

Abstract
Asymptomatic Huntington's disease (HD) patients exhibit memory and cognition deficits that generally worsen with age. Similarly, long-term potentiation (LTP), a form of synaptic plasticity involved in memory encoding, is impaired in HD mouse models well before motor disturbances occur. The reasons why LTP deteriorates are unknown. Here we show that LTP is impaired in hippocampal slices from presymptomatic HdhQ92 and HdhQ111 knock-in mice, describe two factors contributing to this deficit, and establish that potentiation can be rescued with brain-derived neurotrophic factor (BDNF). Baseline physiological measures were unaffected by the HD mutation, but LTP induction and, to a greater degree, consolidation were both defective. The facilitation of burst responses that normally occurs during a theta stimulation train was reduced in HD knock-in mice, as was theta-induced actin polymerization in dendritic spines. The decrease in actin polymerization and deficits in LTP stabilization were reversed by BDNF, concentrations of which were substantially reduced in hippocampus of both HdhQ92 and HdhQ111 mice. These results suggest that the HD mutation discretely disrupts processes needed to both induce and stabilize LTP, with the latter effect likely arising from reduced BDNF expression. That BDNF rescues LTP in HD knock-in mice suggests the possibility of treating cognitive deficits in asymptomatic HD gene carriers by upregulating production of the neurotrophin.
Keywords: neurotrophin, long-term potentiation, polyglutamine, hippocampus, actin, dendritic spines
Introduction
Huntington's disease (HD) is caused by a mutation that expands the number of trinucleotide CAG repeats in the huntingtin protein gene (Vonsattel and DiFiglia, 1998). Clinically, it is associated with severe motor disturbances and cognitive deficits that generally worsen with age (Vonsattel and DiFiglia, 1998). The cognitive deficits include impairments in attention, visuospatial ability, semantic verbal fluency, and short- and long-term memory. Although some debate exists about when cognitive problems first appear, several, especially those involving memory, can be discerned in asymptomatic gene carriers (Lawrence et al., 1998; Kirkwood et al., 2000; Ho et al., 2003; Lemiere et al., 2004). The deficits are typically attributed to disturbances in the cortico-striatal system, but other structures involved in cognition, including amygdala and hippocampus, are affected in early stages of the disease (Rosas et al., 2003).
Impaired learning that occurs before motor symptoms or neuron loss has also been described for mouse models of HD (Lione et al., 1999; Mazarakis et al., 2005; Van Raamsdonk et al., 2005). These behavioral abnormalities are accompanied by deficits in hippocampal long-term potentiation (LTP) (Hodgson et al., 1999; Usdin et al., 1999; Murphy et al., 2000), a form of synaptic plasticity widely regarded as a substrate for memory encoding, and by reductions in mossy fiber potentiation (Gibson et al., 2005) and long-term depression (Milnerwood et al., 2006). Reduced plasticity is evident weeks before the first signs of movement disorders, indicating that it is an early marker for HD rather than a secondary consequence of neurodegeneration. The reasons why LTP deteriorates in HD mice are unknown but are likely to be important for understanding the cognitive problems that accompany the disease. Results from knock-in (72/80 CAG) mice point to a deficit in neurotransmitter mobilization (Usdin et al., 1999), but studies using transgenic (R6/2) mice suggest that processes that normally stabilize potentiation are impaired (Murphy et al., 2000). Importantly, LTP may be a particularly sensitive target for early effects of the HD mutation because baseline physiological measures were normal in both mouse models.
Possibly related to the loss of plasticity is evidence that mutant huntingtin decreases expression of brain-derived neurotrophic factor (BDNF) and its TrkB receptor in neocortex and hippocampus of humans (Ferrer et al., 2000; Zuccato et al., 2001) and mice (Zuccato et al., 2001, 2005; Gines et al., 2003, 2006; Spires et al., 2004a). BDNF is a potent, positive modulator of LTP when the potentiation effect is induced by naturalistic theta burst stimulation (TBS) (Bramham and Messaoud, 2005). The neurotrophin produces its effects, in part, by reducing afterhyperpolarizations that accompany theta burst responses (Kramar et al., 2004) and by facilitating the actin polymerization that occurs in dendritic spines immediately after stimulation (Rex et al., 2007). The first of these actions enhances the depolarization that induces LTP, whereas the second promotes an event essential to the stabilization (or consolidation) of the potentiated state (Kramar et al., 2004, 2006; Bramham and Messaoud, 2005; Lynch et al., 2007). In the present studies, we tested whether LTP induction, consolidation, or both are impaired in HD knock-in mice and whether BDNF can restore normal plasticity.
Materials and Methods
All animal procedures were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and with protocols approved by the Institutional Animal Care and Use Committee of the University of California at Irvine. This includes efforts to minimize animal suffering and the numbers of mice used.
Mice and genotyping.
HdhQ92 and HdhQ111 mice have a chimeric mouse–human exon 1 with 92 and 111 repeats of the trinucleotide CAG, respectively (see next paragraph for actual CAG size determination), inserted into the huntingtin gene (Wheeler et al., 2000). Homozygous HdhQ92 and HdhQ111 breeding pairs were purchased from the Jackson Laboratory (Bar Harbor, ME) and maintained in the laboratory as an inbred colony. Wild-type (WT) mice from the same background strain (C57BL/6J) and vendor were used as controls. Males (N = 27) and females (N = 24) of each genotype were used in these studies. A minimum of four mice was used for each experiment, and the number of mice used for each individual experiment is listed in the methods below and is depicted with an upper case N. The number of slices used for each experiment is provided in the text and in the figures; a lowercase n is used to denote the number of slices.
Genomic DNA was extracted from tail samples at the time of weaning using the Wizard DNA purification kit (Promega, Madison, WI). Periodic genotyping was performed using standard PCR procedures and the following primers: 5′-GGCTGAGGAAGCTGAGGAG-3′, 5′-GTCC-TGACATCGGGAAAGAG-3′, and 5′-GTTCCTCTGCCGGACCTG-3′. In a subset of mice (N = 3 per genotype), repeat stability was determined using a modification of the CAG genotyping assay described by Mangiarini et al. (1997). The PCR primers and conditions are the same as described previously (Mangiarini et al., 1997), but the reverse primer was fluorescently labeled, and the PCR products were sized on an ABI3730XL machine (Applied Biosystems, Foster City, CA). Allele sizes were determined by comparing the PCR products to DNA standards of known CAG repeat size, as determined by DNA sequencing. For the HdhQ92 mice, both alleles had 81.3 ± 0.6 CAGs; for HdhQ111 mice, one allele had 78.3 ± 0.6 CAGs, whereas the other had 96.0 ± 1.0. The onset of HD phenotypes in knock-in mice is determined by the length of the longest CAG allele, as reported in humans, and these lengths are at the high end of CAG size range documented for human juvenile onset cases (Duyao et al., 1993; Stine et al., 1993; Bates, 2002).
Physiology.
Acute hippocampal slices were prepared, as described previously (Kramar et al., 2004), from 2-month-old HdhQ111 (N = 6), 4- to 6-month-old HdhQ92 (N = 5), and age-matched WT mice (N = 4 at 2 months and N = 4 at 6 months) and maintained at 32°C in an interface chamber of local design. Field EPSPs (fEPSPs) were recorded from stratum radiatum of CA1b using a single glass pipette (2–3 MΩ). Baseline stimulation (three pulses per minute) was delivered at two sites in the CA1 region, one stimulating electrode placed on either side of the recording electrode (CA1a and CA1c) in the Schaffer collateral–commissural projections, and the fEPSPs were adjusted to achieve 30% of the maximum spike-free response. The slope of the descending phase of the fEPSP was measured with all values normalized to a 15 min baseline period beginning 1–2 h after slice preparation; the values are reported in the figures as mean ± SEM. LTP was induced using a single train of TBS (10 bursts of four pulses at 100 Hz, interburst interval of 200 ms). BDNF (2 nm, catalog #GF049; Chemicon, Temecula, CA) was prepared and delivered to slices from WT (N = 6) and HdhQ111 (N = 4) mice, as described previously (Kramar et al., 2004).
Whole-cell recordings were performed, using methods standard for our laboratory (Kramar et al., 2004), in WT (N = 3) and HdhQ111 (N = 4) mice. Briefly, CA1 pyramidal neurons were visualized with an infrared microscope in differential interference contrast configuration, and recordings were made with 3–5 MΩ recording pipettes filled with a solution composed of (in mm) 130 Cs gluconate, 0.1 EGTA, 0.5 MgCl2, 10 HEPES, and 2 ATP, pH 7.25 (285 mOsm). Holding potentials were set to −70 mV after correcting for the junction potential. Currents were sampled with a patch amplifier (AxoPatch-200A; Molecular Devices, Palo Alto, CA) with a four-pole low-pass Bessel filter at 2 kHz and digitized at 10 kHz with pClamp 7 acquisition software (Molecular Devices). Patch electrodes were made from borosilicate glass using a Sutter Flaming-Brown horizontal puller (P-87; Sutter Instruments, Novato, CA). IPSCs were recorded from pyramidal cells in the presence of 10 μm DNQX and 100 μm APV with the stimulation electrode placed close the target neuron. Membrane voltage was brought to 0 mV before the delivery of single or double pulses. The recorded responses were entirely blocked by 50 μm picrotoxin.
For all electrophysiology experiments, results are summarized in the text as a percentage of increase in responses from baseline. Data are presented as mean ± SD in the text and mean ± SEM in the figures, unless noted otherwise.
In situ labeling of filamentous actin.
Physiological recordings and delivery of TBS or low-frequency stimulation were performed as described above. Immediately after physiological recording, rhodamine–phalloidin [6 μm/2–4 μl; Sigma (St. Louis, MO) or Invitrogen (Carlsbad, CA)] was applied topically (Kramar et al., 2006) via micropipette three times separated by 3 min. Studies with intracellular or extracellular applications have indicated that this concentration of phalloidin has no effect on baseline physiology, theta burst responses, or LTP (Lin et al., 2005). Note also that post-TBS applications, as used for the experiments reported here, preclude disturbances to the induction and early expression of LTP. To test the effects of BDNF on actin polymerization, human recombinant BDNF (2 nm), or artificial CSF was delivered to slices from WT and HdhQ111 mice via a reperfusion pump system (Cole-Palmer, Vernon Hills, IL) at 1 ml/min 1–2 h before recording. Slices were then fixed in 4% paraformaldehyde in 0.1 m sodium phosphate buffer (PB), pH 7.2, cryoprotected with 20% sucrose in PB, sectioned at 20 μm on a freezing microtome, and coverslipped with Vectashield (Vector Laboratories, Burlingame, CA).
Sections were examined with epifluorescence illumination using an Olympus (Tokyo, Japan) AX70 photomicroscope and an Optronics Microfire CCD camera and, for confirmation purposes, with laser-scanning confocal microscopy using the Bio-Rad (Hercules CA) Radiance 200 or the Zeiss (Thornwood, NY) LSM 510 META laser-scanning system. Quantification of dendritic spines was performed with the CCD camera coupled with parametric estimation signal processing (described below), which had been determined, from preliminary studies, to detect and resolve low-intensity illumination better than the laser-scanning systems. Quantitative analyses were performed on three serial sections situated 20–80 μm below the surface of the original slice. A series of 20–30 high-resolution digital photomicrographs were taken at regular focal steps through each section (Z-stacks). Camera exposure time was adjusted for each experiment so that approximately four to eight spines could be visualized in the sample field of control slices. Subsequent images intended for comparison were then collected with the same illumination and exposure settings. The Z-stacks were collapsed into a single image by extended focal imaging using Microsuite FIVE software (Soft Imaging System, Lakewood, CO). These images were then converted to grayscale, and intensity levels were cropped at values determined for each experiment to visualize low-intensity labeling.
Labeled spine-like structures were measured and counted from a 550 μm2 sampling zone in proximal stratum radiatum between the two stimulating electrodes using in-house software described in detail previously (Lin et al., 2005; Kramar et al., 2006). Counting was done blindly on batches of slices that had been sectioned and stained together. Intensity thresholds (8 bits) were applied to identify spine-like structures at varying levels of label intensity. Digital images of objects included in the counts were overlaid semitransparently with the original photomicrographs to confirm that they were spines. Spine counts from each of the three series of sections were averaged to produce a representative value for each slice.
For double-labeling experiments, slices were labeled with rhodamine–phalloidin (12 μm in pipette, tissue levels will be substantially diluted) and prepared for histology, as described above. Sections were then incubated for 1 h at room temperature with mouse anti-postsynaptic density-95 (PSD-95; MAB1598; Chemicon) at 1:100 in PB containing 4% bovine serum albumin and 0.3% Triton X-100. Slides were rinsed in PB, incubated (45 min at room temperature) in fluorescein anti-mouse IgG (1:200; Vector Laboratories) in PB with 0.3% Triton X-100, and rinsed again. Laser-scanning confocal microscopy was used to assess double labeling. Tissue sections were qualitatively analyzed from image Z-stacks collected at 60× objective magnification. Image processing was performed with Photoshop 6.0 (Adobe Systems, San Jose, CA). Figures show single 1-μm-thick optical sections.
Western blots.
Endogenous BDNF levels were assessed using Western blots with rabbit anti-BDNF (1:1000; catalog #SC546; Santa Cruz Biotechnology, Santa Cruz, CA) that recognizes both mature and pro forms of the protein. Hippocampus of 2-month-old HdhQ111 (N = 7), 2-month-old HdhQ92 (N = 5), and age-matched WT (N = 8 and 7, respectively) mice were homogenized in NP-40 cell lysis buffer (Biosource, Camarillo, CA) with protease inhibitor mixture (catalog #P2714; Sigma) and 1 mm phenylmethanesulfonyl fluoride (Sigma). Protein levels were measured using the Bio-Rad Protein Assay. Protein samples (25 μg/lane) were then separated by 15% PAGE and processed for Western blot analysis as described previously (Kramar et al., 2004) using the enhanced chemiluminescence ECL Plus Detection System (Amersham Biosciences, Buckinghamshire, UK). As a positive control, recombinant human BDNF (Chemicon) was loaded on the same gels as samples. To control for loading variations, blots were stripped and reprobed with anti-actin (1:2000; Sigma). For quanitification, immunoreactive bands were manually outlined, and densities were measured using NIH Image software. For each blot, the densities of BDNF-immunoreactive bands were expressed as a fraction of the actin-immunoreactive band in the same lane; samples were run two to three separate times, and results were averaged. Statistical analyses were run on both raw and actin-normalized values for pro-BDNF and mature BDNF bands. Figure 6 shows the percentage of control mean calculated from the raw density values. Significance of the effect of genotype was evaluated using a one-tailed Student's t test.
Figure 6.

BDNF levels are low in hippocampus of 2-month-old HdhQ111 mice. A, Representative Western blot of hippocampal homogenates shows that both pro-BDNF (pBDNF) and mature BDNF (mBDNF) levels are lower in HdhQ111 mice relative to wild types. Samples for two mice from each genotype are shown. The far left lane shows the migration of recombinant BDNF (rBDNF) protein. The actin immunoband from the stripped and reprobed blot is shown at the bottom. B, Densitometric analyses confirmed that both pBDNF and mBDNF levels are lower in HdhQ111 relative to WT hippocampus (**p = 0.0001 and *p = 0.002, respectively). The decrease in pBDNF in HdhQ111 mice was larger than that for mBDNF (#p = 0.007).
Results
Severe impairment of LTP in two knock-in mouse models of HD
HdhQ92 and HdhQ111 mice, which have 92 or 111 CAG repeats inserted into the huntingtin (Hdh) gene, were selected for these experiments because they closely resemble the genetic component of the human condition and, as with HD patients, have a delayed onset of overt symptoms (Menalled, 2005). Hdh92 and HdhQ111 mice were tested at 2 months and 4–6 months of age, respectively; these ages are months before the onset of motor disturbances.
Low-frequency (baseline) stimulation was delivered to hippocampal slices from HdhQ92, HdhQ111, and age-matched WT mice. fEPSPs were comparable in HD and WT slices with regard to size (slope, amplitude) and waveform. Paired-pulse facilitation, a test for neurotransmitter mobilization, did not detectably differ between the groups with delays of 50 ms (wild type: 64 ± 18%, n = 6; combined HdhQ92, HdhQ111: 70 ± 15%, n = 9; mean ± SEM) or 100 ms (wild type, 52 ± 15%; Hdh, 53 ± 9%). Thus, baseline physiological measures were similar in WT and HD slices.
TBS was applied after a 20–30 min period of baseline recording; low-frequency stimulation (3 min pulses) was resumed after TBS, and responses were collected for an additional 60 min. In WT slices, TBS doubled the size of fEPSPs in the first minute after stimulation (Fig. 1), after which responses decayed to a stable level that was ∼50% greater than the pre-TBS baseline. Slices from HD knock-in mice differed from controls in two ways: (1) the initial potentiation did not reach levels seen in WT mice (p = 0.017 at 20 s after TBS; two-tailed Student's t test) and (2) the responses decayed rapidly (40 min) back to baseline (i.e., stable potentiation was not attained). The latter effect was highly significant (percentage of LTP in WT vs HD mice at 40 min after TBS; p < 0.00001). Figure 1B summarizes the percentage of LTP obtained at 55 min after induction for HdhQ92 and HdhQ111 mice along with each group's age-matched controls. LTP was absent in both mutant strains at this sampling point (−3.3 and −2.8% for HdhQ92 and HdhQ111 slices, respectively), whereas robust potentiation was evident in both age-matched WT groups (Fig. 1B). The differences between HD and WT mice were highly significant for both HD strains (p < 0.00001 and p = 0.00002 for HdhQ92 and HdhQ111, respectively). Combined with previous results from two other strains (Hodgson et al., 1999; Usdin et al., 1999; Murphy et al., 2000), these findings indicate that mutant huntingtin severely impairs the stabilization of LTP across multiple strains and a broad array of experimental conditions.
Figure 1.

LTP is impaired in HdhQ92 and HdhQ111 mice. A, TBS caused an immediate increase in the slope of the fEPSPs in WT mice, after which responses stabilized at a level ∼50% above baseline. In both HdhQ92 and HdhQ111 mice (Hdh), the initial induction of LTP was reduced, and responses decayed steadily back to baseline over 30–40 min. The inset (top right) shows representative fEPSP traces recorded during the baseline period and 2 and 60 min after TBS from a WT and an HdhQ111 mouse. Figures are averages of three consecutive traces. Calibration: 1 mV, 5 ms. B, Summary of the percentage of potentiation observed 55 min after TBS delivery for slices from HdhQ111 (N = 4 mice) and HdhQ92 (N = 5 mice) compared with equal numbers of their age-matched WT controls. Each of the two WT versus mutant comparisons is highly significant (p < 0.00005, two-tailed Student's t test). C, A subgroup of the HD slices that exhibited the highest level of initial potentiation (20–60 s after TBS) is plotted against a subgroup of WT slices with the lowest levels of initial potentiation (fEPSP slope expressed as the percentage of baseline shown). Potentiation in the period immediately following TBS (arrow) is comparable for these two subgroups; nevertheless, LTP decayed over 60 min in the Hdh slices. For this and subsequent graphs, error bars indicate means ± SEM. In A and C, n values are for numbers of slices.
As noted, initial potentiation was reduced in HD mice relative to wild types. With regard to mechanism, it is of interest to consider the extent to which this initial effect might contribute to the stability and ultimate magnitude of LTP. There was essentially no correlation between the magnitude of potentiation at 20 s and 55 min after TBS for HD (r = 0.22) or WT (r = 0.21) mice. Moreover, HdhQ92 and HdhQ111 slices that had relatively high levels of initial potentiation exhibited decremental LTP, whereas WT slices with relatively low initial potentiation had robust and stable LTP (Fig. 1C).
LTP induction and consolidation processes in HdhQ111 mice
Responses to TBS
Events that occur during the 2 s period of theta stimulation can potently influence the threshold, initial expression, magnitude, and stability of LTP (Larson et al., 1986; Arai and Lynch, 1992). In HdhQ111 slices, the size (area) of the composite response to the first of 10 stimulation bursts within a theta train did not differ from wild types (Fig. 2A); group values were 60.9 ± 7.8 mV·ms for HdhQ111 slices and 55.7 ± 25.6 mV·ms for wild types. This result suggests that calcium-dependent enhancement of neurotransmitter release, which normally occurs during TBS (Creager et al., 1980), and the IPSCs that shape the first burst response (Larson et al., 1986; Mott and Lewis, 1991) remain intact in HdhQ111 mice. In marked contrast, the facilitation of the second and subsequent burst responses, each a composite of four closely spaced fEPSPs, was greatly attenuated in HdhQ111 slices (Fig. 2A, burst 4). The facilitation effect was quantified by expressing the areas of burst responses 2–10 as fractions of the area of the first burst response. As is evident (Fig. 2B), within-train facilitation, described in numerous publications (Kramar et al., 2004; Rex et al., 2006), is substantially reduced in HdhQ111 relative to WT slices (mean facilitation across bursts 2–10 was 65.2 ± 20.3% for wild types and 34.4 ± 15.3% for HdhQ111 mice; p = 0.03).
Figure 2.
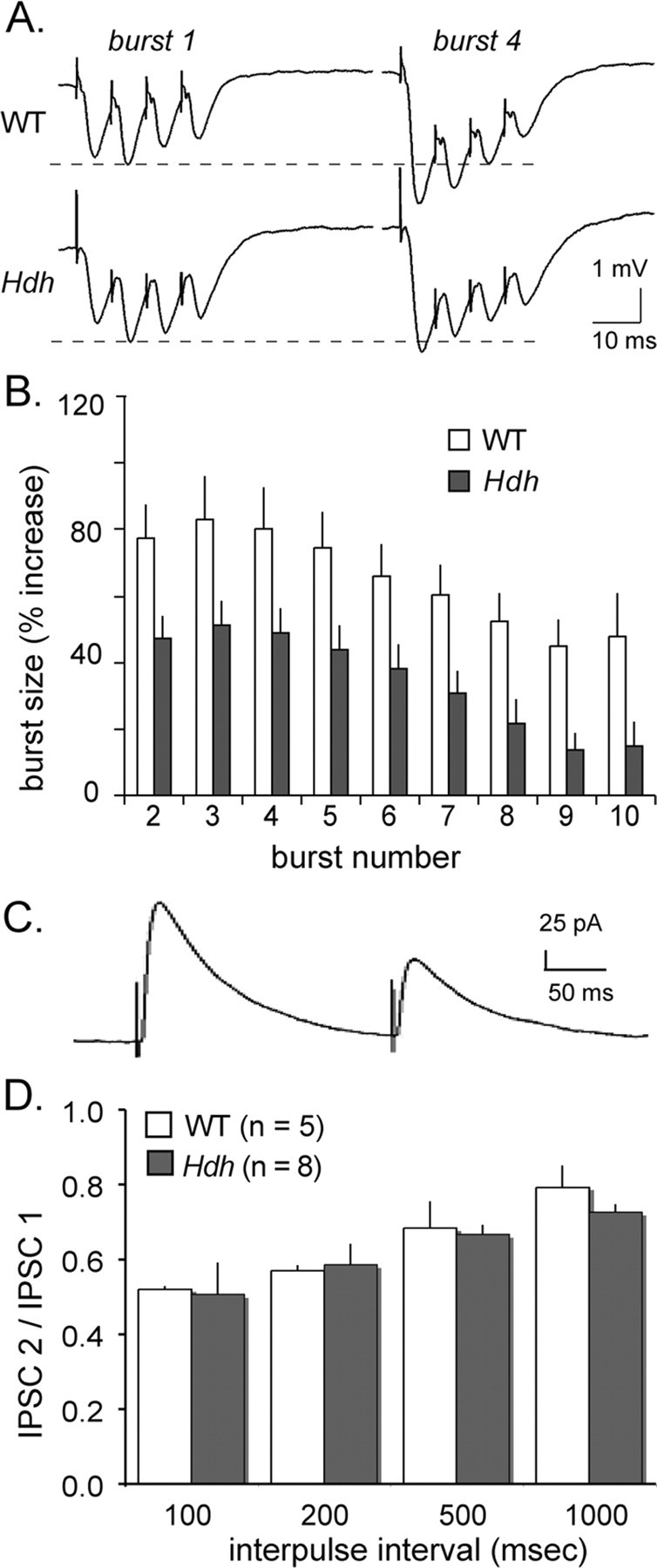
Facilitation of burst responses during TBS is impaired in HdhQ111 mice. A, Shown are responses to the first and fourth stimulation bursts of a theta train from WT (top) and HdhQ111 (bottom) slices. Note that when comparing genotypes, the first burst responses (burst 1) are similar, and in both cases, the fourth burst response (burst 4) is larger than the first; however, between-burst facilitation is more pronounced in the WT slice. B, The graph depicts the sizes of responses to stimulation bursts 2–10 in a theta train with each burst response expressed as a fraction of the area of the composite response to the first burst. The large facilitation of bursts 2–10 seen in slices from WT mice is greatly attenuated in those from HdhQ111 mice (WT vs HdhQ111 mean facilitation across bursts 2–10; p = 0.03). C, The IPSC refractory period is well developed in HdhQ111 slices. Note that the second of two IPSCs induced by a pair of stimulation pulses (interpulse interval, 200 ms) is substantially reduced in HdhQ111 mice, as it is in wild types (not shown). D, Paired stimulation pulses delivered with the indicated interpulse intervals showed that the IPSC refractory effect is time dependent and does not differ between WT and HdhQ111 slices (the size of the second IPSC is plotted as a fraction of the first).
Facilitation of responses during TBS arises because potent feedforward IPSCs, which are activated at the beginning of the first burst, enter a refractory period and therefore exert a smaller current shunting effect on the second and subsequent burst responses (Larson et al., 1986; Mott and Lewis, 1991). Thus, reduced within-train facilitation seen in HdhQ111 mice could be attributable to a change in the refractoriness of IPSCs. We tested this by delivering two single stimulation pulses separated by 100–1000 ms and measuring the size of the second feedforward IPSC relative to the first. The second IPSC in the HdhQ111 slices was markedly reduced when initiated 100 ms after the first response (Fig. 2C), as it was in WT slices. The depression of the second response was time dependent, with the greatest reduction occurring at 100 and 200 ms delays; the decrement at these intervals was 45 ± 15% (n = 8) in HdhQ111 slices and 45 ± 3% (n = 5) in wild types (Fig. 2D). Note that the waveform and refractoriness of these responses resemble those described with other intracellular and extracellular recording methods (Larson et al., 1986). Therefore, IPSCs are unaffected in HD mice and probably do not account for the loss of response facilitation during TBS; other possible contributors to this deficit are discussed below.
Actin polymerization in dendritic spines
TBS causes actin to polymerize in adult spines of dendritic regions containing potentiated synapses (Lin et al., 2005), and this effect is closely related to the stabilization of LTP (Lin et al., 2005; Kramar et al., 2006). The failure of LTP to stabilize in HD knock-in mice prompted us to ask whether actin polymerization is defective. First, we needed to establish whether the in situ method of applying rhodamine-conjugated phalloidin, a toxin that selectively binds to filamentous actin (F-actin) in its polymeric forms, effectively labels polymerized actin in dendritic spines of potentiated synapses in WT mice, as it does in rats (Kramar et al., 2006). After low-frequency stimulation or TBS, rhodamine–phalloidin was applied to hippocampal slices from WT mice. LTP induction with TBS was accompanied by a massive increase in the number of densely labeled puncta compared with slices that received low-frequency stimulation (Fig. 3A,B).
Figure 3.

TBS-induced actin polymerization in dendritic spines. A, B, Photomicrographs showing CA1b stratum radiatum of WT hippocampal slices that received either low-frequency stimulation (A) or TBS (B) to the Schaffer–commissural fibers (pyramidal cell layer is at the top). A, Low-frequency stimulation generated very few structures with intense rhodamine–phalloidin labeling. Faintly labeled puncta can be seen at higher magnification (inset). B, LTP, induced by TBS, was accompanied by the presence of numerous intensely labeled puncta. C, CA1 after TBS at higher magnification: clusters of phalloidin-labeled puncta are evident along the dendrite segment. Some puncta are connected to the dendrite by a lightly labeled, thin neck. D, High-magnification photomicrograph from CA1 of a second slice given TBS. Scale bars: A–D, 10 μm; inset, 5 μm.
Studies using rat slices have shown that densely labeled spines comparable to those in Figure 3 are present after TBS in single neurons clamped with electrodes containing rhodamine–phalloidin (Lin et al., 2005; Kramar et al., 2006). We found that this is also true for WT mice (as illustrated by the example in supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Evidence that the same labeled profiles are seen with intracellular and extracellular phalloidin applications, together with the fact that the marker was applied after TBS in the studies described here, demonstrates that the increase in F-actin labeling in mouse slices is not caused by stimulation effects on transport of the marker across cell membranes (Lin et al., 2005). The distribution of labeled puncta in WT mice, as seen in survey micrographs (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), corresponded to that expected for potentiated synapses (i.e., high concentrations of labeling in the proximal portions of CA1 stratum radiatum with few profiles in stratum pyramidale or more distal portions of stratum radiatum). This pattern of labeling has also been noted in rat hippocampal slices (Rex et al., 2007). The densely labeled puncta seen with intracellular or extracellular applications of phalloidin after LTP induction had the size (∼1 μm in diameter), appearance (bulbous head, thin neck), and distribution (scattered along dendrites) expected for dendritic spines (Fig. 3C,D).
Various lines of evidence, in addition to appearance, indicate that TBS-induced phalloidin staining occurs in spines. The same profiles are found after TBS delivered to the afferents of cells in which phalloidin was applied intracellularly (Lin et al., 2005; Kramar et al., 2006); their appearance is completely blocked by an antagonist of (postsynaptic) NMDA receptors (Lin et al., 2005), and they are routinely double labeled by antibodies against postsynaptic density markers (Lynch et al., 2007; Rex et al., 2007). We confirmed the spine localization of phalloidin labeling in mice by double labeling with an antibody to PSD-95, a constituent of postsynaptic densities at glutamatergic synapses. Both phalloidin (red) and PSD-95 (green) labeling were abundant in the proximal portion of CA1 stratum radiatum in potentiated slices (Fig. 4A; see Fig. 4B,C for higher-magnification images of PSD-95 and phalloidin labeling). The phalloidin-positive puncta typically overlapped PSD-95 immunoreactivity (yellow structures) (Fig. 4D, arrow) or were “capped” by the PSD protein attached to them (arrowhead).
Figure 4.

Polymerized actin colocalizes with synaptic markers. Photomicrographs show in situ phalloidin labeling (red) and PSD-95 immunostaining (green) in CA1b stratum radiatum of WT mice. A, Survey photomicrograph showing double labeling after LTP induction in a WT slice. The arrow and arrowhead point to the same sites shown at higher magnification in subsequent panels. B, C, High-magnification photomicrographs show PSD-95 immunostaining (B) and phalloidin labeling (C) in the field shown in A. D, An overlay of B and C shows that phalloidin and PSD-95 immunoreactivity are colocalized in some puncta (yellow structures; arrow); other phalloidin-labeled puncta are capped by PSD-95 immunostaining (arrowhead). Scale bars: A, 10 μm; B–D, 3 μm.
After establishing that TBS markedly increased F-actin in dendritic spines of WT mice, we tested for similar effects in HdhQ111 slices. As shown in Figure 5, A and B, the number of spines with low-intensity labeling was comparable in slices from HD and WT mice after low-frequency stimulation and TBS. Furthermore, low-frequency stimulation did not affect densely labeled spines in either genotype. However, after TBS, increases in the number of densely labeled spines seen in WT slices (Fig. 5C) were essentially absent in tissue from HdhQ111 mice (Fig. 5D). Quantitative analyses of the incidence of labeled puncta were performed by categorizing fluorescent labeling intensities into nine equal-sized bins ranging from very weak to very strong and counting the number of spines meeting the criteria assigned to each bin (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). It should be noted that the in situ labeling method was developed to detect anatomically distinct spines with high levels of F-actin to test the degree to which theta-induced actin polymerization contributes to the production of stable LTP in adult brain. The concentrations of phalloidin used in the method do not visibly label the majority of spines, as expected if those spines contain modest levels of F-actin. The average number of spines counted across all bins (supplemental Fig. 3, available at www.jneurosci.org as supplemental material) for the low-frequency stimulation WT group was approximately six spines per 100 μm2, whereas estimates of synapse density for stratum radiatum range from 200 to 300/100 μm3 (Sorra and Harris, 1998; Scheff et al., 2005). The presence of a large pool of undetected spines precludes testing for theta-induced transfer of spines from lightly to densely stained bins (i.e., balanced gains and losses).
Figure 5.

Actin polymerization in dendritic spines is greatly reduced in HdhQ111 mice. A, B, Phalloidin labeling of CA1 stratum radiatum of hippocampal slices from WT (A) and HdhQ111 (B) mice after low-frequency stimulation. C, D, Densely labeled spines in WT (C) and Hdh Q111 (D) slices after TBS-induced LTP (pyramidal cell layer is at top left). Images have been thresholded to visualize only the most densely labeled profiles. Scale bar: A–D, 10 μm. E, Quantification of phalloidin-labeled spines in the three lowest (left) and highest (right) intensity labeling bins in WT and HdhQ111 (Hdh) slices that received either low-frequency stimulation (LFS) or TBS [threshold values for low-intensity bins are 180, 170, and 160 pixel intensity units (PIU); those for the high bins are 120, 110, and 100 PIU]. TBS generated a marked increase in the number of intensely labeled spines in WT but not HdhQ111 slices. Note that spine counts for the low-intensity bins are comparable for WT and Hdh slices receiving LFS or TBS. *p = 0.009 compared with WT (LFS); #p = 0.02 compared with WT (TBS).
Although the in situ phalloidin method visibly labels a fraction of the target population, it has provided quantitative measures of relative differences in the effects of experimental treatments on TBS-induced actin polymerization (Kramar et al., 2006), and this proved to be the case for the HD mice. Compared with that seen with low-frequency stimulation, TBS increased the incidence of densely labeled spines (i.e., those in the three highest labeling-intensity bins) in WT slices by ∼10-fold (low frequency, 0.9 ± 4.6 mean ± SEM, n = 10, vs TBS, 10.7 ± 3.6 per 550 μm2, n = 14; p = 0.009), an effect comparable in size to that reported for rats (Lin et al., 2005; Kramar et al., 2006). Although this TBS effect represented a large increase over the number of densely stained spines seen in slices given low-frequency stimulation, it necessarily (given the above cited numbers for excitatory synapses) involved a small percentage of the spine population. This concords with recent estimates that <50 synapses per CA1 neuron are needed to produce EPSPs of the size found in typical slice experiments (Hanse and Gustafsson, 2002). In contrast to effects in slices from WT mice, TBS did not significantly affect the number of densely labeled spines in HdhQ111 slices (low frequency, 0.4 ± 0.2, n = 10 vs TBS, 2.0 ± 4.6, n = 13; p = 0.37). Accordingly, the number of densely labeled spines after TBS was approximately five times lower in HdhQ111 compared with WT slices (p = 0.02, Mann–Whitney U tests, two-tailed). Similarly, the differences between the cumulative probability distributions (frequency vs labeling intensity) after TBS for WT compared with HD data sets were highly significant (p < 0.0001, Kolmogorov–Smirnov test) (supplemental Fig. 3B, available at www.jneurosci.org as supplemental material). In all, measures indicate theta-induced actin polymerization is markedly impaired in HD mutant mice.
BDNF restores LTP in hippocampal slices prepared from HdhQ111 mice
Previous studies showed that BDNF levels were substantially reduced in the neocortex and striatum of 20-week-old HdhQ111 mice (Gines et al., 2003). We tested whether such effects are present in the hippocampus of HdhQ111 mice at 2 months and HdhQ92 mice at 6 months, the ages used for the majority of the electrophysiological studies. Immunoblots of hippocampal samples from HdhQ111 and WT mice showed that BDNF immunoreactivity was distributed across bands representing pro-BDNF and proteolytic fragments including the 14 kDa (Fig. 6A) mature BDNF protein (Mowla et al., 2001). The band at ∼29 kDa was measured as the pro-band because it corresponds to the 30 kDa pro-band described by others (Lee et al., 2001; Pang et al., 2006) and is close to the glycosylated pro-BDNF bands (32 and 28 kDa) seen in cell culture (Mowla et al., 2001). Concentrations of both pro-BDNF and mature BDNF were substantially reduced in HdhQ111 relative to WT mice. Densitometric analyses (Fig. 6B) indicated that mature BDNF levels in hippocampus were 43 ± 7% lower in HdhQ111 (n = 7) than WT (n = 8; p = 0.002, one-tailed t test) mice. The pro-BDNF band, with an approximate mass of 29 kDa, was reduced by 57 ± 11% in these mice (p = 0.0001). Differences between the groups were also significant when values were normalized to within-lane actin bands. Finally, the apparently greater deficit in pro-BDNF versus mature BDNF in HdhQ111 mice, as seen in the group data (Fig. 6B), was statistically significant (p = 0.007, paired Student's t test). Similar results were obtained with an analysis of samples from HdhQ92 mice: hippocampal levels of mature BDNF were 71 ± 24% of the WT mean (n = 7 and 8 for mutant and WT, respectively; p < 0.05, one-tailed t test). The 29 kDa pro-BDNF band in HdhQ92 mice was 68 ± 26% of the WT mean (p < 0.05).
Because BDNF levels were substantially reduced in HdhQ111 mice and the neurotrophin is a potent positive modulator of LTP (Kramar et al., 2004; Bramham and Messaoud, 2005), we tested whether BDNF (2 nm) would reverse LTP deficits in the mutants. Infusing BDNF for 60 min did not detectably change baseline transmission in HdhQ111 or WT slices. Unexpectedly, given previous results for rats (Kang and Schuman, 1995; Figurov et al., 1996; Chen et al., 1999; Kramar et al., 2004), BDNF at 2 nm failed to reliably affect the magnitude of LTP in slices from WT mice. Mean potentiation at 30 min after TBS was 53.2 ± 16.1% above baseline (n = 4), a value that was not meaningfully different from the LTP found in untreated WT slices used in experiments described above (51.8 ± 13.7%; n = 11) (Fig. 1) or for three additional controls (45.0 ± 28.9%). We repeated the experiment using a 10-fold greater BDNF concentration (20 nm) to test whether sensitivity to the neurotrophin varies between the two species; potentiation under these conditions was 76.2 ± 27.0% (n = 3) above baseline, an effect that was significantly different from WT controls (p = 0.04). It thus appears that rats and WT mice are substantially different with regard to the BDNF levels needed to manipulate the magnitude of LTP; it remains to be determined whether the species difference extends to the effects of BDNF on the threshold for inducing LTP (Kramar et al., 2004).
Although BDNF at 2 nm had little effect on WT slices, it potently enhanced LTP in HD mice (Fig. 7A). Slices from HdhQ111 mice that were not treated with BDNF (n = 4) exhibited decremental potentiation (Fig. 7A), replicating the results shown in Figure 1, and were clearly different from WT slices (n = 9) by 20 min after TBS (percentage of potentiation: HdhQ111, 24.3 ± 17.1% vs wild type, 56.5 ± 30.5%; p = 0.04, two-tailed Student's t test) (Fig. 7B). As mentioned above, BDNF at 2 nm did not affect potentiation in WT slices (57.2 ± 6.4% at 20 min after TBS; p = 0.48; n = 4). In contrast, LTP in BDNF-treated HdhQ111 slices (59.9 ± 28.3%; n = 11) was equivalent to that in wild types (p = 0.8) and was significantly greater than that seen in the untreated HD group (p = 0.016) (Fig. 7B). The percentage of potentiation for the subset of slices tested at 45 min after TBS was much greater in the BDNF-treated group than in untreated HdhQ111 slices (43.9 ± 24.3 and 6.0 ± 12.1%, respectively; p < 0.002) (Fig. 7C). Input/output curves constructed in advance of LTP induction were virtually identical for WT and HD slices (supplemental Fig. 4, available at www.jneurosci.org as supplemental material).
Figure 7.

BDNF restores TBS-induced LTP and actin polymerization but not burst response facilitation in HdhQ111 mice. A, LTP in untreated HdhQ111 slices (Hdh; open triangles) was slightly reduced from WT levels in the first minute after TBS and did not stabilize. However, in the presence of BDNF (2 nm), LTP in slices from the same animals (Hdh + BDNF; filled triangles) was not detectably different from wild types (fEPSP slopes, expressed as a percentage of baseline; shown in A and B). B, A summary of the percentage of potentiation observed 20 min after TBS delivery for vehicle-treated slices from HdhQ111 (n = 4 slices) and age-matched WT (n = 9) mice compared those treated with 2 nm BDNF (HdhQ111, n = 11; WT, n = 4 slices). Note that WT slices that received BDNF did not differ from untreated slices, whereas the neurotrophin greatly enhanced potentiation in HdhQ111 slices (*p = 0.016 compared with vehicle-treated Hdh slices, two-tailed Student's t test). C, fEPSPs from a subgroup of BDNF-treated HdhQ111 slices (filled triangles) were recorded for 60 min after TBS. Note that potentiation was still present at the end of recording (data for the untreated HdhQ111 slices from Fig. 1 are shown for comparison). The inset (top right) shows representative fEPSP traces collected at 60 min after TBS. Data are averages of three consecutive traces. Calibration: 1 mV, 5 ms. D, BDNF at the 2 nm concentration, which rescued LTP, did not correct the impaired theta burst facilitation seen in HdhQ111 mice, as evidenced by comparable burst facilitation in BDNF-treated and untreated HdhQ111 slices (sizes of burst responses 2–10 are expressed as a fraction of the first burst response). E, Photomicrographs show phalloidin labeling in CA1 stratum radiatum of TBS-stimulated HdhQ111 slices with (bottom) and without (top) BDNF treatment. Densely labeled spines were essentially absent in untreated slices after TBS (top) but were present in large numbers in BDNF-treated cases (bottom). Scale bar, 10 μm.
BDNF could potentially restore LTP in HdhQ111 slices by enhancing within-train facilitation of theta burst responses, as has been described for rats (Kramar et al., 2004). This idea is particularly relevant given that within-train facilitation is impaired in HdhQ111 mice (Fig. 2B). However, BDNF had no effect on the genotype-specific loss of facilitation from the first to subsequent burst responses (Fig. 7D). Facilitation of the second burst response was 77.3 ± 20.5% for wild types, 46.9 ± 16.8% for untreated HdhQ111 slices, and 46.1 ± 10.2% for BDNF-treated HdhQ111 slices (p < 0.03 for BDNF-treated HdhQ111 compared with WT mice). The loss of facilitation was even more pronounced for the later bursts in the stimulation train (e.g., burst 9 was 45.0 ± 15.4% greater than burst 1 in WT slices and −5.8 ± 9.3% of the burst 1 value for BDNF-treated HdhQ111 slices; p < 0.001). In all, BDNF did not facilitate responses to any stimulation burst in the theta train in HdhQ111 slices.
The failure of BDNF to restore within-train burst facilitation in HdhQ111 mice suggests that it may exert its positive effect on LTP by reversing the deficits in actin polymerization. To test this, rhodamine–phalloidin was applied after physiological recordings were collected from WT and HdhQ111 slices with and without BDNF treatment. Low-frequency stimulation did not elicit changes in phalloidin labeling of BDNF-treated or untreated slices from either genotype. As described above, TBS induced robust LTP in slices from HdhQ111 mice treated with BDNF but not in untreated slices. LTP restoration was accompanied by a marked increase in densely phalloidin-labeled spines in BDNF-treated (Fig. 7E, bottom) compared with untreated (Fig. 7E, top) slices. The number of labeled spines in the high-intensity bins was 40.5 ± 39.3 per 550 μm2 in HdhQ111 slices treated with BDNF (n = 13) and 2.0 ± 4.6 for those without BDNF (n = 13; p < 0.003, Mann–Whitney U test). Thus, the restorative effect of BDNF on TBS-induced actin polymerization may be responsible for the recovery of LTP stabilization in HdhQ111 mice.
Discussion
Cognitive deficits are present in HD gene carriers and early-stage patients well before the onset of the motor symptoms that define the disease (Lawrence et al., 1998; Kirkwood et al., 2000; Ho et al., 2003; Lemiere et al., 2004). A recent longitudinal study concluded that the problems are progressive, even over periods as short as 3 years, and that memory losses are the earliest cognitive manifestations of HD (Lemiere et al., 2004). These findings suggest that HD begins with a discrete disturbance of plasticity and then progresses to motor pathology and neurodegeneration.
Previous studies indicated that LTP is severely impaired in hippocampal field CA1 in HD mouse models (Usdin et al., 1999; Murphy et al., 2000). Paired-pulse facilitation was depressed along with LTP in HD knock-in (72/80 CAG) mice, suggesting that the HD mutation alters release kinetics (Usdin et al., 1999). Although the point was not tested, a presynaptic deficit would presumably disturb frequency facilitation of postsynaptic responses and thus reduce the depolarization needed to trigger LTP. A separate study found that R6/2 and WT mice are comparable in their basic synaptic physiology, including presynaptic neurotransmitter mobilization and release (Murphy et al., 2000). This suggests a postsynaptic locus for the LTP deficit. NMDA receptor-mediated currents appeared normal in R6/2 mice, again indicating that LTP processes downstream of induction (i.e., expression or stabilization) are impaired. In all, previous studies indicate that LTP deficits are present in HD mouse models but disagree as to whether they reflect a presynaptic or postsynaptic problem.
The present experiments used the naturalistic TBS pattern to induce LTP in hippocampal slices prepared from HD knock-in mice. Most of the experiments were performed with 2-month-old HdhQ111 mice to test for deficits that are evident before overt motor disturbances and during the transition from late development to early adulthood. These conditions allow the reasonable assumption that the results are relevant to the early appearance of memory problems in HD patients. LTP was severely impaired in HD mice (HdhQ111 and, to a comparable extent, in HdhQ92) without evidence of presynaptic disturbances or changes in the waveform of postsynaptic responses. Moreover, the size and shape of the composite response to a single burst of afferent stimulation were normal, as were feedforward inhibitory potentials. However, the facilitation of burst responses that normally occurs during a theta train was markedly reduced in HD knock-in mice. Burst facilitation causes greater depolarization that enhances the opening of NMDA receptors triggering LTP (Larson and Lynch, 1988). Thus, impaired burst responses in HdhQ111 mice probably contribute to the LTP deficits.
One possible explanation for modified burst responses is that IPSCs, which accompany individual burst responses (Larson et al., 1986), are altered. IPSCs are reduced during TBS because inhibitory synapses become refractory after they are activated during the first theta burst response, an effect caused by stimulation of presynaptic autoreceptors (Mott and Lewis, 1991). Thus, deficits in processes controlling the strength and duration of this refractory effect could impair burst facilitation. Investigations regarding this idea, however, proved negative: IPSCs were as refractory in HdhQ111 as in WT mice. The remaining factor controlling within-train changes in burst response characteristics is the complex sequence of afterhyperpolarizations triggered by the first burst response (Arai and Lynch, 1992; Sah and Bekkers, 1996); tests for deficits in these responses are in progress. In any event, the pronounced impairment in LTP found in HdhQ111 mice is associated with surprisingly discrete postsynaptic defects.
Possibly related to disturbances in theta burst responses, BDNF levels were reduced in hippocampus of 2-month-old HdhQ111 and 6-month-old HdhQ92 mice. Previous reports found decreased levels of BDNF (Gines et al., 2003) and its TrkB receptor (Gines et al., 2006) in neocortex and striatum of older knock-in mice and in HD patients. There is also evidence that mutant huntingtin adversely affects BDNF transport in dissociated cells and thus disrupts delivery of the neurotrophin to target tissues (Gauthier et al., 2004). These effects, acting alone or in combination, would serve to reduce levels of BDNF normally released by theta bursts (Aicardi et al., 2004). Given that BDNF enhances burst response facilitation during TBS by suppressing afterhyperpolarizations (Kramar et al., 2004) and, as expected from this, promotes the induction of LTP (for review, see Bramham and Messaoud, 2005), these observations suggest a unifying explanation for the pattern of results obtained in HdhQ111 mice (i.e., reduced BDNF release removes a factor that positively modulates theta burst facilitation and thus LTP induction). If so, then infusing BDNF should rescue LTP in HD mice by restoring normal responses to TBS. The first of these predictions was confirmed: HdhQ111 slices exposed to 2 nm BDNF exhibited robust and stable LTP after TBS. This recovery in HD mice did not simply reflect facilitation of LTP but a true rescue, because BDNF at this low concentration did not affect potentiation in wild types. Unexpectedly, however, the rescue of potentiation was not accompanied by the return of normal within-train facilitation. It appears that the dose used to restore LTP is below threshold for affecting the presynaptic frequency facilitation, IPSCs, and afterhyperpolarizations that regulate the size and within-train facilitation of burst responses. The possibility remains, however, that a BDNF effect on these variables, as reported for rat slices (Figurov et al., 1996; Tanaka et al., 1997; Tyler and Pozzo-Miller, 2001), would be obtained at higher concentrations of BDNF. In any event, the absence of any BDNF effect on burst responses indicates that the influences of BDNF on events following TBS are likely to be responsible for the rescue of plasticity in HdhQ111 slices.
The above conclusion led us to investigate whether BDNF affects the second of the two LTP-related processes that were defective in HdhQ111 slices, namely TBS-induced actin polymerization. Tests of this idea were positive: TBS produced a marked increase in the numbers of spines with dense concentrations of F-actin in HdhQ111 slices that had been pretreated with BDNF compared with untreated slices. We assume that these effects are attributable to the triggering of actin polymerization by TBS in the small population of spines innervated by stimulated afferents (i.e., that spines with subthreshold or weak labeling are converted into spines with dense levels of F-actin). An alternative possibility would be that TBS produces new spines and that this process is associated with actin polymerization. However, there are multiple electron microscopic studies (Lee et al., 1980; Chang and Greenough, 1984; Sorra and Harris, 1998; Popov et al., 2004) that have failed to detect an increase in spines after induction of LTP in field CA1 of adult hippocampus. Positive reports have been described for cultured, and thus immature, hippocampal slices (Engert and Bonhoeffer, 1999; Maletic-Savatic et al., 1999). Accordingly, we conclude that the conversion hypothesis is the more conservative.
Multiple lines of evidence indicate that actin polymerization is essential for the stabilization (consolidation) of LTP. For example, agents that disrupt actin polymerization block LTP consolidation (Fukazawa et al., 2003; Kramar et al., 2006), whereas treatments that disrupt consolidation eliminate TBS-induced actin polymerization (Kramar et al., 2006). Moreover, actin polymerization has the same threshold (number of theta bursts) for induction, as does LTP, and becomes resistant to disruption over the same time period that LTP consolidates (Kramar et al., 2006). The absence of TBS-induced actin polymerization can explain why the sizeable initial potentiation found in HdhQ111 slices decays back to baseline rather than stabilizing, whereas recovery of polymerization accounts for the rescue of LTP by BDNF.
The deficits in actin polymerization found in HdhQ111 mice also provide a possible explanation for the abnormal spine morphology found in striatal and cortical neurons of HD transgenic mouse models (Guidetti et al., 2001; Klapstein et al., 2001; Spires et al., 2004b) and in HD patients (Graveland et al., 1985; Ferrante et al., 1991). The actin cytoskeleton regulates the shape of dendritic spines (Fischer et al., 2000; Star et al., 2002), at least in developing tissue, and deficiencies in activity-dependent F-actin assembly would be expected to cause aberrant morphology. Future studies are needed to address the possibility that deficits in TBS-induced actin polymerization and LTP emerge during the juvenile period and are followed in adulthood by significant disturbances to spine morphology.
The restorative effect of BDNF on LTP in HdhQ111 mice raises the question of whether HD-related cognitive impairments can be overcome by upregulating production of this neurotrophin. Various methods for elevating BDNF levels have been reported (e.g., antidepressant drugs, seizures) but most of these have unacceptable side effects for long-term applications. Exercise (Cotman and Berchtold, 2002; Pang et al., 2006) and enriched environments (Spires et al., 2004a) also increase BDNF, but these effects occur in comparison to sedentary or nonenriched controls, and HD patients are not inactive or sensory deprived before emergence of motor disturbances. Alternatively, work from several groups (Lauterborn et al., 2000; Legutko et al., 2001; O'Neill et al., 2005) has shown that BDNF mRNA and protein can be substantially increased in hippocampal neurons by ampakines, compounds that positively modulate AMPA-type glutamate receptors and the fast EPSPs they mediate. Moreover, chronic use of ampakines does not produce significant side effects in rats, monkeys, or humans (Lynch, 2006). Whether ampakines can overcome the HD-associated deficit in BDNF transcription, which is thought to arise from inadequate binding of mutant huntingtin to a neuron-specific silencer element (Zuccato et al., 2003), is an important issue for future research.
Footnotes
This work was supported in part by National Institute of Neurological Disorders and Stroke Grants NS051823 (G.L.) and NS045260 (G.L. and C.M.G.), by a monetary gift from the Huntington's Disease Drug Works Foundation (D.A.S.), and by National Institute on Aging Training Grant 5T32 AG00096 (C.S.R.). We thank Dr. Jihua Liu for assistance with biochemical procedures, Dr. Bin Lin for intracellular phalloidin labeling, and Dr. Malcolm Casale for helpful discussions. We also thank Drs. Leslie Thompson, Barbara Apostol, and Ya-zhen Zhu for help with genotyping and Dr. Marcy Macdonald for determining the CAG repeat length.
References
- Aicardi G, Argilli E, Cappello S, Santi S, Riccio M, Thoenen H, Canossa M. Induction of long-term potentiation and depression is reflected by corresponding changes in secretion of endogenous brain-derived neurotrophic factor. Proc Natl Acad Sci USA. 2004;101:15788–15792. doi: 10.1073/pnas.0406960101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai A, Lynch G. Antagonists of the platelet-activating factor receptor block long-term potentiation in hippocampal slices. Eur J Neurosci. 1992;4:411–419. doi: 10.1111/j.1460-9568.1992.tb00890.x. [DOI] [PubMed] [Google Scholar]
- Bates G. Huntington's disease. Ed 3. Oxford: Oxford UP; 2002. [Google Scholar]
- Bramham C, Messaoud E. BDNF function in adult synaptic plasticity: the synaptic consolidation hypothesis. Prog Neurobiol. 2005;76:99–125. doi: 10.1016/j.pneurobio.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Chang FL, Greenough WT. Transient and enduring morphological correlates of synaptic activity and efficacy change in the rat hippocampal slice. Brain Res. 1984;309:35–46. doi: 10.1016/0006-8993(84)91008-4. [DOI] [PubMed] [Google Scholar]
- Chen G, Kolbeck R, Barde Y, Bonhoeffer T, Kossel A. Relative contribution of endogenous neurotrophins in hippocampal long-term potentiation. J Neurosci. 1999;19:7983–7990. doi: 10.1523/JNEUROSCI.19-18-07983.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotman CW, Berchtold NC. Exercise: a behavioral intervention to enhance brain health and plasticity. Trends Neurosci. 2002;25:295–301. doi: 10.1016/s0166-2236(02)02143-4. [DOI] [PubMed] [Google Scholar]
- Creager R, Dunwiddie T, Lynch G. Paired-pulse and frequency facilitation in the CA1 region of the in vitro rat hippocampus. J Physiol (Lond) 1980;299:409–424. doi: 10.1113/jphysiol.1980.sp013133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyao M, Ambrose C, Myers R, Novelletto A, Persichetti F, Frontali M, Folstein S, Ross C, Franz M, Abbott M, Gray J, Conneally P, Young A, Penney J, Hollingsworth Z, Shoulson I, Lazzarini A, Falek A, Koroshetz W, Sax D, et al. Trinucleotide repeat length instability and age of onset in Huntington's disease. Nat Genet. 1993;4:387–392. doi: 10.1038/ng0893-387. [DOI] [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature. 1999;399:66–70. doi: 10.1038/19978. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Kowall NW, Richardson EP., Jr Proliferative and degenerative changes in striatal spiny neurons in Huntington's disease: a combined study using the section-Golgi method and calbindin D28k immunocytochemistry. J Neurosci. 1991;11:3877–3887. doi: 10.1523/JNEUROSCI.11-12-03877.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Goutan E, Marin C, Rey M, Ribalta T. Brain-derived neurotrophic factor in Huntington disease. Brain Res Rev. 2000;866:257–261. doi: 10.1016/s0006-8993(00)02237-x. [DOI] [PubMed] [Google Scholar]
- Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381:706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- Fischer M, Kaech S, Wagner U, Brinkhaus H, Matus A. Glutamate receptors regulate actin-based plasticity in dendritic spines. Nat Neurosci. 2000;3:887–894. doi: 10.1038/78791. [DOI] [PubMed] [Google Scholar]
- Fukazawa Y, Saitoh Y, Ozawa F, Ohta Y, Mizuno K, Inokuchi K. Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron. 2003;38:447–460. doi: 10.1016/s0896-6273(03)00206-x. [DOI] [PubMed] [Google Scholar]
- Gauthier LR, Charrin BC, Borrell-Pages M, Dompierre JP, Rangone H, Cordelieres FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, Saudou F. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Gibson H, Reim K, Brose N, Morton A, Jones S. A similar impairment in CA3 mossy fibre LTP in the R6/2 mouse model of Huntington's disease and in the complexin II knockout mouse. Eur J Neurosci. 2005;22:1701–1712. doi: 10.1111/j.1460-9568.2005.04349.x. [DOI] [PubMed] [Google Scholar]
- Gines S, Seong I, Fossale E, Ivanova E, Trettel F, Gusella J, Wheeler V, Persichetti F, MacDonald M. Specific progressive cAMP reduction implicates energy deficit in presymptomatic Huntington's disease knock-in mice. Hum Mol Genet. 2003;12:497–508. doi: 10.1093/hmg/ddg046. [DOI] [PubMed] [Google Scholar]
- Gines S, Bosch M, Marco S, Gavalda N, Diaz-Hernandez M, Lucas JJ, Canals JM, Alberch J. Reduced expression of the TrkB receptor in Huntington's disease mouse models and in human brain. Eur J Neurosci. 2006;23:649–658. doi: 10.1111/j.1460-9568.2006.04590.x. [DOI] [PubMed] [Google Scholar]
- Graveland G, Williams R, DiFiglia M. Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington's disease. Science. 1985;227:770–773. doi: 10.1126/science.3155875. [DOI] [PubMed] [Google Scholar]
- Guidetti P, Charles V, Chen EY, Reddy PH, Kordower JH, Whetsell WO, Jr, Schwarcz R, Tagle DA. Early degenerative changes in transgenic mice expressing mutant huntingtin involve dendritic abnormalities but no impairment of mitochondrial energy production. Exp Neurol. 2001;169:340–350. doi: 10.1006/exnr.2000.7626. [DOI] [PubMed] [Google Scholar]
- Hanse E, Gustafsson B. Release dependence to a paired stimulus at a synaptic release site with a small variable pool of immediately releasable vesicles. J Neurosci. 2002;22:4381–4387. doi: 10.1523/JNEUROSCI.22-11-04381.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho AK, Sahakian BJ, Brown RG, Barker RA, Hodges JR, Ane MN, Snowden J, Thompson J, Esmonde T, Gentry R, Moore JW, Bodner T. Profile of cognitive progression in early Huntington's disease. Neurology. 2003;61:1702–1706. doi: 10.1212/01.wnl.0000098878.47789.bd. [DOI] [PubMed] [Google Scholar]
- Hodgson JG, Agopyan N, Gutekunst CA, Leavitt BR, LePiane F, Singaraja R, Smith DJ, Bissada N, McCutcheon K, Nasir J, Jamot L, Li XJ, Stevens ME, Rosemond E, Roder JC, Phillips AG, Rubin EM, Hersch SM, Hayden MR. A YAC mouse model for Huntington's disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 1999;23:181–192. doi: 10.1016/s0896-6273(00)80764-3. [DOI] [PubMed] [Google Scholar]
- Kang HJ, Schuman EM. Neurotrophin-induced modulation of synaptic transmission in the adult hippocampus. J Physiol (Paris) 1995;89:11–22. doi: 10.1016/0928-4257(96)80547-x. [DOI] [PubMed] [Google Scholar]
- Kirkwood SC, Siemers E, Hodes ME, Conneally PM, Christian JC, Foroud T. Subtle changes among presymptomatic carriers of the Huntington's disease gene. J Neurol Neurosurg Psychiatry. 2000;69:773–779. doi: 10.1136/jnnp.69.6.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klapstein GJ, Fisher RS, Zanjani H, Cepeda C, Jokel ES, Chesselet MF, Levine MS. Electrophysiological and morphological changes in striatal spiny neurons in R6/2 Huntington's disease transgenic mice. J Neurophysiol. 2001;86:2667–2677. doi: 10.1152/jn.2001.86.6.2667. [DOI] [PubMed] [Google Scholar]
- Kramar EA, Lin B, Lin C-Y, Arai AC, Gall CM, Lynch G. A novel mechanism for the facilitation of theta-induced long-term potentiation by brain-derived neurotrophic factor. J Neurosci. 2004;24:5151–5161. doi: 10.1523/JNEUROSCI.0800-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramar EA, Lin B, Rex CS, Gall CM, Lynch G. Integrin-driven actin polymerization consolidates long-term potentiation. Proc Natl Acad Sci USA. 2006;103:5579–5584. doi: 10.1073/pnas.0601354103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson J, Lynch G. Role of N-methyl-D-aspartate receptors in the induction of synaptic potentiation by burst stimulation patterned after the hippocampal theta-rhythm. Brain Res. 1988;441:111–118. doi: 10.1016/0006-8993(88)91388-1. [DOI] [PubMed] [Google Scholar]
- Larson J, Wong D, Lynch G. Patterned stimulation at the theta frequency is optimal for the induction of hippocampal long-term potentiation. Brain Res. 1986;368:347–350. doi: 10.1016/0006-8993(86)90579-2. [DOI] [PubMed] [Google Scholar]
- Lauterborn JC, Lynch G, Vanderklish P, Arai A, Gall CM. Positive modulation of AMPA receptors increases neurotrophin expression by hippocampal and cortical neurons. J Neurosci. 2000;20:8–21. doi: 10.1523/JNEUROSCI.20-01-00008.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence A, Hodges J, Rosser A, Kershaw A, French-Constant C, Rubinsztein D, Robbins T, BJ S. Evidence for specific cognitive deficits in preclinical Huntington's disease. Brain Pathol. 1998;121:1329–1341. doi: 10.1093/brain/121.7.1329. [DOI] [PubMed] [Google Scholar]
- Lee KS, Schottler F, Oliver M, Lynch G. Brief bursts of high-frequency stimulation produce two types of structural changes in rat hippocampus. J Neurophysiol. 1980;44:247–258. doi: 10.1152/jn.1980.44.2.247. [DOI] [PubMed] [Google Scholar]
- Lee R, Kermani P, Teng KK, Hempstead BL. Regulation of cell survival by secreted proneurotrophins. Science. 2001;294:1945–1948. doi: 10.1126/science.1065057. [DOI] [PubMed] [Google Scholar]
- Legutko B, Li X, Skolnick P. Regulation of BDNF expression in primary neuron culture by LY392098, a novel AMPA receptor potentiator. Neuropharmacology. 2001;40:1019–1027. doi: 10.1016/s0028-3908(01)00006-5. [DOI] [PubMed] [Google Scholar]
- Lemiere J, Decruyenaere M, Evers-Kiebooms G, Vandenbussche E, Dom R. Cognitive changes in patients with Huntington's disease (HD) and asymptomatic carriers of the HD mutation—a longitudinal follow-up study. J Neurol. 2004;251:935–942. doi: 10.1007/s00415-004-0461-9. [DOI] [PubMed] [Google Scholar]
- Lin B, Kramar EA, Bi X, Brucher FA, Gall CM, Lynch G. Theta stimulation polymerizes actin in dendritic spines of hippocampus. J Neurosci. 2005;25:2062–2069. doi: 10.1523/JNEUROSCI.4283-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lione L, Carter R, Hunt M, Bates G, Morton A, Dunnett S. Selective discrimination learning impairments in mice expressing the human Huntington's disease mutation. J Neurosci. 1999;19:10428–10437. doi: 10.1523/JNEUROSCI.19-23-10428.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch G. Glutamate-based therapeutic approaches: ampakines. Curr Opin Pharmacol. 2006;6:82–88. doi: 10.1016/j.coph.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Lynch G, Rex CS, Gall CM. LTP consolidation: Substrates, explanatory power, and functional significance. Neuropharmacology. 2007;52:12–23. doi: 10.1016/j.neuropharm.2006.07.027. [DOI] [PubMed] [Google Scholar]
- Maletic-Savatic M, Malinow R, Svoboda K. Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science. 1999;283:1923–1927. doi: 10.1126/science.283.5409.1923. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Mahal A, Mott R, Seller M, Bates GP. Instability of highly expanded CAG repeats in mice transgenic for the Huntington's disease mutation. Nat Genet. 1997;15:197–200. doi: 10.1038/ng0297-197. [DOI] [PubMed] [Google Scholar]
- Mazarakis NK, Cybulska-Klosowicz A, Grote H, Pang T, Van Dellen A, Kossut M, Blakemore C, Hannan AJ. Deficits in experience-dependent cortical plasticity and sensory-discrimination learning in presymptomatic Huntington's disease mice. J Neurosci. 2005;25:3059–3066. doi: 10.1523/JNEUROSCI.4320-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menalled L. Knock-in mouse models of Huntington's disease. NeuroRx. 2005;2:465–470. doi: 10.1602/neurorx.2.3.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milnerwood AJ, Cummings DM, Dallerac GM, Brown JY, Vatsavayai SC, Hirst MC, Rezaie P, Murphy KP. Early development of aberrant synaptic plasticity in a mouse model of Huntington's disease. Hum Mol Genet. 2006;15:1690–1703. doi: 10.1093/hmg/ddl092. [DOI] [PubMed] [Google Scholar]
- Mott DD, Lewis DV. Facilitation of the induction of long-term potentiation by GABAB receptors. Science. 1991;252:1718–1720. doi: 10.1126/science.1675489. [DOI] [PubMed] [Google Scholar]
- Mowla SJ, Farhadi HF, Pareek S, Atwal JK, Morris SJ, Seidah NG, Murphy RA. Biosynthesis and post-translational processing of the precursor to brain-derived neurotrophic factor. J Biol Chem. 2001;276:12660–12666. doi: 10.1074/jbc.M008104200. [DOI] [PubMed] [Google Scholar]
- Murphy KP, Carter RJ, Lione LA, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ. Abnormal synaptic plasticity and impaired spatial cognition in mice transgenic for exon 1 of the human Huntington's disease mutation. J Neurosci. 2000;20:5115–5123. doi: 10.1523/JNEUROSCI.20-13-05115.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill MJ, Murray TK, Clay MP, Lindstrom T, Yang CR, Nisenbaum ES. LY503430: pharmacology, pharmacokinetics, and effects in rodent models of Parkinson's disease. CNS Drug Rev. 2005;11:77–96. doi: 10.1111/j.1527-3458.2005.tb00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang TY, Stam NC, Nithianantharajah J, Howard ML, Hannan AJ. Differential effects of voluntary physical exercise on behavioral and brain-derived neurotrophic factor expression deficits in Huntington's disease transgenic mice. Neuroscience. 2006;141:569–584. doi: 10.1016/j.neuroscience.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Popov VI, Davies HA, Rogachevsky VV, Patrushev IV, Errington ML, Gabbott PL, Bliss TV, Stewart MG. Remodelling of synaptic morphology but unchanged synaptic density during late phase long-term potentiation (LTP): a serial section electron micrograph study in the dentate gyrus in the anaesthetised rat. Neuroscience. 2004;128:251–262. doi: 10.1016/j.neuroscience.2004.06.029. [DOI] [PubMed] [Google Scholar]
- Rex CS, Lauterborn JC, Lin CY, Kramar EA, Rogers GA, Gall CM, Lynch G. Restoration of long-term potentiation in middle-aged hippocampus after induction of brain-derived neurotrophic factor. J Neurophysiol. 2006;96:677–685. doi: 10.1152/jn.00336.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex CS, Lin C-Y, Kramer EA, Chen L, Grall CM, Lynch G. Brain-derived neurotrophic factor promotes LTP-related cytoskeletal changes in adult hippocampus. J Neurosci. 2007;27:3017–3029. doi: 10.1523/JNEUROSCI.4037-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas HD, Koroshetz WJ, Chen YI, Skeuse C, Vangel M, Cudkowicz ME, Caplan K, Marek K, Seidman LJ, Makris N, Jenkins BG, Goldstein JM. Evidence for more widespread cerebral pathology in early HD: an MRI-based morphometric analysis. Neurology. 2003;60:1615–1620. doi: 10.1212/01.wnl.0000065888.88988.6e. [DOI] [PubMed] [Google Scholar]
- Sah P, Bekkers JM. Apical dendritic location of slow afterhyperpolarization current in hippocampal pyramidal neurons: implications for the integration of long-term potentiation. J Neurosci. 1996;16:4537–4542. doi: 10.1523/JNEUROSCI.16-15-04537.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Hicks RR, Baldwin SA, Robinson S, Brackney C. Synaptogenesis in the hippocampal CA1 field following traumatic brain injury. J Neurotrauma. 2005;22:719–732. doi: 10.1089/neu.2005.22.719. [DOI] [PubMed] [Google Scholar]
- Sorra KE, Harris KM. Stability in synapse number and size at 2 hr after long-term potentiation in hippocampal area CA1. J Neurosci. 1998;18:658–671. doi: 10.1523/JNEUROSCI.18-02-00658.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires TL, Grote HE, Varshney NK, Cordery PM, van Dellen A, Blakemore C, Hannan AJ. Environmental enrichment rescues protein deficits in a mouse model of Huntington's disease, indicating a possible disease mechanism. J Neurosci. 2004a;24:2270–2276. doi: 10.1523/JNEUROSCI.1658-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires TL, Grote HE, Garry S, Cordery PM, Van Dellen A, Blakemore C, Hannan AJ. Dendritic spine pathology and deficits in experience-dependent dendritic plasticity in R6/1 Huntington's disease transgenic mice. Eur J Neurosci. 2004b;19:2799–2807. doi: 10.1111/j.0953-816X.2004.03374.x. [DOI] [PubMed] [Google Scholar]
- Star EN, Kwiatkowski DJ, Murthy VN. Rapid turnover of actin in dendritic spines and its regulation by activity. Nat Neurosci. 2002;5:239–246. doi: 10.1038/nn811. [DOI] [PubMed] [Google Scholar]
- Stine OC, Pleasant N, Franz ML, Abbott MH, Folstein SE, Ross CA. Correlation between the onset age of Huntington's disease and length of the trinucleotide repeat in IT-15. Hum Mol Genet. 1993;2:1547–1549. doi: 10.1093/hmg/2.10.1547. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Saito H, Matsuki N. Inhibition of GABAA synaptic responses by brain-derived neurotrophic factor (BDNF) in rat hippocampus. J Neurosci. 1997;17:2959–2966. doi: 10.1523/JNEUROSCI.17-09-02959.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler WJ, Pozzo-Miller LD. BDNF enhances quantal neurotransmitter release and increases the number of docked vesicles at the active zones of hippocampal excitatory synapses. J Neurosci. 2001;21:4249–4258. doi: 10.1523/JNEUROSCI.21-12-04249.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usdin M, Shelbourne P, Myers R, Madison D. Impaired synaptic plasticity in mice carrying the Huntington's disease mutation. Hum Mol Genet. 1999;8:839–846. doi: 10.1093/hmg/8.5.839. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Pearson J, Slow EJ, Hossain SM, Leavitt BR, Hayden MR. Cognitive dysfunction precedes neuropathology and motor abnormalities in the YAC128 mouse model of Huntington's disease. J Neurosci. 2005;25:4169–4180. doi: 10.1523/JNEUROSCI.0590-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonsattel J, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- Wheeler VC, White JK, Gutekunst CA, Vrbanac V, Weaver M, Li XJ, Li SH, Yi H, Vonsattel JP, Gusella JF, Hersch S, Auerbach W, Joyner AL, MacDonald ME. Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in HdhQ92 and HdhQ111 knock-in mice. Hum Mol Genet. 2000;9:503–513. doi: 10.1093/hmg/9.4.503. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Ciammola A, Rigamonti D, Leavitt B, Goffredo D, Conti L, MacDonald M, Friedlander R, Silani V, Hayden M, Timmusk T, Sipione S, Cattaneo E. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L, Cataudella T, Leavitt B, Hayden M, Timmusk T, Rigamonti D, Cattaneo E. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet. 2003;35:76–83. doi: 10.1038/ng1219. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Liber D, Ramos C, Tarditi A, Rigamonti D, Tartari M, Valenza M, Cattaneo E. Progressive loss of BDNF in a mouse model of Huntington's disease and rescue by BDNF delivery. Pharmacol Res. 2005;52:133–139. doi: 10.1016/j.phrs.2005.01.001. [DOI] [PubMed] [Google Scholar]