

Visualizing the proliferation marker bromodeoxyuridine (BrdU) requires pretreatment of tissue, typically with dilute hydrochloric acid (HCl). We report here that pretreatment by steam heating of paraformaldehyde-fixed tissue sections covered with citrate buffer yields much brighter labeling of BrdU than HCl pretreatment, allows labeling with many antibodies greatly superior to HCl pretreatment, and allows concurrent high-quality labeling of nuclei with the DNA-binding dyes Hoechst, DAPI (4′,6-diamidino-2-phenylindole), and Syto24. Standard use of antigen retrieval by steamer can facilitate new insights into mechanisms regulating normal progenitor and tumor cell proliferation and novel understandings of protein expression through increased sensitivity of immunohistochemical analysis.
Introduction
A method commonly used to label cells in the S-phase of the cell cycle involves incorporation of the thymidine analog bromodeoxyuridine (BrdU) into replicating DNA and the subsequent immunohistochemical detection of the BrdU (Gratzner, 1982; Miller and Nowakowski, 1988). Administration of BrdU to living animals or addition of BrdU to culture medium can be used to determine relative proliferation rates, the length of the cell cycle, and the percentage of cells in the cell cycle (growth fraction). In BrdU-labeling experiments, it is often desirable to label various antigens in addition to BrdU; such additional labels might, for example, be used to determine whether the proliferating cells express antigens characteristic of a particular cell-type identity (Menezes and Luskin, 1994) and/or to determine total cell number to calculate the percentage of BrdU-labeled cells.
Antibody detection of BrdU incorporated into DNA requires pretreatment of the tissue or cells to expose the BrdU epitope. Commonly, this “antigen retrieval” is done by incubating the slides in warm 2.0 m hydrochloric acid (HCl). However, such treatment abolishes specific labeling by dyes binding to double-stranded DNA, such as Hoechst 33342, Syto 24, and 4′,6-diamidino-2-phenylindole (DAPI), markers that are commonly used for counting total cell number. Furthermore, this HCl pretreatment substantially reduces or abolishes specific labeling by many antibodies, thereby compromising determination of codistribution of BrdU incorporation with various proteins of interest.
We report here that, compared with HCl pretreatment, pretreatment by incubating fixed tissue with sodium citrate buffer heated to near 100°C in an ordinary rice/vegetable steamer results in brighter fluorescent labeling of cells that have incorporated BrdU and at the same time allows high-quality labeling of nuclear DNA and various additional antigens. Although similar protocols for preparation of tissue for BrdU labeling have been reported previously (Dover and Patel, 1994), others have found this type of pretreatment ineffective (Valero et al., 2005), and HCl pretreatment continues to be the standard. We discuss here critical parameters that we have identified for using hot citrate buffer pretreatment and describe a robust protocol for this method.
Materials and Methods
Equipment and reagents
The steamer unit used is described below. Chemicals used included the following: boric acid (catalog #BP168; Fisher, Pittsburgh, PA), BrdU (catalog #B5002; Sigma, St. Louis, MO), citric acid (catalog #C0759; Sigma), HCl (12.1 m; catalog #A144; Fisher), sodium chloride (NaCl; catalog #S271; Fisher), sodium hydroxide (NaOH; catalog #S320; Fisher), sodium phosphate dibasic anhydrous (Na2HPO4; catalog #S374; Fisher), sodium phosphate monobasic monohydrate (NaH2PO4·H2O; catalog #BP330; Fisher), sucrose (catalog #BP220; Fisher), sodium tetraborate decahydrate (catalog #B9876; Sigma), and Tris base (catalog #BP152; Fisher). Antibodies and DNA-binding dyes used are listed in Table 1.
Table 1.
Antibodies and DNA-binding dyes used
| Target (against) or DNA-binding dye | Comment on antibody or dye use/target/target function | Company | Catalog # | Host | mAb/pAb | Final conc (μg/ml) |
|---|---|---|---|---|---|---|
| BrdU | Proliferation marker | Abcam, Cambridge, UK | ab6326 [clone BU1/75 (ICR1)] | Rat | mAb | 2 μg/ml |
| PCNA | Proliferation marker | Santa Cruz Biotechnology, Santa Cruz, CA | sc-56 (clone PC10) | Mouse | mAb | 2 μg/ml |
| Phospho-histone H3 (ser10) | Proliferation marker | Millipore, Bedford, MA | 06-570 | Rabbit | pAb | 5 μg/ml |
| Pax6 | Transcription factor | Covance, Princeton, NJ | PRB-278P | Rabbit | pAb | 1:400 |
| p27Kip1 | Cell-cycle inhibitor | BD Biosciences, San Jose, CA | 610242 (clone 57) | Mouse | mAb | 1.25 μg/ml |
| S100 | Calcium-binding protein expressed in various neural cell types; cytosolic | Dako, Carpinteria, CA | Z0311 | Rabbit | pAb | 1:400 |
| Type III β-tubulin | Neuron-specific tubulin | Promega, Madison, WI | G712A | Mouse | mAb | 1.25 μg/ml |
| Hoechst 33342 | Binds double-stranded DNA; labels all nuclei | Sigma | B2261 | 1 μg/ml | ||
| DAPI | Same as Hoechst 33342 | Sigma | D8417 | 1 μg/ml | ||
| Syto 24 | Same as Hoechst 33342 | Invitrogen, Carlsbad, CA | S7559 | 50 nm | ||
| Mouse IgG | Secondary | Invitrogen | A11029 | Goat | 4 μg/ml | |
| Mouse IgG | Secondary | Invitrogen | A11031 | Goat | 4 μg/ml | |
| Rat IgG | Secondary | Invitrogen | A11077 | Goat | 2 μg/ml | |
| Rabbit IgG | Secondary | Invitrogen | A11008 | Goat | 4 μg/ml |
Primary and secondary antibodies and DNA-binding dyes used in this study are listed. ″Final conc″ is the concentration of the antibody in the solution in which the tissue was incubated. For antibodies for which the vendor stated the stock concentration, final concentration is given in micrograms per milliliter. When the stock concentration was unknown, the dilution is stated. In one experiment (shown in Fig. 1F), the BrdU antibody was used at a final concentration of 0.5 μg/ml; with this single exception, antibodies were used at the concentration shown in the Table. Note that although BrdU is listed in the table as a ″Proliferation Marker″ (and indeed its most common use is in studying proliferative characteristics of cell populations), DNA synthesis (and hence BrdU labeling) can occur in the absence of cell division [Nowakowski and Hayes (2000) and references therein]. mAb, Monoclonal antibody; pAb, polyclonal antibody; PCNA, proliferating cell nuclear antigen.
Buffers
Citric acid.
The standard buffer used in our protocol is 10 mm sodium citrate, pH 6.0, prepared by diluting a 1.0 m, pH 6.0 stock. The 1.0 m citric acid, pH 6.0 stock was prepared as follows: 48.03 g of citric acid was dissolved in 150 ml of water. Water was added to increase the volume to ≈180 ml. The pH was adjusted to 6.0 with 10.0 m NaOH (≈60 ml). Water was added to bring the final volume to 250 ml, the pH was rechecked, and an additional adjustment made, if necessary. For 10 mm sodium citrate, pH 6.0, the 1.0 m stock was diluted 1:100 with water. For experiments assessing the effect of citrate concentration on labeling intensity, we also prepared 100 mm sodium citrate, pH 6.0 by diluting the 1.0 m stock 1:10 with water. For citrate and other buffers used, the pH was not readjusted after dilution of the stock.
PBS.
The composition of the PBS buffer used for immunohistochemistry and also in some pretreatment experiments was 100 mm sodium phosphate and 150 mm NaCl, pH 7.4.
Tris buffer.
A 1.0 m Tris, pH 7.6 stock was prepared by adjusting the pH of Tris base with HCl. Working Tris buffer solutions were prepared by diluting the 1.0 m stock with water.
“Steamer/citrate” pretreatment of sections before immunohistochemistry
The steamer unit used was a food steamer (Oster 6.1 quart; model 5712; available through retail and internet vendors). The “upper steamer bowl” and “rice bowl” that came with the unit were not used. For slide incubation, a polypropylene tube rack (catalog #66023-530; VWR International, West Chester, PA) was placed inside the lower steamer bowl; for incubation, slides were set on this rack. With this configuration, up to eight slides (25 × 75 mm) can be incubated at one time. A thermometer was placed on the rack to allow continuous monitoring of the temperature to which the slides were exposed. After filling the steamer reservoir with clean water, the covered lower steaming bowl containing the tube rack was placed on the steamer base, cooking time was set to >45 min, and the “On” button was pressed to start heating. The temperature in the steamer bowl must be ≈99°C during the entire tissue incubation time; achieving this required preheating the steamer for ≈10–15 min. Slides were washed in PBS three times for 10 min and then placed on the rack in the steamer, and the tissue was covered with buffer solution (800 μl/slide) gently applied using a pipettor. (The standard buffer is 10 mm sodium citrate, pH 6.0, but other buffers were used for experiments shown in Fig. 3.) The lid was placed back on the steamer bowl, and the slides were steamed for 15 min. After the 15 min steaming, the rack was removed from the unit, and the slides, with the tissue still covered by citrate buffer, were allowed to cool at room temperature for 2 min. Immunohistochemical processing was then initiated beginning with a PBS wash of the slides followed by application of blocking solution as described below.
Figure 3.

Ionic strength plays a critical role in determining effectiveness of BrdU labeling when using the steamer/citrate pretreatment method. Parasagittal cryostat sections of P1 rat brain on glass slides were processed and imaged in parallel. All sections were pretreated using the standard steamer method, except that the sections were covered with different buffers, as indicated at the left of each row. A–C, Relative to the standard (10 mm, pH 6.0) citrate buffer pretreatment (A), sections pretreated in 10 mm sodium citrate, pH 6.0, augmented with sucrose to increase the osmolarity (B, C) gave similar labeling results. However, when sections were pretreated using a buffer with 100 mm sodium citrate (D), both BrdU and Pax6 labeling were much reduced. These results suggest pretreatment effectiveness is degraded by increased ionic strength but not by increased osmolarity unaccompanied by increased ionic strength. E, F, Consistent with this interpretation, pretreatment in a PBS solution with 100 mm sodium phosphate and 150 mm sodium chloride gave poor results (E), but good labeling was achieved when this buffer solution was diluted 1:10 (F). Pretreatment in a dilute Tris buffer solution at a higher pH than the citrate also gave good labeling of BrdU and Pax6 (G); however, labeling was much less intense in a 10-fold more concentrated solution of this buffer (data not shown). Hoechst labeling was less intense with the Tris buffer (G) but was entirely satisfactory with a somewhat longer exposure (600 ms; data not shown). In each image, solid lines demarcate the border with the lateral ventricle; dashed lines outline the boundary between the SVZa and adjacent neural tissue; within each row, the solid and dashed lines are identically positioned in each image. Scale bar: G (for A–G), 200 μm.
Alternative heating pretreatment methods
Glass staining dishes sometimes cracked when heated in a water bath or in the microwave; thus, slides were incubated in plastic Coplin jars (catalog #25457-200; VWR International) for both the water bath and microwave protocols. These jars were filled with buffer (60 ml), and for all equilibration and incubation periods, the top was screwed onto the jar. The buffer temperature was measured immediately before and after slide incubation by stirring the buffer with a thermometer. Before incubating the slides in hot buffer, the slides were washed in PBS three times for 10 min.
Water bath.
After equilibration of the buffer to the desired temperature, the slides were immersed in the buffer in the Coplin jar and incubated in the water bath for 15 min. At the end of the incubation, immunohistochemical processing was initiated beginning with a PBS wash of the slides followed by application of blocking solution as described below. For our water bath, when the thermostat was set to 100°C, the temperature of the equilibrated buffer was 97°C; when set to 95°C, the temperature was 95°C; when set to 90°C, the temperature was 90°C.
Microwave.
A 700 W Kenmore brand microwave oven (Model 721) was used. We designed the following “low-energy” and “high-energy” protocols for microwave heating of 60 ml of buffer in a single Coplin jar. (Adjustment of parameters may be required if two or more jars are heated simultaneously.) To prevent boiling of buffer, which could damage the tissue or dislodge it from the slide, we used intermittent rather than continuous heating.
Low-energy protocol.
The buffer was preheated in the microwave for 2 min 30 s at 30% power, which warmed the buffer to ≈96°C. Immediately after this preheating, the slides were immersed in the hot buffer. After 5 min (microwave off during this 5 min period), the solution (with immersed slides) was reheated for 30 s at 30% power. After another 5 min, the solution was again reheated for 30 s at 30% power. After 16 min (5 min, 30 s, 5 min, 30 s, and 5 min), the Coplin jar was removed from the microwave, and the solution (with slides still immersed) was allowed to cool for 20 min at room temperature. Immunohistochemical processing was then initiated beginning with a PBS wash of the slides and then blocking, as described below. During each 5 min cycle, the temperature declined from ≈96°C at the beginning of the cycle to ≈79°C at the end of the cycle.
High-energy protocol.
In this protocol, after preheating the buffer and immersing the slides, the buffer was reheated much more frequently than in the “low-energy protocol.” The buffer was preheated in the microwave for 2 min 30 s at 30% power, which warmed the buffer to ≈96°C. Immediately after this preheating, the slides were immersed in the hot buffer and subjected to a continuous succession of heating cycles in the microwave: 3 s on/1 s off at full power for 15 min (≈225 cycles of 4 s). Using this protocol, the buffer did not boil, but the temperature was maintained at ≈96°C throughout the 15 min incubation period. The Coplin jar was then removed from the microwave, and the solution (with slides still immersed) was allowed to cool for 20 min at room temperature. Immunohistochemical processing was subsequently initiated with a PBS wash of the slides and then blocking, as described below.
HCl pretreatment
A glass Coplin jar was filled with 2.0 m HCl, the cover was screwed on the jar, and the HCl was equilibrated to 40°C in a water bath. Slides were then incubated in the 40°C HCl for 20 min. At the end of this incubation, the slides were washed in 40 mm borate buffer two times for 15 min. This 40 mm borate buffer was prepared by dissolving 7.6 g of sodium tetraborate and 5.0 g of boric acid in 4 L of water and adjusting the pH to 8.5, which required ≈100 μl of 10.0 m NaOH/L buffer. After the borate buffer wash, immunohistochemical processing was initiated with a PBS wash of the slides followed by application of blocking solution, as described below.
BrdU injection and preparation of tissue
All animal procedures were approved by the Institutional Animal Care and Use Committee and the Emory Biosafety Committee of Emory University.
Rat pups (postnatal day 1) were given an intraperitoneal injection of BrdU (200 mg of BrdU per kilogram of body weight; stock is 10 mg/ml BrdU in 0.007 m NaOH solution). Three hours later, the rats were anesthetized (5 min in ice) and then perfused transcardially with PBS (5 min at ≈3 ml/min), followed by cold 4% paraformaldehyde in PBS (5 min at ≈3 ml/min). The brains were then removed and postfixed overnight by immersion in 4% paraformaldehyde in PBS at 4°C. After postfixation, the brains were cryoprotected by incubation overnight in 30% w/v sucrose in PBS (until they sank) and subsequently embedded in OCT compound (catalog #25608-930; VWR International), frozen with liquid nitrogen, and sagittally sectioned at 10 μm with a cryostat. The sections were collected on SuperFrost Plus slides (Fisher Scientific) and stored at −20°C until used.
Immunohistochemical methods and imaging
Before immunohistochemical processing, antigen retrieval was performed through pretreatment using the protocols described in detail above. Primary and secondary antibodies used are listed in Table 1. The sections were washed with PBS three times for 10 min and incubated for 1 h in blocking solution [2% v/v normal goat serum (Vector Laboratories, Burlingame, CA) and 0.3% v/v Triton X-100 in PBS]. The primary antibodies, diluted in blocking solution, were applied, and the sections were incubated with the primary antibodies for 12–18 h at 4°C. For double labeling, antibodies were applied simultaneously. The sections were next rinsed three times for 10 min in PBS and subsequently incubated with the appropriate secondary antibodies diluted in blocking solution for 1 h at room temperature. To label all cell nuclei, the fluorescent nuclear dye Hoechst 33342, DAPI, or Syto24 (Table 1) was included with the secondary antibody solution. After a final rinse three times for 10 min with PBS, the slides were coverslipped with VectaShield (Vector Laboratories). Sections were examined using a Zeiss (Oberkochen, Germany) Axioscope fluorescence microscope equipped with a QImaging (Burnaby, British Columbia, Canada) Retiga 1300 monochrome camera. The images shown in the figures were collected using a 10× Plan Neofluar lens [numerical aperture (NA), 0.30; Zeiss], except that for Figure 1A, a 5× Plan Neofluar lens (NA, 0.15) was used. The captured images were processed using Adobe Photoshop (Adobe Systems, San Jose, CA) and Canvas (ACD Systems International, Victoria, British Columbia, Canada). Within each figure, images showing labeling for the same antigen/target (for example, for BrdU), were all collected with the same exposure time and processed identically. The images accurately represent the visual impression of observers. When tissue from an animal not injected with BrdU was labeled using the BrdU antibody, no nuclear BrdU signal was observed after either “steamer/citrate” or HCl pretreatment (data not shown).
Figure 1.

Intensity of BrdU, Pax6, and DNA label is stronger in tissue pretreated by the steamer/citrate method compared with tissue pretreated with HCl. A, Parasagittal cryostat section of postnatal day 1 (P1) rat forebrain labeled with Hoechst 33342, which labels all cell nuclei. Images in the figures of this study show photomicrographs that include the SVZa as well as part of the adjacent RMS, lateral ventricle (LV), and overlying corpus callosum (for example, to the right of the dashed line in B–F). In Hoechst-labeled sections, the SVZa and RMS, regions of high cell density and therefore high nuclear density, appear quite bright compared with the bordering tissue, which has a much lower density of cells. In the neonate, neuronal progenitors migrate from the SVZa along the RMS to the olfactory bulb, in which they become interneurons. The cells in the SVZa/RMS are highly proliferative, which makes them useful regions for comparing BrdU-labeling techniques. This montage was assembled from images of the same steamer/citrate-pretreated section used for B; the box outlines the field shown in B. B1–D4, Parasagittal sections of P1 rat brain on glass slides that were pretreated by being covered with 10 mm sodium citrate buffer, pH 6.0, and heated in a steamer for 15 min (B1–B4), pretreated by immersion in 2.0 m HCl at 40°C for 20 min (C1–C4), or not pretreated (D1–D4). After pretreatment, all sections were processed and labeled in parallel using the same antibody solutions and procedures. BrdU is incorporated into replicating DNA, Pax6 is a transcription factor protein, and the nuclear dye Hoechst 33342 binds to double-stranded DNA. All three labels are expected to have a principally nuclear distribution. The three sections shown (B–D) are adjacent sections from a single rat brain; these sections have similar histological features and would therefore be expected to exhibit a similar pattern of labeling. For each type of pretreatment tested, a field dominated by the SVZa was sequentially imaged for BrdU (B1, C1, D1; 4 s exposure time), Pax6 (B2, C2, D2; 6 s exposure), and Hoechst (B3, C3, D3; 180 ms exposure). In the merged images (B4, C4, D4), BrdU label is red and Pax6 is green. Solid lines demarcate the border of the lateral ventricle, and dashed lines outline the boundary between the SVZa and adjacent neural tissue. Within each row, the solid and dashed lines are identically positioned in each image. It is noteworthy that the nonspecific rhodamine channel fluorescence was lower for sections pretreated with the steamer/citrate method than for sections pretreated with HCl. BrdU labeling is much brighter for the section that received steamer/citrate pretreatment than for the section pretreated with HCl (compare B1 with C1); no BrdU labeling is detected in the section that received no pretreatment. Pax6 labeling of the steamer/citrate-pretreated section (B2) is much brighter than for the HCl-pretreated (C2) and no pretreatment (D2) sections. Hoechst labeling is appreciably stronger for the steamer/citrate pretreatment (B3) than for no pretreatment (D3), and Hoechst labeling is absent from the HCl-pretreated section (C3). E, F, BrdU label of steamer/citrate-pretreated sections imaged with a shorter exposure time (2 s; E) than in B (4 s) or incubated with more dilute antibody (F). The image in E is the same field as in B1–B4; the image in F is from an adjacent section labeled using the BrdU antibody at a 1:2000 dilution (4-fold more dilute than in B). Note that despite incubation with more dilute antibody and a shorter exposure time, the BrdU label in the steamer/citrate-pretreated section (F) is still brighter than in the HCl-pretreated section (C1). As in B–D, solid lines demarcate the border with the lateral ventricle; dashed lines outline the boundary between the SVZa and adjacent neural tissue. G, Quantitative comparison of density of cells scored as BrdU(+) in steamer/citrate versus HCl-pretreated sections. The density of BrdU(+) cells in the SVZa of three animals was quantified in adjacent sections pretreated either with the steamer/citrate method or HCl. The density of cells scored BrdU(+) is similar, although optimal exposure time for imaging BrdU label in the HCl-pretreated section required an exposure three times longer than that for imaging the steamer/citrate-pretreated slide (3 vs 1 s at 40×). A, Anterior; D, dorsal; LV, lateral ventricle. Scale bars: A, 500 μm; D4 (for B1–F), 200 μm.
BrdU and Hoechst 33342 labeling for steamer/citrate versus HCl pretreatment were directly compared in nine experiments. Relative labeling intensity with all other protocol permutations and for all other antibodies were directly compared with the standard steamer/citrate pretreatment in at least two to three experiments
Quantification of BrdU(+) cells
The detection sensitivity for cells incorporating BrdU was compared between steamer/citrate and HCl-pretreated tissue as follows. For each of the three brains scored, adjacent sections with closely similar neuroanatomy were pretreated by the steamer/citrate or HCl protocols described above and then immunolabeled for BrdU and Pax6. Images were captured and processed as described above, except that a 20× Plan Neofluar lens (NA, 0.50) was used, and images were assembled into photomontages. Based on preliminary experiments, we adopted a longer exposure time for HCl-pretreated sections than for steamer/citrate-pretreated sections so that we could obtain similar BrdU labeling intensity. The boundaries of the rostral migratory stream (RMS) were ascertained by examination of the Pax6 labeling. An area including part of the anterior portion of the subventricular zone (SVZa) and vertical limb of the RMS was chosen for scoring. Each montage was masked, such that only a selected area was visible, and BrdU-labeled cells were scored by an observer blind to the pretreatment applied. The size of the counting area was determined using Canvas; this area varied from 0.14 to 0.38 mm2 between animals but varied ≤15% for the adjacent steamer/citrate- and HCl-pretreated sections of a single brain. From the count and area, the density of BrdU(+) cells was determined.
Results
After fixation, pretreatment of tissue before immunolabeling is required to visualize the commonly used proliferation marker BrdU. The standard pretreatment is incubation of the tissue in dilute HCl, but this pretreatment impedes coimmunolabeling for many other antigens and also blocks labeling by fluorescent DNA-binding dyes such as Hoechst 33342. We have developed a heat-induced epitope recovery method as an alternative to HCl pretreatment for BrdU immunolabeling and defined the conditions required for reproducible high-quality labeling using this method.
Pretreatment of fixed sections using a sodium citrate buffer and a steamer allows high-quality labeling of BrdU with concurrent labeling of other antigens and nuclear DNA
Incubating slides in a commercially available rice/vegetable steamer proved to be a convenient way of exposing tissue to temperatures ∼100°C. For experiments comparing the steamer/citrate method with HCl and other pretreatments, we intraperitoneally injected neonatal rats with BrdU and then 3 h later, perfused these rats with 4% paraformaldehyde fixative and cut 10 μm parasagittal cryostat sections of the forebrain that included the SVZa and the RMS (Fig. 1A). The neonatal SVZa and RMS are particularly suitable for these BrdU-labeling experiments because they have well defined borders (Luskin, 1993) and are comprised of a highly proliferative neuronal progenitor cell population (Menezes et al., 1995; Pencea and Luskin, 2003).
When BrdU labeling of tissue pretreated by our steamer/citrate method was compared with HCl-pretreated tissue, the intensity of the BrdU label was greater for the steamer/citrate-pretreated tissue (Fig. 1B–C). Exposing the BrdU epitope for antibody labeling is believed to require denaturing the double helix of genomic DNA strands, and as expected, no specific BrdU labeling was observed in tissue that received no pretreatment (Fig. 1D).
In the experiment shown in Figure 1B–D, the steamer/citrate, HCl, and no pretreatment protocols were also compared with respect to the quality of labeling by an antibody to the transcription factor Pax6 and to the fluorescent DNA dye Hoechst 33342. In studies ongoing in our laboratory, we have used anti-Pax6 to identify a subpopulation of neuronal progenitor cells in the SVZa and RMS. Pax6 labeling of sections pretreated by steamer/citrate or by HCl or not pretreated showed a similar pattern. The labeling was within the SVZa, stronger in the ventral than the dorsal part of the SVZa, and predominately nuclear labeling was observed. However, the intensity was much greater with the steamer/citrate pretreatment (Fig. 1B) than with the HCl method (Fig. 1C) and no pretreatment (Fig. 1D). Relative to HCl or no pretreatment, the steamer/citrate pretreatment method also substantially improved labeling with a number of other antibodies (Table 2).
Table 2.
Relative labeling intensity for antibodies as a function of pretreatment protocol
| Target (against) | Quality of labeling |
|||
|---|---|---|---|---|
| Steamer/citrate | No pretreatment | HCl | Microwave (low) | |
| BrdU | 4 | – | 2 | – |
| PCNA | 4 | 0–1 | 0–2 | 4 |
| Phospho-H3 | 4 | 0 | 1–2 | 4 |
| Pax6 | 4 | 2–3 | 2–3 | 4 |
| p27Kip1 | 4 | 0 | 0–1 | 3 |
| S100 | 4 | 3–4 | 2–3 | 4 |
| Type III β-tubulin | 4 | 3 | 2–3 | 4 |
| DAPI | 4 | 3–4 | – | 4 |
| Hoechst 33342 | 4 | 3 | – | 4 |
| Syto 24 | 4 | 4 | – | 4 |
4, Equal or almost equal to label intensity with citrate/steamer pretreatment; 3, distinctly weaker than citrate steamer, but still quite satisfactory; 2, quite weak relative to citrate/steamer, but still satisfactory; 1, very weak, but some specific label detectable; 0, no specific label observed (at 10×); –, no specific label detected or marked reduction in labeled cell number, even when viewed at high power. Labeling intensity relative to steamer/citrate pretreatment was subjectively scored by comparison of images collected using a 10× Neofluar lens and an exposure time optimized for the steamer/citrate pretreatment slide of the same experiment. Within each experiment, serial sections of a single brain were labeled; thus, little variability is expected between slides with respect to tissue quality and fixation. For each antibody, labeling intensity was scored for each pretreatment in at least two separate experiments. In every experiment, steamer/citrate was superior to HCl and no pretreatment, but there was some variability between experiments in relative labeling intensity for some antibodies. When the scores varied, the range is indicated. In one experiment, the labeling intensity for Pax6 and S100 was slightly brighter for slides pretreated with the microwave (low) protocol compared with the steamer/citrate protocol. In all other cases, the steamer/citrate protocol produced labeling brighter than or equal to the microwave (low) protocol. PCNA, Proliferating cell nuclear antigen.
Quite unexpectedly, the steamer/citrate-treated tissue also displayed strong labeling with the DNA dye Hoechst 33342 (Fig. 1B). This Hoechst dye binds strongly to double-stranded DNA but only weakly to single-stranded DNA (Drobyshev et al., 1999). The denaturation of the double-stranded genomic DNA resulting from HCl pretreatment entirely ablates specific labeling by Hoechst (Fig. 1C). However, for reasons not yet evident, the steamer/citrate pretreatment effects on the DNA result in both excellent exposure of BrdU epitopes and improved binding of Hoechst.
The BrdU signal remained stronger for steamer/citrate-pretreated tissue compared with HCl-pretreated tissue when the exposure (imaging) time for the steamer/citrate treated tissue was twofold shorter (Fig. 1, compare C1 and E), or the BrdU antibody in the primary antibody solution was fourfold less concentrated for the steamer/citrate than for the HCl-pretreated tissue (Fig. 1, compare C1 and F). When quantified, the density of cells scored as BrdU(+) for steamer/citrate-treated tissue was found to be very similar to the density of cells scored as BrdU(+) in an adjacent section pretreated with HCl (Fig. 1G). However, compared with steamer/citrate-pretreated tissue, for the HCl-pretreated tissue, a much longer exposure was required to provide clear visualization of the BrdU(+) cells for scoring.
The quality of immunolabeling of tissue pretreated with the steamer/citrate method was further evaluated by staining the sections with neuron-specific β-tubulin antibody to assess the cytoarchitecture (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) and whether neural cells known to be postmitotic, based on neuronal-specific nuclear protein (NeuN) labeling, were inappropriately also labeled by the BrdU antibody (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). The cytoarchitecture of neurons was preserved at least as well in steamer/citrate-pretreated sections as in HCl-pretreated sections, and although many NeuN(+) and many BrdU(+) cells were observed in sections immunostained with both antibodies, no double(+) cells were observed.
Temperature is a critical parameter
One previous study described successful BrdU labeling by microwaving slides in citrate buffer (Dover and Patel, 1994). However, another group reported that this method was ineffective in their hands (Valero et al., 2005). Before adopting the steamer, we had performed heat-induced epitope retrieval using a microwave oven to reveal p27Kip1 immunoreactivity (Li et al., 2005). Although the microwave pretreatment protocol used for p27Kip1 (referred to below as “low energy”) was ineffective in revealing BrdU, we serendipitously discovered that more intense microwave pretreatment (referred to below as “high energy”) did reveal BrdU. We hypothesized that a critical parameter might be the intensity of heat treatment and that the failure of Valero et al. (2005) to reproduce the results of Dover and Patel (1994) may have been attributable to differences in the amount of heating of the buffer.
To test this hypothesis, we compared labeling of adjacent slides pretreated using a number of different heating regimens (Fig. 2). For our steamer/citrate method, we always preheat the steamer. With preheating, we found the temperature within the steamer unit (measured with a thermometer placed on the slide rack) to be 99°C throughout the 15 min slide incubation period. Relative to steamer citrate pretreatment (Fig. 2A), pretreatment of slides immersed in citrate buffer heated using the microwave (high) protocol (Fig. 2C) or equilibrated to 97°C in a water bath (Fig. 2E) resulted in BrdU labeling almost as intense as steamer citrate pretreatment (for details of treatment protocols, see Materials and Methods). However, pretreatment by the microwave (low) protocol (Fig. 2D) gave no specific BrdU labeling, and pretreatment by incubation in 95°C citrate buffer (Fig. 2F) yielded BrdU labeling that was substantially less intense than that for the 99°C incubation [although still more intense than HCl (Fig. 2B)]. Pretreatment by incubation in 90°C buffer (Fig. 2G) yielded BrdU labeling that was much less bright than for the 95°C pretreated tissue (Fig. 2F). Collectively, these results indicate that for our tissue, and using the citrate buffer, BrdU labeling intensity falls off sharply as the pretreatment temperature decreases <99°C. However, it is noteworthy that all of the heating protocols (Fig. 2A,C–G) gave excellent Pax6 and Hoechst labeling relative to HCl pretreatment (Fig. 2B). The results of this experiment also suggest that the efficacy of microwave pretreatment is entirely caused by the heating of the buffer and unrelated to other effects of the microwave energy. Stated another way, various methods that achieve a buffer temperature of ∼100°C will give similar, successful results. Finally, our results are consistent with the speculation that the failure of Valero et al. (2005) to reproduce the findings of Dover and Patel (1994) is attributable to Dover and Patel having achieved a buffer temperature of ≈95°C or greater but Valero et al. having only achieved a buffer temperature of <90°C with their microwave treatment.
Figure 2.
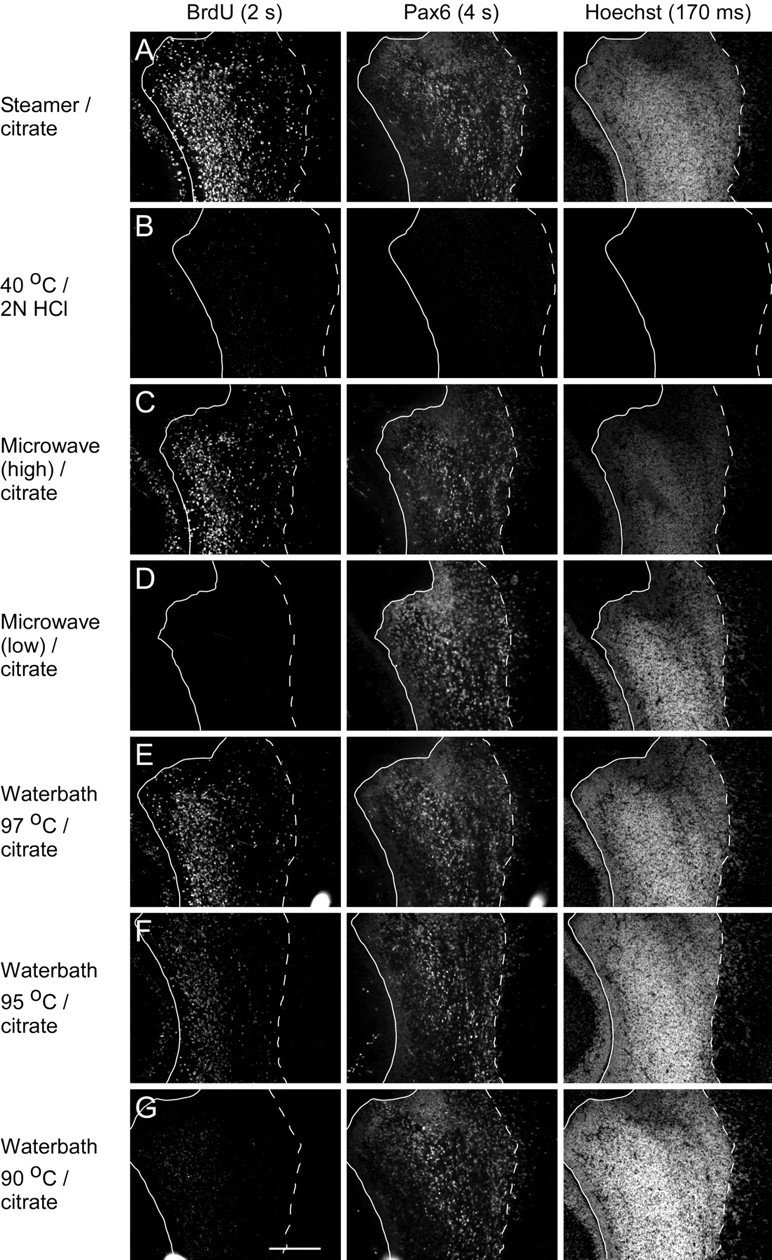
Effective immunolabeling of BrdU using heat pretreatments requires a pretreatment temperature of ∼100°C. A–G, Parasagittal cryostat sections of P1 rat forebrain on glass slides were pretreated as indicated at the left of each row (for details, see Materials and Methods) and then processed and imaged in parallel. Except for the HCl pretreatment (B), the pretreatment solution was 10 mm sodium citrate, pH 6.0, and the duration of the pretreatment was 15 min. Compared with steamer pretreatment (A), pretreatment by immersion in buffer maintained at ∼100°C by microwave heating (C) resulted in BrdU labeling of similar intensity, but a lower energy microwave pretreatment (D) produced no specific BrdU labeling. Pretreatment by immersion in buffer equilibrated to 97°C in a water bath (E), like high-intensity microwave treatment (C), yielded labeling almost as bright as steamer pretreatment. When sections were pretreated by immersion in 95°C buffer (F), BrdU labeling intensity was much reduced compared with steamer pretreatment, and immersion in buffer at 90°C was even weaker (G). For sections pretreated by incubation in buffer at 85°C, no specific BrdU labeling was detected, even with long exposure times (data not shown). All pretreatments, except HCl (B), produced strong Pax6 and Hoechst labeling. Exposure times in this figure (shown at the top of each column) were optimized for the steamer pretreatment. With these exposure times, BrdU and Pax6 labeling are barely detectable for the slide pretreated with HCl. For the HCl pretreatment, no labeling with Hoechst was detectable even with long exposure times. In each image, the solid lines demarcate the border of the lateral ventricle, and dashed lines outline the boundary between the SVZa and adjacent neural tissue. Within each row, the solid and dashed lines are identically positioned in each image. Scale bar: G (for A–G), 200 μm.
Buffer ionic strength is a critical parameter
Pilot experiments led us to suspect that for heat pretreatment to reveal BrdU epitopes, the ionic strength of the buffer in which the tissue is heated is a critical parameter determining labeling effectiveness. To test this, we compared a number of buffers to our standard 10 mm sodium citrate, pH 6.0 buffer. Adding sucrose to our standard buffer, which increases osmotic strength without changing ionic strength, had no effect on labeling intensity (Fig. 3, compare B, C with A). However, increasing the citrate concentration (Fig. 3D) markedly diminished labeling, not only for BrdU, but also for Pax6. Consistent with the results comparing different concentrations of citrate buffer, a diluted PBS solution (Fig. 3F) gave good labeling, but the full strength stock (Fig. 3E) did not. Finally, 10 mm Tris buffer (Fig. 3G) gave excellent labeling, but 100 mm Tris buffer (data not shown) did not. This indicates that other buffers and pH values can be used successfully so long as the ionic concentration of the pretreatment buffer solution is low. It is worth noting that pretreatment with 5 mm sodium citrate, pH 6.0, produced similar BrdU labeling intensity to 10 mm, but that steamer treatment of tissue covered with 1 mm sodium citrate, pH 6.0, and water (0 mm) resulted in severe tissue damage.
Effects on labeling quality of duration of heating in steamer
Based on pilot experiments, we had settled on 15 min incubation in the steamer for our “standard” steamer/citrate protocol. To determine the sensitivity of BrdU labeling effectiveness to variations in steamer time, we compared labeling of slides treated according to the standard protocol (15 min) to slides also pretreated in the standard buffer (10 mm sodium citrate, pH 6.0) but heated in the steamer for shorter (7.5 min) or longer (30 min) times. Label intensity was similar for 7.5, 15, and 30 min incubations. However, the morphology of the tissue heated for 30 min was less sharp than for the tissue heated for 7.5 and 15 min (data not shown). We continue to use 15 min, although this series of experiments suggests the possibility that a shorter incubation time might give reproducible, high-quality labeling.
Application of steamer/citrate method to tissue processed as “floating sections”
Although in our laboratory we currently use only cryostat sections for immunohistochemical analyses, we have developed also a protocol for steamer/citrate pretreatment of sections cut on a sliding microtome and handled as floating sections (see supplemental Fig. 3, available at www.jneurosci.org as supplemental material; legend includes Methods). With the current version of the floating sections protocol, the tissue handling is a bit cumbersome, and labeling is not quite as intense as we obtain with cryostat sections. Nonetheless, it works reasonably well and will be serviceable for situations in which double labeling is needed, but the second antibody is incompatible with HCl pretreatment. Furthermore, this basic protocol may serve as a starting point for others with a strong incentive to develop a higher throughput version.
Discussion
Major criteria for evaluating methods of preparing tissue for BrdU immunohistochemistry are (1) efficacy in revealing BrdU labeling, (2) compatibility with coimmunolabeling for other antigens, (3) compatibility with pan-nuclear labels such as Hoechst 33324, (4) the reproducibility of the procedure, and (5) the ease of the procedure. In this study, we have characterized a steamer/citrate pretreatment protocol as an alternative to the HCl pretreatment method commonly used to reveal BrdU. We find that steamer/citrate pretreatment is superior to HCl for pretreatment of paraformaldehyde-fixed sections with respect to each of these five criteria. Furthermore, compared with HCl pretreatment, steamer/citrate may reduce costs by allowing use of smaller amounts of expensive BrdU antibodies, as well as other antibodies.
Heat-induced epitope retrieval (HIER) methods have “revolutionized the immunohistochemical detection of antigens fixed in cross-linking fixatives (e.g., formaldehyde)” (Ramos-Vara, 2005). However, although the value of HIER methods for labeling a wide range of antigens has been well recognized, such methods have not replaced HCl as the generally accepted method of choice for preparing tissue for BrdU labeling. This may be because the conditions required for robustly obtaining results superior to those achieved with HCl pretreatment have not been defined. HCl pretreatment has been stated to cause “denaturation, dissolution, deamidation, and degradation of the majority of proteins, which has a profound damaging effect on the specimen, resulting in poor morphology and often precluding coimmunostaining for other antigens due to disruption of the epitopes for commonly used antibodies” (Tkatchenko, 2006). These undesirable characteristics have prompted a search for preferable alternatives. Proposed alternatives have included microwave treatment (Dover and Patel, 1994) and DNase treatment (Tkatchenko, 2006).
Dover and Patel (1994) reported that compared with HCl pretreatment, “microwave treatment” of tissue yielded strong BrdU labeling with lower antibody concentration and gave lower background staining when using a mouse monoclonal anti-BrdU antibody. The microwave treatment used in the Dover and Patel study was described as follows: “Samples were placed in a 400 ml bath of 0.1 m sodium citrate buffer, pH 6.0, which was placed in the microwave (700W) for between 5 and 20 min. Microwaving was performed without interruption.” The authors stated “The mechanism by which microwave treatment works is unclear.”; this and other comments in the text suggest that the authors attributed the efficacy to direct effects of the microwaves on the tissue rather than simply the effect of the microwaves in heating the buffer. Although at the time unaware of the Dover and Patel study, we began these experiments using the high energy microwave protocol with immersion of slides in a bath of sodium citrate buffer (as described in Materials and Methods and Results) but switched to using a steamer after noting beautiful examples of BrdU colabeling with other antibodies in a study by Siegenthaler and Miller (2005). In our experience, compared with immersion in hot buffer heated in a microwave oven, the steamer is more convenient and produces slightly superior results.
Although Dover and Patel (1994) and we achieved good results using microwave heat treatment of tissue, Valero et al. (2005), in a study designed to identify “optimized” methods for BrdU and PCNA (proliferating cell nuclear antigen) labeling in the rostral migratory stream of rodents, the same region that we studied, reported that the protocol of Dover and Patel (1994) “proved to be ineffective.” There are two likely reasons for the lack of success Valero et al. had with hot citrate pretreatment: (1) the buffer they used was 100 mm sodium citrate, which we found to yield much weaker staining than 10 mm sodium citrate (Fig. 3, compare A, D), and (2) Dover and Patel (1994) make no mention of the buffer temperature as a function of time for their microwave treatment. It is entirely possible that Valero et al. (2005) did not achieve the necessary ≥95°C temperature, or did not achieve this for sufficient time, to produce effective labeling. Valero et al. (2005) also used Bouin's fixative, which included 1% (w/v) picric acid and 5% (v/v) acetic acid in addition to 4% (w/v) paraformaldehyde, whereas our fixative was only 4% (w/v) paraformaldehyde. We cannot rule out the possibility that the steamer/citrate pretreatment is less effective for Bouin's fixed tissue than for straight formaldehyde-fixed tissue. As an aside, it is worth noting that in Denver (5280 feet/1609 m), water boils at ≈94.5°C; thus, in Denver and other high altitude locations, it may be necessary to use a pressurized device to attain buffer temperatures sufficient for optimal labeling.
In summary, use of the steamer/citrate protocol reported here is expected to facilitate the investigation of characteristics of proliferating cell populations by allowing colabeling with a wider range of antibodies than is possible with HCl pretreatment and simplifying determining total cell number in regions scored for BrdU-labeled cells. The procedure we have developed will be useful in many applications. For example, nuclear labeling in conjunction with BrdU labeling will allow a more precise determination (when using z-stacks) of whether a cell is a neuron or satellite glial cell in contentious cases. More generally, the steamer/citrate method may promote our understanding of neurodevelopment and lead to advances in the identification of mitotically active cancer cells.
Footnotes
Editor's Note: Toolboxes are intended to briefly highlight a new method or a resource of general use in neuroscience or to critically analyze existing approaches or methods. For more information, see http://www.jneurosci.org/misc/itoa.shtml.
This work was supported by National Institute on Deafness and Other Communication Disorders Grant RO1 DC03190 (M.B.L.). We thank Julie Siegenthaler and Dr. Michael Miller (State University of New York, Syracuse, NY) for discussions.
References
- Dover R, Patel K. Improved methodology for detecting bromodeoxyuridine in cultured cells and tissue sections by immunocytochemistry. Histochemistry. 1994;102:383–387. doi: 10.1007/BF00268909. [DOI] [PubMed] [Google Scholar]
- Drobyshev AL, Zasedatelev AS, Yershov GM, Mirzabekov AD. Massive parallel analysis of DNA-Hoechst 33258 binding specificity with a generic oligodeoxyribonucleotide microchip. Nucleic Acids Res. 1999;27:4100–4105. doi: 10.1093/nar/27.20.4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratzner HG. Monoclonal antibody to 5-bromo- and 5-iododeoxyuridine: a new reagent for detection of DNA replication. Science. 1982;218:474–475. doi: 10.1126/science.7123245. [DOI] [PubMed] [Google Scholar]
- Li X, Falls DL, Tang X, Zindy F, Roussel M, Luskin MB. Expression of P27KIP1 along the rostral migratory stream of wild-type and P19INK4D null mice. Soc Neurosci Abstr. 2005;31:143–23. [Google Scholar]
- Luskin MB. Restricted proliferation and migration of postnatally generated neurons derived from the forebrain subventricular zone. Neuron. 1993;11:173–189. doi: 10.1016/0896-6273(93)90281-u. [DOI] [PubMed] [Google Scholar]
- Menezes JR, Luskin MB. Expression of neuron-specific tubulin defines a novel population in the proliferative layers of the developing telencephalon. J Neurosci. 1994;14:5399–5416. doi: 10.1523/JNEUROSCI.14-09-05399.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menezes JR, Smith CM, Nelson KC, Luskin MB. The division of neuronal progenitor cells during migration in the neonatal mammalian forebrain. Mol Cell Neurosci. 1995;6:496–508. doi: 10.1006/mcne.1995.0002. [DOI] [PubMed] [Google Scholar]
- Miller MW, Nowakowski RS. Use of bromodeoxyuridine-immunohistochemistry to examine the proliferation, migration and time of origin of cells in the central nervous system. Brain Res. 1988;457:44–52. doi: 10.1016/0006-8993(88)90055-8. [DOI] [PubMed] [Google Scholar]
- Nowakowski RS, Hayes NL. New neurons: extraordinary evidence or extraordinary conclusion? Science. 2000;288:771. doi: 10.1126/science.288.5467.771a. [DOI] [PubMed] [Google Scholar]
- Pencea V, Luskin MB. Prenatal development of the rodent rostral migratory stream. J Comp Neurol. 2003;463:402–418. doi: 10.1002/cne.10746. [DOI] [PubMed] [Google Scholar]
- Ramos-Vara JA. Technical aspects of immunohistochemistry. Vet Pathol. 2005;42:405–426. doi: 10.1354/vp.42-4-405. [DOI] [PubMed] [Google Scholar]
- Siegenthaler JA, Miller MW. Transforming growth factor beta1 promotes cell cycle exit through the cyclin-dependent kinase inhibitor p21 in the developing cerebral cortex. J Neurosci. 2005;25:8627–8636. doi: 10.1523/JNEUROSCI.1876-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkatchenko AV. Whole-mount BrdU staining of proliferating cells by DNase treatment: application to postnatal mammalian retina. Biotechniques. 2006;40:29–30. 32. doi: 10.2144/000112094. [DOI] [PubMed] [Google Scholar]
- Valero J, Weruaga E, Murias AR, Recio JS, Alonso JR. Proliferation markers in the adult rodent brain: Bromodeoxyuridine and proliferating cell nuclear antigen. Brain Res Brain Res Protoc. 2005;15:127–134. doi: 10.1016/j.brainresprot.2005.06.001. [DOI] [PubMed] [Google Scholar]