

Abstract
The cellular pathways of motor neuronal injury have been investigated in the SOD1 G93A murine model of familial amyotrophic lateral sclerosis (ALS) using laser-capture microdissection and microarray analysis. The advantages of this study include the following: analysis of changes specifically in motor neurons (MNs), while still detecting effects of interactions with neighboring cells; the ability to profile changes during disease progression, an approach not possible in human ALS; and the use of transgenic mice bred on a homogeneous genetic background, eliminating the confounding effects arising from a mixed genetic background. By using this rigorous approach, novel changes in key cellular pathways have been detected at both the presymptomatic and late stages, which have been validated by quantitative reverse transcription-PCR.
At the presymptomatic stage (60 d), MNs extracted from SOD1 G93A mice show a significant increase in expression of genes subserving both transcriptional and translational functions, as well as lipid and carbohydrate metabolism, mitochondrial preprotein translocation, and respiratory chain function, suggesting activation of a strong cellular adaptive response. Mice 90 d old still show upregulation of genes involved in carbohydrate metabolism, whereas transcription and mRNA processing genes begin to show downregulation. Late in the disease course (120 d), important findings include the following: marked transcriptional repression, with downregulation of multiple transcripts involved in transcriptional and metabolic functions; upregulation of complement system components; and increased expression of key cyclins involved in cell-cycle regulation. The changes described in the motor neuron transcriptome evolving during the disease course highlight potential novel targets for neuroprotective therapeutic intervention.
Keywords: amyotrophic lateral sclerosis, microarray, laser-capture microdissection, transcriptome, motor neurons, clinical intervention
Introduction
Amyotrophic lateral sclerosis (ALS) is one of the most common adult-onset neurodegenerative diseases, characterized by progressive and relatively selective degeneration of upper and lower motor neurons, leading to progressive atrophy and weakness of skeletal muscles and eventually death, which usually results from respiratory muscle failure. ALS occurs in both sporadic and familial forms, clinically indistinguishable, with an incidence of 1–2 per 100,000 individuals. Approximately 10% of cases are familial, and 20% of these have been linked to a dominant mutant form of the Cu/Zn superoxide dismutase 1 (SOD1) enzyme (Rosen et al., 1993).
Transgenic mice carrying mutant forms of SOD1 develop a neuromuscular disease very similar to human ALS in both phenotype and histopathological hallmarks, such that mutant SOD1 mice are considered a reliable model for ALS and have been widely used in ALS research. Apart from the 2% of cases carrying an SOD1 mutation, the cause(s) of ALS is still primarily unknown, and the high proportion of sporadic cases suggests that this disease is multifactorial in origin.
Although extensive studies have been undertaken to understand disease pathogenesis in ALS, the causes of selective degeneration of motor neurons are still incompletely understood. Multiple different mechanisms have been shown to contribute to motor neuron injury in ALS, including mitochondrial dysfunction, oxidative stress, glutamate excitotoxicity, disrupted axonal transport, and inflammation (Cleveland and Rothstein, 2001; Dupuis et al., 2004a; Shaw, 2005; Browne et al., 2006). Whether these mechanisms are related, and at which stage of the disease they become operational, is still controversial. The use of murine mutant SOD1 models has indicated that the development of the pathology requires interaction between motor neurons and other cell types (Boillee et al., 2006). In fact, the targeted expression of mutant SOD1 either in motor neurons (Pramatarova et al., 2001; Lino et al., 2002) or in astrocytes (Gong et al., 2000) does not provoke an ALS phenotype. Clearly, it is important to study motor neuron injury in ALS as it occurs in the complex environment of interaction with neighboring cells in the adult CNS. Obtaining a complete picture of which biochemical pathways are altered within degenerating motor neurons and pinpointing at which stage of the disease these changes happen represents an interesting challenge, which may be helpful in the identification of targets for therapeutic intervention at a stage at which dysfunctional motor neurons may still be salvageable.
Microarray analysis has been applied to the study of ALS to investigate gene expression in whole spinal cord homogenates of SOD1 G93A mice and human ALS cases, although the massive presence of glial cells and inflammatory factors has made it difficult to define which gene expression changes were motor neuron specific (Malaspina et al., 2001; Olsen et al., 2001; Yoshihara et al., 2002). Recently, laser-capture microdissection (LCM), combined with microarray analysis, has allowed the identification of motor neuron-specific changes in gene expression in human ALS cases (Jiang et al., 2005).
The aim of the present study is to combine LCM and microarray analysis to study how motor neurons in the spinal cord of transgenic SOD1 G93A mice and transgenic wild-type (WT) SOD1 respond to stimuli determined by the presence of the human mutant protein throughout the evolution of the stages in motor neuron injury, which is impossible when using human postmortem tissue. Evaluation of the mouse model of ALS has key advantages because it allows the CNS to be analyzed at different stages of disease and without the confounding effects of biological alterations attributable to postmortem delay from death until freezing of the tissue. Our mouse model, moreover, compared with the ones used in previous studies, has the advantage of being bred on a homogeneous background. This reduces the variability in the identification of key stages in disease progression by ∼50% compared with results obtained from mice with a mixed genetic background (Heiman-Patterson et al., 2005), giving more consistent and reliable results and reducing the likelihood of spurious gene expression differences emerging from microarray analysis.
In this study, particular interest has been directed to the presymptomatic stage when the earliest transcriptional alterations occur and novel changes are reported in the motor neuron transcriptome before the onset of clinical signs of motor weakness. The presence of both impaired and enhanced functions at this presymptomatic stage indicate potential pathways involved in the earliest stages of motor neuron injury, as well as the attempted compensatory response by motor neurons to the cellular stress induced by the presence of mutant human SOD1 (hSOD1).
Materials and Methods
Murine SOD1 G93A transgenic mouse model.
Transgenic mice B6SJL-Tg (SOD1-G93A) 1 Gur/J high copy number were backcrossed onto C57BL/6J Ola-Hsd (Harlan UK, Bicester, UK) for >20 generations to eliminate the SJL dysferlin mutation (Bittner et al., 1999) and to achieve a homogeneous genetic background more suitable for microarray studies. This homogeneous background reduces the variability in the end point of the disease by 50% compared with the variability published for the mixed background (Heiman-Patterson et al., 2005). Male mice overexpressing the human mutant SOD1 G93A (SOD1 G93A), and their gender-matched nontransgenic littermates (LM) (G93A LM), and transgenic mice overexpressing hSOD1 WT, and their nontransgenic LM (WT LM) were used. Brain and spinal cord were collected at three different stages of disease: presymptomatic (60-d-old mice), symptomatic (90-d-old mice), and late (120-d-old mice). All the experiments on mice were performed according to United Kingdom Home Office regulations.
Sectioning and tissue staining for LCM.
Mice were perfused with a 30% sucrose solution, and the CNS was dissected and frozen. Lumbar spinal cord frozen sections (10 μm) were fixed (70% ethanol, 30 s), washed (DEPC-treated water, 30 s), and stained with toluidine blue (1 min). They were then washed and dehydrated through graded ethanol concentrations (70, 90, and 100%) and xylene.
Microdissection and RNA quality control.
From each animal, 1000 motor neurons were isolated using a PixCell II laser-capture microdissection system (Arcturus Bioscience, Mountain View, CA). RNA was extracted using the PicoPure RNA Extraction kit according to the manufacturer's protocol (Arcturus) and typically yields 60 ng of RNA. The quantity (NanoDrop 1000 spectrophotometer) and the quality (2100 bioanalyzer, RNA 6000 Pico LabChip; Agilent, Palo Alto, CA) of this starting RNA were analyzed, and samples were only taken forward if the quality was satisfactory as indicated by the absence of degradation of the ribosomal RNA (representing 90–95% of the total RNA).
Amplification and microarray hybridization.
Linear amplification of RNA was performed following Eberwine's procedure (Van Gelder et al., 1990) using the RiboAmp Amplification kit (Arcturus). Briefly, this method uses an oligo-dT primer containing a T7 polymerase promoter site. After generation of double-stranded cDNA, copy RNA (cRNA) is transcribed, and this then forms the RNA template for a second round of amplification. At the end of this round, after the synthesis of double-stranded cDNA, in vitro transcription produces biotin-labeled cRNA (GeneChip Expression 3′-Amplification reagents for in vitro transcription labeling; Affymetrix, Santa Clara, CA), which can then be hybridized to the GeneChip. On average, 60 ng of RNA produced 50 μg of labeled cRNA after two rounds of linear amplification. The amplified cRNA was then analyzed for quality (Agilent 2100 Bioanalyzer, RNA 6000 Nano LabChip) and quantity (NanoDrop 1000 Spectrophotometer). Following the manufacturer's protocol, 10 μg of cRNA was fragmented (GeneChip reagents; Affymetrix) and hybridized to Mouse Genome 430A GeneChip (Affy MOE430A). Twelve chips (three SOD1 G93A vs three G93A LM and three SOD1 WT vs three WT LM) were hybridized at each time point according to Affymetrix protocols. After overnight hybridization at 42°C, the GeneChips underwent stringency washes in a GeneChip Fluidics Station 400 (Affymetrix) and were scanned with a laser at high resolution (GeneChip Scanner 3000; Affymetrix). The results were analyzed initially using GeneChip operating software, which automatically acquires and analyzes image data and computes an intensity value for each transcript. The data were subsequently processed using ArrayAssist (Iobion Informatics, La Jolla, CA) to statistically analyze changes in gene expression in the presence of the SOD1 G93A transgene or of the WT hSOD1 transgene. Transcripts were defined as differentially expressed between the transgenic mice and the littermate controls if there was a twofold or greater difference in the gene expression level, plus a p value of <0.05. The statistical test applied by the ArrayAssist program was an unpaired two-tailed t test.
Data analysis.
Differentially expressed probes were classified according to GeneOntology terms (GeneOntology allows classification of genes according to their molecular function, biological process, cellular component, and chromosomal localization). To identify specific pathways involved in the development of the disease, PathwayArchitect (Iobion Informatics) and GenMAPP 1.1 were also used.
Real-time PCR.
RNA was extracted from ∼3500 cells, microdissected as described previously from the spinal cord of six male mice, three SOD1 G93A and three nontransgenic LM, at the presymptomatic stage. Typical yields contained ∼200 ng of RNA. After quantitative and qualitative analysis, 150 ng of RNA was reverse transcribed to cDNA using Superscript II reverse transcriptase according to the manufacturer's protocol (Invitrogen, San Diego, CA). Samples were diluted to the concentration of 12.5 ng/μl and used for quantitative reverse transcription-PCR (Q-PCR). The remaining 50 ng of RNA was amplified using the two-cycle linear amplification protocol from the RiboAmp Amplification kit (Arcturus) as described previously. Further quantitative and qualitative analysis followed the amplification process, and these amplified samples were diluted to the concentration of 12.5 ng/μl for Q-PCR.
Necdin (Ndn), malate dehydrogenase 1 (Mdh1), and cyclin I (Ccni) were chosen to validate the consistency of results obtained by real-time PCR using both amplified and unamplified RNA extracted from motor neurons from G93A mice and LMs at the presymptomatic stage.
Because of the reduced numbers of motor neurons present in the spinal cord of mice at the symptomatic and late stages of the disease, RNA was collected from 1000 motor neurons from six SOD1 G93A mice, three per stage, and their LM, and the RNA was amplified by two cycles of amplification, as described previously, for use in Q-PCR.
Primers were designed to validate significant changes, reported by microarray results, in the expression of selected genes (Table 1). These genes were chosen for further analysis because of their primary role in pathways of particular interest for the development of the disease. Primer concentrations were optimized by testing seven different conditions, combining differing forward and reverse primer concentrations, using 3 μg of mouse universal RNA (Stratagene, La Jolla, CA) reverse transcribed to cDNA using Superscript II reverse transcriptase.
Table 1.
Real-time PCR primer sequences and optimized concentrations
| Gene name | Gene symbol | Primer sequences | Concentrations |
|---|---|---|---|
| ATP synthase δ subunit | Atp5d | F, 5′ CTCCTCTGTGCAGTTACTAGCTGAA 3′ | 600 nm |
| R, 5′ ACTGCGCCTTCTCCAGGTT 3′ | 600 nm | ||
| ATP synthase γ polypeptide 1 | Atp5c1 | F, 5′ GAAGGAGTCCACCACCAGTGA 3′ | 150 nm |
| R, 5′ CGTTCTTGCTGGCGTTGTC 3′ | 150 nm | ||
| Complement component 1 q subcomponent | C1qb | F, 5′ CACCAACGCGAACGAGAACT 3′ | 600 nm |
| β polypeptide | R, 5′ GGCCAGGCACCTTGCA 3′ | 600 nm | |
| Cyclin D2 | Ccnd2 | F, 5′ GCTCTGTGCGCTACCGACTT 3′ | 300 nm |
| R, 5′ CCACGCTTCCAGTTGCAAT 3′ | 150 nm | ||
| Cyclin E2 | Ccne2 | F, 5′ TTTTACCTCCATTGAAGTGGTTAAGA 3′ | 600 nm |
| R, 5′ GCACCATCCAGTCTACACATTCC 3′ | 600 nm | ||
| Cyclin I | Ccni | F, 5′ CACACGGCTACACCATTGGA 3′ | 300 nm |
| R, 5′ AAAAGTAACTGAGGCCTAGCTGACA 3′ | 600 nm | ||
| Dynein cytoplasmic light chain 1 | Dnclc1 | F, 5′ GGCCCATATCAAGAAGGAGTTTG 3′ | 300 nm |
| R, 5′ CATGTGTCACATAACTACCGAAGTTTC 3′ | 300 nm | ||
| Kinesin 1b | Kif1b | F, 5′ GGAAGGTCTGCTGAATGCTTTT 3′ | 300 nm |
| R, 5′ AATAATCTCTCGGCTGACGATCTC 3′ | 150 nm | ||
| Kinesin 5b | Kif5b | F, 5′ GAGTGCAATGTTGATGTCCTAGAAG 3′ | 300 nm |
| R, 5′ ACAGTGAAGACACGAAGGACACA 3′ | 300 nm | ||
| Malate dehydrogenase 1 | Mdh1 | F, 5′ CCATCGCAGACCACATCAGA 3′ | 150 nm |
| R, 5′ AACACCCATCGACACGAACTC 3′ | 150 nm | ||
| Necdin | Ndn | F, 5′ GCAGTGTCTCTGAGGACTAAAAAGGT 3′ | 300 nm |
| R, 5′ GACTCAACCCCACCCTTACACA 3′ | 600 nm | ||
| Plexin domain containing 1 | Plxdc1 | F, 5′ CCACAGAAGATGACACCAAGTTG 3′ | 300 nm |
| R, 5′ ACCCTTGGACTTCGGAGATGA 3′ | 300 nm | ||
| Glyceraldehyde-3-phosphate dehydrogenase | Gapdh | F, 5′ GCTACACTGAGGACCAGGTTGTCT 3′ | 300 nm |
| R, 5′ AGCCCCGGCATCGAA 3′ | 300 nm |
F, Forward; R, reverse.
Q-PCR was performed using 12.5 ng of cDNA, 1× SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA), and concentrations of forward and reverse primers as just described, to a total volume of 20 μl. After an initial denaturation at 95°C for 10 min, templates were amplified by 40 cycles of 95°C for 15 s and 60°C for 1 min, on an MX3000P Real-Time PCR System (Stratagene). At the end, a dissociation curve was generated to ensure amplification of a single product and absence of primer dimers.
After defining the optimal final primer concentration for each of the genes (Table 1), a standard curve was generated to determine the efficiency of the PCR over a wide range of template concentrations, from 25 to 0.3 ng/μl. The efficiency of the PCR for every set of primers was 95 ± 5%, such that gene expression values, normalized to Gapdh expression, could be determined using the ddCt calculation (ABI PRISM 7700 Sequence Detection System protocol; Applied Biosystems). Gene expression levels for each gene in G93A mice are shown as relative concentrations. The unpaired two-tailed t test was used to analyze the data and to determine the statistical significance of any differences in gene expression [GraphPad (San Diego, CA) Prism 5].
Gapdh was chosen as a housekeeping gene to normalize the Q-PCR values because the microarray analysis showed that its expression was stable at every stage of the disease within the transgenic mice, and the expressed values obtained from motor neurons of SOD1 G93A mutant mice were comparable to those obtained for the corresponding control mice.
Results
Mice
Transgenic mice C57BL/6J (SOD1-G93A) carry ∼20 copies of transgenic G93A hSOD1 and develop a neurodegenerative disease reproducing both phenotypic and histopathologic characteristics of human ALS. Briefly, these mice at birth appear identical to their nontransgenic LM and are identified by PCR screening for hSOD1, using DNA extracted from tail biopsies. The presymptomatic stage lasts for ∼75 d, after which the mice start to develop tremors and loss of the hindlimb splaying reflex. Their disease course progresses quickly, leading to reduction in gait length and impairment of motor performance on rotarod, with progressive paralysis initially of the hindlimbs, culminating in total paralysis and death at ∼140 d (SD, ±6). This disease course dictated the time points chosen for this study: 60 d was chosen as the presymptomatic stage, 90 d was chosen as the symptomatic stage, and 120 d was chosen as the late stage of the disease.
Microarray analysis
The transcription profiles of motor neurons isolated using LCM from lumbar spinal cords from three male SOD1 G93A mice and three male nontransgenic G93A LM at 60, 90, and 120 d were generated using the murine GeneChip Mouse Genome 430A (Affy MOE430A). Between 22 and 27% of the 22,000 probes arrayed on the GeneChip were detected as present in the isolated motor neurons of the mice at each time point.
To identify genes differentially expressed in the presence of SOD1 G93A, comparisons were made between the transcription profiles of motor neurons from transgenic mice expressing the mutant protein (G93A) and nontransgenic littermate mice (G93A LM) for each age group using the analysis software ArrayAssist version 4.0. Transcripts were defined as significantly differentially expressed if the expression levels in the SOD1 G93A mice showed a fold change of ≥2 with a p value of <0.05.
To identify genes differentially expressed in the presence of SOD1 WT, the transcription profiles of motor neurons isolated using LCM from lumbar spinal cords from three male SOD1 WT mice and three male WT LM at 60, 90, and 120 d were generated using the same microarray chips used for the SOD1 G93A mice (Affy MOE430A). Between 23 and 30% of the 22,000 probes arrayed on the GeneChip were detected as present in the six groups (SOD1 WT and WT LM for each time point) of isolated motor neurons. During the analysis, the same criteria used for selecting the differentially expressed genes in the SOD1 G93A mice were applied.
Genes showing significantly altered expression levels in SOD1 G93A mice, either at more than one time point or in a key pathway of particular interest for the development of the disease, were selected for further investigation by Q-PCR (Table 2).
Table 2.
Real-time PCR results for validation
| Gene | 60 d |
90 d |
120 d |
|||
|---|---|---|---|---|---|---|
| Microarray fold change | Q-PCR fold change | Microarray fold change | Q-PCR fold change | Microarray fold change | Q-PCR fold change | |
| Atp5d | 2.28 down | 1.2 down (p = 0.01) | 1.9 down | No change | No change | No change |
| Atp5c1 | 5.94 up | 1.78 up (p = 0.0036) | No change | 1.3 down (p = 0.3) | No change | 1.3 down (p = 0.2) |
| C1qb | No change | No change | No change | 7.37 up (p = 0.07) | 5.25 up | 8.03 up (p = 0.0098) |
| Ccnd2 | No change | 2 up (p = 0.01) | No change | 1.6 up (p = 0.1) | 4.3 up | 2 up (p = 0.03) |
| Ccne2 | No change | Up | No change | No change | 1.9 up | Up |
| Ccni | No change | 3.8 up (p = 0.04) | 2.92 up | 1.7 up (p = 0.1) | 2.12 up | 1.5 up (p = 0.03) |
| Dnclc1 | No change | No change | 2 down | 1.4 down (p = 0.6) | 2.61 down | 1.3 down (p = 0.3) |
| Kif1b | 5.16 up | 2 up (p = 0.09) | No change | 1.5 up (p = 0.04) | 3.72 up | 1.6 up (p = 0.05) |
| Kif5b | 2.08 up | 2 up (p = 0.01) | No change | No change | No change | No change |
| Ndn | 4.62 up | 2 up (p = 0.04) | No change | No change | 3.02 down | 1.2 down (p = 0.003) |
| Mdh1 | 2.68 up | 1.32 up (p = 0.3) | No change | 1.8 up (p = 0.2) | 2.93 down | No change |
| Plxdc1 | 2.54 down | 22 down (p = 0.02) | 2.98 down | 260 down (p = 0.003) | 3.04 down | 500 down (p = 0.001) |
Comparison between amplified and unamplified RNA
To validate the consistency of results obtained by real-time PCR using both amplified and unamplified RNA extracted from motor neurons, Necdin (Ndn), malate dehydrogenase 1 (Mdh1), and Cyclin I (Ccni) mRNA levels were investigated before and after the amplification process in G93A and G93A LM at the 60 d time point. The Q-PCR results show consistency of variation in these three probes before and after amplification. At this age, in fact, Ndn and Ccni are significantly upregulated in both amplified (Ndn fold change, 2; p = 0.04; Ccni fold change, 3.8; p = 0.04) and unamplified (Ndn fold change, 1.6; p = 0.01; Ccni fold change, 3; p = 0.05) material, whereas Mhd1 does not reach significance in either sample when analyzed by Q-PCR (p values for amplified and unamplified material are, respectively, 0.37 and 0.31). This consistency of results between amplified and unamplified RNA confirmed the reliability of the amplification process and enabled us to complete the real-time PCR experiments using amplified RNA, thereby overcoming the issue of low RNA yields resulting from depletion of the numbers of motor neurons from the 90 and 120 d mice.
Motor neurons from SOD1 WT mice
The comparison between SOD1 WT mice and the WT LM showed very few changes for each time point, and none of the significant changes in gene expression was present at more than one time point (Table 3). At 60 d, of the 13 genes showing altered expression, three are also altered at the same age in the SOD1 G93A mice. Two of them, F-box and WD-40 domain protein 11 (Fbxw11) and translocation protein 1 (Tloc1), show expression changes in opposite directions in WT compared with SOD1 G93A mice. However, proteolipid protein (myelin) 1 (Plp1) is upregulated in both genotypes and therefore has been excluded from further investigation.
Table 3.
Genes significantly altered at 60, 90, and 120 d in motorneurons expressing the WT SOD1
| Probe identification | Gene | Fold change |
|---|---|---|
| 60 d | ||
| 1416345_at | Translocase of inner mitochondrial membrane 8 homolog a (yeast) | 2.05 |
| 1416500_at | SAC1 (supressor of actin mutations 1, homolog)-like (Saccharomyces cerevisiae) | 2.34 |
| 1424455_at | G-protein-coupled receptor-associated sorting protein 1 | −2.5 |
| 1424918_at | TBC1 domain family, member 19 | 3.01 |
| 1425468_at | Proteolipid protein (myelin) 1 | 2.12 |
| 1427923_at | Zinc metalloproteinase, STE24 homolog (S. cerevisiae) | 2.65 |
| 1435898_x_at | GDP dissociation inhibitor 3 | 2.54 |
| 1437468_x_at | F-box and WD-40 domain protein 11 | −2.07 |
| 1452141_a_at | Selenoprotein P, plasma, 1 | 2.55 |
| 1454697_at | Translocation protein 1 | −2.54 |
| 1455422_x_at | Septin 4 | 2.76 |
| 1456015_x_at | 2.87 | |
| 1456360_at | RIKEN cDNA 1700022C02 gene | 2.45 |
| 90 d | ||
| 1416852_a_at | Neurochondrin | −2.04 |
| 1419738_a_at | Tropomyosin 2 β | −2.11 |
| 1422156_a_at | Ribosomal protein S2 | −2.16 |
| 1449702_at | Zinc finger AN1-type domain 2A | −2.13 |
| 120 d | ||
| 1449434_at | Carbonic anhydrase 3 | 6.87 |
| 1427445_a_at | Titin | 2.45 |
| 1417464_at | Troponin C2 fast | 3.12 |
| 1427026_at | Myosin heavy polypeptide 4 skeletal muscle | 8.90 |
| 1427520_a_at | Myosin heavy polypeptide 1 skeletal muscle adult | 8.29 |
| 1434326_x_at | Coronin actin-binding protein 2B | 2.35 |
| 1437044_a_at | Glucosidase β acid | 2.32 |
| 1439429_x_at | Deltex 2 homolog (Drosophila) | 2.38 |
| 1434893_at | ATPase Na+/K+ transporting α2 polypeptide mRNA (cDNA clone MGC:36347 IMAGE:4955003) | −2.13 |
| 1428289_at | Kruppel-like factor 9 | −2.01 |
| 1433785_at | Myelin-associated oligodendrocytic basic protein | −3.08 |
| 1456084_x_at | Fibromodulin | −2.36 |
| 1424635_at | Eukaryotic translation elongation factor 1 α 1 | −2.72 |
| 1426195_a_at | Cystatin C | −2.03 |
| 1416454_s_at | Actin α2 smooth muscle aorta | −2.11 |
| 1455546_s_at | Splicing factor 3a subunit 2 | 2.36 |
None of the probes whose expression was altered in motor neurons expressing SOD1 WT at 90 and 120 d were altered in motor neurons carrying SOD1 G93A. At 90 d, the number of differentially expressed genes between SOD1 WT mice and their LM is only four, and, categorized by function, they do not belong to any specific class (Table 3).
At 120 d, the number of genes showing altered expression is 16, with an increase in myosin heavy polypeptide 1 and 4 mRNA levels, which are not altered in SOD1 G93A mice (Table 3).
Motor neurons from SOD1 G93A mice at the 60 d (presymptomatic) time point
The analysis of SOD1 G93A mice at 60 d showed a significant change in the transcription profile of 252 genes when comparing SOD1 G93A mutant mice with their nontransgenic LM (Table 4). Two hundred thirty-four transcripts were increased in the presence of mutant SOD1 G93A, and 18 were decreased. Differentially expressed genes have been categorized according to their molecular function and include genes involved in transcriptional regulation, antioxidant and stress responses, apoptosis, protein degradation, and synthesis and metabolism (Table 4).
Table 4.
Genes significantly altered at 60 d in motorneurons expressing the G93A SOD1
| Probe identification | Gene | Fold change |
|---|---|---|
| Apoptosis | ||
| 1422484_at | Cytochrome c, somatic | 2.00 |
| 1424802_a_at | RIKEN cDNA 3300001P08 gene | 3.88 |
| 1456600_a_at | Ring finger protein 7 | 4.87 |
| 1417815_a_at | Tumor differentially expressed 1 | 2.16 |
| 1416642_a_at | Tumor protein translationally controlled 1 | 2.22 |
| Calcium-binding protein | ||
| 1435884_at | Intersectin 1 (SH3 domain protein 1A) | 2.03 |
| 1456379_x_at | Delta/notch-like EGF-related receptor | 2.56 |
| Cell adhesion | ||
| 1427442_a_at | Amyloid β (A4) precursor protein | 2.88 |
| 1434180_at | Pleckstrin homology domain containing, family C (with FERM domain) member 1 | 2.04 |
| 1416003_at | Claudin 11 | 2.21 |
| 1416473_a_at | Neighbor of Punc E11 | 2.59 |
| 1417749_a_at | Tight junction protein 1 | 2.21 |
| 1438928_x_at | Ninjurin 1 | −2.37 |
| Cell cycle/cell growth | ||
| 1439443_x_at | Transketolase | 2.06 |
| 1422313_a_at | Insulin-like growth factor-binding protein 5 | −2.11 |
| 1437711_x_at | Ornithine decarboxylase | 2.60 |
| 1448005_at | SAM and SH3 domain containing 1 | 2.53 |
| 1454610_at | Septin 7 | 2.76 |
| 1417133_at | Peripheral myelin protein | 5.39 |
| 1419663_at | Osteoglycin | 5.57 |
| Cytoskeletal | ||
| 1456312_x_at | Gelsolin | 2.16 |
| 1428608_at | Myosin light chain, regulatory B | 2.12 |
| 1419738_a_at | Tropomyosin 2, β | −3.24 |
| 1416179_a_at | Radixin | 2.25 |
| 1424800_at | Enabled homolog (Drosophila) | 5.77 |
| Ion transport | ||
| 1451674_at | Solute carrier family 12, member 5 | 2.38 |
| 1448771_a_at | Ferritin heavy chain 1 | 2.79 |
| 1416951_a_at | ATPase, H+ transporting, V1 subunit D | 3.22 |
| 1423890_x_at | ATPase, Na+/K+ transporting, β1 polypeptide | 3.85 |
| Kinases/phosphatases | ||
| 1460315_s_at | TANK-binding kinase 1 | 2.01 |
| 1424827_a_at | Casein kinase 1 α1 | 2.37 |
| 1438562_a_at | Protein tyrosine phosphatase, non-receptor type 2 | 2.66 |
| 1417594_at | G kinase anchoring protein 1 | 2.52 |
| 1434935_at | AP2-associated kinase 1 | 4.75 |
| Metabolism | ||
| 1434465_x_at | Very low-density lipoprotein receptor | 2.10 |
| 1416788_a_at | Isocitrate dehydrogenase 3 (NAD+) γ | −2.02 |
| 1437327_x_at | RIKEN cDNA 2310057D15 gene | 2.13 |
| 1455972_x_at | l-3-Hydroxyacyl-coenzyme A dehydrogenase, short chain | 2.18 |
| 1423804_a_at | Isopentenyl-diphosphate δ isomerase | 2.66 |
| 1434642_at | Dehydrogenase/reductase (SDR family) member 8 | 2.39 |
| 1449686_s_at | Sterol carrier protein 2, liver | 2.46 |
| 1448172_at | Malate dehydrogenase 1, NAD (soluble) | 2.68 |
| 1419063_at | UDP-glucuronosyltransferase 8 | 3.36 |
| 1426689_s_at | Succinate dehydrogenase complex, subunit A, flavoprotein | 2.29 |
| 1452005_at | Dihydrolipoamide S-acetyltransferase (E2 component of pyruvate dehydrogenase complex) | 2.51 |
| Mitochondrial proteins | ||
| 1456580_s_at | ATP synthase, H+ transporting, mitochondrial F1 complex, δ subunit | −2.01 |
| 1449710_s_at | ATP synthase, H+ transporting, mitochondrial F1 complex, α subunit, isoform 1 | 2.68 |
| 1435112_a_at | ATP synthase, H+ transporting, mitochondrial F0 complex, subunit d | 2.77 |
| 1416058_s_at | ATP synthase, H+ transporting, mitochondrial F1 complex, γ polypeptide 1 | 5.94 |
| 1424562_a_at | Solute carrier family 25 (mitochondrial carrier, adenine nucleotide translocator), member 4 | 2.31 |
| 1416300_a_at | Solute carrier family 25 (mitochondrial carrier, phosphate carrier), member 3 | 4.08 |
| 1433991_x_at | Diazepam-binding inhibitor | 2.13 |
| 1419657_a_at | RIKEN cDNA C330005L02 gene | 4.50 |
| 1455357_x_at | Translocase of outer mitochondrial membrane 20 homolog (yeast) | 2.75 |
| 1428161_a_at | Coiled–coil-helix–coiled–coil-helix domain containing 2 | 3.59 |
| 1423095_s_at | Cereblon | 4.03 |
| mRNA processing | ||
| 1455696_a_at | PRP4 pre-mRNA processing factor 4 homolog B (yeast) | 2.49 |
| 1437234_x_at | Heterogeneous nuclear ribonucleoprotein methyltransferase-like 1 (S. cerevisiae) | 2.24 |
| 1460547_a_at | Heterogeneous nuclear ribonucleoprotein K | 2.03 |
| 1433829_a_at | Heterogeneous nuclear ribonucleoprotein A2/B1 | 2.88 |
| 1418562_at | Splicing factor 3b subunit 1 | 2.20 |
| 1437838_x_at | G-rich RNA sequence-binding factor 1 | 2.02 |
| Motor activity | ||
| 1418429_at | Kinesin family member 5B | 2.08 |
| 1423994_at | Kinesin family member 1B | 5.16 |
| 1455434_a_at | Kinectin 1 | 2.57 |
| 1456292_a_at | Vimentin | 3.17 |
| 1436836_x_at | Calponin 3, acidic | 3.95 |
| 1439244_a_at | Trinucleotide repeat containing 6a | 4.80 |
| Nuclear protein | ||
| 1434888_a_at | Matrin 3 | 8.12 |
| 1424251_a_at | Heterogeneous nuclear ribonucleoprotein D-like | 4.46 |
| 1423050_s_at | Heterogeneous nuclear ribonucleoprotein U | 2.56 |
| 1452155_a_at | DEAD (Asp-Glu-Ala-Asp) box polypeptide 17 | 2.29 |
| 1416144_a_at | DEAH (Asp-Glu-Ala-His) box polypeptide 15 | 2.05 |
| 1423292_a_at | Periaxin | 2.70 |
| 1437843_s_at | Nucleoporin-like 1 | 2.37 |
| 1434875_a_at | High-mobility group nucleosomal binding domain 3 | 2.60 |
| 1438940_x_at | High-mobility group nucleosomal binding domain 1 | 2.27 |
| 1417073_a_at | Quaking | 2.72 |
| 1417998_at | Telomerase-binding protein, p23 | 2.35 |
| 1415751_at | Heterochromatin protein 1 | 2.24 |
| 1420477_at | Nucleosome assembly protein 1-like 1 | 2.99 |
| 1448358_s_at | Small nuclear ribonucleoprotein polypeptide G | 2.34 |
| 1417351_a_at | Small nuclear ribonucleoprotein polypeptide A′ | 2.32 |
| 1433834_at | Membrane-associated ring finger (C3HC4) 6 | 3.36 |
| 1439012_a_at | Deoxycytidine kinase | 2.23 |
| 1415991_a_at | Kelch domain containing 3 | −2.69 |
| Oxidoreductase activity | ||
| 1415970_at | Cytochrome c oxidase, subunit VIc | 3.79 |
| 1432263_a_at | Cytochrome c oxidase subunit VIIa polypeptide 2-like | 4.38 |
| 1434435_s_at | Cytochrome c oxidase, subunit XVII assembly protein homolog (yeast) | 3.16 |
| 1416185_a_at | Alcohol dehydrogenase 5 (class III), χ polypeptide | 2.52 |
| 1448610_a_at | Superoxide dismutase 2, mitochondrial | 2.35 |
| 1433603_at | NADH dehydrogenase (ubiquinone) Fe-S protein 6 | 2.74 |
| 1422241_a_at | NADH dehydrogenase (ubiquinone) 1 α subcomplex, 1 | 2.86 |
| 1435235_at | Thioredoxin-like 1 | 2.16 |
| 1437143_a_at | Thioredoxin domain containing 1 | 2.64 |
| Protein binding | ||
| 1449172_a_at | Lin 7 homolog b (Caenorhabditis elegans) | −2.82 |
| 1437455_a_at | B-cell translocation gene 1, antiproliferative | 2.10 |
| 1455286_at | BTB (POZ) domain containing 1 | 2.92 |
| 1424821_at | Nedd4 family interacting protein 1 | 4.73 |
| 1452649_at | Reticulon 4 | 4.87 |
| Protein degradation | ||
| 1434853_x_at | Makorin, ring finger protein, 1 | 2.76 |
| 1455825_s_at | Ligand of numb-protein X 1 | 3.66 |
| 1417202_s_at | Ubiquitin-activating enzyme E1C | 2.34 |
| 1437278_a_at | Ubiquitin-like 1 (sentrin) activating enzyme E1B | 3.09 |
| 1417771_a_at | Proteasome (prosome, macropain) 26S subunit, ATPase, 6 | 2.16 |
| 1448591_at | Cathepsin S | 2.74 |
| 1460698_a_at | Sec11-like 3 (S. cerevisiae) | 3.03 |
| 1437468_x_at | F-box and WD-40 domain protein 11 | 3.30 |
| 1415949_at | Carboxypeptidase E | 7.16 |
| Protein folding | ||
| 1438902_a_at | Heat shock protein 90, α | 2.58 |
| 1418503_at | Heat shock protein 9A | 3.18 |
| 1425993_a_at | Heat shock protein 105 | 2.73 |
| 1433447_x_at | Chaperonin subunit 4 (δ) | 2.59 |
| 1437907_a_at | Tubulin cofactor a | 2.28 |
| Signaling | ||
| 1435495_at | Adenosine A1 receptor | 2.11 |
| 1415849_s_at | Stathmin 1 | 2.19 |
| 1425241_a_at | WD repeat and SOCS box-containing 1 | 2.32 |
| 1418189_s_at | Receptor (calcitonin) activity modifying protein 2 | 8.15 |
| 1416636_at | RAS-homolog enriched in brain | 3.22 |
| 1416426_at | RAB5A, member RAS oncogene family | 3.75 |
| 1419246_s_at | RAB14, member RAS oncogene family | 3.69 |
| 1424902_at | Plexin domain containing 1 | −2.54 |
| 1420196_s_at | TBC1 domain family, member 14 | 2.77 |
| Stress response | ||
| 1437715_x_at | Apurinic/apyrimidinic endonuclease 1 | −2.24 |
| Transcription | ||
| 1435382_at | Necdin | 4.62 |
| 1436153_a_at | Zinc finger, MYND domain containing 11 | 3.13 |
| 1455855_x_at | Heterogeneous nuclear ribonucleoprotein A/B | 2.17 |
| 1452774_at | Heterogeneous nuclear ribonucleoprotein A3 | 2.49 |
| 1451770_s_at | DEAH (Asp-Glu-Ala-His) box polypeptide 9 | 2.27 |
| 1454793_x_at | DEAD (Asp-Glu-Ala-Asp) box polypeptide 5 | 2.64 |
| 1436703_x_at | Small nuclear RNA activating complex, polypeptide 2 | −2.36 |
| 1434434_s_at | Transcription elongation regulator 1 (CA150) | 2.49 |
| 1452193_a_at | Wiskott–Aldrich syndrome-like (human) | 2.02 |
| 1415796_at | DAZ-associated protein 2 | 2.23 |
| 1415820_x_at | Non-POU-domain-containing | 2.10 |
| 1416065_a_at | Ankyrin repeat domain 10 | 2.09 |
| 1438992_x_at | Activating transcription factor 4 | 2.21 |
| 1454758_a_at | Transforming growth factor β1 induced transcript 4 | 2.49 |
| 1456250_x_at | Transforming growth factor β induced | 2.97 |
| 1455222_a_at | Upstream-binding protein 1 | 2.50 |
| 1418324_at | Feminization 1 homolog b (C. elegans) | 2.97 |
| 1435362_at | Forkhead box J3 | 3.20 |
| 1433924_at | Paternally expressed 3 | 4.60 |
| 1437035_x_at | Ring finger protein 14 | 2.69 |
| Translation | ||
| 1416276_a_at | Ribosomal protein S4, X-linked | 3.44 |
| 1418062_at | Eukaryotic translation elongation factor 1 α2 | −2.01 |
| 1448252_a_at | Eukaryotic translation elongation factor 1 β2 | 2.39 |
| 1415723_at | Eukaryotic translation initiation factor 5 | 2.86 |
| 1452285_a_at | Eukaryotic translation initiation factor 3, subunit 5 (ϵ) | 2.49 |
| 1439268_x_at | Eukaryotic translation initiation factor 3 subunit 6 | 2.11 |
| 1448425_at | Eukaryotic translation initiation factor 3 subunit 10 (θ) | 3.25 |
| 1448493_at | Polyadenylate-binding protein-interacting protein 2 | 4.19 |
| 1418883_a_at | PolyA-binding protein cytoplasmic 1 | 2.29 |
| 1423665_a_at | Ribosomal protein L5 | 2.08 |
| 1434377_x_at | Ribosomal protein S6 | 3.40 |
| 1438794_x_at | Ribosomal protein S13 | 2.11 |
| 1454859_a_at | Ribosomal protein L23 | 2.14 |
| 1434854_a_at | Ribosomal protein S10 | 2.32 |
| 1436822_x_at | Ribosomal protein L17 | 2.25 |
| 1450925_a_at | Ribosomal protein S27-like | 2.20 |
| Transport | ||
| 1436737_a_at | Sorbin and SH3 domain containing 1 | 2.57 |
| 1436451_a_at | Transmembrane emp24 domain trafficking protein 2 | 4.23 |
| 1437708_x_at | Vesicle-associated membrane protein 3 | 2.55 |
| 1454697_at | Translocation protein 1 | 2.17 |
| 1449003_a_at | Vesicle transport through interaction with t-SNAREs 1B homolog | 2.25 |
| 1436452_x_at | RIKEN cDNA 1110032D12 gene | 2.31 |
| 1416509_at | SM-11044-binding protein | 2.63 |
| 1419027_s_at | Glycolipid transfer protein | 2.71 |
| 1437723_s_at | Der1-like domain family, member 1 | 2.91 |
| 1439368_a_at | Solute carrier family 9 (sodium/hydrogen exchanger) isoform 3 regulator 2 | −2.15 |
| 1426722_at | Solute carrier family 38, member 2 | 3.98 |
| 1439411_a_at | Exportin 7 | 2.02 |
| Miscellaneous | ||
| 1437341_x_at | Cyclic nucleotide phosphodiesterase 1 | 2.01 |
| 1423792_a_at | Chemokine-like factor superfamily 6 | 2.17 |
| 1448233_at | Prion protein | 3.19 |
| 1416009_at | Tetraspanin 3 | 2.24 |
| 1437211_x_at | ELOVL family member 5, elongation of long chain fatty acids (yeast) | 2.46 |
| 1433723_s_at | Small EDRK-rich factor 2 | 2.74 |
| 1437342_x_at | Pituitary tumor-transforming 1 interacting protein | 2.81 |
| 1423328_at | Ganglioside-induced differentiation-associated-protein 1 | 3.31 |
| 1456741_s_at | Glycoprotein m6a | 3.53 |
| 1423091_a_at | Glycoprotein m6b | 4.76 |
| 1454651_x_at | Myelin basic protein | 2.72 |
| 1423253_at | Myelin protein zero | 7.19 |
| 1451718_at | Proteolipid protein (myelin) 1 | 5.51 |
| 1456174_x_at | N-myc downstream regulated gene 1 | 9.46 |
| 1448108_at | Tumor differentially expressed 2 | 4.72 |
| Unknown | ||
| 1452183_a_at | GTL2, imprinted maternally expressed untranslated mRNA | 3.71 |
| 1448504_a_at | Mus musculus, similar to heterochromatin-like protein 1, clone IMAGE:3493144, mRNA | 6.91 |
| 1453651_a_at | Brain protein 44-like | 2.00 |
| 1423939_a_at | Yip1 interacting factor homolog (S. cerevisiae) | −2.61 |
| 1437345_a_at | Bernardinelli-Seip congenital lipodystrophy 2 homolog (human) | −2.48 |
| 1437991_x_at | RUN and SH3 domain containing 1 | −2.28 |
| 1423251_at | LUC7-like 2 (S. cerevisiae) | 2.08 |
| 1439257_x_at | Ribophorin I | 5.62 |
| 1424071_s_at | cDNA sequence BC018507 | 2.12 |
| 1419291_x_at | Growth arrest specific 5 | 2.45 |
| 1439271_x_at | IK cytokine | 2.52 |
| 1456542_s_at | Glutaminyl-tRNA synthase (glutamine-hydrolyzing)-like 1 | 2.73 |
| 1425020_at | UBX domain containing 4, similar to Ubxd4 protein | 4.57 |
| 1433871_at | R3H domain (binds single-stranded nucleic acids) | 2.92 |
| 1448639_a_at | −2.06 | |
| 1450084_s_at | Influenza virus NS1A-binding protein | 3.18 |
| 1455940_x_at | WD repeat domain 6 | 3.27 |
| 1416483_at | Tetratricopeptide repeat domain 3 | 3.46 |
| 1424970_at | Purine-rich element-binding protein G | 3.59 |
| 1433654_at | Meningioma expressed antigen 5 (hyaluronidase) | 4.47 |
| 1428103_at | 3.49 | |
| 1455550_x_at | 2.04 | |
| 1419252_at | 2.25 | |
| 1423758_at | RIKEN cDNA E430034L04 gene | 2.75 |
| 1416911_a_at | RIKEN cDNA 6330407G11 gene | 2.60 |
| 1424707_at | RIKEN cDNA 1110014C03 gene | 2.09 |
| 1448461_a_at | RIKEN cDNA 1500006O09 gene | 2.19 |
| 1416171_at | RIKEN cDNA 2310037I24 gene | 2.27 |
| 1456736_x_at | RIKEN cDNA 5230400G24 gene | 2.28 |
| 1431293_a_at | RIKEN cDNA 1110019C08 gene | 2.42 |
| 1438318_x_at | RIKEN cDNA 1500001L15 gene | 2.91 |
| 1435315_s_at | RIKEN cDNA 2900034E22 gene | 3.35 |
| 1415764_at | RIKEN cDNA 5730454B08 gene | 2.47 |
| 1438171_x_at | RIKEN cDNA 0610012D09 gene | 3.51 |
| 1428288_at | RIKEN cDNA 2310051E17 gene | 3.53 |
| 1450642_at | RIKEN cDNA 3110001I20 gene | 4.78 |
| 1439453_x_at | RIKEN cDNA 1500026D16 gene | −2.84 |
| 1419174_at | RIKEN cDNA 2410004B18 gene | 3.71 |
| 1452167_at | RIKEN cDNA 2810407C02 gene | 4.01 |
| 1434292_at | RIKEN cDNA E130013N09 gene | 2.11 |
The SOD1 G93A mice show a significant increase in genes controlling both transcriptional and translational functions. Of particular interest is the increase in necdin (Ndn), confirmed also by Q-PCR (p = 0.04) (Fig. 1A), and transforming growth factor β1 induced (Tgf1bi), essential in the regulation of the transcription process and involved in the activation of several pathways, including the nuclear factor-kB (Konig et al., 2005) and c-Jun (Parkinson et al., 2004) pathways.
Figure 1.

Q-PCR results. A, Significant upregulation of necdin at 60 d (p = 0.04), which is downregulated at the late stage of the disease (p = 0.003). B, Consistent upregulation of cyclin I throughout the pathology (60 d, p = 0.05; 90 d, p = 0.1; 120 d, p = 0.03). C, Consistent downregulation of plexin domain containing 1 (60 d, p = 0.02; 90 d, p = 0.003; 120 d, p = 0.001). Error bars indicate SD. Tg, Transgenic; N-Tg, nontransgenic. *p ≤ 0.05; **p ≤ 0.01.
At this presymptomatic stage, significant changes occur also in genes involved in lipid and carbohydrate metabolism. As indicated by pathway analysis (GenMAPP 1.1), several upregulated transcripts are key enzymes belonging to the fatty acid degradation pathway (e.g., l-3-hydroxyacyl-coenzyme A dehydrogenase and sterol carrier protein 2) as well as enzymes belonging to the Krebs cycle [e.g., dihydrolipoamide S-acetyltransferase (E2 component of pyruvate dehydrogenase complex) and malate dehydrogenase 1 (Mdh1)]. Q-PCR of Mdh1 showed a trend toward upregulation, but it did not reach statistical significance (p = 0.37) (Table 2). In addition, many transcripts encoding for proteins belonging to complex I, II, and IV of the electron transport chain are strongly upregulated (up to fivefold changes), as well as three subunits of ATP synthase. These data suggest that, at this stage of the pathology, motor neurons have an increased requirement for ATP. Surprisingly, only one of the transcripts involved in the mitochondrial respiratory chain arrayed on the GeneChip was downregulated. This probe encodes for the δ subunit of the mitochondrial F1 complex of ATP synthase (Atp5d), which is responsible for the coupling between the energy produced by proton translocation within the F0 subunit of the ATP synthase and ATP production (Duvezin-Caubet et al., 2003). Downregulation of this transcript has also been observed in the spinal cord gray matter of ALS patients (Dangond et al., 2004). As a key regulator of ATP production, this gene was chosen for further investigation, and its decreased expression was confirmed by Q-PCR (p = 0.01) (Table 2). ATP synthase γ polypeptide 1 (Atp5c1) was also selected for further validation because of its important role in ATP synthesis. This subunit has a central role in proton translocation between F1 and F0 components of the ATP synthase complex, and it directly interacts with Atp5d. Its upregulation was confirmed by Q-PCR (p = 0.0036).
All these mitochondrial proteins are encoded by nuclear genes and then translocated into the inner or outer mitochondrial membranes as preproteins. Specific ligands in the cytosol are responsible for the binding and transport of mitochondrial preproteins (Young et al., 2003), and the protein translocation machinery in the outer mitochondrial membrane transfers the preproteins into the mitochondrion. Key proteins responsible for this translocation are translocase of outer mitochondrial membrane 20 (Tom20) and the heat shock proteins 70 and 90 (Hsp70 and Hsp90) (Young et al., 2003). Both Tom20 and Hsp90 are upregulated at the early stage of the disease (Table 4), supporting the data showing an increase in transcripts encoding for proteins active in the mitochondrial machinery, including proteins belonging to the respiratory chain, Krebs cycle, and lipid metabolism.
Other categories of upregulated genes are those involved in both protein folding (heat shock protein family) and protein degradation (ubiquitin system), suggesting that there is a high turnover of motor neuronal proteins at this presymptomatic stage of the disease. Activation of the ubiquitin system in ALS has been investigated in relation to the presence of SOD1 aggregates in the spinal cord of transgenic mice at various stages of the disease (Wood et al., 2003; Basso et al., 2006). Although SOD1 transgenic mice show the presence of both ubiquitinated inclusions and SOD1 aggregates, their colocalization has not been clearly proven (Basso et al., 2006). Ubiquitination is a physiological mechanism that regulates protein targeting and degradation. Its correct functioning, along with the proteasome and the protein chaperoning system, is fundamental to maintain a balanced level of cellular proteins, which otherwise tend to aggregate into insoluble complexes (Kabashi and Durham, 2006). The compensatory pathological increase of genes involved in the transcriptional process and protein synthesis observed in our study may lead to a dramatic imbalance between protein synthesis and protein degradation in a situation in which the altered solubility of the mutant SOD1 protein has probably perturbed the function of the ubiquitin–proteasome system (Shinder et al., 2001; Kabashi and Durham, 2006).
Vimentin, an intermediate filament, is one of several cytoskeletal proteins differentially expressed. In addition, two members of the kinesin family (Kif1b and Kif5b) and their receptor kinectin, involved in anterograde axonal transport and endoplasmic reticulum development (Vedrenne and Hauri, 2006), are also upregulated. Because of the important involvement that axonal transport has in the development of motor neuron degeneration, Kif1b and Kif5b were selected for confirmation by Q-PCR. The results at 60 d show an increase in both Kif1b (p = 0.09), which is confirmed at 90 d (p = 0.04), and Kif5b (p = 0.01) (Table 2).
Motor neurons from SOD1 G93A mice at the 90 d (symptomatic) time point
At the symptomatic stage of the disease, the comparison between transgenic mice and LM shows less differentially expressed genes, with increases in the expression of 32 transcripts and decreases in the expression of 19 transcripts (Table 5). Categorized by function, alterations occur in multiple functional classes, and, with fold changes not much higher than 2, it is difficult to define a general upregulation or downregulation within specific functional classes (Table 5).
Table 5.
Genes significanlty altered at 90 d in motorneurons expressing the G93A SOD1
| Probe identification | Gene | Fold change |
|---|---|---|
| Calcium-binding protein | ||
| 1452363_a_at | ATPase Ca2+ transporting cardiac muscle slow twitch 2 | −2.00 |
| Cell adhesion | ||
| 1456746_a_at | Cd99 antigen-like 2 | 2.96 |
| 1438928_x_at | Ninjurin 1 | 2.01 |
| Cell cycle/cell growth | ||
| 1416427_at | Cyclin I | 2.92 |
| 1438478_a_at | Protein phosphatase 3 catalytic subunit α isoform | 2.19 |
| Cytoskeletal | ||
| 1422520_at | Neurofilament 3 medium | 2.30 |
| 1448553_at | Myosin heavy polypeptide 7 cardiac muscle β | −2.93 |
| Immune response | ||
| 1456108_x_at | Zinc finger protein 179 | −2.15 |
| Ion transport | ||
| 1451071_a_at | ATPase Na+/K+ transporting α1 polypeptide | 4.12 |
| 1423597_at | ATPase aminophospholipid transporter (APLT) class I type 8A member 1 | 2.01 |
| 1421662_a_at | Tumor suppressor candidate 3 | −2.32 |
| 1437143_a_at | Thioredoxin domain containing 1 | −2.09 |
| Kinases/phosphatases | ||
| 1437100_x_at | Proviral integration site 3 | 2.09 |
| 1437690_x_at | Casein kinase 1 δ (Csnk1d) transcript variant 2 mRNA | 2.01 |
| Metabolism | ||
| 1416561_at | Glutamic acid decarboxylase 1 | −2.68 |
| 1415851_a_at | Inosine 5′-phosphate dehydrogenase 2 | 2.02 |
| 1423294_at | Mesoderm specific transcript | 2.50 |
| mRNA processing | ||
| 1449635_at | PRP19/PSO4 homolog (S. cerevisiae) | 2.52 |
| 1415839_a_at | Nucleophosmin 1 | −2.09 |
| 1416150_a_at | Splicing factor arginine/serine-rich 3 (SRp20) | −2.00 |
| 1417351_a_at | Small nuclear ribonucleoprotein polypeptide A′ | −2.02 |
| 1453928_a_at | Sjogren syndrome antigen B | 2.20 |
| Motor activity | ||
| 1448682_at | Dynein cytoplasmic light chain 1 | −2.00 |
| Nuclear proteins | ||
| 1423132_a_at | RIKEN cDNA 5730427N09 gene, similar to TGFβ-inducible nuclear protein 1 (L-name-related LNR42) | 2.45 |
| 1420476_a_at | Nucleosome assembly protein 1-like 1 | 2.30 |
| 1455035_s_at | Nucleolar protein 5A | 2.12 |
| 1448595_a_at | Reduced expression 3 | −2.05 |
| 1419650_at | Zinc finger RNA-binding protein | −2.15 |
| Oxidoreductase activity | ||
| 1422186_s_at | Cytochrome b5 reductase 3 | 2.01 |
| 1435734_x_at | Dihydrouridine synthase 1-like (S. cerevisiae) | 2.03 |
| 1416417_a_at | NADH dehydrogenase (ubiquinone) 1 β subcomplex 7 | 2.00 |
| Protein degradation | ||
| 1426318_at | Serine (or cysteine) peptidase inhibitor clade B member 1b | −2.68 |
| Signaling | ||
| 1424525_at | Gastrin-releasing peptide | −2.02 |
| 1425241_a_at | WD repeat and SOCS box-containing 1 | −2.40 |
| 1421847_at | WD repeat and SOCS box-containing 2 | −3.21 |
| Transcription | ||
| 1418517_at | Iroquois related homeobox 3 (Drosophila) | 4.39 |
| 1428572_at | Brain abundant membrane attached signal protein 1 | 2.03 |
| 1417026_at | Prefoldin 1 | 2.01 |
| Translation | ||
| 1434328_at | Ribosomal protein L15, similar to ribosomal protein L15 | −2.30 |
| 1416520_x_at | Ribosomal protein L36 | 2.20 |
| 1424635_at | Eukaryotic translation elongation factor 1 α1 | −3.35 |
| Miscellaneous | ||
| 1424902_at | Plexin domain containing 1 | 2.98 |
| Unknown | ||
| 1452597_at | RIKEN cDNA 2310061C15 gene | 2.03 |
| 1437345_a_at | Bernardinelli-Seip congenital lipodystrophy 2 homolog (human) | 2.10 |
| 1416655_at | C1GALT1-specific chaperone 1 | −2.02 |
| 1418710_at | CD59a antigen | 2.04 |
| 1453207_at | Hypothetical gene LOC554362 | 2.95 |
| 1433786_x_at | Small EDRK-rich factor 2 | 2.06 |
| 1438143_s_at | 2.07 | |
| 1460709_a_at | HLA-B-associated transcript 5 | 2.30 |
Nevertheless, one of the categories showing the greatest alteration in gene expression at disease onset is related to transcriptional regulation. Genes belonging to this category still show increased expression, as previously shown at 60 d, although they occur in different transcripts, such as Iroquois-related homeobox 3 (Irx3). Smaller fold changes are also found in genes encoding for proteins involved in signaling (Table 4, Signaling and Kinases/phosphatases) and in nuclear activity, although transcripts are both upregulated and downregulated in both classes. Among the proteins with a role in signaling function, of particular interest is the upregulation of casein kinase 1 δ (Csnk1d). Csnk1d is involved in several pathways, including the DNA damage response, along with p53 and murine double-minute clone 2 protein (MDM2) (Winter et al., 2004), the wingless (WNT) pathway (Price, 2006), and in microtubule dynamics (Wolff et al., 2005).
Interestingly, at this stage, increased expression in two genes involved in the regulation of the cell cycle is observed: one is cyclin I (Ccni), and the other is protein phosphatase 3 catalytic subunit α (Ppp3ca), which is responsible for the transition from the G1 phase to the S phase (Afroze et al., 2003), and it is also involved in the WNT pathway (Kadoya et al., 2000). Cyclin I is constitutively expressed in terminally differentiated cells, and its function is related to the negative control of the cell cycle rather than to its progression (Bennin et al., 2002). Cyclin I showed a threefold increased expression at 90 d and a twofold increase at 120 d. Q-PCR showed a significant increase at 60 d (p = 0.04) and 120 d (p = 0.03), but at 90 d the increase was not significant (p = 0.1) (Fig. 1B).
Motor neurons from SOD1 G93A mice at the 120 d (end-stage) time point
At 120 d, the transcription profile of the SOD1 G93A mutant mice compared with their nontransgenic LM shows a significant change in 167 genes, with 86 transcripts decreased in the presence of mutant SOD1 G93A and 81 transcripts increased (Table 6).
Table 6.
Genes significantly altered at 120 d in motorneurons expressing the G93A SOD1
| Probe identification | Gene | Fold change | |
|---|---|---|---|
| Apoptosis | |||
| 1422484_at | Cytochrome c somatic | −2.51 | |
| 1423534_at | Programmed cell death 2 | −2.25 | |
| 1415910_s_at | Cytokine induced apoptosis inhibitor 1 | −2.48 | |
| 1456071_a_at | NADH dehydrogenase (ubiquinone) Fe-S protein 6 | −2.08 | |
| 1454690_at | Inhibitor of κB kinase γ | −2.16 | |
| 1417516_at | DNA-damage inducible transcript 3 | 3.24 | |
| 1417346_at | PYD and CARD domain containing | 2.44 | |
| Calcium binding | |||
| 1438629_x_at | Granulin | 2.24 | |
| Cell adhesion | |||
| 1456014_s_at | cDNA sequence BC032204 | 4.05 | |
| 1437463_x_at | Transforming growth factor, β induced | 2.18 | |
| 1456133_x_at | Integrin β5 | 3.49 | |
| 1438677_at | Plakophilin 4 | 2.98 | |
| Cell cycle/cell growth | |||
| 1452056_s_at | Protein phosphatase 3 catalytic subunit α isoform | −2.08 | |
| 1436994_a_at | Histone 1 H1c | −2.05 | |
| 1438647_x_at | Centrin 2 | −2.43 | |
| 1439413_x_at | Mortality factor 4-like 2 | −3.28 | |
| 1416427_at | Cyclin I | 2.12 | |
| 1455956_x_at | Cyclin D2 | 3.50 | |
| 1423622_a_at | Cyclin L1 | 2.05 | |
| 1435372_a_at | Proliferation-associated 2G4 | 2.08 | |
| Cytoskeletal | |||
| 1418884_x_at | Tubulin, α1 | −2.47 | |
| 1448991_a_at | Internexin neuronal intermediate filament protein α | 2.7 | |
| 1450732_a_at | Bicaudal D homolog 2 (Drosophila) | 3.31 | |
| Immunity | |||
| 1418021_at | Complement component 4 (within H-2S) | 4.14 | |
| 1437726_x_at | Complement component 1 q subcomponent β polypeptide | 5.25 | |
| 1422903_at | Lymphocyte antigen 86 | 3.36 | |
| 1426808_at | Lectin, galactose binding, soluble 3 | 11.03 | |
| 1439426_x_at | P lysozyme structural | 4.80 | |
| 1423547_at | Lysozyme | 4.48 | |
| 1419873_s_at | Colony stimulating factor 1 receptor | 4.38 | |
| Ion transport | |||
| 1451071_a_at | ATPase, Na+/K+ transporting, α1 polypeptide | −11.03 | |
| 1438578_a_at | RIKEN cDNA 1110056N09 gene | −2.21 | |
| 1426432_a_at | Solute carrier family 4 (anion exchanger), member 4 | 3.35 | |
| 1422557_s_at | Metallothionein 1 | 4.29 | |
| Kinases/phosphatases | |||
| 1422619_at | Phosphatidic acid phosphatase 2a | −3.06 | |
| 1451982_at | Mitogen activated protein kinase kinase 4 | −2.57 | |
| 1436684_a_at | RIO kinase 2 (yeast) | −2.07 | |
| 1424470_a_at | Rap guanine nucleotide exchange factor (GEF) 3 | 3.00 | |
| Metabolism | |||
| 1423804_a_at | Isopentenyl-diphosphate δ isomerase | −4.91 | |
| 1416939_at | Pyrophosphatase | −3.91 | |
| 1424425_a_at | Methylthioadenosine phosphorylase | −3.57 | |
| 1426511_at | Sushi domain containing 2 | −2.61 | |
| 1452005_at | Dihydrolipoamide S-acetyltransferase (E2 component of pyruvate dehydrogenase complex) | −2.38 | |
| 1423564_a_at | Phosphoribosylaminoimidazole carboxylase, phosphoribosylaminoribosylaminoimidazole, succinocarboxamide synthetase | −2.51 | |
| 1448172_at | Malate dehydrogenase 1, NAD (soluble) | −2.93 | |
| 1451537_at | Chitinase 3-like 1 | 2.06 | |
| 1455084_x_at | Serine hydroxymethyl transferase 2 (mitochondrial) | 2.32 | |
| 1424714_at | Aldolase 3, C isoform | 2.71 | |
| 1437874_s_at | Hexosaminidase B | 3.01 | |
| 1449024_a_at | Hexosaminidase A | 2.23 | |
| Motor activity | |||
| 1456292_a_at | Vimentin | 9.58 | |
| 1423995_at | Kinesin family member 1B | 3.72 | |
| 1448682_at | Dynein, cytoplasmic, light chain 1 | −2.61 | |
| 1450053_at | RIKEN cDNA 1500031M22 gene | −3.27 | |
| mRNA processing | |||
| 1451294_s_at | Small nuclear ribonucleoprotein E | −2.20 | |
| 1423758_at | RIKEN cDNA E430034L04 gene | −3.05 | |
| Nuclear protein | |||
| 1456055_x_at | Polymerase (DNA directed), δ1, catalytic subunit | 2.34 | |
| 1424922_a_at | Bromodomain containing 4 | 2.83 | |
| 1423455_at | Prothymosin α | 2.22 | |
| 1456108_x_at | Zinc finger protein 179 | −2.87 | |
| Oxireductase activity | |||
| 1423035_s_at | Thioredoxin-like 5 | −2.67 | |
| 1415970_at | Cytochrome c oxidase, subunit VIc | −3.14 | |
| 1422241_a_at | NADH dehydrogenase (ubiquinone) 1, α subcomplex, 1 | −2.63 | |
| 1428160_at | NADH dehydrogenase (ubiquinone) 1, α/β subcomplex, 1 | −3.17 | |
| Protein binding | |||
| 1417980_a_at | Insulin-induced gene 2 | −2.30 | |
| 1418412_at | Tumor protein D52-like 1 | −3.40 | |
| Protein degradation | |||
| 1437901_a_at | Vacuolar protein sorting 41 (yeast) | 2.07 | |
| 1417869_s_at | Cathepsin Z | 3.24 | |
| 1427718_a_at | Transformed mouse 3T3 cell double minute 2 | 2.07 | |
| 1417490_at | Cathepsin B | 2.04 | |
| 1456094_at | Ubiquitin-specific protease 36 (11 in man) | 2.79 | |
| 1437714_x_at | Ubiquitin-specific peptidase 14 | −4.27 | |
| 1455825_s_at | Ligand of numb-protein X 1 | −4.53 | |
| 1415688_at | Ubiquitin-conjugating enzyme E2G 1 (UBC7 homolog, C. elegans) | −2.45 | |
| Signaling | |||
| 1437502_x_at | CD24a antigen | −4.98 | |
| 1423287_at | Cerebellin 1 precursor protein | −3.31 | |
| 1438839_a_at | Tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein ϵ polypeptide | −3.01 | |
| 1420829_a_at | Tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, θ polypeptide | −3.05 | |
| 1419945_s_at | RAB2, member RAS oncogene family | −2.36 | |
| 1436736_x_at | DNA segment, human D4S114 | −2.88 | |
| 1419811_at | DNA segment, Chr 16, Wayne State University 65, expressed | −2.72 | |
| 1455462_at | Adenylate cyclase 2 | −2.59 | |
| 1419100_at | Serine (or cysteine) proteinase inhibitor, clade A, member 3N | 3.39 | |
| 1450792_at | TYRO protein tyrosine kinase-binding protein | 5.41 | |
| 1460218_at | CD52 antigen | 2.26 | |
| 1424780_a_at | DNA segment Chr 10 University of California at Los Angeles 1 | 3.45 | |
| 1418457_at | Chemokine (C-X-C motif) ligand 14 | 2.78 | |
| 1456212_x_at | Suppressor of cytokine signaling 3 | 2.01 | |
| 1425639_at | Centaurin, α2 | 3.02 | |
| 1418892_at | Ras homolog gene family, member J | 3.60 | |
| 1433757_a_at | Nischarin | 2.36 | |
| 1434020_at | PDGFA-associated protein 1 | 2.30 | |
| 1438403_s_at | Receptor (calcitonin) activity modifying protein 2 | 4.59 | |
| 1439079_a_at | Erbb2 interacting protein | 3.14 | |
| Stress response | |||
| 1416129_at | ERBB receptor feedback inhibitor 1 | −3.09 | |
| 1430979_a_at | Peroxiredoxin 2 | −2.32 | |
| 1416480_a_at | Hypoxia-induced gene 1 | −2.13 | |
| Transcription | |||
| 1418624_at | Nuclease-sensitive element-binding protein 1 | 2.84 | |
| 1415923_at | Necdin | −3.02 | |
| 1427914_a_at | Transcription elongation factor B (SIII), polypeptide 1 | −2.96 | |
| 1425742_a_at | Transforming growth factor β1 induced transcript 4 | −5.96 | |
| 1438250_s_at | TAF9 RNA polymerase II, TATA box-binding protein (TBP)-associated factor | −3.09 | |
| 1433552_a_at | Polymerase (RNA) II (DNA directed) polypeptide B | −3.22 | |
| 1449648_s_at | RNA polymerase 1-1 | −2.23 | |
| 1421292_a_at | RIKEN cDNA A730008L03 gene | −2.14 | |
| Transferase activity | |||
| 1451969_s_at | ADP-ribosyltransferase (NAD+, poly ADP-ribose polymerase)-like 3 | 2.52 | |
| Translation | |||
| 1423220_at | Eukaryotic translation initiation factor 4E | −2.13 | |
| 1415859_at | Eukaryotic translation initiation factor 3, subunit 8 | 3.04 | |
| Transport | |||
| 1450888_at | N-ethylmaleimide-sensitive fusion protein attachment protein β | 2.60 | |
| 1417248_at | ralA-binding protein 1 | 2.40 | |
| 1433774_x_at | Component of oligomeric golgi complex 1 | 2.19 | |
| 1437843_s_at | Nucleoporin-like 1 (Nupl1) mRNA | −3.29 | |
| 1428502_at | ARP6 actin-related protein 6 homolog (yeast) | −2.19 | |
| 1436451_a_at | Transmembrane emp24 domain trafficking protein 2 | −2.99 | |
| Miscellaneous | |||
| 1456438_x_at | Ribophorin I | −3.46 | |
| 1421662_a_at | Tumor suppressor candidate 3 | −2.72 | |
| 1424902_at | Plexin domain containing 1 | −3.04 | |
| 1427351_s_at | Ig heavy chain 6 (heavy chain of IgM) | −5.95 | |
| 1417126_a_at | RIKEN cDNA 3110001N18 gene | −3.23 | |
| 1438178_x_at | ATPase family AAA domain containing 3A | 2.25 | |
| Unknown | |||
| 1436342_a_at | DNA segment Chr 19 ERATO Doi 721 expressed | 5.02 | |
| 1433935_at | Expressed sequence AU020206 | 2.32 | |
| 1418188_a_at | Metastasis-associated lung adenocarcinoma transcript 1 (non-coding RNA) | 2.86 | |
| 1448793_a_at | Syndecan 4 | 6.18 | |
| 1436905_x_at | Lysosomal-associated protein transmembrane 5 | 4.75 | |
| 1424607_a_at | Xanthine dehydrogenase | 3.52 | |
| 1451132_at | Pre-B-cell leukemia transcription factor interacting protein 1 | 2.66 | |
| 1424468_s_at | Pleckstrin homology-like domain, family B, member 1 | 2.16 | |
| 1456319_at | EST X83313 | 7.39 | |
| 1436362_x_at | 2.21 | ||
| 1438069_a_at | RNA-binding motif protein 5 | 2.43 | |
| 1435137_s_at | RIKEN cDNA 1200015M12 gene | 2.37 | |
| 1422861_s_at | RIKEN cDNA 1110001A05 gene | 2.65 | |
| 1451640_a_at | RIKEN cDNA 1500011J06 gene | 2.81 | |
| 1448882_at | RIKEN cDNA 0610009E20 gene | −2.49 | |
| 1450770_at | RIKEN cDNA 3632451O06 gene | −2.74 | |
| 1450904_at | RIKEN cDNA 0610041E09 gene | −2.35 | |
| 1427113_s_at | RIKEN cDNA 2700049H19 gene | −2.48 | |
| 1448179_at | Upregulated during skeletal muscle growth 5 | −3.22 | |
| 1455155_at | cDNA sequence BC040823 | −2.93 | |
| 1434057_at | Similar to NADH dehydrogenase (LOC230075), mRNA | −2.16 | |
| 1418101_a_at | Reticulon 3 | −2.90 | |
| 1423608_at | Integral membrane protein 2A | −3.52 |
PDGFA, Platelet-derived growth factor α.
Consistent with results obtained by the analysis of the motor neuronal NSC34 cell line stably transfected with vector expressing hSOD1 G93A (Kirby et al., 2005), at the late stage of the disease the transgenic mice show a marked degree of transcriptional repression. Key genes such as necdin (Ndn), transcription activation factor 9 RNA polymerase II (Taf9), RNA polymerase 1-1, and transforming growth factor β1-induced transcript 4 (Tgfb1i4) show decreased expression, although Ndn and Tgfb1i4 were upregulated at 60 d. The decreased expression of necdin was also been confirmed by Q-PCR (Fig. 1A).
Of particular interest are the alterations in genes implicated in axonal transport. There is increased expression of vimentin and Kif1b, along with the decreased expression of the light chain of the cytoplasmic dynein (Dnclc1). Kif1b upregulation has been confirmed by Q-PCR at both the symptomatic and the late stages of disease (Table 2), whereas Dnclc1 decrease is not significant at any of the stages analyzed.
At the 120 d stage of the disease, there is increased expression in some genes involved in the immune system, belonging to the first steps of the complement cascade (i.e., C1q and C4), and these changes were not detected by microarray analysis at earlier stages. C1q upregulation was selected for further analysis using Q-PCR, and, with this technique, a trend toward increased expression was detected at 90 d (p = 0.07) and significantly increased expression was observed at 120 d (p = 0.0098) (Table 2).
Interestingly, in support of the hypothesis that oxidative stress may play a role in ALS, genes involved in antioxidant activity and the stress response are significantly decreased, underlining a deficit in this important cellular defense. In contrast, protein degradation pathways are predominantly increased, providing additional evidence of cellular stress.
Surprisingly, because motor neurons are postmitotic cells, multiple cell cycle proteins including cyclins related to the cell cycle re-entry [e.g., cyclin L1 (Ccnl1)] (Redon et al., 2002) and regulating the first steps of the cell cycle [e.g., cyclins E2 and D2 (Ccne2 and Ccnd2)] show increased expression at this late stage of disease. Q-PCR data detected an age-dependent increase in the three cyclins from the presymptomatic stage of the disease, with undetectable expression levels of cyclin E2 in control tissues, as expected. At 90 d, although the increase in the expression of cyclin I and cyclin D2 is still detected, it does not reach statistical significance, which is reached at the late stage of disease as reported in the microarray study (Fig. 1B).
Comparison of gene expression changes at the three time points
Comparing the profiles of significant gene expression changes obtained for the three stages of the disease, it appears that many of the genes, the expression of which is upregulated at 60 d, are downregulated at 120 d (Fig. 2). Interestingly, oxidoreductase activity and transcriptional and translational processes seem to be the most affected by the progression of the disease, showing a complete inversion in the direction of gene expression changes from the 60 d to the 120 d time point.
Figure 2.

Gene ontology classification showing the number of transcripts upregulated and downregulated per category throughout the pathology. Bars on the left indicate the number of upregulated genes, and bars on the right indicate the number of downregulated genes, for each category. The transcripts most affected by the progression of the disease are those encoding for mitochondrial proteins (mito. proteins), proteins presenting oxidoreductase activity, and proteins related to the transcriptional function. These classes undergo a complete inversion in their regulation with the progression of the disease.
Significant changes in the expression level have been observed for 20 transcripts common to the apparent presymptomatic and the late stage of the disease: three of them belong to the carbohydrate metabolism (malate dehydrogenase 1, isopentenyl-diphosphate δ isomerase, E2 component of pyruvate dehydrogenase complex), and two have oxidoreductase activity (cytochrome c oxidase subunit VIc, NADH dehydrogenase (ubiquinone) 1α subcomplex 1). Although the microarray results show a significant increase in the expression of Mdh1 at the presymptomatic stage, Q-PCR detects a trend for increased expression at both presymptomatic and symptomatic stages.
Significant expression changes have been found in Ndn and Tgfb1i4, which were increased, respectively, 4.62- and 2.7-fold at 60 d and decreased 3-fold and 2-fold at 120 d. Ndn alterations have been also confirmed by Q-PCR (Fig. 1A)
Of particular interest are the changes in the expression of genes related to axonal transport. Vimentin and kinesin, upregulated at 60 d, are still both upregulated at 120 d, with an increase in the expression of vimentin with the progression of the disease and a slight decrease in the expression of kinesin (Table 2, Q-PCR results). In contrast, dynein cytoplasmic light chain 1, already downregulated at 90 d, undergoes a further decrease in expression at 120 d.
Also of interest is the striking downregulation of plexin (Plxdc1) throughout the three stages of disease. Q-PCR data confirm these results (Fig. 1C) and suggest that there are undetectable levels of Plxdc1 mRNA in the G93A motor neurons.
Comparing the 90 d with the 120 d results, two genes involved in cell cycle progression altered at the symptomatic stage are still altered at the late stage of the disease. However, whereas cyclin I is still upregulated, in contrast, protein phosphatase 3 catalytic subunit α, responsible for the G1/S transition, is upregulated at 90 d but is downregulated in the late phase of the disease.
Discussion
In this study, microarray technology, combined with LCM, has enabled detection of motor neuron-specific changes in multiple gene classes in response to the presence of the mutant SOD1 protein. The approach we have used has several key advantages over previous gene expression profiling studies in ALS and models of the disease: (1) analysis of gene expression changes specifically in motor neurons, uncontaminated by changes occurring in other cell types such as glia within the CNS; (2) analysis of gene expression profiles in motor neurons that have been extracted from an environment (lumbar spinal cord) where cross talk with neighboring glial cells could take place, which has important advantages over the study of transcriptional changes identified in motor neurons cultured in isolation; (3) the use of the murine model has allowed disease-related gene expression changes to be identified at various time points in the disease course, an approach not possible in human postmortem tissue; and (4) the transgenic murine model used in this study has been bred on a homogeneous genetic background, eliminating the potential confounding effects arising from mice with a mixed genetic background. By using this rigorous approach, we have identified novel and important cellular pathway changes at an early and presymptomatic stage in SOD1-related motor neuron injury and several changes at the late stage of motor neuron injury that have not previously been emphasized.
Previous studies aiming to determine transcriptional changes in transgenic mice using homogenates of whole spinal cord have primarily detected changes in inflammation and apoptosis-related genes (Malaspina et al., 2001; Olsen et al., 2001; Yoshihara et al., 2002). However, these results have not been confirmed by LCM and microarray studies conducted on a motor neuron population (Perrin et al., 2005). Similarly, gene expression profile studies conducted on human tissues using spinal cord gray matter (Dangond et al., 2004) show changes in keeping with astrogliosis that are absent from isolated motor neurons (Jiang et al., 2005). Several factors could account for the lack of detection of significant transcriptional changes in apoptotic genes in the present study. First, one of the most common hallmarks of apoptosis is the activation of the caspase cascade, a class of proteins that are activated by posttranslational proteolytic cleavage that is not detectable by a transcriptional study. Evidence suggests that the apoptotic machinery is constitutively present in the cell and is activated through environmental stimuli (e.g., unfolded protein accumulation and inhibition of survival pathways) (Desagher et al., 2005). An alternative explanation is that a pathway different to apoptosis is responsible for the motor neuronal death observed in the G93A mice. In fact, at the late stages of disease, we detected signs of reactivation of the cell cycle, a mechanism that might lead to cell loss without activation of apoptotic genes. Finally, changes in pro-apoptotic genes less than twofold may induce apoptosis but will not be reported as altered in this study.
In contrast to the previous report analyzing the motor neuronal transcriptome of SOD1 G93A mice (Perrin et al., 2005), we detected multiple transcriptional changes at the very early (presymptomatic) stages of the disease. This crucial difference may be determined by the different genetic background of the mouse models used in the two studies. The homogeneous background used in our study, in fact, greatly reduces the variability in the manifestation of the pathological signs typical of each stage of the disease. The disease onset in our strain (C57bL/6) is 75 ± 3 d, and in the B6SJL-Tg(SOD1-G93A)1Gur mixed background, used in the previous report, the published dataset disease onset was at 80 ± 10 d (Heiman-Patterson et al., 2005). This 12.5% variability in key parameters of the murine disease may lead, in the nonhomogeneous background, to a dispersion of information, because the mice analyzed may be at different stages of motor neuron degeneration and some variation in gene expression may arise as a direct result of the mixed background.
One of the most interesting findings of our study is the increased expression of genes involved in transcriptional and translational processes as well as of those involved in carbohydrate metabolism at 60 d, suggesting the activation of a strong cellular adaptive response. Upregulation of genes involved in Krebs cycle activity and the production of ATP supports the idea that motor neurons are undergoing a massive stress and are trying to compensate with a large increase in the capacity for ATP production. Along with an increase in genes involved in the regulation of carbohydrate metabolism, increased expression of multiple enzymes involved in fatty acid metabolism was identified in the microarray analysis. Upregulation of the mRNA of the two principal proteins involved in the translocation of mitochondrial preproteins (Hsp90 and Tom20) from the cytoplasm to the mitochondrion also suggests that motor neurons, in the presence of mutant SOD1, are attempting to increase the activity of the mitochondrial machinery. Although the primary source of energy of the neuron is glucose, when this supply either is depleted or is insufficient to meet the energy demand, ketone bodies, produced systemically, can be taken up by the CNS and used by neurons in place of glucose. After several weeks of starvation, ketones become the major neuronal energy source (Levin, 2006).
The gene expression changes in metabolic pathways identified in the present study are supported by several reports of functional metabolic studies in mouse models of ALS and human ALS patients. The reported improvement in motor performance obtained with a ketogenic diet tested on SOD1 G93A mice (Zhao et al., 2006) and the age-related reduction in adiposity observed in both SOD1 G93A and G86R mice, along with an increase in the levels of circulating ketone bodies at the early stages of disease (Dupuis et al., 2004b), support our findings indicating activation of the fatty acid metabolism along with the carbohydrate metabolism to satisfy a higher demand for ATP synthesis. These data are also in accordance with the finding that ALS patients have been shown to be hypermetabolic (Kasarskis et al., 1996; Desport et al., 2000, 2005), despite the reduction in muscle mass that occurs during the course of the disease.
Krebs cycle activation, fatty acid degradation, and ATP production are all processes related to the production and consequent detoxification of reactive oxygen species (ROS) (Andreyev et al., 2005). An unbalanced ratio between these processes can lead to the accumulation of free radicals, increasing physiological oxidative stress, one of the hallmarks of ALS (Barber et al., 2006). Previous investigations, conducted by our group in a motor neuronal NSC34 cell line stably transfected with vector expressing hSOD1 G93A, showed a general downregulation in the expression of antioxidant response proteins, the regulation of which is controlled by the nuclear factor erythroid 2-like 2 (Nrf2) (Kirby et al., 2005). It has also been reported that, in the spinal cord of SOD1 G93A rats, the Nrf2 nuclear level is increased in reactive astrocytes, and its activation in in vitro coculture has beneficial effects on motor neuron survival (Vargas et al., 2005). In our study, we could not detect altered expression of Nrf2 or key Nrf2-regulated genes (e.g., quinone oxidoreductase, glutathione S-transferase). However, evidence shows that astrocytes have a higher antioxidant potential compared with neurons (Shih et al., 2003), which may lead to the hypothesis that, to face the increase in the oxidative stress, motor neurons rely on astrocyte activation and support (Pehar et al., 2005). The evident need of motor neurons for molecules capable of responding to the oxidative insult represents another possible therapeutic approach.
Another key finding of the present study is the downregulation of the ATP synthase δ subunit at the presymptomatic stage of the disease. Decreased expression of this gene has also been reported in human ALS (Dangond et al., 2004). The transcription factors and the mechanisms directly involved in the transcriptional regulation of this subunit are still unknown. Nevertheless, one report showed that Atp5d is copper sensitive (Chen et al., 2002) and a low concentration of mitochondrial copper can lead to its downregulation without altering the expression level of other subunits of the respiratory chain. This characteristic might affect Atp5d regulation in SOD1-related cases of ALS, in which altered affinity of mutant SOD1 for copper has been described (Watanabe et al., 2007). Atp5d has a crucial role in the ATP production process at the end of the mitochondrial respiratory chain. In yeast, partial depletion of the ATP synthase δ subunit can lead to a massive proton leak across the mitochondrial inner membrane, without affecting the ATP synthase complex structure (Duvezin-Caubet et al., 2003). This change represents a potential mechanism for the early generation of motor neuronal oxidative stress (Fig. 3A). Because these alterations occur at the very early stages of disease, finding a way to supply energy to motor neurons, to ameliorate their pathological need for increased mitochondrial respiratory chain activity, or to upregulate the expression of the key mitochondrial ATP synthase protein subunit Atp5d may represent logical approaches in the therapeutic protection of motor neurons.
Figure 3.
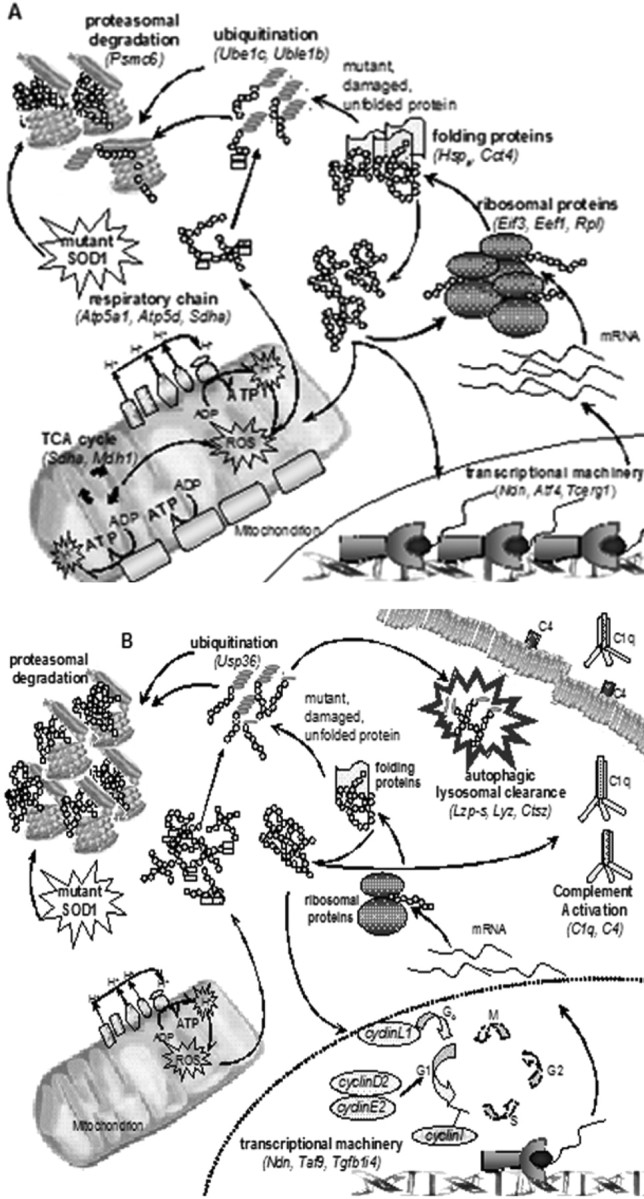
A, Motor neurons isolated from G93A mice at 60 d show upregulation in several classes of genes delineating what is likely to be happening in these cells under stress. There is upregulation of the transcriptional machinery, along with upregulation of translation-related ribosomal and folding proteins, as the motor neuron attempts to compensate for the ongoing cellular stress. All these mechanisms require ATP, provoking a massive increase in the work load of mitochondria, leading to upregulation of carbohydrate metabolism and respiratory chain activity, which in turn causes increased ROS production. The observed imbalance among the subunits forming the ATP synthase complex, shown by downregulation of the δ subunit, will generate additional oxidative stress, with consequent production of oxidized proteins. These are then ubiquitinated and targeted for proteasomal degradation. B, Over time, the accumulation of damaged proteins and ROS is likely to cause a general collapse in cellular functioning, leading to downregulation of the compensatory pathways previously activated and leaving the cell with decreased energy and protein turnover. The cell increases protein degradation functions, with activation of the lysosomal machinery. Production and secretion of some subunits of the complement cascade are important signals of cellular stress for neighboring cells. The final abortive attempt at survival comes through activation of the cell cycle, with upregulation of cyclin L1 (involved in the transition from the quiescent state, G0, to the first phase of the cell cycle, G1) and cyclins D2 and E2 (involved in the progression of the cell cycle through G1 phase). The upregulation of cyclin I suggests that motor neurons are trying to exercise negative control on the transition between the G1 and S phase to prevent the abnormal progression through the cell cycle. Atf4, Activating transcription factor 4; Atp5a1, ATP synthase F1 complex α1 subunit; Atp5d, ATP synthase F1 complex δ subunit; Cct4, chaperonin subunit 4; Ctsz, cathepsin-Z; Eef1, eukaryotic translation elongation factor 1; Eif3, eukaryotic translation initiation factor 3; Hsp, heat shock protein; Lyz, lysozyme; Lzp-s, P-lysozyme structural; Mdh1, malate dehydrogenase 1; Ndn, necdin; Psmc6, proteasome 26S; Rpl, ribosomal protein L; Sdha, succinate dehydrogenase complex subunit A; Taf9, transcription activator factor 9; Tcerg1, transcription elongation regulator 1 (CA150); Tgfb1i4, transforming growth factor β1-induced transcript 4; Ube1c, ubiquitin-activating enzyme E1C; Uble1b, ubiquitin-like activating enzyme E1B; Usp36, ubiquitin-specific preotease 36; H+, hydrogen ion; TCA, tricarboxylic acid.
Our data demonstrate that this observed early upregulation of genes related to mitochondrial ATP production markedly decreases in the late stages of motor neuron injury. This observation is in accordance with previously reported functional studies of the activities of the mitochondrial respiratory chain showing a striking decrease in respiratory chain activity in the spinal cord of ALS patients (Wiedemann et al., 2002), in a motor neuronal cell model stably transfected to express mutant SOD1 at levels equivalent to those seen in human ALS (Menzies et al., 2002) and in a transgenic mutant SOD1 mouse model at the symptomatic and late stages of disease, but not at the presymptomatic stage (Mattiazzi et al., 2002).
Consistent with results obtained by Perrin et al. (2005), vimentin was increased at all stages, including the presymptomatic stage. Vimentin is an intermediate filament playing an important role in the retrograde transport of mitogen-activated protein kinases in the presence of injury signals (Eran Perlson, 2005) and cytoskeletal organization in both physiological and pathological conditions (Helfand et al., 2004). The increase in vimentin, along with the increased expression of kinesin 1b and 5b (responsible for the anterograde transport through the axon), is consistent during the progression of motor neuron injury. In contrast, the cytoplasmic light chain of dynein, which is involved in retrograde transport along the axon, shows a consistent decrease of 2-fold at the symptomatic stage and of 2.6-fold at the late stage of the disease, although these alterations were not confirmed by Q-PCR (Table 2). These gene expression changes could underlie a dysregulation in the balance between anterograde and retrograde axonal transport functions. Upregulation of the KIF family has been reported in other studies (Dupuis et al., 2000) and may occur as a compensatory response to the impairment reported in fast anterograde transport observed from the presymptomatic stage in the G93A mouse model (Warita et al., 1999). The observed downregulation of dynein, on the contrary, is consistent with functional data showing progressive reduction in the activity of this protein, related to consequent muscular weakness in the G93A mouse model (Ligon et al., 2005). Interestingly, cytoplasmic dynein and kinesin are involved in a specific and direct interaction, which may have a role in regulation of the coordination of anterograde and retrograde transport (Ligon et al., 2004). To underline the ongoing axonal impairment, there is the dramatic downregulation of plexin. The plexin family is a very large group of proteins involved in many cellular events, including axonal repulsion, axonal attraction, cell migration, axon pruning, and synaptic plasticity (Waimey and Cheng, 2006). They participate in nervous system development, guiding synaptogenesis and formation of neuromuscular junctions at the embryonic stage through pruning of excessive axon terminal branches (Luo and O'Leary, 2005). After their initial formation, synapses are continuously modified throughout life, and plexins play a fundamental role in synaptic plasticity (Waimey and Cheng, 2006). Downregulation of Plxdc1 throughout the disease, from the presymptomatic stage, may indicate deficient capacity for axonal guidance and limited synaptic plasticity, which could be a cause or consequence of axonal transport impairment.
The relative paucity of gene expression changes observed at the 90 d time point, associated with the onset of motor weakness, may be the result of the presence in the spinal cord of cells at different stages of degeneration, producing an overall balance in the detection of transcripts related to the pathways activated at this stage. After this phase, the late stage of the disease is characterized by the downregulation of many genes that were upregulated at the presymptomatic stage (Fig. 2).
At 120 d, for example, genes regulating translational and transcriptional processes are generally downregulated, as well as genes related to protein folding. The protein degradation system is still partially upregulated with increases in lysosomal-related proteins cathepsin B and Z. However, whereas at the early stage of the disease increased expression of the ubiquitin–proteasome system was detected, at the late stage, the activation of cathepsins is predominant (Fig. 3B). Cathepsins are cysteine proteases involved in the activation of the endosome–lysosome system, which has been reported in the late stages of several neurological diseases (Mantle et al., 1995; Kikuchi et al., 2003).
A striking feature of the 120 d time point is the number of genes differentially expressed that are involved in the immune response. Some of the genes showing increased expression belong to the first steps of the complement activation pathway, supporting findings reported by Goldknopf et al. (2006) of complement subunits present in the serum of ALS patients as biomarkers of disease. Although the activation of this pathway in ALS is already known, our study shows that, surprisingly, motoneurons can actively contribute to the enhancement of the complement cascade through production of the subunits activating the entire pathway (i.e., C1q and C4). Synthesis of complement subunits by neurons has been previously reported in other neurological disorders (Gasque et al., 2000). Nevertheless, this is the first time that this mechanism is reported in ALS. In fact, complement cascade activation has always been attributed exclusively to astrocytes, microglia and T-cells (McGeer and McGeer, 2002). Inhibition of complement could be a potential target to reduce the negative effect of the inflammatory cascade and ameliorate motor neuron injury. In fact, other therapies aiming to reduce the inflammatory cascade have been successful in the G93A mouse model (Kriz et al., 2002; Zhu et al., 2002; Klivenyi et al., 2004)
Also of interest at the 120 d time point is the increased expression of cyclin family members, including cyclins D2, E2, I, and L1 (Fig. 3B). This result is surprising considering that the cell population analyzed is composed of motor neurons that are postmitotic. Nevertheless, there are many reports highlighting cell cycle dysregulation as a common feature associated with neuronal death in diseases such as ataxia-telangiectasia and Alzheimer's disease (Herrup and Busser, 1995; Liu and Greene, 2001; Afroze et al., 2003; Ranganathan and Bowser, 2003; Yang and Herrup, 2005). In ALS, some evidence has been provided of cyclin system dysregulation in postmortem human brain and spinal cord (Nguyen et al., 2003). Within this group of genes, there is early upregulation of cyclin I, D2, and E2, which are significantly differentially expressed from the 60 d time point (as revealed by Q-PCR). In particular, cyclin I, along with cyclins G1 and G2, is expressed in terminally differentiated tissues including brain and muscle, suggesting that its task is to keep the cells in a quiescent state. Its increased expression, along with the increased expression of Ppp3ca at 90 d, seems to be related to G1/S phase cell cycle arrest (Bennin et al., 2002). Cyclins D2 and E2 are involved in progression through the G1 phase. Moreover, cyclin D2 expression has been shown to be specific for the regulation of the cell cycle in adult neurons, compared with cyclins D1 and D3, which are also involved in neonatal neurogenesis (Kowalczyk et al., 2004). The increased expression of the cyclin system at the late stage of disease suggests a possible mechanism for motor neuronal death in ALS, with cell death resulting from an unsuccessful attempt to re-enter the cell cycle (Becker and Bonni, 2004) (Fig. 3B). In this case, drugs directed toward the inhibition of the cell cycle may be of value to counteract the selective motoneuronal death characteristic of this disease, as suggested by in vitro studies on nontransgenic motor neurons (Appert-Collin et al., 2006).
In summary, this gene expression profiling study shows that motor neurons isolated from the spinal cord of SOD1 G93A mice, in the presymptomatic stages, try to respond to the ongoing stress by upregulating several pathways (e.g., metabolism, transcription, and protein synthesis) to compensate for the impairment or damage produced by the toxic gain of function of mutant SOD1. Increased energy (ATP) production is required to sustain all these mechanisms. The compensatory mechanisms to increase ATP production are likely to be associated with a high rate of ROS production with amplification of the oxidative damage. The upregulation of the ubiquitin–proteasome system is in keeping with this proposed pathophysiology. The cellular oxidative stress is likely to be further exacerbated by the observed downregulation of the δ subunit of the ATP synthesis complex, which may result in additional perturbation of mitochondrial function and ROS generation (Fig. 3A). The equilibrium between dysfunctional changes and compensatory changes seems to be balanced in the first stage of the disease, but later in the disease course, the compensatory mechanisms fail. At this stage, there is inability to handle the rising cellular stress and activation of the complement cascade, which is an important signal for neighboring cells. A last desperate attempt to survive is the unsuccessful re-entry into the cell cycle, with activation of a mechanism that will inevitably cause cell death (Fig. 3B).
In conclusion, our study highlights important changes in cellular pathways occurring at specific time points during SOD1-related motor neuron injury. Our findings indicate that several potential therapeutic interventions may be helpful in the neuroprotection of motor neurons at the different stages of the disease. One potential approach could be the provision of a diet designed to supply a balanced combination of substrates for the mitochondrial machinery, to respond to the requirement of motor neurons under stress for ATP. In addition, therapies aimed at supporting the translocation of mitochondrial preproteins to the mitochondrion, enhancing the capacity for ATP production, and supplementation of antioxidant compounds, aiming to reduce mitochondrial oxidative stress, are likely to help the adaptive response of injured motor neurons. Another possible target could be inhibition of the complement cascade, aiming to reduce the inflammatory response activated in the nervous system of ALS patients (Graves et al., 2004). An additional potential approach may be tissue-specific delivery of drugs able to inhibit either the re-entry into or the progression of the cell cycle, which seems to be a molecular signature of late-stage neuronal injury. Because of the complexity of this pathology and the number of mechanisms involved in motor neuron degeneration, a combination of multiple drugs, directed toward different pathways, is likely to represent the most promising neuroprotective strategy.
Footnotes
This work was supported by the Motor Neurone Disease Association (MNDA) and the Wellcome Trust. L.F. and J.K. were supported by the MNDA. P.J.S. was supported by the Wellcome Trust and the MNDA.
References
- Afroze T, Yang LL, Wang C, Gros R, Kalair W, Hoque AN, Mungrue IN, Zhu Z, Husain M. Calcineurin-independent regulation of plasma membrane Ca2+ ATPase-4 in the vascular smooth muscle cell cycle. Am J Physiol Cell Physiol. 2003;285:C88–C95. doi: 10.1152/ajpcell.00518.2002. [DOI] [PubMed] [Google Scholar]
- Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc) 2005;70:200–214. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- Appert-Collin A, Hugel B, Levy R, Niederhoffer N, Coupin G, Lombard Y, Andre P, Poindron P, Gies JP. Cyclin dependent kinase inhibitors prevent apoptosis of postmitotic mouse motoneurons. Life Sci. 2006;79:484–490. doi: 10.1016/j.lfs.2006.01.032. [DOI] [PubMed] [Google Scholar]
- Barber SC, Mead RJ, Shaw PJ. Oxidative stress in ALS: a mechanism of neurodegeneration and a therapeutic target. Biochim Biophys Acta. 2006;1762:1051–1067. doi: 10.1016/j.bbadis.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Basso M, Massignan T, Samengo G, Cheroni C, De Biasi S, Salmona M, Bendotti C, Bonetto V. Insoluble mutant SOD1 is partly oligoubiquitinated in amyotrophic lateral sclerosis mice. J Biol Chem. 2006;281:33325–33335. doi: 10.1074/jbc.M603489200. [DOI] [PubMed] [Google Scholar]
- Becker EB, Bonni A. Cell cycle regulation of neuronal apoptosis in development and disease. Prog Neurobiol. 2004;72:1–25. doi: 10.1016/j.pneurobio.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Bennin DA, Don AS, Brake T, McKenzie JL, Rosenbaum H, Ortiz L, DePaoli-Roach AA, Horne MC. Cyclin G2 associates with protein phosphatase 2A catalytic and regulatory B′ subunits in active complexes and induces nuclear aberrations and a G1/S phase cell cycle arrest. J Biol Chem. 2002;277:27449–27467. doi: 10.1074/jbc.M111693200. [DOI] [PubMed] [Google Scholar]
- Bittner RE, Anderson LV, Burkhardt E, Bashir R, Vafiadaki E, Ivanova S, Raffelsberger T, Maerk I, Hoger H, Jung M, Karbasiyan M, Storch M, Lassmann H, Moss JA, Davison K, Harrison R, Bushby KM, Reis A. Dysferlin deletion in SJL mice (SJL-Dysf) defines a natural model for limb girdle muscular dystrophy 2B. Nat Genet. 1999;23:141–142. doi: 10.1038/13770. [DOI] [PubMed] [Google Scholar]
- Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Browne SE, Yang L, DiMauro JP, Fuller SW, Licata SC, Beal MF. Bioenergetic abnormalities in discrete cerebral motor pathways presage spinal cord pathology in the G93A SOD1 mouse model of ALS. Neurobiol Dis. 2006;22:599–610. doi: 10.1016/j.nbd.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Chen X, Jennings DB, Medeiros DM. Impaired cardiac mitochondrial membrane potential and respiration in copper-deficient rats. J Bioenerg Biomembr. 2002;34:397–406. doi: 10.1023/a:1021258204921. [DOI] [PubMed] [Google Scholar]
- Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2:806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- Dangond F, Hwang D, Camelo S, Pasinelli P, Frosch MP, Stephanopoulos G, Stephanopoulos G, Brown RH, Jr, Gullans SR. Molecular signature of late-stage human ALS revealed by expression profiling of postmortem spinal cord gray matter. Physiol Genomics. 2004;16:229–239. doi: 10.1152/physiolgenomics.00087.2001. [DOI] [PubMed] [Google Scholar]
- Desagher S, Severac D, Lipkin A, Bernis C, Ritchie W, Le Digarcher A, Journot L. Genes regulated in neurons undergoing transcription-dependent apoptosis belong to signaling pathways rather than the apoptotic machinery. J Biol Chem. 2005;280:5693–5702. doi: 10.1074/jbc.M408971200. [DOI] [PubMed] [Google Scholar]
- Desport JC, Preux PM, Truong CT, Courat L, Vallat JM, Couratier P. Nutritional assessment and survival in ALS patients. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:91–96. doi: 10.1080/14660820050515386. [DOI] [PubMed] [Google Scholar]
- Desport JC, Torny F, Lacoste M, Preux PM, Couratier P. Hypermetabolism in ALS: correlations with clinical and paraclinical parameters. Neurodegener Dis. 2005;2:202–207. doi: 10.1159/000089626. [DOI] [PubMed] [Google Scholar]
- Dupuis L, de Tapia M, Rene F, Lutz-Bucher B, Gordon JW, Mercken L, Pradier L, Loeffler JP. Differential screening of mutated SOD1 transgenic mice reveals early up-regulation of a fast axonal transport component in spinal cord motor neurons. Neurobiol Dis. 2000;7:274–285. doi: 10.1006/nbdi.2000.0292. [DOI] [PubMed] [Google Scholar]
- Dupuis L, Muller A, Meininger V, Loeffler JP. Molecular mechanisms of amyotrophic lateral sclerosis: recent contributions from studies in animal models. Rev Neurol (Paris) 2004a;160:35–43. doi: 10.1016/s0035-3787(04)70845-0. [DOI] [PubMed] [Google Scholar]
- Dupuis L, Oudart H, Rene F, Gonzalez de Aguilar JL, Loeffler JP. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: benefit of a high-energy diet in a transgenic mouse model. Proc Natl Acad Sci USA. 2004b;101:11159–11164. doi: 10.1073/pnas.0402026101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvezin-Caubet S, Caron M, Giraud MF, Velours J, di Rago JP. The two rotor components of yeast mitochondrial ATP synthase are mechanically coupled by subunit delta. Proc Natl Acad Sci USA. 2003;100:13235–13240. doi: 10.1073/pnas.2135169100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eran Perlson SH, Keren Ben-Yaakov, Yael Segal-Ruder, Rony Seger, Mike Fainzilber. Vimentin-dependent spatial translocation of an activated MAP kinase in injured nerve. Neuron. 2005;45:715–726. doi: 10.1016/j.neuron.2005.01.023. [DOI] [PubMed] [Google Scholar]
- Gasque P, Dean YD, McGreal EP, VanBeek J, Morgan BP. Complement components of the innate immune system in health and disease in the CNS. Immunopharmacology. 2000;49:171–186. doi: 10.1016/s0162-3109(00)80302-1. [DOI] [PubMed] [Google Scholar]
- Goldknopf IL, Sheta EA, Bryson J, Folsom B, Wilson C, Duty J, Yen AA, Appel SH. Complement C3c and related protein biomarkers in amyotrophic lateral sclerosis and Parkinson's disease. Biochem Biophys Res Commun. 2006;342:1034–1039. doi: 10.1016/j.bbrc.2006.02.051. [DOI] [PubMed] [Google Scholar]
- Gong YH, Parsadanian AS, Andreeva A, Snider WD, Elliott JL. Restricted expression of G86R Cu/Zn superoxide dismutase in astrocytes results in astrocytosis but does not cause motoneuron degeneration. J Neurosci. 2000;20:660–665. doi: 10.1523/JNEUROSCI.20-02-00660.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves MC, Fiala M, Dinglasan LA, Liu NQ, Sayre J, Chiappelli F, van Kooten C, Vinters HV. Inflammation in amyotrophic lateral sclerosis spinal cord and brain is mediated by activated macrophages, mast cells and T cells. Amyotroph Lateral Scler Other Motor Neuron Disord. 2004;5:213–219. doi: 10.1080/14660820410020286. [DOI] [PubMed] [Google Scholar]
- Heiman-Patterson TD, Deitch JS, Blankenhorn EP, Erwin KL, Perreault MJ, Alexander BK, Byers N, Toman I, Alexander GM. Background and gender effects on survival in the TgN(SOD1–G93A)1Gur mouse model of ALS. J Neurol Sci. 2005;236:1–7. doi: 10.1016/j.jns.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Helfand BT, Chang L, Goldman RD. Intermediate filaments are dynamic and motile elements of cellular architecture. J Cell Sci. 2004;117:133–141. doi: 10.1242/jcs.00936. [DOI] [PubMed] [Google Scholar]
- Herrup K, Busser JC. The induction of multiple cell cycle events precedes target-related neuronal death. Development. 1995;121:2385–2395. doi: 10.1242/dev.121.8.2385. [DOI] [PubMed] [Google Scholar]
- Jiang YM, Yamamoto M, Kobayashi Y, Yoshihara T, Liang Y, Terao S, Takeuchi H, Ishigaki S, Katsuno M, Adachi H, Niwa J, Tanaka F, Doyu M, Yoshida M, Hashizume Y, Sobue G. Gene expression profile of spinal motor neurons in sporadic amyotrophic lateral sclerosis. Ann Neurol. 2005;57:236–251. doi: 10.1002/ana.20379. [DOI] [PubMed] [Google Scholar]
- Kabashi E, Durham HD. Failure of protein quality control in amyotrophic lateral sclerosis. Biochim Biophys Acta. 2006;1762:1038–1050. doi: 10.1016/j.bbadis.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Kadoya T, Kishida S, Fukui A, Hinoi T, Michiue T, Asashima M, Kikuchi A. Inhibition of Wnt signaling pathway by a novel axin-binding protein. J Biol Chem. 2000;275:37030–37037. doi: 10.1074/jbc.M005984200. [DOI] [PubMed] [Google Scholar]
- Kasarskis EJ, Berryman S, Vanderleest JG, Schneider AR, McClain CJ. Nutritional status of patients with amyotrophic lateral sclerosis: relation to the proximity of death. Am J Clin Nutr. 1996;63:130–137. doi: 10.1093/ajcn/63.1.130. [DOI] [PubMed] [Google Scholar]
- Kikuchi H, Yamada T, Furuya H, Doh-ura K, Ohyagi Y, Iwaki T, Kira J. Involvement of cathepsin B in the motor neuron degeneration of amyotrophic lateral sclerosis. Acta Neuropathol (Berl) 2003;105:462–468. doi: 10.1007/s00401-002-0667-9. [DOI] [PubMed] [Google Scholar]
- Kirby J, Halligan E, Baptista MJ, Allen S, Heath PR, Holden H, Barber SC, Loynes CA, Wood-Allum CA, Lunec J, Shaw PJ. Mutant SOD1 alters the motor neuronal transcriptome: implications for familial ALS. Brain. 2005;128:1686–1706. doi: 10.1093/brain/awh503. [DOI] [PubMed] [Google Scholar]
- Klivenyi P, Kiaei M, Gardian G, Calingasan NY, Beal MF. Additive neuroprotective effects of creatine and cyclooxygenase 2 inhibitors in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurochem. 2004;88:576–582. doi: 10.1046/j.1471-4159.2003.02160.x. [DOI] [PubMed] [Google Scholar]
- Konig HG, Kogel D, Rami A, Prehn JH. TGF-{beta}1 activates two distinct type I receptors in neurons: implications for neuronal NF-{kappa}B signaling. J Cell Biol. 2005;168:1077–1086. doi: 10.1083/jcb.200407027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczyk A, Filipkowski RK, Rylski M, Wilczynski GM, Konopacki FA, Jaworski J, Ciemerych MA, Sicinski P, Kaczmarek L. The critical role of cyclin D2 in adult neurogenesis. J Cell Biol. 2004;167:209–213. doi: 10.1083/jcb.200404181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriz J, Nguyen MD, Julien JP. Minocycline slows disease progression in a mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2002;10:268–278. doi: 10.1006/nbdi.2002.0487. [DOI] [PubMed] [Google Scholar]
- Levin BE. Metabolic sensing neurons and the control of energy homeostasis. Physiol Behav. 2006;89:486–489. doi: 10.1016/j.physbeh.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Ligon LA, Tokito M, Finklestein JM, Grossman FE, Holzbaur EL. A direct interaction between cytoplasmic dynein and kinesin I may coordinate motor activity. J Biol Chem. 2004;279:19201–19208. doi: 10.1074/jbc.M313472200. [DOI] [PubMed] [Google Scholar]
- Ligon LA, LaMonte BH, Wallace KE, Weber N, Kalb RG, Holzbaur EL. Mutant superoxide dismutase disrupts cytoplasmic dynein in motor neurons. NeuroReport. 2005;16:533–536. doi: 10.1097/00001756-200504250-00002. [DOI] [PubMed] [Google Scholar]
- Lino MM, Schneider C, Caroni P. Accumulation of SOD1 mutants in postnatal motoneurons does not cause motoneuron pathology or motoneuron disease. J Neurosci. 2002;22:4825–4832. doi: 10.1523/JNEUROSCI.22-12-04825.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu DX, Greene LA. Neuronal apoptosis at the G1/S cell cycle checkpoint. Cell Tissue Res. 2001;305:217–228. doi: 10.1007/s004410100396. [DOI] [PubMed] [Google Scholar]
- Luo L, O'Leary DD. Axon retraction and degeneration in development and disease. Annu Rev Neurosci. 2005;28:127–156. doi: 10.1146/annurev.neuro.28.061604.135632. [DOI] [PubMed] [Google Scholar]
- Malaspina A, Kaushik N, de Belleroche J. Differential expression of 14 genes in amyotrophic lateral sclerosis spinal cord detected using gridded cDNA arrays. J Neurochem. 2001;77:132–145. doi: 10.1046/j.1471-4159.2001.t01-1-00231.x. [DOI] [PubMed] [Google Scholar]
- Mantle D, Falkous G, Ishiura S, Perry RH, Perry EK. Comparison of cathepsin protease activities in brain tissue from normal cases and cases with Alzheimer's disease, Lewy body dementia, Parkinson's disease and Huntington's disease. J Neurol Sci. 1995;131:65–70. doi: 10.1016/0022-510x(95)00035-z. [DOI] [PubMed] [Google Scholar]
- Mattiazzi M, D'Aurelio M, Gajewski CD, Martushova K, Kiaei M, Beal MF, Manfredi G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J Biol Chem. 2002;277:29626–29633. doi: 10.1074/jbc.M203065200. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. Inflammatory processes in amyotrophic lateral sclerosis. Muscle Nerve. 2002;26:459–470. doi: 10.1002/mus.10191. [DOI] [PubMed] [Google Scholar]
- Menzies FM, Cookson MR, Taylor RW, Turnbull DM, Chrzanowska-Lightowlers ZM, Dong L, Figlewicz DA, Shaw PJ. Mitochondrial dysfunction in a cell culture model of familial amyotrophic lateral sclerosis. Brain. 2002;125:1522–1533. doi: 10.1093/brain/awf167. [DOI] [PubMed] [Google Scholar]
- Nguyen MD, Boudreau M, Kriz J, Couillard-Despres S, Kaplan DR, Julien JP. Cell cycle regulators in the neuronal death pathway of amyotrophic lateral sclerosis caused by mutant superoxide dismutase 1. J Neurosci. 2003;23:2131–2140. doi: 10.1523/JNEUROSCI.23-06-02131.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen MK, Roberds SL, Ellerbrock BR, Fleck TJ, McKinley DK, Gurney ME. Disease mechanisms revealed by transcription profiling in SOD1–G93A transgenic mouse spinal cord. Ann Neurol. 2001;50:730–740. doi: 10.1002/ana.1252. [DOI] [PubMed] [Google Scholar]
- Parkinson DB, Bhaskaran A, Droggiti A, Dickinson S, D'Antonio M, Mirsky R, Jessen KR. Krox-20 inhibits Jun-NH2-terminal kinase/c-Jun to control Schwann cell proliferation and death. J Cell Biol. 2004;164:385–394. doi: 10.1083/jcb.200307132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehar M, Vargas MR, Cassina P, Barbeito AG, Beckman JS, Barbeito L. Complexity of astrocyte-motor neuron interactions in amyotrophic lateral sclerosis. Neurodegener Dis. 2005;2:139–146. doi: 10.1159/000089619. [DOI] [PubMed] [Google Scholar]
- Perrin FE, Boisset G, Docquier M, Schaad O, Descombes P, Kato AC. No widespread induction of cell death genes occurs in pure motoneurons in an amyotrophic lateral sclerosis mouse model. Hum Mol Genet. 2005;14:3309–3320. doi: 10.1093/hmg/ddi357. [DOI] [PubMed] [Google Scholar]
- Pramatarova A, Laganiere J, Roussel J, Brisebois K, Rouleau GA. Neuron-specific expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor impairment. J Neurosci. 2001;21:3369–3374. doi: 10.1523/JNEUROSCI.21-10-03369.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MA. CKI, there's more than one: casein kinase I family members in Wnt and Hedgehog signaling. Genes Dev. 2006;20:399–410. doi: 10.1101/gad.1394306. [DOI] [PubMed] [Google Scholar]
- Ranganathan S, Bowser R. Alterations in G(1) to S phase cell-cycle regulators during amyotrophic lateral sclerosis. Am J Pathol. 2003;162:823–835. doi: 10.1016/S0002-9440(10)63879-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redon R, Hussenet T, Bour G, Caulee K, Jost B, Muller D, Abecassis J, du Manoir S. Amplicon mapping and transcriptional analysis pinpoint cyclin L as a candidate oncogene in head and neck cancer. Cancer Res. 2002;62:6211–6217. [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Shaw PJ. Molecular and cellular pathways of neurodegeneration in motor neurone disease. J Neurol Neurosurg Psychiatry. 2005;76:1046–1057. doi: 10.1136/jnnp.2004.048652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AY, Johnson DA, Wong G, Kraft AD, Jiang L, Erb H, Johnson JA, Murphy TH. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J Neurosci. 2003;23:3394–3406. doi: 10.1523/JNEUROSCI.23-08-03394.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinder GA, Lacourse MC, Minotti S, Durham HD. Mutant Cu/Zn-superoxide dismutase proteins have altered solubility and interact with heat shock/stress proteins in models of amyotrophic lateral sclerosis. J Biol Chem. 2001;276:12791–12796. doi: 10.1074/jbc.M010759200. [DOI] [PubMed] [Google Scholar]
- Van Gelder RN, von Zastrow ME, Yool A, Dement WC, Barchas JD, Eberwine JH. Amplified RNA synthesized from limited quantities of heterogeneous cDNA. Proc Natl Acad Sci USA. 1990;87:1663–1667. doi: 10.1073/pnas.87.5.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas MR, Pehar M, Cassina P, Martinez-Palma L, Thompson JA, Beckman JS, Barbeito L. Fibroblast growth factor-1 induces heme oxygenase-1 via nuclear factor erythroid 2-related factor 2 (Nrf2) in spinal cord astrocytes: consequences for motor neuron survival. J Biol Chem. 2005;280:25571–25579. doi: 10.1074/jbc.M501920200. [DOI] [PubMed] [Google Scholar]
- Vedrenne C, Hauri HP. Morphogenesis of the endoplasmic reticulum: beyond active membrane expansion. Traffic. 2006;7:639–646. doi: 10.1111/j.1600-0854.2006.00419.x. [DOI] [PubMed] [Google Scholar]
- Waimey KE, Cheng HJ. Axon pruning and synaptic development: how are they per-plexin? Neuroscientist. 2006;12:398–409. doi: 10.1177/1073858406292631. [DOI] [PubMed] [Google Scholar]
- Warita H, Itoyama Y, Abe K. Selective impairment of fast anterograde axonal transport in the peripheral nerves of asymptomatic transgenic mice with a G93A mutant SOD1 gene. Brain Res. 1999;819:120–131. doi: 10.1016/s0006-8993(98)01351-1. [DOI] [PubMed] [Google Scholar]
- Watanabe S, Nagano S, Duce J, Kiaei M, Li QX, Tucker SM, Tiwari A, Brown RH, Jr, Beal MF, Hayward LJ, Culotta VC, Yoshihara S, Sakoda S, Bush AI. Increased affinity for copper mediated by cysteine 111 in forms of mutant superoxide dismutase 1 linked to amyotrophic lateral sclerosis. Free Radic Biol Med. 2007;42:1534–1542. doi: 10.1016/j.freeradbiomed.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Wiedemann FR, Manfredi G, Mawrin C, Beal MF, Schon EA. Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J Neurochem. 2002;80:616–625. doi: 10.1046/j.0022-3042.2001.00731.x. [DOI] [PubMed] [Google Scholar]
- Winter M, Milne D, Dias S, Kulikov R, Knippschild U, Blattner C, Meek D. Protein kinase CK1delta phosphorylates key sites in the acidic domain of murine double-minute clone 2 protein (MDM2) that regulate p53 turnover. Biochemistry. 2004;43:16356–16364. doi: 10.1021/bi0489255. [DOI] [PubMed] [Google Scholar]
- Wolff S, Xiao Z, Wittau M, Sussner N, Stoter M, Knippschild U. Interaction of casein kinase 1 delta (CK1 delta) with the light chain LC2 of microtubule associated protein 1A (MAP1A) Biochim Biophys Acta. 2005;1745:196–206. doi: 10.1016/j.bbamcr.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Wood JD, Beaujeux TP, Shaw PJ. Protein aggregation in motor neurone disorders. Neuropathol Appl Neurobiol. 2003;29:529–545. doi: 10.1046/j.0305-1846.2003.00518.x. [DOI] [PubMed] [Google Scholar]
- Yang Y, Herrup K. Loss of neuronal cell cycle control in ataxia-telangiectasia: a unified disease mechanism. J Neurosci. 2005;25:2522–2529. doi: 10.1523/JNEUROSCI.4946-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara T, Ishigaki S, Yamamoto M, Liang Y, Niwa J, Takeuchi H, Doyu M, Sobue G. Differential expression of inflammation- and apoptosis-related genes in spinal cords of a mutant SOD1 transgenic mouse model of familial amyotrophic lateral sclerosis. J Neurochem. 2002;80:158–167. doi: 10.1046/j.0022-3042.2001.00683.x. [DOI] [PubMed] [Google Scholar]
- Young JC, Hoogenraad NJ, Hartl FU. Molecular chaperones Hsp90 and Hsp70 deliver preproteins to the mitochondrial import receptor Tom70. Cell. 2003;112:41–50. doi: 10.1016/s0092-8674(02)01250-3. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Lange DJ, Voustianiouk A, MacGrogan D, Ho L, Suh J, Humala N, Thiyagarajan M, Wang J, Pasinetti GM. A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC Neurosci. 2006;7:29. doi: 10.1186/1471-2202-7-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Stavrovskaya IG, Drozda M, Kim BY, Ona V, Li M, Sarang S, Liu AS, Hartley DM, Wu DC, Gullans S, Ferrante RJ, Przedborski S, Kristal BS, Friedlander RM. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417:74–78. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]