

Abstract
Protein synthesis is required for persistent forms of synaptic plasticity, including long-term potentiation (LTP). A key regulator of LTP-related protein synthesis is mammalian target of rapamycin (mTOR), which is thought to modulate translational capacity by facilitating the synthesis of particular components of the protein synthesis machinery. Recently, extracellularly regulated kinase (ERK) also was shown to mediate plasticity-related translation, an effect that may involve regulation of the mTOR pathway. We studied the interaction between the mTOR and ERK pathways in hippocampal LTP induced at CA3–CA1 synapses by high-frequency synaptic stimulation (HFS). Within minutes after HFS, the expression of multiple translational proteins, the synthesis of which is under the control of mTOR, increased in area CA1 stratum radiatum. This upregulation was detected in pyramidal cell dendrites and was blocked by inhibitors of the ERK pathway. In addition, ERK mediated the stimulation of mTOR by HFS. The possibility that ERK regulates mTOR by acting at a component further upstream in the phosphatidylinositide 3-kinase (PI3K)–mTOR pathway was tested by probing the phosphorylation of p90-S6 kinase, phosphoinositide-dependent kinase 1 (PDK1), and Akt. ERK inhibitors blocked HFS-induced phosphorylation of all three proteins at sites implicated in the regulation of mTOR. Moreover, a component of basal and HFS-induced ERK activity depended on PI3K, indicating that mTOR-mediated protein synthesis in LTP requires coincident and mutually dependent activity in the PI3K and ERK pathways. The role of ERK in regulating PDK1 and Akt, with their extensive effects on cellular function, has important implications for the coordinated response of the neuron to LTP-inducing stimulation.
Keywords: mammalian target of rapamycin, mitogen-activated protein kinase, dendrites, ERK, hippocampus, LTP, protein synthesis, synaptic plasticity
Introduction
Long-term potentiation (LTP) is a widely studied mechanism for memory formation, and understanding the signaling network that maintains LTP will help to explain why memories persist and how they can be disrupted. At least two phases of LTP can be distinguished: a decremental phase (early LTP) that reflects only posttranslational processes, and a stable phase [late LTP (L-LTP)] that requires de novo protein synthesis (Frey et al., 1988; Huang and Kandel, 1994; Osten et al., 1996; Tsokas et al., 2005). The effect of synthesis inhibitors on LTP can be detected as soon as 15–20 min after induction, suggesting that plasticity-related proteins (PRPs) are produced rapidly and locally after stimulation, and the dendritic expression of some proteins is increased within 5 min after LTP induction (Ouyang et al., 1999; Tsokas et al., 2005). Furthermore, dendrites severed from their cell bodies are competent to express L-LTP (Kang and Schuman, 1996; Cracco et al., 2005; Tsokas et al., 2005; Vickers et al., 2005) (but see Frey et al., 1989), indicating that the local translation of dendritic mRNAs can supply the necessary PRPs.
LTP-related translation is controlled by coordinated mechanisms that regulate mRNA availability and the activity of translation factors (Wu et al., 1998; Scheetz et al., 2000; Kelleher et al., 2004; Atkins et al., 2005; Banko et al., 2005; Sutton and Schuman, 2006). Several studies have pointed to a key role for the protein kinase mammalian target of rapamycin (mTOR), a regulator of cell growth that facilitates translation of the terminal oligopyrimidine (TOP) class of mRNAs (Meyuhas and Hornstein, 2000; Tang et al., 2002; Cammalleri et al., 2003; Tsokas et al., 2005). TOP mRNAs encode components of the translational apparatus, including ribosomal proteins and elongation factors, and the mTOR-mediated synthesis of these components is thought to increase translational capacity, allowing cells to accommodate periods of increased demand for new proteins (Meyuhas and Hornstein, 2000). mTOR is present in the dendrites of hippocampal neurons, and the dendritic expression of the TOP mRNA-encoded elongation factor 1A (eEF1A) rapidly increases in an mTOR-dependent manner after LTP induction; moreover, L-LTP requires a period of mTOR activity (Tang et al., 2002; Cammalleri et al., 2003; Tsokas et al., 2005). These findings agree with the hypothesis that L-LTP-inducing stimulation generates dendritic translational capacity; however, eEF1A is the only TOP mRNA product known to accumulate in neurons undergoing L-LTP.
The canonical pathway for mTOR activation begins with phosphatidylinositide 3-kinase (PI3K), which generates phosphoinositides that recruit and colocalize phosphoinositide-dependent kinase 1 (PDK1) and Akt at the cell membrane. Recent evidence that extracellularly regulated kinase (ERK) also plays a role in the synaptically induced activation of mTOR is of particular interest, because ERK has been implicated in hippocampal LTP and hippocampus-based learning (English and Sweatt, 1997; Atkins et al., 1998; Kelleher et al., 2004).
Here, we examine the mechanism by which ERK regulates the PI3K–mTOR pathway in LTP and its consequences for the expression of multiple TOP mRNA-encoded proteins. Our results support the hypothesis that LTP-inducing stimulation increases dendritic translational capacity by generating new synthetic machinery, and that this process involves convergent effects of PI3K and ERK at the level of PDK1.
Materials and Methods
Electrophysiology.
Each male Sprague Dawley rat (6–8 weeks of age) was deeply anesthetized with halothane and decapitated. The brain was rapidly removed and placed in ice-cold artificial CSF (ACSF) containing the following (in mm): 118 NaCl, 3.5 KCl, 2.5 CaCl2, 1.3 MgSO4, 1.25 NaH2PO4, 24 NaHCO3, and 15 glucose, bubbled with 95% O2/5% CO2. The hippocampus was then quickly dissected out, and 500-μm-thick transverse slices were made on a tissue chopper in a cold room (4°C). The slices were maintained in an interface chamber (ACSF and humidified 95% O2/5% CO2 atmosphere) at room temperature for at least 2 h before removal for electrophysiological recording. Slices were transferred to a submersion chamber preheated to 30–32°C, where they were superfused on a nylon mesh with ACSF. Monophasic, constant-current stimuli (100 μs) were delivered with a bipolar stainless steel electrode placed in stratum radiatum of the CA3 region, and the field EPSP (fEPSP) was recorded in the stratum radiatum of the CA1 region with electrodes filled with ACSF (R e, 2–4 MΩ). The fEPSP was monitored by delivering stimuli at 0.033 Hz, the signal was low-pass filtered at 3 kHz and digitized at 20 kHz, and waveforms were collected and analyzed using either an Axobasic routine or pClamp 9 (Molecular Devices, Foster City, CA). All slices used in the experiments had spike thresholds ≥2.5 mV. LTP was induced by two 1-s-long trains of 100 Hz stimulation [high-frequency synaptic stimulation (HFS)], separated by 20 s, using a stimulus intensity sufficient to evoke 75% of the spike threshold. In all experiments, HFS was delivered at least 30 min after transfer of the slices into the recording chamber, when the basal EPSP had been stable for at least 20 min. Each experimental slice was paired with a sham-stimulated control, which received only test stimuli (0.033 Hz). Drug preincubation was performed at room temperature in submersion maintenance chambers containing ACSF saturated with 95% O2/5% CO2. All drugs, which were prepared as stock solutions in DMSO (final concentration, 0.1%), were applied for 60 min before HFS and for the remainder of the recording (5–30 min after HFS, as indicated).
Western immunoblotting.
Slices were removed from the recording chamber and immediately frozen on glass on dry ice. The CA1 region, or in some experiments the isolated stratum radiatum of CA1, was excised in a cold room (4°C) under a dissection microscope. Samples were then transferred to cold microcentrifuge tubes and stored at −80°C for no more than 3 d before assaying. Each excised region was homogenized in 35–50 μl of ice-cold lysis buffer [in mm (unless indicated otherwise): 25 Tris-HCl, pH 7.4, 150 NaCl, 6 MgCl2, 2 EDTA, 1.25% NP40, 0.125% SDS, 0.625% Na deoxycholate, 4 p-nitrophenyl phosphate, 25 Na fluoride, 2 Na pyrophosphate, 20 DTT, 10 β-glycerophosphate, 1 μm okadaic acid (phosphatase inhibitor cocktail I and II, 2 and 1%, respectively; Calbiochem, La Jolla, CA), 1 phenylmethylsulfonyl fluoride (PMSF), 20 μg/ml leupeptin, and 4 μg/ml aprotinin]. Protein determination was performed using Bio-Rad (Hercules, CA) RC-DC Protein Assay kit. Appropriate volumes of 4× NuPage LDS Sample Buffer (Invitrogen, Carlsbad, CA) and β-mercaptoethanol were added to the homogenates, and samples were boiled for 5 min. Samples were loaded on 8–10% SDS-PAGE gel and resolved by standard electrophoresis. The gels were then transferred onto nitrocellulose membranes (0.2 μm pore size) at 4°C. The membranes were blocked for at least 30 min at room temperature with blocking buffer (BB) [5% nonfat dry milk in TBS containing 0.1% Tween 20 (TBS-T)] and then probed overnight at 4°C using primary antibodies dissolved in BB. Antibodies used for Western blotting were as follows (all were obtained from Cell Signaling Technology, Beverly, MA, unless otherwise indicated): phospho(S218/S222)-MEK (mitogen-activated protein kinase kinase), phospho(T202/Y204)-ERK, phospho(S380)-p90-S6 kinase (RSK), phospho(S241)-PDK1, phospho(T308)-Akt (Upstate Biotechnology, Lake Placid NY), phospho(S2448)-mTOR, phospho(T37/T46)-4EBP1, phospho(Thr421/Ser424)-p70S6K, phospho(Thr389)-p70S6K, eEF1A (Upstate Biotechnology), eEF2, ribosomal protein S6 (rpS6), poly-A binding protein 1 (PABP1), and actin (Sigma-Aldrich, St. Louis, MO).
After washing in TBS-T (three washes, 5 min each), the membranes were incubated with horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG (1:5000; Pierce Biotechnology, Rockford, IL), and proteins were visualized by chemiluminescence (ECL Western Blotting Analysis System; GE Healthcare, Arlington Heights, IL). Densitometric analysis of the bands was performed using NIH Image, and values were normalized to actin. t tests were used to compare samples from experimental slices to their respective controls, and summary data are presented as group means with SE bars.
Immunohistochemistry and confocal microscopy.
Immediately after recording, 500 μm slices were placed in ice-cold 4% paraformaldehyde/0.1% glutaraldehyde in PBS, pH 7.4, and fixed overnight. The slices were then washed with PBS and cut into 40 μm sections using a Leica VT 1000S vibratome. Free-floating sections were blocked with 10% normal goat serum, 1% BSA, and 0.1% Na azide in PBS. The sections then were incubated overnight at 4°C with primary antibody in 1% BSA (in some experiments, two antibodies from different species were incubated together; otherwise, serial immunohistochemistry was performed). Antibodies used for immunohistochemistry included: phospho(Thr421/Ser424)-p70S6K (Santa Cruz Biotechnology, Santa Cruz, CA), phospho(Thr389)-p70S6K (Cell Signaling Technology, Beverly, MA), eEF1A (Upstate Biotechnology), rpS6 (Cell Signaling Technology), PABP (gift from J. Brosius, University of Münster, Münster, Germany), and β-tubulin (Sigma-Aldrich).
Fixed tissue sections were probed with primary antibodies, including β-tubulin to visualize dendrites. After washing in PBS, sections were incubated in 1% BSA with secondary antibodies complexed to either Alexa Fluor 568 or Alexa Fluor 488 (both used at 1:250; Invitrogen). Where multiple monoclonal primary antibodies were used, the slices were washed thoroughly after application of the secondary antibody and sequentially incubated with the next primary antibody. After extensive washing, the sections were mounted and imaged using a Zeiss LSM meta-510 confocal microscope at a scanning depth of 1.9 μm (200×) or 0.9 μm (400×). Alexa Fluor 488 immunofluorescence was detected with an ArKr 488/568 laser set at 505–545 nm bandpass emission. The same laser was used with a 560–615 nm long-pass emission filter to visualize Alexa Fluor 568. All parameters (pinhole, contrast, and brightness) were held constant for all sections from the same experiment. For double-label experiments, scans at different wavelengths were digitally merged. Multiple overlapping fields within the same focal plane were imaged and assembled to construct composites of the CA1 region.
Immunoprecipitation and mTOR kinase assay.
Samples of three pooled CA1 regions were lysed as described previously (Tsokas et al., 2005). The lysates were preincubated for 30 min at 4°C with protein A agarose beads (Roche Products, Welwyn Garden City, UK) to absorb nonspecific binding. After spinning down the beads, antibodies against total mTOR (Upstate Biotechnology) was added to the supernatant and incubated for at least 3 h at 4°C. Next, protein A agarose beads were added and incubated with samples for at least 3 h at 4°C. Immunocomplexes were then spun down and washed five times with ice-cold lysis buffer. Kinase reactions were started by adding 0.9 μg of recombinant human 4EBP1 (gift from N. Sonenberg, McGill University, Montreal, Quebec, Canada), dissolved in kinase assay buffer that included the following (in mm): 50 HEPES, 10 MgCl2, 1 Na fluoride, 2 Na pyrophosphate, 10 β-glycerophosphate, phosphatase inhibitor cocktail I and II (2 and 1%, respectively; Calbiochem), 1 PMSF, 20 μg/ml leupeptin, and 4 μg/ml aprotinin, 1 DTT, and 2 ATP. The tubes were then placed in a 30°C water bath and vortexed every few minutes, and the reaction was stopped after 20 min by adding 20 μl of 4× NuPAGE LDS-PAGE sample buffer (Invitrogen) and heating to 100°C for 5 min to denature the protein. The samples were analyzed by SDS-PAGE and immunoblotting with antibodies against phospho(S65)-4EBP (Cell Signaling Technology).
Results
Induction of late LTP increases the expression of TOP mRNA-encoded proteins in a rapamycin- and anisomycin-sensitive manner
To evaluate whether the hypothesis that increased translational capacity plays a role in LTP, we first tested whether the induction of L-LTP causes a general increase in the expression of TOP mRNA products, as established previously for eEF1A (Tsokas et al., 2005). The proteins that we studied were eEF2, rpS6, and PABP, all of which have been implicated in the response of neurons to plasticity-inducing stimulation (Marin et al., 1997; Wu and Cline, 1998; Scheetz et al., 2000; Huang et al., 2002; Kelleher et al., 2004; Takei et al., 2004; Atkins et al., 2005; Alarcon et al., 2006). Within 30 min after late L-LTP-inducing HFS, immunoreactivity for all of these proteins increased twofold to threefold in homogenates of excised area CA1 (Fig. 1 A). To confirm that these changes required de novo synthesis and were mediated by mTOR, as previously shown for eEF1A, we delivered HFS in the presence of the translation inhibitor anisomycin or the mTOR inhibitor rapamycin. Under these conditions, HFS failed to increase the expression of eEF2, rpS6, or PABP (Fig. 1 A). In contrast, the basal levels of these proteins were not affected by anisomycin or rapamycin (Fig. 1 B), indicating that the inhibitors prevented the de novo translation of the TOP mRNAs. Thus, the induction of L-LTP resulted in a coordinate and mTOR-mediated synthesis of translational machinery, consistent with the proposed role of mTOR in regulating translational capacity.
Figure 1.

HFS induces a general and mTOR-dependent increase in the expression of TOP mRNA-encoded proteins present in dendrites. A, In lysates of CA1 regions from slices frozen 30 min after HFS, immunoreactivity for eEF2 (left), rpS6 (middle), and PABP (right) were elevated relative to controls (Con). All of these increases were mediated by de novo protein synthesis through the mTOR pathway, as shown by their sensitivity to anisomycin (Aniso; 10 μm) and rapamycin (Rapa; 1 μm), respectively. Immunoreactivity, here and in subsequent figures, is expressed relative to sham-stimulated controls. Representative immunoblots are shown above. Asterisks indicate p < 0.05 versus controls; all values of n ≥ 4. B, Anisomycin (10 μm) and rapamycin (1 μm) did not affect the basal expression of eEF2, rpS6, or PABP1 (all values of n = 6 for anisomycin and 4 for rapamycin). C, HFS-induced increases in TOP mRNA products were pronounced in CA1 stratum radiatum. Slices had been frozen 5 min after the delivery of HFS. For each of the three proteins, the increase in immunoreactivity after stimulation was considerably stronger than that seen in complete CA1 regions (compare with A). Asterisks indicate p < 0.05 versus controls; all values of n ≥ 3. D, The dendritic expression of multiple TOP mRNA-encoded proteins increases rapidly after HFS. In area CA1 of slices fixed 5 min after HFS, more intense immunoreactivity for rpS6, PABP, and eEF1A was observed in the apical dendrites and cell bodies of pyramidal neurons in stimulated slices (middle panels) than in controls (left panels), and these effects were blocked by rapamycin (right panels). s.o., s.p., and s.r. indicate strata oriens, pyramidale, and radiatum, respectively. The images are representative of at least four experiments for each condition and antibody. Scale bar, 100 μm.
Expression of TOP mRNA-encoded proteins rapidly increases in the dendrites after HFS
We and others have shown that translation-dependent LTP can be detected within 20 min after HFS (Osten et al., 1996; Kelleher et al., 2004; Tsokas et al., 2005). If the synthesis of LTP-related proteins depends on an mTOR-mediated boost in translational capacity, then increases in the expression of TOP mRNA-encoded proteins should generally be apparent soon after stimulation. We previously reported that eEF1A is upregulated within 5 min after HFS, indicating that increased translational capacity could comprise an early event in the process leading to the synthesis of LTP-related proteins (Tsokas et al., 2005). To determine whether the expression of eEF2, rpS6, and PABP also increases rapidly in stimulated CA1 dendrites, we first performed immunoblots on homogenates of stratum radiatum that had been microdissected from slices frozen 5 min after strong HFS. This excision was conservative, sacrificing some proximal stratum radiatum in the interest of avoiding contamination from pyramidal cell bodies. Homogenates from stimulated slices showed increases in eEF2, rpS6, and PABP immunoreactivity that were considerably more pronounced than those obtained in immunoblots of complete CA1 regions (Fig. 1, compare C and A). The mean eEF2 increase in stratum radiatum was 3.0-fold higher than the increase at 30 min in CA1; for rpS6, 9.1-fold higher; and for PABP, 3.5-fold higher. The stronger upregulation in stratum radiatum suggests that the major component of the increase in protein expression was contributed by the apical dendrites of CA1 neurons. Moreover, the short latency of the increases in eEF2, rpS6, and PABP lends support to the hypothesis that a rapid increase in translational capacity contributes to L-LTP.
To further investigate the cellular location of TOP mRNA translation in LTP, we determined the distribution of the expression of rpS6 and PABP after strong HFS, using immunohistochemical methods. In slices stimulated with HFS and fixed 5 min later, immunoreactivity for both TOP mRNA-encoded proteins was intensified in well-defined dendrites of stratum radiatum relative to slices that had received control stimulation (Fig. 1 D). The enhancement in rpS6 and PABP immunoreactivity extended throughout the proximal-distal extent of the apical dendrites. HFS also increased the expression of both proteins in the cell bodies, where there was substantial immunoreactivity even in control slices, and diffusely in the dendrites of stratum oriens. Thus, the HFS-induced expression of rpS6 and PABP showed a somatodendritic distribution at 5 min that was similar to that reported previously for eEF1A and for phospho-p70S6K (Tsokas et al., 2005) (Figs. 1 D, bottom, 3 B,C). Considering the sparse innervation of the pyramidal cell bodies by excitatory synapses (Megias et al., 2001), the increased somatic expression of TOP mRNA-encoded proteins likely reflects retrograde signaling mediated by active conductances or by transport of these proteins or an upstream signaling component.
Figure 3.

HFS-induced phosphorylation of the mTOR substrate p70S6K is ERK dependent. A, In CA1 regions from slices frozen 30 min after HFS, p70S6K was hyperphosphorylated at the mTOR-dependent site T389 (left) as well as the putative ERK-dependent site T421/S424. Phosphorylation of both sites was blocked by the MEK inhibitor U0126 (20 μm), whereas 1 μm rapamycin (Rapa) blocked phosphorylation only at T389. The asterisks indicate p ≤ 0.05 versus controls (Con), with all group values of n ≥ 5. B, After HFS (middle panel), immunoreactivity for phospho(T389)-p70S6K was elevated in the apical dendrites and cell bodies of CA1 pyramidal neurons, and diffusely in stratum oriens, compared with vehicle-treated control (Veh) (top panel). Preincubation with 20 μm U0126 (bottom panel) prevented phosphorylation at T389. Scale bar, 150 μm. Images are representative of three experiments. C, HFS increased the immunoreactivity for phospho(T421/S424)-p70S6K (left panels) and for eEF1A (right panels) in dendrites and cell bodies of CA1 pyramidal neurons. The increase in T424/S421-p70S6K phosphorylation was sensitive to U0126 (20 μm) but not to rapamycin (Rapa; 1 μm), whereas both inhibitors prevented the HFS-induced rise in eEF1A expression (bottom two images). These images are representative of three independent experiments. Scale bar, 100 μm.
To confirm that the HFS-induced dendritic expression of rpS6 and PABP expression depended on activity in the mTOR pathway, we challenged these effects with rapamycin. In agreement with our immunoblot data, rapamycin blocked the somatodendritic increases in rpS6 and PABP immunoreactivity after strong HFS (Fig. 1 D, right panels). Thus, the mTOR pathway mediates a rapid and general increase in the dendritic expression of TOP mRNA-encoded components of the translational machinery, consistent with previous reports that a protein synthesis-dependent component of LTP emerges within 20 min after HFS (Osten et al., 1996; Tsokas et al., 2005).
ERK mediates the HFS-induced expression of TOP mRNA-encoded proteins
In addition to being a TOP mRNA product, rpS6 is subject to phosphorylation by the mTOR effector p70S6K. Kelleher et al. (2004) found that HFS increased rpS6 phosphorylation at an mTOR-dependent site, and that this effect required an intact ERK pathway, suggesting that ERK regulates the mTOR pathway in LTP. We therefore tested whether inhibitors of the ERK pathway can block the HFS-induced increases in the expression of the TOP mRNA products eEF1A, eEF2, rpS6, and PABP. Immunoblots of CA1 homogenates showed that the ERK kinase (MEK) inhibitors PD98059 and U0126 prevented HFS-induced changes in the expression of all four proteins (Fig. 2 A). This dependence on ERK was not caused by a tonic effect on the expression of these proteins, the basal levels of which were unaffected by ERK inhibition (Fig. S1, available at www.jneurosci.org as supplemental material). We also tested the ability of ERK inhibitors to prevent L-LTP under our conditions and found that synaptic potentiation returned to baseline within 2 h after HFS delivered in the presence of U0126 or PD98059 (Fig. 2 B), in agreement with other studies showing that ERK activity is required for LTP induction (English and Sweatt, 1997; Kelleher et al., 2004). This time course for the ERK-dependent component of LTP is similar to that seen when LTP was induced in the presence of rapamycin or anisomycin, using this HFS protocol or a similar one (Osten et al., 1996; Tsokas et al., 2005). In agreement with Kelleher et al. (2004), these data suggest that ERK controls LTP-related protein synthesis by regulating the mTOR pathway.
Figure 2.
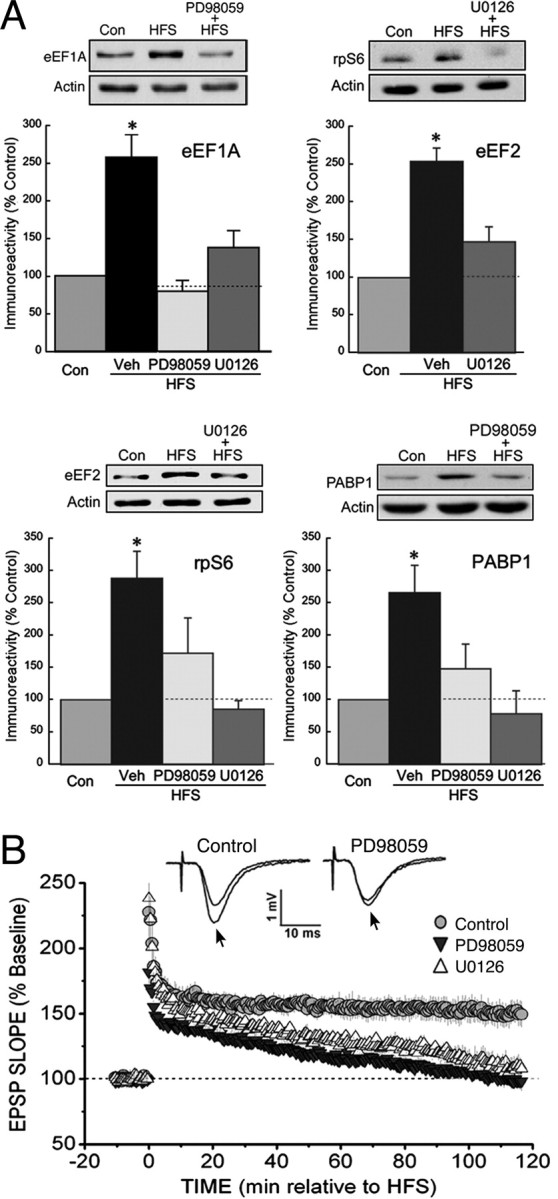
ERK activity is required for the upregulation of translational proteins in LTP. A, Immunoblots of CA1 lysates, from slices frozen 30 min after stimulation, showed that the MEK inhibitors PD98059 (50 μm) and U0126 (20 μm) prevented the HFS-induced increases in the expression of the TOP mRNA products eEF1A, eEF2, rpS6, and PABP1. The asterisks indicate p < 0.05 compared with control (Con), and sample immunoblots are shown above each summary graph. All group values of n were ≥6. B, In the presence of an MEK inhibitor, HFS produced only a decremental potentiation. The rate of decay for slices treated with 50 μm PD98059 or 20 μm U0126 (both values of n = 5) was similar to that observed previously when protein synthesis or mTOR activity was blocked (Tsokas et al., 2005), whereas LTP was stable for 2 h in vehicle-treated controls (Veh) (n = 5). The traces show superimposed fEPSPs recorded during the baseline period and 2 h after HFS (arrows) in a vehicle-treated control slice (left) and a slice treated with PD98059 (right).
ERK activity is required for the HFS-induced phosphorylation of multiple residues in p70S6K
To establish that ERK does in fact mediate the activation of mTOR in LTP, and to elucidate the nature of the interaction between the ERK and mTOR pathways, we began by investigating whether ERK mediates the HFS-induced phosphorylation of p70S6K, which may regulate TOP mRNA translation (Jefferies et al., 1997) (but see Stolovich et al., 2002). There are at least eight phosphorylatable sites in p70S6K, and the activation of this enzyme involves the sequential phosphorylation of residues by multiple protein kinases, including mTOR (Pullen and Thomas, 1997). Among the mTOR-dependent sites is T389 in the hydrophobic linker region. Phosphorylation of this site is often used to indicate p70S6K activation, and we have shown previously that HFS increases phospho-T389 immunoreactivity in CA1 dendrites (Tsokas et al., 2005). The phosphorylation of T389, in turn, requires the previous phosphorylation of T421/S424 in the autoinhibitory region of p70S6K, a site shown to be ERK-dependent in some studies (Zhang et al., 2001; Lehman et al., 2003). To determine whether ERK is required for the phosphorylation of these sites in LTP, we performed immunoblots from CA1 homogenates using phospho-specific antibodies. In vehicle-treated slices, immunoreactivity for both phospho-T389 and phospho-T421/S424 increased after the delivery of HFS (Fig. 3 A), whereas total p70S6K expression was not affected (Fig. S2, available at www.jneurosci.org as supplemental material). As expected, rapamycin pretreatment prevented the HFS-induced phosphorylation of T389 but left the phosphorylation at T421/S424 intact. In contrast, HFS delivered in the presence of U0126 failed to induce phosphorylation at either site. These findings agree with those showing that phosphorylation of T421/S424 is ERK dependent and indicate that the ERK and mTOR pathways might interact at the level of p70S6K through an intramolecular mechanism. However, these data do not rule out the possibility of a more upstream effect of ERK on the mTOR pathway.
To determine whether ERK mediates the HFS-induced stimulation of the mTOR pathway in dendrites, slices were fixed for immunohistochemistry 30 min after HFS. Pretreatment with U0126 blocked the phosphorylation of dendritic p70S6K at both T389 and T421/S424, as well as the increase in dendritic eEF1A expression (Fig. 3 B,C). These findings agree with our immunoblot data, as does the observation that pretreatment with rapamycin prevented the effect on eEF1A without blocking phosphorylation at T421/S424-p70S6K (Fig. 3 C). Thus, ERK is required for the activation of p70S6K in the dendrites after HFS.
The increase in mTOR activity after HFS is ERK dependent
As noted, the data in Figure 3 are consistent with an intramolecular p70S6K mechanism, but they are also compatible with an alternative hypothesis: that ERK controls mTOR at a more upstream component, independent of its role in the phosphorylation of p70S6K at T424/S424 (Fig. 4 A) (Frödin et al., 2000; Roux et al., 2004; Ma et al., 2005). mTOR activity often correlates with its phosphorylation at S2448, which is increased in cells that express constitutively active Akt (Nave et al., 1999; Sekulic et al., 2000) (but see Holz and Blenis, 2005). To examine the possible entry of ERK upstream of mTOR, we tested whether the phosphorylation of S2448-mTOR in response to HFS requires ERK activity. As shown in Figure 4 B, the pretreatment of slices with U0126 prevented HFS-induced mTOR phosphorylation in area CA1 (note that total mTOR expression was unaffected by HFS) (Fig. S2, available at www.jneurosci.org as supplemental material). To confirm the conclusion that ERK regulates mTOR subsequent to the induction of late LTP, we assayed the activity of mTOR immunoprecipitated from CA1 regions, using recombinant eukaryotic initiation factor 4E binding protein 1 (4EBP1) as a substrate. The ability of mTOR to phosphorylate 4EBP1 was found to increase after HFS and to be susceptible to pretreatment with rapamycin (Fig. 4 C). Both U0126 and PD98059 blocked this increase, in agreement with our mTOR phosphorylation results and showing that the activation of mTOR by HFS is ERK dependent.
Figure 4.

HFS activates mTOR in an ERK-dependent manner. A, The ERK and mTOR pathways could interact at multiple levels. A schematic of p70S6K phosphorylation sites is shown, with T229 in the catalytic domain, T389 in the linker domain, and T421/S424 in the inhibitory domain, along with the known obligatory phosphorylation sequence. Phosphorylation at T389 is rapamycin-sensitive, whereas ERK mediates phosphorylation at T421/S424, indicating that ERK might control the mTOR-related activation of p70S6K through an intramolecular mechanism. An alternative explanation for the ERK-dependent phosphorylation of T389 is that mTOR activity depends on ERK (dashed line). B, mTOR phosphorylation by HFS requires ERK activity. Immunoblots for phospho(S2448)-mTOR were performed on lysates of CA1 from slices frozen 30 min after stimulation. The HFS-induced phosphorylation at S2448 was blocked in slices treated with 20 μm U0126. The asterisk indicates p < 0.05 compared with control slices; all values of n ≥ 7. C, HFS causes an ERK-dependent increase in mTOR activity. Slices received either control stimulation in vehicle (Con) or HFS delivered in the presence of vehicle (Veh), 1 μm rapamycin (Rapa), 20 μm U0126, or 50 μm PD98059 (PD). Slices were frozen 10 min after stimulation, and the activity of mTOR purified by immunoprecipitation from CA1 areas was assayed by phosphorylation of recombinant 4EBP1 (detected by immunoblot; see inset, where the arrowhead indicates phospho-4EBP1). The HFS-dependent increase in mTOR activity was completely blocked by the MEK inhibitors, and rapamycin reduced mTOR activity below baseline. The asterisk indicates p < 0.05 compared with control slices; all values of n ≥ 4.
ERK enters the PI3K–mTOR pathway at PDK1 and mediates the HFS-induced phosphorylation of RSK and Akt
In the canonical pathway for mTOR regulation, PI3K-generated phosphoinositides colocalize PDK1 and its substrate, Akt, at the cell membrane. Phosphorylated Akt, in turn, indirectly activates mTOR by phosphorylating and inhibiting the mTOR suppressor TSC2, also known as tuberin (Inoki et al., 2002; Manning et al., 2002; Potter et al., 2002). This pathway is functional in the hippocampus, because PI3K inhibitors block the phosphorylation of Akt and mTOR that is induced by metabotropic glutamate receptor (mGluR) stimulation (Hou and Klann, 2004). Furthermore, PI3K activity is necessary for synaptically induced LTP and for mGluR-mediated long-term depression (Lin et al., 2001; Sanna et al., 2002; Opazo et al., 2003; Hou and Klann, 2004).
The mechanism by which ERK regulates mTOR in LTP is unknown, but studies in other systems have identified two proteins in the PI3K-mTOR pathway where ERK might enter. First, ERK can indirectly phosphorylate and activate PDK1, an effect that is mediated by the ERK substrate RSK (also known as p90S6K) (Frödin et al., 2000). At a more downstream site, ERK can phosphorylate and inactivate TSC2, either directly or through RSK (Roux et al., 2004; Ma et al., 2005). We were particularly interested in the possibility that PDK1 activity might be regulated by ERK, because PDK1 has extensive effects on signal transduction and gene expression (Vanhaesebroeck and Alessi, 2000). PDK1 is stimulated upon phosphorylation by RSK, and this effect requires that RSK be activated by ERK (Frödin et al., 2000). If this mechanism is important in late LTP, then PDK1 activity should be increased by HFS in an ERK-dependent manner. This prediction was tested in several ways. First, we examined the ability of HFS to induce PDK1 autophosphorylation at S241 (Wick et al., 2003). In immunoblots of CA1 homogenates, immunoreactivity for S241-PDK1 was found to increase after HFS (but not the expression of total PDK1) (Fig. S2, available at www.jneurosci.org as supplemental material), and this increase was blocked when HFS was delivered in the presence of ERK pathway inhibitors (Fig. 5 A). Second, we tested for phosphorylation of Akt at T308, which is obligatory for the activation of Akt and directly mediated by PDK1 (Alessi et al., 1997a; Chan et al., 1999). In response to HFS, immunoreactivity to T308-Akt increased more than twofold and in an ERK-dependent manner (Fig. 5 B). Finally, RSK phosphorylation was probed at the ERK-dependent site S386. As shown in Figure 5 C, HFS caused a rapid hyperphosphorylation at S386-RSK, and this effect was blocked by U0126 and PD98059, whereas total RSK expression was not affected by HFS (Fig. S2, available at www.jneurosci.org as supplemental material). In control experiments with unstimulated slices, inhibition of ERK did not affect the basal phosphorylation of RSK, Akt, or PDK1 at these sites (Fig. S1, available at www.jneurosci.org as supplemental material), indicating that HFS-induced phosphorylation of these proteins was specifically blocked by ERK inhibitors under our conditions. Together, these data support the hypothesis that ERK regulates mTOR through the RSK-dependent activation of PDK1 and Akt.
Figure 5.

Reciprocal interactions between ERK and PI3K regulate the mTOR pathway at the level of PDK1. Immunoblots were performed on CA1 regions from slices frozen 5 min after HFS. A, The HFS-induced phosphorylation of PDK1 requires ERK activity. HFS induced the phosphorylation of PDK1 at S241, and this effect was prevented by either 50 μm PD98059 (PD) or 20 μm U0126. B, Phosphorylation of Akt at T308 was increased after HFS, and this effect was blocked by preincubation with 50 μm PD98059 (PD). C, The ERK substrate RSK became hyperphosphorylated after HFS, and this effect was sensitive to the MEK inhibitors U0126 (20 μm) and PD98059 (PD; 50 μm). All asterisks indicate p < 0.05 versus controls (Con); group values of n ≥ 6. D, PI3K activity mediates a component of MEK/ERK phosphorylation. Incubation with the PI3K inhibitor LY294002 (LY; 20 μm) reduced both basal and HFS-induced phosphorylation of MEK1/2 at S218/S222 of MEK1 (black bars) and ERK2 at T202/Y204 (gray bars). Data are from excised CA1 regions, and stimulated slices were frozen 5 min after HFS. Sample immunoblots are shown below. Single and double asterisks indicate p < 0.05 and p < 0.01, respectively; all values of n ≥ 4. E, Schematic of the interactions between the ERK and PI3K–mTOR pathways in regulating TOP mRNA translation, based on the experiments summarized in Figures 1 –5. Components in black are coregulated by the PI3K and ERK pathways (shown in blue and green, respectively). The arrows do not necessarily represent direct interactions [e.g., the connection between mTOR and p70S6K might involve the inhibition of a protein phosphatase (Peterson et al., 1999)], and some known interactions have been excluded for the sake of clarity (e.g., mTOR facilitates cap-dependent translation by phosphorylating 4EBP).
The PI3K–mTOR pathway exerts reciprocal control over ERK
The activation of mTOR requires the colocalization of PDK1 and Akt, mediated by the association of their pleckstrin homology domains with PI3K-generated phosphoinositides (Currie et al., 1999). This requirement is consistent with the ability of PI3K inhibitors to block late LTP in mouse hippocampus (Opazo et al., 2003), and we found that LTP induced by our protocol was similarly PI3K dependent (Fig. S3, available at www.jneurosci.org as supplemental material) (Sanna et al., 2002). Interestingly, Opazo et al. (2003) also observed that PI3K mediated the HFS-induced increase in ERK activity. Coupled with our data, such an effect suggests the possibility of reciprocal control between the ERK and PI3K-mTOR pathways in LTP. To examine this issue, we measured the PI3K-dependent component of ERK pathway activity in area CA1 from control and HFS-stimulated slices (Fig. 5 D). As expected, the PI3K inhibitor LY294002 (20 μm) reduced the HFS-induced increase in phospho-MEK and phospho-ERK. However, the phosphorylation state of MEK and ERK in unstimulated slices also was sensitive to the inhibitor, and the PI3K-dependent fraction of phospho-MEK and phospho-ERK was not significantly changed after stimulation. These findings contrast with the observation that PI3K inhibitors do not affect the level of phospho-ERK in unstimulated mouse hippocampus (Opazo et al., 2003). The basis for this difference is not known, but one possibility is that basal PI3K activity might be higher under our conditions, perhaps reflecting a species-related variation. Substantial PI3K activity in unstimulated slices is likely to support some background colocalization of PDK1 and Akt, which could allow signals in the ERK pathway to propagate to mTOR (see Discussion). It is noteworthy that the twofold increase in ERK phosphorylation that we observed after HFS is nearly identical to that reported by Opazo et al. (2003), suggesting that the contribution of PI3K to basal ERK activity does not reduce the dynamic range of the ERK pathway that is available for stimulation during LTP.
These results, along with our other data on the regulation of the PI3K-mTOR pathway by ERK, indicate that reciprocal interactions between the PI3K and ERK pathways control mTOR-dependent translation at the level of PDK1 (Fig. 5 E).
Discussion
Regulation of the dendritic translational machinery and its role in synapse-specific plasticity
The mTOR-mediated synthesis of translational machinery is observed in many cell types as an early response to stimuli that induce cell growth, and this process depends on translational and transcriptional control over the expression of TOP mRNA-encoded proteins (Meyuhas and Hornstein, 2000). The benefits of such on-demand production of translational machinery would be to reduce the metabolic cost of maintaining excess capacity under conditions where relatively little protein synthesis is needed and to add capacity at specific places in preparation for a period of increased demand. In the case of protein synthesis in the dendrites of a CA1 pyramidal cell, with a total dendritic length of ∼13,500 μm (Ishizuka et al., 1995), the advantages of regulating translational capacity are apparent.
Among the TOP mRNA-encoded proteins studied here, PABP is interesting from the standpoint of LTP. PABP binds to the poly-A tail of mRNA and consequently acts at the initiation step to increase translational efficiency (Wells et al., 2000). Plasticity-inducing stimulation can lengthen poly-A tails (Wells et al., 2001), and under such conditions PABP expression might become rate-limiting for poly-A-dependent translation. Thus, by synthesizing PABP, the neuron could match the supply of this protein to the demands imposed by the dynamics of polyadenylation.
The ability of HFS to increase eEF1A and eEF2 suggests that the process of elongation also is upregulated in LTP. Protein synthesis is commonly regulated at the initiation step; however, in an environment where basal translational capacity is low, it is possible that a general increase in the maximal rate of translation requires upregulation at both the initiation and elongation steps. The dendrite is apparently such a low-capacity environment, based on the sparse appearance of polyribosomes in the dendritic shaft and spines (∼1 per synapse) (Sutton and Schuman, 2006). However, the control of elongation in synaptic plasticity is complex, because some forms of stimulation result in the inhibition of eEF2 (Scheetz et al., 2000).
It is striking that HFS increases the expression of TOP mRNA-encoded proteins occurs throughout the apical dendrites, suggesting that translational capacity is boosted at a distance from the activated synapses. How could a widespread increase in dendritic translational capacity contribute to synapse-specific increases in LTP-related proteins? One possibility is that the increase in capacity causes a similarly widespread synthesis of PRPs, which can be used only at synapses that have been “tagged” by appropriate stimulation (Frey and Morris, 1997; Barco et al., 2002; Govindarajan et al., 2006). An alternative hypothesis postulates that the dendritic transcripts for PRPs are normally translationally repressed and become derepressed specifically at synapses that have been appropriately stimulated (Krichevsky and Kosik, 2001; Blitzer et al., 2005). In this way, a wide distribution of translational capacity would enable a potentially large number of synapses to synthesize PRPs rapidly, locally and on demand, ensuring the input-specificity of synaptic plasticity. Experiments that have addressed the issue of local protein synthesis in LTP have been inconclusive, with some supporting the tagging model (Frey and Morris, 1997, 1998) and others suggesting that LTP depends, at least in part, on local protein synthesis (Barco et al., 2002; Alarcon et al., 2006). Additional studies are needed to resolve the roles of distributed and local protein synthesis in LTP, and how these processes interact.
ERK as a coordinator of regulatory processes in LTP
Multiple regulatory processes at the transcriptional, translational, and posttranslational levels are simultaneously active in LTP, and a major challenge is to understand how these control mechanisms are coordinated by upstream signaling molecules. Among these key signaling components is ERK, which mediates LTP-related protein synthesis and transcription and regulates membrane excitability (Impey et al., 1998; Davis et al., 2000; Yuan et al., 2002; Kelleher et al., 2004). Our finding that ERK enters the PI3K–mTOR pathway at a relatively upstream point in the PI3K–mTOR pathway is significant, because the HFS-induced regulation of PDK1 helps to explain how ERK coordinates the diverse cellular processes that underlie synaptic plasticity. PDK1 has been described as a “master” regulator, because it activates several protein kinases, including some that have been implicated in synaptic plasticity, such as cyclic AMP-dependent protein kinase and protein kinase C (Cheng et al., 1998; Chou et al., 1998; Dutil et al., 1998; Mora et al., 2004). Moreover, the PDK1-dependent Akt similarly regulates multiple translational and transcriptional processes (Impey et al., 1998; Gingras et al., 1999; Meyuhas and Hornstein, 2000; Brazil and Hemmings, 2001; Wang et al., 2001; Banko et al., 2004). The present investigation is the first to demonstrate the importance of ERK in regulating the PDK1 and Akt pathways in LTP.
PDK1 and PI3K as coregulators of the mTOR pathway
There has been some controversy regarding the regulation of PDK1 activity and its role in controlling the mTOR pathway, because previous studies showed the isolated protein to be constitutively active (Alessi et al., 1997b). Both PDK1 and Akt contain pleckstrin homology domains and therefore associate with 3′-phosphoinositides in the cell membrane, so it has been suggested that PI3K regulates the mTOR pathway by bringing constitutively active PDK1 together with Akt. However, studies in intact cells have shown extracellular stimuli to increase PDK1 activity (Wick et al., 2000; Park et al., 2001; Sato et al., 2002), suggesting the presence of regulatory proteins in vivo. Several cellular mechanisms for increasing PDK1 activity have been described, including the binding of PDK1 to a docking site in the hydrophobic domain of RSK, which is generated upon RSK autophosphorylation at S386 by its C-terminal kinase (CTK) domain (Frödin et al., 2000). Importantly, CTK activity is regulated by ERK, which thus controls the interaction between PDK1 and the hydrophobic domain of RSK, an association that should dramatically increase PDK1 activity (Biondi et al., 2000). The very low basal activity of the major RSK isoform in the hippocampus, along with its strong response to stimulation that activates ERK, suggest that the RSK–PDK1 interaction is an important factor in regulating hippocampal mTOR (Zeniou et al., 2002; Dummler et al., 2005). In agreement, our results indicate that this mechanism controls the mTOR pathway in LTP (Fig. 5).
We have shown that ERK is required for the HFS-induced stimulation of the mTOR pathway in the hippocampus; however, ERK is not necessary for mTOR activation under certain other stimulation conditions. For example, the treatment of cultured neurons with brain-derived neurotrophic factor increases Akt phosphorylation even in the presence of PD98059 (Takei et al., 2001; Schratt et al., 2004). These differences in the sensitivity of mTOR to ERK inhibitors are interesting and might reflect the complementary effects of PI3K and ERK on PDK1: PI3K recruits PDK1 to the membrane, where it can phosphorylate Akt and other membrane-associated substrates, whereas the efficacy of this process should be modulated by ERK-dependent changes in the intrinsic activity of PDK1. Thus, any treatment that strongly stimulates PI3K, and thereby causes extensive colocalization of PDK1 and Akt, might allow PDK1 with low intrinsic activity to substantially phosphorylate Akt, and ERK would be unnecessary for the stimulation of the mTOR pathway. Such a cooperative relationship between PI3K and ERK might explain the observation by Opazo et al. (2003) that PI3K inhibitors prevented HFS-induced LTP in mouse hippocampus (Winder et al., 1999), whereas MEK inhibitors were ineffective. Conversely, when PDK1 is stimulated by ERK, even a modest recruitment of PDK1 might be enough to activate mTOR, and this type of regulation would be ERK dependent. This model suggests the intriguing possibility that stimulus-induced changes in ERK activity might affect mTOR even when PI3K activity remains at its basal level, and agrees with the finding that constitutively active Ras or MEK can induce p70S6K phosphorylation without any increase in PI3K activity (Ma et al., 2005). Furthermore, PI3K is subject to regulation by βγ subunits and tyrosine kinase activity (Murga et al., 1998; Patapoutian and Reichardt, 2001); thus, there could be a dynamic relationship between PI3K and ERK in which neurotransmitters and growth factors set the level of PI3K activity, and this in turn determines the ability of ERK signaling to propagate to mTOR. The concept of ERK playing an executive role in regulating mTOR is clearly distinct from the canonical mechanism, and it will be interesting to identify conditions where neuronal mTOR is activated by stimuli that specifically stimulate ERK. Moreover, by associating ERK activity with the pleiotropic effects of PDK1 and its substrate Akt on gene expression, the present findings help to explain how ERK can orchestrate the complex cellular response that underlies LTP.
Footnotes
This work was supported by National Institutes of Health Grants DA015863 (E.M.L.) and GM54508 (R.I.) and by a Veterans Administration Merit Award (E.M.L.). P.T. is a recipient of an Alexander S. Onassis Public Benefit Foundation award. We thank Nahum Sonenberg for recombinant 4EBP, Jürgen Brosius for PABP antibodies, and Elizabeth Grace and Vinay Patel for their help with immunohistochemistry and Western blots.
References
- Alarcon JM, Barco A, Kandel ER. Capture of the late phase of long-term potentiation within and across the apical and basilar dendritic compartments of CA1 pyramidal neurons: synaptic tagging is compartment restricted. J Neurosci. 2006;26:256–264. doi: 10.1523/JNEUROSCI.3196-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr Biol. 1997a;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Deak M, Casamayor A, Caudwell FB, Morrice N, Norman DG, Gaffney P, Reese CB, MacDougall CN, Harbison D, Ashworth A, Bownes M. 3-Phosphoinositide-dependent protein kinase-1 (PDK1): structural and functional homology with the Drosophila DSTPK61 kinase. Curr Biol. 1997b;7:776–789. doi: 10.1016/s0960-9822(06)00336-8. [DOI] [PubMed] [Google Scholar]
- Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. The MAPK cascade is required for mammalian associative learning. Nat Neurosci. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- Atkins CM, Davare MA, Oh MC, Derkach V, Soderling TR. Bidirectional regulation of cytoplasmic polyadenylation element-binding protein phosphorylation by Ca2+/calmodulin-dependent protein kinase II and protein phosphatase 1 during hippocampal long-term potentiation. J Neurosci. 2005;25:5604–5610. doi: 10.1523/JNEUROSCI.5051-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banko JL, Hou L, Klann E. NMDA receptor activation results in PKA- and ERK-dependent Mnk1 activation and increased eIF4E phosphorylation in hippocampal area CA1. J Neurochem. 2004;91:462–470. doi: 10.1111/j.1471-4159.2004.02734.x. [DOI] [PubMed] [Google Scholar]
- Banko JL, Poulin F, Hou L, DeMaria CT, Sonenberg N, Klann E. The translation repressor 4E-BP2 is critical for eIF4F complex formation, synaptic plasticity, and memory in the hippocampus. J Neurosci. 2005;25:9581–9590. doi: 10.1523/JNEUROSCI.2423-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barco A, Alarcon JM, Kandel ER. Expression of constitutively active CREB protein facilitates the late phase of long-term potentiation by enhancing synaptic capture. Cell. 2002;108:689–703. doi: 10.1016/s0092-8674(02)00657-8. [DOI] [PubMed] [Google Scholar]
- Biondi RM, Cheung PC, Casamayor A, Deak M, Currie RA, Alessi DR. Identification of a pocket in the PDK1 kinase domain that interacts with PIF and the C-terminal residues of PKA. EMBO J. 2000;19:979–988. doi: 10.1093/emboj/19.5.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitzer RD, Iyengar R, Landau EM. Postsynaptic signaling networks: cellular cogwheels underlying long-term plasticity. Biol Psychiatry. 2005;57:113–119. doi: 10.1016/j.biopsych.2004.02.031. [DOI] [PubMed] [Google Scholar]
- Brazil DP, Hemmings BA. Ten years of protein kinase B signalling: a hard Akt to follow. Trends Biochem Sci. 2001;26:657–664. doi: 10.1016/s0968-0004(01)01958-2. [DOI] [PubMed] [Google Scholar]
- Cammalleri M, Lutjens R, Berton F, King AR, Simpson C, Francesconi W, Sanna PP. Time-restricted role for dendritic activation of the mTOR-p70S6K pathway in the induction of late-phase long-term potentiation in the CA1. Proc Natl Acad Sci USA. 2003;100:14368–14373. doi: 10.1073/pnas.2336098100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan TO, Rittenhouse SE, Tsichlis PN. AKT/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Annu Rev Biochem. 1999;68:965–1014. doi: 10.1146/annurev.biochem.68.1.965. [DOI] [PubMed] [Google Scholar]
- Cheng X, Ma Y, Moore M, Hemmings BA, Taylor SS. Phosphorylation and activation of cAMP-dependent protein kinase by phosphoinositide-dependent protein kinase. Proc Natl Acad Sci USA. 1998;95:9849–9854. doi: 10.1073/pnas.95.17.9849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou MM, Hou W, Johnson J, Graham LK, Lee MH, Chen CS, Newton AC, Schaffhausen BS, Toker A. Regulation of protein kinase C ζ by PI 3-kinase and PDK-1. Curr Biol. 1998;8:1069–1077. doi: 10.1016/s0960-9822(98)70444-0. [DOI] [PubMed] [Google Scholar]
- Cracco JB, Serrano P, Moskowitz SI, Bergold PJ, Sacktor TC. Protein synthesis-dependent LTP in isolated dendrites of CA1 pyramidal cells. Hippocampus. 2005;15:551–556. doi: 10.1002/hipo.20078. [DOI] [PubMed] [Google Scholar]
- Currie RA, Walker KS, Gray A, Deak M, Casamayor A, Downes CP, Cohen P, Alessi DR, Lucocq J. Role of phosphatidylinositol 3,4,5-trisphosphate in regulating the activity and localization of 3-phosphoinositide-dependent protein kinase-1. Biochem J. 1999;337:575–583. [PMC free article] [PubMed] [Google Scholar]
- Davis S, Vanhoutte P, Pages C, Caboche J, Laroche S. The MAPK/ERK cascade targets both Elk-1 and cAMP response element-binding protein to control long-term potentiation-dependent gene expression in the dentate gyrus in vivo . J Neurosci. 2000;20:4563–4572. doi: 10.1523/JNEUROSCI.20-12-04563.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dummler BA, Hauge C, Silber J, Yntema HG, Kruse LS, Kofoed B, Hemmings BA, Alessi DR, Frodin M. Functional characterization of human RSK4, a new 90-kDa ribosomal S6 kinase, reveals constitutive activation in most cell types. J Biol Chem. 2005;280:13304–13314. doi: 10.1074/jbc.M408194200. [DOI] [PubMed] [Google Scholar]
- Dutil EM, Toker A, Newton AC. Regulation of conventional protein kinase C isozymes by phosphoinositide-dependent kinase 1 (PDK-1) Curr Biol. 1998;8:1366–1375. doi: 10.1016/s0960-9822(98)00017-7. [DOI] [PubMed] [Google Scholar]
- English JD, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- Frey U, Morris RG. Synaptic tagging and long-term potentiation. Nature. 1997;385:533–536. doi: 10.1038/385533a0. [DOI] [PubMed] [Google Scholar]
- Frey U, Morris RG. Weak before strong: dissociating synaptic tagging and plasticity-factor accounts of late-LTP. Neuropharmacology. 1998;37:545–552. doi: 10.1016/s0028-3908(98)00040-9. [DOI] [PubMed] [Google Scholar]
- Frey U, Krug M, Reymann KG, Matthies H. Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro. Brain Res. 1988;452:57–65. doi: 10.1016/0006-8993(88)90008-x. [DOI] [PubMed] [Google Scholar]
- Frey U, Krug M, Brodemann R, Reymann K, Matthies H. Long-term potentiation induced in dendrites separated from rat's CA1 pyramidal somata does not establish a late phase. Neurosci Lett. 1989;97:135–139. doi: 10.1016/0304-3940(89)90152-3. [DOI] [PubMed] [Google Scholar]
- Frödin M, Jensen CJ, Merienne K, Gammeltoft S. A phosphoserine-regulated docking site in the protein kinase RSK2 that recruits and activates PDK1. EMBO J. 2000;19:2924–2934. doi: 10.1093/emboj/19.12.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras AC, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoekstra MF, Aebersold R, Sonenberg N. Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes Dev. 1999;13:1422–1437. doi: 10.1101/gad.13.11.1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindarajan A, Kelleher RJ, Tonegawa S. A clustered plasticity model of long-term memory engrams. Nat Rev Neurosci. 2006;7:575–583. doi: 10.1038/nrn1937. [DOI] [PubMed] [Google Scholar]
- Holz MK, Blenis J. Identification of S6 kinase 1 as a novel mammalian target of rapamycin (mTOR)-phosphorylating kinase. J Biol Chem. 2005;280:26089–26093. doi: 10.1074/jbc.M504045200. [DOI] [PubMed] [Google Scholar]
- Hou L, Klann E. Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2004;24:6352–6361. doi: 10.1523/JNEUROSCI.0995-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YS, Jung MY, Sarkissian M, Richter JD. N-methyl-d-aspartate receptor signaling results in Aurora kinase-catalyzed CPEB phosphorylation and αCaMKII mRNA polyadenylation at synapses. EMBO J. 2002;21:2139–2148. doi: 10.1093/emboj/21.9.2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Kandel ER. Recruitment of long-lasting and protein kinase A-dependent long-term potentiation in the CA1 region of hippocampus requires repeated tetanization. Learn Mem. 1994;1:74–82. [PubMed] [Google Scholar]
- Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR. Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron. 1998;21:869–883. doi: 10.1016/s0896-6273(00)80602-9. [DOI] [PubMed] [Google Scholar]
- Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- Ishizuka N, Cowan WM, Amaral DG. A quantitative analysis of the dendritic organization of pyramidal cells in the rat hippocampus. J Comp Neurol. 1995;362:17–45. doi: 10.1002/cne.903620103. [DOI] [PubMed] [Google Scholar]
- Jefferies HB, Fumagalli S, Dennis PB, Reinhard C, Pearson RB, Thomas G. Rapamycin suppresses 5′TOP mRNA translation through inhibition of p70s6k . EMBO J. 1997;16:3693–3704. doi: 10.1093/emboj/16.12.3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Schuman EM. A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science. 1996;273:1402–1406. doi: 10.1126/science.273.5280.1402. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ, III, Govindarajan A, Jung HY, Kang H, Tonegawa S. Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell. 2004;116:467–479. doi: 10.1016/s0092-8674(04)00115-1. [DOI] [PubMed] [Google Scholar]
- Krichevsky AM, Kosik KS. Neuronal RNA granules: a link between RNA localization and stimulation-dependent translation. Neuron. 2001;32:683–696. doi: 10.1016/s0896-6273(01)00508-6. [DOI] [PubMed] [Google Scholar]
- Lehman JA, Calvo V, Gomez-Cambronero J. Mechanism of ribosomal p70S6 kinase activation by granulocyte macrophage colony-stimulating factor in neutrophils: cooperation of a MEK-related, THR421/SER424 kinase and a rapamycin-sensitive, m-TOR-related THR389 kinase. J Biol Chem. 2003;278:28130–28138. doi: 10.1074/jbc.M300376200. [DOI] [PubMed] [Google Scholar]
- Lin CH, Yeh SH, Lu KT, Leu TH, Chang WC, Gean PW. A role for the PI-3 kinase signaling pathway in fear conditioning and synaptic plasticity in the amygdala. Neuron. 2001;31:841–851. doi: 10.1016/s0896-6273(01)00433-0. [DOI] [PubMed] [Google Scholar]
- Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121:179–193. doi: 10.1016/j.cell.2005.02.031. [DOI] [PubMed] [Google Scholar]
- Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10:151–162. doi: 10.1016/s1097-2765(02)00568-3. [DOI] [PubMed] [Google Scholar]
- Marin P, Nastiuk KL, Daniel N, Girault JA, Czernik AJ, Glowinski J, Nairn AC, Premont J. Glutamate-dependent phosphorylation of elongation factor-2 and inhibition of protein synthesis in neurons. J Neurosci. 1997;17:3445–3454. doi: 10.1523/JNEUROSCI.17-10-03445.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megias M, Emri Z, Freund TF, Gulyas AI. Total number and distribution of inhibitory and excitatory synapses on hippocampal CA1 pyramidal cells. Neuroscience. 2001;102:527–540. doi: 10.1016/s0306-4522(00)00496-6. [DOI] [PubMed] [Google Scholar]
- Meyuhas O, Hornstein E. Translational control of TOP mRNAs. In: Sonenberg N, Hershey JWB, Mathews MB, editors. Translational control of gene expression. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 2000. pp. 671–693. [Google Scholar]
- Mora A, Komander D, van Aalten DM, Alessi DR. PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol. 2004;15:161–170. doi: 10.1016/j.semcdb.2003.12.022. [DOI] [PubMed] [Google Scholar]
- Murga C, Laguinge L, Wetzker R, Cuadrado A, Gutkind JS. Activation of Akt/protein kinase B by G protein-coupled receptors. A role for alpha and beta gamma subunits of heterotrimeric G proteins acting through phosphatidylinositol-3-OH kinasegamma. J Biol Chem. 1998;273:19080–19085. doi: 10.1074/jbc.273.30.19080. [DOI] [PubMed] [Google Scholar]
- Nave BT, Ouwens M, Withers DJ, Alessi DR, Shepherd PR. Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem J. 1999;344:427–431. [PMC free article] [PubMed] [Google Scholar]
- Opazo P, Watabe AM, Grant SG, O'Dell TJ. Phosphatidylinositol 3-kinase regulates the induction of long-term potentiation through extracellular signal-related kinase-independent mechanisms. J Neurosci. 2003;23:3679–3688. doi: 10.1523/JNEUROSCI.23-09-03679.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osten P, Valsamis L, Harris A, Sacktor TC. Protein synthesis-dependent formation of protein kinase Mζ in long-term potentiation. J Neurosci. 1996;16:2444–2451. doi: 10.1523/JNEUROSCI.16-08-02444.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang Y, Rosenstein A, Kreiman G, Schuman EM, Kennedy MB. Tetanic stimulation leads to increased accumulation of Ca2+/calmodulin-dependent protein kinase II via dendritic protein synthesis in hippocampal neurons. J Neurosci. 1999;19:7823–7833. doi: 10.1523/JNEUROSCI.19-18-07823.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Hill MM, Hess D, Brazil DP, Hofsteenge J, Hemmings BA. Identification of tyrosine phosphorylation sites on 3-phosphoinositide-dependent protein kinase-1 and their role in regulating kinase activity. J Biol Chem. 2001;276:37459–37471. doi: 10.1074/jbc.M105916200. [DOI] [PubMed] [Google Scholar]
- Patapoutian A, Reichardt LF. Trk receptors: mediators of neurotrophin action. Curr Opin Neurobiol. 2001;11:272–280. doi: 10.1016/s0959-4388(00)00208-7. [DOI] [PubMed] [Google Scholar]
- Peterson RT, Desai BN, Hardwick JS, Schreiber SL. Protein phosphatase 2A interacts with the 70-kDa S6 kinase and is activated by inhibition of FKBP12-rapamycin associated protein. Proc Natl Acad Sci USA. 1999;96:4438–4442. doi: 10.1073/pnas.96.8.4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter CJ, Pedraza LG, Xu T. Akt regulates growth by directly phosphorylating Tsc2. Nat Cell Biol. 2002;4:658–665. doi: 10.1038/ncb840. [DOI] [PubMed] [Google Scholar]
- Pullen N, Thomas G. The modular phosphorylation and activation of p70s6k . FEBS Lett. 1997;410:78–82. doi: 10.1016/s0014-5793(97)00323-2. [DOI] [PubMed] [Google Scholar]
- Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci USA. 2004;101:13489–13494. doi: 10.1073/pnas.0405659101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanna PP, Cammalleri M, Berton F, Simpson C, Lutjens R, Bloom FE, Francesconi W. Phosphatidylinositol 3-kinase is required for the expression but not for the induction or the maintenance of long-term potentiation in the hippocampal CA1 region. J Neurosci. 2002;22:3359–3365. doi: 10.1523/JNEUROSCI.22-09-03359.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Fujita N, Tsuruo T. Regulation of kinase activity of 3-phosphoinositide-dependent protein kinase-1 by binding to 14-3-3. J Biol Chem. 2002;277:39360–39367. doi: 10.1074/jbc.M205141200. [DOI] [PubMed] [Google Scholar]
- Scheetz AJ, Nairn AC, Constantine-Paton M. NMDA receptor-mediated control of protein synthesis at developing synapses. Nat Neurosci. 2000;3:211–216. doi: 10.1038/72915. [DOI] [PubMed] [Google Scholar]
- Schratt GM, Nigh EA, Chen WG, Hu L, Greenberg ME. BDNF regulates the translation of a select group of mRNAs by a mammalian target of rapamycin-phosphatidylinositol 3-kinase-dependent pathway during neuronal development. J Neurosci. 2004;24:7366–7377. doi: 10.1523/JNEUROSCI.1739-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekulic A, Hudson CC, Homme JL, Yin P, Otterness DM, Karnitz LM, Abraham RT. A direct linkage between the phosphoinositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells. Cancer Res. 2000;60:3504–3513. [PubMed] [Google Scholar]
- Stolovich M, Tang H, Hornstein E, Levy G, Cohen R, Bae SS, Birnbaum MJ, Meyuhas O. Transduction of growth or mitogenic signals into translational activation of TOP mRNAs is fully reliant on the phosphatidylinositol 3-kinase-mediated pathway but requires neither S6K1 nor rpS6 phosphorylation. Mol Cell Biol. 2002;22:8101–8113. doi: 10.1128/MCB.22.23.8101-8113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Schuman EM. Dendritic protein synthesis, synaptic plasticity, and memory. Cell. 2006;127:49–58. doi: 10.1016/j.cell.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Takei N, Kawamura M, Hara K, Yonezawa K, Nawa H. Brain-derived neurotrophic factor enhances neuronal translation by activating multiple initiation processes. J Biol Chem. 2001;276:42818–42825. doi: 10.1074/jbc.M103237200. [DOI] [PubMed] [Google Scholar]
- Takei N, Inamura N, Kawamura M, Namba H, Hara K, Yonezawa K, Nawa H. Brain-derived neurotrophic factor induces mammalian target of rapamycin-dependent local activation of translation machinery and protein synthesis in neuronal dendrites. J Neurosci. 2004;24:9760–9769. doi: 10.1523/JNEUROSCI.1427-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci USA. 2002;99:467–472. doi: 10.1073/pnas.012605299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsokas P, Grace EA, Chan P, Ma T, Sealfon SC, Iyengar R, Landau EM, Blitzer RD. Local protein synthesis mediates a rapid increase in dendritic elongation factor 1A after induction of late long-term potentiation. J Neurosci. 2005;25:5833–5843. doi: 10.1523/JNEUROSCI.0599-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Alessi DR. The PI3K-PDK1 connection: more than just a road to PKB. Biochem J. 2000;346:561–576. [PMC free article] [PubMed] [Google Scholar]
- Vickers CA, Dickson KS, Wyllie DJ. Induction and maintenance of late-phase long-term potentiation in isolated dendrites of rat hippocampal CA1 pyramidal neurones. J Physiol (Lond) 2005;568:803–813. doi: 10.1113/jphysiol.2005.092924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Paulin FE, Campbell LE, Gomez E, O'Brien K, Morrice N, Proud CG. Eukaryotic initiation factor 2B: identification of multiple phosphorylation sites in the ε-subunit and their functions in vivo. EMBO J. 2001;20:4349–4359. doi: 10.1093/emboj/20.16.4349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells DG, Richter JD, Fallon JR. Molecular mechanisms for activity-regulated protein synthesis in the synapto-dendritic compartment. Curr Opin Neurobiol. 2000;10:132–137. doi: 10.1016/s0959-4388(99)00050-1. [DOI] [PubMed] [Google Scholar]
- Wells DG, Dong X, Quinlan EM, Huang YS, Bear MF, Richter JD, Fallon JR. A role for the cytoplasmic polyadenylation element in NMDA receptor-regulated mRNA translation in neurons. J Neurosci. 2001;21:9541–9548. doi: 10.1523/JNEUROSCI.21-24-09541.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick MJ, Dong LQ, Riojas RA, Ramos FJ, Liu F. Mechanism of phosphorylation of protein kinase B/Akt by a constitutively active 3-phosphoinositide-dependent protein kinase-1. J Biol Chem. 2000;275:40400–40406. doi: 10.1074/jbc.M003937200. [DOI] [PubMed] [Google Scholar]
- Wick MJ, Ramos FJ, Chen H, Quon MJ, Dong LQ, Liu F. Mouse 3-phosphoinositide-dependent protein kinase-1 undergoes dimerization and trans-phosphorylation in the activation loop. J Biol Chem. 2003;278:42913–42919. doi: 10.1074/jbc.M304172200. [DOI] [PubMed] [Google Scholar]
- Winder DG, Martin KC, Muzzio IA, Rohrer D, Chruscinski A, Kobilka B, Kandel ER. ERK plays a regulatory role in induction of LTP by theta frequency stimulation and its modulation by β-adrenergic receptors. Neuron. 1999;24:715–726. doi: 10.1016/s0896-6273(00)81124-1. [DOI] [PubMed] [Google Scholar]
- Wu GY, Cline HT. Stabilization of dendritic arbor structure in vivo by CaMKII. Science. 1998;279:222–226. doi: 10.1126/science.279.5348.222. [DOI] [PubMed] [Google Scholar]
- Wu L, Wells D, Tay J, Mendis D, Abbott MA, Barnitt A, Quinlan E, Heynen A, Fallon JR, Richter JD. CPEB-mediated cytoplasmic polyadenylation and the regulation of experience-dependent translation of α-CaMKII mRNA at synapses. Neuron. 1998;21:1129–1139. doi: 10.1016/s0896-6273(00)80630-3. [DOI] [PubMed] [Google Scholar]
- Yuan LL, Adams JP, Swank M, Sweatt JD, Johnston D. Protein kinase modulation of dendritic K+ channels in hippocampus involves a mitogen-activated protein kinase pathway. J Neurosci. 2002;22:4860–4868. doi: 10.1523/JNEUROSCI.22-12-04860.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeniou M, Ding T, Trivier E, Hanauer A. Expression analysis of RSK gene family members: the RSK2 gene, mutated in Coffin-Lowry syndrome, is prominently expressed in brain structures essential for cognitive function and learning. Hum Mol Genet. 2002;11:2929–2940. doi: 10.1093/hmg/11.23.2929. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Dong Z, Nomura M, Zhong S, Chen N, Bode AM. Signal transduction pathways involved in phosphorylation and activation of p70S6K after exposure to UVA irradiation. J Biol Chem. 2001;276:20913–20923. doi: 10.1074/jbc.M009047200. [DOI] [PubMed] [Google Scholar]