

Abstract
The underlying cause of sporadic Alzheimer disease (AD) is unknown, but a number of environmental and genetic factors are likely to be involved. One environmental factor that is increasingly being recognized as contributing to brain aging is diet, which has evolved markedly over modern history. Here we show that dietary supplementation with docosahexaenoic acid (DHA), an n-3 polyunsaturated fatty acid, in the 3xTg-AD mouse model of AD reduced the intraneuronal accumulation of both amyloid-β (Aβ) and tau. In contrast, combining DHA with n-6 fatty acids, either arachidonic acid or docosapentaenoic acid (DPAn-6), diminished the efficacy of DHA over a 12 month period. Here we report the novel finding that the mechanism accounting for the reduction in soluble Aβ was attributable to a decrease in steady-state levels of presenilin 1, and not to altered processing of the amyloid precursor protein by either the α- or β-secretase. Furthermore, the presence of DPAn-6 in the diet reduced levels of early-stage phospho-tau epitopes, which correlated with a reduction in phosphorylated c-Jun N-terminal kinase, a putative tau kinase. Collectively, these results suggest that DHA and DPAn-6 supplementations could be a beneficial natural therapy for AD.
Keywords: tau, Alzheimer's, docosahexanoic acid, docosapentaenoic acid, presenilin, amyloid, transgenic
Introduction
Alzheimer disease (AD) is a progressive neurodegenerative disorder of the elderly. Two well defined pathologies of the disease have been studied extensively. The first is the accumulation of the 4 kDa peptide amyloid-β (Aβ), which deposits to form amyloid plaques but can also accumulate intraneuronally. The second is the somatodendritic buildup of hyperphosphorylated tau, which forms neurofibrillary tangles (NETs). The relationship between these two pathologies is not yet fully understood, but for a therapy to be successful, it is likely that it will need to alleviate both pathologies. Mouse models of AD have been very useful for investigating potential novel therapies, with the hope that the findings will be translatable to the human disease. Nutraceuticals represent a facile means of ameliorating or preventing the disease.
There is mounting evidence that alterations in dietary intake could be beneficial for preventing and treating AD, which could provide an alternative or supplementary therapy. Other natural therapies have already shown promise in transgenic mouse models of AD such as blueberry-containing diets (Joseph et al., 2003), environmental enrichment (Lazarov et al., 2005), exercise (Adlard et al., 2005), green tea (Rezai-Zadeh et al., 2005), and melatonin (Lahiri et al., 2004). Docosahexanoic acid (DHA) is an omega n-3 polyunsaturated fatty acid (PUFA) found in fish and certain algae. This PUFA is obtained mainly though dietary intake because biosynthesis from precursors in mammals does not readily occur. Sixty percent of the fatty acids that make up neuronal cell membranes consist of DHA, and it is particularly concentrated in synaptic membranes (Bazan and Scott, 1990) and in myelin sheaths (Ansari and Shoeman, 1990). DHA is essential for prenatal brain development as well as normal healthy brain functioning.
Intriguingly, AD patients have decreased serum and brain DHA compared with age-matched, nondemented subjects, suggesting that a deficiency in this PUFA could play a role in the disease (Tully et al., 2003). Furthermore, greater consumption of fish (and in particular, DHA) significantly reduced the likelihood of developing AD (Morris et al., 2003; Schaefer et al., 2006). Loss of DHA in AD could be accounted for by oxidation of DHA converting it into F4-neuroprostanes, which accumulate in AD patients (Reich et al., 2001). Because DHA makes up such a large proportion of the fatty acids in the brain, and plays roles in neuronal excitability, it is possible that DHA reduction contributes to memory impairments in AD. To illustrate, dietary DHA treatment simultaneously with Aβ infusion into rat brain protected against memory impairments induced by Aβ alone, suggesting that DHA can prevent or protect from the downstream effects of Aβ that cause memory loss (Hashimoto et al., 2005). DHA depletion in a mouse model of AD exacerbated dendritic and synaptic pathology caused by the amyloid pathology (Calon et al., 2004), suggesting that DHA, which is normally present in mouse chow, might partially protect against secondary amyloid pathologies. Recent data have shown that DHA supplementation of mouse chow (0.6%) can reverse some amyloid pathology in Tg2576 mice (Lim et al., 2005), but a mechanism has yet to be elucidated.
Materials and Methods
Immunoblotting.
Protein extracts were prepared from cells using M-per (Pierce, Rockford, IL) extraction buffer and Complete Mini Protease Inhibitor Tablets (Roche, Indianapolis, IN). Protein extracts were prepared from whole-brain samples by homogenizing in T-per (Pierce) extraction buffer and Complete Mini Protease Inhibitor Tablets (Roche), followed by high-speed centrifugation at 100,000 × g for 1 h. The supernatant was taken as the protein extract. Protein concentrations were determined by the Bradford method. Equal amounts of protein (20–50 μg depending on protein of interest) were separated by SDS-PAGE on a 10% Bis/Tris gel (Invitrogen, Carlsbad, CA), transferred to 0.45 μm polyvinylidene difluoride membranes, blocked for 1 h in 5% (v/v) nonfat milk in Tris-buffered saline, pH 7.5, supplemented with 0.2% Tween 20 (TBS-T), and processed as described. Antibodies and dilutions used in this study include 6E10 (1:1000; Signet, Dedham, MA), CTF20 (1:5000; Calbiochem, La Jolla, CA), HT7 (1:3000; Innogenetics, Gent, Belgium), AT8 (1:1000; Pierce), AT180 (1:1000; Pierce), AT270 (1:1000; Pierce), anti-β-APP-cleaving enzyme (BACE) (1:1000; Calbiochem), anti-insulin degrading enzyme (IDE; 1:1000; a kind gift from Dr. D. Selkoe, Harvard Medical School, Boston, MA), anti-presenilin 1 (PS1; 1:500; Cell Signaling Technology, Danvers, MA), anti-ADAM10 (1:1000; Chemicon, Temecula, CA), prealbumin [for transthyretin (TTR), 1:1000; Santa Cruz Biotechnology, Santa Cruz, CA], anti-apolipoprotein E (ApoE; 1:1000; Abcam, Cambridge, MA), anti-cdk5 (1:3000; Calbiochem), anti-p23 C' terminal (for p25 and p35, 1:500; Santa Cruz Biotechnology), anti-glycogen synthase kinase-3α/β (GSK3α/β; 1:2000; Calbiochem), anti-GSK3β ser9 (1:3000; Cell Signaling Technology), anti-phosphorylated c-Jun N-terminal kinase (phospho-JNK; 1:1000; Cell Signaling Technology), and anti-actin (1:10,000; Sigma-Aldrich, St. Louis, MO). Quantitative densiometric analyses were performed on digitized images of immunoblots with Scion (Frederick, MD) Image 4.0.
Dot blot.
Ten micrograms of protein were made up to 10 μl in H2O, pipetted onto 0.45 μm nitrocellulose membrane (Pierce), and allowed to dry. The membrane was blocked for 45 min in 5% powder milk in TBS-T and incubated in A11 (a generous gift from C. Glabe, University of California, Irvine, CA) at 1:1000 overnight at 4°C. The membrane was then washed five times in TBS-T and incubated for 1 h in HRP goat anti-rabbit antibody (1:10,000; Sigma-Aldrich). After an additional five washes, the membrane was coated with ECL Plus (Amersham Biosciences, Piscataway, NJ) and developed on photographic film. Quantitative densiometric analyses were performed on digitized images of immunoblots using Scion Image 4.0 software.
Aβ ELISA.
Aβ1–40 and Aβ1–42 were measured using a sensitive sandwich ELISA system. Soluble and insoluble Aβ was isolated from whole-brain homogenates using T-per extraction buffer (Pierce) and 70% formic acid (FA), respectively. Soluble fractions were loaded directly onto ELISA plates, and FA fractions were diluted 1:20 in neutralization buffer (1 m Tris base and 0.5 m NaH4PO4) before loading. MaxiSorp immunoplates (Nunc, Rochester, NY) were coated with mAB20.1 antibody (a kind gift from Dr. W. Van Nostrand, Stony Brook University, Stony Brook, NY) at a concentration of 25 μg/ml in coating buffer (0.1 m NaCO3 buffer, pH 9.6) and blocked with 3% BSA. Standards of both Aβ40 and Aβ42 were made in antigen capture buffer [20 mm NaH2PO4, 2 mm EDTA, 0.4 m NaCl, 0.5 g of CHAPS (3-[(3-cholamido-propyl)dimethylammonio]-1-propanesulfonate), and 1% BSA, pH 7.0] and loaded onto ELISA plates in duplicate. Samples were then loaded in duplicate and incubated overnight at 4°C. Plates were washed and probed with either HRP-conjugated anti-Aβ35–40 (MM32-13.1.1, for Aβ1–40) or anti-Aβ35–42 (MM40-21.3.4, for Aβ1–42) overnight at 4°C. 3,3′,5,5′-Tetramethylbenzidine was used as the chromagen, and the reaction was stopped by 30% O-phosphoric acid and read at 450 nm on a plate reader (Molecular Dynamics, Sunnyvale, CA). Aβ readings were then normalized to protein concentrations of the samples loaded or to the protein concentration of the cell layer that the medium was incubated with, in the case of the in vitro assays. This takes into account any variations of cell numbers or protein concentrations that may otherwise affect Aβ readings.
Immunostaining.
Light-level immunohistochemistry was performed using an avidin–biotin immunoperoxidase technique (ABC kit; Vector Laboratories, Burlingame, CA) and was visualized with diaminobenzidine (DAB) as described previously (Oddo et al., 2003). The following antibodies were used: anti-Aβ, 6E10, anti-Tau HT7, and MC1 (a kind gift from Dr. P. Davies, Albert Einstein College of Medicine, Bronx, NY). Primary antibodies were applied at dilutions of 1:1000 for 6E10, 1:200 for HT7, and 1:200 for MC1. Quantification of DAB staining was performed by taking three adjacent images from each hippocampus at 20×. These images were loaded in Scion Image 4.0, and the DAB pixel count was measured by setting the threshold to the same value for each section. Pixel counts were averaged from the three adjacent sections for at least three animals per group, and the data were plotted.
Cell culture.
SHSY5Y cells were maintained in DMEM (Invitrogen) supplemented with 10% FBS. Cells were passaged at 1:40 when 70% confluency had been achieved and discarded after 20 passages. For experiments, equal numbers of cells were plated down in six-well plates. Wells were treated 24 h later by removing the medium and replacing with 2 ml of fresh medium containing either DHA (Nu-Chek Prep, Elysian, MN) complexed to BSA (1:3) or the equivalent BSA alone.
Real-time PCR.
After treatment, RNA was extracted from cells using an Aurum RNA extraction kit (Bio-Rad, Hercules, CA) as per the manufacturer's instructions. RNA (1 μg) from each sample was converted to cDNA, using the iScript cDNA synthesis kit (Bio-Rad), as per the manufacturer's instructions. DNA (1 μg) was taken and diluted 1:100. This was mixed with 500 nm of each primer and 10 μl of SYBERGREEN supermix (Bio-Rad). The volume was made up to 20 μl with nuclease-free water. Real-time PCR was done on a MyCycler IQ system (Bio-Rad) with the following parameters: 1 time at 95°C for 5 min; 40 times at 95°C for 30 s, 62°C for 30 s, and 72°C for 30 s; followed by melt-curve analysis to ensure single products.
Primers were designed as follows: actin left, 5-ACTGTGTTGGCATAGAGGTCTTTA-3; actin right, 5-CTAGACTTCGAGCAGGAGATGG-3; mouse PS1 left, 5-CTCGCCATTTTCAAGAAAGC-3; mouse PS1 right, 5-GGGCTTGCTCTCTCTGTTTTTG-3.
Brain total lipids extraction.
The brains were maintained at −80°C until analysis. The brains were freeze-dried, and the lipids were extracted in 4 ml of 2:1 (v/v) chloroform/methanol with 0.5% butylated hydroxytoluene (BHT) as an antioxidant. The mixture was sonicated for 10 min and centrifuged to pellet out the solids.
Total brain lipid analysis.
The brain lipid extract (1.2 mg) was analyzed for brain total fatty acids. The brain total lipids were converted to fatty acid methyl esters (FAMEs) with 14% BF3/methanol at 100°C for 30 min (Morrison and Smith, 1964). BHT was added before saponification, and all samples were purged with N2 throughout the process to minimize oxidation. Tricosanoic free fatty acid (23:0) was added to each sample as an internal standard before FAME analysis.
Brain phospholipid analysis.
Brain phosphatidylcholine (PC), phosphatidylserine (PS), and phosphatidylethanolamine (PE) were separated using the methods of Gilfillan et al. (1983). K silica gel plates (0.25 mm thick, 20 × 20 cm; Whatman, Clifton, NJ) were activated for 60 min in a 100°C oven. A sample (0.6 mg) of total brain extract was spotted on the plate and developed in a TLC chamber using chloroform/methanol/petroleum ether/acetic acid/boric acid 40:20:30:10:1.8 (v/v/v/v/w). The plate was developed to within 1 cm of the top of the plate. The plate was sprayed with cupric acetate to visualize the bands. The PC, PS, and PE bands were scraped into test tubes, and the lipids were converted to FAMEs with 14% BF3/methanol at 100°C for 30 min (Morrison and Smith, 1964). BHT was added before saponification, and all samples were purged with N2 throughout the process to minimize oxidation. Tricosanoic free fatty acid (23:0) was added to each sample as an internal standard before FAME analysis.
Red blood cell analysis.
Total lipids were extracted from 400 μl of packed red blood cells (RBCs) using the methods of Bligh and Dyer (1959). Tricosanoic free fatty acid (23:0) was added to each sample as an internal standard before extraction. The RBC lipids were saponified with 0.5N methanolic sodium hydroxide, and the fatty acids were converted to methyl esters with 14% BF3/methanol at 100°C for 30 min (Morrison and Smith, 1964). BHT was added before saponification, and all samples were purged with N2 throughout the process to minimize oxidation.
Gas chromatogram analysis.
FAMEs were analyzed by gas–liquid chromatography using a Hewlett Packard (Palo Alto, CA) 6890 equipped with a flame ionization detector. The FAMEs were separated on a 30 m FAMEWAX capillary column (0.25 mm diameter, 0.25 μm coating thickness; Restek, Bellefonte, PA) using helium at a flow rate of 2.1 ml/min with split ratios of 48:1 and 20:1. The chromatographic run parameters included an oven starting temperature of 130°C that was increased at 6°C/min to 225°C, where it was held for 20 min before increasing to 250°C at 15°C/min, with a final hold of 5 min. The injector and detector temperatures were constant at 220°C and 230°C, respectively. Peaks were identified by comparison of retention times with external FAME standard mixtures from Nu-Chek Prep. The fatty acid profiles were expressed as a percentage of the total microgram of fatty acid (weight percentage).
Results
Our laboratory developed a transgenic mouse model that exhibits both Aβ and tau pathologies and is referred to as the 3xTg-AD mice (Oddo et al., 2003). To investigate the influence of dietary PUFA on AD-related pathologies, 3-month-old 3xTg-AD mice were fed one of the following four dietary regimens: (1) a control diet containing an n-6/n-3 ratio of 10:1; (2) a diet containing DHA and an n-3/n-6 ratio of 1:1; (3) a diet containing DHA and docosapentaenoic acid n-6 (DHA–DPA) and an n-3/n-6 ratio of 1:1; and (4) a diet containing both DHA and arachidonic acid (DHA–ARA) and an n-3/n-6 ratio of 1:1. Investigators were double blinded to the diet compositions or contents throughout the study, and diets were identified to investigators by color only. The diet formulation was the AIN-76 rodent diet containing 5% total fat. The target fatty acid composition of each diet is described in Table 1 and was achieved by blending a combination of vegetable and microalgal oils as outlined in Table 2.
Table 1.
n-3 and n-6 fatty acid content of rodent diets containing corn/soy, DHASCO, DHA-S, and DHASCO/ARASCO oils
| Fatty acid (g/100 g diet) | Dietary oil tested |
|||
|---|---|---|---|---|
| Control (corn/soy) | DHA (DHASCO) | DHA-DPA (DHA-S) | DHA-ARA (DHASCO/ARASCO) | |
| n-6 series | ||||
| 18:2 linoleic acid | 2.34 | 1.28 | 0.68 | 0.84 |
| 20:4 ARA | 0 | 0 | 0.03 | 0.48 |
| 22:5 DPA | 0 | 0.01 | 0.51 | 0.01 |
| n-3 series | ||||
| 18:3 linolenic acid | 0.23 | 0.01 | 0.01 | 0.05 |
| 20:5 EPA | 0 | 0 | 0.08 | 0 |
| 22:6 DHA | 0 | 1.27 | 1.25 | 1.27 |
| % SAT | 27.4% | 27.3% | 26.4% | 26.7% |
| % MONO | 21.0% | 20.8% | 20.1% | 19.9% |
| % POLY | 51.6% | 51.9% | 53.5% | 53.3% |
| n-6-to-n-3 ratio | 10.10 | 1.00 | 0.91 | 1.01 |
SAT, Saturated; MONO, monounsaturated; POLY, polyunsaturated.
Table 2.
Composition of the oil mixture used for the experimental diets
| Oil type | Control diet (corn/soy) (g/100 g diet) | DHA DHASCO (g/100 g diet) | DHA + DPA DHAS (g/100 g diet) | DHA + ARA ARASCO/DHASCO (g/100 g diet) |
|---|---|---|---|---|
| DHASCO | 0 | 30 | 0 | 30 |
| Corn | 15 | 9 | 0 | 0 |
| Soybean | 27 | 0 | 0 | 2 |
| Safflower | 0 | 9 | 8 | 8 |
| Coconut | 8 | 2 | 1 | 0 |
| DHA-S | 0 | 0 | 31 | 0 |
| ARASCO | 0 | 0 | 0 | 11 |
| Sunflower | 0 | 0 | 10 | 0 |
| Sum | 50 | 50 | 50 | 51 |
All diets contained 5 g of fat/100 g chow; each of the DHA-containing diets had 1.3 g of DHA/100 g chow and had a 1:1 ratio of n-6 to n-3 fatty acids compared with the control diet, which had a 10:1 ratio. The ratio of 10:1 (n-6/n-3) was constructed to represent the current typical intakes of these fatty acids in the United States diet. The Institute of Medicine estimates that the average intake of linoleic and α-linolenic acid (ALA) in the adult United States population to be ∼15 and 1.4 g per day, respectively (ratio of 10:1). The percentage of saturated, monounsaturated, and polyunsaturated fatty acids were equivalent across the diets. The amount of total protein (20%) and carbohydrate (66%) as well as total energy (3.9 kcal/g) were also equivalent across all diets. The test diets were designed to provide a constant level of DHA, while maintaining a constant percentage of saturated, monounsaturated, and polyunsaturated fatty acids. Furthermore, the test diets were constructed to maintain a constant total n-6 (linoleic acid plus ARA plus DPAn-6) and n-3 [α-linolenic plus DHA plus eicosapentaenoic acid (EPA)] PUFA level at an n-6/n-3 PUFA ratio of 1:1. All of the diets were isocaloric and provided 5% total fat. To maintain all of the above variables constant, different amounts of soybean, corn, safflower, coconut, sunflower, DHASCO, DHA-S, and ARASCO oils were added to each diet. The oils contain mixed tocopherols (including α-tocopherol) in the processing at a range of 250–500 ppm, ascorbyl palmitate at 250 ppm, as well as tBHQ (tert-butyl hydroxy-quinone) at 0.02 gm%, which are antioxidants, although the antioxidant content was not varied between diets.
Supplementation of diets continued for 3, 6, or 9 months with separate cohorts of animals killed after each treatment period for analyses. Mean body weights of male and female mice did not differ across the diets after 3, 6, or 9 months of treatment, as shown in Table 3. Mice continued to grow and remained healthy on all diets over the course of the study.
Table 3.
Mean body weights
| Male | +/− | Female | +/− | |
|---|---|---|---|---|
| After 3 month diet | ||||
| Control | 38.50 | 1.88 | 29.00 | 2.32 |
| DHA | 32.08 | 0.68 | 28.43 | 0.33 |
| DHA–DPA | 40.54 | 2.31 | 32.10 | 1.42 |
| DHA–ARA | 31.23 | 1.23 | 27.43 | 0.73 |
| After 6 month diet | ||||
| Control | 39.60 | 5.42 | 26.46 | 0.81 |
| DHA | 47.10 | 5.27 | 32.50 | 1.79 |
| DHA–DPA | 50.13 | 1.74 | 34.53 | 4.22 |
| DHA–ARA | 39.26 | 3.77 | 32.83 | 2.82 |
| After 9 month diet | ||||
| Control | 39.68 | 1.688 | 35.40 | 3.15 |
| DHA | 45.81 | 6.16 | 46.44 | 3.95 |
| DHA–DPA | 46.56 | 1.69 | 37.53 | 6.08 |
| DHA–ARA | 47.51 | 2.89 | 32.45 | 0 |
Fatty acid profiles in treated mice
Brain and RBC fatty acids were altered in the 6-, 9-, and 12-month-old mice as a result of the PUFA-enriched diets (Fig. 1; 9- and 12-month-old animal data not shown). RBC DHA (22:6 n-3) weight percentage levels were more than double the control levels with all of the PUFA-supplemented diets at all time points. The total brain DHA levels were also increased one to three weight percentages across all PUFA-supplemented diets, but not as great as in the RBCs. Mice fed the DHA-alone diet had the largest changes in DHA weight percentages in both the RBCs and the brain total lipids. As a weight percentage of fatty acids, DHA and ARA (20:4 n-6) are the most abundant long-chain PUFAs in both the brain and RBC total lipids. EPA (20:5 n-3) levels in the brain and RBCs are low, and DHA weight percentage levels in the brain are typically ∼15 times higher than EPA and ∼5 times higher in the RBCs.
Figure 1.

Whole-brain and RBC fatty acid profiles in 3xTg-AD mice after PUFA dietary supplementation. a, Whole-brain homogenates and RBC fatty acid profiles of the four groups after 3 months (n = 6) of supplementation with various PUFAs. Values are expressed as a weight percentage of total brain (a) or RBC (b) fatty acids. The four diets are shown as control, DHA, DHA–DPA, and DHA–ARA. Error bars indicate SEM. DMA, Dimethylacetals; LA, linoleic acid (n-6); ADRENIC, adrenic acid (n-6).
Although DHA levels increased, there was a subsequent decrease in total brain lipid ARA with all three PUFA-enriched diets compared with the control diet (which had an n-6-to-n-3 ratio of 10:1). Both DHA and ARA brain fatty acid levels were maintained throughout the supplemented period with both the supplemented and control diets. In the RBCs, the DHA-alone diet greatly lowered ARA levels by 11 weight percentages compared with the control group across all time points. As expected, the mice on the DHA–ARA diet had ARA RBC levels closest to the control mice.
RBC and total brain lipid DPA (22:5 n-6) levels in the DHA–DPA diet were more than double the control levels across all time points. Brain and RBC DPA levels were very low or nondetectable in the mice fed the DHA-alone or DHA–ARA diets. There is minimal ALA (18:3 n-3) in the brain and RBC lipids, and very small fatty acid changes were seen in either tissue type for this precursor of long-chain PUFAs. Linolenic acid (18:2 n-6) levels are ∼15 times higher in RBCs compared with the brain. RBC linoleic acid weight percentage levels were lower than the controls with all PUFA-enriched diets across the time points. These changes are reflective of the fatty acid compositions of the diets administered. RBC EPA weight percentage levels were higher across all PUFA-supplemented diets versus control levels, although these diets did not contain appreciable amounts of EPA. This may signify the retroconversion of DHA to EPA in blood cells. No appreciable amounts of EPA were detected in brain lipids across the various PUFA-supplemented diets and time periods.
PS, PE, and the PC brain phospholipid fatty acids were also altered in the 6-, 9-, and 12-month-old mice with the PUFA-enriched diets (Fig. 2; 9- and 12-month-old mouse data not shown). As a weight percentage, DHA is five times more abundant in the PS and PE fractions compared with the PC fraction, and ARA is most abundant in the PE fraction. EPA, linoleic acid, and ALA levels in the brain phospholipids are very low. The DHA-alone diet led to the largest increase from control in the brain phospholipid DHA weight percentages for all three phospholipids. There was also a corresponding decrease in ARA levels with the DHA-alone diet compared with controls in all three brain phospholipids. As seen in the RBCs and total brain lipids, the DPA weight percentage levels in all brain phospholipid fractions increased most in the mice fed the DHA–DPA diet.
Figure 2.

Whole-brain phospholipid fatty acid profiles in 3xTg-AD mice after dietary supplementation. Whole-brain phospholipid fatty acid profiles of the four groups after 3 months of dietary supplementation (n = 6) are shown. Values are expressed as a weight percentage of total brain fatty acids. The four diets are shown as control, DHA, DHA–DPA, and DHA–ARA. Error bars indicate SEM. DMA, Dimethylacetals; LA, linoleic acid (n-6); ADRENIC, adrenic acid (n-6).
In summary, the ratios of n-6-to-n-3 fatty acid compositions of the diets were reflected in RBC and brain fatty acid levels. Increasing DHA content in the diets led to significant increases in DHA levels in both RBC and brain levels. With increased levels of DHA in the diet, there was a subsequent decrease in n-6 fatty acids, an effect well characterized in the nutritional literature. The addition of other fatty acids (i.e., DPA and ARA) to the diets caused their corresponding fatty acid levels to increase in the tissues as well. Overall, brain fatty acid levels were well maintained throughout the supplementation period for each diet.
Dietary DHA supplementation reduces soluble Aβ accumulation
After 3 months of dietary PUFA supplementation, mice were killed, and brain Aβ levels were assessed by sandwich ELISA. Both soluble Aβ40 and Aβ42 were significantly reduced in all three treatment diets (∼50%; p < 0.05, Tukey test) but were insignificant from one another (Fig. 3 a). Insoluble Aβ levels were unaffected by dietary supplementation and, as expected, very low because of the age of the mice (6 months old) (Fig. 3 b). Using immunohistochemistry, we observed a widespread reduction in intraneuronal Aβ-like staining in the cell bodies of the hippocampus (Fig. 3 g,h) and amygdala (data not shown), two regions affected in AD. Reductions in intraneuronal Aβ staining correlated well with reductions in soluble Aβ. All three treatments prevented Aβ accumulation equally, making it likely that it was the presence of DHA in the diet that was causing the effect, because all three treatment diets contained the same amount of DHA.
Figure 3.
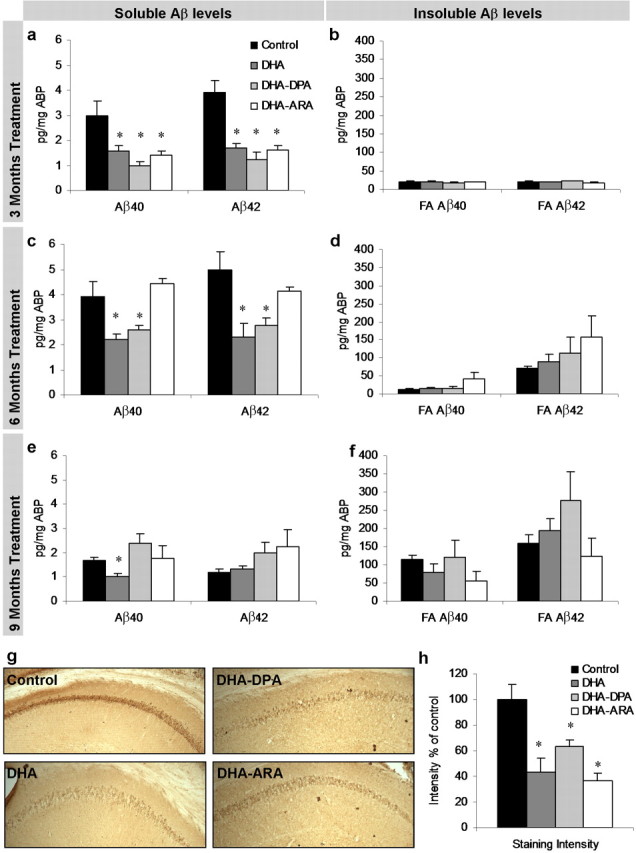
Aβ levels in 3xTg-AD mice after PUFA dietary supplementation. a–f, Soluble and insoluble Aβ40 and Aβ42 levels were measured from 3xTg-AD whole-brain homogenates after 3 months (a, b; n = 6), 6 months (c, d; n = 6), and 9 months (e, f; n = 6) of dietary supplementation. Soluble Aβ40 and Aβ42 levels were significantly decreased in the treatment diets after 3 months of supplementation (p < 0.05), in the DHA and DHA–DPA diets after 6 months of treatment (p < 0.05), and only Aβ40 in the DHA group after 9 months of supplementation (p < 0.05). Insoluble Aβ40 and Aβ42 levels were unaffected but demonstrated a gradual increase in levels as time progressed. g, DAB staining with 6E10 shows Aβ-like immunoreactivity in 40 μm sections from 3 month diet-treated mice. The hippocampus region is shown (original magnification, 5×). DAB Aβ-like immunoreactivity was elevated in cell bodies of the hippocampus and amygdale, corresponding to the decreased soluble Aβ40 and Aβ42 as measured by ELISA. h, Quantification of g. Error bars indicate SEM.
After 6 months of dietary supplementation, soluble Aβ40 and Aβ42 levels were significantly reduced for the DHA-alone and DHA–DPA diets, whereas Aβ levels were unchanged in mice receiving the DHA–ARA diet, which contained ARA in addition to DHA, and were comparable to those on the control diet (Fig. 3 c). Consistent with the first time point, insoluble Aβ levels were not altered by the treatment diets, and at this age, we can observe an elevation in Aβ42 (Fig. 3 d) corresponding to the initial seeding of plaques.
At the final time point, after 9 months of supplementation, only the DHA-alone diet significantly reduced levels of soluble Aβ, whereas both the DHA–DPA and DHA–ARA diets have similar levels to the control diet (Fig. 3 e). Insoluble Aβ levels are unchanged by the diet, and here we now observe an increase in Aβ40 (Fig. 3 f).
Although we cannot completely rule out the possibility that small amounts of high-molecular-weight Aβ aggregates could contribute to the soluble fraction because of the detergent used, we tried to minimize this by comparing multiple detergents and using one that gives lower soluble levels than stronger homogenization buffers. The soluble fraction also contains low-molecular-weight Aβ aggregates, or oligomers. Dot blot analysis of these with the oligomer-specific antibody A11 (Kayed et al., 2003) revealed no significant differences between dietary groups at any treatment time point, but levels did increase with age, as expected (Fig. 4 a,b).
Figure 4.

Aβ oligomer, APP fragments, and IDE, TTR, and ApoE steady-state levels in 3xTg-AD mice after DHA dietary supplementation. Western blot analyses of protein extracts from whole-brain homogenates of 3 month diet-treated 3xTg-AD mice (n = 6) are shown as alternating lanes: C, control; D, DHA; DP, DHA–DPA; DA, DHA–ARA. a, Dot blot analyses for oligomer levels in brain homogenates from 6-, 9-, and 12-month-old 3xTg-AD mice treated with diets for 3, 6, and 9 months, respectively (n = 6 per dietary group). b, Quantification of a. c, Steady-state levels of APP and APP CTFs C83 and C99. A longer exposure of APP is also shown to expose a band at 56 kDa corresponding to the Aβ 12mer oligomer (APP56). d, Quantification of protein blots from c shown normalized to β-actin levels as a loading control. e, Steady-state levels of IDE, TTR, and ApoE. f, Quantification of protein blots from e shown normalized to β-actin levels as a loading control. Error bars indicate SEM.
These results indicate that dietary supplementation with DHA is able to reduce the levels of soluble Aβ in a preventative manner, but that this treatment loses efficacy over time when other n-6 fatty acids are also added. Importantly, a dietary regimen containing DHA was able to prevent accumulation of Aβ, indicating that its utilization could represent a potential treatment option to prevent or delay AD.
Dietary DHA does not affect α- or β-amyloid precursor protein processing or Aβ degradation
Aβ levels can be reduced by a variety of enzymes and proteins that could either decrease production or increase degradation. To elucidate the mechanism by which DHA reduced Aβ levels, we focused on the first time point following 3 months of dietary supplementation when all three PUFA-enriched diets decreased soluble Aβ levels equally. We first measured steady-state levels of the amyloid precursor protein (APP), the parent protein of Aβ. Increases in this membrane protein lead to increases in Aβ production, whereas the opposite is also true. Because the human APP transgene in the 3xTg-AD mice is under the control of the Thy1.2 promoter, it is unlikely that APP transcription is affected. Steady-state levels of APP were unaltered across all four diets (Fig. 4 c,d), and although APP steady-state levels exhibited a lower trend in the DHA group, it did not reach significance (p = 0.1). Analysis of homogenates with 6E10 also reveals a recently identified Aβ 12mer (Lesne et al., 2006), with longer exposures (Fig. 4 c,d). Steady-state levels of this putative oligomer were not altered between treatment groups, in accordance with the A11 dot blot analysis (Fig. 4 a,b). We previously showed that this band selectively dissipates with hexafluoroisopropanol treatment and is detectable by multiple Aβ antibodies (Oddo et al., 2006; Billings et al., 2007) in the 3xTg-AD mice. We next measured the C'-terminal fragments (CTFs) of APP, C99 and C83, because these represent the products of both α- and β-secretase processing pathways and changes in these reflect alterations in APP processing. Unexpectedly, given the reductions in Aβ levels, no difference was seen with dietary treatment for either C83 or C99 (Fig. 4 c,d).
Aβ levels are also modulated by degradation of the peptide. Several Aβ-degrading enzymes have been identified including IDE (Kurochkin and Goto, 1994) and neprilysin (Iwata et al., 2000), of which steady-state levels of IDE and neprilysin are reduced in the AD brain (Caccamo et al., 2005). Steady-state levels of IDE were unaffected by the dietary treatments (Fig. 4 e,f) and, although steady-state levels do not necessarily equate to activity, it is probable that degradation was also not affected. We also measured steady-state levels of neprilysin but found no differences between the four groups (data not shown). However, we cannot rule out the possibility of other Aβ-degrading enzymes being involved. Because serum and brain fatty acid profiles were altered with each treatment group, we examined steady-state levels of ApoE because it is a well characterized lipoprotein transporter, which is also a significant risk factor for AD (Travis, 1993). ApoE has been postulated to act as an Aβ transporter and thus could facilitate Aβ clearance from cells or from the brain (Koistinaho et al., 2004). However, we found that ApoE steady-state levels were unchanged between dietary groups (Fig. 4 e,f). We also investigated whether levels of TTR, another Aβ carrier protein found in serum and CSF, was altered. TTR exists as a 55 kDa tetramer made of two dimers and has been shown to be protective against development of Aβ plaques (Stein and Johnson, 2002), by sequestering Aβ (Tsuzuki et al., 2000). It has also been shown to be upregulated with dietary DHA treatment (Puskas et al., 2003) and could account for reductions in Aβ levels. Steady-state analysis of TTR dimers and tetramers showed no significant differences between dietary treatment groups in the brain homogenates (Fig. 4 e,f), although there was a trend for higher levels in the DHA group. Although we cannot rule out altered dynamics of Aβ degradation/sequestration by any of these Aβ-binding proteins, we find no changes in steady-state levels that could account for the reductions in soluble Aβ measured with dietary DHA treatment.
Dietary DHA reduces PS1 steady-state levels
Dietary DHA reduces Aβ levels, but the mechanism behind this reduction is puzzling and did not appear to be attributable to increased degradation of the peptide or altered processing of APP. Consequently, we next evaluated the steady-state levels of the three secretases that process APP and control the production of Aβ. ADAM10 is the leading candidate as the constitutively active α-secretase and cleaves APP to release sAPPα and C83, precluding Aβ formation (Lammich et al., 1999). Levels of mature ADAM10 were unaltered by the dietary supplementation (Fig. 5 a,b), in concurrence with unaltered levels of C83 (Fig. 4 c,d). Furthermore, BACE levels were unaltered (Fig. 5 a,b), in agreement with C99 levels (Fig. 4 c,d). Release of Aβ is liberated by additional proteolysis of C99 by the γ-secretase complex. This complex consists of the catalytic core of PS1 or PS2 and an additional three proteins: nicastrin, PEN2, and Aph1 (Marlow et al., 2003). It usually cleaves C99 40–42 amino acids from the N' terminus, liberating the Aβ peptide and releasing the C' terminus–APP intracellular domain. We found that all three dietary supplementations significantly reduced PS1 levels relative to controls (Fig. 5 c,d) (p < 0.05). This reduction correlates with the decreased Aβ levels (Fig. 3) and accounts for the lack of alterations on APP processing. Notably, nicastrin steady-state levels were unaffected (Fig. 5 c,d), showing a specific effect on PS1 rather than the γ-secretase complex as a whole. Although a reduction in PS1 steady-state levels does not necessarily correspond to reduced γ-secretase activity, small interfering RNA knockdown of PS1 results in lower Aβ production in vitro (Luo et al., 2004), and the reduction in PS1 levels correlates strongly with the reduction in Aβ. We next examined PS1 steady-state levels after 6 and 9 months of dietary treatment accordingly, because soluble Aβ was also reduced at these time points in the DHA plus DHA–DPA and DHA groups, respectively. After 6 months of dietary treatment, steady-state levels of PS1 were significantly decreased in the DHA group only, whereas after 9 months of treatment, no significant differences were found (Fig. 5 e,f). Thus, it seems as with soluble Aβ levels, changes in PS1 levels also decrease with treatment duration and that changes in PS1 levels occur before changes in soluble Aβ.
Figure 5.

Secretase steady-state levels in 3xTg-AD mice after DHA dietary treatment. Western blot analyses of protein extracts from whole-brain homogenates of 3 month diet-treated 3xTg-AD mice (n = 6) are shown as alternating lanes: C, control; D, DHA; DP, DHA–DPA; DA, DHA–ARA. a, Steady-state levels of ADAM10 and BACE. b, Quantification of protein blots from a shown normalized to β-actin levels as a loading control. c, Steady-state levels of PS1 and nicastrin. d, Quantification of protein blots from c shown normalized to β-actin levels as a loading control. e, Steady-state levels of PS1 from 6 and 9 months of dietary treatment brain homogenates. f, Quantification of protein blots from e shown normalized to β-actin levels as a loading control. g, SHSY5Y cells treated for 48 h with either 0.3 μg/ml DHA complexed 3:1 to BSA (n = 3) or with the equivalent BSA alone (n = 3). mRNA was extracted, and real-time PCR was used to quantify PS1 mRNA levels. The graph shows PS1 mRNA levels normalized to β-actin mRNA as a loading control. DHA significantly reduced PS1 mRNA. The asterisk indicates significance (p < 0.05). Error bars indicate SEM.
To further elucidate the mechanism by which DHA treatment reduces PS1 levels, we evaluated its effects on a human neuroblastoma cell line, SHSY5Y. Treatment of these cells for 48 h with 0.3 μg/ml DHA complexed 3:1 to BSA, or with the equivalent BSA alone as a control, resulted in a twofold decrease in PS1 mRNA with DHA, as measured by real-time PCR (Fig. 5 g). This finding shows that DHA, at least in vitro, is able to modulate PS1 mRNA levels and nicely complements our animal studies in which we found that DHA also led to a reduction in the steady-state levels of the PS1 protein. Linoleic acid has been shown previously to affect PS1 levels and activity in vitro (Liu et al., 2004), showing that fatty acids can modulate presenilin. Because the test diets also contain linoleic acid as well as DHA, it is possible that it is the linoleic acid modulating presenilin levels rather than DHA. However, brain levels of linoleic acid (Fig. 1 a) are comparable between groups and are very low compared with the other brain fatty acids. This level is typical for this n-6 fatty acid and likely reflects its function as a precursor that is highly metabolized to the longer-chain PUFAs. The linoleic acid brain levels in the control and DHA groups did not differ. This is in contrast to brain DHA levels that were significantly different in the DHA group from control, making it unlikely that the effects on presenilin in brain are attributable to changes in linoleic acid levels.
DHA treatment reduces somatodendritic accumulation of tau
After 3 months, all dietary DHA treatments significantly reduced steady-state levels of tau protein (∼37%; p < 0.05), as observed by Western blot relative to mice on the control diet (Fig. 6 a,b). Likewise, the reduction in tau staining could also be visualized in hippocampal sections, where HT7 staining is observed abundantly in the somatodendritic compartment and throughout the processes in the control diet (Fig. 6 c,d, representative control and DHA–DPA treatments, respectively, from the 3 month treatment time point). After 6 months of supplementation, tau accumulation is significantly decreased in both the DHA-alone and DHA–DPA groups compared with controls (Fig. 6 e,f). At the final time point, after 9 months of supplementation, when soluble Aβ is only decreased in the DHA-alone group, human tau is similarly affected (Fig. 6 g,h). Therefore, it seems that the effects of DHA on tau accumulation lose efficacy over time, as also seen with soluble Aβ levels, strongly suggesting a direct relationship between Aβ and tau buildup.
Figure 6.

Tau steady-state levels in 3xTg-AD mice after DHA dietary enhancement. Western blot analyses of protein extracts from whole-brain homogenates of 3 month dietary-treated 3xTg-AD mice (n = 6) are shown as alternating lanes: C, control; D, DHA; DP, DHA–DPA; DA, DHA–ARA. a, Steady-state levels of total human tau from 3 month PUFA-treated 3xTg-AD mice. b, Quantification of protein blots from a shown normalized to β-actin levels as a loading control. c, d, Representative HT7 DAB staining of hippocampus from control and DHA–DPA-treated animals. e, Steady-state levels of total human tau from 6 month PUFA-treated 3xTg-AD mice. f, Quantification of protein blots from e shown normalized to β-actin levels as a loading control. g, Steady-state levels of total human tau from 9 month PUFA-treated 3xTg-AD mice. h, Quantification of protein blots from g shown normalized to β-actin levels as a loading control. The asterisk denotes significance (p < 0.05). Error bars indicate SEM.
Dietary DHA–DPA reduces phosphorylation and fragmentation of tau
Tau pathology progresses in a hierarchical manner, initially manifesting with its somatodendritic accumulation, followed by changes in conformation and phosphorylation patterns, and eventually developing into mature NFTs. All three treatment diets were able to reduce somatodendritic accumulation of tau, but this effect was transient and no longer apparent after 3 months. Presumably, this effect on tau buildup was attributable to the presence of DHA in the diets affecting Aβ levels, because all diets initially affected Aβ in the same manner. In the 3xTg-AD mice, we usually observe conformationally altered tau around 6 months of age, which can be visualized with immunostaining using antibody MC1 (Oddo et al., 2003). Staining of hippocampal sections from the control, DHA-alone, and DHA–DPA groups revealed reduced MC1-positive neurons in the DHA–DPA group and, to a lesser extent, the DHA-only group, relative to controls (Fig. 7 a, representative staining shown, quantified in Fig. 7 b). These data suggest that the presence of DHA in the diet is able to reduce soluble Aβ and somatodendritic accumulation of tau, but the additional presence of DPA prevented additional pathological tau progression.
Figure 7.
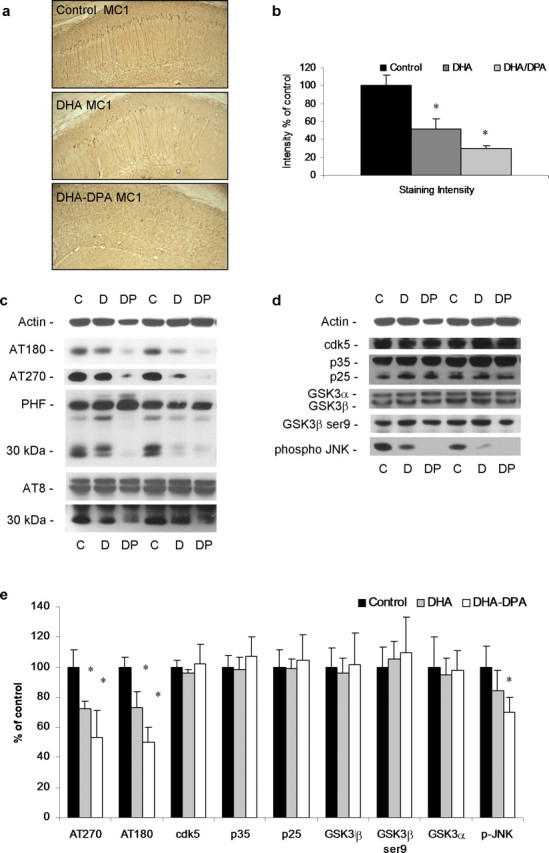
DPA reduces conformationally altered and phosphorylated tau in 3xTg-AD mice after dietary supplementation. a, DAB staining with MC1 shows conformationally altered tau immunoreactivity in 40 μm sections from 3 month diet-treated mice (control diet, DHA diet, and DHA–DPA diet). The hippocampus region is shown (magnification, 5×). DAB tau immunoreactivity was elevated in cell bodies of the hippocampus. b, Quantification of a. c, Steady-state levels of phospho-tau epitopes from 9 month PUFA-treated 3xTg-AD mice. d, Steady-state levels of putative tau kinases from 9 month PUFA-treated 3xTg-AD mice. e, Quantification of protein blots from c and d shown normalized to β-actin levels as a loading control. The asterisk denotes significance (p < 0.05). Error bars indicate SEM.
After 9 months, the effects of the DHA–DPA diet supplementation on Aβ and tau accumulation had dissipated, whereas DHA alone still had efficacy. At this age, we can examine the effects of the DHA–DPA diet on phosphorylation of tau, somewhat independently from its effects on Aβ, because there were no changes in the Aβ load at this time point (Fig. 3 e,f), although we cannot be certain that the same is true for events downstream of the Aβ pathology. Because steady-state levels of human tau (Fig. 6 g,h), the most well defined downstream pathology of Aβ in these mice, had returned to control levels, this seems likely. Interestingly early phospho-tau epitopes (AT270 and AT180) were significantly reduced in the DHA–DPA group compared with controls (Fig. 7 c,e). Levels of phospho-tau were also reduced in the DHA group, albeit to a lesser extent (Fig. 7 c,e), which may be attributable to the effects of DHA on tau phosphorylation or because the DHA group also had less total tau accumulation at this time point (Fig. 6 g,h), whereas the DHA–DPA group did not. Later phospho-tau epitopes, such as PHF and AT8, were not altered with the dietary treatments (Fig. 7 c,e), but a ∼30 kDa fragment was reduced with DPA treatment (Fig. 7 c).
We also examined the two major tau kinases to determine whether their steady-state levels or active forms varied. GSK3β and cdk5 have been shown to hyperphosphorylate tau and are sometimes referred to as tau kinase 1 and 2 (Kobayashi et al., 1993; Flaherty et al., 2000). We found no differences in cdk5 or in its activator p25 (Fig. 7 d,e). Likewise, there were no differences in GSK3β or its inactive form, which is phosphorylated at ser9 (Fig. 7 d,e). n-3 PUFAs (EPA and DHA) have previously been shown to reduce phospho-JNK both in vivo and in vitro (Moon and Pestka, 2003; Xue et al., 2006), and in agreement with this, we found significant reductions in phospho-JNK in the DHA–DPA group compared with controls (Fig. 7 d,e). These reductions correlate with the reductions in phosho-tau seen with this dietary treatment. This mitogen-activated protein (MAP) kinase has been shown to be activated around NFTs in AD (Zhu et al., 2001).
Discussion
Here we report the novel findings that dietary DHA-mediated reductions in soluble Aβ correlate with decreased steady-state levels of PS1. Thus, the influence of DHA on Aβ levels is most likely mediated by a reduction in γ-secretase activity. If APP processing by α- or β-secretases was altered, then we would expect to observe differences in the APP holoprotein or the CTFs C83 and C99, which we did not. An increase in CTFs may have been expected with a decrease in PS1 levels; however, we did not find one, perhaps because of the chronic nature of the treatment or sensitivity of the assay (Western blot for the CTFs vs the more sensitive sandwich ELISA for Aβ measurements), or perhaps because the CTFs are more rapidly degraded, because they have been shown previously to be degraded by the endosomal/lysosomal systems (Agiostratidou et al., 2006). Supporting this, PS1 is known to stablize APP CTFs such that similar increases are seen in both Aβ and C99 with PS1 overexpression (Pitsi and Octave, 2004) rather than a concurrent decrease in C99 levels. This finding corroborates recent studies showing that dietary DHA supplementation could reverse Aβ pathology in mouse models of AD, which also found no differences in APP processing with increased DHA (Lim et al., 2005; Oksman et al., 2006). Hence, DHA seems to act as a natural γ-secretase “suppressor.” How it does this is not clear, but fatty acids have previously been shown to affect PS1 levels and activity (Liu et al., 2004). Because the steady-state levels of nicastrin were unaltered, the effects of DHA appear to be specific for PS1, rather than the entire γ-secretase complex, although we cannot rule out effects on other members of the γ-secretase complex such as Pen2 or Aph-1.
Intriguingly, dietary DHA treatment reduced soluble and intraneuronal Aβ but not the insoluble Aβ pool. The most likely reason is that only a certain amount of soluble Aβ is able to aggregate sufficiently into high-molecular-weight assemblies and that dietary PUFA treatment does not affect these dynamics, or that there are multiple pools of Aβ (Skovronsky et al., 1998) and PUFA treatment does not affect the pool that is destined to become high-molecular-weight aggregates. It may be that the aged brain allows more Aβ to aggregate into these high-molecular-weight assemblies, which may explain why transgenic mice overexpressing APP develop increasing amounts of Aβ plaques with age, despite constant levels of APP expression.
Although the treatment diets contained DHA, they also contained varying amounts of other PUFAs, such as DPA and ARA. Clearly, the treatment diets all increased DHA levels in the brain and RBCs, but effects on other PUFAs were also notable. One striking observation was that the presence of DHA in the diets appeared to reduce the amount of ARA in both brain and RBCs, although this effect was somewhat negated by the additional presence of DPA. This reduction in ARA may also have effects on Aβ processing through its breakdown products, because prostaglandin E2 has been shown to increase γ-secretase activity resulting in more Aβ (Qin et al., 2003). Reducing ARA, with DHA treatment, would lead to a subsequent reduction in prostaglandins.
Our study also revealed that dietary DHA treatment reduces intraneuronal Aβ accumulation in the 3xTg-AD mice. The relevance of this Aβ pool to AD and to cognition is still not completely understood. Although Aβ-laden plaques are the most visible and well characterized amyloid pathology in the AD brain, plaque load correlates poorly with cognitive decline (Braak and Braak, 1997), with many elderly showing Aβ plaque pathology postmortem without cognitive decline before death (Benzing et al., 1993). One argument is that Aβ in plaques is merely an end point of the amyloidosis and, moreover, may provide a protective mechanism to sequester soluble oligomeric Aβ species and diminish the influence of these more toxic Aβ species on cognition function. In support of this, levels of soluble Aβ correlate well with cognitive decline (Lue et al., 1999; McLean et al., 1999; Naslund et al., 2000). Furthermore, the 3xTg-AD mouse develops intracellular Aβ accumulation in the cell bodies of neurons in the hippocampus before any plaque pathology develops, and at an age at which both synaptic dysfunction and cognitive impairments are observed (Oddo et al., 2003; Billings et al., 2005). Therefore, it could be argued that intracellular accumulation of Aβ in AD is a critical factor in the disease progression and any therapy that reduces or prevents it would be beneficial.
In addition to the reduction in soluble and intraneuronal Aβ, dietary DHA also reduces steady-state levels of tau, which accumulate in the somatodendritic compartments of neurons. Our previous studies clearly demonstrated a hierarchical relationship between Aβ and tau pathologies, with Aβ causing tau to accumulate and to undergo phosphorylation. Removal of intraneuronal Aβ via immunotherapy leads to tau clearance from neurons several days after the Aβ itself has cleared (Oddo et al., 2004). Therefore, the reduction we observe here in steady-state levels of tau with DHA seems to be closely related to the reduction seen in soluble Aβ. In fact, the correlation between soluble Aβ levels and steady-state levels of tau is very strong, at all treatment time points. At the point when the n-6 PUFA-containing diets lose efficacy at reducing soluble Aβ, we also noted that they no longer reduced tau steady-state levels, strengthening the relationship between the two pathologies.
One surprising and novel aspect of our finding is that although we found that dietary DHA supplementation was effective at preventing accumulation of both Aβ and tau, it should be noted that the efficacy of the DHA–ARA and DHA–DPA diets declined with time and, after 6 and 9 months of treatment, respectively, had lost all efficacy completely. DHA alone was still effective, however, up to the last time point evaluated, which was after 9 months of treatment. This finding is impressive given the fact that it had to overcome transgene expression rather than the gradual increase in Aβ in AD that takes many years to form. Therefore, despite the fact that the omega-3 and omega-6 combined diets lost efficacy with time, increased dietary DHA had long-lasting effects on AD pathology, and it remains to be determined whether DHA will be effective in AD patients, where the pathology presumably develops more gradually, compared with transgenic mice, where it develops more acutely.
Perhaps the most novel aspect of our findings is that the presence of DPAn-6 in the diet had dramatic effects on the progression of tau pathology, as seen after 9 months of dietary treatment when the animals had reached 12 months of age, preventing both conformational changes and phosphorylation of tau. These effects were independent from any effects on Aβ because they occurred at time points when soluble Aβ were no longer reduced with DPAn-6 (after 9 months of treatment). However, the role that phospho-tau and neurofibrillary tangles actually play in AD is unclear, including whether or not they contribute to cognitive decline and neuronal loss, or whether it is a different soluble tau species that is toxic. As additional research is conducted, this issue will be resolved, but the accumulation of phospho-tau in the AD brain is a highly visible and defined event that represents a potential therapeutic avenue, albeit a harder and less defined one than preventing production of the Aβ peptide. Our results also suggest that these striking reductions in tau phosphorylation are not mediated by effects on GSK3β and cdk5. However, we did find a significant reduction in levels of activated (phosphorylated) JNK, a member of the stress-activated MAP kinase family, with DHA–DPA treatment, which correlated with this reduction in phospho-tau. JNK has been shown to hyperphosphorylate tau (Reynolds et al., 1997; Yoshida et al., 2004; Tatebayashi et al., 2006) and is found in tau-laden neurons in AD (Pei et al., 2001; Zhu et al., 2001). Furthermore, n-3 PUFAs (EPA and DHA) have been shown to reduce phospho-JNK both in vivo and in vitro (Moon and Pestka, 2003; Xue et al., 2006), although DPA has not been tested previously.
DHA-DPA is the first treatment paradigm described that effectively ameliorates tau pathology, and because it is a combination of PUFAs (omega-3 and omega-6 fatty acids) rather than a pharmacological compound, it is a well characterized, safe, and viable treatment option for AD. Notably, the presence of DPAn-6 in the diets prevented the appearance of a ∼30 kDa fragment of PHF and AT8 phospho-tau. This size fragment corresponds to a previously described cleavage fragment of tau, mediated by thrombin (Arai et al., 2005; Khlistunova et al., 2006), which prefers to cleave extensively phosphorylated tau rather than dephosphorylated tau. Although the relevance of this fragment to AD pathology and cognition remains unknown, it may prove to be critical for aggregation and/or toxicity of tau making DHA–DPA an attractive therapy that seems to target this tau fragment as well as decrease early phospho-tau epitopes.
Overall, additional studies in aged humans should be conducted to assess how well PUFA supplementation translates from AD mouse models, because DHA and DHA–DPA look very promising as potential therapeutic agents.
Footnotes
This work was supported by grants from Martek Biosciences Corporation. We thank Drs. Matthew Blurton-Jones and Salvatore Oddo for helpful discussions and Dennis Selkoe for the anti-IDE antibody.
References
- Adlard PA, Perreau VM, Pop V, Cotman CW. Voluntary exercise decreases amyloid load in a transgenic model of Alzheimer's disease. J Neurosci. 2005;25:4217–4221. doi: 10.1523/JNEUROSCI.0496-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agiostratidou G, Muros RM, Shioi J, Marambaud P, Robakis NK. The cytoplasmic sequence of E-cadherin promotes non-amyloidogenic degradation of A beta precursors. J Neurochem. 2006;96:1182–1188. doi: 10.1111/j.1471-4159.2005.03616.x. [DOI] [PubMed] [Google Scholar]
- Ansari KA, Shoeman DW. Arachidonic and docosahexanoic acid content of bovine brain myelin: implications for the pathogenesis of multiple sclerosis. Neurochem Res. 1990;15:7–11. doi: 10.1007/BF00969177. [DOI] [PubMed] [Google Scholar]
- Arai T, Guo JP, McGeer PL. Proteolysis of non-phosphorylated and phosphorylated tau by thrombin. J Biol Chem. 2005;280:5145–5153. doi: 10.1074/jbc.M409234200. [DOI] [PubMed] [Google Scholar]
- Bazan NG, Scott BL. Dietary omega-3 fatty acids and accumulation of docosahexaenoic acid in rod photoreceptor cells of the retina and at synapses. Ups J Med Sci Suppl. 1990;48:97–107. [PubMed] [Google Scholar]
- Benzing WC, Mufson EJ, Armstrong DM. Immunocytochemical distribution of peptidergic and cholinergic fibers in the human amygdala: their depletion in Alzheimer's disease and morphologic alteration in non-demented elderly with numerous senile plaques. Brain Res. 1993;625:125–138. doi: 10.1016/0006-8993(93)90145-d. [DOI] [PubMed] [Google Scholar]
- Billings L, Green KN, McGaugh JL, LaFerla FM. Learning decreases Abeta*56, tau pathology and ameliorates behavioral decline in 3xTg-AD mice. J Neurosci. 2007;27:751–761. doi: 10.1523/JNEUROSCI.4800-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Diagnostic criteria for neuropathologic assessment of Alzheimer's disease. Neurobiol Aging. 1997;18:S85–S88. doi: 10.1016/s0197-4580(97)00062-6. [DOI] [PubMed] [Google Scholar]
- Caccamo A, Oddo S, Sugarman MC, Akbari Y, LaFerla FM. Age- and region-dependent alterations in Abeta-degrading enzymes: implications for Abeta-induced disorders. Neurobiol Aging. 2005;26:645–654. doi: 10.1016/j.neurobiolaging.2004.06.013. [DOI] [PubMed] [Google Scholar]
- Calon F, Lim GP, Yang F, Morihara T, Teter B, Ubeda O, Rostaing P, Triller A, Salem N, Jr, Ashe KH, Frautschy SA, Cole GM. Docosahexaenoic acid protects from dendritic pathology in an Alzheimer's disease mouse model. Neuron. 2004;43:633–645. doi: 10.1016/j.neuron.2004.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty DB, Soria JP, Tomasiewicz HG, Wood JG. Phosphorylation of human tau protein by microtubule-associated kinases: GSK3beta and cdk5 are key participants. J Neurosci Res. 2000;62:463–472. doi: 10.1002/1097-4547(20001101)62:3<463::AID-JNR16>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Gilfillan AM, Chu AJ, Smart DA, Rooney SA. Single plate separation of lung phospholipids including disaturated phosphatidylcholine. J Lipid Res. 1983;24:1651–1656. [PubMed] [Google Scholar]
- Hashimoto M, Tanabe Y, Fujii Y, Kikuta T, Shibata H, Shido O. Chronic administration of docosahexaenoic acid ameliorates the impairment of spatial cognition learning ability in amyloid beta-infused rats. J Nutr. 2005;135:549–555. doi: 10.1093/jn/135.3.549. [DOI] [PubMed] [Google Scholar]
- Iwata N, Tsubuki S, Takaki Y, Watanabe K, Sekiguchi M, Hosoki E, Kawashima-Morishima M, Lee HJ, Hama E, Sekine-Aizawa Y, Saido TC. Identification of the major Abeta1–42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat Med. 2000;6:143–150. doi: 10.1038/72237. [DOI] [PubMed] [Google Scholar]
- Joseph JA, Denisova NA, Arendash G, Gordon M, Diamond D, Shukitt-Hale B, Morgan D. Blueberry supplementation enhances signaling and prevents behavioral deficits in an Alzheimer disease model. Nutr Neurosci. 2003;6:153–162. doi: 10.1080/1028415031000111282. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Khlistunova I, Biernat J, Wang Y, Pickhardt M, von Bergen M, Gazova Z, Mandelkow E, Mandelkow EM. Inducible expression of Tau repeat domain in cell models of tauopathy: aggregation is toxic to cells but can be reversed by inhibitor drugs. J Biol Chem. 2006;281:1205–1214. doi: 10.1074/jbc.M507753200. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Ishiguro K, Omori A, Takamatsu M, Arioka M, Imahori K, Uchida T. A cdc2-related kinase PSSALRE/cdk5 is homologous with the 30 kDa subunit of tau protein kinase II, a proline-directed protein kinase associated with microtubule. FEBS Lett. 1993;335:171–175. doi: 10.1016/0014-5793(93)80723-8. [DOI] [PubMed] [Google Scholar]
- Koistinaho M, Lin S, Wu X, Esterman M, Koger D, Hanson J, Higgs R, Liu F, Malkani S, Bales KR, Paul SM. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat Med. 2004;10:719–726. doi: 10.1038/nm1058. [DOI] [PubMed] [Google Scholar]
- Kurochkin IV, Goto S. Alzheimer's beta-amyloid peptide specifically interacts with and is degraded by insulin degrading enzyme. FEBS Lett. 1994;345:33–37. doi: 10.1016/0014-5793(94)00387-4. [DOI] [PubMed] [Google Scholar]
- Lahiri DK, Chen D, Ge YW, Bondy SC, Sharman EH. Dietary supplementation with melatonin reduces levels of amyloid beta-peptides in the murine cerebral cortex. J Pineal Res. 2004;36:224–231. doi: 10.1111/j.1600-079X.2004.00121.x. [DOI] [PubMed] [Google Scholar]
- Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M, Haass C, Fahrenholz F. Constitutive and regulated alpha-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci USA. 1999;96:3922–3927. doi: 10.1073/pnas.96.7.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarov O, Robinson J, Tang YP, Hairston IS, Korade-Mirnics Z, Lee VM, Hersh LB, Sapolsky RM, Mirnics K, Sisodia SS. Environmental enrichment reduces Abeta levels and amyloid deposition in transgenic mice. Cell. 2005;120:701–713. doi: 10.1016/j.cell.2005.01.015. [DOI] [PubMed] [Google Scholar]
- Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Lim GP, Calon F, Morihara T, Yang F, Teter B, Ubeda O, Salem N, Jr, Frautschy SA, Cole GM. A diet enriched with the omega-3 fatty acid docosahexaenoic acid reduces amyloid burden in an aged Alzheimer mouse model. J Neurosci. 2005;25:3032–3040. doi: 10.1523/JNEUROSCI.4225-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Yang L, Conde-Knape K, Beher D, Shearman MS, Shachter NS. Fatty acids increase presenilin-1 levels and γ-secretase activity in PSwt-1 cells. J Lipid Res. 2004;45:2368–2376. doi: 10.1194/jlr.M400317-JLR200. [DOI] [PubMed] [Google Scholar]
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo HM, Deng H, Xiao F, Gao Q, Weng W, Zhang PF, Li XG. Down-regulation amyloid beta-protein 42 production by interfering with transcript of presenilin 1 gene with siRNA. Acta Pharmacol Sin. 2004;25:1613–1618. [PubMed] [Google Scholar]
- Marlow L, Canet RM, Haugabook SJ, Hardy JA, Lahiri DK, Sambamurti K. APH1, PEN2, and Nicastrin increase Abeta levels and gamma-secretase activity. Biochem Biophys Res Commun. 2003;305:502–509. doi: 10.1016/s0006-291x(03)00797-6. [DOI] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Moon Y, Pestka JJ. Deoxynivalenol-induced mitogen-activated protein kinase phosphorylation and IL-6 expression in mice suppressed by fish oil. J Nutr Biochem. 2003;14:717–726. doi: 10.1016/j.jnutbio.2003.08.009. [DOI] [PubMed] [Google Scholar]
- Morris MC, Evans DA, Bienias JL, Tangney CC, Bennett DA, Wilson RS, Aggarwal N, Schneider J. Consumption of fish and n-3 fatty acids and risk of incident Alzheimer disease. Arch Neurol. 2003;60:940–946. doi: 10.1001/archneur.60.7.940. [DOI] [PubMed] [Google Scholar]
- Morrison WR, Smith LM. Preparation of fatty acid methyl esters and dimethylacetals from lipids with boron fluoride–methanol. J Lipid Res. 1964;5:600–608. [PubMed] [Google Scholar]
- Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA. 2000;283:1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43:321–332. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Oddo S, Vasilevko V, Caccamo A, Kitazawa M, Cribbs DH, Laferla FM. Reduction of soluble Abeta and tau, but not soluble Abeta alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J Biol Chem. 2006;281:39413–39423. doi: 10.1074/jbc.M608485200. [DOI] [PubMed] [Google Scholar]
- Oksman M, Iivonen H, Hogyes E, Amtul Z, Penke B, Leenders I, Broersen L, Lutjohann D, Hartmann T, Tanila H. Impact of different saturated fatty acid, polyunsaturated fatty acid and cholesterol containing diets on beta-amyloid accumulation in APP/PS1 transgenic mice. Neurobiol Dis. 2006;23:563–572. doi: 10.1016/j.nbd.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Pei JJ, Braak E, Braak H, Grundke-Iqbal I, Iqbal K, Winblad B, Cowburn RF. Localization of active forms of C-jun kinase (JNK) and p38 kinase in Alzheimer's disease brains at different stages of neurofibrillary degeneration. J Alzheimers Dis. 2001;3:41–48. doi: 10.3233/jad-2001-3107. [DOI] [PubMed] [Google Scholar]
- Pitsi D, Octave JN. Presenilin 1 stabilizes the C-terminal fragment of the amyloid precursor protein independently of gamma-secretase activity. J Biol Chem. 2004;279:25333–25338. doi: 10.1074/jbc.M312710200. [DOI] [PubMed] [Google Scholar]
- Puskas LG, Kitajka K, Nyakas C, Barcelo-Coblijn G, Farkas T. Short-term administration of omega 3 fatty acids from fish oil results in increased transthyretin transcription in old rat hippocampus. Proc Natl Acad Sci USA. 2003;100:1580–1585. doi: 10.1073/pnas.0337683100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin W, Ho L, Pompl PN, Peng Y, Zhao Z, Xiang Z, Robakis NK, Shioi J, Suh J, Pasinetti GM. Cyclooxygenase (COX)-2 and COX-1 potentiate beta-amyloid peptide generation through mechanisms that involve gamma-secretase activity. J Biol Chem. 2003;278:50970–50977. doi: 10.1074/jbc.M307699200. [DOI] [PubMed] [Google Scholar]
- Reich EE, Markesbery WR, Roberts LJ, II, Swift LL, Morrow JD, Montine TJ. Brain regional quantification of F-ring and D-/E-ring isoprostanes and neuroprostanes in Alzheimer's disease. Am J Pathol. 2001;158:293–297. doi: 10.1016/S0002-9440(10)63968-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds CH, Nebreda AR, Gibb GM, Utton MA, Anderton BH. Reactivating kinase/p38 phosphorylates tau protein in vitro. J Neurochem. 1997;69:191–198. doi: 10.1046/j.1471-4159.1997.69010191.x. [DOI] [PubMed] [Google Scholar]
- Rezai-Zadeh K, Shytle D, Sun N, Mori T, Hou H, Jeanniton D, Ehrhart J, Townsend K, Zeng J, Morgan D, Hardy J, Town T, Tan J. Green tea epigallocatechin-3-gallate (EGCG) modulates amyloid precursor protein cleavage and reduces cerebral amyloidosis in Alzheimer transgenic mice. J Neurosci. 2005;25:8807–8814. doi: 10.1523/JNEUROSCI.1521-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer EJ, Bongard V, Beiser AS, Lamon-Fava S, Robins SJ, Au R, Tucker KL, Kyle DJ, Wilson PW, Wolf PA. Plasma phosphatidylcholine docosahexaenoic acid content and risk of dementia and Alzheimer disease: the Framingham Heart Study. Arch Neurol. 2006;63:1545–1550. doi: 10.1001/archneur.63.11.1545. [DOI] [PubMed] [Google Scholar]
- Skovronsky DM, Doms RW, Lee VM. Detection of a novel intraneuronal pool of insoluble amyloid beta protein that accumulates with time in culture. J Cell Biol. 1998;141:1031–1039. doi: 10.1083/jcb.141.4.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein TD, Johnson JA. Lack of neurodegeneration in transgenic mice overexpressing mutant amyloid precursor protein is associated with increased levels of transthyretin and the activation of cell survival pathways. J Neurosci. 2002;22:7380–7388. doi: 10.1523/JNEUROSCI.22-17-07380.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatebayashi Y, Planel E, Chui DH, Sato S, Miyasaka T, Sahara N, Murayama M, Kikuchi N, Yoshioka K, Rivka R, Takashima A. c-jun N-terminal kinase hyperphosphorylates R406W tau at the PHF-1 site during mitosis. FASEB J. 2006;20:762–764. doi: 10.1096/fj.05-4362fje. [DOI] [PubMed] [Google Scholar]
- Travis J. New piece in Alzheimer's puzzle. Science. 1993;261:828–829. doi: 10.1126/science.8346434. [DOI] [PubMed] [Google Scholar]
- Tsuzuki K, Fukatsu R, Yamaguchi H, Tateno M, Imai K, Fujii N, Yamauchi T. Transthyretin binds amyloid beta peptides, Abeta1–42 and Abeta1–40 to form complex in the autopsied human kidney - possible role of transthyretin for abeta sequestration. Neurosci Lett. 2000;281:171–174. doi: 10.1016/s0304-3940(00)00834-x. [DOI] [PubMed] [Google Scholar]
- Tully AM, Roche HM, Doyle R, Fallon C, Bruce I, Lawlor B, Coakley D, Gibney MJ. Low serum cholesteryl ester-docosahexaenoic acid levels in Alzheimer's disease: a case-control study. Br J Nutr. 2003;89:483–489. doi: 10.1079/BJN2002804. [DOI] [PubMed] [Google Scholar]
- Xue H, Wan M, Song D, Li Y, Li J. Eicosapentaenoic acid and docosahexaenoic acid modulate mitogen-activated protein kinase activity in endothelium. Vascul Pharmacol. 2006;44:434–439. doi: 10.1016/j.vph.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Hastie CJ, McLauchlan H, Cohen P, Goedert M. Phosphorylation of microtubule-associated protein tau by isoforms of c-Jun N-terminal kinase (JNK) J Neurochem. 2004;90:352–358. doi: 10.1111/j.1471-4159.2004.02479.x. [DOI] [PubMed] [Google Scholar]
- Zhu X, Raina AK, Rottkamp CA, Aliev G, Perry G, Boux H, Smith MA. Activation and redistribution of c-jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer's disease. J Neurochem. 2001;76:435–441. doi: 10.1046/j.1471-4159.2001.00046.x. [DOI] [PubMed] [Google Scholar]