

Abstract
Implementation in Europe of large-scale testing to detect bovine spongiform encephalopathy (BSE)-infected cattle and prevent the transmission of this prion disease to humans has recently led to the discovery of novel types of bovine prions. We characterized atypical isolates called BSE L-type by analyzing their molecular and neuropathological properties during transmission to several mouse lines transgenic for the prion protein (PrP). Unexpectedly, such isolates acquired strain features closely similar to those of BSE-type agents when propagated in mice expressing ovine PrP, although they retained phenotypic traits distinct from BSE in other lines, including bovine PrP mice. These findings further underline the relationship between the crossing of species barrier and prion strain diversification, and, although the origin of the epidemic BSE agent has only been speculative until now, they provide new insight into the nature of the events that could have led to the appearance of this agent.
Keywords: bovine prion, BSE, atypical BSE, strain evolution, degeneration, transgenic
Introduction
Bovine spongiform encephalopathy (BSE) is a prion epidemic that has caused the death of ∼200 000 cattle in Europe since its emergence in 1985. Its transmission to humans through contaminated food is believed to be responsible for the variant form of Creutzfeldt-Jakob disease (vCJD) (Prusiner, 1997; Aguzzi, 2006), which has resulted in the death of almost 200 patients to date (http://www.cjd.ed.ac.uk/vcjdworld.htm). When transmitted to mice or macaques, BSE and vCJD prions share similar strain-specific properties, including a unique molecular signature based on the size and glycoform ratio of the disease-associated prion protein (PrP) fragments generated by proteolytic digestion (PrPres) (Collinge et al., 1996; Lasmezas et al., 1996; Bruce et al., 1997; Scott et al., 1999). Although feeding cattle with contaminated foodstuff after changes in the rendering practice has been clearly incriminated in the massive extension of the BSE epidemic, the origin of its causative agent has remained enigmatic (Wilesmith and Wells, 1991).
In recent years, two atypical, presumably sporadic forms of BSE have been recognized in European and other countries through intensive livestock surveillance that uses rapid tests detecting PrPres. H-type isolates are characterized by PrPres fragments of significantly higher molecular sizes compared with classical BSE. Such isolates have been transmitted to both wild-type and transgenic (tg) mice and shown to retain strain-specific features distinct from that of BSE or vCJD agents transmitted to the same mice (Biacabe et al., 2004; Baron et al., 2006; Beringue et al., 2006). Another group of isolates designated L-type or BASE (bovine amyloidotic spongiform encephalopathy) depending on the authors, present a protease-resistant core of a slightly lower size than PrPBSE together with a noticeable different glycopattern (Casalone et al., 2004; Buschmann et al., 2006). In addition, affected cattle brains exhibit distinct histopathological features compared with BSE, including the presence of abundant amyloid plaques (Casalone et al., 2004).
Here we report the characterization of L-type and BASE isolates by transmission to several transgenic mouse lines. We found that, although retaining unique characteristics compared with BSE-type agents during transmission to bovine and human PrP transgenic mice, such isolates acquired phenotypic traits intriguingly similar to those of the BSE agent when propagated on mice expressing ovine PrP. L-type and BASE isolates will hereafter be collectively referred to as BSE-L for the sake of clarity.
Materials and Methods
Isolates.
The Italian isolates of classical BSE and L-type BSE [designated BASE by the authors (Casalone et al., 2004)] came from the National Reference Centre for Spongiform Encephalopathies (Istituto Zooprofilattico Spermentale, Torino, Italy). The French BSE-L, goat BSE [CH636 case (Eloit et al., 2005)], and experimental sheep BSE (Crozet et al., 2001) samples came from the National TSE Reference Laboratory (Afssa, Lyon, France). French, German, and Belgian BSE samples were provided by the Institut National de la Recherche Agronomique (Toulouse, France), the Institute for Novel and Emerging Infectious Diseases (Insel Reims, Germany), and the National Reference Laboratorium for Veterinary TSEs (Ukkel, Belgium), respectively. CH1641 sheep scrapie strain (Foster and Dickinson, 1988) was from the Institute for Animal Health (Edinburgh, UK). The vCJD isolate was obtained from the National Institute for Biological Standards and Control (Potters Bar, UK).
Transgenic mice and prion transmission.
The bovine (tg540 line), ovine (tg338 line; Val136Arg154Gln171 allele), and tga20 lines have been described previously (Fischer et al., 1996; Vilotte et al., 2001; Le Dur et al., 2005; Beringue et al., 2006). The human PrP transgenic mouse (tgHu) used expresses the Met129 allele (tg650 line; to be published in the future). All of the lines overexpress (fivefold to eightfold) the heterologous PrP on a mouse PrP null background. All experiments were performed according to national guidelines. Each inoculum was prepared extemporarily in a class II microbiological cabinet according to a strict protocol to avoid any cross-contamination. Individually identified 6- to 10-week-old mice were inoculated intracerebrally with 20 μl of a 10% (w/v) brain homogenate, monitored daily, and killed in extremis, as described previously (Le Dur et al., 2005).
Analysis of PrPres molecular pattern.
Brain tissue was homogenized at 20% (w/v) in 5% glucose with a Rybolyser (Hybaid, Middlesex, UK). PrPres was extracted according to the Bio-Rad (Hercules, CA) test protocol, by using 200 μg/ml proteinase K (Euromedex, Mundolsheim, France) for 10 min at 37°C. After denaturation in Laemmli buffer, the samples were run on 12% NuPAGE gels (Invitrogen, Cergy Pontoise, France), electrotransferred onto nitrocellulose membranes, and immunoblotted with 0.1 μg/ml biotinylated anti-PrP antibody Sha31 (Feraudet et al., 2005). The amount of brain tissue loaded onto the gels varied between 0.05 and 3 mg. Immunoreactivity was visualized by chemiluminescence (GE Healthcare, Little Chalfont, UK). Glycoform ratios and apparent molecular mass were determined by the GeneTools software after acquisition of chemiluminescent signals with a GeneGnome digital imager (Syngene, Frederick, MD).
Histopathology.
Histoblot analyses were performed on two to three brains per infection. Ten-micrometer-thick frozen sections were cut and transferred onto Superfrost slides and then onto nitrocellulose membranes soaked in lysis buffer (0.5% sodium deoxycholate, 0.5% NP-40, 100 mm NaCl, 10 mm EDTA, and 10 mm Tris, pH 7.8). After drying and rehydration in TBST (10 mm Tris, pH 7.8, 100 mm NaCl, and 0.2% Tween 20), membranes were digested by 25 μg/ml proteinase K at 37°C for 1 h. After washes with TBST, the membranes were denatured with 3 m guanidinium thiocyanate in 20 mm Tris HCl, pH 7.8, for 5–10 min at room temperature. After being washed with TBST, the membranes were blocked with TBST–5% skimmed milk before a 2 h incubation at room temperature with the mouse anti-PrP 12F10 antibody (1:3000 in TBST; provided by J. Grassi, CEA, Saclay, France). After TBST washes, the membranes were incubated with alkaline phosphatase-conjugated anti-mouse IgG antibody for 1 h (1:2500 in TBST; Sigma, St. Quentin Fallavier, France). After washing in TBS, alkaline phosphatase activity was revealed with nitroblue-tetrazolium-chloride/5-bromo-4-chloro-indolyl-phosphate substrate (Invitrogen). Two-micrometer-thick Formalin-fixed brain sections were stained with hematoxylin–eosin for assessment of spongiform changes. Vacuolation profiles were established following the standard method described previously (Fraser and Dickinson, 1968), using two to eight brains per isolate and per passage (at least five for BSE-L and cattle BSE infections). The standard gray (G1–G9) and white (W1–W3) matter areas used to construct the profile were as follows: G1, dorsal medulla; G2, cerebellar cortex; G3, superior colliculus; G4, hypothalamus; G5, medial thalamus; G6, hippocampus; G7, septum; G8, medial cerebral cortex at the level of the thalamus; G9, medial cerebral cortex at the level of the septum; W1, cerebellar white matter; W2, white matter of the mesencephalic tegmentum; and W3, pyramidal tract.
Results
Efficient transmission of BSE-L to ovine PrP mice
Three French and one Italian BSE-L isolates were inoculated intracerebrally to various transgenic mouse lines (see Materials and Methods). The transmission data available to date, together with those obtained comparatively with cattle BSE or BSE-related agents, are shown in Table 1. BSE-L isolates induced a typical neurological disease with a 100% attack rate on mice expressing bovine (tgBov) or ovine (tgOv) PrP. Remarkably, the observed survival times, ∼200 and 400 d, respectively, were consistently shorter than those produced by BSE-type agents (including sheep and goat BSE and vCJD), even compared with tgBov secondary passed BSE. Secondary transmission of BSE-L and BSE agents on tgBov mice produced no major change of the survival time. Conversely, BSE-L failed to transmit the disease to mouse PrP mice (tga20) (Fischer et al., 1996), in sharp contrast to BSE-type agents. Altogether, the above data supported the view that BSE-L isolates involve an infectious agent distinct from BSE. Subpassaging of the BSE-L agent onto tgOv mice, however, led to the intriguing observation that the incubation time (∼140 d) (Table 1) became similar to that of BSE and related agents serially passed onto the same mice.
Table 1.
Transmission of BSE-L isolates to transgenic mice expressing bovine, ovine, or murine prion protein
| Isolate | Origin (case number) | Mean ± SEM survival time in days (n/n0)a |
|||||
|---|---|---|---|---|---|---|---|
| tgBov mice |
tgOv mice |
tga20 |
|||||
| 1st passage | 2nd passage | 1st passage | 2nd passage | 3rd passage | 1st passage | ||
| BSE-L | Fr (7) | 212 ± 15 (7/7) | 237 ± 10 (6/6) | 432 ± 19 (6/6) | 141 ± 2 (7/7) | ||
| Fr (10) | 216 ± 15 (10/10) | 218 ± 10 (8/8) | na | ||||
| Fr (11) | 236 ± 12 (7/7) | na | |||||
| It (1088) | 228 ± 10 (9/9) | 423 ± 7 (9/9) | 140 ± 2 (10/10) | 143 ± 2 (6/6) | 654 ± 25 (0/10) | ||
| Cattle BSE | Fr (3) | 298 ± 7 (9/9)b | 283 ± 10 (5/5) | 704 ± 36 (6/7) | na | 447 ± 39 (6/6) | |
| Gec | 269 ± 11 (5/5) | 485, 555 (2/4) | 146 ± 8 (5/5) | 135 ± 3 (7/7) | |||
| It (128204) | 694 ± 16 (1/7) | na | 477 ± 53 (6/6) | ||||
| Be | 360 ± 20 (6/6) | 698 ± 25 (0/10) | |||||
| Sheep BSEd | Fr (ARQ1) | 278 ± 2 (6/6) | 263 ± 6 (6/6) | 560 ± 60 (5/5) | 178 ± 2 (4/4) | 133 ± 2 (6/6) | 317 ± 15 (8/8) |
| Fr (ARQ1)e | 244 ± 8 (4/4) | 530 ± 32 (6/6) | 393 ± 50 (6/6)f | 133 ± 1 (6/6) | 284 ± 9 (6/6) | ||
| Goat BSEg | Fr (CH636) | 253 ± 9 (6/6) | 291 ± 27 (5/5) | 590 ± 43 (4/4) | 426 ± 53 (6/6)f | 135 ± 1 (6/6) | |
| vCJD | UK (NHBY0/0003) | 343 ± 8 (5/5) | 293 ± 11 (6/6) | 792 ± 22 (6/6) | 195 ± 9 (6/6) | 139 ± 1 (6/6) | 364 ± 38 (5/5) |
na, Not available (experiments still ongoing); Fr, French; It, Italian, Ge, German; Be, Belgian.
aIntracerebral inoculation with 2 mg of brain tissue equivalent; n/n0, diseased, PrPres-positive/inoculated animals.
bData from Beringue et al. (2006) appear in italic.
cOne passage on tgBov mice.
dExperimental case.
eOne passage on tgOv (ARQ allele) mice (see Materials and Methods).
fMice inoculated with 0.2 mg of brain tissue equivalent.
gField case.
Molecular shift of L-BSE agent in tgOv mice
The brains of inoculated mice were examined for the presence of PrPres by immunoblot analysis. PrPres was readily detected from the first passage in terminally diseased, BSE-L infected tgBov and tgOv mice but not in BSE-L inoculated tga20 mice. In tgBov mice, the BSE-L and BSE agents produced clearly distinct PrPres signatures (Fig. 1). As observed for the BSE H-type agent (Beringue et al., 2006), BSE-L showed a PrPres profile resembling that in cattle (Fig. 1A); this profile, which is characterized by an equilibrated glycoform ratio whereas diglycoforms strongly predominate in PrPBSE (Fig. 1C), remained unchanged on subsequent transmission (Fig. 1B). PrPres could also be readily detected in the brains of tgHu mice killed while healthy at 400 d after inoculation with the BSE-L agent (Fr-7 isolate). This was much earlier than in the brains of cattle BSE-infected tgHu mice, in which PrPres was only detected in a minority (approximately one-fifth) of terminally diseased animals (Fig. 2A). Again, PrPBSE-L exhibited a glycopattern similar to that in cattle and distinct from that of PrPBSE (Fig. 2B). Quite a different situation was observed in tgOv mice, in which the BSE-L agent produced a PrPres profile uniformly distinct from that in cattle (up to three passages, 33 brains analyzed). Moreover, this profile was indistinguishable from the PrPres profile of BSE and BSE-related agents propagated onto tgOv mice (Fig. 3A–C). It is noteworthy that the tgOv-passed BSE agent retained a typical PrPres profile but with a lower predominance of diglycoforms. Reverse transmission of tgOv-adapted BSE agent to tgBov mice, however, restored the original glycoform ratio (Fig. 3D), essentially similar to that in cattle.
Figure 1.
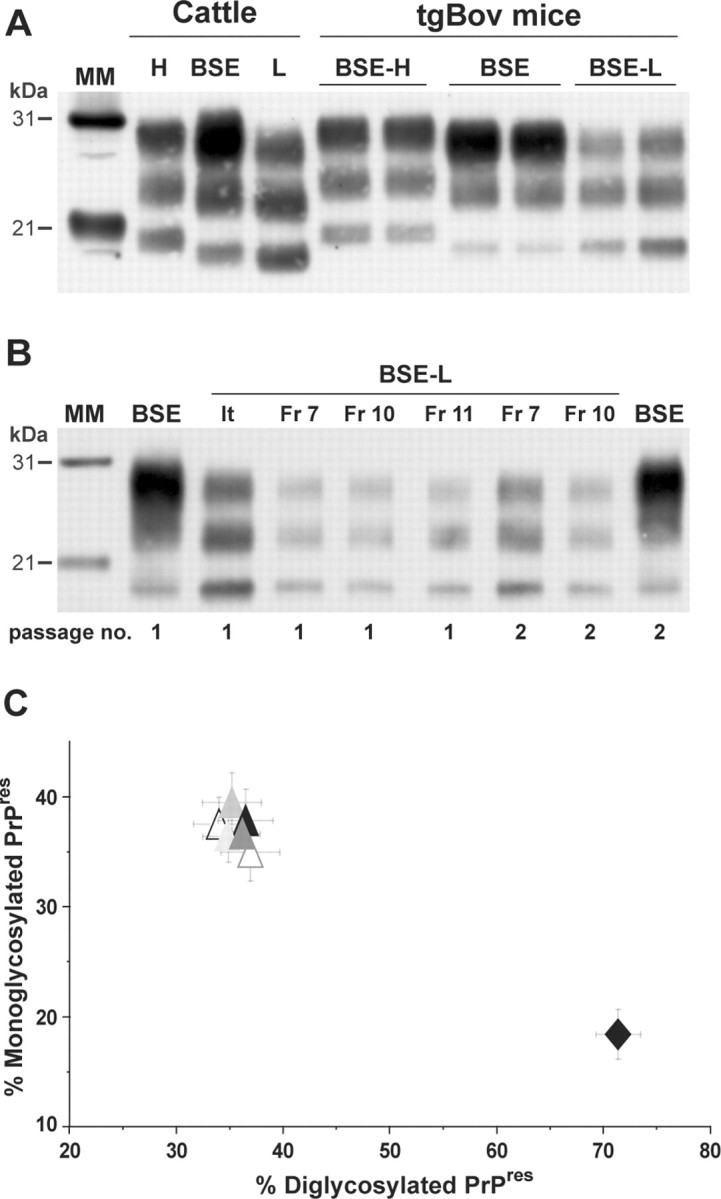
Brain PrPres in bovine PrP transgenic mice infected with BSE-L or BSE agents. A Western blot analysis of proteinase K-digested brain homogenates from diseased mice is presented. A, The distinct PrPres profiles of BSE H-type, BSE-L, and BSE agents are essentially preserved during cattle-to-mice transmission (2 mouse brains shown for each agent). B, The Italian (It) and French (Fr) BSE-L isolates have undistinguishable PrPres profiles that are conserved on second passage. C, Ratio of diglycosylated and monoglycosylated PrPres species (data plotted as means ± SEM). The BSE-L isolates are represented by triangles from different colors (open, first passage; filled, second passage) and BSE by a black diamond. MM, Molecular markers.
Figure 2.

Brain PrPres in human PrP transgenic mice infected with BSE-L or BSE agents. Western blots of proteinase K-digested brain homogenates are shown. A, Mice were killed healthy (up to 550 d; vertical dotted line) or in extremis after primary inoculation with either BSE-L (square; Fr-7 isolate, ongoing experiment) or BSE agent (circle; data from 3 isolates). Brains positive for PrPres in Western blot are visualized by filled symbols. B, PrPres profile in BSE-L (2 brains) and BSE-infected mice. The profile produced by human CJD (low type) is shown for comparison.
Figure 3.

Brain PrPres in ovine PrP transgenic mice infected with BSE-L or BSE agents. Western blots of proteinase K-digested brain homogenates are shown. The profiles in duplicate or triplicate are from different mice. A, Primary transmission to tgOv mice. The PrPres profile of BSE-L is similar to that observed with tgOv-passed BSE and differs from that in mice inoculated with BSE H-type or sheep scrapie (e.g., CH1641 strain). B, The BSE-like profile of BSE-L PrPres is maintained on subpassaging in tgOv mice. BSE-related agents are sheep BSE (left) and vCJD (right). C, Ratio of diglycoform and monoglycoform PrPres (data plotted as means ± SEM). BSE-L passages are visualized by triangles [Italian (It) first passage, black; second and third passage, dark and light gray; French (Fr-7) first passage; white] and tgOv-passed BSE (third passage) by a black diamond. D, PrPres in tgBov mice inoculated with BSE serially passed in either tgBov or tgOv mice.
Similar neuropathology of BSE-L and BSE in tgOv mice
Analyzing the spatial distribution and intensity of the PrP deposits and of the spongiform changes in the brain are two well established methods to discriminate between prion strains in mice (Fraser and Dickinson, 1968; Hecker et al., 1992). We thus compared the neuropathological phenotypes of the BSE-L and BSE agents by PrPres histoblotting and by vacuole scoring. The PrP deposition patterns of BSE-L and BSE in tgBov mice were clearly distinct. As illustrated in Figure 4, fine PrP deposits were abundant throughout the cerebrum, particularly in the cortex, several layers of the hippocampus, and rostral and ventral thalamic nuclei with BSE-L, whereas PrPBSE deposits were much more granular and rather confined to the corpus callosum and ventral nuclei of the thalamus. Lesion profiling also revealed clear differences: BSE-L induced a pronounced vacuolation in several gray matter areas, whereas BSE-induced vacuolation was discrete overall (Fig. 4D–F). It is worth noting that the neuropathological features observed on secondary passage of the BSE-L agent onto tgBov mice were essentially preserved (the main change being a moderate increase of the vacuolation intensity) and again clearly distinct from those associated to the BSE agent (Fig. 4D and data not shown). Applying the same comparative analysis to tgOv mice led to the opposite conclusion, i.e., an intriguing similarity of the PrP deposition and vacuolation patterns induced by BSE-L and BSE-type agents. In both cases, PrPres staining predominantly involved several nuclei (habenular, laterodorsal, geniculate, and submedius) of the thalamus (Fig. 5A–D), with other regions, such as the septum, external cortex of the inferior colliculus, and tegmentum being slightly less decorated (Fig. 5E–J). Only subtle differences were noted, such as in the fasciculus retroflexus, more strongly stained with BSE-L (Fig. 5H). Overall, such a distribution pattern was unique compared with the BSE-H agent and distinct scrapie strains propagated onto these mice (Le Dur et al., 2005; Beringue et al., 2006), including CH1641 (Fig. 5D) whose incubation time (130 d) (our unpublished data) compared with those of tgOv-adapted BSE-L and BSE agents. The spongiform changes observed with either type of agent were discrete in primary inoculated mice, unlike those produced by BSE-H, and became more severe on secondary passage. Again, they involved essentially the same areas of the gray and white matters (Fig. 5K–N).
Figure 4.

Regional distribution of PrPres and vacuolation in the brain of bovine PrP transgenic mice infected with BSE-L or BSE agents. A–D, Histoblots of representative coronal sections at the level of the hippocampus are shown. Note the different distribution and nature of PrP deposits between BSE (A) and BSE-L [B, Italian (It); C, French (Fr); D, French second passage]. E, F, Lesion profiles in the brain of tgBov mice infected with the BSE-L (E, open circles, Italian; filled circles, French isolates) or BSE (F, open circles, cattle BSE; open squares, goat BSE) agent. The intensity of vacuolation was scored as means ± SEM in standard gray (G1–G9) and white (W1–W3) matter areas (see Materials and Methods). The BSE-L agent induced a severe vacuolation in several regions of the gray matter compared with BSE.
Figure 5.

Regional distribution of PrPres and vacuolation in the brain of ovine PrP transgenic mice infected with BSE-L or BSE-type agents. A–D, Histoblots of representative coronal sections of mouse brains at the level of the hippocampus are shown. The patterns produced by cattle BSE (A, third pass) and BSE-L (B, second pass; C, third pass) are similar. The habenular (H), laterodorsal (L), geniculate (G), and submedius (S) nuclei of the thalamus are more particularly involved. The pattern produced by the CH1641 sheep scrapie strain is shown for comparison (D, second pass). E–J, Histoblots showing the PrP deposits in mice (third passage) infected with vCJD (E), cattle BSE (G), sheep BSE (I), or BSE-L (F, H, J) in the septum (E, F), hippocampus/thalamus (G, H), and midbrain at the level of the inferior colliculus (I, J). Note the similar PrPres regional distribution between the agents, except in the fasciculus retroflexus (arrowhead). K–N, Lesion profiles in tgOv mice infected with BSE-L or BSE agents. Mean ± SEM scores reflecting the vacuolation intensity on primary (open symbols) and subsequent (filled symbols) passages are shown for BSE-L (K, solid line, Italian; broken line, French), BSE-H (L), cattle BSE (M), sheep BSE (N, circles) and vCJD (N, squares). Standard brain areas are as in Figure 4.
Discussion
We examined the strain behavior of a class of cattle prion isolates newly identified by their atypical PrPres signature compared with the classical BSE agent (Casalone et al., 2004; Buschmann et al., 2006), designated BSE-L in the present study. During transmission to transgenic mice, the different isolates analyzed shared identical molecular and biological features, supporting the view that a unique infectious agent is actually involved in the cases named L-type BSE and BASE, depending on the authors (see Introduction). The main finding emerging from this study is that the BSE-L agent, although displaying phenotypic traits distinct from those of BSE and BSE-related agents on the other transgenic mouse lines studied, acquired a BSE-like strain phenotype during transmission to mice expressing ovine PrP.
When serially transmitted to mice expressing the same PrP sequence as the natural host (tgBov), BSE-L (four cases) and BSE (three cases) maintained separate, stable phenotypes. Thus, BSE-L isolates showed (1) faster disease transmission than cattle BSE and related agents, as was reported recently for a German case (Buschmann et al., 2006), (2) a distinctive PrPres signature, as in cattle, and (3) distinguishable PrP deposition and vacuolation patterns in the brain. Primary transmission to mouse or human PrP transgenic mice also revealed striking differences between the two agents. Thus, unlike that seen with various sources of BSE or the vCJD agent, BSE-L provoked no overt disease or detectable PrPres accumulation in tga20 mice. Inversely, it induced an early PrPres accumulation in the brain of tgHu mice relative to cattle BSE, with either agent retaining its distinctive PrPres profile. A worrying implication of this latter result is that the BSE-L agent might be transmissible to and possibly more virulent in humans than classical BSE, an issue that deserves to be investigated further.
In contrast, the BSE-L agent appeared to undergo a strain phenotype shift during transmission to tgOv mice, despite an incubation period during primary inoculation (∼400 d) much shorter than with cattle BSE (∼700 d) or BSE-H (∼600 d) (Beringue et al., 2006), suggesting a lower transmission barrier from cattle to tgOv for BSE-L. Surprisingly, this novel strain phenotype appeared very similar to that of cattle BSE and related agents propagated onto the same mice, according to various criteria. First, the survival times observed after stabilization on tgOv differed by ∼5% at most between BSE-L and BSE-type agents, which is unlikely to be significant. Second, the spatial distribution of PrPres in the brain showed only a few, minor differences, although being clearly distinct from the patterns observed with several classes of transmissible spongiform encephalopathy (TSE) agents characterized on these mice, including the BSE-H agent (Le Dur et al., 2005; Beringue et al., 2006) (our unpublished data). Third, the vacuolation profiles observed from the second passage on, once the spongiform changes became substantial, essentially overlapped. Fourth, the PrPres molecular profiles of BSE-L and BSE agents on tgOv mice were indistinguishable.
The available typing methods, relying on the comparison of neuropathological features or a PrPres signature (Fraser and Dickinson, 1968; Hecker et al., 1992), arguably have inherent limitations and may be considered of higher value in differentiating two strains than in looking for a potential strain identity. Another issue is the number and the diversity of isolates propagated on the recipient host. In this regard, it is worth emphasizing that 136 prion infectious sources from various species, including ∼100 natural sheep or goat scrapie isolates, have been transmitted successfully to tgOv mice to date (supplemental Table S1, available at www.jneurosci.org as supplemental material). Remarkably, a PrPres profile with unglycosylated species of intermediate molecular size (∼20 kDa) and prominent diglycoforms has been uniquely and consistently observed with cattle BSE or BSE-related agents, until BSE-L isolates were analyzed. All other sources generated unglycosylated species with either higher or lower (e.g., CH1641 scrapie isolate) mobility (Vilotte et al., 2001; Le Dur et al., 2005; Beringue et al., 2006). Therefore, the observed phenotypic convergence between BSE-L and BSE during transmission on tgOv mice appears as an uncommon event, so far involving only these two strains, both of bovine origin.
Our findings may represent a novel, striking example of the evolutionary potential of prion agents during transmission to a foreign host, which can promote strain shift and emergence of unprecedented properties in a yet unpredictable manner (Kimberlin et al., 1987; Scott et al., 1997; Bartz et al., 2000; Wadsworth et al., 2004). It is unclear in the present state of our investigations which type of mechanism, selection or de novo emergence, may account for the apparent conversion of BSE-L toward the classical BSE strain. Preferential selection in these mice of a classical-type BSE agent that would preexist as a minor component in the brain tissue of BSE-L-infected cattle is one possibility. Indeed, recent observations have questioned the strain homogeneity of the agent present in cattle BSE infectious sources. Thus, transmission of BSE isolates to C57BL and SJL mice expressing the same mouse PrP allele was reported to result in a divergent strain phenotype among the two lines (Asante et al., 2002; Lloyd et al., 2004). Also, biochemical analyses revealed a dual PrPres signature in BSE (and vCJD) brain homogenates (Yull et al., 2006), which may suggest, albeit not exclusively, the presence of a secondary, minor strain component. However, favored propagation of a BSE-like component preexisting in BSE-L-infected brain tissues is unlikely to explain our observation because, as mentioned above, the BSE agent produces much prolonged incubation periods compare with BSE-L in tgOv mice. A still missing piece of information is whether the observed convergence truly reflects a permanent strain shift of the BSE-L agent, because both reversible and irreversible changes have been reported to occur during heterologous transmission of a biologically cloned prion strain (Kimberlin et al., 1987; Scott et al., 1997). Experiments are underway to determine whether the “ovinized” BSE-L agent would retain BSE phenotypic traits during reinoculation to tgBov mice, as we found it to be the case for the ovinized BSE agent. It would also be of interest to learn whether BSE-L would behave similarly on mice expressing an ovine PrP allotype other than Val136Arg154Gln171 as in the present study, because even single amino acid differences in the PrP sequence might be crucial in promoting a strain phenotype shift (Bruce, 2003; Wadsworth et al., 2004).
In conclusion, the findings reported in the present study provide new insight into the nature of the events that could have contributed to the emergence of the BSE epidemic. The various theories currently proposed regarding the origin of the BSE agent invoke two kinds of mechanisms (Colchester and Colchester, 2005; Baron and Biacabe, 2006): an intrinsic cause, i.e., a spontaneously diseased cattle attributable for instance to somatic or germ-line mutation of PrP, or an extrinsic cause, i.e., the infection of cattle by a prion from another species, involving a strain change or not. Our observation is consistent with the view that the epidemic BSE agent could have originated from an endogenous, cattle prion. It also points to the theoretical possibility of a multiple causative event, in which a prion sporadically present in cattle may have “mutated” through passage on an intermediary host such as a sheep.
During the review process of this paper, a study performed on conventional mice has been published that reports converging features of BASE-subpassaged mice toward BSE-inoculated mice (Capobianco et al., 2007), thus strengthening the notion that the BSE-L agent tends to acquire epidemic BSE-like properties during transmission to a heterologous host.
Footnotes
This work was supported by a joint grant from Institut National de la Recherche Agronomique (INRA)–Agence Française de Sécurité Sanitaire des Aliments. We thank M. Fontes (Institut National de la Santé et de la Recherche Médicale Unité 491, Marseille, France) for authorizing us to include the tgHu mice in this study, C. Weissmann for the tga20 mouse line, N. Hunter (Institute for Animal Health, Edinburgh, UK) for the CH1641 scrapie strain, J. Grassi (Commissariat à l'Energie Atomique, Saclay, France) for Sha31 and 12F10 antibodies, and the staff from Laboratoire de Génétique Biochimique et de Cytogénétique and Unité Expérimentale Animalerie Rongeurs (INRA, Jouy-en-Josas, France) for excellent mouse care. Parts of this paper have been presented previously at the Neuroprion Symposium (Turin, Italy, September 2006).
References
- Aguzzi A. Prion diseases of humans and farm animals: epidemiology, genetics, and pathogenesis. J Neurochem. 2006;97:1726–1739. doi: 10.1111/j.1471-4159.2006.03909.x. [DOI] [PubMed] [Google Scholar]
- Asante EA, Linehan JM, Desbruslais M, Joiner S, Gowland I, Wood AL, Welch J, Hill AF, Lloyd SE, Wadsworth JD, Collinge J. BSE prions propagate as either variant CJD-like or sporadic CJD-like prion strains in transgenic mice expressing human prion protein. EMBO J. 2002;21:6358–6366. doi: 10.1093/emboj/cdf653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron T, Biacabe AG. Origin of bovine spongiform encephalopathy. Lancet. 2006;367:297–298. doi: 10.1016/S0140-6736(06)68060-4. author reply 298–299. [DOI] [PubMed] [Google Scholar]
- Baron TG, Biacabe AG, Bencsik A, Langeveld JP. Transmission of new bovine prion to mice. Emerg Infect Dis. 2006;12:1125–1128. doi: 10.3201/eid1207.060107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartz JC, Bessen RA, McKenzie D, Marsh RF, Aiken JM. Adaptation and selection of prion protein strain conformations following interspecies transmission of transmissible mink encephalopathy. J Virol. 2000;74:5542–5547. doi: 10.1128/jvi.74.12.5542-5547.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beringue V, Bencsik A, Le Dur A, Reine F, Lai TL, Chenais N, Tilly G, Biacabe AG, Baron T, Vilotte JL, Laude H. Isolation from cattle of a prion strain distinct from that causing bovine spongiform encephalopathy. PLoS Pathog. 2006;2:e112. doi: 10.1371/journal.ppat.0020112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biacabe AG, Laplanche JL, Ryder S, Baron T. Distinct molecular phenotypes in bovine prion diseases. EMBO Rep. 2004;5:110–115. doi: 10.1038/sj.embor.7400054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce ME. TSE strain variation. Br Med Bull. 2003;66:99–108. doi: 10.1093/bmb/66.1.99. [DOI] [PubMed] [Google Scholar]
- Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A, McCardle L, Chree A, Hope J, Birkett C, Cousens S, Fraser H, Bostock CJ. Transmissions to mice indicate that “new variant” CJD is caused by the BSE agent. Nature. 1997;389:498–501. doi: 10.1038/39057. [DOI] [PubMed] [Google Scholar]
- Buschmann A, Gretzschel A, Biacabe AG, Schiebel K, Corona C, Hoffmann C, Eiden M, Baron T, Casalone C, Groschup MH. Atypical BSE in Germany: proof of transmissibility and biochemical characterization. Vet Microbiol. 2006;117:103–116. doi: 10.1016/j.vetmic.2006.06.016. [DOI] [PubMed] [Google Scholar]
- Capobianco R, Casalone C, Suardi S, Mangieri M, Miccolo C, Limido L, Catania M, Rossi G, Fede GD, Giaccone G, Bruzzone MG, Minati L, Corona C, Acutis P, Gelmetti D, Lombardi G, Groschup MH, Buschmann A, Zanusso G, Monaco S, et al. Conversion of the BASE prion strain into the BSE strain: the origin of BSE? PLoS Pathog. 2007;3:e31. doi: 10.1371/journal.ppat.0030031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casalone C, Zanusso G, Acutis P, Ferrari S, Capucci L, Tagliavini F, Monaco S, Caramelli M. Identification of a second bovine amyloidotic spongiform encephalopathy: molecular similarities with sporadic Creutzfeldt-Jakob disease. Proc Natl Acad Sci USA. 2004;101:3065–3070. doi: 10.1073/pnas.0305777101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colchester AC, Colchester NT. The origin of bovine spongiform encephalopathy: the human prion disease hypothesis. Lancet. 2005;366:856–861. doi: 10.1016/S0140-6736(05)67218-2. [DOI] [PubMed] [Google Scholar]
- Collinge J, Sidle KC, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the aetiology of “new variant” CJD. Nature. 1996;383:685–690. doi: 10.1038/383685a0. [DOI] [PubMed] [Google Scholar]
- Crozet C, Bencsik A, Flamant F, Lezmi S, Samarut J, Baron T. Florid plaques in ovine PrP transgenic mice infected with an experimental ovine BSE. EMBO Rep. 2001;2:952–956. doi: 10.1093/embo-reports/kve204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eloit M, Adjou K, Coulpier M, Fontaine JJ, Hamel R, Lilin T, Messiaen S, Andreoletti O, Baron T, Bencsik A, Biacabe AG, Beringue V, Laude H, Le Dur A, Vilotte JL, Comoy E, Deslys JP, Grassi J, Simon S, Lantier F, et al. BSE agent signatures in a goat. Vet Rec. 2005;156:523–524. doi: 10.1136/vr.156.16.523-b. [DOI] [PubMed] [Google Scholar]
- Feraudet C, Morel N, Simon S, Volland H, Frobert Y, Creminon C, Vilette D, Lehmann S, Grassi J. Screening of 145 anti-PrP monoclonal antibodies for their capacity to inhibit PrPSc replication in infected cells. J Biol Chem. 2005;280:11247–11258. doi: 10.1074/jbc.M407006200. [DOI] [PubMed] [Google Scholar]
- Fischer M, Rulicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 1996;15:1255–1264. [PMC free article] [PubMed] [Google Scholar]
- Foster JD, Dickinson AG. The unusual properties of CH1641, a sheep-passaged isolate of scrapie. Vet Rec. 1988;123:5–8. doi: 10.1136/vr.123.1.5. [DOI] [PubMed] [Google Scholar]
- Fraser H, Dickinson AG. The sequential development of the brain lesion of scrapie in three strains of mice. J Comp Pathol. 1968;78:301–311. doi: 10.1016/0021-9975(68)90006-6. [DOI] [PubMed] [Google Scholar]
- Hecker R, Taraboulos A, Scott M, Pan KM, Yang SL, Torchia M, Jendroska K, DeArmond SJ, Prusiner SB. Replication of distinct scrapie prion isolates is region specific in brains of transgenic mice and hamsters. Genes Dev. 1992;6:1213–1228. doi: 10.1101/gad.6.7.1213. [DOI] [PubMed] [Google Scholar]
- Kimberlin RH, Cole S, Walker CA. Temporary and permanent modifications to a single strain of mouse scrapie on transmission to rats and hamsters. J Gen Virol. 1987;68:1875–1881. doi: 10.1099/0022-1317-68-7-1875. [DOI] [PubMed] [Google Scholar]
- Lasmezas CI, Deslys JP, Demaimay R, Adjou KT, Lamoury F, Dormont D, Robain O, Ironside J, Hauw JJ. BSE transmission to macaques. Nature. 1996;381:743–744. doi: 10.1038/381743a0. [DOI] [PubMed] [Google Scholar]
- Le Dur A, Beringue V, Andreoletti O, Reine F, Lai TL, Baron T, Bratberg B, Vilotte JL, Sarradin P, Benestad SL, Laude H. A newly identified type of scrapie agent can naturally infect sheep with resistant PrP genotypes. Proc Natl Acad Sci USA. 2005;102:16031–16036. doi: 10.1073/pnas.0502296102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd SE, Linehan JM, Desbruslais M, Joiner S, Buckell J, Brandner S, Wadsworth JD, Collinge J. Characterization of two distinct prion strains derived from bovine spongiform encephalopathy transmissions to inbred mice. J Gen Virol. 2004;85:2471–2478. doi: 10.1099/vir.0.79889-0. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. Prion diseases and the BSE crisis. Science. 1997;278:245–251. doi: 10.1126/science.278.5336.245. [DOI] [PubMed] [Google Scholar]
- Scott MR, Groth D, Tatzelt J, Torchia M, Tremblay P, DeArmond SJ, Prusiner SB. Propagation of prion strains through specific conformers of the prion protein. J Virol. 1997;71:9032–9044. doi: 10.1128/jvi.71.12.9032-9044.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott MR, Will R, Ironside J, Nguyen HO, Tremblay P, DeArmond SJ, Prusiner SB. Compelling transgenetic evidence for transmission of bovine spongiform encephalopathy prions to humans. Proc Natl Acad Sci USA. 1999;96:15137–15142. doi: 10.1073/pnas.96.26.15137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilotte JL, Soulier S, Essalmani R, Stinnakre MG, Vaiman D, Lepourry L, Da Silva JC, Besnard N, Dawson M, Buschmann A, Groschup M, Petit S, Madelaine MF, Rakatobe S, Le Dur A, Vilette D, Laude H. Markedly increased susceptibility to natural sheep scrapie of transgenic mice expressing ovine prp. J Virol. 2001;75:5977–5984. doi: 10.1128/JVI.75.13.5977-5984.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadsworth JD, Asante EA, Desbruslais M, Linehan JM, Joiner S, Gowland I, Welch J, Stone L, Lloyd SE, Hill AF, Brandner S, Collinge J. Human prion protein with valine 129 prevents expression of variant CJD phenotype. Science. 2004;306:1793–1796. doi: 10.1126/science.1103932. [DOI] [PubMed] [Google Scholar]
- Wilesmith JW, Wells GA. Bovine spongiform encephalopathy. Curr Top Microbiol Immunol. 1991;172:21–38. doi: 10.1007/978-3-642-76540-7_2. [DOI] [PubMed] [Google Scholar]
- Yull HM, Ritchie DL, Langeveld JP, van Zijderveld FG, Bruce ME, Ironside JW, Head MW. Detection of type 1 prion protein in variant Creutzfeldt-Jakob disease. Am J Pathol. 2006;168:151–157. doi: 10.2353/ajpath.2006.050766. [DOI] [PMC free article] [PubMed] [Google Scholar]