

Abstract
A large body of pharmaco-behavioral data implicates the basolateral nucleus of the amygdala (BLA) in the facilitation of memory consolidation by emotions. Overall, this evidence suggests that stress hormones released during emotional arousal increase the activity of BLA neurons. In turn, this increased BLA activity would facilitate synaptic plasticity elsewhere in the brain, to which the BLA projects. However, the direct effects of glucocorticoids on BLA neurons are incompletely understood. In the present study, we examined the direct effects of corticosterone (CORT) on principal neurons of the rat BLA in vitro using whole-cell patch-clamp recordings. We found that application of a stress level of CORT for 20 min caused significant changes in the passive properties and responsiveness of BLA cells measured 1–2 h later. Indeed, CORT application produced a depolarization of the resting potential, an increase in input resistance, and a dramatic decrease in spike-frequency adaptation. In addition, GABAA IPSPs evoked by stimulation of the external capsule were significantly reduced by CORT application. This effect of CORT was not attributable to a reduction in the amount of GABA released because GABAB IPSPs were unchanged and the resistance drop associated with GABAA IPSPs was not altered. Rather, we found that this effect of CORT resulted from a positive shift of the GABAA reversal potential. Overall, these results suggest that, in agreement with previous behavioral findings, glucocorticoids enhance the excitability of principal BLA cells by increasing their intrinsic excitability and decreasing the impact of GABAA IPSPs.
Keywords: amygdala, corticosterone, emotions, GABA, learning, memory
Introduction
Usually, stressful and emotionally arousing experiences are more vividly remembered than mundane events (Christianson, 1992). Considerable behavioral evidence suggests that the basolateral nucleus of the amygdala (BLA) plays a critical role in the facilitation of memory by emotional arousal. For instance, local intra-BLA infusions of drugs that enhance or reduce the excitability of BLA neurons, respectively, facilitate or impair memory formation when performed within 2 h after training (Dickinson-Anson et al., 1993; Packard et al., 1994; Ferry and McGaugh, 1999; Hatfield and McGaugh, 1999). Consistent with this, the firing rate of BLA neurons increases for hours after training on an aversive memory task (Pelletier et al., 2005). These and other findings suggest that emotional arousal enhances memory consolidation by increasing BLA activity. In turn, the BLA would then modulate memory formation for numerous learning tasks through its widespread projections to other brain regions (for review, see McGaugh, 2002, 2004).
How do stress and emotional arousal increase the activity of BLA neurons? Considerable evidence suggests that adrenal stress hormones play a crucial role (de Kloet et al., 1999; Roozendaal, 1999; McGaugh and Roozendaal, 2002). Indeed, they are released in high concentrations during stress (Sapolsky et al., 1986), and posttraining infusions of glucocorticoid receptor (GR) antagonists in the BLA impair memory consolidation (Roozendaal and McGaugh, 1997). Moreover, peripheral or intra-BLA injections of GR agonists after training facilitate memory formation in a variety of learning paradigms (Roozendaal and McGaugh, 1997; Sandi et al., 1997; Akirav et al., 2004; Hui et al., 2004).
Consistent with this data, it is known that, once released into the bloodstream, glucocorticoids readily pass the blood–brain barrier and bind to two types of intracellular steroid receptors: high-affinity mineralocorticoid receptors (MRs), which are occupied at basal glucocorticoid levels, and low-affinity GRs, which require stress levels of glucocorticoids to be fully activated (for review, see de Kloet et al., 1990). Moreover, through these receptors, glucocorticoids classically exert late-onset and long-lasting effects by modulating gene expression (Reichardt and Schutz, 1998). In keeping with this, the genomic effects of these receptors have been implicated in the memory-enhancing effects of glucocorticoids. For instance, a point mutation in the GR gene that selectively prevents DNA binding by GRs impairs spatial memory formation (Oitzl et al., 2001).
However, despite overwhelming behavioral evidence indicating that glucocorticoids modulate memory consolidation via the BLA, the direct effects of glucocorticoids on BLA neurons are incompletely understood. The sole study that addressed this question focused on voltage-gated Ca2+ currents in BLA neurons (Karst et al., 2002). Thus, the present study aimed to investigate the direct effects of corticosterone (CORT) (the glucocorticoid in rodents) on principal BLA cells in vitro. Our results indicate that CORT enhances the excitability of the principal BLA cells by increasing their intrinsic excitability and decreasing the impact of GABAA inhibition.
Materials and Methods
Preparation of slices.
Coronal brain slices containing the amygdala were obtained from Sprague Dawley rats (3–5 weeks old). Experiments were performed with the approval of the Institutional Animal Care and Use Committee of Rutgers University and in accordance with the National Institutes of Health Guide to the Care and Use of Laboratory Animals. A total of 95 rats were used. Each rat was acclimated to handling for 2 d before the experiment. On the day of the experiment, the animal was deeply anesthetized with pentobarbital (60 mg/kg, i.p.), ketamine (80 mg/kg, i.p.), and xylazine (12 mg/kg, i.p.) and decapitated. Then, the brain was rapidly removed and placed in an ice-cold oxygenated artificial CSF (ACSF) containing the following (in mm): 126 NaCl, 2.5 KCl, 1.25 NaH2PO4, 1 MgCl2, 2 CaCl2, 26 NaHCO3, and 10 glucose. The pH was adjusted to 7.2 (when oxygenated, the pH increases to physiological levels) and osmolarity to ∼300 mOsm. Brain slices (400-μm-thick) were cut using a vibrating microtome and kept for at least 1 h in an oxygenated holding chamber at 23°C before the experiment. One slice was then transferred to a recording chamber perfused with oxygenated ACSF at a rate of 4 ml/min. The temperature of the chamber was gradually increased to 32°C before the recordings began.
Drugs.
All drugs were obtained from Sigma (St. Louis, MO). Corticosterone (catalog #C2505) was initially dissolved in DMSO and then diluted 1:100 with ACSF. This stock solution was then diluted 2:100 or 2:10,000 with ACSF, resulting in final concentrations of 100 or 1 nm, respectively. The MR antagonist spironolactone (catalog #S3378) was dissolved in DMSO and diluted 1:1000 with ACSF to yield a final concentration of 5 μm. The GR antagonist RU38486 (11β, 17β-11[4-(dimethylamino)phenyl]-17-hydroxy-17-(1-(propynyl)-estra-4,9-dien-3-one) (catalog #M8046) was dissolved in HCl and diluted 1:1000 with ACSF to yield a final concentration of 5 μm, followed by readjustment of the pH to 7.2 with NaOH. The pure GABAA agonist isoguvacine (catalog #G002) was dissolved in ACSF (200 μm). CNQX (20 μm; catalog #C239), AP-5 (100 μm; catalog #A5282), and CGP-35348 (p-3-aminopropyl-p-diethoxymethyl phosphoric acid) (100 μm; catalog #C5851) were dissolved in ACSF.
Data acquisition.
Unless otherwise noted, stimulating electrodes were placed in the external capsule (EC), and a neuron was recorded in the BLA as shown in Figure 1A. The positions of stimulating and recording electrodes were kept uniform to minimize variability between cells. Current-clamp recordings were obtained under visual guidance with pipettes (4–6 MΩ) pulled from borosilicate glass capillaries and filled with a solution containing the following (in mm): 130 K-gluconate, 10 HEPES, 10 KCl, 2 MgCl2, 2 ATP-Mg, and 0.2 GTP-Tris. The pH was adjusted to 7.2 with KOH and osmolarity to ∼280 mOsm. The liquid junction potential was 10 mV with this solution, and the membrane potential (Vm) was corrected accordingly. Recordings with series resistance >15 MΩ were discarded. Current-clamp recordings were obtained with an Axoclamp 2B amplifier under visual guidance using differential interference contrast and infrared video microscopy and digitized at 10 kHz with a Digidata 1200 interface controlled by the software pClamp 8.1 (Molecular Devices, Palo Alto, CA).
Figure 1.

Experimental approach and identification of principal BLA neurons. A, Scheme showing the position of the recording (pipette, Rec.) and stimulation (dots, Stim.) sites on a coronal section of the rat amygdala. The stimulation electrodes were positioned in the external capsule. B, Superimposed voltage responses of three BLA cells to a series of negative and positive current pulses (insets). The cells displayed a continuum of spike-frequency adaptation (B1–B3). C, Graphs plotting the number of current-evoked spikes (y-axes) as a function of spike amplitude (C1) or resting potential (C2). To control for intercell variations in Rin, we measured the number of evoked spikes using current pulses that brought the Vm to approximately −45 mV. AB, Accessory basal nucleus; BL, basolateral amygdala; CE, central amygdala; IC, internal capsule; LA, lateral amygdala; ME, medial amygdala; OT, optic tract; rh, rhinal fissure; ST, striatum.
Electroresponsive properties were investigated by applying graded series of depolarizing and hyperpolarizing current pulses in 10 pA increments (500 ms duration) from a membrane potential of −75 mV, as determined by intracellular current injection. Note that little current had to be applied to reach this value because it was close to the resting potential of BLA neurons (see Results). The input resistance (Rin) of the cells was estimated in the linear portion of current–voltage plots.
The amplitude of the slow afterhyperpolarizations (AHPs) was measured in two ways: after 0.5 s depolarizing current pulses that brought the Vm to −45 mV or after spikes evoked by these current pulses. In the former case, the AHP was measured at its peak after the offset of the current pulse. However, qualitatively identical results were obtained when we used a constant interval based on measurements of time-to-peak of the AHP in vehicle-treated cells. In the case of spike AHPs, the same postspike interval was used to measure AHP amplitudes in both cell groups, as determined from the average time to AHP peak in vehicle-treated cells. Note that we only considered the second current-evoked spike because many vehicle-treated BLA cells did not generate more than two spikes. We ignored the first action potential because it often occurred at an interval shorter than the membrane time constant (i.e., the first AHP was contaminated by the passive rise of the Vm as a result of current injection). It should be noted that the interval between the first and second spike was statistically indistinguishable in CORT (58.2 ± 7.3 ms) versus vehicle-treated cells (56.5 ± 5.1 ms; t test, p = 0.48). Also, qualitatively identical results were obtained when we measured the AHP that followed the third spike.
To investigate the responsiveness of the cells to synaptically released GABA, the external capsule was stimulated (0.1 ms pulses of 0.1–1 mA stimuli in 0.1 mA increments) with tungsten electrodes (80 μm in diameter) from a membrane potential of −65 mV. To determine the reversal potential of GABA-mediated IPSPs, evoked responses were examined at membrane potentials of −65, −75, −85, and −95 mV. To examine GABAA responses in isolation, local applications of the pure GABAA agonist isoguvacine were performed by applying air pressure pulses (5 ms) to a patch pipette filled with ACSF containing 200 μm isoguvacine.
Data analysis.
All analyses were performed off-line with pClamp 8.2 (Molecular Devices) and Igor Pro 4.03 (WaveMetrics, Lake Oswego, OR). For statistical analyses, the level of significance was defined as p < 0.05, and data were analyzed with ANOVAs followed by post hoc t tests with correction of the significance level for multiple comparisons with the Bonferroni's method.
Results
Database
This study aimed to examine the direct effect of CORT on principal BLA cells. The BLA is known to contain two main cell types (for review, see McDonald, 1992). Most BLA neurons are glutamatergic projection cells, and a minority of them are GABAergic local-circuit neurons with generally smaller somata (McDonald, 1985). Thus, by chance alone, our sample of BLA neurons should mainly comprise projection (principal) neurons. Nevertheless, to further restrict our attention to principal BLA cells, recording pipettes were aimed to neurons with large-diameter cell bodies. Moreover, we only considered cells that displayed the characteristic electroresponsive properties of principal BLA cells, including a lack of spontaneous discharges at rest, generation of spike trains exhibiting frequency adaptation during membrane depolarization, broad action potentials (>0.7 ms at half-amplitude), and various degrees of inward rectification in the hyperpolarizing direction (Washburn and Moises, 1992a; Rainnie et al., 1993; Pare et al., 1995; Pape and Driesang, 1998; Faber et al., 2001).
Using these criteria, a total of 156 principal BLA neurons having a resting Vm negative to −60 mV (average of 75.1 ± 0.2 mV) and generating overshooting action potentials (average of 93.7 ± 0.5 mV) were recorded in the whole-cell configuration. Consistent with previous observations (Faber et al., 2001; Faber and Sah, 2002), these neurons exhibited varying degrees of spike-frequency adaptation (Fig. 1B), ranging from neurons that fired only a few spikes at the onset of the depolarizing current pulses (Fig. 1B1,B2), to cells that fired throughout the depolarizing current injection (Fig. 1B3). To test whether the high level of spike-frequency adaptation seen in some BLA neurons was a recording artifact, we correlated the number of spikes evoked by current pulses that brought the Vm to approximately −45 mV with various parameters known to be sensitive to neuronal health and recording quality. At odds with this possibility, however, the number of current-evoked spikes did not correlate significantly with spike amplitude (r = 0.17) (Fig. 1C1), resting Vm (r = −0.2) (Fig. 1C2), Rin (r = 0.14), and spike duration (r = −0.12). Thus, these results suggest that the continuum of spike-frequency adaptation seen in BLA cells does not reflect variations in recording quality but in the intrinsic properties of BLA neurons, as suggested previously (Faber et al., 2001; Faber and Sah, 2002).
Approach used to study the impact of corticosterone on principal BLA cells
Glucocorticoids are known to act via two main intracellular receptors: the high-affinity MRs that are occupied at basal glucocorticoid levels and the low-affinity GRs that require stress levels of glucocorticoids to be activated (Reul and de Kloet, 1985). Thus, to examine the differential effects of activating MRs versus MRs and GRs, we used two different concentrations of CORT: a low one (1 nm) known to activate only MRs (Joels and de Kloet, 1991) and a high one (100 nm) that was shown to activate both MRs and GRs and mimics the effects of stress (Karst et al., 2000; Verkuyl et al., 2005).
The main difficulty in studying the effects of glucocorticoids is that they have a long latency and a protracted time course. Indeed, once activated, MRs and GRs translocate to the nucleus in which they act as transcription factors and affect gene expression (Reichardt and Schutz, 1998). As a result, using within-cell comparisons to study these slow effects would involve prolonged dialysis of neurons by the pipette solution, potentially interfering with the CORT effects. Thus, to avoid this confound, we opted for a between-cell design. Namely, in each slice, a control neuron was first recorded to examine the baseline properties of principal BLA cells. This was followed by a brief (20 min) application of CORT or vehicle. One to 2 h later, another neuron was recorded in the same slice (Fig. 2A).
Figure 2.

Effect of CORT on the passive membrane properties of principal BLA cells. A, Scheme of the protocol used in this experiment. B, Effect of low and high doses of CORT (red open and filled bars, respectively) on the resting potential of BLA neurons (y-axis). Both doses of CORT produced a significant depolarization of the resting Vm. C, Effect of low and high doses of CORT (red open and filled bars, respectively) on the Rin of BLA neurons (y-axis). Only the high dose of CORT (filled red bar) resulted in a significant increase in Rin compared with the other three cell groups. Group means ± SEM. * p < 0.05.
Effect of corticosterone on the passive membrane properties of principal BLA cells
We first tested the effects of CORT on the passive membrane properties of principal BLA cells. Although CORT-treated BLA cells continued to lack spontaneous discharges at rest, both CORT1 nm- and CORT100 nm-treated cells exhibited a slightly more depolarized resting membrane potential (−73.5 ± 0.7 mV, n = 17; and −72.6 ± 0.7 mV, n = 26, respectively) than vehicle (−75.6 ± 0.5 mV; n = 26) and control (−76 ± 0.4 mV; n = 50) cells (Fig. 2B). This was confirmed with a one-way ANOVA, which revealed a significant effect of group (F(3,115) = 7.28; p < 0.001). A post hoc test further revealed that CORT-treated cells were significantly different from both control (CORT1 nm, p < 0.05; and CORT100 nm, p < 0.05) and vehicle (CORT1 nm, p < 0.05; and CORT100 nm, p < 0.05) cells, whereas the latter two groups did not differ (p > 0.05). Because this was the only significant effect of the low dose of CORT seen in this study, for simplicity, the results obtained with CORT1 nm-treated cells will not be mentioned below. Nevertheless, this group was included in the statistical analyses and figures.
Next, we measured the effect of CORT on Rin. In this analysis, Rin was estimated in the linear portion of current–voltage plots. To identify possible voltage-dependent effects of CORT, we also compared estimates of Rin from voltage responses to small negative versus positive current pulses. However, the three methods yielded identical results. Thus, we only report the averages obtained with the first method. CORT100 nm-treated cells had a higher Rin (195.3 ± 20.6 MΩ; n = 26) than vehicle-treated (131.8 ± 8.8 MΩ; n = 26) and control (135.5 ± 7.2 MΩ; n = 50) cells (Fig. 2C). A one-way ANOVA revealed a significant effect of group (F(3,115) = 5.84; p < 0.001), and post hoc t tests further confirmed that CORT100 nm-treated cells had a significantly higher Rin than vehicle (p < 0.01) and control (p < 0.01) cells, whereas the latter two groups were comparable (p > 0.05).
Additional analysis revealed that the effects of CORT100 nm on Rin were not homogeneously distributed in our sample. Indeed, the CORT100 nm group included a subset of cells (15% of CORT100 nm neurons) with very high Rin (405 ± 32 MΩ) and a second one (85% CORT100 nm) with lower Rin (160.6 ± 11.2 MΩ). Nevertheless, the Rin of this second subset of BLA neurons was significantly higher than seen in vehicle-treated cells (t test, p < 0.05).
There are two possible explanations for the heterogeneous effects of CORT100 nm on Rin. The first one is that this heterogeneity reflects differences in the levels of GR expression or coupling of GRs to effector mechanisms. The second is that this variability is the consequence of a sampling artifact: the inadvertent inclusion of interneurons in our sample of CORT100 nm-treated cells. However, various factors suggest that the subset of CORT100 nm-treated cells with extremely high Rin were not interneurons. First, these cells generated action potentials >0.7 ms in duration and generated accommodating trains of action potentials, as is typical of principal BLA neurons. Second, the highest Rin seen in our samples of vehicle and control cells was 280 MΩ, and it seems unlikely that, by chance, as many as 15% of our CORT100 nm recordings (n = 26) would have been obtained from BLA interneurons compared with 0% in our samples of vehicle (n = 26) and control (n = 50) neurons. Thus, we suspect that the heterogeneous effects of CORT100 nm on Rin reflect genuine cell-to-cell variations in the levels of GR expression or coupling of GRs to effector mechanisms (Morimoto et al., 1996).
Corticosterone increases the intrinsic excitability of principal BLA cells
A more depolarized membrane potential and an increased Rin are expected to increase neuronal excitability. Therefore, we next aimed to test this prediction. To this end, we first measured the threshold current to spiking by applying current pulses increasing in steps of 10 pA from a Vm of −75 mV. Consistent with the analysis of CORT effects on Rin, CORT100 nm-treated cells required lower current injections (118.4 ± 9.5 pA; n = 26) to reach firing threshold compared with vehicle-treated (161.5 ± 11 pA; n = 26) and control (158.3 ± 8 pA; n = 49) cells. A one-way ANOVA revealed an effect of group (F(3,114) = 3.94; p < 0.05), and post hoc tests further confirmed that CORT100 nm-treated cells required a significantly lower amount of depolarizing current injection compared with control (p < 0.05) and vehicle-treated (p < 0.05) cells. In contrast, the spike threshold was similar in all groups (F < 1). Qualitatively identical results were obtained when the subset of CORT100 nm cells with high Rin were excluded from the analysis.
To test whether CORT affects the firing pattern of principal BLA cells, we next examined its impact on spike-frequency adaptation. As mentioned above, principal BLA cells exhibited a continuum of spike-frequency adaptation. Thus, based on the speed of adaptation during 0.5 s current pulses that brought the Vm to approximately −45 mV, BLA cells were arbitrarily divided in three groups: (1) cells that fired at least three spikes at the onset of the current injection and remained silent for the rest of the current pulse [rapidly adapting (RA) cells] (Fig. 3A1); (2) cells that fired four to six spikes with gradually increasing interspike intervals [intermediately adapting (IA) cells] (Fig. 3A2); and (3) cells that fired more than six spikes with slower spike-frequency adaptation [slowly adapting (SA) cells] (Fig. 3A3). The proportion of RA cells observed after CORT100 nm treatment (3.8%; n = 25) was much lower than in vehicle and control groups (47.2%; n = 74) (Fig. 3B). Conversely, there was a marked increase in the proportion of IA and SA cells in the CORT100 nm group (IA, 53%; SA, 42.3%) compared with vehicle and control groups (IA, 31.9%; SA, 20.8%; χ2 test, p < 0.001) (Fig. 3B).
Figure 3.
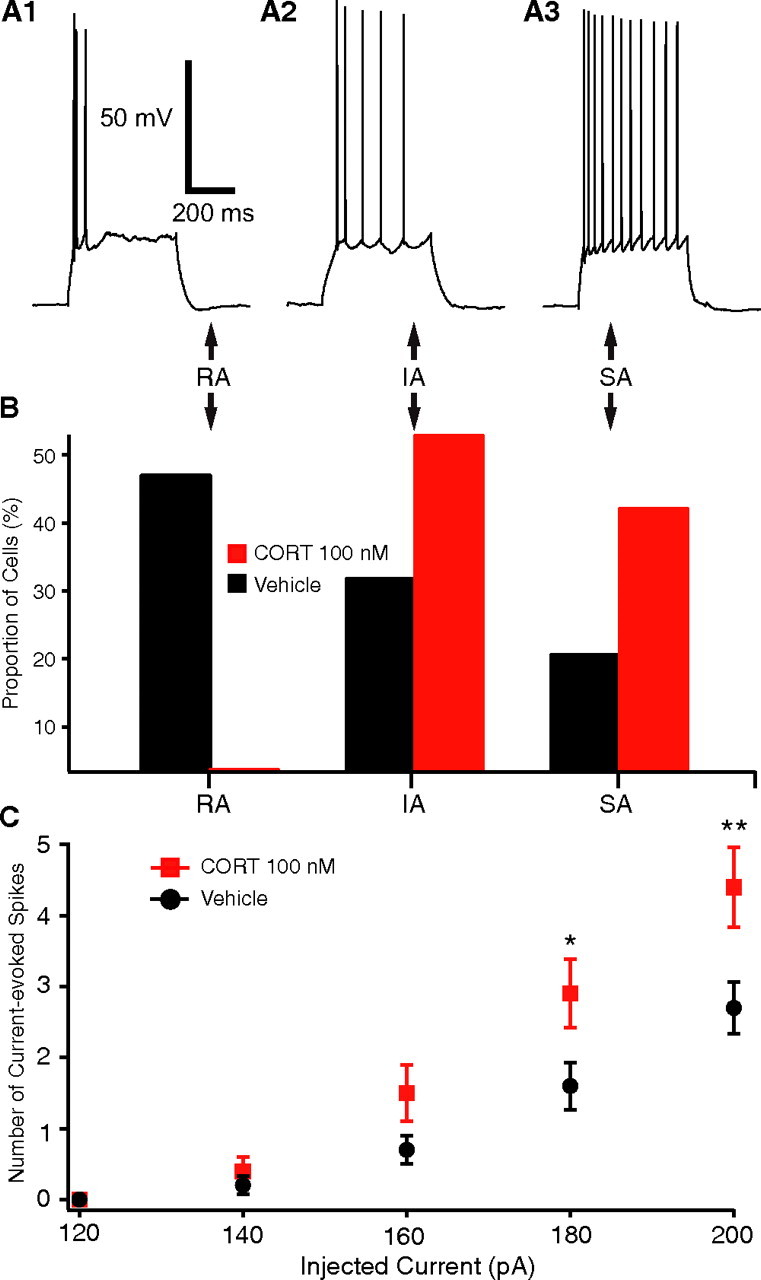
High dose of CORT (100 nm) increases the intrinsic excitability of principal BLA cells. A, Based on spike-frequency adaptation, the cells were divided into three groups: RA (A1), IA (A2), and SA (A3) cells. B, Proportion of BLA cells falling in the RA, IA, and SA classes among CORT-treated (red bars) versus vehicle-treated (black bars) cells. C, Graph plotting the number of evoked spikes (y-axis) as a function of injected current (x-axis) in subsets of CORT-treated (red) and vehicle-treated (black) cells matched for Rin. Even after matching the CORT versus vehicle cells for Rin, the CORT-treated cells fired significantly more spikes in response to 160–200 pA current injections. Group means ± SEM. * p < 0.05; ** p < 0.001.
To determine whether the effects of CORT on spike-frequency adaptation were only a consequence of the higher Rin of CORT-treated cells, we selected cells from the CORT100 nm and vehicle groups with comparable Rin and measured the number of spikes elicited by graded series of depolarizing current injections (120–200 pA, 500 ms). As shown in Figure 3C, even after controlling for Rin, CORT100 nm-treated cells (n = 10) fired more spikes compared with vehicle cells (n = 23) in response to 160–200 pA current injections. A repeated-measures ANOVA revealed a main effect of drug treatment (F(1,124) = 4.86; p < 0.05) and current (F(4,124) = 77.8; p < 0.001), as well as a significant interaction (F(4,124) = 4.28; p < 0.05). Additional post hoc tests confirmed that CORT100 nm-treated cells fired significantly more spikes compared with vehicle cells in response to 180 pA (p < 0.05) and 200 pA (p < 0.001) current pulses but not to current pulses of lower amplitude. This result therefore suggests that the increase in Rin is not sufficient to account for the decreased spike-frequency adaptation seen at the highest current levels with CORT100 nm.
To shed light on the mechanisms responsible for the decreased spike-frequency adaptation seen in CORT-treated cells, we then compared the amplitude of the AHPs that followed by depolarizing current pulses or current-evoked spikes. In the former case, we only considered current pulses that brought the Vm to approximately −45 mV. The AHP was measured as the difference between the prepulse potential and the AHP peak, after the offset of the current pulse (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material). Measured in this way, the AHP amplitude was significantly lower in CORT-treated (−2.18 ± 0.13 mV) than vehicle-treated (−2.91 ± 0.23 mV; t test p < 0.05) cells despite the fact that current pulses evoked many more spikes in CORT-treated cells (see above).
For AHPs that followed spikes, two components could be recognized (supplemental Fig. S1B, available at www.jneurosci.org as supplemental material). A fast component, hereafter termed fast AHP (fAHP) (Fig. S1B, arrowhead, available at www.jneurosci.org as supplemental material), that peaked 4.5 ± 0.3 ms after spike initiation, and a slower component, hereafter termed medium AHP (mAHP) (Fig. S1B, arrow, available at www.jneurosci.org as supplemental material), that peaked 27.1 ± 2.9 ms after spike initiation. Both components were measured by subtracting the voltage at the peak of the hyperpolarization from the threshold potential for spike initiation. The AHPs that followed the first action potential were ignored because they were often distorted by the passive Vm rise that resulted from current injection. Also, because some vehicle-treated BLA cells stopped firing after two spikes, we focused on the AHPs that followed the second current-evoked spike. Measured in this way, the mAHP amplitude was significantly lower in CORT-treated (−4.94 ± 0.49 mV) than vehicle-treated (−6.47 ± 0.51 mV; t test p < 0.05) cells, although the interval between the first and second spikes was similar in the two groups (CORT, 58.2 ± 7.3 ms; vehicle, 56.5 ± 5.1 ms; t test, p = 0.48). Qualitatively identical results were obtained when we measured the mAHP that followed the third spike (CORT, −5.84 ± 0.41 mV; vehicle, −7.61 ± 0.78 mV; t test, p < 0.05). In contrast, no difference was found between the fAHP amplitude of CORT- and vehicle-treated cells for both the second (respectively, 6.69 ± 0.68 and 6.22 ± 0.61 mV; t test, p > 0.05) and third (respectively, 7.32 ± 0.65 and 7.27 ± 0.67 mV; t test, p > 0.05) spikes.
Overall, these findings suggest that the high dose of CORT increases the intrinsic excitability of principal BLA neurons as measured with threshold current to spiking, spike-frequency adaptation, and AHP amplitudes.
Corticosterone decreases the GABAergic responsiveness of principal BLA cells
Using field potential recordings in vitro, a recent study has shown that the facilitating effects of stress on memory are accompanied by an attenuation of GABAergic inhibition in the BLA (Rodriguez Manzanares et al., 2005). Therefore, we sought to investigate whether direct CORT application had a similar effect in the BLA. To test this, we examined the effect of CORT on GABAergic responses evoked in principal BLA cells by stimulation of the EC from a Vm of −65 mV. EC stimuli (0.1 ms duration) of gradually increasing intensity (range of 0.1–1 mA; 0.1 mA increments) were applied at 0.05 Hz. Consistent with previous findings in the BLA (Rainnie et al., 1991; Washburn and Moises, 1992b), EC stimuli of sufficient intensity (see below) evoked an early EPSP that was followed by a biphasic IPSP. Previous studies have repeatedly established that the early phase of this IPSP is mediated by GABAA receptors, whereas the late one is dependent on GABAB receptor activation (Rainnie et al., 1991; Washburn and Moises, 1992b; Danober and Pape, 1998; Martina et al., 2001). Thus, for simplicity, the early and late phases of EC-evoked IPSPs will hereafter be termed GABAA and GABAB IPSPs, respectively.
The amplitude of EC-evoked GABAA IPSPs was lower in CORT100 nm-treated cells compared with the other three groups (Fig. 4A,B1). A repeated-measures ANOVA revealed a main effect of drug treatment (F(3,840) = 5.28; p < 0.01) and current intensity (F(30,840) = 1.97; p < 0.01) and a significant interaction (F(10,840) = 78.5; p < 0.0001). Additional post hoc tests confirmed that CORT100 nm-treated cells (n = 21) generated GABAA IPSPs of lower amplitude in response to EC stimuli of 0.6–1 mA compared with control (n = 32; p < 0.05), vehicle-treated (n = 18; p < 0.05), and CORT1 nm-treated (n = 17; p < 0.05) cells, whereas the latter three groups did not differ (p > 0.05).
Figure 4.

CORT decreases GABAA IPSPs in principal BLA cells. A, Responses of representative vehicle-treated (A1) and CORT-treated (A2) cells to EC stimuli of gradually increasing intensity (numbers on the right in milliamperes). Insets in top right of A1 and A2 show the same data with an expanded time base so that the EPSPs are visible. B, Graphs plotting amplitude (y-axes) of GABAA (B1) or GABAB (B2) IPSPs as a function of the intensity (x-axes) of electrical stimuli delivered to the external capsule. GABAA and GABAB IPSPs were measured at a time corresponding to the peak of the IPSPs elicited by the most intense stimulus tested (left and right arrows in A, respectively). B1 and B2 show group means ± SEM.
The attenuation of GABAA IPSPs observed in CORT100 nm-treated cells could result from a reduction in the amount of GABA released or an alteration of postsynaptic properties. Reduced GABA release could result from a diminished excitatory drive to BLA interneurons and/or a lowered probability of GABA release by their axon terminals. However, in both cases, one would expect to observe a parallel reduction in the amplitude of the GABAB IPSPs. At odds with this prediction, however, we found that CORT100 nm-treated cells exhibited GABAB IPSPs that were comparable in amplitude with those of the other three groups (Fig. 4A,B2). A repeated-measures ANOVA confirmed that there was no main effect of drug treatment (F(3,850) = 1.48; p > 0.05) and no significant interaction (F(30,850) = 1.26; p > 0.05), indicating that the different current intensities had the same effect on all groups. This suggested that the amount of GABA released was comparable between all groups and that the effect of CORT100 nm was likely mediated postsynaptically.
To test this possibility, we first compared the Rin drop at the peak of the GABAA IPSPs in the vehicle and CORT100 nm groups (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Either using absolute values or after normalization to the baseline Rin, all cell groups exhibited a similar reduction in Rin at the peak of EC-evoked GABAA IPSPs (the Rin dropped to 53.3 vs 52.8% of prestimulus values in vehicle vs CORT cells, respectively). A one-way ANOVA revealed that there was no significant difference between the groups (F < 1), suggesting that the effect of CORT100 nm on GABAA IPSPs was not attributable to a change in the postsynaptic GABAA conductance.
If CORT does not affect the amount of GABA released or the postsynaptic GABAA conductance, then we reasoned that it likely modified chloride homeostasis. This could change the driving force acting on chloride ions, resulting in the observed reduction in GABAA IPSPs. In support of this hypothesis, we found that the GABAA reversal potential was more depolarized by ∼6 mV in CORT100 nm-treated cells (−68.6 ± 2.2 mV; n = 18) compared with vehicle-treated cells (−74.8 ± 0.7 mV; n = 22; t test, p < 0.05), with no obvious change in the slope IPSP amplitude versus Vm curves (Fig. 5A). Moreover, consistent with the finding that CORT100 nm did not affect the amplitude of evoked GABAB responses, there was no significant difference between the GABAB reversal potential of the groups (CORT100 nm, −97.4 ± 1.3 mV; vehicle, −96.1 ± 0.9 mV; t test, p > 0.05).
Figure 5.
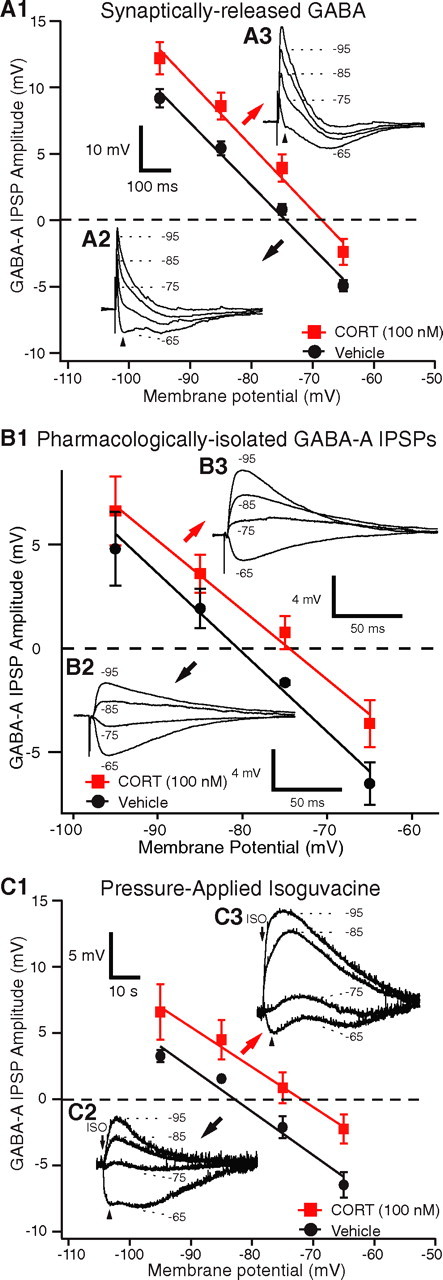
CORT produces a positive shift of the GABAA reversal potential. A1, GABAA IPSP amplitudes (y-axis) as a function of Vm (x-axis) for synaptically released GABA. A2, A3, EC-evoked IPSPs at different membrane potentials (numbers on the right) in representative BLA cells exposed to vehicle (A2) or CORT (100 nm; A3). B1, Reversal potential of pharmacologically isolated GABAA IPSPs evoked by stimulation of the basolateral nucleus in the presence of CNQX, AP-5, and CGP-35348. The graph plots GABAA IPSP amplitudes (y-axis) as a function of Vm (x-axis). B2, B3, Basolateral-evoked GABAA IPSPs at different membrane potentials (numbers) in representative BLA cells exposed to vehicle (B2) or CORT (100 nm; B3). C1, GABAA IPSP amplitudes (y-axis) as a function of Vm (x-axis) for pressure-applied isoguvacine. C2, C3, Isoguvacine-evoked IPSPs at different membrane potentials (numbers on the right) in representative BLA cells exposed to vehicle (C2) or CORT (100 nm; C3). A1, B1, and C1 show group means ± SEM.
Importantly, the same result was obtained when we considered pharmacologically isolated GABAA IPSPs (Fig. 5B) that were evoked by electrical stimuli delivered in the basolateral nucleus, 1 mm from the recorded cells, but in the presence of the GABAB receptor antagonist CGP-35348 (100 μm) and the glutamate receptor antagonists CNQX (20 μm) and AP-5 (100 μm). As was observed with EC stimuli, the reversal potential of these pharmacologically isolated GABAA IPSPs (Fig. 5B) was significantly more depolarized in the CORT100 nm-treated cells (−74.4 ± 1.6 mV; n = 6) than in the vehicle group (−80.3 ± 1.2 mV; n = 5; t test, p < 0.05). Note that these averages are more negative than in the above experiments. The difference presumably results from a contamination of the GABAA IPSPs by the preceding EPSPs evoked by EC stimulation.
To further control for possible presynaptic effects of CORT, we next compared the GABAA reversal potentials of CORT- versus vehicle-treated cells in response to local pressure application of the pure GABAA agonist isoguvacine (200 μm, 5 ms). Consistent with the results obtained with electrical stimuli, isoguvacine-evoked GABAA currents reversed at a more depolarized Vm in CORT100 nm-treated cells (−73 ± 2.4; n = 11) compared with vehicle-treated cells (−82.2 ± 0.6; n = 11) (Fig. 5C).
To summarize, these results suggested that the high dose of CORT reduces the inhibitory action of GABA by producing a positive shift in the GABAA reversal potential.
Impact of CORT on EC-evoked EPSPs
We also examined the impact of CORT on EC-evoked EPSPs (supplemental Fig. S3, available at www.jneurosci.org as supplemental material) using the same approach and data as for GABAergic IPSPs. However, these analyses revealed no significant differences between amplitudes of EC-evoked EPSPs as a function of the stimulation intensity in the different groups (supplemental Fig. S3A, available at www.jneurosci.org as supplemental material). A repeated-measures ANOVA revealed that there was no main effect of drug treatment (F(3,850) = 1.084; p > 0.05) and no significant interaction (F(10,850) = 1.397; p > 0.05). Similarly, the extrapolated reversal potential of EC-evoked EPSPs did not differ significantly between the various groups (CORT100 nm, −27.5 ± 3.0 mV; vehicle, −28.4 ± 4.7 mV; t test, p = 0.9) (Fig. S3B, available at www.jneurosci.org as supplemental material). Note that these reversal potentials are much more negative than the expected value (∼0 mV), indicating that these EPSPs were contaminated by inhibition.
Does CORT have short-latency effects on principal BLA neurons?
In the experiments described so far, an interval of 1–2 h elapsed between the CORT application and experimental measurements. However, it is also possible that CORT has short-latency effects. To address this possibility, we tested the effects of a 10 min application of CORT100 nm (n = 6) using a within-cell design. In addition, in a separate group of interleaved BLA cells (n = 5) recorded in different slices, we applied vehicle instead of CORT to control for possible time-dependent effects. In both groups, every 30 s, we applied a hyperpolarizing current pulse (500 ms, 40 pA), followed, 1 s later, by an EC stimulus (0.1 ms, 0.5 mA, from a Vm of −65 mV). After a 5 min baseline, we switched the ACSF to one containing either vehicle or CORT100 nm. We then followed the fate of synaptic and current-evoked responses for 10 min. In addition, immediately before and after the conclusion of these tests, we studied the voltage response of BLA cells to current pulses of gradually increasing amplitude and measured the IPSP reversal potentials as above.
As shown in supplemental Figure S4 (available at www.jneurosci.org as supplemental material), CORT had no short-latency effects. All of the variables we monitored remained unchanged (paired t tests, p > 0.05). For instance, the amplitude of EPSPs (supplemental Fig. S4A, available at www.jneurosci.org as supplemental material), as well as that of the early (supplemental Fig. S4B, available at www.jneurosci.org as supplemental material) and late (supplemental Fig. S4C, available at www.jneurosci.org as supplemental material) IPSPs, remained unchanged. In addition, the reversal potential of the early and late IPSPs did not change (average changes of 0.533 ± 0.448 and 0.004 ± 0.758 mV, respectively), the Vm was unaffected (average change of −0.5 ± 1.74 mV from baseline), and the Rin was unchanged (average change of 2.6 ± 3.2% from baseline). Similarly, the amount of spike-frequency adaptation, as assessed by number of spikes evoked by current pulses that brought the Vm to approximately −45 mV, was unaffected by CORT application (from 5.3 ± 0.42 to 5.6 ± 0.33 spikes).
Are the effects of CORT mediated by classical glucocorticoid and mineralocorticoid receptors?
The amygdala facilitates memory consolidation for emotionally arousing, stressful experiences (McGaugh, 2002, 2004). Behavioral studies suggest that low-affinity GRs in the BLA are involved in the memory-enhancing effects of stress (for review, see McGaugh, 2004). For instance, intra-BLA infusions of the GR agonist RU28362 [11,17-dihydroxy-6-methyl-17-(1-propynyl)androsta-1,4,6-triene-3-one] after inhibitory avoidance training have been shown to enhance memory consolidation, whereas intra-BLA infusions of the GR antagonist RU38486 impair the consolidation of memory for a spatial task (Roozendaal and McGaugh, 1997). Hence, it is important to determine whether the effects of CORT observed here are mediated by GRs in a manner similar to the effects of stress on memory consolidation.
To address this question, we applied a GR antagonist and tested which effects of the high dose of CORT100 nm, which activates both MRs and GRs, could be blocked. To this end, we applied RU38486 (5 μm) throughout the experiment via the perfusate. First, in the presence of this antagonist, we obtained a baseline control cell. This was followed by 20 min application of CORT100 nm. Another cell was recorded in the same slice 1–2 h later (Fig. 6A). We found that CORT100 nm + RU38486-treated cells (n = 14) behaved comparably with controls and vehicle-treated cells in all of the measured parameters except Vm (Vm of −73.3 ± 1.0, p < 0.05; Rin of 124.1 ± 13.1, p > 0.05; percentage of RA cells, 42%; percentage of SA cells, 21%; GABAA reversal, −73.4 ± 2; p > 0.05) (Fig. 6B). These results suggest that the effects of CORT100 nm on the intrinsic excitability and GABAA reversal potential, but not the resting Vm, are mediated by GRs. Consistent with this finding, the only significant effect of CORT1 nm was a depolarization of the resting Vm, and this effect was not observed when the experiments were performed in the presence of the MR antagonist spironolactone [5 μm (as seen in Fig. 6A); CORT1 nm + spironolactone, −75.6 ± 0.6 mV; p > 0.05; n = 8].
Figure 6.

GR antagonist prevents the increased intrinsic excitability and positive shift in GABAA reversal potential produced by CORT (100 nm). A, Scheme of the protocol used in this experiment. The GR antagonist RU38486 (5 μm) was added to the ACSF before, during, and after the CORT (100 nm) application. B, Graph plotting the normalized change in, from left to right, Vm, Rin, GABAA reversal potential, and proportion of SA BLA neurons in slices exposed to CORT alone (red bars; 100%) versus CORT and RU38486 (gray bars). RU38486 prevented all of the effects of CORT (100 nm), with the exception of the shift in resting potential.
Discussion
The present study was undertaken to determine the direct effects of CORT on principal BLA cells. The interest of this question stems from pharmaco-behavioral studies demonstrating that GR activation in the BLA during stress and emotional arousal causes an increase in the firing rate of BLA neurons, ultimately leading to a facilitation of memory consolidation. Consistent with these findings, our results indicate that CORT enhances the excitability of principal BLA cells by increasing their intrinsic excitability and decreasing the impact of GABAA inhibition. Below, we consider our findings in light of previous studies and discuss their significance for the role of the amygdala in consolidation of emotional memories.
Pharmaco-behavioral evidence suggesting that glucocorticoids constitute the link between emotional arousal and enhanced BLA activity
Glucocorticoids released during emotionally arousing experiences play an important role in the facilitation of memory consolidation by emotions (McGaugh and Roozendaal, 2002). Considerable evidence implicates the GRs in the amygdala in the memory-enhancing effects of glucocorticoids. Posttraining intra-BLA administration of GR agonists results in the enhancement of memory formation for the inhibitory avoidance task. Conversely, posttraining intra-BLA infusions of GR antagonist impairs memory consolidation (Roozendaal and McGaugh, 1997). It is thought that glucocorticoids exert their effects by increasing the activity of BLA neurons, a contention supported by a recent study in which it was shown that inhibitory avoidance learning causes the firing rate of BLA neurons to increase in a sustained manner (Pelletier et al., 2005).
Consistent with this, in the present study, CORT enhanced the excitability of principal BLA neurons through multiple parallel mechanisms. First, their Rin was significantly increased after CORT treatment. In line with this, the threshold current to spiking was significantly decreased in CORT-treated cells with no change in spike threshold. Second, CORT decreased spike-frequency adaptation. However, the Rin increase does not fully account for this effect because the number of current-evoked spikes was significantly higher in CORT-treated compared with vehicle-treated cells even after controlling for Rin. Third, there was a significant reduction in GABAA responses resulting from a positive shift in the GABAA reversal potential. Coupled with the observation that these effects of CORT could be blocked with application of the GR-specific antagonist, our findings suggest that CORT enhances the excitability of principal BLA cells through several parallel mechanisms resulting from the binding glucocorticoids to GRs.
Mechanisms underlying the excitatory effects of glucocorticoids on principal BLA neurons
Both high and low doses of CORT caused a modest depolarization of BLA neurons. Several mechanisms might account for this effect, including the inhibition of the sodium–potassium pump, of the resting K+ conductance, or of a voltage-dependent K+ current activated near rest. Because the membrane depolarization was observed without a significant increase in Rin in the low-dose CORT group, it is more likely that the effect of CORT is mediated by an inhibition of the sodium–potassium pump.
In contrast, the increase in Rin seen with the high dose of CORT probably involved the inhibition of one or more K+ currents such as the K+ leak or of a K+ conductance that is active near rest. Moreover, the fact that the high CORT dose markedly reduced spike-frequency adaptation suggests that CORT also inhibited one or more subtype(s) of voltage-gated or Ca2+-dependent K+ currents that are activated in the suprathreshold range of membrane potentials. In support of this hypothesis, previous studies have revealed that the BLA cells display varying degrees of spike-frequency adaptation (Faber et al., 2001), which appears to reflect differences in the magnitude of the apamin-resistant Ca2+-dependent K+ current they express (Faber and Sah, 2002). Consistent with this, it was shown that a rapidly adapting cell could be converted into a slowly adapting cell type by blockade of an apamin-resistant Ca2+-dependent K+ current (Faber and Sah, 2002). Thus, these considerations suggest that one possible effect of CORT on BLA neurons is the inhibition of this apamin-insensitive Ca2+-dependent K+ conductance (Faber and Sah, 2002) and/or of a voltage-dependent K+ conductance (Faber and Sah, 2005). It is unlikely that this effect depends on a reduction of Ca2+ influx because previous work has revealed that glucocorticoids actually enhance high-voltage-activated Ca2+ currents (Karst et al., 2002). Thus, the effect of CORT on spike-frequency adaptation may depend on a decrease in the expression of K+ channels or an inhibitory regulation of existing channels.
Another major effect of CORT seen in the present study was a reduction in the amplitude of GABAA IPSPs. Several factors suggest that this effect was not mediated presynaptically. First, this reduction was not accompanied by a parallel reduction of GABAB IPSPs. Second, the Rin drop associated to GABAA IPSPs was not altered by CORT. Third, CORT produced a positive shift in the reversal potential for GABAA IPSPs whether they were evoked by synaptically released GABA or by pressure application of the pure GABAA agonist isoguvacine. In light of previous findings, these results are consistent with the possibility that CORT affects chloride homeostasis. Indeed, chloride is the main permeant ion of GABAA receptors (Bormann et al., 1987; Kaila and Voipio, 1987; Kaila et al., 1989; Kaila, 1994). A change in the driving force for Cl− currents, through a positive shift in ECl, would result in reduction in the amplitude of GABAA IPSPs. Because the trans-membrane chloride gradient is mainly determined by the action of two types of chloride pumps, potassium–chloride cotransporter (KCC) mediating chloride extrusion (Misgeld et al., 1986; Thompson et al., 1988) and Na-K-2Cl cotransporter (NKCC) mediating chloride uptake (Rohrbough and Spitzer, 1996; Kakazu et al., 1999), our results suggest that CORT affects chloride homeostasis by either inhibiting KCCs or by enhancing the activity of NKCCs.
The fact that we were able to observe a difference in GABAA reversal potential between CORT- versus vehicle-treated cells is inconsistent with the widespread belief that the composition of the ACSF and pipette solution completely determines ionic gradients in whole-cell recordings. With our solutions, the calculated chloride equilibrium potential was −61 mV, yet the GABAA reversal potential of vehicle-treated cell was approximately −75 for synaptically released GABA and −82 mV for responses evoked by isoguvacine. This is unlikely to reflect an incorrect estimation of the junction or membrane potentials because, in a previous study using the same solutions, we observed that the GABAA reversal potential of fast-spiking interneurons and principal BLA cells deviated from ECl, but in opposite directions. Moreover, gramicidin perforated-patch recordings yielded the same results (Martina et al., 2001). Thus, these observations suggest that, despite dialysis of the cells by the pipette solution, the intracellular chloride concentration is regulated by strong homeostatic mechanisms intrinsic to BLA neurons. Presumably, the rate of chloride diffusion from the pipette is insufficient to overwhelm KCCs.
Effects of CORT outside the amygdala
The CORT-induced increase in BLA neuronal excitability observed here contrasts with the effects of CORT in the hippocampus. Although low concentrations of CORT, which preferentially activate MRs, have excitatory effects on hippocampal neurons (for review, see Joels and de Kloet, 1992; de Kloet et al., 1999), high concentrations of CORT, which activate both MRs and GRs, have inhibitory effects in the hippocampus (for review, see Joels and de Kloet, 1992; de Kloet et al., 1999). For example, in CA1 pyramidal neurons, low and high doses of CORT, respectively, cause a decrease and an increase in spike-frequency adaptation (Joels and de Kloet, 1991). This contrasts with the effects of CORT in the BLA, which are excitatory at low and high concentrations. Nevertheless, consistent with our findings in vitro, a recent in vivo study showed that systemic CORT injection increases the spontaneous firing rate of BLA neurons (Kavushansky and Richter-Levin, 2006).
The differential effects of CORT in the hippocampus and the amygdala are consistent with the opposite effects of behavioral stress on activity-dependent synaptic plasticity in the two regions. For instance, stress blocks long-term potentiation (LTP) of Schaffer collaterals and perforant-path inputs in the hippocampus but enhances LTP of cortical afferents in the BLA (Alfarez et al., 2002; Yamada et al., 2003; Vouimba et al., 2004; Rodriguez Manzanares et al., 2005). Furthermore, whereas stress can lead to dendritic atrophy of hippocampal neurons, it increases dendritic branching in the amygdala (Vyas et al., 2002). Consistent with these findings, stress has been shown to impair memories that are dependent on the hippocampus but facilitate memories that are dependent on the amygdala (for review, see Kim et al., 2006).
Together, these findings indicate that CORT plays different roles in the hippocampus and the amygdala. Although the underlying mechanisms remain unclear, they may result from differences in the proportions of MRs to GRs in the two areas (Patel et al., 2001) and/or in the coupling of GRs to effector mechanisms.
Conclusions
Our results indicate that glucocorticoids enhance the excitability of principal BLA neurons via GRs. This increase in excitability involved several parallel mechanisms and was observed up to 2 h after CORT application. Together, our findings strengthen the view that glucocorticoids exert their memory-enhancing effects by increasing the activity of principal BLA neurons.
Footnotes
This work was supported by the National Science Foundation under Grant 0208712 and by National Institute of Mental Health Grant RO1 MH073610.
References
- Akirav I, Kozenicky M, Tal D, Sandi C, Venero C, Richter-Levin G. A facilitative role for corticosterone in the acquisition of a spatial task under moderate stress. Learn Mem. 2004;11:188–195. doi: 10.1101/lm.61704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfarez DN, Wiegert O, Joels M, Krugers HJ. Corticosterone and stress reduce synaptic potentiation in mouse hippocampal slices with mild stimulation. Neuroscience. 2002;115:1119–1126. doi: 10.1016/s0306-4522(02)00483-9. [DOI] [PubMed] [Google Scholar]
- Bormann J, Hamill OP, Sakmann B. Mechanism of anion permeation through channels gated by glycine and gamma-aminobutyric acid in mouse cultured spinal neurones. J Physiol (Lond) 1987;385:243–286. doi: 10.1113/jphysiol.1987.sp016493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson SA. Handbook of emotion and memory: current research and theory. Hillsdale, NJ: Erlbaum; 1992. [Google Scholar]
- Danober L, Pape HC. Mechanisms and functional significance of a slow inhibitory potential in neurons of the lateral amygdala. Eur J Neurosci. 1998;10:853–867. doi: 10.1046/j.1460-9568.1998.00092.x. [DOI] [PubMed] [Google Scholar]
- de Kloet ER, Reul JM, Sutanto W. Corticosteroids and the brain. J Steroid Biochem Mol Biol. 1990;37:387–394. doi: 10.1016/0960-0760(90)90489-8. [DOI] [PubMed] [Google Scholar]
- de Kloet ER, Oitzl MS, Joels M. Stress and cognition: are corticosteroids good or bad guys? Trends Neurosci. 1999;22:422–426. doi: 10.1016/s0166-2236(99)01438-1. [DOI] [PubMed] [Google Scholar]
- Dickinson-Anson H, Mesches MH, Coleman K, McGaugh JL. Bicuculline administered into the amygdala blocks benzodiazepine-induced amnesia. Behav Neural Biol. 1993;60:1–4. doi: 10.1016/0163-1047(93)90638-x. [DOI] [PubMed] [Google Scholar]
- Faber ES, Sah P. Physiological role of calcium-activated potassium currents in the rat lateral amygdala. J Neurosci. 2002;22:1618–1628. doi: 10.1523/JNEUROSCI.22-05-01618.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber ES, Sah P. Independent roles of calcium and voltage-dependent potassium currents in controlling spike frequency adaptation in lateral amygdala pyramidal neurons. Eur J Neurosci. 2005;22:1627–1635. doi: 10.1111/j.1460-9568.2005.04357.x. [DOI] [PubMed] [Google Scholar]
- Faber ES, Callister RJ, Sah P. Morphological and electrophysiological properties of principal neurons in the rat lateral amygdala in vitro. J Neurophysiol. 2001;85:714–723. doi: 10.1152/jn.2001.85.2.714. [DOI] [PubMed] [Google Scholar]
- Ferry B, McGaugh JL. Clenbuterol administration into the basolateral amygdala post-training enhances retention in an inhibitory avoidance task. Neurobiol Learn Mem. 1999;72:8–12. doi: 10.1006/nlme.1998.3904. [DOI] [PubMed] [Google Scholar]
- Hatfield T, McGaugh JL. Norepinephrine infused into the basolateral amygdala posttraining enhances retention in a spatial water maze task. Neurobiol Learn Mem. 1999;71:232–239. doi: 10.1006/nlme.1998.3875. [DOI] [PubMed] [Google Scholar]
- Hui GK, Figueroa IR, Poytress BS, Roozendaal B, McGaugh JL, Weinberger NM. Memory enhancement of classical fear conditioning by post-training injections of corticosterone in rats. Neurobiol Learn Mem. 2004;81:67–74. doi: 10.1016/j.nlm.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Joels M, de Kloet ER. Effect of corticosteroid hormones on electrical activity in rat hippocampus. J Steroid Biochem Mol Biol. 1991;40:83–86. doi: 10.1016/0960-0760(91)90170-a. [DOI] [PubMed] [Google Scholar]
- Joels M, de Kloet ER. Control of neuronal excitability by corticosteroid hormones. Trends Neurosci. 1992;15:25–30. doi: 10.1016/0166-2236(92)90345-9. [DOI] [PubMed] [Google Scholar]
- Kaila K. Ionic basis of GABAA receptor channel function in the nervous system. Prog Neurobiol. 1994;42:489–537. doi: 10.1016/0301-0082(94)90049-3. [DOI] [PubMed] [Google Scholar]
- Kaila K, Voipio J. Postsynaptic fall in intracellular pH induced by GABA-activated bicarbonate conductance. Nature. 1987;330:163–165. doi: 10.1038/330163a0. [DOI] [PubMed] [Google Scholar]
- Kaila K, Pasternack M, Saarikoski J, Voipio J. Influence of GABA-gated bicarbonate conductance on potential, current and intracellular chloride in crayfish muscle fibres. J Physiol (Lond) 1989;416:161–181. doi: 10.1113/jphysiol.1989.sp017755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakazu Y, Akaike N, Komiyama S, Nabekura J. Regulation of intracellular chloride by cotransporters in developing lateral superior olive neurons. J Neurosci. 1999;19:2843–2851. doi: 10.1523/JNEUROSCI.19-08-02843.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karst H, Karten YJ, Reichardt HM, de Kloet ER, Schutz G, Joels M. Corticosteroid actions in hippocampus require DNA binding of glucocorticoid receptor homodimers. Nat Neurosci. 2000;3:977–978. doi: 10.1038/79910. [DOI] [PubMed] [Google Scholar]
- Karst H, Nair S, Velzing E, Rumpff-van Essen L, Slagter E, Shinnick-Gallagher P, Joels M. Glucocorticoids alter calcium conductances and calcium channel subunit expression in basolateral amygdala neurons. Eur J Neurosci. 2002;16:1083–1089. doi: 10.1046/j.1460-9568.2002.02172.x. [DOI] [PubMed] [Google Scholar]
- Kavushansky A, Richter-Levin G. Effects of stress and corticosterone on activity and plasticity in the amygdala. J Neurosci Res. 2006;84:1580–1587. doi: 10.1002/jnr.21058. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Song EY, Kosten TA. Stress effects in the hippocampus: synaptic plasticity and memory. Stress. 2006;9:1–11. doi: 10.1080/10253890600678004. [DOI] [PubMed] [Google Scholar]
- Martina M, Royer S, Pare D. Cell-type-specific GABA responses and chloride homeostasis in the cortex and amygdala. J Neurophysiol. 2001;86:2887–2895. doi: 10.1152/jn.2001.86.6.2887. [DOI] [PubMed] [Google Scholar]
- McDonald AJ. Immunohistochemical identification of gamma-aminobutyric acid-containing neurons in the rat basolateral amygdala. Neurosci Lett. 1985;53:203–207. doi: 10.1016/0304-3940(85)90186-7. [DOI] [PubMed] [Google Scholar]
- McDonald AJ. Cell types and intrinsic connections of the amygdala. In: Aggleton JP, editor. The amydala: neurobiological aspects of emotion, memory, and mental dysfunction. New York: Wiley-Liss; 1992. pp. 69–76. [Google Scholar]
- McGaugh JL. Memory consolidation and the amygdala: a systems perspective. Trends Neurosci. 2002;25:456. doi: 10.1016/s0166-2236(02)02211-7. [DOI] [PubMed] [Google Scholar]
- McGaugh JL. The amygdala modulates the consolidation of memories of emotionally arousing experiences. Annu Rev Neurosci. 2004;27:1–28. doi: 10.1146/annurev.neuro.27.070203.144157. [DOI] [PubMed] [Google Scholar]
- McGaugh JL, Roozendaal B. Role of adrenal stress hormones in forming lasting memories in the brain. Curr Opin Neurobiol. 2002;12:205–210. doi: 10.1016/s0959-4388(02)00306-9. [DOI] [PubMed] [Google Scholar]
- Misgeld U, Deisz RA, Dodt HU, Lux HD. The role of chloride transport in postsynaptic inhibition of hippocampal neurons. Science. 1986;232:1413–1415. doi: 10.1126/science.2424084. [DOI] [PubMed] [Google Scholar]
- Morimoto M, Morita N, Ozawa H, Yokoyama K, Kawata M. Distribution of glucocorticoid receptor immunoreactivity and mRNA in the rat brain: an immunohistochemical and in situ hybridization study. Neurosci Res. 1996;26:235–269. doi: 10.1016/s0168-0102(96)01105-4. [DOI] [PubMed] [Google Scholar]
- Oitzl MS, Reichardt HM, Joels M, de Kloet ER. Point mutation in the mouse glucocorticoid receptor preventing DNA binding impairs spatial memory. Proc Natl Acad Sci USA. 2001;98:12790–12795. doi: 10.1073/pnas.231313998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packard MG, Cahill L, McGaugh JL. Amygdala modulation of hippocampal-dependent and caudate nucleus-dependent memory processes. Proc Natl Acad Sci USA. 1994;91:8477–8481. doi: 10.1073/pnas.91.18.8477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape HC, Driesang RB. Ionic mechanisms of intrinsic oscillations in neurons of the basolateral amygdaloid complex. J Neurophysiol. 1998;79:217–226. doi: 10.1152/jn.1998.79.1.217. [DOI] [PubMed] [Google Scholar]
- Pare D, Pape HC, Dong J. Bursting and oscillating neurons of the cat basolateral amygdaloid complex in vivo: electrophysiological properties and morphological features. J Neurophysiol. 1995;74:1179–1191. doi: 10.1152/jn.1995.74.3.1179. [DOI] [PubMed] [Google Scholar]
- Patel PD, Lopez JF, Lyons DM, Burke S, Wallace M, Schatzberg AF. Glucocorticoid and mineralocorticoid receptor mRNA expression in squirrel monkey brain. J Psychiatr Res. 2001;34:383–392. doi: 10.1016/s0022-3956(00)00035-2. [DOI] [PubMed] [Google Scholar]
- Pelletier JG, Likhtik E, Filali M, Pare D. Lasting increases in basolateral amygdala activity after emotional arousal: implications for facilitated consolidation of emotional memories. Learn Mem. 2005;12:96–102. doi: 10.1101/lm.88605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainnie DG, Asprodini EK, Shinnick-Gallagher P. Inhibitory transmission in the basolateral amygdala. J Neurophysiol. 1991;66:999–1009. doi: 10.1152/jn.1991.66.3.999. [DOI] [PubMed] [Google Scholar]
- Rainnie DG, Asprodini EK, Shinnick-Gallagher P. Intracellular recordings from morphologically identified neurons of the basolateral amygdala. J Neurophysiol. 1993;69:1350–1362. doi: 10.1152/jn.1993.69.4.1350. [DOI] [PubMed] [Google Scholar]
- Reichardt HM, Schutz G. Glucocorticoid signaling–multiple variations of a common theme. Mol Cell Endocrinol. 1998;146:1–6. doi: 10.1016/s0303-7207(98)00208-1. [DOI] [PubMed] [Google Scholar]
- Reul JM, de Kloet ER. Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology. 1985;117:2505–2511. doi: 10.1210/endo-117-6-2505. [DOI] [PubMed] [Google Scholar]
- Rodriguez Manzanares PA, Isoardi NA, Carrer HF, Molina VA. Previous stress facilitates fear memory, attenuates GABAergic inhibition, and increases synaptic plasticity in the rat basolateral amygdala. J Neurosci. 2005;25:8725–8734. doi: 10.1523/JNEUROSCI.2260-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrbough J, Spitzer NC. Regulation of intracellular Cl− levels by Na+-dependent Cl− cotransport distinguishes depolarizing from hyperpolarizing GABAA receptor-mediated responses in spinal neurons. J Neurosci. 1996;16:82–91. doi: 10.1523/JNEUROSCI.16-01-00082.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roozendaal B. Glucocorticoids and the regulation of memory consolidation. Psychoneuroendocrinology. 1999;25:213–238. doi: 10.1016/s0306-4530(99)00058-x. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, McGaugh JL. Glucocorticoid receptor agonist and antagonist administration into the basolateral but not central amygdala modulates memory storage. Neurobiol Learn Mem. 1997;67:176–179. doi: 10.1006/nlme.1996.3765. [DOI] [PubMed] [Google Scholar]
- Sandi C, Loscertales M, Guaza C. Experience-dependent facilitating effect of corticosterone on spatial memory formation in the water maze. Eur J Neurosci. 1997;9:637–642. doi: 10.1111/j.1460-9568.1997.tb01412.x. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM, Krey LC, McEwen BS. The neuroendocrinology of stress and aging: the glucocorticoid cascade hypothesis. Endocr Rev. 1986;7:284–301. doi: 10.1210/edrv-7-3-284. [DOI] [PubMed] [Google Scholar]
- Thompson SM, Deisz RA, Prince DA. Outward chloride/cation co-transport in mammalian cortical neurons. Neurosci Lett. 1988;89:49–54. doi: 10.1016/0304-3940(88)90479-x. [DOI] [PubMed] [Google Scholar]
- Verkuyl JM, Karst H, Joels M. GABAergic transmission in the rat paraventricular nucleus of the hypothalamus is suppressed by corticosterone and stress. Eur J Neurosci. 2005;21:113–121. doi: 10.1111/j.1460-9568.2004.03846.x. [DOI] [PubMed] [Google Scholar]
- Vouimba RM, Yaniv D, Diamond D, Richter-Levin G. Effects of inescapable stress on LTP in the amygdala versus the dentate gyrus of freely behaving rats. Eur J Neurosci. 2004;19:1887–1894. doi: 10.1111/j.1460-9568.2004.03294.x. [DOI] [PubMed] [Google Scholar]
- Vyas A, Mitra R, Shankaranarayana Rao BS, Chattarji S. Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J Neurosci. 2002;22:6810–6818. doi: 10.1523/JNEUROSCI.22-15-06810.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn MS, Moises HC. Electrophysiological and morphological properties of rat basolateral amygdaloid neurons in vitro. J Neurosci. 1992a;12:4066–4079. doi: 10.1523/JNEUROSCI.12-10-04066.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn MS, Moises HC. Inhibitory responses of rat basolateral amygdaloid neurons recorded in vitro. Neuroscience. 1992b;50:811–830. doi: 10.1016/0306-4522(92)90206-h. [DOI] [PubMed] [Google Scholar]
- Yamada K, McEwen BS, Pavlides C. Site and time dependent effects of acute stress on hippocampal long-term potentiation in freely behaving rats. Exp Brain Res. 2003;152:52–159. doi: 10.1007/s00221-003-1519-0. [DOI] [PubMed] [Google Scholar]