM, Malhotra R, McDowell JH, Smith KA, Krebs MP, Kaushal S. Retinoids assist the cellular folding of the autosomal dominant retinitis pigmentosa opsin mutant P23H. J Biol Chem. 2004;279:16278–16284. doi: 10.1074/jbc.M312101200. [DOI] [PubMed] [Google Scholar]
- Organisciak DT, Darrow RM, Barsalou L, Kutty RK, Wiggert B. Susceptibility to retinal light damage in transgenic rats with rhodopsin mutations. Invest Ophthalmol Vis Sci. 2003;44:486–492. doi: 10.1167/iovs.02-0708. [DOI] [PubMed] [Google Scholar]
- Paskowitz DM, Lavail MM, Duncan JL. Light and inherited retinal degeneration. Br J Ophthalmol. 2006;90:1060–1066. doi: 10.1136/bjo.2006.097436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao RV, Bredesen DE. Misfolded proteins, endoplasmic reticulum stress and neurodegeneration. Curr Opin Cell Biol. 2004;16:653–662. doi: 10.1016/j.ceb.2004.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samardzija M, Wenzel A, Naash M, Reme CE, Grimm C. Rpe65 as a modifier gene for inherited retinal degeneration. Eur J Neurosci. 2006;23:1028–1034. doi: 10.1111/j.1460-9568.2006.04639.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam BM, Moritz OL. Characterization of rhodopsin P23H-induced retinal degeneration in a Xenopus laevis model of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2006;47:3234–3241. doi: 10.1167/iovs.06-0213. [DOI] [PubMed] [Google Scholar]
- Tam BM, Xie G, Oprian DD, Moritz OL. Mislocalized rhodopsin does not require activation to cause retinal degeneration and neurite outgrowth in Xenopus laevis. J Neurosci. 2006;26:203–209. doi: 10.1523/JNEUROSCI.3849-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Sakata N, Lele KM, Zhou M, Jiang H, Ginsberg HN. A two-site model for ApoB degradation in HepG2 cells. J Biol Chem. 1997;272:11575–11580. doi: 10.1074/jbc.272.17.11575. [DOI] [PubMed] [Google Scholar]