

Abstract
Mature mammals are uniquely vulnerable to permanent auditory and vestibular deficits, because the cell proliferation that produces replacement hair cells in other vertebrates is limited in mammals. To investigate the cellular mechanisms responsible for that difference, we created excision lesions in the sensory epithelium of embryonic and 2-week-old mouse utricles. Lesions in embryonic utricles closed in <24 h via localized expansion of supporting cells, which then reentered the cell cycle. Pharmacological treatments combined with time-lapse microscopy demonstrated that the healing depended on Rho-mediated contraction of an actin ring at the leading edge of the lesion. In contrast, lesions in utricles from 2-week-old and older mice remained open even after 48 h. Supporting cells in those utricles remained compact and columnar and had significantly stouter cortical actin belts than those in embryonic sensory epithelia. This suggests that cytoskeletal changes may underlie the age-related loss of proliferation in mammalian ears by limiting the capacity for mature supporting cells to change shape. In mature utricles, exogenous stimulation with lysophosphatidic acid overcame this maturational block and induced closure of lesions, promoting supporting cell expansion and subsequent proliferation. After lysophosphatidic acid treatment, 85% of the mature supporting cells that had spread to a planar area >300 μm2 entered S-phase, whereas only 10% of those cells that had a planar area <100 μm2 entered S-phase. Together, these results indicate that cellular shape change can overcome the normal postnatal cessation of supporting cell proliferation that appears to limit regeneration in mammalian vestibular epithelia.
Keywords: hair cell, regeneration, lysophosphatidic acid, LPA, vestibular, wound healing, cytoskeleton
Introduction
Mammals appear to be unique in their susceptibility to permanent auditory and vestibular deficits that arise through loss of sensory hair cells. After damage to the sensory epithelia of birds, amphibians, and fish, supporting cells dedifferentiate, reenter the cell cycle, and produce replacement hair cells that restore hearing and balance function (for review, see Corwin et al., 1991). After damage to mature mammalian vestibular epithelia, some supporting cells will proliferate (Warchol et al., 1993). Such damage also may evoke nonproliferative mechanisms of recovery (Li and Forge, 1997), but the clinical permanence of hearing and vestibular deficits in mammals suggests that the low level of the proliferative response may preclude significant functional recovery. Thus, there is considerable interest in understanding the control of proliferation in the mammalian inner ear.
In mammals, supporting cell proliferation can be enhanced by growth factor or pharmacological treatment (Lambert, 1994; Gu et al., 1996; Zheng et al., 1997; Montcouquiol and Corwin, 2001a, b). The intracellular cascades that control this proliferation appear similar in young rodents and chickens (Witte et al., 2001), but proliferative response to growth factors decreases sharply during the first postnatal weeks in mammals (Gu et al., 1997, 2007; Hume et al., 2003).
Two cyclin-dependent kinase inhibitors, p27Kip1 and p19Ink4, and the tumor-suppressor retinoblastoma (Rb1) provide downstream regulation of cell-cycle entry within mammalian hair cell epithelia (Chen and Segil, 1999; Lowenheim et al., 1999; Chen et al., 2003; White et al., 2006), yet the fundamental upstream changes that underlie the loss of proliferative responsiveness in mammals remain unknown, as do the mechanisms in other vertebrates that are responsible for initiating proliferation during hair cell regeneration.
One candidate trigger for proliferation in hair cell epithelia is change in supporting cell shape (Corwin et al., 1996). Hair cell loss in birds is followed by significant expansion of the apical surfaces of supporting cells near the sites of damage and subsequent passage through the cell cycle (Cotanche, 1987; Cotanche and Dopyera, 1990; Marsh et al., 1990). A flattened and spread shape is necessary for endothelial cell proliferation in vitro (Folkman and Moscona, 1978; Chen et al., 1997), and cell density and spreading correlate with S-phase entry in cultured sensory epithelia isolated from avian utricles (Warchol, 1995, 2002; Witte et al., 2001).
To determine whether changes in supporting cell shape would trigger S-phase entry in mammalian sensory epithelia that remained in situ, we made surgical lesions in mouse utricles. The precise timing and reproducibility of this method for producing damage in organ culture allowed direct observation of the early cellular responses to damage, which often are obscured by variability in the timing, location, and extent of hair cell death caused by acoustic overstimulation and aminoglycoside toxicity. In embryonic utricles, supporting cells spread and covered over the lesions in <24 h via Rho-mediated contraction of an actin purse string, and, by 72 h, the spread supporting cells entered S-phase. In contrast, lesions in utricles from 14-d-old mice remained open for at least 48 h, with the supporting cells retaining compact and columnar shapes. To activate Rho, such older utricles were treated with lysophosphatidic acid (LPA), which resulted in spreading of supporting cells, closure of lesions, and S-phase entry at levels not normally observed in mature mammalian ears. These results suggest that a decreased capacity for supporting cells to change shape may underlie the loss of regenerative proliferation in the vestibular organs of mature mammals, but substantial levels of proliferation can be restored by stimulating cellular shape change.
Materials and Methods
Dissection of utricles.
All animal experiments were performed according to protocols approved by the Animal Care and Use Committee at the University of Virginia. Swiss Webster mice were obtained from Taconic Farms (Germantown, NY) and Charles River Laboratories (Wilmington, MA), and Tg(GFPU)5Nagy mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Animals were killed by CO2 asphyxiation and decapitated. Labyrinths were removed aseptically from temporal bones in ice-cold DMEM/F-12 (Invitrogen, Carlsbad, CA), the utricles were isolated, and the roof and the otoconia were removed.
In vitro culture and lesioning. Embryonic utricles were adhered with the nerve side down on poly-l-lysine-coated glass-bottom dishes (MatTek, Ashland, MA). Utricles from postnatal animals were adhered nerve side down in MatTek dishes treated with CellTak (2 μl, air-dried onto the glass and washed three times with medium; BD Biosciences, San Jose, CA). For time-lapse experiments, embryonic utricles were adhered nerve side down to a 42 mm round cover glass treated with CellTak. No difference in healing was observed between embryonic utricles attached via poly-l-lysine versus CellTak.
Micropunches were fabricated by electrolytic etching of blunt 29 gauge stainless steel hypodermic needles (nominal internal diameter of 180 μm; Hamilton, Reno, NV) in a solution of 34% sulfuric acid and 42% phosphoric acid at 4.5 V, 500 mA. Rough edges were removed with 1 μm grit polishing paper (3M, Minneapolis, MN), and etching and polishing were repeated until the tip had a sharp, circular edge.
The micropunches were pressed into utricles to make circular lesions in the hair cell epithelium, and cells from within the lesion were removed with a sharpened tungsten needle. Lesioned utricles were then cultured in DMEM/F-12 with 5% FBS (HyClone, Logan, UT), 3 μg/ml bromodeoxyuridine (BrdU; Sigma, St. Louis, MO), 0.25 μg/ml Fungizone (Invitrogen), and 10 μg/ml Ciprofloxacin (Bayer, Berlin, Germany), and fixed in Glyofixx (for immunocytochemistry), 4% paraformaldehyde in PBS (for phalloidin visualization of actin), or 2.5% glutaraldehyde/2% paraformaldehyde in 0.1 m sodium cacodylate (for electron microscopy). Utricles fixed for electron microscopy were made conductive by osmium–thiocarbohydrazide impregnation (Kelley et al., 1973; Malick and Wilson, 1975).
Time-lapse microscopy.
Lesioned utricles were adhered to a 42-mm-diameter cover glass with CellTak (BD Biosciences) for time-lapse microscopy. The cover glass was sealed into a POC-R imaging chamber (LaCon, Staig, Germany) containing DMEM/F-12 with 5% FBS, 3 μg/ml BrdU, 0.25 μg/ml Fungizone, and 10 μg/ml Ciprofloxacin. The chamber was placed on an Axiovert 200M microscope (Zeiss, Oberkochen, Germany) with the epithelia oriented downward for imaging. A microscope stage incubator (PeCon, Erbach-Bach, Germany) was maintained at 5% CO2 and 37°C for the duration of the experiment. A motorized stage that was under MetaMorph software control (Molecular Devices, Sunnyvale, CA) allowed sequential imaging of responses occurring concurrently in 2–12 utricles throughout each time-lapse recording. Every 10–30 min, images were recorded at each of 10 focal levels extending through the depth of the sensory epithelium for each utricle in the chamber.
For time-lapse tests of pharmacologic agents, utricles were divided into equal groups and preincubated for 30 min at 37°C in either the control medium or medium supplemented with the drug. Then all of the utricles were lesioned with a micropunch, and the utricles in each pool (control and experimental) were adhered to a single 12-mm-diameter cover glass coated with CellTak. The control and experimental cover glasses were each fitted with a custom-made ring of 250-μm-thick silicone gasket material (McMaster-Carr, Atlanta GA), and either control medium or medium containing a pharmacological agent was added to the small well formed by the gasket. The two gasketed cover glasses were then inverted onto a 42 mm circular cover glass, creating two individually sealed compartments within the same POC-R imaging chamber.
To block actin polymerization, utricles were cultured in 1 μm cytochalasin D or 1 μm cytochalasin B (Sigma), and controls were cultured in 0.1% DMSO as a vehicle control. To inhibit Rho-mediated kinases, utricles were cultured in 50 μm (R)-(+)-trans-N-(4-pyridyl)-4-(1-aminoethyl)-cyclohexanecarboxamide (Y-27632; EMD Biosciences, La Jolla, CA). To inhibit myosin light chain kinase (MLCK), utricles were cultured in 1 or 20 μm 1-(5-iodonaphthalene-1-sulfonyl)homopiperazine (ML-7; EMD Biosciences). To washout media containing drugs, the gasketed coverslips were lifted out of the POC-R chamber, and the cover glass was washed in four changes of fresh drug-free culture medium over 20 min before the chambers were reassembled with fresh culture medium without drugs for continued imaging.
Time-lapse of green fluorescent protein-expressing utricles.
To visualize changes in individual cell shapes, time-lapse recordings were made using utricles removed at embryonic day 18 (E18) (n = 11) or on the day of birth [postnatal day 0 (P0)] (n = 12) from transgenic mice that expressed green fluorescent protein (GFP) under control of the β-actin promoter [Tg(GFPU)5Nagy] (Hadjantonakis et al., 1998). Variations in the levels of GFP fluorescence among the cells in the sensory epithelium allowed individual cells to be tracked during the course of the recording (see Fig. 2). Images over 10 focal planes, each 10 μm apart, were acquired every hour for 24 h using a Zeiss 510 confocal LSM with an incubation chamber as described above. The positions of individual bright or dim cells were then tracked via automated object tracking using MetaMorph software, in which the center of each cell was determined in each frame based on an object-specific template defined by differences in signal intensity and shape. The template was updated with each frame to adjust for changes during the time-lapse recordings.
Figure 2.
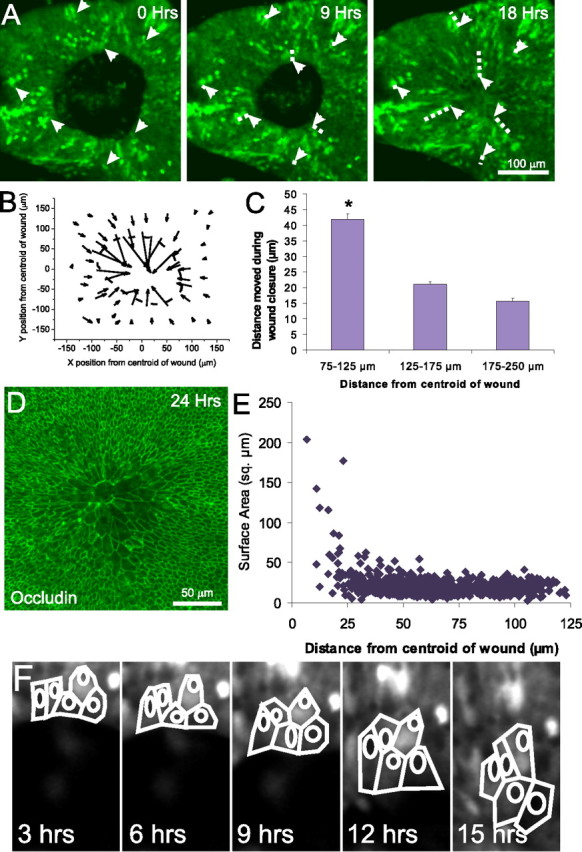
Closure of lesions in embryonic utricles occurs via movement and cell shape change of cells at the edge of the lesion. A, Frames from a time-lapse recording of a closing of a lesion in the utricle from an E18 transgenic mouse that expresses GFP under control of the β-actin promoter. The GFP is expressed in a mosaic pattern, allowing tracking of individual cell movements (arrows). The trajectory of the cells in the 9 and 18 h images is marked with a dashed line. Cells well outside the lesion moved little during closure of lesions, whereas cells at the leading edge of the lesion moved into the center of the lesion. B, A vector plot showing the distance and direction of movement of 59 cells whose positions were tracked over 15 h of closure of a lesion in a P0 GFP mouse utricle. The cells began at the back of the arrows and moved to the tip of the arrowheads over the 15 h, with each arrow representing the movement of a single cell in the epithelium. The dashed line indicates the initial location of the lesion. Cells at the edge of the lesion moved substantially inward, whereas cells distant from the lesion moved less. C, Histogram of distance moved from 75 cells from each of six E18 GFP mosaic utricles (tracked as in B) plotted based on the original distance from the centroid of the lesion (mean ± SEM). Cells at the edge of the lesion, between 75 and 125 μm of the centroid of the lesion, move significantly farther than cells >125 μm from the lesion (*p < 0.005). D, Fluorescent micrograph showing occludin labeling of the tight junctions between the epithelial cells in an E18 utricle fixed 24 h after lesioning. The occludin staining demarcates the lateral edge of the apical surface of the cells, revealing the expansion of supporting cells at the center of the lesion. E, Comparison of planar surface area versus distance to the center of the lesion for 627 cells in an E18 utricle, showing that cells that moved into center of the lesion site took on greatly expanded apical surface area, whereas more distant cells retained a compact shape. F, Tracings of five cells at the leading edge of the lesion of an E18 GFP mosaic utricle taken every 3 h showing their expansion as the lesion closes.
LPA stimulation of mature utricles.
To exogenously stimulate Rho, 24 mature utricles were cultured in the presence of 10 μm lysophosphatidic acid (Avanti Polar Lipids, Alabaster, AL). A 1 mm stock of LPA was made in 3% fatty-acid-free bovine serum albumin (FAF-BSA; Sigma) in water; 21 controls were cultured in FAF-BSA without LPA. LPA is released from platelets in the standard harvesting of serum (Eichholtz et al., 1993), so we sought to reduce potential variability by maintaining both the experimental and control cultures in charcoal–dextran-filtered FBS (HyClone). Half of the media was changed every 12 h.
LPA stimulation of delaminated sensory epithelia.
Cultures of isolated utricular epithelium were obtained as by Montcouquiol and Corwin (2001b). Briefly, 2-week-old utricles were incubated in thermolysin (0.5 mg/ml; Sigma) for 45 min at 37°C, and the sensory epithelium was removed and plated on glass-bottom dishes (MatTek) coated with poly-l-lysine. Sensory epithelia were cultured in DMEM/F-12 with 5% charcoal–dextran-filtered FBS containing 10 μm LPA. Images of the epithelia were obtained after plating and every 24 h for 3 d.
Immunohistochemistry.
Cultures for phalloidin labeling were fixed in 4% paraformaldehyde in PBS overnight at 4°C, washed in PBS containing 0.2% Triton X-100 (PBST) three times over 15 min, followed by a 15 min incubation in AlexaFluor-labeled phalloidin (5 U/ml; Invitrogen) in PBST and then washed three times over 15 min in PBST.
Cultures to be labeled with anti-BrdU were treated with 1N HCl for 20 min to denature the DNA before Triton X-100 extraction. All cultures for immunohistochemistry were preincubated 30 min at room temperature (RT) in PBST containing 10% normal goat serum (NGS) (Vector Laboratories, Burlingame, CA). Then they were incubated in the appropriate primary antibodies in PBST with 2% NGS for 1 h, followed by cyanine (Cy) 2-, Cy3-, and Cy5-conjugated secondary antibodies (1:200; Jackson ImmunoResearch, West Grove, PA) in PBST for 1 h at RT. Utricles were mounted in SlowFade (Invitrogen), and images were taken with a confocal microscope (Zeiss LSM 510).
Primary antibodies and concentrations.
Mouse anti-occludin (2.5 μg/ml; Zymed, San Francisco, CA) to label tight junctions (Furuse et al., 1993), rabbit anti-calretinin (1:1000; Chemicon, Temecula, CA) to label hair cells (Rogers, 1989), and rat anti-BrdU (25 μg/ml; Abcam, Cambridge, MA) to label cells that entered or had passed through S-phase.
Quantification of labeling within junctional complexes.
A Zeiss 510 LSM was used to capture stacks of images in the z-axis at the interface between the sensory epithelium and nonsensory epithelium in whole-mount utricles labeled with anti-occludin or phalloidin. The average pixel intensities along lines that crossed the cell–cell junctions perpendicularly and the lateral widths of the bands of junctional labeling were measured for 20 randomly selected junctions (10 in the sensory epithelium, 10 in the nonsensory epithelium) for each utricle. Within each image, the labeling in the nonsensory epithelium was normalized to 1, and the labeling in the sensory epithelium was expressed as the fraction of nonsensory labeling. The mean ± SEM for each group was then calculated and plotted, and an ANOVA was used to test for significance.
Quantification of apical surface area and BrdU.
Whole-mount utricles labeled with anti-occludin and anti-BrdU were imaged on a Zeiss 510 LSM. Two parallel lines were drawn across each image, both running through the lesioned region, and outlines of all cells along those lines were drawn in MetaMorph using anti-occludin labeling to mark the extent of the apical surface. MetaMorph was then used to make an automated decision about whether there was nuclear BrdU labeling within that cell border and for calculating the planar area and position of the cell. t tests were used to determine statistical significance of the differences in area between the samples of BrdU-positive and BrdU-negative cells.
Results
Lesions in embryonic utricles heal rapidly
To investigate potential differences between embryonic and mature mammals in the ability for the sensory epithelium to recover from injury and to test whether supporting cell spreading would trigger proliferation, we fabricated micropunches and used them to make small excision lesions in mouse utricles in organ culture. The circular lesions averaged 2.6 × 104 ± 0.1 × 104 μm2 (n = 7) (Fig. 1A,B), removing 13% of the 2 × 105 μm2 area of the utricular macula (Desai et al., 2005). We estimate that each lesion removed ∼780 cells, ∼422 hair cells, and ∼358 supporting cells, based on the morphometric data of Desai et al. (2005), removing 13% of the 3246 hair cells and 2754 supporting cells in the sensory epithelium.
Figure 1.

Excision lesions in embryonic utricles close rapidly. A, Scanning electron micrographs of utricles from E18 mice that received excision lesions and were fixed after the indicated time in culture. At 1 h after lesioning, there was a large open lesion exposing the underlying stromal matrix. The lesion contracted by 10 h after lesioning and was fully closed within 24 h, with the location of the lesion covered over by supporting cells with greatly expanded apical surfaces. B, Individual micrographs from a time-lapse recording of an E18 mouse utricle after lesioning. The lesion rapidly contracted and completely closed by 18 h after the lesioning. C, Closure of lesions measured from simultaneous time-lapse recordings of seven E18 lesioned mouse utricles. The lesion area is plotted every 2 h for each of the utricles, and the average of all seven utricles is plotted (thick black line). Closure of lesions took an average of 17.5 ± 1.5 h.
Scanning electron microscopy showed that supporting cells completely covered lesions made in utricles from E18 and E19 mice in <24 h and restored epithelial integrity (Fig. 1A). To observe the events underlying this re-epithelialization, we made synchronous time-lapse recordings of cellular changes occurring in several lesioned utricles at a time. These showed that the lesion in utricles from E18 and E19 mice closed completely in 17 ± 1.5 h (all data presented are mean ± SEM; n = 7) (Fig. 1B,C) (supplemental Video 1, available at www.jneurosci.org as supplemental material).
To determine which cells were moving to close the lesions, we used time-lapse recordings of lesioned utricles from E18 and P0 mice expressing a mosaic pattern of GFP under control of the β-actin promoter [Tg(GFPU)5Nagy; n = 11 E18; n = 12 P0] (Fig. 2A). Analysis of those movies suggested that cells at the leading edge of the lesion moved substantially farther than cells farther away from the lesion (Fig. 2A). Tracking of individual cells showed that the cells within a ring bordering the lesion, which initially resided between 75 and 125 μm from the centroid of the lesion, moved 41.9 ± 1.7 μm (Fig. 2B,C). In contrast, cells located within concentric rings extending between 125 and 175 μm from the centroid and between 175 and 250 μm from the centroid of the lesion moved an average of 21.1 ± 0.8 and 15.6 ± 0.9 μm, respectively (n = 75 cells from each of 6 E18 utricles; 177 cells between 75 and 125 μm, 200 cells between 125 and 175 μm, and 73 cells between 175 and 250 μm) (Fig. 2C). Thus, the cells closest to the lesion moved a significantly greater distance than cells farther from the lesion (p < 0.005 by ANOVA).
SEM and confocal microscope images of specimens with healed lesions revealed that the sites of the former lesions were covered over by relatively small numbers of cells. The supporting cells that covered the former lesions had undergone radical shape changes, taking on a significantly expanded profile, changing from their normal tight columnar packing to a spread, nearly squamous shape (Figs. 1A, 2D,E). The average planar surface area of cells that came to lie at the middle of the former lesions, <25 μm from the centroid of the lesion, was 63.1 ± 9.9 μm2 (range of 4.6–203.2; n = 214). Those cells were significantly larger than the cells that were >50 μm from the centroid, which had an average planar area of 18.3 ± 0.4 μm2 (range of 3.3–54.2; n = 412; p < 0.005 by t test). Tracing individual cells from the β-actin/GFP time-lapse movies allowed direct visualization of the expansion of cells at the leading edge of the healing lesion as they covered over the opening (Fig. 2F). Thus, during closure, the supporting cells near the leading edge of the wounded epithelium quickly covered over the lesion by taking on significantly expanded shapes and moving the edge of the epithelium inward.
Quiescent supporting cells enter S-phase
Cell spreading has been correlated with proliferation in isolated endothelial cells and epithelial sheets from avian utricles (Folkman and Moscona, 1978; Warchol, 1995, 2002; Chen et al., 1997), so we sought to determine whether the lesion-induced expansion of supporting cells would trigger their reentry into the cell cycle. To label cells passing through S-phase of the cell cycle, lesioned and control utricles from E18 and E19 mice were cultured for 72 h in the presence of BrdU. In lesioned utricles, labeling resulted in 114 ± 16 BrdU-positive cells in a circular 30,000 μm2 region centered over the lesion and 35 ± 8 BrdU-positive nuclei in the 30,000 μm2 peripheral region concentric to the lesion (n = 7) (Fig. 3B,C). In contrast, in unlesioned control sensory epithelia, 9 ± 3 cells within a circular 30,000 μm2 region centered in the macula were BrdU positive and 9 ± 2 cells were BrdU positive in an equivalent concentric 30,000 μm2 region (n = 5) (Fig. 3A,C). Thus, there was a significant increase in the number of BrdU-positive cells at the site of the lesion compared with the surrounding tissue or with unlesioned epithelia (p < 0.05 by ANOVA). Although there was a measured increase in the number of BrdU-positive cells in the sample from the peripheral region outside the lesion, it did not reach statistical significance (p > 0.05 by ANOVA).
Figure 3.
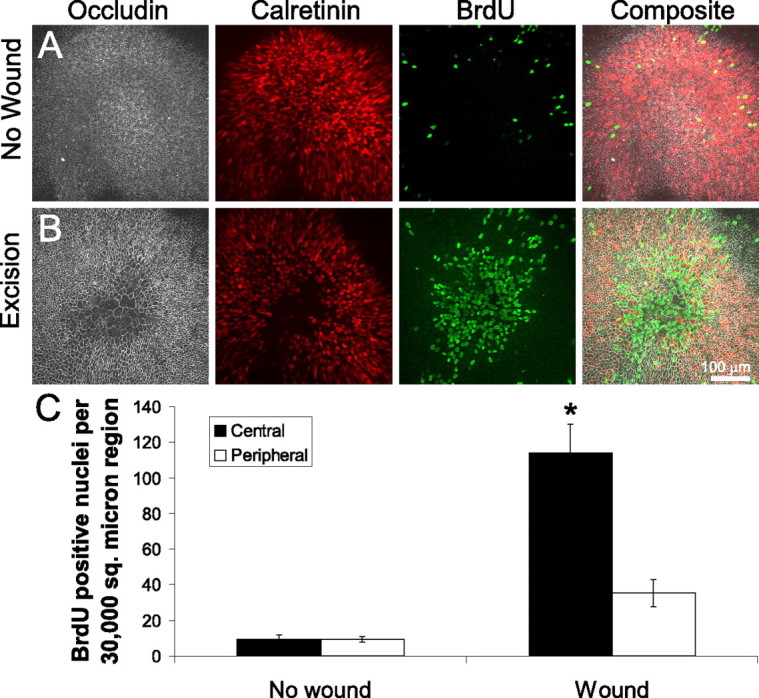
Cells at the lesion site proliferate after wound closure in embryonic mouse utricles. A, Immunolabeling of an unlesioned E19 mouse utricle cultured for 72 h. Anti-occludin shows the apical surface area of the cells, and anti-calretinin labels the hair cells. Anti-BrdU labels the nuclei of cells that have passed through S-phase during the 72 h of culturing. Few BrdU-positive cells are found within the sensory epithelium. B, Immunolabeling of an E19 mouse utricle cultured for 72 h after excision lesioning. The lesion region in the center of the epithelium shows supporting cells with expanded surface area and extensive BrdU incorporation. C, Quantification of BrdU-positive nuclei in two 30,000 μm2 regions for unlesioned and lesioned utricles: a central circular region over the lesion or center of the unlesioned utricle, and a concentric peripheral region (mean ± SEM). There are few BrdU-positive nuclei in the unlesioned utricles in either the central or peripheral regions, but there is a significant increase in the number of BrdU-positive nuclei in the central region of lesioned utricles (*p < 0.05).
In these experiments, BrdU was present in the culture medium from the time the lesions were made, so cycling cells could have been labeled from immediately after the lesions up until the cultures were fixed 72 h later. However, parallel time-lapse experiments that began immediately after lesioning and continued for 24 h showed cells changing position and shape without dividing (Fig. 2E). Those observations suggest that the proliferative response follows the cellular shape change. The rapid speed of lesion closure also makes it unlikely that mitotic divisions of previously quiescent cells would contribute to the wound closure, consistent with models in which epithelial cell proliferation follows wound closure (Ihara and Motobayashi, 1992; Nodder and Martin, 1997; Jacinto et al., 2001). Thus, the lesion-induced spreading of supporting cells reversed their normal quiescence and led to significantly increased passage into S-phase.
Lesions in embryonic utricles close by contraction of an actin ring
Because lesion-induced spreading of supporting cells appeared to stimulate proliferation, we wanted to identify the mechanism that was driving this cellular shape change. Two processes have been proposed to account for epithelial wound healing: purse-string contraction of a leading-edge actin ring and lamellipodial crawling (for review, see Woolley and Martin, 2000). Six hours after excisional lesioning in an E18 utricle, phalloidin labeling revealed a pronounced filamentous actin ring along the leading edge around the lesion (n = 6) (Fig. 4A), and, at that point, the edge appeared smooth (Figs. 1A,B, 4A) (supplemental Video 1, available at www.jneurosci.org as supplemental material). This suggested that contractile pulling of an actin purse string was the primary mechanism driving the cellular shape change. To test this hypothesis, we adapted our time-lapse protocol to allow simultaneous visualization of lesion closure in control utricles and utricles treated with cytochalasin, a drug that inhibits actin polymerization. Although the lesions in utricles in control medium (with 0.1% DMSO as vehicle control) closed completely within 24 h (n = 3) (Fig. 4B,D), the lesions in utricles cultured with 1 μm cytochalasin D (n = 5; data not shown) or 1 μm cytochalasin B remained open even after 24 h, leaving a hole 77 ± 6% of the initial lesion size (n = 5) (Fig. 4C,D) (supplemental Video 2, available at www.jneurosci.org as supplemental material).
Figure 4.
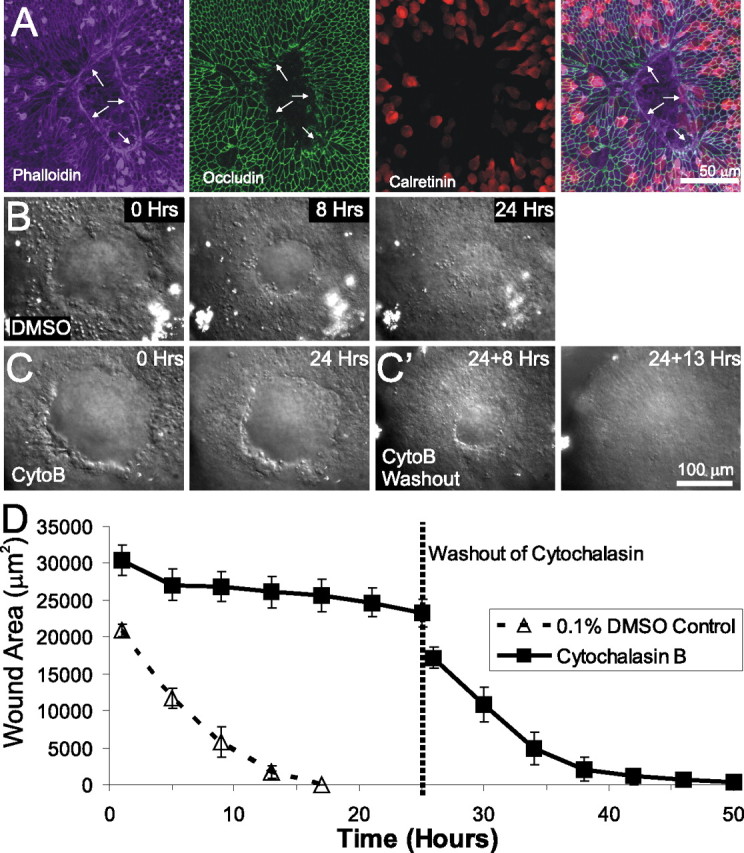
Lesion closure in embryonic utricles occurs via actin purse-string contraction. A, An E18 utricle fixed 6 h after lesioning and triple labeled for phalloidin to label actin, anti-occludin to label tight junctions, and anti-calretinin to label hair cells. An actin purse string runs along the leading edge of the lesion (arrows). B, Individual frames from a time-lapse series showing closure of lesions in a utricle cultured in control media containing 0.1% DMSO. The lesion closed within 24 h. C, Frames from a time-lapse series showing lesion closure in a utricle cultured in media containing 1 μm cytochalasin B (CytoB). After 24 h, the lesion had neither rounded up nor closed. C', After washout of cytochalasin B containing media and replacement with control media, the lesion shown in C contracted and closed 13 h after washout. D, Quantification of lesion size from simultaneous time-lapse recordings of utricles cultured in DMSO control media (dashed line) or cytochalasin B (solid line). The mean ± SEM are plotted every 4 h, showing that lesion closure is inhibited by cytochalasin B but recovers after washout of cytochalasin B.
The micropunch produced lesions with slightly irregular edges (Figs. 1A, 4B,C). Under control conditions, the irregular edges became more circular and smooth within 8 h (Figs. 1A,B, 4B, 5A) (supplemental Video 1, available at www.jneurosci.org as supplemental material). In contrast, the edges of lesions in utricles treated with cytochalasin remained irregular for at least 24 h (Figs. 4C, 5B) (supplemental Video 2, available at www.jneurosci.org as supplemental material). This suggests that the actin ring is responsible for pulling the leading edge into the circular and smooth shape. To control for potential toxicity of the cytochalasin treatment, we washed out the cytochalasin after the initial 24 h recording and continued the time-lapse recordings of the recovery of the epithelium in control medium. In each case, the recordings showed that the inhibitory effects of cytochalasin B were fully reversible. After washout, changes at the leading edges of the lesions in the cytochalasin-treated utricles followed nearly the same time course of closure as untreated controls as the edges became smooth and the lesions closed completely within 18 ± 3 h (n = 5) (Fig. 4C',D) (supplemental Video 2, available at www.jneurosci.org as supplemental material). Thus, the formation of an actin ring along the leading edge of utricular lesions appears to be necessary for the supporting cell shape change that underlies closure of the excision lesions.
Figure 5.

Y-27632 inhibits contraction but not formation of the actin ring. A, A control E18 utricle fixed 10 h after lesioning and labeled with phalloidin, demonstrating formation of a leading edge actin ring. B, An E18 utricle treated with 1 μm cytochalasin D and fixed 10 h after lesioning. Phalloidin labeling shows that cytochalasin blocked formation of the actin ring, leaving an irregular-shaped lesion. C, An E18 utricle treated with 50 μm Y-27632 and fixed 10 h after lesioning. Phalloidin shows that an actin ring formed and that the edge of the lesion rounded up and became smooth.
Contraction of the actin ring is dependent on Rho kinase
Because the contractions of actin purse strings that close lesions in other tissues appear to be mediated by signaling via the small GTPase Rho (Brock et al., 1996; Bement et al., 1999; Wood et al., 2002), we sought to determine whether Rho controlled the contraction of utricular lesions. To test whether Rho activity was required, we cultured lesioned E18 and E19 utricles in the presence of the protein kinase inhibitor Y-27632 (50 μm). Y-27632 blocks signaling through Rho kinase (ROCK), inhibits other Rho-associated kinases [protein kinase C-related kinase 1 (PRK1) and PRK2], and less effectively inhibits mitogen- and stress-activated protein kinase 1, mitogen-activated protein kinase 2, and phosphorylase kinase (Davies et al., 2000). Simultaneous time-lapse imaging of utricles in control media showed full closure of the lesions within 24 h (n = 5), whereas utricles cultured with Y-27632 still had open lesions 81 ± 12% of initial lesion size after 24 h (n = 4) (Fig. 6A–C) (supplemental Video 3, available at www.jneurosci.org as supplemental material). To test whether the inhibition of wound closure was attributable to toxicity, Y-27632 was washed out, and continuing time-lapse recordings showed reversibility with closure of lesions complete within 13 h (Fig. 6B',C) (supplemental Video 3, available at www.jneurosci.org as supplemental material).
Figure 6.
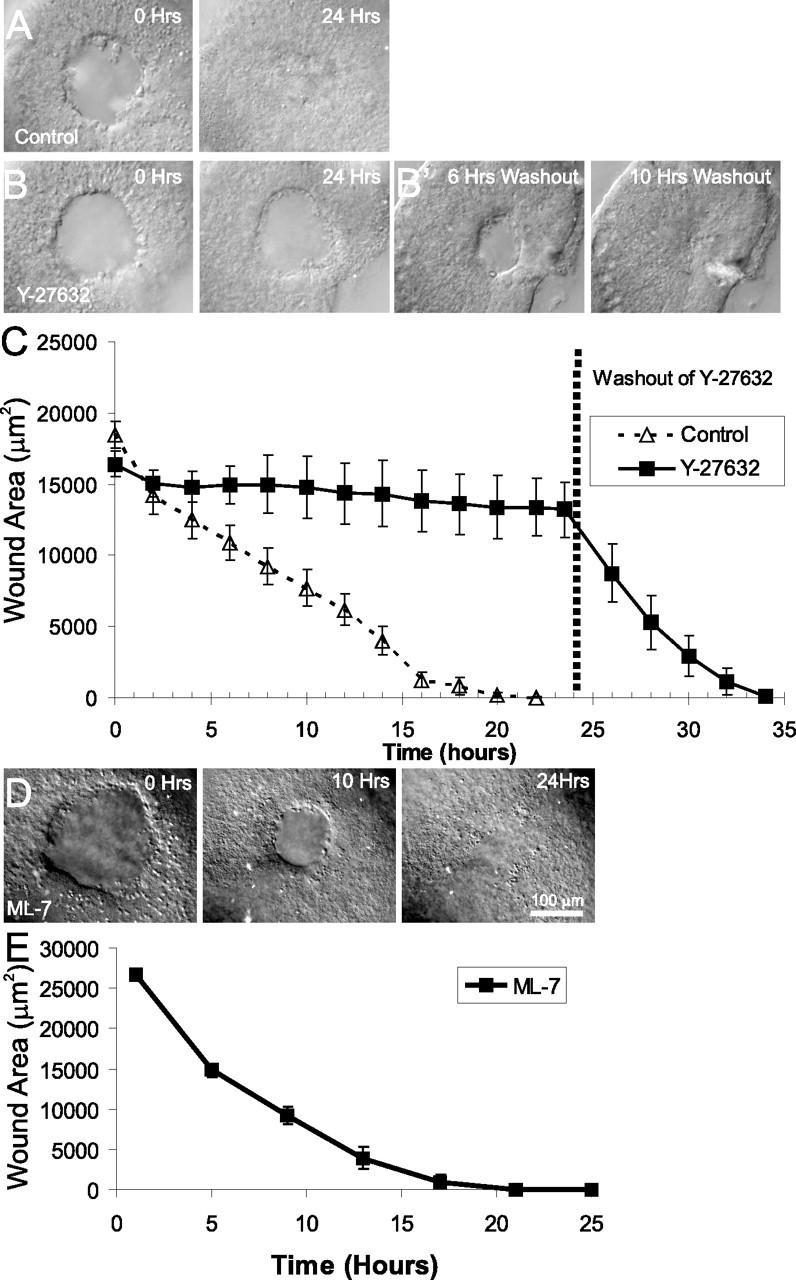
Closure of lesions in embryonic utricles requires activity of ROCK but not activity of MLCK. A, Individual frames from a time-lapse recording of closure of lesions in an E18 utricle cultured in control media. The lesion closes completely within 24 h. B, Individual frames from a time-lapse recording of closure of lesions in an E18 utricle cultured in the presence of 50 μm of the ROCK inhibitor Y-27632 for 24 h. Lesion healing is inhibited in the presence of Y-27632, although the lesion edge becomes smooth and round. Cyto D, Cytochalasin D. B', Frames from the time-lapse shown in B after washout of Y-27632 and replacement with control media. The effects of Y-27632 are completely reversible, with closure occurring within 10 h of washout. C, Quantification of closure of lesions from simultaneous time-lapse recordings from E18 utricles cultured in control media or media containing 50 μm Y-27632 for 24 h; mean ± SEM is plotted every 2 h. Although the control utricles healed normally and fully closed within 24 h, utricles cultured in Y-27632 showed little closure of lesions but healed rapidly after washout of the drug. D, Frames from a time-lapse recording of an E18 utricle cultured in 20 μm ML-7. The lesion rounds up and contracts normally, fully closing within 24 h. E, Quantification of closure of lesions from simultaneous time-lapse recordings of E18 utricles cultured in 20 μm ML-7 for 24 h; mean ± SEM are plotted every 4 h showing normal closure of the lesions.
Although cytochalasin and Y-27632 both inhibited closure of the lesions, the leading edges of the lesions in the cytochalasin-treated utricles remained irregular for the duration of the exposure to that drug, but the leading edges of the lesions in the utricles that were exposed to Y-27632 became circular and smooth (Figs. 5C, 6B) (supplemental Video 3, available at www.jneurosci.org as supplemental material). Also, after washout, the Y-27632-treated epithelia closed at a faster rate than occurred in controls and after cytochalasin washout. We interpret these differences as evidence that the polymerization of new actin filaments or the action of motor proteins not inhibited by Y-27632 at the edges of the epithelial lesion produces tension that is sufficient to draw the leading edges into a smooth, round shape, preparing the lesion for closure, but that tension generated via Rho-mediated contraction is necessary for the supporting cells to spread and close the lesions.
To determine whether the activity of MLCK was necessary for this contraction, we cultured lesioned sensory epithelia in the MLCK inhibitor ML-7 (Ki of 300 nm) (Saitoh et al., 1987). Neither 1 μm (n = 4; data not shown) nor 20 μm (n = 4) ML-7 inhibited closure of lesions in utricles (Fig. 6D,E). At concentrations above 20 μm, ML-7 can inhibit other kinases, including PKC and cyclic-AMP-dependent kinase (Saitoh et al., 1987), so we did not test higher concentrations. Thus, contraction of the leading edge actin ring appears to be independent of MLCK activity. The mechanism of wound closure in the utricle is consistent with that reported for embryonic mouse and chick epidermis (Martin and Lewis, 1992; Brock et al., 1996; Redd et al., 2004) and several other epithelia (Bement et al., 1993; Brock et al., 1996; Heath, 1996; Danjo and Gipson, 1998; Wood et al., 2002). In particular, the change in supporting cell shape that leads to re-epithelialization after lesioning requires actin polymerization into a leading edge ring and the activity of a Y-27632-sensitive kinase, such as the Rho-associated kinases ROCK or PRK1/2, to trigger contraction of the ring and pull the columnar supporting cells into a spread shape.
Lesions in mature utricles remain open for at least 2 d
Because mammalian utricular supporting cells lose proliferative capacity as they mature during the first postnatal weeks (Gu et al., 1997, 2007; Hume et al., 2003), we explored the potential for coincident changes in the capacity for cellular shape change in response to lesions in more mature utricles. In contrast to the utricles from embryonic mice, in which all lesions closed completely in <24 h, lesions made in epithelia from mice between postnatal days 13 and 21 remained open at least 48 h after lesioning (Fig. 7A) (n = 13). At 12 and 24 h after lesioning, the leading edge of the lesion was smooth, with actin cables detectable at the leading edge (n = 7 for each time point) (Fig. 7A,B). This indicates that actin polymerization at the leading edge was not the limiting step in the loss of closure of lesions in mature utricles and suggested that subsequent contraction might be impaired.
Figure 7.

Cells from the sensory macula of 2-week-old mouse utricles lose the capacity to close lesions, corresponding with a change in actin concentration. A, Phalloidin labeling of a P15 utricle cultured for 48 h after lesioning, demonstrating that the lesion had not closed. The sensory epithelium (SE) and the nonsensory epithelium (NSE) are labeled. B, Higher-magnification view of phalloidin labeling of actin in a P19 utricle cultured 24 h after lesioning showing that, although there was little contraction, there was a buildup of actin along the leading edge of the lesion (arrows). C, Phalloidin labeling of 2-week-old utricles cultured for 36 h after a lesion that extended out of the sensory epithelium into the nonsensory epithelium. Although the sensory epithelium had not closed in on the lesion, cells from the nonsensory epithelium migrated into the lesion (arrow). D–H, Cells in the sensory epithelium have enhanced cortical actin belts compared with cells in the nonsensory epithelium in mature utricles. D, A P0 utricle labeled with phalloidin, showing that the immature utricular epithelium had a comparable level of actin and thickness of the cortical belts between the sensory epithelium and the nonsensory epithelium. E, A P17 utricle labeled with anti-occludin shows that the concentration of occludin at the tight junction and thickness of the tight junctions are also comparable for cells in the sensory epithelium and nonsensory epithelium. F, A P17 utricle labeled with phalloidin reveals the sharp increase in labeling intensity and thickness of the cortical actin belts for the cells within the sensory epithelium compared with the cells in the nonsensory epithelium. G, Quantification of the ratio of labeling intensity for sensory epithelium compared with nonsensory epithelium for P0 phalloidin labeling, P17 occludin labeling, and P17 phalloidin labeling. H, Quantification of the junctional width for cells in the sensory epithelium and nonsensory epithelium for 10 cell junctions in the sensory epithelium and 10 cell junctions in the nonsensory epithelium per utricle for P0 phalloidin labeling (n = 5 utricles), P17 occludin labeling (n = 7 utricles), and P17 phalloidin labeling (n = 6 utricles). The width of the actin belts in the P17 sensory epithelium is significantly greater than the other junctional complexes (*p < 0.005).
Loss of spreading into lesions is limited to sensory epithelium
When lesions cut into the nonsensory epithelium that surrounds the sensory epithelium, the cells of the nonsensory epithelium migrated over 100 μm into the lesions within 36 h, whereas the cells of the adjacent sensory epithelium did not close over the lesion (Fig. 7C) (n = 5). The nonsensory epithelium cells that migrated into the lesion did not maintain the smooth concave edge indicative of purse-string contraction but rather developed a convex leading edge more consistent with active lamellipodial-based migration into the lesion (Fig. 7C).
Along with this difference in cell motility, the phalloidin labeling of mature utricles revealed a higher level of staining within the sensory epithelium compared with the nonsensory epithelium (Fig. 7A–C,F). Phalloidin labeling of filamentous actin in the cortical actin belts of supporting cells in the mature sensory epithelium was 3.9 ± 0.3 times brighter than the labeling of cortical actin in cells from the nonsensory epithelium (n = 60 sensory epithelium junctions and 60 nonsensory epithelium junctions, 10 each from 6 utricles) (Fig. 7F,G). In embryonic utricles, in which supporting cells readily change shape and cover over lesions, the F-actin labeling intensity in the sensory epithelium was only 1.9 ± 0.3 times brighter than in the nonsensory epithelium (n = 50 each sensory and nonsensory epithelium junctions, 10 each from 5 utricles) (Fig. 7D,G). To test whether the difference in actin labeling in the mature utricle represented a generalized augmentation of the junctional complexes that link the epithelial cells, we examined occluding-labeled tight junctions of P17 utricles. Occludin labeling in the sensory epithelium was only 1.5 ± 0.1 times brighter than that in the nonsensory epithelium (n = 70 sensory and 70 nonsensory epithelium junctions, 10 each from 7 utricles) (Fig. 7E,G). The ratio of sensory to nonsensory junctional phalloidin labeling was significantly higher for P17 utricles than it was for the P0 utricles. It also was significantly greater than the ratio of sensory to nonsensory occludin labeling in the tight junctions of the same P17 utricles (p < 0.005 by ANOVA).
To determine whether this difference in labeling represented a decreased level of actin in the nonsensory epithelium or an augmentation of the cortical actin belts in the sensory epithelium, we measured the lateral width of the cell–cell junctions labeled with either phalloidin or occludin. The width of phalloidin-labeled junctions in P0 utricles was 1.3 ± 0.1 μm for nonsensory epithelium cells (n = 50 junctions) and 1.4 ± 0.1 μm for sensory epithelium cells (n = 50 junctions). The width of tight junctional complexes labeled with occludin in P17 utricles was similar, at 1.1 ± 0.1 μm for nonsensory epithelium cells and 1.2 ± 0.1 μm for sensory epithelium cells (n = 70 junctions each). The width of phalloidin-labeled junctions in the nonsensory epithelium of mature utricles was also comparable at 1.2 ± 0.1 μm (n = 60 junctions each). In contrast, phalloidin-labeled junctions within the sensory epithelium of P17 utricles were significantly thicker, at 2.7 ± 0.1 μm (p < 0.0005 by ANOVA; n = 60 junctions) (Fig. 7H). Therefore, the cytoskeletal alteration underlying the differential phalloidin labeling appears to be attributable to augmentation of the cortical actin belts that develop postnatally within the sensory epithelium but not within the nonsensory epithelium.
LPA stimulates closure of lesions in postnatal epithelia
We next tested the hypothesis that exogenous activation of the Rho pathway, which controls lesion-induced supporting cell shape change in embryonic utricles, would promote supporting cell shape change in mature utricles. LPA can bind to high-affinity LPA receptors, thereby activating Rho and leading to cytoskeletal remodeling and myosin-mediated contraction (Moolenaar, 1999). LPA has been reported to speed closure of lesions in epithelia (Sturm et al., 1999; Balazs et al., 2000; Lee et al., 2000), so we sought to determine whether LPA would promote wound closure and cellular shape change in mature utricles. To test this, we made lesions in utricles from 2-week-old mice and then cultured controls in the standard medium and experimentals in medium containing 10 μm LPA. In six lesioned P15 utricles cultured with 10 μm LPA, the lesions closed completely within 48 h, whereas five P15 utricles cultured for 48 h in control medium had open lesions that averaged 40 ± 11% of the initial lesion size (Fig. 8A,B). Confocal microscopy showed that the LPA-mediated lesion closure occurred concurrently with spreading of the supporting cells that bordered the lesion (Fig. 8B). The cellular shape change that occurred when lesions in mature LPA-treated utricles closed resembled the shape changes that occur during wound closure in embryonic utricles. In contrast, the supporting cells in the lesioned utricles that were cultured in control medium retained compact columnar shapes (Fig. 8A). Thus, the treatment with LPA led to responses that restored the capacity for rapid wound closure in mature utricles and was sufficient to either evoke or permit shape change in the mature supporting cells.
Figure 8.
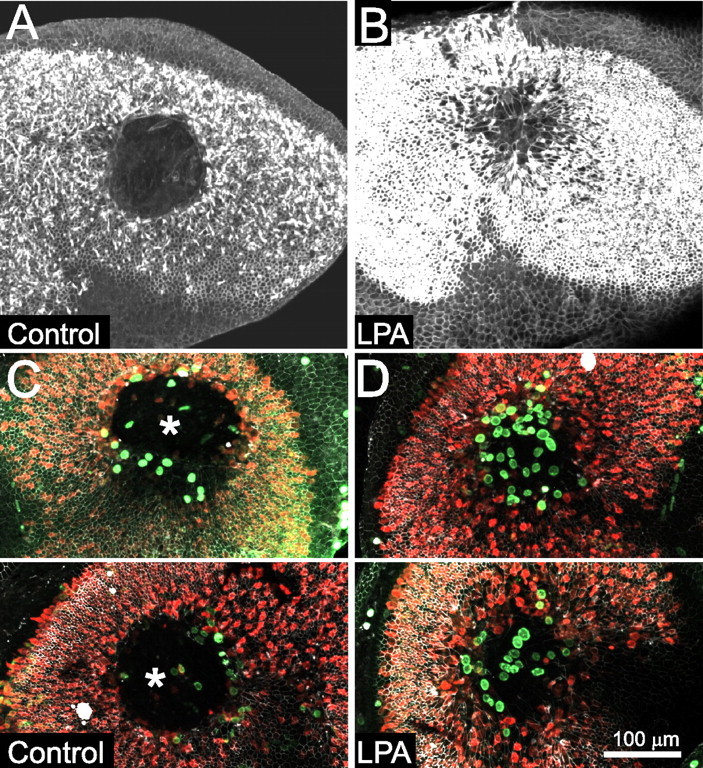
Closure of lesions and reentry into the cell cycle in mature utricles is stimulated by LPA. A, Phalloidin labeling of actin in a utricle from a P15 mouse cultured for 48 h after lesioning in control media. The lesion has not closed. B, Phalloidin labeling of actin in a utricle from a P15 mouse cultured in media containing 10 μm LPA for 48 h after lesioning. LPA stimulated closure of the lesion and resulted in supporting cells with expanded apical surfaces covering over the lesion site. C, Two utricles from P16 mice, lesioned and cultured for 4 d control media with BrdU, labeled with anti-occludin (white), anti-BrdU (green), and anti-calretinin (red). Large open lesions are still visible (asterisks), although a few of the supporting cells at the lesion edge that spread have taken up BrdU. D, Two utricles from P16 mice, lesioned and cultured for 4 d in BrdU and 10 μm LPA, labeled as in C. The lesions have fully closed, and many of the spread cells have reentered the cell cycle, as indicated by BrdU labeling.
Mature supporting cells reenter the cell cycle when lesions close
Because supporting cell shape change is strongly correlated with proliferation in embryonic utricles, we sought to determine whether the LPA-induced changes in supporting cell shape that occurred during lesion closure would stimulate cell-cycle reentry in mature utricles. Thirty-three utricles from 2-week-old mice were lesioned and cultured for 4 d in medium containing 10 μm LPA and BrdU or vehicle control medium containing BrdU. Lesioned utricles cultured in control medium for 4 d showed limited spreading of supporting cells around the open lesions and had 25 ± 4 BrdU-positive nuclei in the sensory epithelium (n = 15; range of 6–60) (Fig. 8C). When closure of lesions was stimulated by culturing in the presence of 10 μm LPA, there were 44 ± 8 BrdU-positive nuclei in the lesion area after 4 d in culture (n = 18; range of 19–128) (Fig. 8D). Thus, the stimulation of lesion closure and cellular shape change by LPA brought about a significant increase in the number of mature supporting cells that reentered the cell cycle (p < 0.05, t test).
To test whether LPA itself was having an effect on cell morphology or proliferation, we examined unlesioned utricles from 16-d-old mice cultured in the presence of LPA and BrdU for 4 d. These undamaged utricles had compact cells with 0–3 BrdU-positive nuclei in the sensory epithelium (n = 6; data not shown). Treatment of thermolysin-isolated, mature utricular epithelia with LPA in culture also did not promote spreading on a fibronectin substrate (data not shown). Thus, treatment with LPA does not appear sufficient to stimulate proliferation or shape change in mature supporting cells in the absence of lesions.
LPA-mediated proliferation occurs specifically in spread supporting cells
Because the BrdU-positive nuclei were localized to the former lesion site and appeared to be associated with supporting cells that had large apical surfaces, we next investigated the relationship between cellular shape and the incidence of S-phase entry for mature supporting cells (Fig. 8C,D). To test this relationship, we cultured lesioned utricles in the continuous presence of 10 μm LPA and BrdU for 7d to label all cells that entered S-phase after lesioning. On average, the sensory epithelia in these utricles contained 98 ± 18 BrdU-positive nuclei (range of 56–119; n = 4) (Fig. 9A). For each cell in a line that ran across the epithelium through the lesion, we measured the planar area bounded by α-occludin labeling of the tight junction and determined whether the nucleus was BrdU positive (Fig. 9B,C). Measurements from 303 cells showed that the average planar area of BrdU-positive cells was 300.5 ± 8.5 μm2, whereas the average planar area of BrdU-negative cells was only 51.3 ± 15.2 μm2 (p < 0.005, t test; 73 BrdU-positive and 230 BrdU-negative cells, from 3 epithelia). Only 10% of the cells with <100 μm2 planar area (n = 231) were BrdU positive, whereas 85% of the cells that had a planar area >300 μm2 (n = 39) and 100% of the cells with a planar area >400 μm2 (n = 22) were BrdU positive (Fig. 9D). Of the cells that had a planar area between 100 and 300 μm2, 46% were BrdU positive (n = 39). Thus, the probability of a mature mammalian supporting cell passing into the cell cycle appears to be directly correlated with whether that cell has changed from a columnar to a spread shape. In mature supporting cells, such shape change normally appears to be restricted by maturational changes such as the strengthened cortical actin belt that develops between P0 and P17. After lesioning, however, the effects of LPA are sufficient to overcome these limitations and promote supporting cell shape change that leads to re-epithelialization and increased cell-cycle reentry.
Figure 9.

In mature utricles that have been treated with LPA, proliferation is strongly correlated with whether the cells have an expanded planar area. A, A utricle from a P16 mouse, lesioned and cultured for 7 d in 10 μm LPA, labeled with anti-occludin (white), anti-BrdU (green), and anti-calretinin (red). Many of the cells at the center of the lesion had reentered the cell cycle. B, Representative tracing of cell surface areas based on occludin labeling for a single line of cells from a P16 mouse utricle cultured as in A. Filled cells had a BrdU-positive nucleus, whereas outlined cells did not have a BrdU-positive nucleus. C, Composite plot of cell surface area versus distance along the x-axis for 303 cells from three utricles traced as in B, revealing that BrdU-positive cells were predominantly the large cells covering over the lesion (filled circles represent BrdU-positive cells, and × signs represent BrdU-negative cells). D, A histogram showing the percentage of cells that are BrdU-positive for three bins of cell area. A total of 90% cells with a surface area <100 μm2 were BrdU negative, whereas 85% cells with a surface area >300 μm2 were BrdU positive.
Discussion
Our results demonstrate that supporting cells in the embryonic mammalian utricle change from columnar to spread shapes as they close excision lesions, and this leads to cell-cycle reentry. This remodeling capacity is lost during the first weeks of postnatal development coincident with thickening of the cortical actin belts that are near the cell junctions, but it can be reinitiated in mature utricles by treatment with LPA. Thus, the development of proliferative quiescence in the mammalian inner ear may be attributable to pharmacologically reversible cytoskeletal and contractile alterations that normally limit the capacity of the supporting cells to change shape and reenter the cell cycle.
The actin ring formation and contraction that occurs in response to excision lesions in the embryonic utricle is similar to the formation of actin rings that occurs after acoustic overstimulation and aminoglycoside poisoning in the ear (Raphael and Altschuler, 1991; Meiteles and Raphael, 1994; Gale et al., 2002) and the formation and contraction that occurs in wounds in embryonic epidermis (Martin and Lewis, 1992; Brock et al., 1996; Redd et al., 2004). This contraction results in supporting cells changing from columnar to spread shapes with greatly expanded planar areas (Figs. 2D,E, 8B, 9A–C), a change reminiscent of the spreading of supporting cell surfaces that occurs after overstimulation and before regeneration of hair cells in the avian basilar papilla (Cotanche, 1987; Cotanche and Dopyera, 1990; Marsh et al., 1990). Therefore, although our excision lesions do not replicate damage seen clinically, the apparent consistency of the processes in these wound-healing responses suggests that similar mechanisms may drive repair of different types of injury in these inner ear epithelia.
Maturation of the utricle limits cell shape change and lesion closure
Actin ring contraction is an important wound-healing mechanism in embryos, but there is a change to lamellipodial migration in some adult epithelia (Martin and Lewis, 1992; Redd et al., 2004), whereas other adult epithelia retain actin purse-string contraction (Danjo and Gipson, 1998). In the mature utricular sensory epithelium, supporting cells developed an actin ring but did not move substantially (Fig. 7A). Nonsensory cells, however, invaded the lesions in adult utricles (Fig. 7C) in a manner more consistent with lamellipodial-based migration than purse-string contraction.
Although nonsensory epithelium cells may use a different mechanism for their movement, they are able spread into lesions, similar to embryonic supporting cells. This suggests that the changes that limit spreading in mature supporting cells are specific to the sensory epithelium. A notable difference between the sensory epithelium and nonsensory epithelium was the abrupt change in thickness of cortical actin belts (Fig. 7). The stouter actin belts that develop as supporting cells mature between P0 and P17 may contribute to increased cytoskeletal rigidity that limits the closure of lesions and thereby restricts regenerative proliferation in the mature mammalian utricle. Maturational changes in the capacity for supporting cells to change shape and proliferate also appear to correlate with changes in integrin expression and hemidesmosome development, which both affect cytoskeletal anchorage (Davies et al., 2007). If cytoskeletal anchorage and rigidity are responsible for limiting proliferation in the mature ears of mammals, then similar cytoskeletal and anchorage changes may not occur during the maturation of nonmammalian vertebrates, in which supporting cells retain the capacity to change shape, proliferate, and replace hair cells after damage.
LPA stimulates lesion closure and supporting cell shape change
After utricular supporting cells mature to a state that limits their ability to change shape and proliferate, treatment with LPA can overcome that limit and stimulate or permit cellular spreading, closure of lesions, and proliferation. Two candidate mechanisms could be affected by the LPA treatment: cytoskeletal remodeling leading to cell migration (Cook et al., 1998; Palazzo et al., 2001; Zhang et al., 2003) and activation of Rho/ROCK-mediated actinomyosin contractility (Hirose et al., 1998; Ramakers and Moolenaar, 1998; Kranenburg et al., 1999; Parizi et al., 2000; Fringer and Grinnell, 2001). In unlesioned utricles from 2-week-old mice, LPA treatment had little effect, and it did not stimulate the outward lamellipodial-mediated spreading of supporting cells from cultures of thermolysin-isolated epithelia from 2-week-old utricles. Thus, the effect of LPA on the utricle is more consistent with stimulation of contraction than promotion of lamellipodial-mediated migration. Although LPA promoted spreading of the supporting cells that bordered the lesion, the other supporting cells in the LPA-treated utricles retained the compact, columnar shapes and the stout actin belts typical of mature sensory epithelia (Fig. 8B). Therefore, the results appear consistent with the hypothesis that LPA treatment results in wound closure, cellular shape change, and proliferation by virtue of its action in stimulating or permitting the contraction of the actin ring at the leading edge of the wound.
Cell shape controls cell-cycle reentry in the utricle
After lesions closed, the spread supporting cells reentered the cell cycle, whereas the compact, columnar supporting cells were considerably less likely to enter the cell cycle. The results indicate that lesion-evoked cellular shape change may be a critical trigger for proliferation. Even in mature epithelia, nearly all of the supporting cells that underwent significant shape change also entered S-phase (Fig. 9). This suggests that the majority of mature supporting cells retain the capacity for proliferation but that capacity is limited by their reduced tendency to change shape.
Analytical investigations of isolated endothelial cells in vitro have shown that cell shape change can limit proliferation (Chen et al., 1997). Endothelial cells cultured on extracellular matrix islands of 400 μm2 or greater passed through S-phase, whereas those cultured on islands <100 μm2 in area were not proliferative despite the presence of appropriate mitogenic growth factors. Similarly, in the present study, with the utricular sensory epithelium in situ, 85% of the mature supporting cells that spread to >300 μm2 entered S-phase, whereas only 10% of those with a planar area <100 μm2 did (Fig. 9D).
Lesion-mediated cell shape change occurs quickly and cell volume should remain nearly constant, so supporting cells must flatten as they spread. A conservatively modeled cylindrical supporting cell with a planar surface area of 50 μm2, near the measured mean for BrdU-negative cells, and a height of 30 μm (Wersäll and Flock, 1965; Desai et al., 2005), would flatten to <3.75 μm tall when spread to an area of >400 μm2, above which all of the supporting cells were BrdU positive. This is consistent with the findings of Folkman and Moscona (1978), that few 20-μm-tall endothelial cells would pass through S-phase, but cells that flattened to <4 μm tall reentered the cell cycle.
Thus, our results provide quantitative evidence consistent with the conclusion that supporting cell spreading is an important regulator for cell-cycle passage in hair cell epithelia. The close quantitative match between the changes in the measured shapes of supporting cells in sensory epithelia that healed in situ and the dimensions of isolated endothelial cells that proliferate in culture could indicate that shape-mediated control of proliferation is similar in diverse epithelial cell types. Wound-healing mechanisms appear to be conserved across many epithelia, so shape-dependent control of cell proliferation may underlie regeneration in the olfactory system, the retina, and other neuroepithelia. Cellular shape may also play a critical role in the glial proliferation that follows injury in the CNS, because isolated astroglia take on a flattened cell shape before proliferating and a compact, elongate form when differentiating in the presence of neurons (Hatten, 1985). It remains to be determined whether the speed or extent of supporting cell shape changes that occur after acoustic overstimulation and after aminoglycoside-induced hair cell loss in mammals are normally insufficient to promote proliferation. Alternatively, those shape changes may be comparable with those that follow damage in avian ears, but there may be additional controls that limit proliferation in mammals.
Role of cellular shape in control of proliferation
Our results suggest that stimulating shape change is sufficient to overcome other controls that limit proliferation in mature mammalian utricles. p27Kip1, p19Ink4, and Rb1 are known to regulate proliferation in the inner ear (Chen and Segil, 1999; Lowenheim et al., 1999; Chen et al., 2003; Sage et al., 2005; White et al., 2006), but cellular shape may be upstream and able to reverse such cell-cycle regulation, because nearly all of the mature supporting cells that spread reentered the cell cycle. Shape-dependent cytoskeletal tension has been reported to influence passage through the G1/S checkpoint by controlling the levels of cyclin D1, p27Kip1, and phosphorylated Rb1 (Huang et al., 1998). Activation of Rho and ROCK also have been tied to control of p27Kip1 at the G1/S transition as well as regulation of cyclin D (Roovers et al., 2003; Mammoto et al., 2004). Columnar-shaped supporting cells may use these regulators as checkpoints to prevent cell-cycle passage, changing expression or phosphorylation after lesion-induced spreading and thereby allowing passage into S-phase. It remains to be determined whether the shape change we explored in the inner ear provides a permissive or an instructive signal for proliferation.
To summarize, we report that the development of the proliferative quiescence that appears to leave mammals vulnerable to permanent sensory deficits that arise from hair cell loss may be attributable to cytoskeletal and contractile alterations that limit the ability of the supporting cells to change shape. Spreading of supporting cells in utricular epithelia that were wounded and healed in situ reversed the quiescence that normally limits proliferation. Overcoming supporting cell quiescence could be the critical step for initiating proliferative hair cell regeneration in mature mammals, because the progeny of supporting cell divisions in nonmammalian vertebrates readily differentiate into replacement hair cells that contribute to functional recovery. We found one pharmacological method for reversing quiescence by promoting cell shape changes that increase proliferation in mature mammalian utricles. Combining such methods with pharmacological promotion of hair cell differentiation could contribute to the development of treatments for clinically important forms of hearing and balance deficit.
Footnotes
This work was supported by a Howard Hughes Medical Institute predoctoral fellowship (J.R.M.) and National Institute on Deafness and Other Communication Disorders Award R01-00200 (J.T.C.). We thank Dawn Davies and Kevin Lynch for discussions about this manuscript.
References
- Balazs L, Okolicany J, Ferrebee M, Tigyi G. Topical application of LPA accelerates wound healing. Ann NY Acad Sci. 2000;905:270–273. doi: 10.1111/j.1749-6632.2000.tb06558.x. [DOI] [PubMed] [Google Scholar]
- Bement WM, Forscher P, Mooseker MS. A novel cytoskeletal structure involved in purse string wound closure and cell polarity maintenance. J Cell Biol. 1993;121:565–578. doi: 10.1083/jcb.121.3.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bement WM, Mandato CA, Kirsch MN. Wound-induced assembly and closure of an actomyosin purse string in Xenopus oocytes. Curr Biol. 1999;9:579–587. doi: 10.1016/s0960-9822(99)80261-9. [DOI] [PubMed] [Google Scholar]
- Brock J, Midwinter K, Lewis J, Martin P. Healing of incisional wounds in the embryonic chick wing bud: characterization of the actin purse-string and demonstration of a requirement for Rho activation. J Cell Biol. 1996;135:1097–1107. doi: 10.1083/jcb.135.4.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CS, Mrksich M, Huang S, Whitesides GM, Ingber DE. Geometric control of cell life and death. Science. 1997;276:1425–1428. doi: 10.1126/science.276.5317.1425. [DOI] [PubMed] [Google Scholar]
- Chen P, Segil N. p27(Kip1) links cell proliferation to morphogenesis in the developing organ of Corti. Development. 1999;126:1581–1590. doi: 10.1242/dev.126.8.1581. [DOI] [PubMed] [Google Scholar]
- Chen P, Zindy F, Abdala C, Liu F, Li X, Roussel MF, Segil N. Progressive hearing loss in mice lacking the cyclin-dependent kinase inhibitor Ink4d. Nat Cell Biol. 2003;5:422–426. doi: 10.1038/ncb976. [DOI] [PubMed] [Google Scholar]
- Cook TA, Nagasaki T, Gundersen GG. Rho guanosine triphosphatase mediates the selective stabilization of microtubules induced by lysophosphatidic acid. J Cell Biol. 1998;141:175–185. doi: 10.1083/jcb.141.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corwin JT, Jones JE, Katayama A, Kelley MW, Warchol ME. Hair cell regeneration: the identities of progenitor cells, potential triggers and instructive cues. Ciba Found Symp. 1991;160:103–120. doi: 10.1002/9780470514122.ch6. discussion 120–130. [DOI] [PubMed] [Google Scholar]
- Corwin JT, Warchol ME, Saffer LD, Finley JE, Gu R, Laber PR. Growth factors as potential drugs for the sensory epithelia of the ear. Ciba Found Symp. 1996;196:167–182. doi: 10.1002/9780470514863.ch12. discussion 182–167. [DOI] [PubMed] [Google Scholar]
- Cotanche DA. Regeneration of hair cell stereociliary bundles in the chick cochlea following severe acoustic trauma. Hear Res. 1987;30:181–195. doi: 10.1016/0378-5955(87)90135-3. [DOI] [PubMed] [Google Scholar]
- Cotanche DA, Dopyera CE. Hair cell and supporting cell response to acoustic trauma in the chick cochlea. Hear Res. 1990;46:29–40. doi: 10.1016/0378-5955(90)90137-e. [DOI] [PubMed] [Google Scholar]
- Danjo Y, Gipson IK. Actin “purse string” filaments are anchored by E-cadherin-mediated adherens junctions at the leading edge of the epithelial wound, providing coordinated cell movement. J Cell Sci. 1998;111:3323–3332. doi: 10.1242/jcs.111.22.3323. [DOI] [PubMed] [Google Scholar]
- Davies D, Magnus C, Corwin JT. Developmental changes in cell-extracellular matrix interactions limit proliferation in the mammalian inner ear. Eur J Neurosci. 2007;25:985–998. doi: 10.1111/j.1460-9568.2007.05355.x. [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai SS, Zeh C, Lysakowski A. Comparative morphology of rodent vestibular periphery. I. Saccular and utricular maculae. J Neurophysiol. 2005;93:251–266. doi: 10.1152/jn.00746.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichholtz T, Jalink K, Fahrenfort I, Moolenaar WH. The bioactive phospholipid lysophosphatidic acid is released from activated platelets. Biochem J. 1993;291:677–680. doi: 10.1042/bj2910677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkman J, Moscona A. Role of cell shape in growth control. Nature. 1978;273:345–349. doi: 10.1038/273345a0. [DOI] [PubMed] [Google Scholar]
- Fringer J, Grinnell F. Fibroblast quiescence in floating or released collagen matrices: contribution of the ERK signaling pathway and actin cytoskeletal organization. J Biol Chem. 2001;276:31047–31052. doi: 10.1074/jbc.M101898200. [DOI] [PubMed] [Google Scholar]
- Furuse M, Hirase T, Itoh M, Nagafuchi A, Yonemura S, Tsukita S. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol. 1993;123:1777–1788. doi: 10.1083/jcb.123.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale JE, Meyers JR, Periasamy A, Corwin JT. Survival of bundleless hair cells and subsequent bundle replacement in the bullfrog's saccule. J Neurobiol. 2002;50:81–92. doi: 10.1002/neu.10002. [DOI] [PubMed] [Google Scholar]
- Gu R, Marchionni M, Corwin JT. Glial growth factor enhances supporting cell proliferation in rodent vestibular epithelia cultured in isolation. Soc Neurosci Abstr. 1996;21:520. [Google Scholar]
- Gu R, Marchionni M, Corwin JT. Age-related decreases in proliferation within isolated mammalian vestibular epithelia cultured in control and glial growth factor 2 medium. Assoc Res Otolaryngol Abstr. 1997;20:98. [Google Scholar]
- Gu R, Montcouquiol M, Marchionni M, Corwin JT. Proliferative responses to growth factors decline rapidly during postnatal maturation of mammalian hair cell epithelia. Eur J Neurosci. 2007;25:1363–1372. doi: 10.1111/j.1460-9568.2007.05414.x. [DOI] [PubMed] [Google Scholar]
- Hadjantonakis AK, Gertsenstein M, Ikawa M, Okabe M, Nagy A. Generating green fluorescent mice by germline transmission of green fluorescent ES cells. Mech Dev. 1998;76:79–90. doi: 10.1016/s0925-4773(98)00093-8. [DOI] [PubMed] [Google Scholar]
- Hatten ME. Neuronal regulation of astroglial morphology and proliferation in vitro. J Cell Biol. 1985;100:384–396. doi: 10.1083/jcb.100.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath JP. Epithelial cell migration in the intestine. Cell Biol Int. 1996;20:139–146. doi: 10.1006/cbir.1996.0018. [DOI] [PubMed] [Google Scholar]
- Hirose M, Ishizaki T, Watanabe N, Uehata M, Kranenburg O, Moolenaar WH, Matsumura F, Maekawa M, Bito H, Narumiya S. Molecular dissection of the Rho-associated protein kinase (p160ROCK)-regulated neurite remodeling in neuroblastoma N1E-115 cells. J Cell Biol. 1998;141:1625–1636. doi: 10.1083/jcb.141.7.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Chen CS, Ingber DE. Control of cyclin D1, p27(Kip1), and cell cycle progression in human capillary endothelial cells by cell shape and cytoskeletal tension. Mol Biol Cell. 1998;9:3179–3193. doi: 10.1091/mbc.9.11.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume CR, Kirkegaard M, Oesterle EC. ErbB expression: the mouse inner ear and maturation of the mitogenic response to heregulin. J Assoc Res Otolaryngol. 2003;4:422–443. doi: 10.1007/s10162-002-3008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihara S, Motobayashi Y. Wound closure in foetal rat skin. Development. 1992;114:573–582. doi: 10.1242/dev.114.3.573. [DOI] [PubMed] [Google Scholar]
- Jacinto A, Martinez-Arias A, Martin P. Mechanisms of epithelial fusion and repair. Nat Cell Biol. 2001;3:E117–E123. doi: 10.1038/35074643. [DOI] [PubMed] [Google Scholar]
- Kelley RO, Dekker RA, Bluemink JG. Ligand-mediated osmium binding: its application in coating biological specimens for scanning electron microscopy. J Ultrastruct Res. 1973;45:254–258. doi: 10.1016/s0022-5320(73)80051-6. [DOI] [PubMed] [Google Scholar]
- Kranenburg O, Poland M, van Horck FP, Drechsel D, Hall A, Moolenaar WH. Activation of RhoA by lysophosphatidic acid and Galpha12/13 subunits in neuronal cells: induction of neurite retraction. Mol Biol Cell. 1999;10:1851–1857. doi: 10.1091/mbc.10.6.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert PR. Inner ear hair cell regeneration in a mammal: identification of a triggering factor. Laryngoscope. 1994;104:701–718. doi: 10.1288/00005537-199406000-00010. [DOI] [PubMed] [Google Scholar]
- Lee H, Goetzl EJ, An S. Lysophosphatidic acid and sphingosine 1-phosphate stimulate endothelial cell wound healing. Am J Physiol Cell Physiol. 2000;278:C612–C618. doi: 10.1152/ajpcell.2000.278.3.C612. [DOI] [PubMed] [Google Scholar]
- Li L, Forge A. Morphological evidence for supporting cell to hair cell conversion in the mammalian utricular macula. Int J Dev Neurosci. 1997;15:433–446. doi: 10.1016/s0736-5748(96)00102-5. [DOI] [PubMed] [Google Scholar]
- Lowenheim H, Furness DN, Kil J, Zinn C, Gultig K, Fero ML, Frost D, Gummer AW, Roberts JM, Rubel EW, Hackney CM, Zenner HP. Gene disruption of p27(Kip1) allows cell proliferation in the postnatal and adult organ of corti. Proc Natl Acad Sci USA. 1999;96:4084–4088. doi: 10.1073/pnas.96.7.4084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malick LE, Wilson RB. Modified thiocarbohydrazide procedure for scanning electron microscopy: routine use for normal, pathological, or experimental tissues. Stain Technol. 1975;50:265–269. doi: 10.3109/10520297509117069. [DOI] [PubMed] [Google Scholar]
- Mammoto A, Huang S, Moore K, Oh P, Ingber DE. Role of RhoA, mDia, and ROCK in cell shape-dependent control of the Skp2–p27kip1 pathway and the G1/S transition. J Biol Chem. 2004;279:26323–26330. doi: 10.1074/jbc.M402725200. [DOI] [PubMed] [Google Scholar]
- Marsh RR, Xu LR, Moy JP, Saunders JC. Recovery of the basilar papilla following intense sound exposure in the chick. Hear Res. 1990;46:229–237. doi: 10.1016/0378-5955(90)90004-9. [DOI] [PubMed] [Google Scholar]
- Martin P, Lewis J. Actin cables and epidermal movement in embryonic wound healing. Nature. 1992;360:179–183. doi: 10.1038/360179a0. [DOI] [PubMed] [Google Scholar]
- Meiteles LZ, Raphael Y. Scar formation in the vestibular sensory epithelium after aminoglycoside toxicity. Hear Res. 1994;79:26–38. doi: 10.1016/0378-5955(94)90124-4. [DOI] [PubMed] [Google Scholar]
- Montcouquiol M, Corwin JT. Intracellular signals that control cell proliferation in mammalian balance epithelia: key roles for phosphatidylinositol-3 kinase, mammalian target of rapamycin, and S6 kinases in preference to calcium, protein kinase C, and mitogen-activated protein kinase. J Neurosci. 2001a;21:570–580. doi: 10.1523/JNEUROSCI.21-02-00570.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montcouquiol M, Corwin JT. Brief treatments with forskolin enhance S-phase entry in balance epithelia from the ears of rats. J Neurosci. 2001b;21:974–982. doi: 10.1523/JNEUROSCI.21-03-00974.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moolenaar WH. Bioactive lysophospholipids and their G protein-coupled receptors. Exp Cell Res. 1999;253:230–238. doi: 10.1006/excr.1999.4702. [DOI] [PubMed] [Google Scholar]
- Nodder S, Martin P. Wound healing in embryos: a review. Anat Embryol (Berl) 1997;195:215–228. doi: 10.1007/s004290050041. [DOI] [PubMed] [Google Scholar]
- Palazzo AF, Cook TA, Alberts AS, Gundersen GG. mDia mediates Rho-regulated formation and orientation of stable microtubules. Nat Cell Biol. 2001;3:723–729. doi: 10.1038/35087035. [DOI] [PubMed] [Google Scholar]
- Parizi M, Howard EW, Tomasek JJ. Regulation of LPA-promoted myofibroblast contraction: role of Rho, myosin light chain kinase, and myosin light chain phosphatase. Exp Cell Res. 2000;254:210–220. doi: 10.1006/excr.1999.4754. [DOI] [PubMed] [Google Scholar]
- Ramakers GJ, Moolenaar WH. Regulation of astrocyte morphology by RhoA and lysophosphatidic acid. Exp Cell Res. 1998;245:252–262. doi: 10.1006/excr.1998.4224. [DOI] [PubMed] [Google Scholar]
- Raphael Y, Altschuler RA. Scar formation after drug-induced cochlear insult. Hear Res. 1991;51:173–183. doi: 10.1016/0378-5955(91)90034-7. [DOI] [PubMed] [Google Scholar]
- Redd MJ, Cooper L, Wood W, Stramer B, Martin P. Wound healing and inflammation: embryos reveal the way to perfect repair. Philos Trans R Soc Lond B Biol Sci. 2004;359:777–784. doi: 10.1098/rstb.2004.1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers JH. Two calcium-binding proteins mark many chick sensory neurons. Neuroscience. 1989;31:697–709. doi: 10.1016/0306-4522(89)90434-x. [DOI] [PubMed] [Google Scholar]
- Roovers K, Klein EA, Castagnino P, Assoian RK. Nuclear translocation of LIM kinase mediates Rho-Rho kinase regulation of cyclin D1 expression. Dev Cell. 2003;5:273–284. doi: 10.1016/s1534-5807(03)00206-5. [DOI] [PubMed] [Google Scholar]
- Sage C, Huang M, Karimi K, Gutierrez G, Vollrath MA, Zhang DS, Garcia-Anoveros J, Hinds PW, Corwin JT, Corey DP, Chen ZY. Proliferation of functional hair cells in vivo in the absence of the retinoblastoma protein. Science. 2005;307:1114–1118. doi: 10.1126/science.1106642. [DOI] [PubMed] [Google Scholar]
- Saitoh M, Ishikawa T, Matsushima S, Naka M, Hidaka H. Selective inhibition of catalytic activity of smooth muscle myosin light chain kinase. J Biol Chem. 1987;262:7796–7801. [PubMed] [Google Scholar]
- Sturm A, Sudermann T, Schulte KM, Goebell H, Dignass AU. Modulation of intestinal epithelial wound healing in vitro and in vivo by lysophosphatidic acid. Gastroenterology. 1999;117:368–377. doi: 10.1053/gast.1999.0029900368. [DOI] [PubMed] [Google Scholar]
- Warchol ME. Supporting cells in isolated sensory epithelia of avian utricles proliferate in serum-free culture. NeuroReport. 1995;6:981–984. doi: 10.1097/00001756-199505090-00008. [DOI] [PubMed] [Google Scholar]
- Warchol ME. Cell density and N-cadherin interactions regulate cell proliferation in the sensory epithelia of the inner ear. J Neurosci. 2002;22:2607–2616. doi: 10.1523/JNEUROSCI.22-07-02607.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warchol ME, Lambert PR, Goldstein BJ, Forge A, Corwin JT. Regenerative proliferation in inner ear sensory epithelia from adult guinea pigs and humans. Science. 1993;259:1619–1622. doi: 10.1126/science.8456285. [DOI] [PubMed] [Google Scholar]
- Wersäll J, Flock A. Functional anatomy of the vestibular and lateral line organs. Contrib Sens Physiol. 1965;14:39–61. doi: 10.1016/b978-1-4831-6746-6.50007-9. [DOI] [PubMed] [Google Scholar]
- White PM, Doetzlhofer A, Lee YS, Groves AK, Segil N. Mammalian cochlear supporting cells can divide and trans-differentiate into hair cells. Nature. 2006;441:984–987. doi: 10.1038/nature04849. [DOI] [PubMed] [Google Scholar]
- Witte MC, Montcouquiol M, Corwin JT. Regeneration in avian hair cell epithelia: identification of intracellular signals required for S-phase entry. Eur J Neurosci. 2001;14:829–838. doi: 10.1046/j.0953-816x.2001.01695.x. [DOI] [PubMed] [Google Scholar]
- Wood W, Jacinto A, Grose R, Woolner S, Gale J, Wilson C, Martin P. Wound healing recapitulates morphogenesis in Drosophila embryos. Nat Cell Biol. 2002;4:907–912. doi: 10.1038/ncb875. [DOI] [PubMed] [Google Scholar]
- Woolley K, Martin P. Conserved mechanisms of repair: from damaged single cells to wounds in multicellular tissues. BioEssays. 2000;22:911–919. doi: 10.1002/1521-1878(200010)22:10<911::AID-BIES6>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Zhang XF, Schaefer AW, Burnette DT, Schoonderwoert VT, Forscher P. Rho-dependent contractile responses in the neuronal growth cone are independent of classical peripheral retrograde actin flow. Neuron. 2003;40:931–944. doi: 10.1016/s0896-6273(03)00754-2. [DOI] [PubMed] [Google Scholar]
- Zheng JL, Helbig C, Gao WQ. Induction of cell proliferation by fibroblast and insulin-like growth factors in pure rat inner ear epithelial cell cultures. J Neurosci. 1997;17:216–226. doi: 10.1523/JNEUROSCI.17-01-00216.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]