

Abstract
Extensive epidemiological data in humans and studies in animal models of Parkinson's disease (PD) suggest that sporadic forms of the disorder are not strictly genetic in nature but most likely because of combined environmental exposures over the period of the life-span coupled with increased genetic susceptibilities. Environmental paraquat and neonatal iron exposure have both been separately suggested as potential risk factors for sporadic forms of the disease. In this study, we demonstrate that combined environmental exposure to these two agents results in accelerated age-related degeneration of nigrostriatal dopaminergic neurons. Furthermore, pretreatment with the synthetic superoxide dismutase/catalase mimetic, EUK-189, significantly attenuated neuronal death mediated by combined paraquat and iron treatment. These findings support the notion that environmental PD risk factors may act synergistically to produce neurodegeneration associated with the disorder and that iron and paraquat may act via common oxidative stress-mediated mechanisms.
Keywords: dopaminergic neurons, iron, paraquat, Parkinsonism, risk factors, sporadic
Introduction
Parkinson's disease (PD) is a progressive neurodegenerative disease that is pathologically characterized by preferential degeneration of dopaminergic neurons of the substantia nigra pars compacta (SNpc) (Forno, 1996; Lang and Lozano, 1998). Earlier epidemiological evidence from an extensive twin study and more recently published genome-wide single nucleotide polymorphism analyses of a population cohort of Parkinsonian patients versus neurologically normal controls strongly suggests that the common idiopathic form of the disease is not because of strict genetic heritability (Tanner et al., 1999; Fung et al., 2006). This, along with geographic variations in PD incidence, suggests that sporadic PD is most likely because of environmental factors, perhaps in combination with genetic susceptibilities.
Elevated substantia nigra (SN) iron levels have also been reported to be associated with sporadic PD (Sofic et al., 1991; Riederer et al., 1992; Jellinger et al., 1993; Griffiths et al., 1999). The high concentration of iron within the SN may act to catalyze the conversion of H2O2 produced during breakdown of dopamine to highly reactive hydroxyl radicals resulting in increased oxidative damage in the region (Jellinger et al., 1993). Markers of lipid peroxidation (Dexter et al., 1994), protein oxidation (Yoritaka et al., 1996), and DNA oxidation (Alam et al., 1997; Shimura-Miura et al., 1999) have all been reported to be elevated within the Parkinsonian SN. We reported recently that administration of iron during the neonatal period results in SN iron elevation and oxidative stress that correlates with neurodegeneration of dopaminergic SN neurons at late stages in the murine life-span (Kaur et al., 2007).
Exposure to agricultural chemicals has widely been postulated as a potential environmental risk factor for the disease, and this is supported by extensive epidemiological evidence demonstrating increased PD incidence in persons living in a rural environment, drinking well water, and experiencing occupational exposure to such agents (Hertzman et al., 1990; Jimenez-Jimenez et al., 1992; Semchuk et al., 1992; Hubble et al., 1993; Liou et al., 1997; Gorell et al., 1998; Stephenson, 2000). The widely used herbicide 1,1′-dimethyl-4,4′-bipyridium (paraquat; PQ) is considered a prime risk factor in this category based on both epidemiological evidence of increased incidence of PD after exposure and its chemical similarity to the parkinsonism-inducing agent 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (Hertzman et al., 1990; Liou et al., 1997).
Because PD appears to be a multifactorial disorder, the use of animal models to investigate additive or synergistic effects of environmental risk factors are of great importance, especially in the context of aging, which is the single major risk factor for the disease. In this study, we assessed the combined effects of neonatal iron feeding and environmental paraquat exposure on age-related nigrostriatal degeneration. We report here that increased oral intake of iron in the neonatal period leads to a progressive age-related enhancement of dopaminergic neurodegeneration associated with paraquat neurotoxicity. Furthermore, neurodegeneration associated with this combined environmental exposure could be attenuated by systemic treatment with the bioavailable antioxidant compound EUK-189 that also represses c-Jun N-terminal kinase (JNK) pathway activation.
Materials and Methods
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), 1,1′-dimethyl-4,4′-bipyridium dichloride (paraquat), and carbonyl iron were purchased from Sigma (St. Louis, MO). Rabbit anti-phospho-SAPK/JNK (Thr183/Tyr185), anti-phospho-c-Jun (Ser63), and anti-cleaved caspase-3 antibodies were obtained from Cell Signaling Technology (Beverly, MA). Rabbit anti-3-nitrotyrosine (3-NT) polyclonal antibody was from Invitrogen (San Diego, CA). Rabbit and sheep anti-tyrosine hydroxylase (TH) polyclonal antibodies were from Millipore (Bedford, MA). Media and sera were purchased from Invitrogen. Osmotic minipumps (2004; Alzet, Cupertino, CA) were obtained from Alza Scientific Products (Mountain View, CA). The salen manganese complex EUK-189 was a gift from Proteome Systems (Woburn, MA); CEP-11004 was a gift from Cephalon (West Chester, PA).
Cell culture.
The rat dopaminergic cell line 1RB3AN27 (N27) was grown in RPMI 1640 medium supplemented with 10% fetal calf serum (Invitrogen), 100 U/ml penicillin, and 100 μg/ml streptomycin. Cell viability was determined by MTT incorporation (Peng et al., 2002). DNA fragmentation was examined by terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeled (TUNEL) analysis with an in situ cell death detection kit (Roche Molecular Biochemicals, Welwyn Garden City, UK) according to the manufacturer's instructions (Peng et al., 2002). For immunocytochemical analysis of cleaved caspase-3, cells were fixed with 4% paraformaldehyde in PBS and permeabilized with 0.3% Triton X-100 in PBS as described previously (Peng et al., 2002). Briefly, cells were then incubated with rabbit polyclonal anti-cleaved caspase-3 (1:100) in PBS containing 10% normal goat serum and 0.3% Triton X-100 overnight at 4°C. Cells were washed with PBS and incubated with fluorochrome-conjugated secondary antibody (1:200; Jackson ImmunoResearch, West Grove, PA) for 2 h at room temperature. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (Vector Laboratories, Burlingame, CA). Control experiments were performed in which the primary antibody was omitted. No staining was observed under these conditions. Stained cells were counted in 10 randomly chosen microscopic fields (≥500 cells). Data were expressed as the mean ± SEM of the percentage of total cells that showed positive staining. To evaluate the effect of the salen manganese complex, EUK-189, or specific JNK inhibitors, anthra[1,9-cd]pyrazol-6(2H)-one (SP600125) and CEP-11004, these compounds were added 1 h before paraquat combined with iron on cell death.
Primary mesencephalic cultures.
Primary mesencephalic cell cultures were prepared from gestation day 15 mouse embryos as described previously (Peng et al., 2004). Briefly, dissociated cells were seeded at 7 × 105 cells per well onto poly-d-lysine-coated 24-well culture plates. Cultures were maintained at 37°C in a humidified atmosphere containing 95% air and 5% carbon dioxide in Neurobasal medium (Invitrogen) containing 2% B27 supplement, 2 mm glutamate, 100 U/ml penicillin, and 100 μg/ml streptomycin. After 4 d, one-half of the medium was replaced with fresh medium. Cells were grown an additional 2 d and then treated with 30 μm paraquat with or without 8 μm FeCl2 for 18 or 24 h. The number of TH-positive neurons in mesencephalic cultures was determined as described previously (Peng et al., 2004). The specificity of neurotoxicity was analyzed by double-label immunostaining with antibodies against TH and cleaved caspase-3 as described previously (Peng et al., 2004).
Drug administration.
C57BL/6 mice were obtained from the Jackson Laboratory (Bar Harbor, ME). Carbonyl iron was administered to male mice from postnatal days 10–17 via oral gavage at a dosage of 120 mg/kg. Control animals were fed equal volumes of sterile saline. Mice were aged to 2, 6, 12, and 24 months of age and were intraperitoneally injected with saline, 10 mg/kg paraquat (2, 6, and 12 months of age), or 8 mg/kg paraquat (24 months of age) twice per week for 3 weeks. Animals were killed at day 4 or 7 after final administration. Administration of the salen manganese complex, EUK-189, was performed as described previously (Zhang et al., 2004; Peng et al., 2005). Briefly, mice were anesthetized with 4% isoflurane in 70% N2O/30% O2 and subcutaneously implanted with an osmotic minipump containing either 5% mannitol (as vehicle control) or 15 mm EUK-189 (dissolved in 5% mannitol). Pumps delivered EUK-189 at a rate of 0.25 μl/h for a 28 d period. Experimental protocols were in accordance with the National Institutes of Health guidelines for use of live animals and were approved by the Animal Care and Use Committee at the Buck Institute of Age Research.
TH staining and stereology.
Mice were fixed by perfusion as described previously (Peng et al., 2004). Cryostat-cut sections were taken through the entire midbrain. TH-positive neurons were immunolabeled by incubating the tissue sections successively with a rabbit polyclonal anti-TH antibody (1:500) and biotinylated horse anti-rabbit IgG (1:200; Vector Laboratories) and after the staining procedure outlined by the manufacturers of the Vectastain ABC kit (Vector Laboratories) in combination with DAB reagents. The total number of TH-positive neurons in the substantia nigra pars compacta was counted from four to five mice per group by using the optical fractionator method, an unbiased stereological technique of cell counting (West, 1999) as described previously (Peng et al., 2004).
Quantitative analysis of double-labeled neurons in the SNpc.
The accuracy of counting double-labeled neurons was determined by analysis of systematically sampled candidate neurons from saline- or paraquat-treated brains. Sections were fixed with 4% paraformaldehyde in PBS, and incubated in blocking buffer (2% horse serum/0.2% Triton X-100/0.1% BSA in PBS) for 1 h at room temperature as described previously (Peng et al., 2004). Primary antibodies used in this study were as follows: sheep polyclonal anti-TH (1:500), rabbit polyclonal anti-phospho-JNK (1:100), rabbit polyclonal anti-phospho-c-Jun (1:100), and rabbit polyclonal anti-3-nitrotyrosine (1:1000). Primary antibodies were added in blocking buffer and incubated with sections at 4°C overnight. The secondary antibodies were Alexa Fluro 488- or 594-conjugated donkey anti-sheep or anti-rabbit IgG (1:200; Invitrogen). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) using proLong Gold anti-fade reagent (Invitrogen), and fluorescence signals were detected with an LSM 510 NLO Confocal Scanning System mounted on an Axiovert 200 inverted microscope (Carl Zeiss, Thornwood, NY) equipped with a two-photon Chameleon laser (Coherent, Santa Clara, CA). Three-color images were scanned using Argon, 543 HeNe, and Chameleon (750–780 nm for DAPI) lasers. IMARIS (Bitplane, Zurich, Switzerland) imaging software was used for three-dimensional image reconstruction. Images were acquired using LSM 510 Imaging Software (Carl Zeiss). The specificity of each label was first verified using single-channel scans that were then merged into multiple-channel views. Neurons were considered double-labeled if colabeling with relevant morphology was seen throughout the extent of the nucleus for nuclear markers or if a cytoplasmic marker surrounds a nuclear marker when viewed in x–y cross-section as well as in x–z and y–z cross-sections produced by orthogonal reconstructions from z-stacks taken at 400× magnification. TH single-labeled neurons and neurons double labeled for TH and phospho-JNK or phospho-c-Jun were recorded in three 50 μm sections per animal. Controls included omitting or preabsorbing primary antibodies or omitting secondary antibodies.
Statistical analysis.
All data are expressed as mean ± SEM for the number of independent experiments performed. Differences among the means for all experiments described were analyzed using one- or two-way ANOVA. Newman–Keuls post hoc analysis was used when differences were observed by ANOVA testing (p < 0.05).
Results
Iron exacerbates paraquat-induced neurotoxicity in vitro
The rat dopaminergic cell line 1RB3AN27 (N27) produces dopamine and expresses the dopamine-synthesizing enzyme TH and the dopamine transporter (Prasad et al., 1994). This cell line has been used previously by our laboratory to study the role of paraquat-mediated oxidative stress on the JNK signaling pathway as a model for PD (Peng et al., 2004, 2005). To study the potential neurotoxic synergism between paraquat and redox-active iron, we treated N27 cells with combined paraquat and FeCl2 and measured cell viability via measurement of MTT reduction capacity. As shown previously (Peng et al., 2004), paraquat alone reduced cell viability in a dose-dependent manner (Fig. 1A). Although exposure to 80 μm FeCl2 was nontoxic on its own, FeCl2 at this concentration significantly increased vulnerability to neuronal toxicity induced by paraquat (Fig. 1A). This coincided with increased apoptosis as assessed by cleaved caspase-3 immunoreactivity and DNA fragmentation (Fig. 1B–F) as observed previously in cells treated with paraquat alone (Peng et al., 2004, 2005).
Figure 1.

Iron enhances paraquat-induced dopaminergic N27 cell apoptosis in vitro. A, N27 cells were treated with increasing concentrations of paraquat and 80 μm FeCl2 for 24 h. Cell viability was measured via the MTT assay and expressed as a percentage of viability in control. B–F, Cells were treated with EUK-189 1 h before the addition of 80 μm FeCl2 and 350 μm paraquat and cell viability at 24 h (B), cleaved caspase-3-positive cells at 18 h (C, E), and TUNEL-positive cells at 24 h (D, F) were measured. Error bars indicate mean ± SEM. n = 5. #p < 0.01, significantly from control; *p < 0.01, significantly from paraquat; **p < 0.01, significantly from paraquat alone; *** p < 0.01, significantly from paraquat plus FeCl2 alone. White bar, Paraquat; black bar, paraquat plus FeCl2.
The salen manganese complexes (EUKs), synthetic superoxide dismutase, and catalase mimetics have been shown to be neuroprotective in several animal models including systemic PQ exposure (Malfroy et al., 1997; Baker et al., 1998; Rong et al., 1999; Jung et al., 2001; Peng et al., 2005). To test whether EUK-189 was protective against exacerbated neurotoxicity induced by paraquat combined with FeCl2, EUK-189 (20 or 40 μm) was added 1 h before cotreatment with paraquat and FeCl2. As shown previously (Peng et al., 2005), pretreatment with EUK-189 protected cells from paraquat-mediated neuronal toxicity (Fig. 1B–F). It was also efficacious in preventing additional apoptosis generated by iron treatment of the paraquat-treated cells in a dose-dependent manner (Fig. 1B–F). EUK-189 alone at the concentrations used was neither stimulatory nor inhibitory in terms of neuronal survival (data no shown).
JNK signaling pathway involved in cell death induced by paraquat combination with iron
We demonstrated previously that paraquat-generated superoxide leads to activation of the JNK signaling pathway, resulting in subsequent dopaminergic neuronal apoptosis both in vitro and in vivo (Peng et al., 2004, 2005). To determine whether JNK signal transduction pathways are involved in the exacerbation of dopaminergic neurodegeneration with paraquat administration in combination with FeCl2, we treated N27 cells with the JNK inhibitors SP600125 (8 μm) or CEP-11004 (200 nm) beginning 1 h before paraquat and/or FeCl2 cotreatment. As previously shown (Peng et al., 2004), pretreatment with JNK inhibitors protected N27 cells from neuronal toxicity induced by paraquat alone (Fig. 2A–C). Furthermore, pretreatment with SP600125 and CEP-11004 protected cells from additional apoptosis induced by paraquat in combination with FeCl2 (Fig. 2A). As demonstrated in Figure 2B,C, SP600125 and CEP-11004 both separately reduced the numbers of cleaved caspase-3- or TUNEL-positive cells to the same level in the presence of paraquat alone or paraquat and iron combined, suggesting that it is efficacious in preventing additional apoptosis elicited in the presence of iron.
Figure 2.

Inhibition of JNK kinase attenuates additional dopaminergic apoptosis induced by combined paraquat and iron in both N27 and primary mesencephalic cultures in vitro. N27 cells were treated with the indicated inhibitors 1 h before the addition of 350 μm paraquat or 350 μm paraquat plus 80 μm FeCl2. A–C, Cleaved caspase-3-positive cells at 18 h (A), TUNEL-positive cells at 24 h (B), and cell viability at 24 h (C) were measured. Mesencephalic cultures were treated with the indicated inhibitors 1 h before the addition of paraquat or paraquat plus FeCl2. D, E, Cleaved caspase-3-positive cells at 18 h (D) and cell viability at 24 h (E) were measured. Error bars indicate mean ± SEM. n = 4–5. #p < 0.05, significantly from paraquat alone; *p < 0.01, significantly from paraquat alone; **p < 0.001, significantly from paraquat plus FeCl2 alone. Gray bars, Without inhibitors; white bars, SP600125; black bars, CEP-11004.
To also investigate the relationship between paraquat in combination with iron on the induction of the JNK signaling pathway on midbrain dopaminergic neurons, we examined the effects of the JNK inhibitor administration on apoptosis associated with either paraquat alone or paraquat plus FeCl2-treated in primary mesencephalic cultures via dual immunofluorescence with antibodies specific for TH and cleaved caspase-3 coupled with 4′, 6-diamidino-2-phenylindole staining. Cultures were pretreated with SP600125 (5 μm) or CEP-11004 (100 nm) 1 h before treatment with paraquat alone or paraquat plus FeCl2. As shown in Figure 2D,E, both SP600125 or CEP-11004 were able to significantly reduce colocalization of activated caspase-3 within TH-positive neurons and in addition attenuated TH-positive neuron loss to the same levels in cells treated with combined paraquat and iron versus those treated with paraquat alone. These data suggest that paraquat and iron treatment synergistically lead to the activation of the JNK signaling pathway in these neurons and subsequent apoptotic cell death.
Neonatal iron leads to a progressive age-related exacerbation of dopaminergic neurodegeneration elicited by systemic paraquat administration
To assess whether neonatal iron administration acts to exacerbate paraquat-induced nigral dopaminergic neuronal death in vivo, pups were fed iron daily from postnatal days 10–17 as described previously (Kaur et al., 2007). They were housed with their mothers until weaning at week three and aged to 2, 6, 12, or 24 months of age. At the end of each time period, a subset of iron versus vehicle-fed mice were intraperitoneally injected with saline or paraquat as described previously (Thiruchelvam et al., 2003) and killed at day 7. No signs of acute systemic toxicity were observed in the iron-fed pups at the dosages used in this study (Kaur et al., 2007). No changes in body weights were observed in the mice at the dosage of paraquat used in the experiments (Thiruchelvam et al., 2003). Although dopaminergic SN neuronal numbers remained unchanged in saline-treated animals, paraquat exposure reduced dopaminergic neurons in all four age groups to the same extent, suggesting that paraquat toxicity is age independent (Fig. 3). This is, to our knowledge, the first study performed assessing the impact of paraquat administration with age on this parameter and demonstrates that young animals are as susceptible as older animals to exposure to this particular neurotoxin alone.
Figure 3.
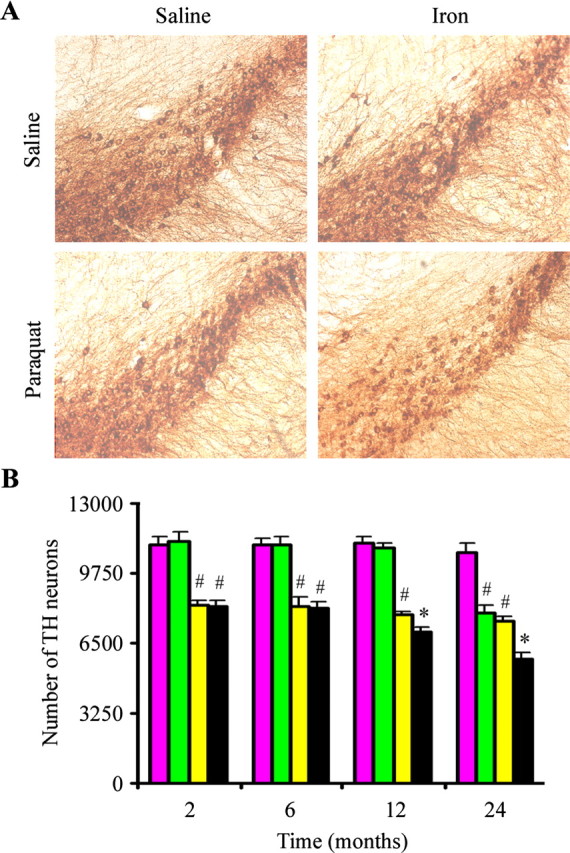
Effects of combined neonatal iron and systemic paraquat administration on dopaminergic neurodegeneration in vivo with increasing age. A, Photomicrographs of SNpc TH-immunostained sections from 24-month-old mice. Original magnification, 10×. B, Quantitative stereological analysis of the number of TH-stained profiles from saline plus saline (purple bars), neonatal iron fed plus saline (green bars), saline plus paraquat (yellow bars), and neonatal iron plus paraquat (black bars). Error bars indicate mean ± SEM. n = 4–5. #p < 0.01, significantly from saline plus saline group;*p < 0.05, significantly from either neonatal iron fed plus saline group or saline plus paraquat group.
We demonstrated previously that neonatal iron feeding results not only in higher basal iron content in the adult SNpc but also progressive age-related dopaminergic neurodegeneration in this brain region culminating in actual cell loss by 24 months of age (Kaur et al., 2007). In this current study, we assessed the combined effects of neonatal iron feeding followed by systemic administration of paraquat at various ages on SN dopaminergic cell loss. By 12 months of age, paraquat-induced loss in dopaminergic neuron numbers was exacerbated in those animals that had previously received elevated oral iron during the neonatal period, and this effect was exacerbated at 24 months of age (Fig. 3). This demonstrates that older iron-fed animals are more susceptible to exposure to the herbicide paraquat than younger iron-fed animals and that paraquat administration accelerates SN dopaminergic cell loss in these animals as a consequence of this early exposure to iron.
EUK-189 reduces neonatal iron and paraquat-induced dopaminergic neuronal death
To investigate whether EUK-189 inhibited the increased dopaminergic neuronal death after paraquat administration in the 12-month-old iron-fed mice previously fed iron versus those treated with paraquat alone, we implanted mice with osmotic minipumps containing either 5% mannitol (as vehicle control) or 15 mm EUK-189 3 d before paraquat treatment (Zhang et al., 2004; Peng et al., 2005). At this age, neonatal iron exposure alone does not result in dopaminergic SN cell loss (Kaur et al., 2007). In comparison with unlesioned controls, however, subcutaneous administration of EUK-189 attenuated the additional loss of nigral dopamine neurons at day 7 elicited by neonatal iron feeding versus paraquat treatment alone (Fig. 4).
Figure 4.
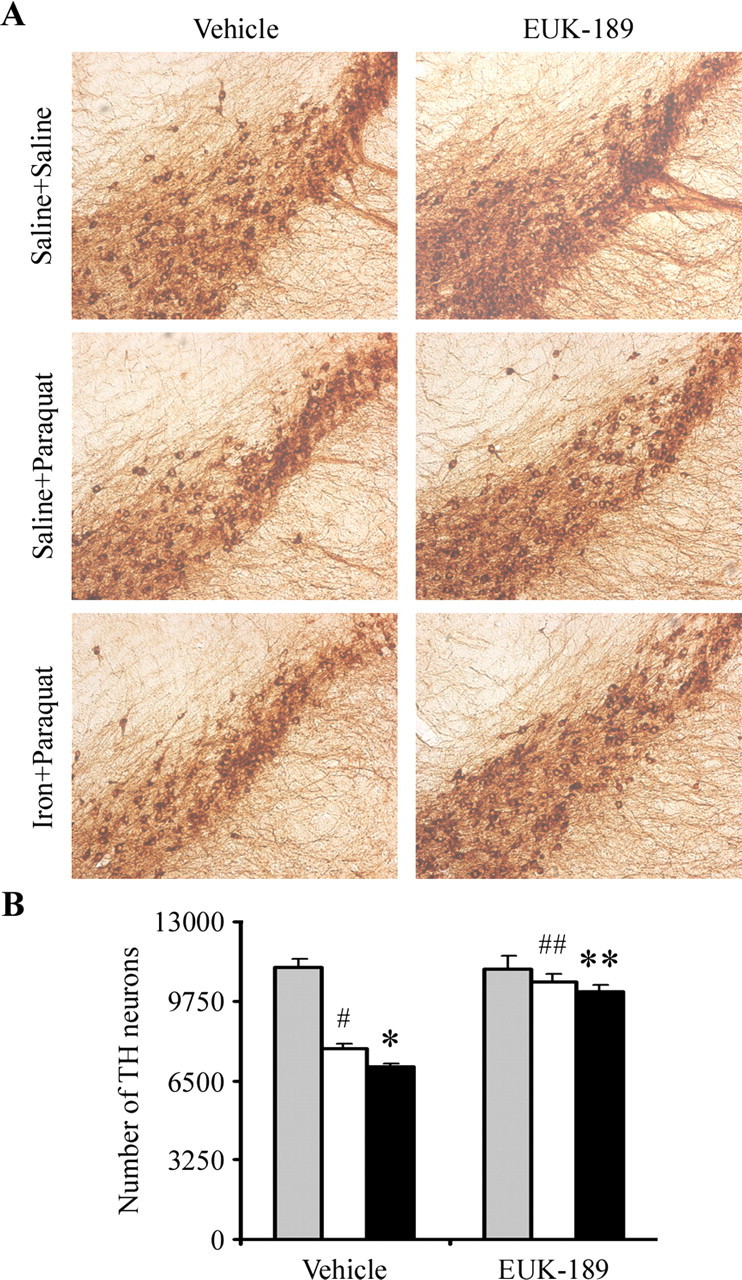
Administration of EUK-189 attenuates combined neonatal iron and paraquat-induced dopaminergic neuronal death at 12 months of age. A, Photomicrographs of SNpc TH-immunostained sections. Original magnification, 10×. B, Quantitative stereological analysis of the number of TH-stained profiles from saline plus saline (gray bars), saline plus paraquat (white bars), and neonatal iron fed plus paraquat (black bars). Error bars indicate mean ± SEM. n = 4. #p < 0.01, significantly from saline plus saline plus vehicle group; *p < 0.05, significantly from saline plus paraquat plus vehicle group; ##p < 0.01, significantly from saline plus paraquat plus vehicle group; **p < 0.01, significantly from neonatal iron fed plus paraquat plus vehicle group.
EUK-189 prevents increases in oxidative stress in the SNpc elicited by combined iron and paraquat exposures
Systemic administration of paraquat in young (2-month-old) mice was reported previously to increase levels of 3-NT oxidation in dopaminergic neurons of the SNpc (McCormack et al., 2005). Therefore, to determine whether additional loss of SNpc neurons elicited by combined iron and paraquat exposures is because of increased levels of oxidative stress, we performed immunofluorescent double-labeling experiments using TH as a dopaminergic marker and 3-NT as a biomarker for protein oxidation. Neonatal iron alone does not result in increased levels of oxidative damage within dopaminergic SNpc neurons at 2 months of age, but by 12 months of age, there is a significant increase in this parameter (Fig. 5) (Kaur et al., 2007), although we demonstrated previously that this is not associated with an increase in dopaminergic cell loss in this brain region (Kaur et al., 2007). As demonstrated previously, paraquat administration at 2 months results in increases in 3-NT levels (McCormack et al., 2005) that are further increased by 12 months of age (although not in association with an increase in dopaminergic SNpc cell death) (Fig. 3) and are exacerbated in the presence of neonatal iron exposure (Fig. 5). Treatment with EUK-189 was found to attenuate the age-related increases in protein oxidation induced not only by either agent alone but also as a consequence of combined iron and paraquat exposure where an increase in dopaminergic SNpc cell loss occurs (Fig. 5).
Figure 5.
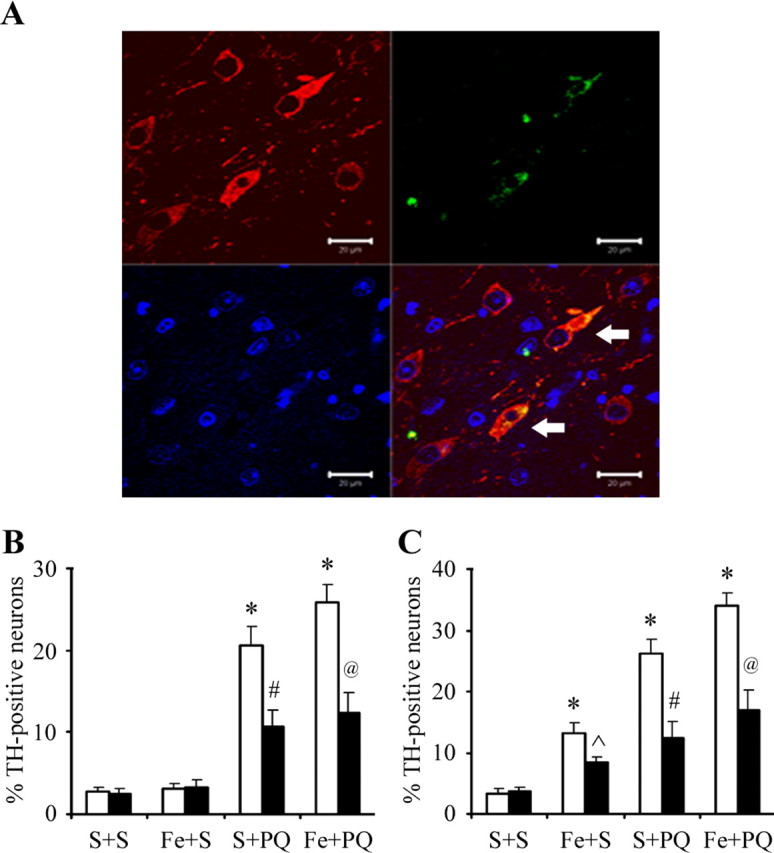
3-Nitrotyrosine immunopositive cells count in the SNpc. A, An example of localization (arrows) of 3-nitrotyrosine-immunopositive staining (green) within dopaminergic neurons (red). 4′,6-Diamidino-2-phenylindole (blue) was used to counterstain nuclei. Scale bars, 20 μm. B, C, Quantitative analysis of double labeling for TH with 3-nitrotyrosine in the SNpc of 2-month-old (B) and 12-month-old (C) mice. White bars, Vehicle; black bars, EUK-189 treated. Error bars indicate mean ± SEM. n = 3. *p < 0.001, significantly from saline plus saline plus vehicle group; p̂ < 0.05, significantly from neonatal iron fed plus saline plus vehicle group; #p < 0.001, significantly from saline plus paraquat plus vehicle group; @p < 0.001, significantly from neonatal iron fed plus paraquat plus vehicle group.
Increased activation of the JNK signaling pathway occurs in the presence of neonatal iron exposure versus paraquat administration alone, which is attenuated by EUK administration
To determine whether oxidative activation of the JNK signaling pathway by paraquat (Peng et al., 2004, 2005) is also exacerbated in SNpc dopaminergic neurons by neonatal iron exposure, we performed immunofluorescent double-labeling experiments for TH with phospho-JNK or phospho-c-Jun. Double-labeling revealed qualitatively that phospho-JNK and phospho-c-Jun immunoreactivity was weak and diffusely cytoplasmic in nigral dopaminergic neurons in midbrain tissues isolated from untreated mouse brains (Fig. 6A,B). In contrast, in tissues from mice treated with paraquat, cytoplasmic immunolocalization within dopamine neurons exhibited a bright speckled appearance that was more intense in tissues isolated from iron-fed mice versus saline control animals (Fig. 6A,B). Furthermore, treatment with EUK-189 appeared to reduce intensity (Fig. 6A,B) and colocalization of phospho-JNK or phospho-c-Jun within TH-positive neurons induced by not only paraquat alone but also in combination with iron (Fig. 6C–F). Colocalization of phospho-JNK and phospho-Jun has found to be elevated in the TH-positive SN neurons of paraquat-treated animals previously fed iron versus those controls previously fed only saline (Fig. 6C–F). Moreover, EUK-189 administration reduced the colocalization of phospho-JNK and phospho-c-Jun within TH-positive neurons in the SNpc to a similar extent in animals treated with paraquat alone or in combination with iron (Fig. 6C–F). Together, our data demonstrate that EUK-189 attenuates both the exacerbation in paraquat-elicited JNK activation induced by neonatal iron feeding as well as age-related increases in nigrostriatal dopaminergic cell death.
Figure 6.

Administration of EUK-189 inhibits increased activation of the JNK signaling pathway elicited by combined paraquat and iron in SNpc versus paraquat alone. A, B, Phospho-JNK (A) and phospho-c-Jun (green; B) localized (yellow) within TH-positive SNpc neurons (red). 4′,6-Diamidino-2-phenylindole (blue) was used to counterstain nuclei. Original magnification, 10×. C, D, Representative higher-magnification images demonstrating localization of phospho-JNK (C) and phospho-c-Jun (green; D) within dopaminergic neurons (red; arrow). 4′,6-Diamidino-2-phenylindole (blue) was used to counterstain nuclei. Original magnification, 100×. E, F, Quantitative analysis of double labeling of TH with phospho-JNK (E) and phospho-c-Jun (F). Purple bars, Saline plus saline; green bars, neonatal iron fed plus saline; yellow bars, saline plus paraquat; black bars, neonatal iron fed plus paraquat. Error bars indicate mean ± SEM. n = 3. #p < 0.01, significantly from saline plus saline plus vehicle group; *p < 0.05, significantly from saline plus paraquat plus vehicle group; ##p < 0.001, significantly from saline plus paraquat plus vehicle group; **p < 0.001, significantly from neonatal iron fed plus paraquat plus vehicle group.
Discussion
Epidemiological evidence from an extensive twin study indicates that idiopathic PD in individuals with an age of onset after the age of 50 is not explainable by strict genetic heritability (Tanner et al., 1999). This, along with variations in incidence of PD by geographic region, suggests that sporadic late-onset PD may be because of environmental factors, perhaps in combination with genetic susceptibility (Lanska, 1997; Liou et al., 1997). As PD is likely to be a multifactorial disorder, the use of in vivo models to explore the additive or synergistic effects of combined risk factors, especially in the context of aging, are likely to be of great importance in understanding sporadic disease etiology. In this study, we explored the impact of increased oral iron administration during the neonatal period on nigrostriatal neurodegeneration associated with the herbicide paraquat. Both agents have separately been suggested to be potential risk factors for the disease. Here, we demonstrate that older animals are more susceptible to the combined effects of these agents.
Paraquat administration at the same or similar dosage regimes used in this current study have been shown previously not only to cross the blood–brain barrier through the neutral amino acid transporter (McCormack and Di Monte, 2003) but also to selectively damage the nigrostriatal dopaminergic system in mice (McCormack et al., 2002; Peng et al., 2004, 2005). Epidemiological evidence in humans, although not definitive in terms of defining the exact relationship between age of exposure, effects of multiple exposures, or exposure concentrations to rates of PD occurrence have nonetheless strongly linked paraquat exposure to increased incidence of PD after prolonged contact (Brown et al., 2006; Dinis-Oliveira et al., 2006).
The mechanism of paraquat neurotoxicity appears to be mediated via oxidative stress. Superoxide anion radicals are generated by paraquat through both redox cycling via reaction with molecular oxygen and electron transfer reactions with NADH-dependent oxireductases (Bus and Gibson, 1984; Clejan and Cederbaum, 1989; Burkitt et al., 1993). We reported previously evidence that suggests paraquat mediates dopaminergic cell death via oxidative stress-mediated activation of the JNK signaling pathway (Peng et al., 2004, 2005). Recent data from Drosophila demonstrates that paraquat administration results in selective loss of a subset of dopaminergic neurons and affects on motor behavior (Chaudhuri et al., 2007). Furthermore, flies mutant in dopamine metabolizing genes were found to have altered paraquat sensitivity, suggesting an interaction between paraquat and genetic factors in this invertebrate Parkinsonian model. Together, these data suggest that additional epidemiological analyses in which the interrelationship between environmental and genetic parameters in human PD incidence are warranted to be more thoroughly explored.
Iron levels in the SN have been reported to be elevated in PD patients (Sofic et al., 1991; Riederer et al., 1992; Jellinger et al., 1993; Griffiths et al., 1999). Redox-available iron has been detected in Lewy bodies within the SNpc of postmortem parkinsonian brains (Castellani et al., 2000), and the oxidation state of iron has been found to change from ferrous to ferric ion in SNpc dopaminergic neurons during PD progression (Yoshida et al., 2001). This alteration in redox state has been postulated to be because of the capacity of iron to catalyze oxidative reactions. In dopaminergic neurons, accessible ferrous iron can react with H2O2 produced during oxidative deamination of dopamine to produce highly reactive hydroxyl radicals, which in turn can damage proteins, nucleic acids, and membrane phospholipids leading to cellular degeneration (Jellinger et al., 1993; Jenner, 2003; Zecca et al., 2004). Furthermore, elevated iron is able to catalyze Parkinsonism-inducing neurotoxin MPTP to MPP+ (1-methyl-4-phenylpyridium ion) in solution producing reactive oxygen species as by-products of the reaction (Poirier et al., 1985) and sequentially lead to lipid peroxidation and dopaminergic neuron death within the SN, suggesting that high a concentration of iron can contribute to the toxicity of the compound (Lan and Jiang, 1997). However, iron chelation via either genetic expression of the iron-binding protein ferritin or oral administration of the bioavailable metal chelator clioquinol attenuates MPTP-mediated biochemical, neuropathological, and behavioral deficits (Kaur et al., 2003). Similarly, rats pretreated with the iron chelator desferrioxamine are resistant to parkinsonian-inducing agent 6-OHDA (6-hydroxydopamine) neurotoxicity (Ben-Shachar et al., 1991; Glinka et al., 1996). These studies all suggest that increased levels in midbrain iron may be upstream of neurodegeneration associated with PD. The regimen of neonatal iron feeding used in this current study was based on previous work from our laboratory in which neonatal iron absorption into the blood was adjusted to mimic the amount of absorption after ingestion of iron fortified infant formula in a human infant during a similar developmental time period (Kaur et al., 2007). Data from this previous study suggested that such a regimen resulted in a progressive, age-related degeneration of dopaminergic neurons of the SN in mice. Neonatal iron intake, therefore, may be an important risk factor rendering the nigrostriatal system more susceptible to subsequent insults, such as exposure to environmental toxins including paraquat, or the aging process itself (Kaur et al., 2007).
In addition to their direct neurotoxic effects, paraquat and iron may induce secondary effects that can contribute to neurodegeneration, including endogenous cellular iron release. Oxidative stress, such as that produced by paraquat and iron, can result in activation of inducible factors such as heme oxygenase (HO-1). HO-1 has been demonstrated to be induced in dopaminergic neurons both selectively in the SN of idiopathic Parkinsonian patients and after treatment with polychlorobiphenyls, such as dieldrin, exposure of which has been epidemiologically linked to increased susceptibility to the disease and results in PD-like pathology in animals (Schipper et al., 1998; Kim et al., 2005; Lee et al., 2006). Although largely thought of as being cytoprotective, its activation has also been demonstrated to result in cellular iron release (Keyse and Tyrrell, 1990). In addition, proteins that contain iron-sulfur clusters, including mitochondrial complex I and aconitase, are major targets for oxidative damage, resulting in displacement of iron from the cluster and protein malfunction (Drapier, 1997; Gardner et al., 1997; Kennedy et al., 1997; Rogers et al., 2003). Superoxide produced by in vivo MPTP treatment has been demonstrated to result in oxidative inactivation of mitochondrial aconitase and iron release (Liang and Patel, 2004). Specifically, this involves oxidation of the [4Fe-4S]+ center to an unstable cluster that spontaneously degrades to a [3Fe-4S]+ form with loss of a catalytic iron atom (Imlay, 2006).
Aging is the number one risk factor for Parkinson's disease. Our data demonstrates that older animals are more susceptible to the effects of increased levels of superoxide and hydrogen produced by paraquat combined with increased reactive iron that can convert these species to highly reactive and toxic hydroxyl radical. Levels of antioxidants that normally act to reduce levels of superoxide and hydrogen peroxide (superoxide dismutase-1, catalase, glutathione peroxidase) have been reported to be reduced in the aging brain (Meng et al., 2007), but levels of oxidant damage and midbrain cell loss were not found to be increased following PQ alone in an age-dependent manner even up to 24 months of age. This suggests that it is not the production of these oxidative species by paraquat alone but it could be the reaction of these species with iron that is primarily responsible for the observed age-related increase in oxidant damage and cellular toxicity first observed at 12 months. In keeping with this, elevation in basal iron levels achieved via neonatal iron feeding in the adult brain have been found to result in a significant age-related increase in oxidative damage in the dopaminergic SNpc neurons (Kaur et al., 2007, present study). Reported age-related increases in brain iron may also contribute to basal iron elevation in young animals (de Lima et al., 2007). This increase in oxidative damage was found to correlate in this current study with an elevation in paraquat-induced loss of dopaminergic SNpc neurons by 12 months of age in animals that received oral iron neonatally versus those that did not; although, iron alone did not cause age-related cell loss, and there was no increase in paraquat-induced dopaminergic cell loss at 12 versus 2 months of age. The increase in paraquat-induced cell loss in the presence of previous neonatal iron feeding was preventable by pre-EUK-189 treatment, which would remove reactive oxygen species that would normally interact with elevated SNpc iron levels. Alternatively, the additive age-related effects of neonatal iron and paraquat at 12 months of age may be because of combined increases in oxidative damage.
The JNK signal pathway has been shown to be induced by oxidative stress in numerous cell types, including dopaminergic SNpc neurons after in vivo paraquat exposure, and to result in increased apoptosis (Davis, 2000; Kyriakis and Avruch, 2001). Accumulating evidence suggested that the JNK signal transduction pathway may be involved in neurodegeneration associated with PD and that blocking the activity of JNK protein kinases may prevent disease-related neuropathology (Peng and Andersen, 2003). In previous studies from our laboratory, we have demonstrated that paraquat induces sequential phosphorylation of JNK and c-Jun and activation of caspase-3 (Peng et al., 2004). Furthermore, the JNK inhibitors SP600125 and CEP-11004 not only blocked increased paraquat-induced phosphorylation of the JNK signaling pathway elicited by neonatal iron feeding in vivo but also abrogated in vitro iron/paraquat-induced dopaminergic apoptosis. This suggests that oxidative activation of the JNK signaling cascade may be involved in increased dopaminergic SNpc cell death associated with combined iron/paraquat exposure.
The salen manganese EUK complexes catalytically eliminate both superoxide and hydrogen peroxide (Baudry et al., 1993; Gonzalez et al., 1995; Baker et al., 1998) and have been shown to be neuroprotective in several animal models of PD (Doctrow et al., 1997; Peng et al., 2005). The EUK-189 compound used in this study is more lipophilic than earlier generations of salen-manganese complexes, thus improving intracellular and intraorganellar delivery (Melov et al., 2001). Although we have not ruled out that EUK acts by peripherally impacting on entry of paraquat and/or iron into the brain, EUK compounds have been demonstrated to ameliorate brain-specific injury in several in vivo model systems indicating its ability to cross the blood–brain barrier and elicit protective antioxidant effects (Baker et al., 1998; Melov et al., 2001; Liu et al., 2003; Browne et al., 2004). The present findings support a pre-eminent role for oxidative stress in neurodegeneration and impairment of dopaminergic function in an experimental model of PD involving combined environmental exposure and suggest that bioavailable antioxidants may be a viable therapeutic approach for neurodegenerative diseases associated with oxidative stress such as PD.
Footnotes
This work was supported by National Institutes of Health Grant U54 ES12077 (J.K.A.).
References
- Alam ZI, Daniel SE, Lees AJ, Marsden DC, Jenner P, Halliwell B. A generalised increase in protein carbonyls in the brain in Parkinson's but not incidental Lewy body disease. J Neurochem. 1997;69:1326–1329. doi: 10.1046/j.1471-4159.1997.69031326.x. [DOI] [PubMed] [Google Scholar]
- Baker K, Marcus CB, Huffman K, Kruk H, Malfroy B, Doctrow SR, Gonzalez PK, Zhuang J, Benson PF, Menconi MJ, Fink MP, Baudry M, Etienne S, Bruce A, Palucki M, Jacobsen E. Synthetic combined superoxide dismutase/catalase mimetics are protective as a delayed treatment in a rat stroke model: a key role for reactive oxygen species in ischemic brain injury. J Pharmacol Exp Ther. 1998;284:215–221. [PubMed] [Google Scholar]
- Baudry M, Etienne S, Bruce A, Palucki M, Jacobsen E, Malfroy B. Salen-manganese complexes are superoxide dismutase-mimics. Biochem Biophys Res Commun. 1993;192:964–968. doi: 10.1006/bbrc.1993.1509. [DOI] [PubMed] [Google Scholar]
- Ben-Shachar D, Eshel G, Finberg JP, Youdim MB. The iron chelator desferrioxamine (Desferal) retards 6-hydroxydopamine-induced degeneration of nigrostriatal dopamine neurons. J Neurochem. 1991;56:1441–1444. doi: 10.1111/j.1471-4159.1991.tb11444.x. [DOI] [PubMed] [Google Scholar]
- Brown TP, Rumsby PC, Capleton AC, Rushton L, Levy LS. Pesticides and Parkinson's disease–is there a link? Environ Health Perspect. 2006;114:156–164. doi: 10.1289/ehp.8095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne SE, Roberts LJ, II, Dennery PA, Doctrow SR, Beal MF, Barlow C, Levine RL. Treatment with a catalytic antioxidant corrects the neurobehavioral defect in ataxia-telangiectasia mice. Free Radic Biol Med. 2004;36:938–942. doi: 10.1016/j.freeradbiomed.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Burkitt MJ, Kadiiska MB, Hanna PM, Jordan SJ, Mason RP. Electron spin resonance spin-trapping investigation into the effects of paraquat and desferrioxamine on hydroxyl radical generation during acute iron poisoning. Mol Pharmacol. 1993;43:257–263. [PubMed] [Google Scholar]
- Bus JS, Gibson JE. Paraquat: model for oxidant-initiated toxicity. Environ Health Perspect. 1984;55:37–46. doi: 10.1289/ehp.845537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellani RJ, Siedlak SL, Perry G, Smith MA. Sequestration of iron by Lewy bodies in Parkinson's disease. Acta Neuropathol (Berl) 2000;100:111–114. doi: 10.1007/s004010050001. [DOI] [PubMed] [Google Scholar]
- Chaudhuri A, Bowling K, Funderburk C, Lawal H, Inamdar A, Wang Z, O'Donnell JM. Interaction of genetic and environmental factors in a Drosophila parkinsonism model. J Neurosci. 2007;27:2457–2467. doi: 10.1523/JNEUROSCI.4239-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clejan L, Cederbaum AI. Synergistic interactions between NADPH-cytochrome P-450 reductase, paraquat, and iron in the generation of active oxygen radicals. Biochem Pharmacol. 1989;38:1779–1786. doi: 10.1016/0006-2952(89)90412-7. [DOI] [PubMed] [Google Scholar]
- Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- de Lima MN, Dias CP, Torres JP, Dornelles A, Garcia VA, Scalco FS, Guimaraes MR, Petry RC, Bromberg E, Constantino L, Budni P, Dal-Pizzol F, Schroder N. Reversion of age-related recognition memory impairment by iron chelation in rats. Neurobiol Aging. 2007 doi: 10.1016/j.neurobiolaging.2007.02.006. in press. [DOI] [PubMed] [Google Scholar]
- Dexter DT, Holley AE, Flitter WD, Slater TF, Wells FR, Daniel SE, Lees AJ, Jenner P, Marsden CD. Increased levels of lipid hydroperoxides in the parkinsonian substantia nigra: an HPLC and ESR study. Mov Disord. 1994;9:92–97. doi: 10.1002/mds.870090115. [DOI] [PubMed] [Google Scholar]
- Dinis-Oliveira RJ, Remiao F, Carmo H, Duarte JA, Navarro AS, Bastos ML, Carvalho F. Paraquat exposure as an etiological factor of Parkinson's disease. Neurotoxicology. 2006;27:1110–1122. doi: 10.1016/j.neuro.2006.05.012. [DOI] [PubMed] [Google Scholar]
- Doctrow SR, Huffman K, Marcus CB, Musleh W, Bruce A, Baudry M, Malfroy B. Salen-manganese complexes: combined superoxide dismutase/catalase mimics with broad pharmacological efficacy. Adv Pharmacol. 1997;38:247–269. doi: 10.1016/s1054-3589(08)60987-4. [DOI] [PubMed] [Google Scholar]
- Drapier JC. Interplay between NO and [Fe-S] clusters: relevance to biological systems. Methods. 1997;11:319–329. doi: 10.1006/meth.1996.0426. [DOI] [PubMed] [Google Scholar]
- Forno LS. Neuropathology of Parkinson's disease. J Neuropathol Exp Neurol. 1996;55:259–272. doi: 10.1097/00005072-199603000-00001. [DOI] [PubMed] [Google Scholar]
- Fung HC, Chen CM, Hardy J, Singleton AB, Lee-Chen GJ, Wu YR. Analysis of the PINK1 gene in a cohort of patients with sporadic early-onset parkinsonism in Taiwan. Neurosci Lett. 2006;394:33–36. doi: 10.1016/j.neulet.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Gardner PR, Costantino G, Szabo C, Salzman AL. Nitric oxide sensitivity of the aconitases. J Biol Chem. 1997;272:25071–25076. doi: 10.1074/jbc.272.40.25071. [DOI] [PubMed] [Google Scholar]
- Glinka Y, Tipton KF, Youdim MB. Nature of inhibition of mitochondrial respiratory complex I by 6-hydroxydopamine. J Neurochem. 1996;66:2004–2010. doi: 10.1046/j.1471-4159.1996.66052004.x. [DOI] [PubMed] [Google Scholar]
- Gonzalez PK, Zhuang J, Doctrow SR, Malfroy B, Benson PF, Menconi MJ, Fink MP, Baudry M, Etienne S, Bruce A, Palucki M, Jacobsen E. EUK-8, a synthetic superoxide dismutase and catalase mimetic, ameliorates acute lung injury in endotoxemic swine. J Pharmacol Exp Ther. 1995;275:798–806. [PubMed] [Google Scholar]
- Gorell JM, Johnson CC, Rybicki BA, Peterson EL, Richardson RJ. The risk of Parkinson's disease with exposure to pesticides, farming, well water, and rural living. Neurology. 1998;50:1346–1350. doi: 10.1212/wnl.50.5.1346. [DOI] [PubMed] [Google Scholar]
- Griffiths PD, Dobson BR, Jones GR, Clarke DT. Iron in the basal ganglia in Parkinson's disease. An in vitro study using extended X-ray absorption fine structure and cryo-electron microscopy. Brain. 1999;122:667–673. doi: 10.1093/brain/122.4.667. [DOI] [PubMed] [Google Scholar]
- Hertzman C, Wiens M, Bowering D, Snow B, Calne D. Parkinson's disease: a case-control study of occupational and environmental risk factors. Am J Ind Med. 1990;17:349–355. doi: 10.1002/ajim.4700170307. [DOI] [PubMed] [Google Scholar]
- Hubble JP, Cao T, Hassanein RE, Neuberger JS, Koller WC. Risk factors for Parkinson's disease. Neurology. 1993;43:1693–1697. doi: 10.1212/wnl.43.9.1693. [DOI] [PubMed] [Google Scholar]
- Imlay JA. Iron-sulphur clusters and the problem with oxygen. Mol Microbiol. 2006;59:1073–1082. doi: 10.1111/j.1365-2958.2006.05028.x. [DOI] [PubMed] [Google Scholar]
- Jellinger KA, Kienzl E, Rumpelmaier G, Paulus W, Riederer P, Stachelberger H, Youdim MB, Ben-Shachar D. Iron and ferritin in substantia nigra in Parkinson's disease. Adv Neurol. 1993;60:267–272. [PubMed] [Google Scholar]
- Jenner P. Oxidative stress in Parkinson's disease. Ann Neurol. 2003;53:S26–S36. doi: 10.1002/ana.10483. [DOI] [PubMed] [Google Scholar]
- Jimenez-Jimenez FJ, Mateo D, Gimenez-Roldan S. Exposure to well water and pesticides in Parkinson's disease: a case-control study in the Madrid area. Mov Disord. 1992;7:149–152. doi: 10.1002/mds.870070209. [DOI] [PubMed] [Google Scholar]
- Jung C, Rong Y, Doctrow S, Baudry M, Malfroy B, Xu Z. Synthetic superoxide dismutase/catalase mimetics reduce oxidative stress and prolong survival in a mouse amyotrophic lateral sclerosis model. Neurosci Lett. 2001;304:157–160. doi: 10.1016/s0304-3940(01)01784-0. [DOI] [PubMed] [Google Scholar]
- Kaur D, Yantiri F, Rajagopalan S, Kumar J, Mo JQ, Boonplueang R, Viswanath V, Jacobs R, Yang L, Beal MF, DiMonte D, Volitaskis I, Ellerby L, Cherny RA, Bush AI, Andersen JK. Genetic or pharmacological iron chelation prevents MPTP-induced neurotoxicity in vivo: a novel therapy for Parkinson's disease. Neuron. 2003;37:899–909. doi: 10.1016/s0896-6273(03)00126-0. [DOI] [PubMed] [Google Scholar]
- Kaur D, Peng J, Chinta SJ, Rajagopalan S, Di Monte DA, Cherny RA, Andersen JK. Increased murine neonatal iron intake results in Parkinson-like neurodegeneration with age. Neurobiol Aging. 2007;28:907–913. doi: 10.1016/j.neurobiolaging.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Kennedy MC, Antholine WE, Beinert H. An EPR investigation of the products of the reaction of cytosolic and mitochondrial aconitases with nitric oxide. J Biol Chem. 1997;272:20340–20347. doi: 10.1074/jbc.272.33.20340. [DOI] [PubMed] [Google Scholar]
- Keyse SM, Tyrrell RM. Induction of the heme oxygenase gene in human skin fibroblasts by hydrogen peroxide and UVA (365 nm) radiation: evidence for the involvement of the hydroxyl radical. Carcinogenesis. 1990;11:787–791. doi: 10.1093/carcin/11.5.787. [DOI] [PubMed] [Google Scholar]
- Kim DK, Kim JS, Kim JE, Kim SJ, Lee JS, Kim DJ, Son JH, Chun HS. Heme oxygenase-1 induction by dieldrin in dopaminergic cells. NeuroReport. 2005;16:509–512. doi: 10.1097/00001756-200504040-00018. [DOI] [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- Lan J, Jiang DH. Excessive iron accumulation in the brain: a possible potential risk of neurodegeneration in Parkinson's disease. J Neural Transm. 1997;104:649–660. doi: 10.1007/BF01291883. [DOI] [PubMed] [Google Scholar]
- Lang AE, Lozano AM. Parkinson's disease. First of two parts. N Engl J Med. 1998;339:1044–1053. doi: 10.1056/NEJM199810083391506. [DOI] [PubMed] [Google Scholar]
- Lanska DJ. The geographic distribution of Parkinson's disease mortality in the United States. J Neurol Sci. 1997;150:63–70. doi: 10.1016/s0022-510x(97)05371-9. [DOI] [PubMed] [Google Scholar]
- Lee DW, Gelein RM, Opanashuk LA. Heme-oxygenase-1 promotes polychlorinated biphenyl mixture aroclor 1254-induced oxidative stress and dopaminergic cell injury. Toxicol Sci. 2006;90:159–167. doi: 10.1093/toxsci/kfj052. [DOI] [PubMed] [Google Scholar]
- Liang LP, Patel M. Iron-sulfur enzyme mediated mitochondrial superoxide toxicity in experimental Parkinson's disease. J Neurochem. 2004;90:1076–1084. doi: 10.1111/j.1471-4159.2004.02567.x. [DOI] [PubMed] [Google Scholar]
- Liou HH, Tsai MC, Chen CJ, Jeng JS, Chang YC, Chen SY, Chen RC. Environmental risk factors and Parkinson's disease: a case-control study in Taiwan. Neurology. 1997;48:1583–1588. doi: 10.1212/wnl.48.6.1583. [DOI] [PubMed] [Google Scholar]
- Liu R, Liu IY, Bi X, Thompson RF, Doctrow SR, Malfroy B, Baudry M. Reversal of age-related learning deficits and brain oxidative stress in mice with superoxide dismutase/catalase mimetics. Proc Natl Acad Sci USA. 2003;100:8526–8531. doi: 10.1073/pnas.1332809100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malfroy B, Doctrow SR, Orr PL, Tocco G, Fedoseyeva EV, Benichou G. Prevention and suppression of autoimmune encephalomyelitis by EUK-8, a synthetic catalytic scavenger of oxygen-reactive metabolites. Cell Immunol. 1997;177:62–68. doi: 10.1006/cimm.1997.1091. [DOI] [PubMed] [Google Scholar]
- McCormack AL, Di Monte DA. Effects of L-dopa and other amino acids against paraquat-induced nigrostriatal degeneration. J Neurochem. 2003;85:82–86. doi: 10.1046/j.1471-4159.2003.01621.x. [DOI] [PubMed] [Google Scholar]
- McCormack AL, Thiruchelvam M, Manning-Bog AB, Thiffault C, Langston JW, Cory-Slechta DA, Di Monte DA. Environmental risk factors and Parkinson's disease: selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat. Neurobiol Dis. 2002;10:119–127. doi: 10.1006/nbdi.2002.0507. [DOI] [PubMed] [Google Scholar]
- McCormack AL, Atienza JG, Johnston LC, Andersen JK, Vu S, Di Monte DA. Role of oxidative stress in paraquat-induced dopaminergic cell degeneration. J Neurochem. 2005;93:1030–1037. doi: 10.1111/j.1471-4159.2005.03088.x. [DOI] [PubMed] [Google Scholar]
- Melov S, Doctrow SR, Schneider JA, Haberson J, Patel M, Coskun PE, Huffman K, Wallace DC, Malfroy B. Lifespan extension and rescue of spongiform encephalopathy in superoxide dismutase 2 nullizygous mice treated with superoxide dismutase-catalase mimetics. J Neurosci. 2001;21:8348–8353. doi: 10.1523/JNEUROSCI.21-21-08348.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Q, Wong YT, Chen J, Ruan R. Age-related changes in mitochondrial function and antioxidative enzyme activity in fischer 344 rats. Mech Ageing Dev. 2007;128:286–292. doi: 10.1016/j.mad.2006.12.008. [DOI] [PubMed] [Google Scholar]
- Peng J, Andersen JK. The role of c-Jun N-terminal kinase (JNK) in Parkinson's disease. IUBMB Life. 2003;55:267–271. doi: 10.1080/1521654031000121666. [DOI] [PubMed] [Google Scholar]
- Peng J, Wu Z, Wu Y, Hsu M, Stevenson FF, Boonplueang R, Roffler-Tarlov SK, Andersen JK. Inhibition of caspases protects cerebellar granule cells of the weaver mouse from apoptosis and improves behavioral phenotype. J Biol Chem. 2002;277:44285–44291. doi: 10.1074/jbc.M207407200. [DOI] [PubMed] [Google Scholar]
- Peng J, Mao XO, Stevenson FF, Hsu M, Andersen JK. The herbicide paraquat induces dopaminergic nigral apoptosis through sustained activation of the JNK pathway. J Biol Chem. 2004;279:32626–32632. doi: 10.1074/jbc.M404596200. [DOI] [PubMed] [Google Scholar]
- Peng J, Stevenson FF, Doctrow SR, Andersen JK. Superoxide dismutase/catalase mimetics are neuroprotective against selective paraquat-mediated dopaminergic neuron death in the substantial nigra: implications for Parkinson disease. J Biol Chem. 2005;280:29194–29198. doi: 10.1074/jbc.M500984200. [DOI] [PubMed] [Google Scholar]
- Poirier J, Donaldson J, Barbeau A. The specific vulnerability of the substantia nigra to MPTP is related to the presence of transition metals. Biochem Biophys Res Commun. 1985;128:25–33. doi: 10.1016/0006-291x(85)91639-0. [DOI] [PubMed] [Google Scholar]
- Prasad KN, Carvalho E, Kentroti S, Edwards-Prasad J, Freed C, Vernadakis A. Establishment and characterization of immortalized clonal cell lines from fetal rat mesencephalic tissue. In Vitro Cell Dev Biol Anim. 1994;30A:596–603. doi: 10.1007/BF02631258. [DOI] [PubMed] [Google Scholar]
- Riederer P, Dirr A, Goetz M, Sofic E, Jellinger K, Youdim MB. Distribution of iron in different brain regions and subcellular compartments in Parkinson's disease. Ann Neurol. 1992;32:S101–104. doi: 10.1002/ana.410320717. [DOI] [PubMed] [Google Scholar]
- Rogers PA, Eide L, Klungland A, Ding H. Reversible inactivation of E. coli endonuclease III via modification of its [4Fe-4S] cluster by nitric oxide. DNA Repair (Amst) 2003;2:809–817. doi: 10.1016/s1568-7864(03)00065-x. [DOI] [PubMed] [Google Scholar]
- Rong Y, Doctrow SR, Tocco G, Baudry M. EUK-134, a synthetic superoxide dismutase and catalase mimetic, prevents oxidative stress and attenuates kainate-induced neuropathology. Proc Natl Acad Sci USA. 1999;96:9897–9902. doi: 10.1073/pnas.96.17.9897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schipper HM, Liberman A, Stopa EG. Neural heme oxygenase-1 expression in idiopathic Parkinson's disease. Exp Neurol. 1998;150:60–68. doi: 10.1006/exnr.1997.6752. [DOI] [PubMed] [Google Scholar]
- Semchuk KM, Love EJ, Lee RG. Parkinson's disease and exposure to agricultural work and pesticide chemicals. Neurology. 1992;42:1328–1335. doi: 10.1212/wnl.42.7.1328. [DOI] [PubMed] [Google Scholar]
- Shimura-Miura H, Hattori N, Kang D, Miyako K, Nakabeppu Y, Mizuno Y. Increased 8-oxo-dGTPase in the mitochondria of substantia nigral neurons in Parkinson's disease. Ann Neurol. 1999;46:920–924. [PubMed] [Google Scholar]
- Sofic E, Paulus W, Jellinger K, Riederer P, Youdim MB. Selective increase of iron in substantia nigra zona compacta of parkinsonian brains. J Neurochem. 1991;56:978–982. doi: 10.1111/j.1471-4159.1991.tb02017.x. [DOI] [PubMed] [Google Scholar]
- Stephenson J. Exposure to home pesticides linked to Parkinson disease. JAMA. 2000;283:3055–3056. doi: 10.1001/jama.283.23.3055. [DOI] [PubMed] [Google Scholar]
- Tanner CM, Ottman R, Goldman SM, Ellenberg J, Chan P, Mayeux R, Langston JW. Parkinson disease in twins: an etiologic study. JAMA. 1999;281:341–346. doi: 10.1001/jama.281.4.341. [DOI] [PubMed] [Google Scholar]
- Thiruchelvam M, McCormack A, Richfield EK, Baggs RB, Tank AW, Di Monte DA, Cory-Slechta DA. Age-related irreversible progressive nigrostriatal dopaminergic neurotoxicity in the paraquat and maneb model of the Parkinson's disease phenotype. Eur J Neurosci. 2003;18:589–600. doi: 10.1046/j.1460-9568.2003.02781.x. [DOI] [PubMed] [Google Scholar]
- West MJ. Stereological methods for estimating the total number of neurons and synapses: issues of precision and bias. Trends Neurosci. 1999;22:51–61. doi: 10.1016/s0166-2236(98)01362-9. [DOI] [PubMed] [Google Scholar]
- Yoritaka A, Hattori N, Uchida K, Tanaka M, Stadtman ER, Mizuno Y. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc Natl Acad Sci USA. 1996;93:2696–2701. doi: 10.1073/pnas.93.7.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida S, Ektessabi A, Fujisawa S. XANES spectroscopy of a single neuron from a patient with Parkinson's disease. J Synchrotron Radiat. 2001;8:998–1000. doi: 10.1107/s0909049500017726. [DOI] [PubMed] [Google Scholar]
- Zecca L, Youdim MB, Riederer P, Connor JR, Crichton RR. Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci. 2004;5:863–873. doi: 10.1038/nrn1537. [DOI] [PubMed] [Google Scholar]
- Zhang HJ, Doctrow SR, Xu L, Oberley LW, Beecher B, Morrison J, Oberley TD, Kregel KC. Redox modulation of the liver with chronic antioxidant enzyme mimetic treatment prevents age-related oxidative damage associated with environmental stress. FASEB J. 2004;18:1547–1549. doi: 10.1096/fj.04-1629fje. [DOI] [PubMed] [Google Scholar]