

Abstract
In vitro long-term facilitation (ivLTF) is a novel form of activity-independent postsynaptic enhancement of AMPA receptor function in hypoglossal (XII) motoneurons that can be induced by intermittent activation of 5-HT2 receptors. In vivo respiratory long-term facilitation (LTF) is characterized by a persistent 5-HT2 receptor-dependent increase in respiratory motor output or ventilation after episodic exposures to hypoxia in adult rats. Here, we demonstrate that ivLTF can also be induced by episodic but not continuous stimulation of α1-adrenergic receptors that requires protein kinase C (PKC), but not PKA (protein kinase A), activation. Additionally, we show that in vivo respiratory LTF is also α1-adrenergic receptor dependent. We suggest that, in vivo, concurrent episodic activation of 5-HT2 and α1-adrenergic receptors is necessary to produce long-lasting changes in the excitability of respiratory motoneurons, possibly involving PKC activation via the Gαq–PLC (phospholipase C) signaling pathway common to both receptor subtypes. Such plasticity of XII motor output may increase upper airway muscle (innervated by XII nerve) tone and improve the likelihood that airway patency will be maintained. Elucidating the mechanism underlying LTF can be of clinical importance to the patients suffering from sleep-disordered breathing.
Keywords: long-term facilitation (LTF), plasticity, respiration, PKC, motoneuron, adrenergic
Introduction
Synapses can undergo persistent changes in strength based on experience. Studies on such synaptic plasticity in mammals have focused on glutamatergic synapses in hippocampal, cortical, and cerebellar neurons. Phrenic and XII motoneurons innervating the diaphragm and the upper airway muscles, respectively, receive respiratory-modulated glutamate-mediated synaptic inputs (Rekling et al., 2000), with AMPA receptors as a principal postsynaptic target (Liu et al., 1990; Funk et al., 1993). Because persistent changes in respiratory motor output are critical for adaptive changes in breathing appropriate for blood gas regulation (Feldman et al., 2003; Mitchell and Johnson, 2003), understanding the conditions and mechanisms underlying respiratory motoneuron plasticity per se is of interest.
Persistent compensatory increases in ventilation or respiratory motor output, referred to as respiratory long-term facilitation (LTF), are induced by episodic but not continuous exposures to hypoxia in adult rats (Baker and Mitchell, 2000; Fuller et al., 2000; Mitchell et al., 2001) and in adult humans during sleep (Babcock et al., 2002; Shkoukani et al., 2002). In vivo, LTF is characterized by a persistent increase in phrenic and XII nerve discharge after episodic (three cycles of 5 min on/5 min off) hypoxia in anesthetized adult (Bach and Mitchell, 1996; Baker and Mitchell, 2000) or neonatal (McKay et al., 2004) rats. LTF initiation, but not maintenance, is blocked by previous administration of 5-HT2 receptor antagonists (Kinkead and Mitchell, 1999; Fuller et al., 2001; Baker-Herman and Mitchell, 2002).
In vitro, persistent increases in XII motoneuronal activity are induced by episodic activation of 5-HT2 receptors (Bocchiaro and Feldman, 2004). Specifically, episodic (three cycles of 3 min on/5 min off) but not continuous (9 min on) 5-HT2 receptor activation increases both XII nerve output and postsynaptic AMPA receptor conductance in XII motoneurons. This effect is blocked by group 1 metabotropic glutamate receptor (mGluR1) antagonists.
Protein kinases and phosphatases modulate postsynaptic AMPA receptor-mediated currents in XII motoneurons (Bocchiaro et al., 2003). Stimulation of a cAMP-dependent pathway increases XII nerve innervated genioglossus muscle activity in rats (Aoki et al., 2006). Because in vitro long-term facilitation (ivLTF) involves increases in postsynaptic AMPA receptor currents (Bocchiaro and Feldman, 2004), we hypothesized that the activation of protein kinases, such as protein kinase A (PKA) or protein kinase C (PKC), were essential for its induction and/or maintenance.
5-HT2 (Roth et al., 1998) and mGluR1 (Hermans and Challiss, 2001) receptors act principally via the Gαq–phospholipase C (PLC) pathway where PKC is activated, as do α1-adrenoreceptors (Garcia-Sainz et al., 1999). In the intact mammalian brain, the XII nucleus receives multiple noradrenergic projections from pontine areas, including locus subceruleus, as well as locus ceruleus and A5 (Moore and Bloom, 1979; Aldes et al., 1992), that are activated by hypoxia, similarly to medullary serotonergic neurons (Guyenet et al., 1993; Erickson and Millhorn, 1994). In addition, α1-adrenoreceptors are abundantly expressed in XII motoneurons (Volgin et al., 2001). Therefore, we hypothesized that ivLTF could be induced by episodic α1-adrenoreceptor activation via a pathway involving PKC and/or PKA. Furthermore, we hypothesized that similar mechanisms are operative in vivo and, thus, that α1-adrenoreceptor antagonists would inhibit in vivo LTF after intermittent hypoxia.
Materials and Methods
In vitro slice preparation experiments
All in vitro protocols were approved by the Office for the Protection of Research Subjects, University of California Research Committee. Experiments were performed on medullary slice preparations from neonatal Sprague Dawley rats[postnatal day 0 (P0) to P3] that retain functional respiratory networks generating a respiratory-related rhythm (Smith et al., 1991). Neonatal rats were anesthetized with isoflurane, decerebrated, and the brainstem was isolated by dissection in a bath containing artificial CSF (ACSF) (in mm: 128.0 NaCl, 3.0 KCl, 1.5 CaCl2, 1.0 MgCl2, 23.5 NaHCO3, 0.5 NaH2PO4, 30.0 d-glucose), pH 7.4, bubbled with 95% O2–5% CO2 at room temperature. The dura, superficial blood vessels, and cerebellum were then removed. After that, the brainstem was sectioned with a vibratome (Vibratome, St. Louis, MO) in the transverse plane starting from the rostral medulla until the identifiable landmarks of the compact formation of the nucleus ambiguous and the inferior olive could be seen. A single transverse 700-μm-thick slice that included the XII nerve rootlets and the pre-Bötzinger complex (Smith et al., 1991) was then cut. The slice was transferred to a recording chamber and continuously superfused (5 ml/min) with ACSF containing elevated K+ (9 mm) to maintain rhythmic output (Funk et al., 1993). The slice was maintained at 27°C.
Drug application.
(R)-(−)-Phenylephrine hydrochloride[phenylephrine (PE)] (10 μm; Sigma, St. Louis, MO), methysergide maleate salt (methysergide) (10 μm; Sigma), chelerythrine chloride (chelerythrine) (10 μm; Sigma), (S)-(+)-α-amino-4-carboxy-2-methylbenzeneacetic acid (LY367385) (100 μm; Tocris, Bristol, UK), prazosin hydrochloride (prazosin) (10 μm; Sigma), α-methyl-5-hydroxytryptamine maleate (α-Me-5-HT) (3 μm; Tocris), tetrodotoxin (TTX) (10 μm; Sigma), R-cyclic 3′,5′-hydrogen phosphorothioate adenosine triethylammonium salt (Rp-cAMP) (100 μm; Sigma), and AMPA (10 μm; Sigma) were dissolved in ACSF for bath application and local injection. Methysergide, prazosin, chelerythrine, Rp-cAMP, and LY367385 were bath applied 20 min before the ivLTF protocol and remained in the bath until the end of each experiment.
Protocol.
The slice was placed in the recording chamber (the chamber volume was 1 ml, the dead space between the pump and the chamber was 2 ml, and the flow rate was 10 ml/min). XII nerve output was allowed to reach a stable value and then was recorded for at least 20 min to assure stable amplitude. For experiments described in Figures 2 and 3, the antagonists were bath applied and XII activity was recorded for at least 20 more minutes. ivLTF protocols were then performed: three, 3 min bath applications of the agonist, spaced by 5 min. Agonists included the following: 10 μm PE, 3 μm α-Me-5-HT, or ACSF containing 15 mm K+. XII nerve activity or motoneuronal currents were recorded for an additional 60 min.
Figure 1.

Episodic but not continuous PE (10 μm; bath applied) induced ivLTF. A, Three applications of PE (ivLTF protocol) produced a persistent increase in amplitude of integrated XII nerve output (∫XIIn). B, From A, on expanded timescale, ∫XIIn and corresponding endogenous respiratory-related XII motoneuronal drive currents (iXII MN) during the control period and at 60 min after the ivLTF protocol (iXII MN was 131 ± 22% at 60 min; n = 6; p < 0.05, paired t test). C, An example of several ∫XIIn bursts before and 60 min after the ivLTF protocol. D, Normalized peak amplitude of ∫XIIn before and after ivLTF protocol (n = 7). Post-ivLTF protocol increase was statistically significant (***p < 0.0001, **p < 0.001, and *p < 0.01, one-way ANOVA, Dunnett's post hoc test), 151 ± 21% of control at 60 min. E, Whole-cell currents from respiratory-modulated XII motoneurons under TTX (1 μm) in response to local AMPA (10 μm) ejections. Left, Control recordings showing the stability of the current over a period of 60 min. Right, Currents before (black line) and 60 min after (gray line) the ivLTF protocol. F, Normalized peak amplitude of AMPA-induced currents before and after ivLTF protocol (n = 5). The increase at 60 min after ivLTF protocol was 146 ± 10% of control (p < 0.001, one-way ANOVA, Dunnett's post hoc test). G, A single 9 min application of PE produced only short-term, but no long-term increase of ∫XIIn. H, Normalized peak amplitude of ∫XIIn before and after one 9 min PE application (n = 5). Initial increase of ∫XIIn amplitude was not statistically significant (ns; one-way ANOVA, Dunnett's post hoc test). Error bars indicate SD in B and SE in D, F, and H
Figure 3.
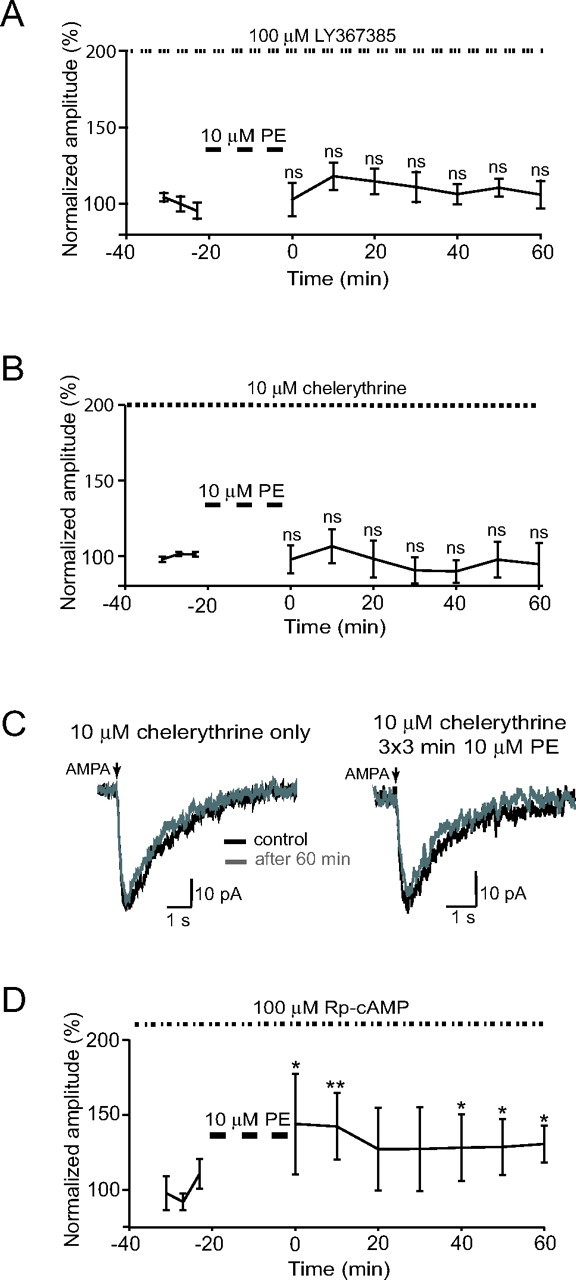
PE-induced ivLTF is blocked by mGluR1 and PKC antagonists. A, Bath-applied mGluR1a blocker LY367385 (100 μm) occluded PE-induced ivLTF (106 ± 9% of control at 60 min; ns; one-way ANOVA, Dunnett's post hoc test; n = 5). B, Bath-applied PKC blocker chelerythrine (10 μm) occluded PE-induced ivLTF (95 ± 14% of control at 60 min; ns; one-way ANOVA, Dunnett's post hoc test; n = 6). C, Bath-applied chelerythrine occluded ivLTF of AMPA-mediated postsynaptic currents recorded from XII motoneurons under TTX. Left, Control recording showing the stability of current over a period of 60 min in the presence of 10 μm chelerythrine (n = 3). Right, Currents before (black line) and 60 min after (gray line) ivLTF protocol in the presence of chelerythrine. The current amplitude 60 min after ivLTF protocol was 98 ± 3% (n = 4). D, PKA blocker Rp-cAMP (10 μm) diminished but did not occlude PE-induced ivLTF (131 ± 12% of control at 60 min; n = 4). Post-ivLTF protocol increase was statistically significant (*p < 0.01, **p < 0.001, one-way ANOVA, Dunnett's post hoc test). Error bars indicate SE.
XII nerve recordings.
XII motor output was recorded from the cut ends of XII nerve rootlets with a suction electrode, amplified and filtered (5000×; 0.3–1 kHz). Population discharges of the XII nerve rootlets were rectified and integrated (time constant τ ≈ 100 ms). Signals were digitized and stored on computer hard disk using pClamp 8.0 software (Molecular Devices, Sunnyvale, CA).
Whole-cell recordings from XII motoneurons.
Respiratory XII motoneurons were visualized using infrared-differential interference contrast microscopy and identified according to the criteria described previously (Funk et al., 1993). Whole-cell voltage-clamp recordings (holding potential, Vh, −70 mV) were made from XII motoneurons using borosilicate glass electrodes pulled on a horizontal patch electrode puller (model P-87 CA; Sutter Instruments, Novato, CA). The electrodes (4–8 MΩ; 2 μm tip diameter) contained a patch solution of the following composition (in mm): 140 K+ gluconate, 1.1 EGTA, 5 NaCl, 0.1 CaCl2, 10 HEPES, 2 ATP (Mg2+ salt), pH 7.3 adjusted using KOH. Access resistance was continuously monitored throughout the experiment and was always <30 MΩ, according to the criteria set previously (Funk et al., 1993); cells with large or unstable access resistances varying >5% of the control values were rejected from analysis. Individual XII motoneurons exhibited periodic inward synaptic drive currents occurring in phase with integrated motoneuron population discharge (Funk et al., 1993). Currents were recorded with an Axopatch 1D amplifier (Molecular Devices), filtered using a 1 dB Bessel filter and digitized at 10 kHz via a Digidata 1200 interface with a software filter (bandpass, 2 Hz to 5 kHz) in pClamp 8.0 software (Molecular Devices).
In experiments involving recordings of exogenous AMPA-mediated currents, a XII motoneuron was patched and identified as respiratory in voltage-clamp mode (i.e., the motoneuron had rhythmic inward drive currents in synchrony with XII nerve bursts). Then, an ejection pipette (2 μm tip diameter) filled with AMPA (10 μm in ACSF) was positioned over the motoneuronal soma. AMPA was pressure ejected onto the patch-clamped XII motoneuron in 40 ms pulses at 10 psi every 90 s; this interval was chosen to avoid receptor desensitization. After the reliable exogenous AMPA-mediated currents were established, TTX (1 μm) was added to the bath for the remainder of the experiment.
Analysis of in vitro data.
Integrated XII nerve discharges were analyzed using DataView 4.7c (W. J. Heitler, University of St. Andrews, St. Andrews, UK). Every 5 min before and every 10 min after ivLTF protocol, 10 consecutive respiratory-related bursts were averaged, and the peak amplitude was measured. These averaged data points were normalized within each experiment using Origin 7.0 (OriginLab, Northampton, MA). Then, these normalized data points were compared with data points at corresponding times from other experiments of the same experimental group, and the amplitude mean ± SE for each time point was calculated for each group of experiments. The statistical significance of the difference between the pre- (control) and post-ivLTF protocol values was assessed using one-way ANOVA; p values were obtained by post hoc analyses based on the Dunnett's test for multiple comparisons of treatment groups to the control group (SAS 9.1; SAS Institute, Cary, NC).
The averages of endogenous respiratory-related XII motoneuronal currents were constructed in DataView 4.7c (W. J. Heitler); 10 consecutive XII nerve bursts and concurrent inspiratory drive currents were averaged immediately before and 60 min after ivLTF protocol application. Averaged peak currents were normalized to the current amplitude before the ivLTF protocol. The exogenous AMPA-mediated currents were recorded and measured in Clampfit 8.0 (Molecular Devices). The data were transferred to Origin 7.0, where AMPA-mediated currents were analyzed. The statistical significance of the difference between the control and post-ivLTF peak amplitude values was assessed using a paired t test and expressed as mean ± SD..
In vivo experiments
All in vivo experimental procedures were approved by the Animal Care and Use Committee, School of Veterinary Medicine, University of Wisconsin, Madison. Experiments were performed on Sprague Dawley rats (3–5 months of age; colony P04; Charles River Laboratories, Wilmington, MA).
Surgical preparation.
Rats were anesthetized with isoflurane in 50% O2–50% N2, and then slowly converted to urethane anesthesia (1.6 g/kg, i.v.). The adequacy of anesthesia was assessed periodically by testing blood pressure and respiratory nerve responses to toe pinch; supplemental doses of urethane were given if necessary. After conversion to urethane anesthesia, an intravenous infusion of a 1:11 solution of NaHCO3 and standard lactated Ringer's solution was initiated to maintain an acid–base balance (4 ml · kg−1 · h−1). A tracheal cannula was placed and the animals were ventilated. The animals were vagotomized and paralyzed with pancuronium bromide (2.5 mg/kg) to prevent pump entrainment of respiratory outflow and breathing movements, respectively.
Measurements.
The left XII nerve was isolated using a dorsal approach, cut, desheathed, and placed on bipolar silver recording electrodes submerged in mineral oil. Nerve activity was amplified (gain, 10,000; A-M Systems, Everett, WA), bandpass filtered (100 Hz to 10 kHz), and integrated (time constant, 50 ms; 821 Paynter filter; CWE, Ardmore, PA). The signal was then digitized, recorded, and analyzed using WINDAQ data acquisition system (DATAQ Instruments, Akron, OH).
After the experimental preparation was complete, at least 1 h was allowed for stabilization of the preparation. Before the establishment of baseline conditions, intravenous injections of prazosin were made, or blood was withdrawn, as required by the experimental protocol, and an additional 20 min were allowed before establishing baseline conditions.
Baseline conditions.
To establish steady baseline nerve activity, rats were ventilated with moderate hyperoxic gas mixtures (50% O2–50% N; PaO2 > 120 mmHg), with CO2 added to the inspired gas so that PaCO2 was 2–3 mmHg above the CO2 apneic threshold (i.e., CO2 levels below which breathing movements disappear) (Bach and Mitchell, 1996). End-tidal CO2 was monitored throughout the protocol using a flow-through capnograph (Novametrix, Wallingford, CT) with sufficient response time to measure end-tidal CO2 in rats. Blood gases and arterial pressure were monitored and corrected as necessary to ensure that posthypoxia values were maintained near baseline levels. Arterial CO2 was maintained ±1 mmHg from the initial baseline value by adjusting inspired CO2 and/or ventilator frequency.
LTF protocol.
sPeak amplitudes of integrated XII nerve activity were measured before, during, and after three, 5 min hypoxic episodes (11% O2) with intervening 5 min intervals of baseline hyperoxia. Measurements were made in the following groups of rats (each n = 3): (1) pretreatment with prazosin (150 μg/kg, i.v.) with exposure to intermittent hypoxia; (2) pretreatment with prazosin (150 μg/kg, i.v.) without exposure to intermittent hypoxia; (3) sham experiments with intermittent hypoxia, but without drug injections; and (4) control experiments for reduced blood pressure caused by prazosin. In the latter experiments, we slowly withdrew blood from the arterial catheter without fluid replacement to reduce blood volume. Blood withdrawal was continued until blood pressure stabilized at a level approximating values seen in prazosin-treated rats.
Statistical analysis.
Peak burst amplitudes and frequency of integrated XII nerve activity were averaged in 1 min bins at each recorded data point (baseline, during the final minute of the first hypoxic episode, and 15, 30, and 60 min after hypoxia). XII burst amplitudes were normalized as a percentage of the baseline value. Burst frequency responses were expressed as a change from baseline values in bursts per minute. Statistical comparisons were made among experimental groups at 15, 30, and 60 min after episodic hypoxia (or equivalent time in sham experiments) using a two-way ANOVA with a repeated-measures design. The Fisher least significant difference (LSD) method was used as a post hoc test to make individual comparisons. Differences were considered significant if p < 0.05. All values are expressed as mean ± SE.
Results
In vitro studies
In the in vitro slice preparation, persistent increases in inspiratory-modulated XII nerve activity lasting longer than 60 min (and up to 14 h) (data not shown) were observed after episodic bath application of PE (“ivLTF protocol”) (Fig. 1A–D). At 60 min after the ivLTF protocol, integrated XII nerve output increased to 151 ± 21% of control (n = 7; p < 0.001, one-way ANOVA, Dunnett's post hoc test) (Fig. 1D), and peak intracellular currents increased to 131 ± 22% of control (n = 6; p < 0.05, paired t test) (Fig. 1B). PE application produced transient increases in tonic activity that rapidly declined after drug washout (Fig. 1A,G; note the changes in the amplitude directly under the bars indicating three PE applications). A single 9 min PE application failed to generate a persistent increase in XII nerve activity[n = 5; nonsignificant (ns)] (Fig. 1G,H).
To exclude a possible component of ivLTF presynaptic to motoneurons, they were patch clamped and then synaptically isolated with TTX (1 μm; bath applied). AMPA (10 μm) was pressure ejected in short (40 ms) pulses onto the motoneuron. The induced AMPA currents were recorded before and for at least 60 min after the ivLTF protocol. Although there was no change in the peak AMPA current over the course of 1 h in control conditions (i.e., no ivLTF protocol) (Fig. 1E, left), AMPA current amplitude increased after the ivLTF protocol to 146 ± 10% with respect to the baseline at 60 min (n = 5; p < 0.001, one-way ANOVA, Dunnett's post hoc test) (Fig. 1 E, right, F).
To control for actions of PE on other receptors, slices in normal ACSF (i.e., without TTX) were preincubated with the α1-adrenergic receptor antagonist prazosin (10 μm), and then the ivLTF protocol was performed. Both ivLTF and short-term increases in tonicity usually observed during PE application were completely abolished (101 ± 5% of control at 60 min after PE administration; n = 3; ns; one-way ANOVA, Dunnett's post hoc test) (Fig. 2 A). Incubation of the slice with prazosin alone did not affect the baseline XII nerve activity for at least 2 h.
Figure 2.

ivLTF requires specific stimulation of either α1-adrenergic or 5-HT2 receptors. A, α1-adrenergic antagonist prazosin (10 μm) completely blocked induction of ivLTF by PE (101 ± 5% of control at 60 min; ns; one-way ANOVA, Dunnett's post hoc test; n = 3). B, Broad-spectrum 5-HT receptor antagonist methysergide (10 μm) attenuated but did not block PE-induced ivLTF (124 ± 10% of control at 60 min; n = 6; **p < 0.001, ***p < 0.0001, one-way ANOVA, Dunnett's post hoc test). C, ivLTF is induced with α-Me-5-HT (3 μm; 5-HT2 receptor agonist). Normalized peak ∫XIIn was 142 ± 23% of control at 60 min (n = 5; *p < 0.01, **p < 0.001, one-way ANOVA, Dunnett's post hoc test). D, α1-adrenergic antagonist prazosin (10 μm) attenuated but did not block α-Me-5-HT-induced ivLTF (127 ± 12% of control at 60 min; n = 5; *p < 0.01, **p < 0.001, ***p < 0.0001, one-way ANOVA, Dunnett's post hoc test). E, Episodic stimulation with ACSF containing 15 mm K+ did not produce ivLTF of ∫XIIn (103 ± 4% of control at 60 min; ns; one-way ANOVA, Dunnett's post hoc test; n = 3). Error bars indicate SE.
To test whether the PE-induced changes in XII motoneurons required activation of 5-HT receptors (Bocchiaro and Feldman, 2004), the ivLTF protocol was performed in the presence of the 5-HT receptor antagonist methysergide. When methysergide was applied to the bath, ∫XII nerve burst amplitude decreased immediately by 23 ± 5% and remained stable for at least 2 h (data not shown). In a different set of slices, after methysergide was applied to the bath, integrated ∫XII nerve amplitude was allowed to stabilize at the newly established lower baseline, and the ivLTF protocol was performed with episodic PE. Methysergide reduced, but did not abolish, PE-induced ivLTF; XII nerve burst amplitude was 124 ± 10% (at 60 min after ivLTF protocol; n = 6; p < 0.0001, one-way ANOVA, Dunnett's post hoc test) (Fig. 2 B) of the lower baseline established after methysergide application. The difference between the control ivLTF response (Fig. 1D) and the diminished ivLTF response in the presence of methysergide (Fig. 2B) was statistically significant (p < 0.01; two-way repeated-measures ANOVA).
Conversely, ivLTF elicited by episodic 5-HT2 receptor activation with α-Me-5-HT (3 μm; ∫XII nerve burst amplitude was 142 ± 23% of control at 60 min after ivLTF protocol; n = 5; p < 0.001, one-way ANOVA, Dunnett's post hoc test) (Fig. 2C) persisted in the presence of 10 μm prazosin, where ∫XII nerve burst amplitude was 127 ± 12% of baseline at 60 min after ivLTF protocol (n = 5; p < 0.0001, one-way ANOVA, Dunnett's post hoc test) (Fig. 2 D). 5-HT2 receptor-dependent ivLTF in the presence of prazosin was reduced compared with 5-HT2 receptor-dependent ivLTF alone[127 ± 12% (Fig. 2 D) vs 142 ± 23% (Fig. 2C)], and the difference was not statistically significant (ns; two-way repeated-measures ANOVA). Thus, although serotonergic antagonists may reduce ivLTF induced by PE, and noradrenergic antagonists may reduce ivLTF induced by α-Me-5-HT, substantial ivLTF can result from exclusive activation of either α1-adrenergic or 5-HT2 receptors.
Because both 5-HT2 and α1-adrenergic receptor-induced ivLTF produces short-term increases in excitability, we considered the possibility that, rather than resulting from activation of receptor-specific pathways, ivLTF could simply be attributable to generic episodic increases in excitability or activity. We therefore applied to the bath three, 3 min episodes (spaced by 5 min) of ACSF containing elevated K+ (15 mm); such high levels of K+ induce short-term increases in XII nerve activity. After the elevated K+ was removed, no persistent increase of XII nerve amplitude was detected (103 ± 4% of control at 60 min; n = 3; ns; one-way ANOVA, Dunnett's post hoc test) (Fig. 2 E).
5-HT2 receptor-induced ivLTF is abolished by antagonism of mGluR1s (Bocchiaro and Feldman, 2004). To determine whether mGluR1s underlie similar metamodulation of PE-induced ivLTF, slices were preincubated with the selective type 1a mGluR1 antagonist LY367385 (100 μm). No effect was observed on the baseline activity or on the acute response to PE, but PE-induced ivLTF was abolished (106 ± 9% at 60 min; n = 5; ns; one-way ANOVA, Dunnett's post hoc test) (Fig. 3A). Thus, the effect of mGluR1 antagonism is the same for either 5-HT2 or α1-adrenergic receptor-mediated ivLTF.
PKC is a downstream element in the Gαq–PLC intracellular signaling pathway common to both α1-adrenergic and 5-HT2 receptor activation. To determine whether PKC is required for ivLTF induction, slices were preincubated with the membrane-permeable PKC inhibitor chelerythrine (10 μm). Although there was no effect on the baseline control activity or on the acute response to PE, PE-induced ivLTF was abolished (95 ± 14% at 60 min; n = 6; ns; one-way ANOVA, Dunnett's post hoc test) (Fig. 3B). The effect of chelerythrine was postsynaptic because chelerythrine also abolished ivLTF of the exogenous AMPA currents in synaptically isolated XII motoneurons (98 ± 3% at 60 min; n = 4) (Fig. 3C, right) (compare with Fig. 1 E, right), whereas chelerythrine alone did not affect exogenous AMPA currents (Fig. 3C, left).
Because PKA is constitutively active in XII motoneurons (Bocchiaro et al., 2003), we also tested the possibility that PKA is involved in the induction of ivLTF. Preincubation of the slice with the membrane-permeable PKA inhibitor Rp-cAMP (10 μm) had no effect on baseline activity or the acute response to PE administration, but did not prevent ivLTF (131 ± 12% at 60 min; n = 4; p < 0.01, one-way ANOVA, Dunnett's post hoc test) (Fig. 3 D). The difference between the control ivLTF response (Fig. 1D) and the ivLTF response in the presence of Rp-cAMP (Fig. 3 D) was statistically significant (p < 0.001; two-way repeated-measures ANOVA).
In vivo studies
Prazosin blocked in vivo XII LTF induced by episodic hypoxia (Fig. 4). In all rats exposed to episodic hypoxia, integrated XII burst amplitude increased during each episode, consistent with many previous reports (Mitchell et al., 2001). Shortly after episodic hypoxia, integrated XII burst amplitude returned to baseline levels in all experimental groups. However, in control rats receiving episodic hypoxia without prazosin (n = 3), XII burst amplitude exhibited a progressive, ramping increase in burst amplitude beginning at 15 min, becoming statistically significant at 60 min after hypoxia (177 ± 26% of baseline; p < 0.001) (Fig. 4 A, top trace; B). This ramping increase is characteristic of XII nerve LTF (Bach and Mitchell, 1996) (for review, see Mitchell et al., 2001).
Figure 4.

In vivo XII LTF is blocked by prazosin, but not decreased blood pressure. A, In vivo XII LTF results after intermittent hypoxia (IH) (top trace) in untreated rats and blood pressure control rats (IH BP Control) (bottom trace), but not in rats pretreated with prazosin (150 μg/kg, i.v.) before IH (IH with prazosin) (middle trace). In each tracing, recordings of ∫XIIn are shown during baseline conditions, three 5 min hypoxic episodes with 5 min intervals in between, and then a return to baseline conditions. The hypoxic episodes produce three increases in peak ∫XIIn; an arrow below the tracings marks the second hypoxic episode. The horizontal dashed line indicates peak ∫XIIn during baseline conditions; any increase in peak ∫XIIn above this level 60 min after hypoxia indicates in vivo LTF. B, Normalized peak ∫XIIn in various conditions. Mean values are expressed at 15, 30, and 60 min after the final hypoxic episode. Experimental groups include rats with IH (three 5 min hypoxic episodes spaced by 5 min) but without drug (open circles; n = 3), hypovolemic control rats receiving IH without drug (solid circles; n = 3), rats receiving prazosin and IH (solid diamonds; n = 3), and rats receiving prazosin, but without IH (open diamonds; n = 3). IH elicited XII LTF in control and hypovolemic rats, and this response was not significantly different. Conversely, rats that received prazosin before IH no longer exhibited XII LTF, nor did rats receiving prazosin without IH. Statistical inferences were made with two-way, repeated-measures ANOVA followed by individual comparisons with the Fisher LSD method. *Significant difference from baseline (p < 0.001). #Significant difference between rats receiving prazosin and IH-treated control rats at the corresponding time point (p < 0.05). Error bars indicate SE.
Because prazosin induces hypotension as a result of its action on the peripheral vasculature, we performed control experiments to determine whether hypotension alone could affect the induction of in vivo XII LTF. In rats made hypotensive by blood withdrawal (mean arterial blood pressure, 57 ± 6 mmHg, vs 77 ± 5 mmHg in control rats) that did not receive prazosin (n = 3), XII burst amplitude increased to 213 ± 25% of baseline (60 min after episodic hypoxia; p < 0.001) (Fig. 4 A, bottom trace, B). Thus, hypotension alone did not impair in vivo XII LTF.
In rats pretreated with prazosin (mean arterial blood pressure, 53 ± 8 mmHg) and then exposed to episodic hypoxia (n = 3), XII burst amplitude was only 84 ± 17% of baseline at 60 min after hypoxia (Fig. 4 A, middle trace, B), a response significantly lower than that observed in episodic hypoxia-treated rats without prazosin (p < 0.001) or blood pressure controls (p < 0.001). Thus, episodic hypoxia-induced in vivo XII LTF was not inducible in the presence of prazosin.
In rats pretreated with prazosin but without exposure to episodic hypoxia (n = 3), no increase in integrated XII amplitude was observed at 60 min after hypoxia (103 ± 3% of baseline; mean arterial pressure, 55 ± 2 mmHg) (Fig. 4 B); this response was significantly less than that in rats without prazosin exposed to episodic hypoxia (p < 0.01) or hypovolemic blood pressure controls (p < 0.002), and was not significantly different from rats pretreated with prazosin receiving episodic hypoxia (p = 0.43).
Mean arterial pressures were higher in untreated control rats (77 ± 5 mmHg; p < 0.05) versus rats receiving prazosin and episodic hypoxia (53 ± 8 mmHg; p < 0.02), prazosin alone (55 ± 2 mmHg; p < 0.03), or hypovolemic blood pressure controls (57 ± 6 mmHg; p < 0.03). Thus, blood pressure decrements caused by systemic administration of prazosin did not impair in vivo respiratory LTF.
Discussion
Episodic but not continuous stimulation of α1-adrenoceptors in vitro led to a persistent increase in postsynaptic AMPA receptor-mediated inspiratory-modulated XII motoneuronal activity. Similar changes are observed after episodic 5-HT2 receptor activation, in which increased AMPA receptor function also underlies the long-term change in excitability (Bocchiaro and Feldman, 2004). Both forms of ivLTF require active mGluR1s, which are linked to PLC (Hermans and Challiss, 2001) as are 5HT2 receptors and α1-adrenoceptors. In vivo, blockade of α1-adrenergic receptors abolished respiratory LTF after episodic hypoxia, suggesting that both adrenergic and serotonergic receptors are necessary, possibly by interactions between signaling pathways or even convergence on a given protein effector, such as PKC, in a shared pathway. Indeed, PKC activation is necessary for α1-adrenoceptor-induced ivLTF, suggesting common downstream mechanisms. Conversely, PKA activation is not necessary for α1-adrenoceptor-induced in vitro LTF, suggesting that this kinase is an unlikely point of convergence between the respective receptors.
Because ivLTF can be induced by activation of different receptor classes each producing increased activity, it could be a nonspecific excitability-dependent phenomenon. However, increasing activity in vitro by episodic elevation of[K+] failed to induce ivLTF, suggesting that the mechanism is specifically linked to signaling pathways associated with 5-HT2 or α1-adrenergic receptor activation. Furthermore, because synaptic isolation with TTX did not block PE-induced (Fig. 1 E,F) or 5-HT2 receptor-induced (Fig. 2C) (Bocchiaro and Feldman, 2004) ivLTF, network activity is not necessary for these changes in motoneuronal excitability. In vivo LTF is also not simply the consequence of transient excitation, because it is induced by intermittent hypoxia but not intermittent hypercapnia, each a powerful, independent stimulus that increase breathing (Baker et al., 2001).
PE-induced ivLTF is specific to α1-adrenoceptor activation because it was completely blocked by prazosin, a specific α1-adrenoceptor antagonist (Fig. 2 A). The possibility that ivLTF could be attributable to the persistent α1-adrenoreceptor occupancy is highly unlikely because one continuous 9 min PE application produced a transient XII nerve amplitude increase but not a persistent one. PE-induced ivLTF does not require 5-HT receptor activation, because it was attenuated but not blocked by methysergide (Figs. 1D, 2B). This attenuation might reflect the following: (1) a contributory role of 5-HT receptors in PE-induced ivLTF, (2) a reduction of overall neuronal excitability as evidenced by the ∼25% decrease in baseline activity when methysergide was added to the bath, or (3) nonspecific effects of methysergide on adrenergic receptors. Conversely, 5-HT2 receptor-dependent ivLTF persisted in the presence of prazosin (Fig. 2C,D), which did not alter the XII nerve activity baseline in the absence of ivLTF induction protocol.
Based on our observations, we suggest that episodic activation of either 5-HT2 or α1-adrenergic receptors on XII motoneurons initiates Gαq–PLC-mediated signaling cascades leading to PKC activation. We suggest that PKC activation induced subsequent mechanisms that underlie long-term increases in the AMPA receptor function (Li et al., 1999; Feldman et al., 2003; Sutton et al., 2004) (q.v., McGuire and Ling, 2004).
Stimulation of 5-HT2 and α1-adrenergic receptors could initiate cascades that converge at several points: (1) at membrane-bound PLC, which can be activated through the interaction of Gαq and α1-adrenoreceptors, mGluR1s, or 5-HT2 receptors; (2) at a second messenger activating the same protein kinase: for example, Ca2+ released from the intracellular stores activating PKC; or (3) via different pathways acting independently of each other, but converging at the AMPA receptor.
Although PKA antagonism did not block ivLTF (Fig. 3D), suggesting that active PKA is not necessary for the induction and/or maintenance of ivLTF, it did diminish ivLTF amplitude. Thus, PKA may contribute to ivLTF expression, possibly by mediating phosphorylation of AMPA receptors, influencing AMPA channel properties, and/or by maintaining AMPA receptor insertion in the postsynaptic membrane (Carvalho et al., 2000).
The fact that episodic stimulation of either α1-adrenoreceptors or 5-HT2 receptors elicits ivLTF, whereas one-time stimulation produces no long-lasting effect, suggests a critical time-dependent pattern sensitivity in the downstream signal transduction mechanisms. The critical point in the signal transduction pathway that requires episodicity is presently unknown.
Because antagonism of mGluR1s abolishes both 5-HT2-dependent (Bocchiaro and Feldman, 2004) and PE-induced (Fig. 3A) ivLTF, mGluR1s serve a permissive role for ivLTF. Thus, mGluR1s on XII motoneurons are either constitutively active under our experimental conditions, or the glutamate concentration at the synapse even in the presence of TTX is sufficiently high to activate mGluR1s (Bocchiaro and Feldman, 2004). We suggest that the active state of mGluR1s may be necessary to enable one or more steps in the signal transduction pathway linking α1-adrenoreceptors or 5-HT2 receptors to increased postsynaptic AMPA receptor function. mGluR1s are linked to the Gαq–PLC signaling pathway (Hermans and Challiss, 2001), the main pathway of action for both α1-adrenoceptors and 5-HT2 receptors. We suspect that mGluR1 convergence onto this same pathway or direct receptor–receptor interactions between mGluR1s and either AMPA or α1-adrenergic receptors underlie this interaction, as seen with other receptor types (Rocheville et al., 2000).
In vivo respiratory LTF induced by episodic hypoxia in adult rats is blocked by antagonists to 5-HT2 (Bach and Mitchell, 1996; Kinkead and Mitchell, 1999; Fuller et al., 2001) or α1-adrenergic (Fig. 4) receptors. We suggest two possible mechanisms. (1) Both α1-adrenergic and 5-HT2 receptors are permissive for the actions of each other in a similar manner to that proposed for mGluR1s. (2) There is a summation effect whereby the separate actions of noradrenaline and 5-HT acting on their respective receptors is insufficient to induce LTF, but their concurrent action in individual (moto)neurons converging on the same Gαq–PLC-dependent intracellular cascade is sufficient. In this second possibility, the induction of ivLTF by activating either receptor class alone may be possible because the concentrations of bath-applied drugs may be considerably higher than that induced by episodic hypoxia at synapses in vivo.
There are many possible mechanisms by which antagonism of either of these receptors could block in vivo LTF. (1) Hypoxia-induced activation of both serotonergic and noradrenergic cell groups is necessary for in vivo LTF after episodic hypoxia. (2) Basal activation of α1-adrenoceptors (and other Gαq–PLC-coupled receptors) is necessary to maintain critical levels of essential intracellular signaling molecules, such as activated PKC. In this scheme, if PKC activation fell below a certain level, 5-HT2 receptor activation caused by episodic hypoxia may be insufficient to raise PKC activity to levels necessary for the expression of in vivo LTF. The existence of such a PKC activation threshold would suggest that all Gαq–PLC-coupled receptors have the potential to elicit LTF if sufficiently activated, and that multiple Gαq–PLC-coupled receptors may be necessary for the successful expression of LTF induced by any other receptor.
In contrast to in vivo LTF in which both 5-HT2 and α1-adrenergic receptor activation are necessary, 5-HT antagonism does not prevent, but only attenuates, PE-induced ivLTF; similarly, α1-adrenergic antagonism does not prevent, but only attenuates, 5-HT2 receptor-induced ivLTF. This result could occur if the effective concentrations of PE and α-Me-5-HT used to induce ivLTF are higher than the physiological levels of endogenous 5-HT and noradrenaline induced by episodic hypoxia in vivo.
Noradrenegic and serotonergic neurons increase activity under conditions in which the induction of LTF may be of physiological usefulness. In the intact mammalian brain, the XII motor nucleus receives both serotonergic (Manaker and Tischler, 1993) and noradrenergic (Moore and Bloom, 1979; Aldes et al., 1992; Guyenet et al., 1993; Kinkead et al., 2001) inputs. Genioglossus muscle activity is under both serotonergic and noradrenergic state-dependent, especially sleep–wake, modulation (Aston-Jones and Bloom, 1981; Siegel, 1994; Kubin et al., 1998). The withdrawal of serotonergic (Jacobs and Azmitia, 1992; Kubin et al., 1994) and noradrenergic (Fenik et al., 2005) inputs depresses XII nerve activity during sleep or REM sleep-like states induced by pontine carbachol injections. Moreover, when serotonin is applied to the XII nucleus during sleep in behaving rats, genioglossus muscle activity increases (Jelev et al., 2001). The loss of serotonergic and/or noradrenergic inputs could lead to failure of XII motoneurons to produce appropriate output during sleep; this can result in loss of tone in tongue muscles leading to airway blockage, as seen in humans who suffer from obstructive sleep apnea (OSA).
Thus, our results might model a mechanism whereby repetitive arousal caused by airway obstruction during sleep (as seen in OSA) and resulting hypoxia causes episodic noradrenaline (and serotonin) release, which in turn could contribute to persistent increases in XII motoneuron activity. The result of such facilitation would increase upper airway muscle tone and improve the likelihood that airway patency could be maintained.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant NS24742, Roman Reed Spinal Cord Injury Research Fund of California, and NIH Training Grant HL07654. We thank Patrice Guyenet for pointing out the behavioral link between serotonin and noradrenaline pharmacology.
References
- Aldes LD, Chapman ME, Chronister RB, Haycock JW. Sources of noradrenergic afferents to the hypoglossal nucleus in the rat. Brain Res Bull. 1992;29:931–942. doi: 10.1016/0361-9230(92)90168-w. [DOI] [PubMed] [Google Scholar]
- Aoki CR, Liu H, Downey GP, Mitchell J, Horner RL. Cyclic nucleotides modulate genioglossus and hypoglossal responses to excitatory inputs in rats. Am J Respir Crit Care Med. 2006;173:555–565. doi: 10.1164/rccm.200509-1469OC. [DOI] [PubMed] [Google Scholar]
- Aston-Jones G, Bloom F. Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J Neurosci. 1981;1:876–886. doi: 10.1523/JNEUROSCI.01-08-00876.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock M, Shkoukani M, Aboubakr SE, Badr MS. Determinants of long-term facilitation in humans during NREM sleep. J Appl Physiol. 2002;94:53–59. doi: 10.1152/japplphysiol.00476.2002. [DOI] [PubMed] [Google Scholar]
- Bach KB, Mitchell GS. Hypoxia-induced long-term facilitation of respiratory activity is serotonin dependent. Respir Physiol. 1996;104:251–260. doi: 10.1016/0034-5687(96)00017-5. [DOI] [PubMed] [Google Scholar]
- Baker TL, Mitchell GS. Episodic but not continuous hypoxia elicits long-term facilitation of phrenic motor output in rats. J Physiol (Lond) 2000;529:215–219. doi: 10.1111/j.1469-7793.2000.00215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker TL, Fuller DD, Zabka AG, Mitchell GS. Respiratory plasticity: differential actions of continuous and episodic hypoxia and hypercapnia. Respir Physiol. 2001;129:25–35. doi: 10.1016/s0034-5687(01)00280-8. [DOI] [PubMed] [Google Scholar]
- Baker-Herman TL, Mitchell GS. Phrenic long-term facilitation requires spinal serotonin receptor activation and protein synthesis. J Neurosci. 2002;22:6239–6246. doi: 10.1523/JNEUROSCI.22-14-06239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocchiaro CM, Feldman JL. Synaptic activity-independent persistent plasticity in endogenously active mammalian motoneurons. Proc Natl Acad Sci USA. 2004;101:4292–4295. doi: 10.1073/pnas.0305712101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocchiaro CM, Saywell SA, Feldman JL. Dynamic modulation of inspiratory drive currents by protein kinase A and protein phosphatases in functionally active motoneurons. J Neurosci. 2003;23:1099–1103. doi: 10.1523/JNEUROSCI.23-04-01099.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho AL, Duarte CB, Carvalho AP. Regulation of AMPA receptors by phosphorylation. Neurochem Res. 2000;25:1245–1255. doi: 10.1023/a:1007644128886. [DOI] [PubMed] [Google Scholar]
- Erickson JT, Millhorn DE. Hypoxia and electrical stimulation of the carotid sinus nerve induce Fos-like immunoreactivity within catecholaminergic and serotoninergic neurons of the rat brainstem. J Comp Neurol. 1994;348:161–182. doi: 10.1002/cne.903480202. [DOI] [PubMed] [Google Scholar]
- Feldman JL, Mitchell GS, Nattie EE. Breathing: rhythmicity, plasticity, chemosensitivity. Annu Rev Neurosci. 2003;26:239–266. doi: 10.1146/annurev.neuro.26.041002.131103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenik VB, Davies RO, Kubin L. REM sleep-like atonia of hypoglossal (XII) motoneurons is caused by loss of noradrenergic and serotonergic inputs. Am J Respir Crit Care Med. 2005;172:1322–1330. doi: 10.1164/rccm.200412-1750OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller DD, Bach KB, Baker TL, Kinkead R, Mitchell GS. Long-term facilitation of phrenic motor output. Respir Physiol. 2000;121:135–146. doi: 10.1016/s0034-5687(00)00124-9. [DOI] [PubMed] [Google Scholar]
- Fuller DD, Zabka AG, Baker TL, Mitchell GS. Phrenic long-term facilitation requires 5-HT receptor activation during but not following episodic hypoxia. J Appl Physiol. 2001;90:2001–2006. doi: 10.1152/jappl.2001.90.5.2001. [DOI] [PubMed] [Google Scholar]
- Funk GD, Smith JC, Feldman JL. Generation and transmission of respiratory oscillations in medullary slices: role of excitatory amino acids. J Neurophysiol. 1993;70:1497–1515. doi: 10.1152/jn.1993.70.4.1497. [DOI] [PubMed] [Google Scholar]
- Garcia-Sainz JA, Vazquez-Prado J, Villalobos-Molina R. [alpha]1-Adrenoceptors: subtypes, signaling, and roles in health and disease. Arch Med Res. 1999;30:449–458. doi: 10.1016/s0188-0128(99)00059-7. [DOI] [PubMed] [Google Scholar]
- Guyenet PG, Koshiya N, Huangfu D, Verberne AJ, Riley TA. Central respiratory control of A5 and A6 pontine noradrenergic neurons. Am J Physiol. 1993;264:R1035–R1044. doi: 10.1152/ajpregu.1993.264.6.R1035. [DOI] [PubMed] [Google Scholar]
- Hermans E, Challiss RAJ. Structural, signalling and regulatory properties of the group I metabotropic glutamate receptors: prototypic family C G-protein-coupled receptors. Biochem J. 2001;359:465–484. doi: 10.1042/0264-6021:3590465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs BL, Azmitia EC. Structure and function of the brain serotonin system. Physiol Rev. 1992;72:165–229. doi: 10.1152/physrev.1992.72.1.165. [DOI] [PubMed] [Google Scholar]
- Jelev A, Sood S, Liu H, Nolan P, Horner RL. Microdialysis perfusion of 5-HT into hypoglossal motor nucleus differentially modulates genioglossus activity across natural sleep–wake states in rats. J Physiol (Lond) 2001;532:467–481. doi: 10.1111/j.1469-7793.2001.0467f.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinkead R, Mitchell GS. Time-dependent hypoxic ventilatory responses in rats: effects of ketanserin and 5-carboxamidotryptamine. Am J Physiol. 1999;227:R658–R666. doi: 10.1152/ajpregu.1999.277.3.R658. [DOI] [PubMed] [Google Scholar]
- Kinkead R, Bach KB, Johnson SM, Hodgeman BA, Mitchell GS. Plasticity in respiratory motor control: intermittent hypoxia and hypercapnia activate opposing serotonergic and noradrenergic modulatory systems. Comp Biochem Physiol A Physiol. 2001;130:207–218. doi: 10.1016/s1095-6433(01)00393-2. [DOI] [PubMed] [Google Scholar]
- Kubin L, Reignier C, Tojima H, Taguchi O, Pack AI, Davies RO. Changes in serotonin level in the hypoglossal nucleus region during carbachol-induced atonia. Brain Res. 1994;645:291–302. doi: 10.1016/0006-8993(94)91663-2. [DOI] [PubMed] [Google Scholar]
- Kubin L, Davies RO, Pack AI. Control of upper airway motoneurons during REM sleep. News Physiol Sci. 1998;13:91–97. doi: 10.1152/physiologyonline.1998.13.2.91. [DOI] [PubMed] [Google Scholar]
- Li P, Kerchner GA, Sala C, Wei F, Huettner JE, Sheng M, Zhuo M. AMPA receptor-PDZ interactions in facilitation of spinal sensory synapses. Nat Neurosci. 1999;2:972–977. doi: 10.1038/14771. [DOI] [PubMed] [Google Scholar]
- Liu G, Feldman JL, Smith JC. Excitatory amino acid-mediated transmission of inspiratory drive to phrenic motoneurons. J Neurophysiol. 1990;64:423–436. doi: 10.1152/jn.1990.64.2.423. [DOI] [PubMed] [Google Scholar]
- Manaker S, Tischler LJ. Origin of serotoninergic afferents to the hypoglossal nucleus in the rat. J Comp Neurol. 1993;334:466–476. doi: 10.1002/cne.903340310. [DOI] [PubMed] [Google Scholar]
- McGuire M, Ling L. Activation of protein kinase C near/in phrenic motoneurons is required for phrenic long-term facilitation in rats. Am J Respir Crit Care Med. 2004;169:A433. [Google Scholar]
- McKay LC, Janczewski WA, Feldman JL. Episodic hypoxia evokes long-term facilitation of genioglossus muscle activity in neonatal rats. J Physiol (Lond) 2004;557:13–18. doi: 10.1113/jphysiol.2004.064006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell GS, Johnson SM. Plasticity in respiratory motor control: invited review: neuroplasticity in respiratory motor control. J Appl Physiol. 2003;94:358–374. doi: 10.1152/japplphysiol.00523.2002. [DOI] [PubMed] [Google Scholar]
- Mitchell GS, Baker TL, Nanda SA, Fuller DD, Zabka AG, Hodgeman BA, Bavis RW, Mack KJ, Olson EBJ. Invited review: intermittent hypoxia and respiratory plasticity. J Appl Physiol. 2001;90:2466–2475. doi: 10.1152/jappl.2001.90.6.2466. [DOI] [PubMed] [Google Scholar]
- Moore RY, Bloom FE. Central catecholamine neuron systems: anatomy and physiology of the norepinephrine and epinephrine systems. Annu Rev Neurosci. 1979;2:113–168. doi: 10.1146/annurev.ne.02.030179.000553. [DOI] [PubMed] [Google Scholar]
- Rekling JC, Funk GD, Bayliss DA, Dong X-W, Feldman JL. Synaptic control of motoneuronal excitability. Physiol Rev. 2000;80:767–852. doi: 10.1152/physrev.2000.80.2.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocheville M, Lange DC, Kumar U, Sasi R, Patel RC, Patel YC. Subtypes of the somatostatin receptor assemble as functional homo- and heterodimers. J Biol Chem. 2000;275:7862–7869. doi: 10.1074/jbc.275.11.7862. [DOI] [PubMed] [Google Scholar]
- Roth BL, Willins DL, Kristiansen K, Kroeze WK. 5-Hydroxytryptamine2-family receptors (5-hydroxytryptamine2A, 5-hydroxytryptamine2B, 5-hydroxytryptamine2C): where structure meets function. Pharmacol Ther. 1998;79:231–257. doi: 10.1016/s0163-7258(98)00019-9. [DOI] [PubMed] [Google Scholar]
- Shkoukani M, Babcock MA, Badr MS. Effect of episodic hypoxia on upper airway mechanics in humans during NREM sleep. J Appl Physiol. 2002;92:2565–2570. doi: 10.1152/japplphysiol.00938.2001. [DOI] [PubMed] [Google Scholar]
- Siegel JM. Brainstem mechanisms generating REM sleep. In: Kryger MH, Roth T, Dement WC, editors. Principles and practice of sleep medicine. Ed 2. Philadelphia: Saunders; 1994. pp. 125–144. [Google Scholar]
- Smith JC, Ellenberger HH, Ballanyi K, Richter DW, Feldman JL. Pre-Bötzinger complex: a brainstem region that may generate respiratory rhythm in mammals. Science. 1991;254:726–729. doi: 10.1126/science.1683005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Bagnall MW, Sharma SK, Shobe J, Carew TJ. Intermediate-term memory for site-specific sensitization in Aplysia is maintained by persistent activation of protein kinase C. J Neurosci. 2004;24:3600–3609. doi: 10.1523/JNEUROSCI.1134-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volgin DV, Mackiewicz M, Kubin L. [alpha]1B receptors are the main postsynaptic mediators of adrenergic excitation in brainstem motoneurons, a single-cell RT-PCR study. J Chem Neuroanat. 2001;22:157–166. doi: 10.1016/s0891-0618(01)00124-7. [DOI] [PubMed] [Google Scholar]