

Abstract
The therapeutic management of methamphetamine (METH)-induced psychoses often involves treatment with the typical antipsychotic drug and dopamine D2 receptor antagonist haloperidol. We report here that subchronic haloperidol administration after a high-dose regimen of METH produces a heretofore unrecognized toxicity to GABAergic cells, as reflected by GAD67 mRNA expression histochemistry, in the rat substantia nigra pars reticulata (SNr) through an acute and persistent augmentation of glutamate release, NMDA receptor activation, and DNA fragmentation. The dopaminergic cells in the substantia nigra pars compacta were unaffected by METH or haloperidol alone or the combination of METH and haloperidol. These findings suggest that the current therapeutic management of METH-induced psychoses with haloperidol may be contraindicated because of a resultant GABAergic cell death in the SNr, which may predispose some individuals to the development of hyperkinetic movement disorders and seizures.
Keywords: antipsychotics, glutamate, dopamine, microdialysis, basal ganglia, neurodegeneration
Introduction
Methamphetamine (METH) is a psychostimulant drug that is abused worldwide and has accounted for 102,843 emergency room admissions during 2004 in the United States (Drug Abuse Warning Network statistics). METH induces acute psychotic episodes and persistent paranoid psychoses (Sato et al., 1992; Ellison, 1994; Cretzmeyer et al., 2003) that are often treated with typical antipsychotics such as the dopamine D2 receptor antagonist haloperidol (HAL) (Sato et al., 1983; Derlet et al., 1989; Richards et al., 1998; Pittman, 2005). This treatment strategy presumably counteracts the effects of acute METH-induced increases in dopamine (DA) release (Schmidt et al., 1987) and a host of psychiatric symptoms typically associated with increased DA transmission (Lieberman et al., 1990).
In contrast to the acute effects of METH, the long-term effects of METH are characterized by decreases in DA transmission as reflected by decreased DA tissue content, tyrosine hydroxylase (TH) activity, and DA transporter levels in DA terminal regions such as the striatum of animals and humans (Gibb and Kogan, 1979; Ricaurte et al., 1982; Stephans and Yamamoto, 1996; Volkow et al., 2001). Nevertheless, there is no evidence of DA cell body loss in the substantia nigra (SN) (Brunswick et al., 1992; Harvey et al., 2000; Thomas et al., 2004).
Relatively few studies have investigated basal ganglia (BG) structures extrinsic to the striatum that could be affected by METH, such as the SN. The SN not only provides DAergic input to the striatum from DA cells located in the pars compacta (SNc) but is also a convergence point of information flow from the “direct” and “indirect” BG pathways (Bevan et al., 1994) and also in the outflow of information from GABAergic cells located in the pars reticulata (SNr) projecting to the ventral thalamus and other motor areas (Oertel and Mugnaini, 1984). Recent evidence illustrates that DA enhances METH-induced glutamate (GLU) release from corticostriatal terminals through the “direct pathway” involving D1 receptors on striatonigral GABAergic projections (Mark et al., 2004). However, no studies have examined the impact of METH on the “indirect pathway” of the BG involving the D2 receptor-mediated inhibition of glutamatergic input to the SNr from the subthalamic nucleus (STN) (Hatzipetros and Yamamoto, 2006). This dopaminergic inhibition of the indirect pathway is particularly pertinent because D2 antagonists are used to treat METH-induced psychoses.
Glutamatergic innervation of the SN is provided by afferents arising from the STN, medial prefrontal cortex, and pedunculopontine nucleus (Smith et al., 1996; Bezard and Gross, 1998), as well as ionotropic and metabotropic GLU receptors in the SN (Overton and Clark, 1991; Meltzer et al., 1997; Tse and Yung, 2000). To elucidate the role of DA in mediating the effects of METH on GLU in the SN, the present study examined the effects of D2 receptor antagonism, a strategy typically used to treat METH overdose, on glutamatergic transmission and cell viability in the SN. It is hypothesized that treatment of high-dose METH exposure with the D2 antagonist haloperidol will produce a heretofore unrecognized cell loss in the SN via disinhibition of GLU release, activation of GLU receptors, and excitotoxicity that otherwise would not be observed by METH alone.
Materials and Methods
Animals.
Adult male Sprague Dawley rats (200–300 g; Harlan Sprague Dawley, Indianapolis, IN) were used in all experiments. Rats were housed four per cage before surgery/dialysis and were maintained on a 12 h light/dark cycle in a temperature- and humidity-controlled environment. Food and water were available ad libitum. Animal care was in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, which was approved and monitored by the Institutional Animal Care and Use Committee of Boston University.
Drugs and drug regimens.
METH was supplied by the National Institute on Drug Abuse (Research Triangle Park, NC). Haloperidol and Dulbecco's PBS were purchased from Sigma (St. Louis, MO). MK-801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate] was purchased from Tocris Bioscience (Ellisville, MO). Haloperidol was prepared by dissolving 0.5 mg of the drug in 50 μl of 1.0 m tartaric acid. The volume was then raised to 800 μl with distilled water, and the pH was adjusted to 5.8 with 1.0 m NaOH. More distilled water was added to make the total volume 1000 μl. The vehicle solution was prepared in a similar manner but without any drug.
The following treatment regimen was used for all experiments, except the microdialysis studies: day 0, four injections of METH (10 mg/kg) or saline (1 ml/kg) were administered intraperitoneally every 2 h; days 3–7, haloperidol (0.5 mg/kg) or vehicle was administered intraperitoneally once daily; day 14, the rats were killed by decapitation, and their brains were removed and quickly frozen with dry ice. The brains were stored at −80°C until they were sectioned. MK-801 (0.1 mg/kg) was administered by a single intraperitoneal injection 15 min before haloperidol in certain experiments.
Surgical probe implantation.
Dialysis probes were constructed as described previously (Yamamoto and Pehek, 1990). They were of a concentric flow design with a dialysis membrane length of 1.5 mm. Rats were anesthetized with a combination of xylazine (6 mg/kg, i.p.) and ketamine hydrochloride (70 mg/kg, i.p.) and were mounted into a stereotaxic apparatus (David Kopf Instruments, Tujunga, CA) for the implantation of the microdialysis probes into the SN. The skull was exposed, holes were drilled, and probes were slowly lowered to the following coordinates aimed at the SN (at a 15° lateral angle): anteroposterior, −5.8 mm; mediolateral, ±3.8 mm as referenced from bregma; dorsoventral, −7.4 mm as referenced from dura (Paxinos and Watson, 1986). The probes were secured to the skull with three stainless steel machine screws and cranioplastic cement. Rats were transferred into the dialysis cages and were allowed to recover overnight. Dialysis was conducted on the following day.
In vivo microdialysis.
The microdialysis probes were perfused with modified Dulbecco's PBS (in mm: 138 NaCl, 2.7 KCl, 0.5 MgCl2, 1.5 KH2PO4, 1.2 CaCl2, and 5.0 d-glucose, pH 7.4) at a constant perfusion rate of 1.5 μl/min using a microinfusion pump (Harvard Apparatus, Holliston, MA). After a 2 h prebaseline perfusion period, dialysate samples (45 μl) were collected every 30 min. Once stable baseline concentrations of GLU were achieved in the dialysate, the experiments proceeded with the drug administrations. All drugs were administered intraperitoneally. At the end of each experiment, the rats were killed by decapitation, and the probes were manually perfused with green dye before they were extracted. The brains were removed and frozen for later sectioning in 40-μm-thick coronal slices for probe placement verification. The green dye was used to determine the location of the dialysis membrane and the exact area that was perfused. Only rats with probe placement in the SN were included in the statistical analysis.
Measurement of extracellular glutamate by HPLC.
Glutamate was assayed from 20 μl dialysate aliquots by HPLC coupled to fluorescence detection after precolumn derivatization with o-pthaldialdehyde as described previously (Donzanti and Yamamoto, 1988) with slight modifications. A 10 μl aliquot of the derivatization reagent was automatically added to the 20 μl dialysate sample by an ESA (Chelmsford, MA) model 542 autosampler, mixed, and allowed to react for exactly 90 s before injection onto the column. Separation was achieved with a 3 μm C18 column and a mobile phase consisting of 0.1 m sodium phosphate, 0.1 mm EDTA, and 10% methanol, pH 6.65. Fluorescence detection was performed with a Waters (Milford, MA) 474 Fluorescence Detector. The excitation and emission wavelengths were set at 340 and 440 nm, respectively. Data were collected using EZChrom Elite application software (Scientific Software, Pleasanton, CA).
Dopamine tissue content.
SN tissue was dissected out bilaterally from two consecutive 400-μm-thick coronal brain slices sectioned on a Microm HM500 cryostat (Richard-Allan Scientific, Kalamazoo, MI). The tissue was sonicated in 200 μl of cold 0.1 m perchloric acid and centrifuged at 12,000 × g for 6 min. DA was assayed from 20 μl aliquots of the supernatant by HPLC coupled to electrochemical detection. Separation was achieved on a C18 column (100 × 2.0 mm, 3 μm particle size; Phenomenex, Torrance, CA) and eluted with a mobile phase consisting of 32 mm citric acid, 54.3 mm sodium acetate, 0.074 mm EDTA (Na2-EDTA), 0.215 mm octyl sodium sulfate, and 3% methanol, pH 3.8. Detection was achieved with an LC-4C amperometric detector (Bioanalytical Systems, West Lafayette, IN) with a glassy carbon working electrode maintained at a potential of +0.670 V relative to an Agv̄/AgCl reference electrode. Data were recorded using the EZChrom Elite application software. The pellet, which was dissolved in 1 m NaOH, was used to determine protein concentrations using the Bradford method (Bio-Rad, Hercules, CA). Final DA concentrations were expressed as picograms of DA per microgram of protein.
Immunohistochemistry of TH and neuronal nuclei.
Immunohistochemistry was performed using a standard peroxidase-based method (Vectastain Elite ABC kit; Vector Laboratories, Burlingame, CA) on 10-μm-thick coronal sections through the SN obtained from fresh-frozen brains sectioned on a Microm HM500 cryostat (Richard-Allan Scientific). The slide-mounted sections were fixed in 4% paraformaldehyde for 5 min, quickly washed in 0.2 m potassium PBS (KPBS) plus Triton X-100, and incubated for 1 h in 0.2 m KPBS with 5% normal goat serum (NGS) at room temperature. Sections were then incubated overnight at 4°C with a rabbit anti-TH polyclonal antibody (1:250; Chemicon) or a mouse anti-neuronal-specific nuclear protein (NeuN) monoclonal antibody (1:500; Chemicon) in 0.2 m KPBS, 1% NGS, and 0.2% Triton X-100. The next day, sections were washed in 0.2 m KPBS plus 0.2% Triton X-100 and were incubated for 1 h at room temperature in affinity-purified biotinylated goat anti-rabbit IgG for TH (1:200; Chemicon) or in affinity-purified biotinylated mouse anti-rabbit IgG for NeuN (1:200; Chemicon). Sections were washed in 0.2 m KPBS and processed with the avidin–biotin complex (ABC; Vector Laboratories). Immunolabeling was revealed after incubation of the sections in a DAB/Metal Concentrate (Pierce, Rockford, IL). Sections were dehydrated and mounted in Eukitt mounting medium.
Quantitative analysis of TH immunolabeling in the SNc was conducted on digitized pictures by computerized densitometry using NIH Image 1.61 software, AF Micro Nikon (Tokyo, Japan) 60 mm f/2.8D lens, and a Sony (Tokyo, Japan) CCD video camera connected to a Macintosh computer (Apple Computers, Seattle, WA). The SNc was analyzed for each rat, and the value of TH immunolabeling per rat was the average of two consecutive sections taken at 5.3 mm posterior from bregma. No other subregions were analyzed because of the use of slices for the subregional analyses of NeuN.
Quantitative analysis of NeuN immunolabeling in the SNc and SNr was conducted on photomicrographs taken by light microscopy with a 10× objective on a Nikon Eclipse 600 microscope connected to a monochrome SPOT Insight digital camera (Diagnostic Instruments, Sterling Heights, MI). NeuN-positive neurons were manually counted by an observer blind to the experimental groups and were expressed as NeuN-positive profiles/area. Three frames per section were analyzed, all in the same locations. The number of NeuN-positive profiles per frame was counted (each frame had an area of 33,000 μm2), and the total number of NeuN-positive profiles per section was calculated. The value of NeuN-positive neurons per rat was the average of three consecutive sections within each of three regions (rostral, middle, and caudal regions beginning at 4.8, 5.3, and 5.8 mm posterior from bregma, respectively). These coordinates were based on the reported differential innervation by glutamatergic afferents to the SNr (Gonzalez-Hernandez and Rodriguez, 2000).
Glutamic acid decarboxylase 67 in situ hybridization and autoradiography.
In situ hybridization histochemistry with a riboprobe was performed on 20-μm-thick coronal sections from the SN as described previously (Nielsen and Soghomonian, 2004). Briefly, sections were quickly dried and fixed in 4% paraformaldehyde followed by prehybridization washes in triethanolamine and Tris-glycine. Sections were then hybridized for 4 h at 52°C with 4.0 ng in 20 μl of radiolabeled cRNA probe per section (average specific activity, 4.3 × 105 cpm/ng). The 35S-labeled cRNA probe was synthesized from feline glutamic acid decarboxylase 67 (GAD67) cDNA with rat cross-reactivity, inserted into a Bluescript SK plasmid, which was linearized with HindIII. The cRNA probe was diluted in hybridization solution containing 40% formamide, 10% dextran sulfate, 1× SSC, 10 mm dithiothreitol, 1.0% sheared salmon sperm DNA, 1.0% yeast tRNA, and 1× Denhardt's solution. The sections were subsequently washed in 50% formamide at 52°C first for 5 min and then for 20 min, in RNase A (100 μg/ml; Sigma) for 30 min at 37°C, and in 50% formamide for 5 min at 52°C. Sections were then dehydrated in ethanol and xylene. After dehydration, slides were dipped in Kodak NTB3 nuclear emulsion (diluted 1:1 with water containing ammonium acetate; Eastman Kodak, Rochester, NY), dried for 3 h, and stored in light-tight boxes for 10 d at room temperature. Slides were developed in Kodak D-19 for 3.5 min at 14°C, lightly counterstained with hematoxylin and eosin (H&E), and mounted with Eukitt mounting medium. Photomicrographs were taken by light microscopy with a 20× objective on a Nikon Eclipse 600 microscope connected to a monochrome SPOT Insight digital camera (Diagnostic Instruments). The numbers of H&E-labeled neuronal profiles covered by reduced silver grains representing GAD67 mRNA on emulsion autoradiographs were manually counted by an observer blind to the experimental groups. The value of GAD67 mRNA-positive neurons per rat was the average of three consecutive sections taken at 5.3 mm posterior from bregma.
Fluorescein FragEL DNA fragmentation assay.
The DNA fragmentation assay (Calbiochem, San Diego, CA) was performed on 20-μm-thick coronal sections through the SN obtained from fresh-frozen brains sectioned on a Microm HM500 cryostat (Richard-Allan Scientific). Sections were dried and fixed in 4% paraformaldehyde for 15 min, washed in 1× TBS for 15 min, and incubated with Proteinase K for 10 min at room temperature to permeabilize. Slides were rinsed in 1× TBS and incubated in 1× TdT Equilibration Buffer for 30 min at room temperature. Each specimen was covered with 60 μl of the Fluorescein FragEL TdT Reaction mixture, coverslipped with Parafilm, and incubated at 37°C for 1 h. The Parafilm coverslip was removed, and the slides were washed in 1× TBS and mounted with a glass coverslip using Fluorescein FragEL mounting media. The labeled DNA fragments were analyzed using a standard fluorescein filter (465–495 nm), an Olympus Optical (Thornwood, NY) DP70 camera with the accompanied DP Manager software, and a Nikon Eclipse TE2000-S camera connected to a Dell computer (Dell Computer Company, Round Rock, TX). Fluorescence was quantified on an arbitrary scale (0, no fluorescence; 1, low fluorescence; 2, intermediate fluorescence; 3, high fluorescence) and was presented in arbitrary units. The average fluorescence per rat was the average of two consecutive sections that were taken at 5.3 mm posterior from bregma.
Data analysis.
Microdialysis data are presented as means of percentage baseline ± SEM, and statistical significance was determined by a two-way ANOVA with repeated measures coupled to Bonferroni's post hoc test. For the DA tissue content data, significance was determined by a two-way ANOVA, and for the NeuN data, significance was determined by a one-way ANOVA coupled to Bonferroni's post hoc test. For the TH immunohistochemistry and GAD67 mRNA in situ hybridization data, significance was determined by a one-way ANOVA. For the Fluorescein FragEL DNA Fragmentation assay data, significance was determined by a Kruskal–Wallis one-way ANOVA on ranks. For all analyses, significance values were set at p < 0.05.
Results
D2 antagonism during or after METH increases GLU in the SN
In vivo microdialysis experiments were performed to examine the effects of acute METH administration in the presence or absence of D2 antagonism on GLU release in the SN of freely moving rats (Fig. 1). Basal GLU levels in the SN were 1518 ± 494 pg/20 μl. METH administration alone (four times, 10 mg/kg, i.p., every 2 h) did not affect extracellular GLU concentrations in the SN over the course of 8 h. When the D2 receptor antagonist HAL (0.5 mg/kg, i.p.) was administered 15 min before the first and third METH injections, there was a significant increase in the concentrations of extracellular GLU in the SN. The maximum effect was 545 ± 100% of baseline and took place 7 h after the first METH injection. HAL by itself also produced an increase in extracellular GLU in the SN that reached maximum concentrations of 385 ± 99% of baseline (Fig. 1). The highly specific D2 receptor antagonist raclopride also produced a similarly significant increase in the concentrations of extracellular GLU in the SN when it was administered 15 min before the first and third METH injections (data not shown).
Figure 1.

D2 antagonism with haloperidol during acute METH administration. Dialysate glutamate levels are expressed as percentage of baseline concentrations. Dialysate samples were collected from the SN every 30 min, during the 1.5 h baseline period, and for the 8 h drug treatment period. Data shown are mean ± SEM. METH (10 mg/kg) or 0.9% saline (SAL; 1 ml/kg) were given intraperitoneally once every 2 h for a total of four injections (indicated by black arrows). HAL (0.5 mg/kg) or tartaric acid vehicle (VEH) were given intraperitoneally 15 min before the first and the third METH injections (indicated by gray arrows). The METH–HAL and SAL–HAL treatment groups showed significant increases in extracellular GLU concentrations compared with the METH–VEH or SAL–VEH groups. Significance was determined via a two-way ANOVA with repeated measures; * p < 0.05, significance compared with SAL–VEH; #p < 0.05, significance compared with METH–VEH. n = 6 for SAL–VEH; n = 7 for METH–VEH; n = 4 for SAL–HAL; n = 5 for METH–HAL.
Another microdialysis experiment was performed to investigate the long-term effects of METH on basal extracellular GLU concentrations in the SN, 2 d after its administration. METH-pretreated rats had approximately twofold higher basal extracellular GLU concentrations in the SN compared with saline-pretreated controls (Fig. 2, inset). The basal GLU concentrations in the METH-pretreated rats were 2687 ± 413 pg/20 μl, whereas the basal GLU concentrations in the saline-pretreated rats were 1331 ± 327 pg/20 μl. Furthermore, the METH-pretreated rats were significantly more responsive to a challenge injection of the D2 antagonist HAL (0.5 mg/kg, i.p.) compared with the saline-pretreated controls (Fig. 2). In the METH-pretreated rats, HAL further enhanced GLU concentrations in the SN by 1402 ± 290 pg/20 μl, which resulted in maximum GLU concentrations of 4089 ± 326 pg/20 μl, 2.5 h after the HAL challenge injection. In the saline-pretreated rats, HAL enhanced GLU concentrations in the SN by only 936 ± 218 pg/20 μl, resulting in maximum GLU concentrations of 2267 ± 530 pg/20 μl at 2.5 h after the HAL challenge injection (Fig. 2).
Figure 2.

METH pretreatment on basal GLU concentrations in the SN: effect of haloperidol challenge. Rats were administered METH (10 mg/kg 4 times, every 2 h) or 0.9% saline (1 ml/kg 4 times, every 2 h) 2 d before the dialysis experiment. Basal GLU concentrations are expressed as picograms per 20 μl of dialysate. Data shown are mean ± SEM. METH pretreatment caused a significant increase in basal GLU concentrations in the SN compared with saline pretreatment (inset; significance was determined via a t test; *p < 0.05; n = 4 for both groups). Dialysate GLU concentrations are expressed in picograms per 20 μl as the absolute difference from baseline concentrations. Data shown are mean ± SEM. A haloperidol challenge injection (0.5 mg/kg, i.p.) was administered after baseline collection (indicated by a black arrow). HAL produced an overall greater increase in extracellular GLU concentrations in the METH-pretreated group compared with the saline-pretreated group. Significance was determined via a two-way ANOVA with repeated measures. *p < 0.05, significant overall treatment effect; #p < 0.05, significant difference between the METH and saline groups. n = 4 for both groups.
Subchronic haloperidol after METH does not damage DA cells in the SNc
In different groups of rats, HPLC and immunohistochemical techniques were used to examine whether DA cells in the SNc are lost after subchronic HAL and METH. Figure 3 illustrates that HAL alone, METH alone, or the combination of subchronic HAL and METH did not affect DA tissue content in the SN, 1 week after the last HAL or vehicle injection. The DA tissue content of saline controls animals was 7.75 ± 1.13 pg/μg protein.
Figure 3.

Subchronic HAL treatment after METH on DA tissue content in the SN. Rats were administered METH (10 mg/kg 4 times, every 2 h) or 0.9% saline (1 ml/kg 4 times, every 2 h) on day 0 and then HAL (0.5 mg/kg) or vehicle (VEH) once daily on days 3–7. Rats were killed on day 14, and brains were assayed. DA tissue content was determined by HPLC and expressed as picograms per microgram of protein. Data shown are mean ± SEM. There were no significant group differences in DA content. Significance was determined via a two-way ANOVA; n = 8 for SAL–VEH; n = 7 for SAL–HAL; n = 15 for METH–VEH; n = 13 for METH–HAL.
Similarly, there was no drug treatment effect on TH immunostaining as quantified by computerized densitometry (Fig. 4). In addition, counts of NeuN-positive profiles in the SNc region detected no differences between control rats and rats that received HAL, METH, or subchronic HAL after METH (Fig. 5).
Figure 4.

Subchronic HAL treatment after METH on TH immunostaining in the SNc. Rats were administered METH (10 mg/kg 4 times, every 2 h) or 0.9% saline (1 ml/kg 4 times, every 2 h) on day 0 and then HAL (0.5 mg/kg) or vehicle once daily on days 3–7 after METH administration. Rats were killed on day 14, and the brains were assayed. Quantitative analysis of TH immunolabeling is expressed as relative optical units and is the average of two consecutive slices from each rat. Data shown are mean ± SEM. TH immunostaining in the SNc revealed no significant group differences. Significance was determined via a one-way ANOVA; n = 5 for all groups.
Figure 5.
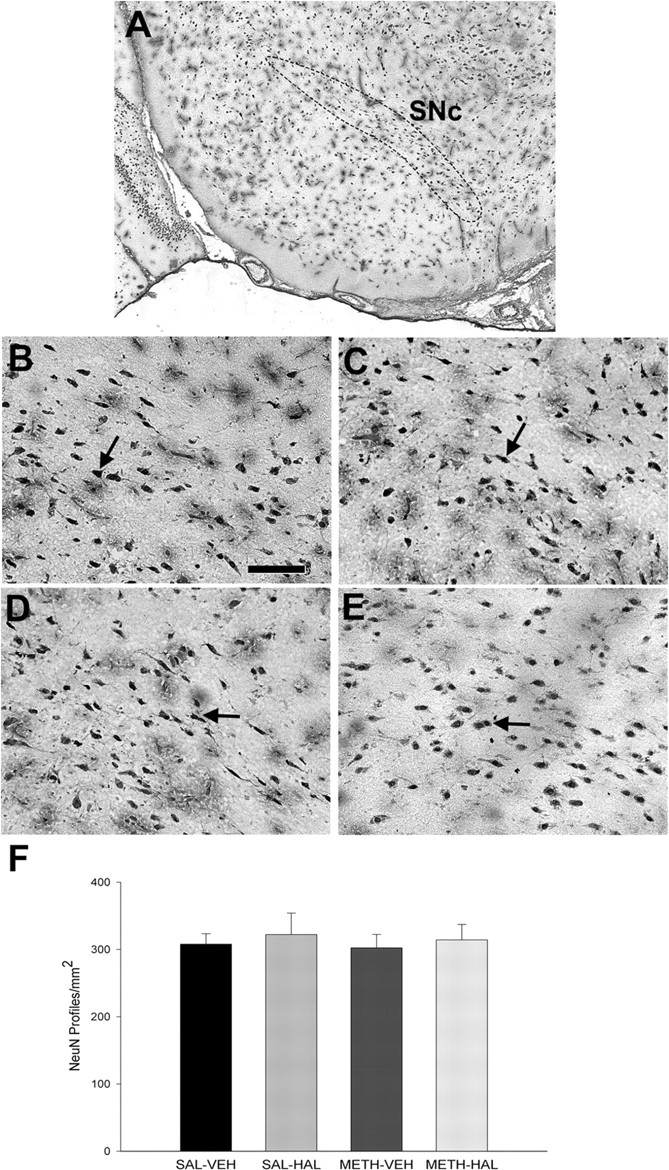
Subchronic HAL treatment after METH on NeuN immunostaining in the SNc. Rats were administered METH (10 mg/kg 4 times, every 2 h) or 0.9% saline (1 ml/kg 4 times, every 2 h) on day 0, and then HAL (0.5 mg/kg) or vehicle (VEH) once daily on days 3–7. Rats were killed on day 14, and the brains were assayed. NeuN-positive profiles were counted by an observer blind to the treatment conditions and are expressed as NeuN-positive profiles per square millimeter. Data shown represent the average of three consecutive slices and are mean ± SEM. A, Coronal view illustrating the SNc region that was sampled (dotted lines; −5.3 mm from bregma). Representative, high-magnification images from the SNc of rats treated with SAL–VEH (B), SAL–HAL (C), METH–VEH (D), and METH–HAL (E) and quantification of NeuN-positive profiles in the SNc (F) revealed no significant group differences. Arrows indicate representative NeuN-labeled nuclei. Scale bar: B–E, 100 μm. Significance was determined via a one-way ANOVA; n = 9 for SAL–VEH; n = 4 for SAL–HAL; n = 9 for METH–VEH; n = 10 for METH–HAL.
Subchronic HAL after METH results in GABAergic cell death in the SNr via glutamatergic mechanisms
NeuN immunostaining experiments showed a significant 34% decrease in the number of NeuN-positive profiles in the middle part of SNr in rats that received subchronic HAL and METH compared with saline control rats, 1 week after the last HAL injection (Fig. 6). There was a nonsignificant 20% decrease in NeuN profiles in the caudal SNr and a 7% decrease in the rostral SNr in the METH–HAL group (data not shown). Rats that received subchronic HAL alone or METH alone did not show any significant differences in the numbers of NeuN-positive profiles in the SNr at either the rostral, middle, or caudal sections.
Figure 6.
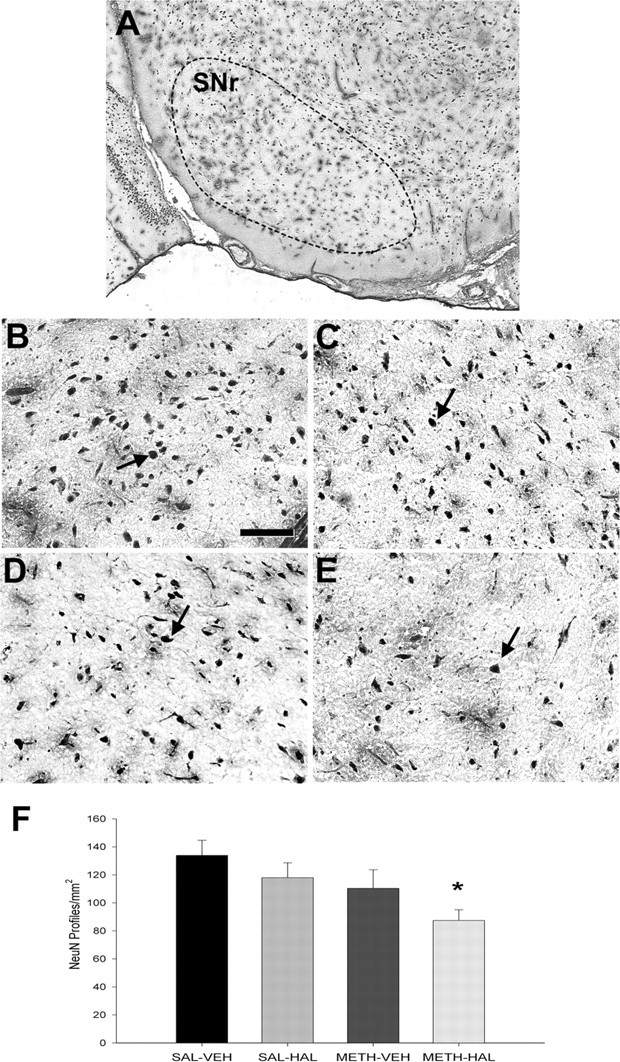
Subchronic HAL treatment after METH on NeuN immunostaining in the SNr. Rats were administered METH (10 mg/kg 4 times, every 2 h) or 0.9% saline (1 ml/kg 4 times, every 2 h) on day 0 and then HAL (0.5 mg/kg) or vehicle (VEH) once daily on days 3–7. Rats were killed on day 14. NeuN-positive profiles, which were counted by an observer blind to the treatment conditions, are expressed as NeuN-positive profiles per square millimeter. Data shown represent the average of three consecutive slices and are mean ± SEM. A, Coronal view of a mature brain illustrating the SNr region that was sampled (dotted lines; −5.3 mm from bregma). Representative high-magnification images are shown from the SNr of rats treated with SAL–VEH (B), SAL–HAL (C), METH–VEH (D), and METH–HAL (E). F, NeuN-positive profile counts in the SNr showed that there was a significant decrease in the SAL–HAL treatment group compared with the SAL–VEH group. Arrows indicate representative NeuN-labeled cells. Scale bar: B–E, 100 μm. Significance was determined via a one-way ANOVA; *p < 0.05 compared with SAL–VEH. n = 9 for SAL–VEH; n = 4 for SAL–HAL; n = 9 for METH–VEH; n = 10 for METH–HAL.
To determine the phenotype of the lost NeuN profiles in the METH–HAL group, in situ hybridization histochemistry of GAD67 mRNA expression was performed. The rats that received subchronic HAL and METH had 24% less GAD67 mRNA-expressing profiles in the SNr compared with saline controls, whereas no differences were observed in the other treatment groups (Fig. 7). In a separate group of rats, the NMDA receptor antagonist MK-801 (0.1 mg/kg, i.p.) blocked the decrease in GAD67 mRNA-expressing profiles when administered 15 min before each HAL injection (Fig. 7).
Figure 7.

Subchronic HAL treatment after METH on GAD 67 mRNA-labeled neuronal profiles in the SNr. Rats were administered METH (10 mg/kg 4 times, every 2 h) or 0.9% saline (1 ml/kg 4 times, every 2 h) on day 0 and then HAL (0.5 mg/kg) or vehicle (VEH) once daily on days 3–7. One group of rats received MK-801 (0.1 mg/kg, i.p.) daily, 15 min before each HAL during days 3–7. Rats were killed on day 14. Coronal sections were processed for in situ hybridization histochemistry with an 35S-labeled GAD67, followed by emulsion autoradiography. GAD67-labeled neurons were manually counted. The average of three consecutive slices is expressed as the percentage of saline–vehicle controls. Data shown are mean ± SEM. Representative, bright-field photomicrographs illustrating GAD67 mRNA expression in neurons in the SNr of SAL–VEH (A), SAL–HAL (B), METH–VEH (C), METH–HAL (D), and METH–MK-801–HAL (E). F, Counts of GAD67 neurons showed a significant decrease in the METH–HAL treatment group compared with the SAL–VEH group. Arrows indicate representative GAD67 mRNA-labeled cells. Scale bars: A–E, 50 μm. Significance was determined via a one-way ANOVA between the SAL–VEH, METH–HAL, and METH–MK-801–HAL groups; *p < 0.05 compared with SAL–VEH. n = 8 for SAL–VEH; n = 4 for SAL–HAL; n = 8 for METH–VEH; n = 8 for METH–HAL; n = 4 for METH–MK-801–HAL.
To investigate whether the decreases in NeuN-positive and GAD67 mRNA-expressing profiles represented cell death and not an underexpression of NeuN and GAD67 mRNA, we used a fluorescent histochemical approach that specifically stained DNA fragments. The rats that received subchronic HAL and METH had increased fluorescence labeling indicative of DNA fragmentation in the SNr (Fig. 8). In a separate group of rats, the NMDA receptor antagonist MK-801 blocked the increase in DNA fragmentation.
Figure 8.

Subchronic HAL treatment after METH on DNA fragmentation in the SNr. Rats were administered METH (10 mg/kg 4 times, every 2 h) or 0.9% saline (1 ml/kg 4 times, every 2 h) on day 0 and then HAL (0.5 mg/kg) or vehicle (VEH) once daily on days 3–7. One group received MK-801 (0.1 mg/kg, i.p.) daily, 15 min before each HAL on days 3–7. Rats were killed on day 14. Coronal sections were processed for fluorescence histochemistry for DNA fragment end labeling. Fluorescence was quantified on an arbitrary scale (0, no fluorescence; 1, low fluorescence; 2, intermediate fluorescence; 3, high fluorescence) and is presented in arbitrary units. Data shown are mean ± SEM. Representative fluorescence photos illustrating DNA fragment end labeling in the SNr are shown from rats treated with SAL–VEH (A), SAL–HAL (B), METH–VEH (C), METH–HAL (D), and METH–MK-801–HAL (E). F, Immunofluorescence intensity showed a significant increase in DNA fragment end labeling in the METH–HAL treatment group compared with the SAL–VEH control group. Scale bars, 200 μm. Significance was determined via Kruskal–Wallis one-way ANOVA on ranks followed by Dunn's post hoc test; *p < 0.05 compared with SAL–VEH. n = 6 for SAL–VEH; n = 5 for SAL–HAL; n = 5 for METH–VEH; n = 5 for METH–HAL; n = 5 for METH–MK-801–HAL.
Discussion
The present findings demonstrate that subchronic treatment with HAL after high doses of METH produces death of GABAergic neurons in the SNr through GLU and NMDA receptor activation but spares dopaminergic neurons in the SNc. These results are clinically important because HAL is commonly administered to human METH abusers for the treatment of METH-induced psychoses (Sato et al., 1983; Richards et al., 1998; Srisurapanont et al., 2001; Pittman, 2005).
The mechanisms mediating SNr cell death were defined by the collective results from several experiments. The increase in extracellular GLU within the SN after administration of the D2 antagonist haloperidol during multiple dosing with METH (Fig. 1) is presumably attributable to D2 antagonism on striatal GABAergic output neurons and on subthalamonigral GLU terminals in the SN (Hatzipetros and Yamamoto, 2006), the combination of which results in the disinhibition of GLU release from subthalamonigral terminals. These results also explain why METH alone, which increases DA release in the SN (Bustamante et al., 2002), does not increase GLU release acutely caused by inhibition by the D2 receptor in the SN (Zhang et al., 2001) (Fig. 1). Acute challenges with HAL alone increased extracellular GLU in the SN (Fig. 1), probably because of D2 receptor antagonism on striatal GABAergic output neurons rather than D2 antagonism locally within the SN (Hatzipetros and Yamamoto, 2006). Two days later, basal GLU concentrations remained elevated in METH-pretreated rats (Fig. 2). In contrast, the acute increases in GLU in the saline-pretreated group with or without HAL appear to be transient and presumably decline to predrug levels during the next 2 d (Fig. 2, inset). The prolonged elevation of GLU 2 d after METH exposure was further augmented in response to a HAL challenge (Fig. 2). Therefore, the persistent elevation in basal GLU levels after METH combined with the augmented GLU responses after five daily challenges with HAL could explain the cell loss at 14 d after METH exposure (Fig. 6).
The increased GLU concentration 2 d after METH exposure (Fig. 2, inset) is most likely the consequence of METH-induced decreases in DAergic tone in the striatum that persists for at least 12 h after METH (B. K. Yamamoto, unpublished findings). The diminished DAergic tone in the striatum at late time points after METH could disinhibit the basal ganglia indirect pathway to increase the activity of the subthalamonigral projection and increase GLU release in the SN. The enhanced elevations of basal GLU produced by HAL in METH-pretreated rats is most likely attributable to the D2-mediated disinhibition of GLU release from the indirect pathway via heteroreceptors on subthalamic axon terminals in the SN. Saline-pretreated rats were less responsive to a HAL challenge than METH-treated rats as evidenced by a significantly smaller increase in extracellular GLU (Fig. 2). It is unclear whether the presence of D2 receptors is restricted to subthalamonigral versus corticonigral glutamatergic afferents that preferentially innervate the SNr (Carter, 1982), but this finding is consistent with the observation that the D2 modulation of GLU release from subthalamonigral terminals is activity dependent (Hatzipetros and Yamamoto, 2006).
The consequence of the augmented and persistent glutamatergic responsiveness to HAL was evaluated by examining the possible loss of cells in the SN. METH alone, subchronic HAL alone, or subchronic HAL after METH did not alter DA tissue content in the SN when compared with vehicle controls (Fig. 3). Furthermore, these treatments did not alter TH immunostaining (Fig. 4) or the number of NeuN-positive profiles (Fig. 5) in the SNc and are consistent with previous studies that showed that METH does not damage DA cell bodies in the SNc (Ricaurte et al., 1982; Wilson et al., 1996; Harvey et al., 2000). In addition, METH–HAL did not change DA content or DA transporter immunoreactivity in the striatum (data not shown). Similar to the lack of changes in NeuN-positive profiles in the SNc, the number of NeuN-positive profiles in the SNr after METH alone or subchronic HAL alone were unchanged (Fig. 6). In marked contrast, METH–HAL decreased the number of NeuN-positive profiles in the middle portion along the rostrocaudal extent of the SNr (Fig. 6), a subregion of the SN that receives a robust glutamatergic innervation from the STN (Kita and Kitai, 1987) and contains a high density of neurons (Gonzalez-Hernandez and Rodriguez, 2000). In contrast, no changes were observed after METH–HAL in the more rostral and caudal portions of the SNr, areas with fewer GABA cells and less glutamatergic innervation than the middle sections. Thus, although METH or subchronic HAL alone did not alter the number of NeuN-positive profiles in the SNc or SNr along the rostrocaudal extent, the combined drug treatment selectively decreased NeuN-positive profiles in the middle portion of the SNr.
To ascertain the phenotype of the cells lost in the SNr, GAD67 mRNA expression was measured to identify GABA cells known to represent the majority of neurons in the reticulata (Chesselet et al., 1987; Smith et al., 1987). The number of neurons expressing GAD67 mRNA was significantly less in the METH–HAL group (Fig. 7), suggesting that the decrease in NeuN profiles is primarily attributable to a decrease in the number of GABA neurons. The decrease in the number of NeuN profiles, however, was slightly greater than the decrease in the number of GAD 67 mRNA-expressing profiles (34 vs 24%), suggesting that a small number of non-GABAergic neurons (presumably dopaminergic soma) in the SNr may also be lost after METH–HAL treatment. As with the NeuN staining, no changes in GAD67 mRNA-expressing cells were observed in the SNr after treatment with METH or HAL alone (Fig. 7). Despite the decreases in GAD67 mRNA-expressing and NeuN-positive profiles in the SNr of METH–HAL-treated rats, decreases in these markers do not unequivocally demonstrate cell death. Therefore, additional experiments were performed to determine more definitively whether cell death occurs in the SNr in response to the serial treatment with METH and HAL.
Additional studies assessed DNA fragmentation as an unbiased marker of cell death (Higuchi, 2003). METH–HAL treatment produced more intense fluorescence labeling in the SNr indicative of DNA fragmentation and cell death (Fig. 8). The magnitude of this increase appeared to be greater compared with the loss of GABA cells at the same time point, suggesting that ongoing DNA loss may be a precursor event to a potentially even greater GABA cell loss at later time points. Although this finding alone does not directly reveal the phenotype of the dying cells, it provides the first evidence that combined HAL and METH treatment causes neuronal cell loss and damage in the SNr that is absent in rats treated with METH or HAL alone.
To evaluate the mechanism underlying cell death in the SNr based on the elevated and persistent increase in extracellular GLU (Fig. 2, inset) and the presence of NMDA receptors in the SNr (Chatha et al., 2000), the effect of the NMDA receptor antagonist MK-801 was evaluated. Administration of MK-801 before each daily injection of HAL in rats previously exposed to METH blocked the increase in DNA fragmentation and the decrease in GAD67 mRNA-expressing cells (Figs. 7, 8). Because MK-801 was administered systemically, the localization of its effect in a select brain region cannot be determined. Regardless, the cell loss observed in the SNr after the combined treatment of METH and HAL appears to be mediated by GLU and NMDA receptors to produce excitotoxicity, DNA fragmentation, and a loss of GABA cells.
In conclusion, the current findings have several important implications with regard to the mechanism of METH action, the functional consequences of the activation of the subthalamonigral glutamatergic pathway, a novel toxicity associated with the frequent combined exposure to these drugs, and the therapeutic management of METH overdose and psychoses. In contrast to the evidence that increased extracellular GLU concentrations mediate the toxicity of METH to DA terminals in the striatum (Abekawa et al., 1994; Stephans and Yamamoto, 1994), the lack of an increase in GLU transmission in the SN attributable to the inhibitory effects of DA may explain the lack of DA cell death in SN to METH or HAL alone. Conversely, the findings of decreased inhibitory tone and disinhibition of glutamatergic transmission in the SNr produced by D2 receptor antagonism demonstrate a previously unrecognized, subregional, and selective toxic effect to the SN. The consequence of this aberrant modulation of the basal ganglia indirect pathway is evidenced by death of GABAergic cells in the SNr with sparing of DA cells in the SNc. Consequently, the acute therapeutic management of METH-induced psychoses with HAL may be contraindicated as a result of GABAergic cell death in the SNr. The potential loss GABAergic cells of the SNr may predispose such treated individuals to the development of hyperkinetic movement disorders and seizures (Deransart et al., 1998) rather than the traditional association of METH abuse with hypokinetic disorders such as Parkinson's disease. Along therapeutic lines for the treatment of METH overdose, consideration for the use of atypical antipsychotic medications with a lower D2 affinity profile appears warranted.
Footnotes
This work was supported by National Institutes of Health Grants DA07606 and DA16866 and a gift from Hitachi America. We thank Jennifer Amato for assistance with the cell counts in the substantia nigra.
References
- Abekawa T, Ohmori T, Koyama T. Effects of repeated administration of a high dose of methamphetamine on dopamine and glutamate release in rat striatum and nucleus accumbens. Brain Res. 1994;643:276–281. doi: 10.1016/0006-8993(94)90033-7. [DOI] [PubMed] [Google Scholar]
- Bevan MD, Bolam JP, Crossman AR. Convergent synaptic input from the neostriatum and the subthalamus onto identified nigrothalamic neurons in the rat. Eur J Neurosci. 1994;6:320–334. doi: 10.1111/j.1460-9568.1994.tb00275.x. [DOI] [PubMed] [Google Scholar]
- Bezard E, Gross CE. Compensatory mechanisms in experimental and human parkinsonism: towards a dynamic approach. Prog Neurobiol. 1998;55:93–116. doi: 10.1016/s0301-0082(98)00006-9. [DOI] [PubMed] [Google Scholar]
- Brunswick DJ, Benmansour S, Tejani-Butt SM, Hauptmann M. Effects of high-dose methamphetamine on monoamine uptake sites in rat brain measured by quantitative autoradiography. Synapse. 1992;11:287–293. doi: 10.1002/syn.890110404. [DOI] [PubMed] [Google Scholar]
- Bustamante D, You ZB, Castel MN, Johansson S, Goiny M, Terenius L, Hokfelt T, Herrera-Marschitz M. Effect of single and repeated methamphetamine treatment on neurotransmitter release in substantia nigra and neostriatum of the rat. J Neurochem. 2002;83:645–654. doi: 10.1046/j.1471-4159.2002.01171.x. [DOI] [PubMed] [Google Scholar]
- Carter CJ. Topographical distribution of possible glutamatergic pathways from the frontal cortex to the striatum and substantia nigra in rats. Neuropharmacology. 1982;21:379–383. doi: 10.1016/0028-3908(82)90019-3. [DOI] [PubMed] [Google Scholar]
- Chatha BT, Bernard V, Streit P, Bolam JP. Synaptic localization of ionotropic glutamate receptors in the rat substantia nigra. Neuroscience. 2000;101:1037–1051. doi: 10.1016/s0306-4522(00)00432-2. [DOI] [PubMed] [Google Scholar]
- Chesselet MF, Weiss L, Wuenschell C, Tobin AJ, Affolter HU. Comparative distribution of mRNAs for glutamic acid decarboxylase, tyrosine hydroxylase, and tachykinins in the basal ganglia: an in situ hybridization study in the rodent brain. J Comp Neurol. 1987;262:125–140. doi: 10.1002/cne.902620110. [DOI] [PubMed] [Google Scholar]
- Cretzmeyer M, Sarrazin MV, Huber DL, Block RI, Hall JA. Treatment of methamphetamine abuse: research findings and clinical directions. J Subst Abuse Treat. 2003;24:267–277. doi: 10.1016/s0740-5472(03)00028-x. [DOI] [PubMed] [Google Scholar]
- Deransart C, Vercueil L, Marescaux C, Depaulis A. The role of basal ganglia in the control of generalized absence seizures. Epilepsy Res. 1998;32:213–223. doi: 10.1016/s0920-1211(98)00053-9. [DOI] [PubMed] [Google Scholar]
- Derlet RW, Albertson TE, Rice P. The effect of haloperidol in cocaine and amphetamine intoxication. J Emerg Med. 1989;7:633–637. doi: 10.1016/0736-4679(89)90011-5. [DOI] [PubMed] [Google Scholar]
- Donzanti BA, Yamamoto BK. An improved and rapid HPLC-EC method for the isocratic separation of amino acid neurotransmitters from brain tissue and microdialysis perfusates. Life Sci. 1988;43:913–922. doi: 10.1016/0024-3205(88)90267-6. [DOI] [PubMed] [Google Scholar]
- Ellison G. Stimulant-induced psychosis, the dopamine theory of schizophrenia, and the habenula. Brain Res Brain Res Rev. 1994;19:223–239. doi: 10.1016/0165-0173(94)90012-4. [DOI] [PubMed] [Google Scholar]
- Gibb JW, Kogan FJ. Influence of dopamine synthesis on methamphetamine-induced changes in striatal and adrenal tyrosine hydroxylase activity. Naunyn Schmiedebergs Arch Pharmacol. 1979;310:185–187. doi: 10.1007/BF00500283. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Hernandez T, Rodriguez M. Compartmental organization and chemical profile of dopaminergic and GABAergic neurons in the substantia nigra of the rat. J Comp Neurol. 2000;421:107–135. doi: 10.1002/(sici)1096-9861(20000522)421:1<107::aid-cne7>3.3.co;2-6. [DOI] [PubMed] [Google Scholar]
- Harvey DC, Lacan G, Tanious SP, Melega WP. Recovery from methamphetamine induced long-term nigrostriatal dopaminergic deficits without substantia nigra cell loss. Brain Res. 2000;871:259–270. doi: 10.1016/s0006-8993(00)02439-2. [DOI] [PubMed] [Google Scholar]
- Hatzipetros T, Yamamoto BK. Dopaminergic and GABAergic modulation of glutamate release from rat subthalamic nucleus efferents to the substantia nigra. Brain Res. 2006;1076:60–67. doi: 10.1016/j.brainres.2006.01.015. [DOI] [PubMed] [Google Scholar]
- Higuchi Y. Chromosomal DNA fragmentation in apoptosis and necrosis induced by oxidative stress. Biochem Pharmacol. 2003;66:1527–1535. doi: 10.1016/s0006-2952(03)00508-2. [DOI] [PubMed] [Google Scholar]
- Kita H, Kitai ST. Efferent projections of the subthalamic nucleus in the rat: light and electron microscopic analysis with the PHA-L method. J Comp Neurol. 1987;260:435–452. doi: 10.1002/cne.902600309. [DOI] [PubMed] [Google Scholar]
- Lieberman JA, Kinon BJ, Loebel AD. Dopaminergic mechanisms in idiopathic and drug-induced psychoses. Schizophr Bull. 1990;16:97–110. doi: 10.1093/schbul/16.1.97. [DOI] [PubMed] [Google Scholar]
- Mark KA, Soghomonian JJ, Yamamoto BK. High-dose methamphetamine acutely activates the striatonigral pathway to increase striatal glutamate and mediate long-term dopamine toxicity. J Neurosci. 2004;24:11449–11456. doi: 10.1523/JNEUROSCI.3597-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meltzer LT, Christoffersen CL, Serpa KA. Modulation of dopamine neuronal activity by glutamate receptor subtypes. Neurosci Biobehav Rev. 1997;21:511–518. doi: 10.1016/s0149-7634(96)00030-9. [DOI] [PubMed] [Google Scholar]
- Nielsen KM, Soghomonian JJ. Normalization of glutamate decarboxylase gene expression in the entopeduncular nucleus of rats with a unilateral 6-hydroxydopamine lesion correlates with increased GABAergic input following intermittent but not continuous levodopa. Neuroscience. 2004;123:31–42. doi: 10.1016/j.neuroscience.2003.08.010. [DOI] [PubMed] [Google Scholar]
- Oertel WH, Mugnaini E. Immunocytochemical studies of GABAergic neurons in rat basal ganglia and their relations to other neuronal systems. Neurosci Lett. 1984;47:233–238. doi: 10.1016/0304-3940(84)90519-6. [DOI] [PubMed] [Google Scholar]
- Overton P, Clark D. N-methyl-d-aspartate increases the excitability of nigrostriatal dopamine terminals. Eur J Pharmacol. 1991;201:117–120. doi: 10.1016/0014-2999(91)90332-k. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. Sydney: Academic; 1986. The rat brain in stereotaxic coordinates. [DOI] [PubMed] [Google Scholar]
- Pittman HJ. Methamphetamine overdose. Nursing. 2005;35:88. doi: 10.1097/00152193-200504000-00053. [DOI] [PubMed] [Google Scholar]
- Ricaurte GA, Guillery RW, Seiden LS, Schuster CR, Moore RY. Dopamine nerve terminal degeneration produced by high doses of methylamphetamine in the rat brain. Brain Res. 1982;235:93–103. doi: 10.1016/0006-8993(82)90198-6. [DOI] [PubMed] [Google Scholar]
- Richards JR, Derlet RW, Duncan DR. Chemical restraint for the agitated patient in the emergency department: lorazepam versus droperidol. J Emerg Med. 1998;16:567–573. doi: 10.1016/s0736-4679(98)00045-6. [DOI] [PubMed] [Google Scholar]
- Sato M, Chen CC, Akiyama K, Otsuki S. Acute exacerbation of paranoid psychotic state after long-term abstinence in patients with previous methamphetamine psychosis. Biol Psychiatry. 1983;18:429–440. [PubMed] [Google Scholar]
- Sato M, Numachi Y, Hamamura T. Relapse of paranoid psychotic state in methamphetamine model of schizophrenia. Schizophr Bull. 1992;18:115–122. doi: 10.1093/schbul/18.1.115. [DOI] [PubMed] [Google Scholar]
- Schmidt CJ, Levin JA, Lovenberg W. In vitro and in vivo neurochemical effects of methylenedioxymethamphetamine on striatal monoaminergic systems in the rat brain. Biochem Pharmacol. 1987;36:747–755. doi: 10.1016/0006-2952(87)90729-5. [DOI] [PubMed] [Google Scholar]
- Smith Y, Charara A, Parent A. Synaptic innervation of midbrain dopaminergic neurons by glutamate-enriched terminals in the squirrel monkey. J Comp Neurol. 1996;364:231–253. doi: 10.1002/(SICI)1096-9861(19960108)364:2<231::AID-CNE4>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Smith Y, Parent A, Seguela P, Descarries L. Distribution of GABA-immunoreactive neurons in the basal ganglia of the squirrel monkey (Saimiri sciureus) J Comp Neurol. 1987;259:50–64. doi: 10.1002/cne.902590105. [DOI] [PubMed] [Google Scholar]
- Srisurapanont M, Jarusuraisin N, Kittirattanapaiboon P. Treatment for amphetamine withdrawal. Cochrane Database Syst Rev. 2001;4:CD003021. doi: 10.1002/14651858.CD003021. [DOI] [PubMed] [Google Scholar]
- Stephans S, Yamamoto B. Methamphetamines pretreatment and the vulnerability of the striatum to methamphetamine neurotoxicity. Neuroscience. 1996;72:593–600. doi: 10.1016/0306-4522(95)00587-0. [DOI] [PubMed] [Google Scholar]
- Stephans SE, Yamamoto BK. Methamphetamine-induced neurotoxicity: roles for glutamate and dopamine efflux. Synapse. 1994;17:203–209. doi: 10.1002/syn.890170310. [DOI] [PubMed] [Google Scholar]
- Thomas DM, Walker PD, Benjamins JA, Geddes TJ, Kuhn DM. Methamphetamine neurotoxicity in dopamine nerve endings of the striatum is associated with microglial activation. J Pharmacol Exp Ther. 2004;311:1–7. doi: 10.1124/jpet.104.070961. [DOI] [PubMed] [Google Scholar]
- Tse YC, Yung KK. Cellular expression of ionotropic glutamate receptor subunits in subpopulations of neurons in the rat substantia nigra pars reticulata. Brain Res. 2000;854:57–69. doi: 10.1016/s0006-8993(99)02292-1. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Chang L, Wang GJ, Fowler JS, Franceschi D, Sedler M, Gatley SJ, Miller E, Hitzemann R, Ding YS, Logan J. Loss of dopamine transporters in methamphetamine abusers recovers with protracted abstinence. J Neurosci. 2001;21:9414–9418. doi: 10.1523/JNEUROSCI.21-23-09414.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JM, Kalasinsky KS, Levey AI, Bergeron C, Reiber G, Anthony RM, Schmunk GA, Shannak K, Haycock JW, Kish SJ. Striatal dopamine nerve terminal markers in human, chronic methamphetamine users. Nat Med. 1996;2:699–703. doi: 10.1038/nm0696-699. [DOI] [PubMed] [Google Scholar]
- Yamamoto BK, Pehek EA. A neurochemical heterogeneity of the rat striatum as measured by in vivo electrochemistry and microdialysis. Brain Res. 1990;506:236–242. doi: 10.1016/0006-8993(90)91256-g. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Loonam TM, Noailles PA, Angulo JA. Comparison of cocaine- and methamphetamine-evoked dopamine and glutamate overflow in somatodendritic and terminal field regions of the rat brain during acute, chronic, and early withdrawal conditions. Ann NY Acad Sci. 2001;937:93–120. doi: 10.1111/j.1749-6632.2001.tb03560.x. [DOI] [PubMed] [Google Scholar]