

Abstract
Huntingtin is a protein that is mutated in Huntington's disease (HD), a dominant inherited neurodegenerative disorder. We previously proposed that, in addition to the gained toxic activity of the mutant protein, selective molecular dysfunctions in HD may represent the consequences of the loss of wild-type protein activity. We first reported that wild-type huntingtin positively affects the transcription of the brain-derived neurotrophic factor (BDNF) gene, a cortically derived survival factor for the striatal neurons that are mainly affected in the disease. Mutation in huntingtin decreases BDNF gene transcription. One mechanism involves the activation of repressor element 1/neuron-restrictive silencer element (RE1/NRSE) located within the BDNF promoter. We now show that increased binding of the RE1 silencing transcription factor/neuron-restrictive silencer factor (REST/NRSF) repressor occurs at multiple genomic RE1/NRSE loci in HD cells, in animal models, and in postmortem brains, resulting in a decrease of RE1/NRSE-mediated gene transcription. The same molecular phenotype is produced in cells and brain tissue depleted of endogenous huntingtin, thereby directly validating the loss-of-function hypothesis of HD. Through a ChIP (chromatin immunoprecipitation)-on-chip approach, we examined occupancy of multiple REST/NRSF target genes in the postmortem HD brain, providing the first example of the application of this technology to neurodegenerative diseases. Finally, we show that attenuation of REST/NRSF binding restores BDNF levels, suggesting that relief of REST/NRSF mediated repression can restore aberrant neuronal gene transcription in HD.
Keywords: BDNF, genome, Huntington's disease, neuronal death, REST/NRSF, transcription
Introduction
Huntingtin is a 348 kDa protein containing a polyglutamine (polyQ) tract in its N terminus that when expanded beyond 35 glutamines causes Huntington's disease (HD), an autosomal dominant neurodegenerative disorder (The Huntington's Disease Research Collaborative Group, 1993). Although wild type and polyQ-expanded huntingtin are ubiquitously expressed, HD is associated with selective neuronal loss, especially in the striatum and cerebral cortex (Reiner et al., 1988). The dominant inheritance of the mutation causes disease predominantly by a gain-of-function mechanism, but there is considerable evidence that loss of normal huntingtin function(s) in postmitotic neurons may also contribute to HD (Cattaneo et al., 2001, 2005).
In the search for a more direct role of wild-type huntingtin in neuronal activity, we have demonstrated that the normal but not the mutant protein promotes cortical brain-derived neurotrophic factor (BDNF) gene transcription and that BDNF mRNA and protein levels are reduced in HD (Zuccato et al., 2001), pointing at BDNF loss as one of the potential noncell-autonomous effectors of striatal vulnerability in HD (for review, see Zuccato and Cattaneo, 2007).
In previous studies, we have shown that the stimulatory effect of wild-type huntingtin on BDNF transcription is mediated through the repressor element 1/neuron restrictive silencer element (RE1/NRSE) at the BDNF promoter II (Zuccato et al., 2003). RE1 silencing transcription factor/neuron-restrictive silencer factor (REST/NRSF) is a transcription factor (Chong et al.,1995; Schoenherr and Anderson, 1995) that is recruited to RE1/NRSE sites, repressing or silencing neuron-specific genes in both neural (Palm et al., 1998, 1999; Wood et al., 2003) and non-neural (Belyaev et al., 2004) cells. We reported that wild-type huntingtin sequesters REST/NRSF in the cytoplasm, thereby permitting activated transcription of the BDNF gene. In contrast, in the presence of mutant huntingtin, REST/NRSF accumulates in the nucleus and represses BDNF gene transcription, resulting in a decrease in BDNF synthesis (Zuccato et al., 2003).
Because bioinformatic studies have shown that there are >1300 RE1/NRSE sites in the human genome (Bruce et al., 2004; Johnson et al., 2006), we reasoned that increased nuclear localization of REST/NRSF will likely lead to repression of other genes in addition to BDNF. We have therefore examined the direct interaction of REST/NRSF with potential target genes in cells and mice expressing mutant huntingtin and depleted by endogenous huntingtin. We also evaluated the impact of REST/NRSF rescue in HD cell models after infection with a dominant-negative form of REST/NRSF. Finally, we examined occupancy of multiple REST/NRSF target genes in postmortem HD brains by using a “ChIP-on-chip” approach.
Our results indicate that an increase in RE1/NRSE genomic binding is found in HD, resulting in repression of REST/NRSF-regulated gene transcription. We also found that the abnormal RE1/NRSE phenotype observed in HD is strictly dependent on the level/activity of wild-type huntingtin, implying that a loss-of-function mechanism is in operation in HD at the level of the RE1/NRSEs. Finally, we show that attenuation of REST/NRSF binding can restore REST/NRSF aberrant neuronal gene transcription in HD.
Materials and Methods
Cell lines and tissues.
Knock-in cells were generated by immortalization of primary embryonic day 14 (E14) brain cells from wild-type littermate mice (Hdh7/7 cells) and from homozygous knock-in mice, having two copies of the huntingtin allele, each bearing 109 CAG repeats (Hdh109/109 cells) (Trettel et al., 2000). Cells were propagated in DMEM supplemented with 10% fetal calf serum at 33°C in a 5% CO2 atmosphere.
Hdh+/Hdh+ (ES+/+), Hdhexon4/5/Hdh+ (ES+/−), and Hdhexon4/5/Hdhexon4/5 (ES−/−) cells are routinely propagated without feeders on gelatin-coated plastic in Glasgow's modified Eagle's medium, supplemented with 1 mm sodium pyruvate, 100 μm nonessential amino acids, 2 mm l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 0.1 mm β-mercaptoethanol, 10% FBS (EuroClone, Milano, Italy), and 1000 U/ml leukemia inhibitory factor (LIF; Millipore, Billerica, MA).
We analyzed frozen brain tissues obtained from R6/2 transgenic mice (Mangiarini et al., 1996) mutant huntingtin knock-in mice (Wheeler et al., 1999) and huntingtin conditional knock-out mice (Dragatsis et al., 2000).
Human postmortem brain tissues (parietal cortical tissues, Brodmann's area 7) were obtained by the Massachusetts General Hospital (Charlestown, MA) and by the Harvard Brain Tissue Resource Center (Belmont, MA). See supplemental Table 1 (available at www.jneurosci.org as supplemental material) for the list of human brain tissues used in this study.
RNA isolation and reverse transcription.
We isolated total RNA from cells and tissues with Trizol Reagent (Invitrogen, Carlsbad, CA). Genomic DNA was digested with DNA-free (Applied Biosystems, Foster City, CA) at 37°C for 15 min. Total RNA (1 μg) was reverse-transcribed to single-stranded cDNA using Superscript III RNaseH̄ reverse transcriptase (Invitrogen) and random primers in a volume of 20 μl, according to the manufacturer's instructions.
Chromatin immunoprecipitation.
Cells (20 × 106) were harvested with trypsin treatment and washed three times with PBS (1×) and then treated with 1% formaldehyde in PBS by rotation for 8–10 min at 4°C. Fixation was stopped by addition of glycine to a final concentration of 125 mm. Cells were washed two times with PBS, and pellets were suspended in chromatin immunoprecipitation (ChIP) lysis buffer [10 mm Tris-HCl, pH 8, 140 mm NaCl, 1 mm EDTA, 1% Triton X-100, 0.1% sodium deoxycholate supplemented with 1 mm phenylmethylsulfonyl fluoride, and protease inhibitors mixture MINI (Roche, Basel, Switzerland)] and passed through 25–21 ga needles.
For ChIP on mouse tissue, a whole cortex and liver were used. The tissue was chopped into small pieces in 5 ml PBS and then treated with 1% formaldehyde with rotation for 10 min at 4°C. Fixation was stopped by addition of glycine to a final concentration of 125 mm. Fixed tissues were washed three times in PBS and homogenized in 2 ml PBS in a Dounce homogenizer and spun at 400 × g for 10 min at 4°C. Pellets were resuspended in 250 μl cell lysis buffer plus 1% SDS (final concentration) and passed through 25–21 ga needles. An additional 2 ml of lysis buffer without SDS was then added to each sample before sonication.
ChIP on human parietal cortex tissue was performed using 500–700 mg frozen tissue. The tissue was frozen in liquid nitrogen, diced into small pieces in 5 ml PBS containing 1% formaldehyde, and agitated at 4°C for 10 min. A 125 mm final concentration of glycine was used to quench fixation, and the tissue was transferred to a Falcon tube and centrifuged at 400 × g for 5 min at 4°C. The pellet was washed three times in PBS and centrifuged again at 400 × g for 5 min at 4°C and homogenized in 3 ml PBS in a Dounce homogenizer. Homogenized tissue was centrifuged at 400 × g for 5 min at 4°C and resuspended in 300 μl of cell lysis buffer plus 1% SDS (final concentration). An additional 3 ml of lysis buffer without SDS was then added to each sample before sonication.
Sonication of the cell and tissue extract was performed four times with 15 s pulses with the microprobe at 40–50% output and 70% duty cycle. To minimize foaming of the solution, sonications were performed in glass tubes. Under these conditions, DNA fragments with an average size of 200–700 bp were obtained. Sonicated extracts were centrifuged, and chromatin yield has been evaluated by UV spectrometry. Equal amounts of chromatin had been precleared with blocked protein G-Sepharose and were incubated by overnight rotation with 1 μg primary antibodies anti-REST/NRSF N2174 (raised in rabbit to CYFLEEAEEQE) (Belyaev et al., 2004) and preimmune serum as a negative control for the IP. As a positive control, 1 μg of anti-histone H3 and a corresponding amount of rabbit IgG (Millipore) were used to immunoprecipitate the same chromatin. Anti-Sp1 (s.c.-59) and anti-Sp4 (s.c.-645) are from Santa Cruz Biotechnology (Santa Cruz, CA).
Protein G-Sepharose (GE Healthcare, Little Chalfont, UK) was added and followed by 1 h of incubation with rotation. Beads were spun at 10,000 × g for 30 s and washed sequentially with increasing concentrations of salts and nonionic detergents: a first wash was done by using a solution composed of 10 mm Tris-HCl, pH 8, 500 mm NaCl, 1 mm EDTA, 1% Triton X-100, 0.1% sodium deoxycholate and a second wash with 10 mm Tris-HCl, pH 8, 1 mm EDTA, 250 mm LiCl, 0.5% NP40, 0.5% sodium deoxycholate. Finally, beads were eluted with 1% SDS in 0.1 m NaHCO3. Bound fractions were de-cross-linked by adding 200 mm NaCl and by incubation at 65°C for 6–8 h. De-cross-linked samples were treated with RNase and Proteinase K, and DNA was purified by phenol-chloroform, precipitated with 2 volumes of absolute ethanol, washed two times with 70% ethanol, and pellets were resuspended in 50 μl of HPLC water.
Before PCR amplification, DNA content of input and output DNA were evaluated by using the PicoGreen system (Invitrogen) as recommended by the manufacturer.
Quantitative real-time PCR (iCycler Thermal Cycler with Multicolor Real-time PCR Detection System; Bio-Rad, Hercules, CA) using SYBR Green incorporation was used to quantitatively assess REST/NRSF occupancy. Three independent PCR experiments were performed for each RE1-containing site.
PCR was performed in a total volume of 20 μl containing equal amounts of input and immunoprecipitated DNA, 50 mm KCl, 20 mm Tris-HCl, pH 8.4, 0.2 mm dNTPs, 25 U/ml iTaq DNA polymerase, 3 mm MgCl2, SYBR Green I, 10 nm fluorescein, stabilizers (iQ SYBR Green Supermix; Bio-Rad), and 0.2 μm of forward and reverse primers. Input from chromatin that had been cross-linked reversed similar to the analyzed samples represents a control for PCR effectiveness.
Primers flanking the NRSE of the following genes were used: mBdnf S, 5′-GGATTTGTCCGAGGTGGTAG-3′; mBdnf AS, 5′-CGGAAAAGACGCTTTTTAAGG-3′; hBDNF S, 5-GTAAAGCCAACCCTGTGTCG-3′; hBDNF AS, 5′-TCCGCTCCAAAATCTGACTC-3′; mSyn1 S, 5′-GCACACTCAGAGGGGAACAT-3′; mSyn1 AS, 5′-CTCTTTTCTTTGCCCGACAG-3′; hSYN1 S, 5′-CAACACTACAAACCGAGTATCTGC-3′; hSYN1 AS, 5′-GCCTCATCCTGGTCCTAAAA-3′; mChrm4 S, 5′-AGGAGACTCGTGCCATCTTC-3′; mChrm4 AS, 5′-CGAGCATCTACCCTGGCTAA-3′; hCHRM4 S, 5′-GGCCTGTAACCCCAAATTC-3′; hCHRM4 AS, 5′-GGGGAGGGTCTTGAGTTGTT-3′; mDrd3 S, 5′-GGTCCCTCCCAAATCCTCTA-3′; mDrd3 AS, 5′-TCATAGCTGCTTTCCCAAGG-3′; mPenk1 S, 5′-AATTCCCGC-TATTTGCCAGT-3′; mPenk1 AS, 5′-AGTGGAGGGACAGCTCGTTA-3′; mChrnb2 S, 5′-AATAAGCCGGCAGACTCTTG-3′; mChrnb2 AS, 5′-CTCCAGCCACCACTTGAGAT-3′; hCHRNB2 S, 5′-GTTCTCCTACCCCAGCCAAC-3′; hCHRNB2 AS, 5′-GCAGACTCCCCCACCTCTA-3′; hDRD2 S, 5′-ATGATGATCTGGAGAGGCAG-3′; hDRD2 AS, 5′-GTTGCCGAAGACGATGACAG-3′.
In parallel, anti-REST/NRSF immunoprecipitated genomic DNA was also assessed by real-time PCR using primers corresponding to genomic regions distal to any RE1/NRSE, including the M4 muscarinic cholinergic receptor (Chrm4): mChrm4 S, 5′-TCCTCACCTGGACACCCTAC-3′; mChrm4 AS, 5′-ACGTAGCAGAGCCAGTAGCC-3′. β-actin was also included (which is not regulated by REST/NRSF): mβ-actin S, 5′-AGGTATCCTGACCCTGAAG-3′; mβ-actin AS, 5′-GCTCATTGTAGAAGGTGTGG-3′; hβ-actin S, 5′-TGCCTAGGTCACCCACTAATG-3′; hβ-actin AS, 5′-GTGGCCCGTGATGAAGGCTA-3′.
The following PCR amplification protocol was applied: 95°C for 3 min, followed by 45 cycles of 30 s at 95°C, 30 s at 60°C, and 30 s at 72°C. Fluorescence was quantified during the 60°C annealing step and product formation confirmed by melting curve analysis (55–94°C). For the amplification of hDRD2 Sp1 responsive site, the annealing step had been performed at 57°C for 30 s.
Immunoprecipitated DNA for the different analyzed RE1/NRSE loci was quantified using a standard curve (nanograms of DNA). Binding of REST/NRSF at the RE1/NRSEs was calculated as nanograms of DNA IP REST antibody per nanogram of DNA IP preimmune serum. Similarly, binding of histone H3 at each of the analyzed RE1/NRSE, representing the positive control of the ChIP assay, was calculated as nanograms of DNA IP H3 per nanogram of DNA IP IgG.
ChIP Scanning Assay.
For the scanning ChIP, we followed the ChIP protocol described above. Cross-linked REST/NRSF-DNA complexes were precipitated with anti REST/NRSF antibody N2174. The assay was performed on the mouse and human BDNF locus.
For the mouse Bdnf locus (GenBank accession number AY057907), quantitative real-time PCR was performed on the precipitated DNA fragments using five pairs of oligonucleotide primers designed to produce amplicons covering the RE1/NRSE site in the BDNF gene (see above) and flanking sequences, located 477 and 3426 bp upstream and 383, 1123, 3353, and 17393 bp downstream of the RE1/NRSE (for details on primers positions, see supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
A parallel approach was used to analyze REST/NRSF binding at the human BDNF locus (GenBank accession number AF411339) using primers for flanking sequences located 758 bp upstream and 431, 1222, 2279, and 19350 bp downstream to the RE1/NRSE (for details on primers positions, see Fig. 4a).
Figure 4.
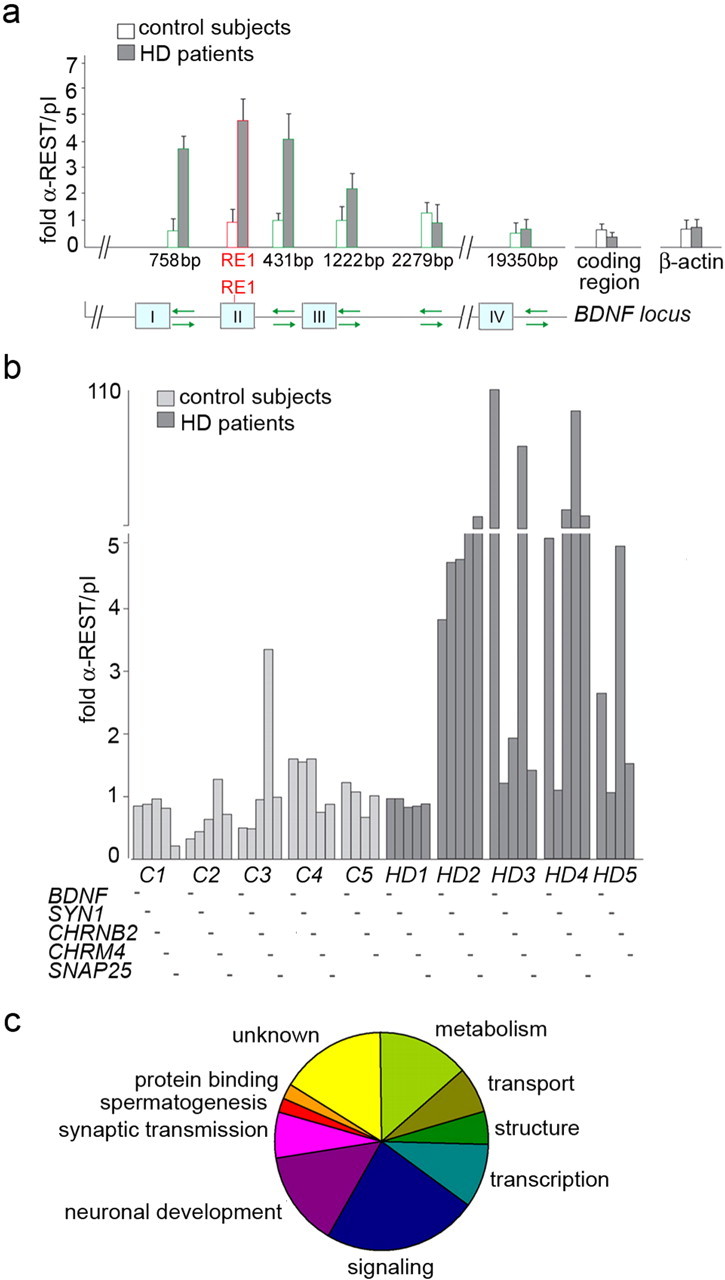
REST/NRSF binding at the RE1/NRSE is increased in HD patients. a, ChIP scanning at the human BDNFlocus. ChIP analysis of cortical tissue from human HD (3723 and 2866) and control (3899 and 3688) samples. Anti-REST/NRSF-immunoprecipitated genomic DNA was assessed by real-time PCR using six pairs of oligonucleotide primers designed to produce amplicons covering the RE1/NRSE site in the BDNF gene and flanking sequences located 758 bp upstream the RE1/NRSE and 431, 1222, 2279, and a distal 19350 bp downstream of the RE1/NRSE. Relative positions of these primer pairs are indicated in the schematic diagram (green arrows). White columns represent the average of REST/NRSF enrichment in controls. Gray columns indicate the average of REST/NRSF enrichment in HD subjects. The scanning ChIP assay shows a peak of REST/NRSF occupancy centered at the RE1/NRSE site of the BDNF gene. No enrichment is seen at distal regions, and no enrichment is seen at the coding region of BDNF and β-actin gene, which is not regulated by REST/NRSF. b, REST/NRSF was immunoprecipitated from chromatin extracted from the cerebral cortices of five HD patients (from left to right: 3484, 3723, 3176, 2866, 6062) and five age-matched controls (from left to right: 3899, 3688, 3932, 3888, 3746). REST/NRSF enrichment over a sham antibody (preimmune serum) was calculated by quantitative PCR at the RE1/NRSEs of five REST/NRSF target genes, BDNF, SYN1, CHRM4, CHRNB2 and SNAP25. c, Conventional ChIP and ChIP on chip indicated that REST/NRSF target genes showing increased occupancy in HD subjects can be assigned to nine functional groups.
The PCR was performed in a total volume of 20 μl containing equal amounts of input and immunoprecipitated DNA, 50 mm KCl, 20 mm Tris-HCl, pH 8.4, 0.2 mm dNTPs, 25 U/ml iTaq DNA polymerase, 3 mm MgCl2, SYBR Green I, 10 nm fluorescein, stabilizers (iQ SYBR Green Supermix; Bio-Rad), and 0.2 μm of forward and reverse primers.
The primers used in the assay were comprised of the following: m477 S, 5′-GAATCGGGTTTACCCACTGCAAG-3′; m477 AS, 5′-GACGGTTGTCAGACAAGCATC-3′; m3246 S, 5′-TGGCCAGAAACTGTTGACAAAGTC-3′; m3246 AS, 5′-GGGAGGATGGTCTGTATGCGAA-3′; m383 S, 5′-AAAGGACGCGTAGTGGAGAGG-3′; m383 AS, 5′-CCCAGGTTCTCACCTAGGTC-3′; m1123 S, 5′-GGCCGGATGCTTCCTTGAGC-3′; m1123 AS, 5′-GAAAGGACCTTCCACTCCGG-3′; m3353 S, 5′-TCAACCTGTGTAAGCCGCTGC-3′; m3353 AS, 5′-GTTTCCCCTAGAGGACAGGC-3′; m17393 S, 5′-GCAAGGAAAAGGCGCGTCGT-3′; m17393 AS, 5′-AGCTCTTCGGTTGAGCTTCGATTGG-3′; h758 S, 5′-CACTTGAGTCTCCAGGACAGCA-3′; h758 AS, 5′-TCTTTGGCGTGTGAAGTGCTAGG-3′; h431 S, 5′-GGTAGAG-GGAGAGCATGAGAG-3′; h431 AS, 5′-TATCCGCAGGAAGACTGAGTTTC-3′; h1222 S, 5′-GGAGTAGAAGGTCCTTTCCGG-3′; h1222 AS, 5′-CAGCTGATTGGTGGCTCTGTC-3′; h2279 S, 5′-GTCACGTCATCCTCTACACACAC-3′; h2279 AS, 5′-CATAAACAGTGAATATCAGGTGCACC-3′; h19350 S, 5′-GCAGCTGCCTTGATGGTTACTTTG-3′; h19350 AS, 5′-CCAGGCAATGACAGACCTCG-3′.
DNA from the same chromatin immunoprecipitation underwent quantitative real-time PCR for β-actin, a gene not regulated by REST/NRSF and not proximal to any RE1/NRSEs. Primers close to the β-actin promoter have been used and are as follows: mβ-actin S, 5′-AGGTATCCTGACCCTGAAG-3′; mβ-actin AS, 5′-GCTCATTGTAGAAGGTGTGG-3′; hβ-actin S, 5′-TGCCTAGGTCACCCACTAATG-3′; hβ-actin AS, 5′-GTGGCCCGTGATGAAGGCTA-3′.
The primers used for the BDNF coding region are as follows: mBDNF S, 5′-TCGTTCCTTTCGAGTTAGCC-3′; mBDNF AS, 5′-TTGGTAAACGGCACAAAAC-3′; hBDNF S, 5′-GTAAAGCCAACCCTGTGTCG-3′; hBDNF AS, 5′-TCCGCTCCAAAATCTGACTC-3′.
The following PCR amplification protocol was applied: 95°C for 3 min, followed by 45 cycles of 30 s at 95°C, 30 s at 60°C, and 30 s at 72°C. Fluorescence was quantified during the 60°C annealing step, and product formation was confirmed by a melting curve analysis (55–94°C). Immunoprecipitated DNA for the different analyzed RE1/NRSE loci was quantified using a standard curve (nanograms of DNA). Binding of REST/NRSF at the RE1/NRSEs was calculated as nanograms of DNA IP REST antibody per nanogram of DNA IP preimmune serum.
Real-time PCR for gene expression.
Three total RNA preparations were used, and two independent reverse transcription (RT) reactions were set up for every RNA stock. Real-time PCR analyses were done in triplicate for each of the analyzed genes to obtain replicates for statistical analyses.
iCycler Thermal Cycler with Multicolor Real-time PCR Detection System (Bio-Rad) was used. All reactions were performed in a total volume of 25 μl containing 50 ng of cDNA, 50 mm KCl, 20 mm Tris-HCl, pH 8.4, 0.2 mm dNTPs, 25 U/ml iTaq DNA polymerase, 3 mm MgCl2, SYBR Green I, 10 nm fluorescein, stabilizers (iQ SYBR Green Supermix; Bio-Rad), and 0.2 μm of forward and reverse primers.
Amplification cycles consisted of an initial denaturing cycle at 95°C for 3 min, followed by 45 cycles of 30 s at 95°C, 30 s at 60°C, and 30 s at 72°C for all of the genes analyzed. Fluorescence was quantified during the 60°C annealing step, and product formation was confirmed by a melting curve analysis (55–94°C). Amounts of target gene mRNA were normalized to a reference gene (β-actin) according to Pfaffl (2001). Primer sequences used are as follows: mBDNF S, 5′-TCGTTCCTTTCGAGTTAGCC-3′; mBDNF AS, 5′-TTGGTAAACGGCACAAAAC-3′; mSyn1 S, 5′-GAGCAGATTGCCATGTCTGA-3′; mSyn1 AS, 5′-CACTGCGCAGATGTCAAGTC-3′; mChrm4 S, 5′-TCCTCACCTGGACACCCTAC-3′; mChrm4 AS, 5′-ACGTAGCAGAGCCAGTAGCC-3′; mDrd3 S, 5′-GCTGTGATGTTTTTGTCACCC-3′; mDrd3 AS, 5′-GGCATGACCACTGCTGTGTA-3′; mPenk1 S, 5′-AATTCCCGCTATTTGCCAGT-3′; mPenk1 AS, 5′-AGTGGAGGGACAGCTCGTTA-3′; mChrnb2 S, 5′-AGAGGTGAAGCACTTCCCATTT-3′; mChrnb2 AS, 5′-GCCA-CATCGCTTTTGAGCAC-3′; mβ-actin S, 5′-AGTGTGACGTTGACATCCGTA-3′; mβ-actin AS, 5′-GCCAGAGCAGTAATCTCCTTCT-3′; mREST/NRSF S, 5′-CGAACTCACACAGGAGAACG-3′; mREST/NRSF AS, 5′-GAGGCCACATAATTGCACTG-3; mGAPDH S, 5′-AATGGTGAAGGTCGGTGTGAAC-3′; mGAPDH AS, 5′-TCGCTC-CTGGAAGATGGTGATG-3′.
Radioactive RT-PCR.
Radioactive PCR was performed in a total volume of 50 μl containing cDNA made from 0.25 μg of RNA, 20 mm Tris-HCl, pH 8.4, 50 mm KCl, 1.5 mm MgCl2, 0.2 mm dNTPs, 1.7 μCi[α32P]dCTP, 0.4 μm of each primer, and 2 U Taq polymerase (Invitrogen). All BDNF mRNA isoforms were amplified with a multiple cycle number (25–30 cycles) to determine the appropriate conditions for obtaining semiquantitative differences in their expression levels. Cycle steps and primer sequences are described in Zuccato et al. (2001). PCR products were separated by nondenaturing 6% PAGE and visualized by autoradiography. In each PCR experiment, BDNF levels were quantified and normalized relative to β-actin mRNA level.
DN:REST delivery.
Sequences encoding the DNA binding domain of REST/NRSF (amino acid residues 234–437) fused to a myc epitope were excised from pMTDomNeg12 and inserted into pAdTrack-CMV (Wood et al., 2003). Plaque-forming virus particles (109 U/ml) were used to infect Hdh7/7 and Hdh109/109 cells. RNA and proteins were harvested 48 h later as described in Wood et al. (2003). The same assay was performed with similar results by transfecting cells with Lipofectamine PLUS (Invitrogen) according to manufacturer's instructions.
RE1/NRSE microarray.
A bioinformatic search for consensus RE1/NRSE motifs were compiled into a searchable database (RE1db) (Bruce et al., 2004). There are 537 putative RE1/NRSEs (containing no mismatches) in the human genome. Primers were designed to sequences <250 bp distal from each of these potential RE1s, with amplicon sizes between 200 and 350 bp. Sense primers were designed with an 8 bp linker at the 5′ of the sequence TGACCATG, which was required to immobilize the sequences to the Codelink amine binding slides (GE Healthcare). The PCRs were performed in two rounds of amplification. These reactions were set up in 96-well plates (Costar, Cambridge, MA) by adding 5 μl of 100 ng/μl human genomic DNA, 6 μl of 10× buffer (500 mm KCl, 50 mm Tris, pH 8.5, 25 mm MgCl2), 3 μl of 10 mm dNTP, 1.5 μl of 200 ng/μl sense primer, 1.5 μl of 200 ng/μl antisense primer, and 0.375 μl of 5 U/μl Taq polymerase (Promega, Madison, WI) in a total reaction volume of 60 μl, under the following conditions: 95°C for 10 min, followed by 30 cycles of 95°C for 60 s, 58°C for 60 s, and 72°C for 1 min and 30 s, with a final extension step of 10 min at 72°C. These PCR products were diluted 1:10, and 2 μl was used in the second round of PCR, in a reaction containing 6 μl amino-linking buffer (500 mm KCl, 25 mm MgCl2, 50 mm Tris, pH 8.5), 6 μl of 10 mm dNTPs, 3 μl of 200 ng/μl 5′ amino-modified universal primer (GCTGAACAGTATGACCATG, the underlined 8 bp of which recognizes the linker from the first round of PCR), 3 μl of 200 ng/μl antisense primer (same as the antisense primer from the first round), and 0.6 μl of 5 U/μl Taq polymerase in a final volume of 60 μl. PCR products were filtered through Multiscreen-GV 96-well filter plates (Millipore, Billerica, MA) and added to 15 μl of 4× spotting buffer (1 m sodium phosphate, pH 8.5, 0.001% Sarkosyl). The products were arrayed in quadruplicate at a constant temperature of 25°C and a relative humidity of 45% and incubated in a humidity chamber at 80% relative humidity for 36 h. The slides were washed in 1% ammonium hydroxide, then rinsed in 0.1% SDS, followed by HPLC grade water and were submerged in 95°C water for 2 min to remove the antisense strands. They were then rinsed twice in water at room temperature and dried by centrifugation at 800 × g for 5 min. Control spots on the array included PCR products of 38 (7% array coverage) sequences that do not contain REST/NRSF binding sites to avoid nonspecific hybridization with other genomic RE1/NRSE sites, negative spots included sequences from Arabidopsis genes, and blank spots contained only spotting buffer. The RE1/NRSEs present on the microarray and the closest genes to these sequences have been included in the RE1/NRSE database (http://www.bioinformatics.leeds.ac.uk/cgi-bin/RE1db/nrse.cgi).
Five hundred nanograms of anti-REST/NRSF ChIP DNA and input DNA were labeled by direct incorporation of cyanine 3 (Cy3) and Cy5, respectively. 60 μl of 2.5× random primers (Invitrogen) were added to 70 μl of input or ChIP DNA. The samples were denatured for 10 min in a 100°C heat block and cooled on ice, then incubated at 37°C overnight with 1.5 μl of 1 mm Cy5- or Cy3-labeled dCTP (New England Biolabs, Ipswich, MA), 15 μl of dNTPs (1 mm dCTP and 2 mm dATP, dGTP, and dTTP), and 3 μl Klenow (120 U) (Invitrogen). Unincorporated nucleotides were removed from the reactions by spinning the samples through micro-spin Sepharose G50 microcolumns (GE Healthcare). The Cy3- and Cy5-labeled DNAs were mixed together and precipitated with 135 μg of human Cot1 DNA. The DNA was pelleted and resuspended in a 2× saline sodium citrate (SSC) hybridization buffer containing 10 mm Tris-HCl, pH 7.4, 50% formamide, 5% dextran sulfate, and 0.01% Tween 20. Samples were denatured in 70°C heat block for 10 min, and 30 μg yeast tRNA was added. Slides were incubated with the DNA at 37°C for 48 h with agitation, then washed in PBS/0.05% Tween 20 at 37°C six times, followed by 3 × 10 s in 0.2× SSC at 52°C. The slides were washed again in PBS/0.05% Tween 20 at 23°C six times. Each wash consisted of a 20 s flow time followed by a 10 s wash time. Slides were rinsed in HPLC-grade water at room temperature for 30 s, dried, and scanned.
A GMS418 array scanner (Affymetrix; Genetic Microsystems, Santa Clara, CA) scanned the two wavelengths compatible with excitation for Cy5 and Cy3, and the images acquired were analyzed with BlueFuse software to yield background-subtracted intensity values. The channel intensities were normalized using the LOWESS function. Per array, mean Cy3/Cy5 (REST/NRSF ChIP DNA/input DNA) ratios for each probe were derived from their constituent quadruplicate spots and data-points with SEM values >25% excluded from subsequent analysis. The mean fold enrichment of the 38 RE1/NRSE-devoid regions (7% array coverage) was used to derive a corrective baseline for the data set. Those corrected RE1/NRSE containing probe ratios that deviated from this reference mean by a value greater than the mean plus two SDs (non-RE1/NRSE mean, 1.043; SD, 0.095; mean plus 2 SDs, 1.23), were considered enriched (corresponding to a 95% confidence interval, p < 0.05). This provided every sequence on the microarray with a relative REST/NRSF enrichment. Relative REST/NRSF enrichments were averaged from three independent HD patient samples and two independent control patient (non-HD) samples and the SEM calculated. For each putative RE1/NRSE locus, relative REST/NRSF enrichments for the HD samples were divided by those of the non-HD samples to show the increase in REST/NRSF recruitment in HD (Table 1).
Table 1.
ChIP-on-chip analyses on the human HD cortex
| Gene name | Protein name | Ontology | HD/NORM |
|---|---|---|---|
| SNN | Stannin | Signaling | 3.1 |
| OAT | Ornithine aminotransferase mitochondrial precursor | Metabolism | 2.5 |
| NPC2 | Epididymal secretory protein E1 precursor | Unknown | 2.3 |
| PCSK1 | Neuroendocrine convertase 1 precursor | Signaling | 2.2 |
| SLC13A1 | Solute carrier family 13, member 1 | Transport | 2.1 |
| CNTNAP | Contactin-associated protein-like 2 precursor | Structure | 2.0 |
| CACNA1H | Voltage-dependent T-type calcium channel subunit alpha-1H | Signaling | 2.0 |
| CREB3L3 | CREB/ATF family transcription factor | Transcription | 1.9 |
| GRIK3 | Glutamate receptor, ionotropic kainate 3 precursor | Signaling | 1.9 |
| FAM105A | Unknown | Unknown | 1.9 |
| CMKLR1 | Chemokine receptor-like 1 | Immune response, development, signaling | 1.9 |
| FUT9 | α-(1,3)-Fucosyltransferase | Metabolism | 1.8 |
| OSBP2 | Oxysterol-binding protein 2 | Metabolism | 1.7 |
| LRRTM4 | Leucine rich repeat transmembrane neuronal 4 | Protein binding | 1.7 |
| ARRDC3 | Arrestin domain-containing protein 3 | Unknown | 1.7 |
| EPHA10 | Ephrin type-A receptor 10 precursor | Signaling | 1.7 |
| VIP | Vasoactive intestinal peptide | Signaling | 1.7 |
| ENSG00000110427 | G2 protein | Unknown | 1.6 |
| TRPC7 | Short transient receptor potential channel 7 | Signaling | 1.6 |
| PHKB | Phosphorylase b kinase regulatory subunit beta | Metabolism, signaling | 1.6 |
| HNRPUL1 | Heterogeneous nuclear ribonucleoprotein U-like protein 1 | Transcription, RNA processing | 1.6 |
| NELL1 | Protein kinase C-binding protein NELL1 precursor | Structure, neurogenesis, metabolism | 1.5 |
| CBLN1 | Cerebellin-like glycoprotein 1 precursor | Structure | 1.5 |
| B3GAT1 | Galactosylgalactosylxylosyl protein 3-beta-glucuronosyltransferase 1 | Metabolism | 1.5 |
| DDX25 | ATP-dependent RNA helicase DDX25 | Spermatogenesis, RNA helicase | 1.5 |
| SYT4 | Synaptotagmin-4 | Synaptic activity | 1.5 |
| LHX5 | LIM/homeobox protein Lhx5 | Transcription, neuronal development | 1.5 |
| NCAM2 | Neural cell adhesion molecule 2 precursor | Neuronal development, adhesion | 1.4 |
| XKR4 | XK-related protein 4 | Unknown | 1.4 |
| TCBA1 | T-cell lymphoma breakpoint associated target protein 1 | Unknown | 1.4 |
| USP31 | Ubiquitin-specific proteinase 31, putative | Metabolism | 1.4 |
| FGF14 | Fibroblast growth factor 14 | Neuronal development, signaling | 1.3 |
| KCNJ6 | G-protein-activated inward rectifier potassium channel 2 | Signaling | 1.3 |
| SLC7A14 | Solute carrier family 7 (cationic amino acid transporter, y + system), member 14 | Amino acid transport | 1.3 |
| KCNAB2 | Voltage-gated potassium channel subunit beta-2 | Transport, signaling | 1.3 |
| FGF12 | Fibroblast growth factor 12 | Neuronal development, signaling | 1.3 |
| BDNF | Brain-derived neurotrophic factor (BDNF) | Growth factor, neurodevelopment | 1.3 |
| TSPAN14 | Tetraspanin-14 | Unknown | 1.3 |
| GENSCAN00000062697 | Presynaptic cytomatrix protein Piccolo homolog | Synaptic transmission | 1.3 |
| POU4F1 | POU domain, class 4, transcription factor 1 | Neuronal development, transcription | 1.3 |
| EFNA5 | Ephrin-A5 precursor | Neuronal development, signaling | 1.3 |
| SYN1 | Synapsin I | Synaptic transmission | 1.2 |
| HPGD1 | 15-Hydroxyprostaglandin dehydrogenase [NAD+] | Metabolism | 1.2 |
Relative REST/NRSF enrichments were averaged from three independent HD patient samples (3932, 2866, 5570) and two independent control patient (non-HD) samples (5919, 6002). Values indicated as HD/NORM mean relative enrichment seen in HD tissue when normalized to control tissue. Forty-three RE1/NRSE containing genes are enriched in the HD cortex versus control (corresponding to a 95% confidence interval, p < 0.05). RE1/NRSE location and gene ontology information were extracted from Ensembl (http://www.ensembl.org), UniProt (http://www.expasy.uniprot.org), and the RE1/NRSE database (Bruce et al., 2004). CREB, cAMP response element-binding protein; ATF, activating transcription factor.
REST/NRSF target genes were characterized according to the function of their protein products using information from the scientific literature and the UniProt (www.uniprot.org) and Ensembl (www.ensembl.org) databases. A full list of these genes, proteins, and ontologies can be viewed in Table 1 and Figure 4c (pie chart).
ELISA assays.
Cell lysates were prepared in lysis buffer consisting of 10% glycerol, 25 mm Tris HCl, pH 7.5, 150 mm NaCl, 1% Triton X100, 5 mm EDTA, and 1 mm EGTA supplemented with 1:100 Protease Inhibitor Mixture (Sigma-Aldrich, St. Louis, MO). Samples were homogenized, sonicated, and centrifuged (15 min at 4°C max speed Biofuge). The supernatants were collected and stored at −30°C. Samples were assayed for BDNF by using the ImmunoAssay System (Promega), as described by the manufacturer.
Statistical analysis.
For ChIP-PCR, gene expression, and ELISA studies, we compared data using one-way ANOVA.
Results
REST/NRSF binding at the endogenous RE1/NRSE in HD cells and animal models
To test whether HD is characterized by an increase of nuclear binding activity of REST/NRSF at the RE1/NRSE of endogenous chromosomal target genes, we used ChIP and assessed occupancy of a cohort of REST/NRSF target genes including Bdnf, synapsin 1 (Syn1), Chrm4, dopamine receptor D3 (Drd3), proenkephalin 1 (Penk1), and cholinergic receptor nicotinic β polypeptide2 (Chrnb2) using an N-terminal REST/NRSF antibody in HD samples. Specificity of REST/NRSF immunoprecipitation in our ChIP assays was assessed by evaluating REST/NRSF occupancy at non-REST/NRSF target genes and also by using scanning ChIP across the BDNF locus (supplemental Figs. 1, 2, available at www.jneurosci.org as supplemental material).
ChIP analyses were initially performed on chromatin extracted from neural cell lines derived from homozygous mutant huntingtin knock-in mice in which an expanded CAG has been inserted into the endogenous mouse Hdh gene (Hdh109/109) and their wild-type counterparts (Hdh7/7) (Trettel et al., 2000). Figure 1a (left) shows that no REST/NRSF could be detected at RE1/NRSE-containing regions of any of the target genes in Hdh7/7 cells, whereas, in contrast, the equivalent loci were all occupied by REST/NRSF in Hdh109/109 cells. Under these conditions, levels of total REST/NRSF protein and mRNA were identical in both Hdh109/109 cells and Hdh7/7 cells (Zuccato et al., 2003) (see Fig. 3a). Next, we performed real-time PCR to see whether this increased occupancy in Hdh109/109 cells was accompanied by a corresponding decrease in gene expression. Consistent with previous semiquantitative analyses (Zuccato et al., 2003), these experiments showed a significant decrease in the expression levels of RE1/NRSE genes in Hdh109/109 cells compared with Hdh7/7 cells (Fig. 1a, right). These data demonstrate a direct correlation between increased REST/NRSF occupancy and REST/NRSF-mediated transcriptional repression in the presence of endogenous mutant huntingtin.
Figure 1.

Endogenous REST/NRSF binding at RE1/NRSEs is increased in HD leading to repression of RE1/NRSE gene transcription. a, Left, ChIP analysis on six RE1/NRSE selected loci in neural cells established from homozygous (Hdh109/109) knock-in mice and wild-type littermates (Hdh7/7). Data represent the average of three different experiments. Right, mRNA levels of the same RE1/NRSE-controlled genes in Hdh7/7 and Hdh109/109 cells by real-time PCR. Data are the average of three different experiments. *p < 0.05; **p < 0.01 versus Hdh7/7; ANOVA test. b, Left, ChIP on cortical lysates from R6/2 transgenic mice at 12 weeks of age and corresponding littermates. Right, mRNA levels by real-time PCR of the same RE1/NRSE genes in R6/2 transgenics and controls at the same age. ChIP and mRNA data represent the average of five R6/2 transgenics and five controls. *p < 0.05; ** p < 0.01 versus littermates, ANOVA test. c, Left, ChIP for REST/NRSF on cortical lysates from Hdh109/109 at 5 months. Data represent the average of three Hdh109/109 and three Hdh7/7 mice. *p < 0.05 versus Hdh7/7 mice; ANOVA test. Right, REST/NRSF binding at the NRSEbdnf in Hdh109/109 at 1 and 5 months. Data are the average of three Hdh109/109 and three Hdh7/7 mice at 1–5 months. *p < 0.05 versus Hdh7/7 mice; ANOVA test. d, ChIP for REST/NRSF on liver lysates from three R6/2 mice and three controls at 12 weeks of age. *p < 0.05 versus wild-type mice; ANOVA test.
Figure 3.

Expression of DN:REST derepressed RE1/NRSE-regulated transcription in HD. a, Bdnf, synapsin-1, and M4 muscarinic cholinergic receptor mRNA levels were measured in native Hdh7/7 and Hdh109/109 cells after delivery of DN:REST or empty vector. Bdnf, synapsin-1, and M4 muscarinic cholinergic receptor mRNA levels were restored in Hdh109/109 cells infected with DN:REST compared with untransfected cells and cells transfected with empty vector (− and E, respectively). mRNA levels of REST/NRSF and glyceraldehyde-3-phosphate dehydrogenase (GAPDH), genes whose transcription is not regulated by the RE1/NRSE silencer, are not affected by DN:REST expression. Data are expressed as percentage of controls (100% was assigned to Hdh7/7 cells) and represent the average of four independent experiments. *p < 0.05 versus Hdh7/7 untreated cells (−); #p < 0.05 versus Hdh109/109 untreated cells and cell with empty vector (− and E, respectively). b, Left, Bdnf exon II, III, and IV mRNA levels in Hdh7/7 and Hdh109/109 cells by semiquantitative radioactive RT-PCR. Right, Quantitative analyses of the BDNF mRNA levels. The peak densitometric areas relative to Bdnf II, III, and IV were normalized over the peak densitometric area of the β-actin band. Data are expressed as percentage of controls (100% was assigned to Hdh7/7 untreated cells) and represent the average of three independent experiments. *p < 0.05 versus Hdh7/7 cells; #p < 0.05 versus Hdh109/109 untreated cells and cells with empty vector (− and E, respectively). c, Bdnf protein levels determined by ELISA on lysates from Hdh7/7 and Hdh109/109 cells and cells infected with DN:REST and empty vector. Data are the average of three independent experiments. *p < 0.05 versus Hdh7/7 untreated cells (−). #p < 0.05 versus Hdh109/109 untreated cells and cells with empty vector (− and E, respectively).
Next, we proceeded to see whether this correlation could be observed in the cerebral cortices of 12-week-old symptomatic R6/2 transgenic mice expressing the human huntingtin exon 1 fragment bearing a 150 glutamine expansion (Mangiarini et al., 1996). Increased REST/NRSF binding was seen in symptomatic mice at the RE1/NRSE of the Bdnf, Chrm4, and Penk1 genes, whereas no differences were observed at the Syn1, Drd3, and Chrnb2 RE1/NRSE loci (Fig. 1b, left). Real-time PCR analyses of the corresponding mRNA levels demonstrated that increased REST/NRSF binding in R6/2 mice is accompanied by a significant decrease in expression of the same RE1/NRSE-controlled neuronal genes (Fig. 1b, right). These data demonstrate that at some, but not all, RE1/NRSE neuronal genes mutant huntingtin causes a selective increase in REST/NRSF occupancy and a concomitant decrease in transcription.
We extended these studies by analyzing cortical samples from homozygous Hdh109/109 mutant huntingtin knock-in mice because they represent a more accurate genetic model of HD with expression of the full-length endogenous mutant huntingtin allele driven from the endogenous Hdh promoter. Compared with R6/2 mice, which exhibit a rapid and severe neurological phenotype, Hdh109/109 mice display a slower progressive phenotype recapitulating earlier stages of human HD (Wheleer et al., 1999). ChIP experiments performed on cortical lysates from 5-month-old mice showed increased occupancy by REST/NRSF at its target RE1/NRSE loci, compared with controls (Fig. 1c, left). Importantly, this phenotype is progressive because REST/NRSF binding at the Bdnf locus is greater at 5 months compared with 1-month-old mice (Fig. 1c, right).
This evidence indicates for the first time a direct engagement of REST/NRSF at selected RE1/NRSE genomic loci in HD mouse models and that, at least for the Bdnf locus, a correlate exists between REST/NRSF enrichment and disease evolution in knock-in mice, possibly indicating that this molecular phenotype of increased RE1/NRSE occupancy and repressed gene transcription may be a general feature of disease progression. Furthermore, it can be seen that discrete changes in REST/NRSF occupancy signature are evident in different animal and cellular HD models effecting that disruption of target gene expression may be a composite of common and individual changes.
An enigma of HD is that whereas both REST/NRSF and huntingtin are also widely expressed outside the brain, HD pathology is largely restricted to the nervous system. We have previously shown that many REST/NRSF targets that are silent in non-neural cells are inaccessible to REST/NRSF (Wood et al., 2003; Belyaev et al., 2004). Accordingly, we postulated that such targets would be refractory to increases in nuclear REST/NRSF consequent to expression of mutant huntingtin, thus conferring brain specificity to the dysfunction of the REST/NRSF–NRSE signaling pathway. To test this notion, we examined the interaction of REST/NRSF with neural and hepatic target genes in the liver. We chose three silent neural target genes, Bdnf, Penk1, and Chrm4 and one active hepatic REST/NRSF target gene, hepatic nuclear factor (Hnf-1a) (Bruce et al., 2004). Our data showed that in liver chromatin from wild-type mice, no REST/NRSF could be detected at any of these targets, but that in R6/2 mice, REST/NRSF could be detected at the active Hnf-1a locus but not at the silent Bdnf, Penk1, and Chrm4 loci (Fig. 1d). These data confirm our proposal that expression of mutant huntingtin leads to an increase in nuclear REST/NRSF, which in turn leads to an increase in occupancy, and potentially dysfunction of actively transcribed genes, therefore affecting neural tissues and also selective genes in non-neural tissues.
REST/NRSF interactions in systems depleted by endogenous huntingtin
Although HD is predominantly seen as a “gain-of-function” disorder, there is increasing evidence for a “loss-of-function” component (Cattaneo et al., 2001, 2005). Based on the demonstration of increased activity of an RE1/NRSEBdnf reporter in response to overexpression of wild-type huntingtin (Zuccato et al., 2003), and together with the present data, we propose that the abnormal RE1/NRSE phenotype observed in HD is strictly dependent on the level/activity of the wild-type protein, implying that a loss-of-function mechanism is in operation at the level of the RE1/NRSEs in neurons that express limited but detectable levels of REST/NRSF. To test this notion, we examined the interaction of REST/NRSF with its target genes in embryonic stem (ES) cells in which one or two alleles of the Hdh gene have been inactivated via removal of exon 4 and 5 of the endogenous mouse huntingtin gene (Duyao et al., 1995), indicated as Hdh+/Hdhexon4/5 (ES+/−) and Hdhexon4/5/Hdhexon4/5 (ES−/−), respectively (a Western blot showing huntingtin protein levels in these cells is reported in supplemental Fig. 3, available at www.jneurosci.org as supplemental material). REST/NRSF binding at the Bdnf locus was incrementally increased in ES+/− cells and ES−/− cells compared with control ES+/+ cells (Fig. 2a, left). This increase in occupancy is not resulting from increased total levels of REST/NRSF, because levels of REST/NRSF mRNA were the same in ES+/+, ES+/− cells, and ES−/− cells (Fig. 2a, right). In concordance with the increased levels of REST/NRSF occupancy, total Bdnf expression was reciprocally reduced in ES+/− cells and ES−/− cells compared with ES+/+ cells (Fig. 2b, left). Also, this reduction in Bdnf mRNA is attributable to a specific loss of Bdnf mRNA II (Fig. 2b, right), whose transcription is directly regulated by a RE1/NRSE in exon II. Consistent with these data, transcription of Bdnf III and IV, which are not REST/NRSF regulated, are not affected by dramatic huntingtin deficiency (Fig. 2b, right).
Figure 2.

Loss of wild-type huntingtin increases REST/NRSF binding at the RE1/NRSE. a, Left, ChIP for REST/NRSF binding at the RE1/NRSEBdnf on chromatin lysates from Hdh+/+, Hdh+/−and Hdh−/− ES cells. Right, REST/NRSF mRNA levels in Hdh+/+, Hdh+/− and Hdh−/− ES cells. Data are the average from three independent experiments. *p < 0.05 versus Hdh+/+, #p < 0.05 versus Hdh+/−; ANOVA test. b, Left, Real-time PCR for total Bdnf mRNA levels in Hdh+/+, Hdh +/−, and Hdh−/− embryonic stem cells. Data are the average from three independent experiments. *p < 0.05 versus Hdh+/+; #p < 0.05 versus Hdh+/−; ANOVA test. Right, Semiquantitative radioactive RT-PCR for Bdnf exon II, III, and IV mRNA levels. β-actin was used as control. Graphs show densitometric analyses of the expression levels of Bdnf mRNA II, III, and IV compared with levels of β-actin. Values represent the percentage of arbitary units (a.u.) relative to Hdh+/+ cells to which 100% was assigned. Data are the average from eight independent PCR experiments. *p < 0.05 versus Hdh+/+; #p < 0.05 versus Hdh+/−; ANOVA test. c, ChIP for REST/NRSF on cortical samples from three null Hdh conditional knock-out mice cre Hdh flox/− (indicated as Hdh−/−) and three Hdh flox/− (indicated as Hdh+/−) and wild-type mice. REST/NRSF binding was analyzed at the RE1/NRSE of Bdnf, Syn1, Chrm4, Drd3, Penk1, and Chrnb2. *p < 0.05 versus Hdh+/+ mice; #p < 0.05 versus Hdh+/− mice; ANOVA test. d, Bdnf levels in cortical samples from null Hdh conditional knock-out mice. Left, Total Bdnf mRNA levels by real-time PCR. Data are the average from three independent experiments. *p < 0.05; **p < 0.01 versus Hdh+/+ mice; #p < 0.05 versus Hdh+/− ANOVA test. Right, Semiquantitative radioactive RT-PCR for Bdnf exon II, III, and IV mRNA levels. β-actin was used as control. A quantitative evaluation of Bdnf mRNA II, III, and IV is shown in supplemental Figure 7 (available at www.jneurosci.org as supplemental material).
Next, we extended these experiments to examine REST/NRSF interactions in the brains of Hdh+/+, Hdh+/−, and Hdh−/− mice. Conditional ablation of Hdh in the adult mouse forebrain is accompanied by apoptosis, progressive neurodegeneration, and behavioral abnormalities resembling the HD phenotype (Dragatsis et al., 2000). Similar to what is seen in mice carrying the mutant protein, ChIP analyses in the cortex from Hdh+/− and Hdh−/− mice showed a proportional increase in REST/NRSF occupancy at the RE1/NRSE of the Bdnf gene, compared with their wild-type Hdh+/+ counterparts (Fig. 2c). Under these conditions, levels of total REST/NRSF mRNA were identical in Hdh+/+, Hdh+/−, and Hdh−/− mice (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). Furthermore, the increase in REST/NRSF occupancy at the Bdnf locus was accompanied by a reciprocal decrease in Bdnf expression (Fig. 2d, left), clearly indicating that expression of Bdnf in vivo is affected by reduced endogenous wild-type huntingtin. Consistent with our results obtained with ES cells, the reduction in Bdnf transcript levels could be largely attributed to a specific loss of Bdnf mRNAII transcripts in Hdh+/− and Hdh−/− mice (Fig. 2d, right). In addition, we found that REST/NRSF occupancy at Syn1, Drd3, and Chrnb2 genes was increased in Hdh−/− relative to Hdh+/− or Hdh+/+ mice. REST/NRSF occupancy was undetectable in Hdh+/− and Hdh−/− mice, but real-time PCR indicated a proportional decrease in Syn1 and Drd3 mRNA levels in Hdh+/− and Hdh+/+ mice with respect to wild-type mice (supplemental Fig. 5, available at www.jneurosci.org as supplemental material).
We conclude that loss of wild-type huntingtin causes a selective increase in REST/NRSF binding at the RE1/NRSEs, leading to generalized transcriptional repression of RE1/NRSE bearing neuronal genes. Because this molecular phenotype is seen in the presence of mutant huntingtin, we would also suggest that the expanded polyQ tract in HD might abrogate huntingtin normal activity in REST/NRSF-mediated gene repression. Thus, reduced activity of wild-type huntingtin in HD may contribute to disease pathogenesis.
Expression of dominant-negative REST/NRSF derepresses neuronal gene transcription in HD
We reasoned that if a role of normal huntingtin was to facilitate expression of RE1/NRSE-bearing genes and that mutant huntingtin attenuated this function, then it may also follow that attenuation of REST/NRSF function should derepress those genes repressed in the presence of the mutant protein. Accordingly, we used a dominant-negative REST/NRSF construct (DN:REST) comprising the DNA binding domain (Wood et al., 2003; Belyaev et al., 2004; Bruce et al., 2004) to attenuate REST/NRSF binding to its cognate RE1/NRSE sites in Hdh109/109 mutant huntingtin knock-in cells. These experiments showed that 48 h after delivery of DN:REST into Hdh109/109 cells, Bdnf mRNA was increased to similar levels to that seen in untreated wild-type Hdh7/7 cells (Fig. 3a). Cells treated with empty virus or untreated cells showed no variation in total Bdnf mRNA level. In addition, because DN:REST acts at many RE1/NRSEs (Bruce et al., 2004), then it follows that it is likely that transcription of other neuronal genes whose expression is decreased in the disease state could be similarly restored. Figure 3a shows indeed that synapsin1 and M4 muscarinic cholinergic receptor mRNA levels are also rescued by DN:REST in Hdh109/109 cells. Furthermore, mRNA levels of genes, whose transcription is not controlled by REST/NRSF, are unchanged after DN:REST expression, indicating that DN:REST acts specifically on RE1/NRSE targets (Fig. 3a).
In addition, the change in Bdnf RNA levels could be attributed specifically to changes in Bdnf mRNA II transcripts (Fig. 3b). This rescue of Bdnf mRNA was mirrored by an increase in Bdnf protein levels as judged by ELISA assays (Fig. 3c). Our results therefore indicate that it is possible to restore Bdnf protein levels in HD cells by attenuation of REST/NRSF binding at the Bdnf locus.
A global analysis of endogenous REST/NRSF binding in HD human brain
The repertoire of potential REST/NRSF-regulated genes is extensive and includes those encoding growth factors and hormones, neuronal transcription factors, ion channels, proteins involved in axonal guidance, neurotransmitters, proteins involved in vesicle trafficking, and fusion and synaptic transmission (Bruce et al., 2004). To test whether REST/NRSF shows increased binding at genomic RE1/NRSE loci in the human HD brain, we performed ChIP assays on chromatin lysates from cortical specimens obtained from five individuals with HD (grade III-IV) and five age-matched controls. First, specificity of the ChIP assay was validated using a scanning ChIP of the BDNF locus, and two controls and two HD subjects were analyzed (Fig. 4a). In the ChIP-scanning assay, the anti-REST/NRSF immunoprecipitated genomic DNA was assessed by real-time PCR using five pairs of oligonucleotide primers designed to produce amplicons covering the RE1/NRSE site in the BDNF gene and flanking sequences located upstream and downstream of the RE1/NRSE up to maximum distance of ∼20,000 bp. In addition, primers corresponding to the BDNF coding region, which lies in excess of 50,000 bp distant from the RE1/NRSE, were used. The scanning ChIP assays show a peak of REST/NRSF occupancy centered at the RE1/NRSE site of the BDNF gene (Fig. 4a). In addition, REST/NRSF occupancy is significantly greater in HD samples than in control tissue. No enrichment is observed at distal regions located >1000 bp from the RE1/NRSE site. We also show that REST/NRSF is not detected at the coding region of the β-actin gene that is not regulated by REST/NRSF, also indicating that REST/NRSF immunoprecipitation specifically enriches for REST/NRSF-associated genes.
Then, conventional ChIP was performed on chromatin lysates from cortical specimens obtained from five individuals with HD (grade III-IV) and five age-matched controls. These data show that REST/NRSF binding at five selected RE1/NRSE loci represented by BDNF, SYN1, CHRM4, CHRNB2, and synaptosomal-associated protein 25 kDa (SNAP25), is increased in four of the HD samples compared with controls (Fig. 4b). Variation in binding was seen at equivalent loci among different individuals and among different loci from the same individual, but the overall trend of increased occupancy at multiple loci was nevertheless clear. Interestingly, tissue from HD1 showed no changes in REST/NRSF occupancy of the selected target genes, despite the fact that this patient had a similar CAG expansion and grade of HD progression to the other patients. Changes in REST/NRSF were specific and were not accompanied by parallel changes in occupancy by other transcription factors as can be seen by the unchanged enrichments seen with anti-Sp1 and anti-Sp4 antibodies (supplemental Fig. 6, available at www.jneurosci.org as supplemental material). Although we cannot know the reason for this variance, it is important to realize that changes in gene occupancy and gene expression do show individual variance in a similar way that we have shown for different animal and cell models of HD.
Subsequently, to provide a more global view of REST/NRSF dysfunction in HD, we used a ChIP-on-chip approach. Genomic microarrays containing the flanking sequence from 537 consensus RE1 sites were hybridized with the ChIP output of anti-REST/NRSF-precipitated chromatin obtained from postmortem cortical tissues from three individuals with HD (grade III-IV) and age-matched controls. These assays have been performed on a different set of postmortem tissues from those used for the conventional ChIP-PCR because of the limiting amount of available tissue. An amount of 500–700 mg wet weight of tissue is required for an extraction that gives a suitable yield of chromatin for ChIP-on-chip assays, so it was not possible to prepare replicates of the same material used in both ChIP-PCR and ChIP-on-chip analyses. To establish REST/NRSF occupancy of target genes across different samples, we used anti-REST/NRSF ChIP DNA isolated from the HD and non-HD cortex tissue and hybridized it to our genomic RE1/NRSE microarrays. Control regions on the array were represented by genomic sequences that lay distal to any RE1/NRSE (as defined by a position-specific scoring matrix cutoff score of >0.91) (Johnson et al., 2006). Examination of these control regions showed an average REST/NRSF ChIP DNA/input DNA ratio of 1.04 ± 0.02 SEM for the HD samples and 1.05 ± 0.03 SEM for the non-HD samples. Because these sequences are not proximal to any REST/NRSF binding sites, their REST/NRSF ChIP DNA/input DNA ratio represents background enrichment. Accordingly, those targets that show REST/NRSF ChIP DNA/input DNA ratios in excess of two SDs plus the control mean were considered significantly enriched (confidence interval 95%; p < 0.05). Relative REST/NRSF enrichments were averaged from three independent HD patient samples and two independent control patient (non-HD) samples and the SEM calculated. This allows identification of those RE1/NRSEs that show greater enrichment in HD versus non-HD samples, supporting the notion of a general dysfunction in REST/NRSF-regulated genes in the HD postmortem cortex. The list of genes in which an increased occupancy by REST/NRSF is seen in HD compared with control tissue is presented in Table 1.
Discussion
Transcriptional dysregulation is a central pathogenic mechanism in HD and several transcription factors have been implicated in this pathology (Sugars and Rubinsztein, 2003). More recently, in vitro studies have demonstrated that wild-type huntingtin associates with REST/NRSF in the cytoplasm, thereby restricting its access to the nucleus, and consequently permits transcription of the BDNF gene. In the presence of mutant huntingtin, this association is disrupted and REST/NRSF translocates to the nucleus where it represses transcription of this neurotrophin (Zuccato et al., 2003).
There are >1300 RE1/NRSE sites in the human and murine genomes, and the majority encodes for proteins that are fundamental for neuronal development and differentiation (Bruce et al., 2004; Johnson et al., 2006). In the present study, using chromatin immunoprecipitation assays, we analyzed REST/NRSF nuclear occupancy on a selection of RE1/NRSE genes, in addition to brain-derived neurotrophic factor that includes synapsin 1, M4 muscarinic cholinergic receptor, dopamine receptor D3, proenkephalin 1, and cholinergic receptor nicotinic β polypeptide2. We show here that in HD there is an increase in REST/NRSF occupancy of the cognate RE1/NRSEs resulting in repression of gene transcription. No increase in cellular levels of REST/NRSF was found in the presence of mutant huntingtin. In particular, we found that REST/NRSF occupancy of RE1/NRSE sites is increased in mutant huntingtin knock-in cells, in the cortex from asymptomatic knock-in mice and from symptomatic R6/2 transgenic mice. Moreover, increased REST/NRSF occupancy occurs early in knock-in mice before the development of a neuropathological phenotype, suggesting that the increased binding of REST/NRSF at the RE1/NRSE could be an early molecular event and that preventing REST/NRSF binding from early stages might be beneficial in HD.
Our results in HD mice also indicate that an increased occupancy of REST/NRSF occurs at the RE1/NRSE of active genes encoding for non-neuronal proteins in non-neural tissues in HD. This can be seen at the hepatic transcription factor HNF-1a locus in liver in which REST/NRSF occupancy is increased at the RE1/NRSE in symptomatic R6/2 transgenic mice compared with controls. This result opens up the possibility to exploit the REST/NRSF genomic occupancy profile in peripheral cells in the search for biomarkers to monitor disease progression in HD patients. Such markers may provide clues to the state of HD and may be of predictive value in clinical trials.
Importantly, we also report for the first time a novel application of ChIP-on-Chip technology to generate a genome-wide perspective on those REST/NRSF targets that are affected in the HD postmortem brain. Genes showing significant changes in REST/NRSF occupancy in HD cortical specimens (parietal cortex, Brodmann's area 7) include those that encode ion channels, adhesion molecules, proteins involved in synaptic activity, signal transduction, metabolism, and neurotrophins such as BDNF. Because increased occupancy by REST/NRSF might be expected to result in lower expression of the corresponding target genes in HD tissue, we examined the results of earlier studies that had performed expression array analyses of HD versus control cortex (Hodges et al., 2006). Such comparisons are confounded by several factors, principally use of different tissues, differential representations on arrays, and correlation of occupancy and expression. Nevertheless, we found that of 958 genes listed as downregulated in the motor cortex (Brodmann's area 4) from 19 HD subjects (Vonsattel grade 2) versus 17 age-matched controls, (Hodges et al., 2006), 27 contained an RE1/NRSE (supplemental Table 2, available at www.jneurosci.org as supplemental material). Furthermore, we found that genes, such as SNAP25 and FGF12, stannin (SNN), PCSK1 (neuroendocrine convertase 1 precursor), KCNAB2 (voltage-gated potassium channel b subunit 2), BDNF, and SYN1, all of which show increased REST/NRST occupancy in the present study, were also downregulated in the HD cortex (Zuccato et al., 2003; Hodges et al., 2006). Thus, despite the potential confounds of such comparisons, these analyses indicate that HD brain dysregulation of REST/NRSF function can lead to parallel changes in both occupancy and expression of REST/NRSE target genes. Moreover, because samples analyzed by Hodges et al. (2006) were obtained from grade 0–II HD subjects, whereas our data were derived from grade III and IV subjects, it is possible that dysregulation of the REST/NRSF regulon is an early event in human HD disease progression. Newly identified targets include SNN, a highly conserved protein implicated in mediating organotin toxicity (Davidson et al., 2004) and B3GAT1 (β-1,3 glucuronyltransferase-1), implicated as a candidate for schizophrenia-like psychosis (Jeffries et al., 2003) and in control of spatial memory and learning (Yamamoto et al., 2002). In addition, the ornithine aminotransferase (OAT) mitochondrial precursor gene was identified in our assay, and the activity of this protein is reduced in the HD brain (Wong et al., 1982). Because this protein is involved in the synthesis of glutamate, a reduction of OAT mRNA level is in agreement with the evidence of a deterioration of the corticostriatal glutamatergic pathway in HD (Zeron et al., 2002, 2004). In addition, an increase in REST/NRSF binding has been found in the promoter region of the OSBP2(oxysterol binding protein 2) gene, a member of a family of sterol binding proteins implicated in vesicle transport, lipid metabolism, and signal transduction (Wang et al., 2005) and which may possibly contribute to the observed defects in lipid metabolism in HD (Sipione et al., 2002; Valenza et al., 2005). Although often overlooked, HD also results in peripheral abnormalities (Luthi-Carter et al., 2002; Borovecki et al., 2005; Papalexi et al., 2005; Strand et al., 2005; Valenza et al., 2005). One such example is the testicular atrophy and reduced fertility observed in transgenic mouse models for HD (Papalexi et al., 2005; Van Raamsdonk et al., 2005) together with defects in spermatogenesis seen in mice depleted of wild-type huntingtin (Dragatsis et al., 2000). Interestingly, we detected increased REST/NRSF occupancy of the RE1/NRSE associated with the gonadotropin-regulated testicular RNA helicase (GRTH/Ddx25) gene, a protein essential for spermatid development and completion of spermatogenesis (Tsai-Morris et al., 2004). Together, these data clearly indicate a widespread neuronal dysfunction in HD caused by specific disruption of the REST/NRSF regulon.
Finally, to test whether wild-type huntingtin is an essential protein to maintain RE1/NRSE gene transcription and if loss of wild-type huntingtin function might contribute to pathogenesis of HD, we investigated the effect of reduced huntingtin expression on REST/NRSF occupancy. Here, we provide direct functional evidence that both mutant huntingtin and reduced levels of wild-type huntingtin both result in the same effects on RE1/NRSE occupancy and REST/NRSF target gene expression, clearly indicating that some aspects of HD may be attributed to a loss of function of wild-type huntingtin, in addition to gain of function of the mutant allele. We also found that changes in REST/NRSF occupancy and BDNF gene transcription were significantly exacerbated in homozygous Hdh−/− compared with heterozygous Hdh+/− cells, suggesting a dose-dependent effect of wild-type huntingtin allele. In addition, because the REST/NRSF regulon is disrupted when wild-type huntingtin is reduced to <50% of its normal level, we also suggest that the polyQ tract acts as a dominant negative mutation in the adult brain, ultimately resulting in the release of REST/NRSF from control by wild-type huntingtin. Indeed, it is well documented that wild-type huntingtin is sequestered into mutant huntingtin aggregates (Dyer and McMurray, 2001; Busch et al., 2003) and possibly cleaved by proteolytic events triggered by the mutant protein (Zhang et al., 2003). The fact that a reduction of wild-type huntingtin exacerbates the pathological behavior of HD mice in the absence of a clear striatal pathology also suggests that a simple increase in the wild-type huntingtin allelic dose may not be sufficient to ameliorate disease symptoms (Van Raamsdonk et al., 2005).
Novel therapeutic strategies should therefore be aimed at restoring the activity of the downstream targets of wild-type huntingtin. We have indeed shown that inhibition of REST/NRSF function by using a dominant negative form of REST/NRSF restores Bdnf mRNA and protein levels in mutant huntingtin cells. In particular, the BDNF increase is mainly resulting from the increased activity of BDNF promoter exon II that is directly regulated by the RE1/NRSE. Moreover, synapsin-1 and M4 muscarinic cholinergic receptor gene transcription, which are driven by the RE1/NRSE silencer, are increased in HD after treatment with a dominant negative form of REST/NRSF, suggesting that transcription of several neuronal genes, repressed in the disease state, can be reactivated by means of this approach. This opens up the possibility that genetic or chemical inactivation of the aberrant silencing activity of REST/NRSF may ultimately restore global neuronal gene transcription in HD.
Footnotes
This work was supported by the Huntington's Disease Society of America Coalition for the Cure (E.C., M.M.), Telethon (Italy), Fondazione Cariplo (Italy), Ministero dell'Istruzione dell'Università e della Ricerca (Italy), and NeuroNE (European Union's sixth Framework Program) (E.C.); Conferenza dei Rettori delle Università Italiane (C.Z.); the Welcome Trust (N.Bu., E.C.); and National Institutes of Health–National Institute of Neurological Disorders and Stroke Grants NS32765 and NS16367 (M.M.). E.C. and M.M. are members of the Huntingtin Function Team, Coalition for the Cure (Huntington's Disease Society of America). We are very grateful to Alberto Clivio (University of Milan) for his generous hospitality and collaboration. We thank Dave Vetrie and Alexander Bruce (Sanger Institute, Hinxton, UK) for assistance with array construction. We also thank Ruth Luthi-Carter (Ecole Polytechnique Fédérale de Lausanne, Lausanne, Switzerland) for kindly providing microarray raw data.
References
- Belyaev ND, Wood IC, Bruce AW, Street M, Trinh JB, Buckley NJ. Distinct RE-1 silencing transcription factor-containing complexes interact with different target genes. J Biol Chem. 2004;279:556–561. doi: 10.1074/jbc.M310353200. [DOI] [PubMed] [Google Scholar]
- Borovecki F, Lovrecic L, Zhou J, Jeong H, Then F, Rosas HD, Hersch SM, Hogarth P, Bouzou B, Jensen RV, Krainc D. Genome-wide expression profiling of human blood reveals biomarkers for Huntington's disease. Proc Natl Acad Sci USA. 2005;102:11023–11028. doi: 10.1073/pnas.0504921102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce AW, Donaldson IJ, Wood IC, Yerbury SA, Sadowski MI, Chapman M, Gottgens B, Buckley NJ. Genome-wide analysis of repressor element 1 silencing transcription factor/neuron-restrictive silencing factor (REST/NRSF) target genes. Proc Natl Acad Sci USA. 2004;101:10458–10463. doi: 10.1073/pnas.0401827101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch A, Engemann S, Lurz R, Okazawa H, Lehrach H, Wanker EE. Mutant huntingtin promotes the fibrillogenesis of wild-type huntingtin: a potential mechanism for loss of huntingtin function in Huntington's disease. J Biol Chem. 2003;278:41452–41461. doi: 10.1074/jbc.M303354200. [DOI] [PubMed] [Google Scholar]
- Cattaneo E, Rigamonti D, Goffredo D, Zuccato C, Squitieri F, Sipione S. Loss of normal huntingtin function: new developments in Huntington's disease research. Trends Neurosci. 2001;24:182–188. doi: 10.1016/s0166-2236(00)01721-5. [DOI] [PubMed] [Google Scholar]
- Cattaneo E, Zuccato C, Tartari M. Normal Huntingtin function: an alternative approach to Huntington's disease. Nat Rev Neurosci. 2005;6:919–930. doi: 10.1038/nrn1806. [DOI] [PubMed] [Google Scholar]
- Chong JA, Tapia-Ramirez J, Kim S, Toledo-Aral JJ, Zheng Y, Boutros MC, Altshuller YM, Frohman MA, Kraner SD, Mandel G. REST: a mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell. 1995;80:949–957. doi: 10.1016/0092-8674(95)90298-8. [DOI] [PubMed] [Google Scholar]
- Davidson CE, Reese BE, Billingsley ML, Yun JK. Stannin, a protein that localizes to the mitochondria and sensitizes NIH-3T3 cells to trimethyltin and dimethyltin toxicity. Mol Pharmacol. 2004;66:855–863. doi: 10.1124/mol.104.001719. [DOI] [PubMed] [Google Scholar]
- Dragatsis I, Levine MS, Zeitlin S. Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat Genet. 2000;26:300–306. doi: 10.1038/81593. [DOI] [PubMed] [Google Scholar]
- Duyao MP, Auerbach AB, Ryan A, Persichetti F, Barnes GT, McNeil SM, Ge P, Vonsattel JP, Gusella JF, Joyner AL, et al. Inactivation of the mouse Huntington's disease gene homolog Hdh. Science. 1995;269:407–410. doi: 10.1126/science.7618107. [DOI] [PubMed] [Google Scholar]
- Dyer RB, McMurray CT. Mutant protein in Huntington disease is resistant to proteolysis in affected brain. Nat Genet. 2001;29:270–278. doi: 10.1038/ng745. [DOI] [PubMed] [Google Scholar]
- Hodges A, Strand AD, Aragaki AK, Kuhn A, Sengstag T, Hughes G, Elliston LA, Hartog C, Goldstein DR, Thu D, Hollingsworth ZR, Collin F, Synek B, Holmans PA, Young AB, Wexler NS, Delorenzi M, Kooperberg C, Augood SJ, Faull RL, et al. Regional and cellular gene expression changes in human Huntington's disease brain. Hum Mol Genet. 2006;15:965–977. doi: 10.1093/hmg/ddl013. [DOI] [PubMed] [Google Scholar]
- Jeffries AR, Mungall AJ, Dawson E, Halls K, Langford CF, Murray RM, Dunham I, Powell JF. beta-1,3-Glucuronyltransferase-1 gene implicated as a candidate for a schizophrenia-like psychosis through molecular analysis of a balanced translocation. Mol Psychiatry. 2003;8:654–663. doi: 10.1038/sj.mp.4001382. [DOI] [PubMed] [Google Scholar]
- Johnson R, Gamblin RJ, Ooi L, Bruce AW, Donaldson IJ, Westhead DR, Wood IC, Jackson RM, Buckley NJ. Identification of the REST regulon reveals extensive transposable element-mediated binding site duplication. Nuc Acids Res. 2006;34:3862–3877. doi: 10.1093/nar/gkl525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthi-Carter R, Hanson SA, Strand AD, Bergstrom DA, Chun W, Peters NL, Woods AM, Chan EY, Kooperberg C, Krainc D, Young AB, Tapscott SJ, Olson JM. Dysregulation of gene expression in the R6/2 model of polyglutamine disease: parallel changes in muscle and brain. Hum Mol Genet. 2002;11:1911–1926. doi: 10.1093/hmg/11.17.1911. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- Palm K, Belluardo N, Metsis M, Timmusk T. Neuronal expression of zinc finger transcription factor REST/NRSF/XBR gene. J Neurosci. 1998;18:1280–1296. doi: 10.1523/JNEUROSCI.18-04-01280.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm K, Metsis M, Timmusk T. Neuron-specific splicing of zinc finger transcription factor REST/NRSF/XBR is frequent in neuroblastomas and conserved in human, mouse and rat. Brain Res Mol Brain Res. 1999;72:30–39. doi: 10.1016/s0169-328x(99)00196-5. [DOI] [PubMed] [Google Scholar]
- Papalexi E, Persson A, Bjorkqvist M, Petersen A, Woodman B, Bates GP, Sundler F, Mulder H, Brundin P, Popovic N. Reduction of GnRH and infertility in the R6/2 mouse model of huntington's disease. Eur J Neurosci. 2005;22:1541–1546. doi: 10.1111/j.1460-9568.2005.04324.x. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e 45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A, Albin RL, Anderson KD, D'Amato CJ, Penney JB, Young AB. Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci USA. 1988;85:5733–5735. doi: 10.1073/pnas.85.15.5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenherr CJ, Anderson DJ. The neuron-restrictive silencer factor (NRSF): a coordinate repressor of multiple neuron-specific genes. Science. 1995;267:1360–1363. doi: 10.1126/science.7871435. [DOI] [PubMed] [Google Scholar]
- Sipione S, Rigamonti D, Valenza M, Zuccato C, Conti L, Pritchard J, Kooperberg C, Olson JM, Cattaneo E. Early transcriptional profiles in huntingtin-inducible striatal cells by microarray analyses. Hum Mol Genet. 2002;11:1953–1965. doi: 10.1093/hmg/11.17.1953. [DOI] [PubMed] [Google Scholar]
- Strand AD, Aragaki AK, Shaw D, Bird T, Holton J, Turner C, Tapscott SJ, Tabrizi SJ, Schapira AH, Kooperberg C, Olson JM. Gene expression in Huntington's disease skeletal muscle: a potential biomarker. Hum Mol Genet. 2005;14:1863–1876. doi: 10.1093/hmg/ddi192. [DOI] [PubMed] [Google Scholar]
- Sugars KL, Rubinsztein DC. Transcriptional abnormalities in Huntington disease. Trends Genet. 2003;19:233–238. doi: 10.1016/S0168-9525(03)00074-X. [DOI] [PubMed] [Google Scholar]
- The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, Cattaneo E, MacDonald ME. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet. 2000;9:2799–2809. doi: 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- Tsai-Morris CH, Sheng Y, Lee E, Lei KJ, Dufau ML. Gonadotropin-regulated testicular RNA helicase (GRTH/Ddx25) is essential for spermatid development and completion of spermatogenesis. Proc Natl Acad Sci USA. 2004;101:6373–6378. doi: 10.1073/pnas.0401855101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenza M, Rigamonti D, Goffredo D, Zuccato C, Fenu S, Jamot L, Strand A, Tarditi A, Woodman B, Racchi M, Mariotti C, Di Donato S, Corsini A, Bates G, Pruss R, Olson JM, Sipione S, Tartari M, Cattaneo E. Dysfunction of the cholesterol biosynthetic pathway in Huntington's disease. J Neurosci. 2005;25:9932–9939. doi: 10.1523/JNEUROSCI.3355-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Pearson J, Rogers DA, Bissada N, Vogl AW, Hayden MR, Leavitt BR. Loss of wild-type huntingtin influences motor dysfunction and survival in the YAC128 mouse model of Huntington disease. Hum Mol Genet. 2005;14:1379–1392. doi: 10.1093/hmg/ddi147. [DOI] [PubMed] [Google Scholar]
- Wang PY, Weng J, Anderson RG. OSBP is a cholesterol-regulated scaffolding protein in control of ERK 1/2 activation. Science. 2005;307:1472–1476. doi: 10.1126/science.1107710. [DOI] [PubMed] [Google Scholar]
- Wheeler VC, Auerbach W, White JK, Srinidhi J, Auerbach A, Ryan A, Duyao MP, Vrbanac V, Weaver M, Gusella JF, Joyner AL, MacDonald ME. Length-dependent gametic CAG repeat instability in the Huntington's disease knock-in mouse. Hum Mol Genet. 1999;8:115–122. doi: 10.1093/hmg/8.1.115. [DOI] [PubMed] [Google Scholar]
- Wong PT, McGeer PL, Rossor M, McGeer EG. Ornithine aminotransferase in Huntington's disease. Brain Res. 1982;231:466–471. doi: 10.1016/0006-8993(82)90385-7. [DOI] [PubMed] [Google Scholar]
- Wood IC, Belyaev ND, Bruce AW, Jones C, Mistry M, Roopra A, Buckley NJ. Interaction of the repressor element 1-silencing transcription factor (REST) with target genes. J Mol Biol. 2003;12:863–874. doi: 10.1016/j.jmb.2003.10.017. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Oka S, Inoue M, Shimuta M, Manabe T, Takahashi H, Miyamoto M, Asano M, Sakagami J, Sudo K, Iwakura Y, Ono K, Kawasaki T. Mice deficient in nervous system-specific carbohydrate epitope HNK-1 exhibit impaired synaptic plasticity and spatial learning. J Biol Chem. 2002;277:27227–27231. doi: 10.1074/jbc.C200296200. [DOI] [PubMed] [Google Scholar]
- Zeron MM, Hansson O, Chen N, Wellington CL, Leavitt BR, Brundin P, Hayden MR, Raymond LA. Increased sensitivity to N-methyl-D-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington's disease. Neuron. 2002;33:849–860. doi: 10.1016/s0896-6273(02)00615-3. [DOI] [PubMed] [Google Scholar]
- Zeron MM, Fernandes HB, Krebs C, Shehadeh J, Wellington CL, Leavitt BR, Baimbridge KG, Hayden MR, Raymond LA. Potentiation of NMDA receptor-mediated excitotoxicity linked with intrinsic apoptotic pathway in YAC transgenic mouse model of Huntington's disease. Mol Cell Neurosci. 2004;25:469–479. doi: 10.1016/j.mcn.2003.11.014. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Li M, Drozda M, Chen M, Ren S, Mejia Sanchez RO, Leavitt BR, Cattaneo E, Ferrante RJ, Hayden MR, Friedlander RM. Depletion of wild-type huntingtin in mouse models of neurologic disease. J Neurochem. 2003;87:101–106. doi: 10.1046/j.1471-4159.2003.01980.x. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Cattaneo E. Role of brain-derived neurotrophic factor in Huntington's disease. Prog Neurobiol. 2007;81:294–330. doi: 10.1016/j.pneurobio.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L, Cataudella T, Leavitt BR, Hayden MR, Timmusk T, Rigamonti D, Cattaneo E. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet. 2003;35:76–83. doi: 10.1038/ng1219. [DOI] [PubMed] [Google Scholar]