

Abstract
Increased brain deposition of amyloid β protein (Aβ) and cognitive deficits are classical signals of Alzheimer's disease (AD) that have been highly associated with inflammatory alterations. The present work was designed to determine the correlation between tumor necrosis factor-α (TNF-α)-related signaling pathways and inducible nitric oxide synthase (iNOS) expression in a mouse model of AD, by means of both in vivo and in vitro approaches. The intracerebroventricular injection of Aβ1–40 in mice resulted in marked deficits of learning and memory, according to assessment in the water maze paradigm. This cognition impairment seems to be related to synapse dysfunction and glial cell activation. The pharmacological blockage of either TNF-α or iNOS reduced the cognitive deficit evoked by Aβ1–40 in mice. Similar results were obtained in TNF-α receptor 1 and iNOS knock-out mice. Aβ1–40 administration induced an increase in TNF-α expression and oxidative alterations in prefrontal cortex and hippocampus. Likewise, Aβ1–40 led to activation of both JNK (c-Jun-NH2-terminal kinase)/c-Jun and nuclear factor-κB, resulting in iNOS upregulation in both brain structures. The anti-TNF-α antibody reduced all of the molecular and biochemical alterations promoted by Aβ1–40. These results provide new insights in mouse models of AD, revealing TNF-α and iNOS as central mediators of Aβ action. These pathways might be targeted for AD drug development.
Keywords: Alzheimer's disease, amyloid β, inflammation, TNF-α, iNOS, cognitive deficits
Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disorder associated with memory loss, spatial disorientation, and weakness of intellectual capacity. Histopathological studies of AD brain have revealed the presence of classical hallmarks including neurofibrillary tangles and senile plaques. The deposition of amyloid β protein (Aβ) in brain areas involved in cognitive functions is assumed to initiate a pathological cascade that results in synaptic dysfunction, synaptic loss, and neuronal death (Walsh and Selkoe, 2004).
Recent evidence suggests an inflammatory component in AD, which is characterized by astrogliosis, microgliosis, cytokine elevation, and changes in acute phase proteins (Walsh and Selkoe, 2004; Wyss-Coray, 2006). Tumor necrosis factor-α (TNF-α) is a cytokine thought to play a central role in the self-propagation of neuroinflammation (Perry et al., 2001). TNF-α regulates many cellular processes, including inflammation, differentiation, and cell death through activation of TNF receptor 1 (TNFR1) or TNFR2 (Wajant et al., 2003). The transduction pathways activated by TNF-α include mitogen-activated protein kinases (MAPKs) and IκB kinase, which control gene expression through transcriptional factors such as activator protein-1 (AP-1) and nuclear factor-κB (NF-κB) (Wajant et al., 2003). Regarding the CNS, microglia and astrocytes are believed to be the primary sources of TNF-α. Evidence indicates the presence of increased levels of TNF-α in the brain and plasma of AD patients and an upregulation of TNFR1 have been detected in the AD brain (Fillit et al., 1991; Li et al., 2004). In addition, TNF-α has been implicated recently as a critical mediator of long-term potentiation (LTP) reduction by Aβ (Wang et al., 2005). However, the mechanisms through which TNF-α promotes its pathological actions in AD are poorly understood.
Brain oxidative stress seems to exert an important role in cognitive impairment observed in AD. Inducible nitric oxide synthase (iNOS) generates nitric oxide (NO) and NO-derived reactive nitrogen species such as peroxynitrite. Accumulation of highly reactive molecules induces lipid peroxidation, tyrosine nitrosylation, DNA oxidative damage, and neuronal disruption, which are common characteristics of the AD brain (Butterfield et al., 2001). Interestingly, these events seem to be modulated by TNF-α. Also, Aβ has been shown to interact in a synergistic manner with cytokines to induce neuronal damage via reactive oxygen species (ROS)- and NO-dependent pathways (Goodwin et al., 1995; Meda et al., 1995).
Numerous animal models have been used to evaluate the role of inflammation in the course of AD. An experimental model that mimics the progression of AD was developed using an intracerebroventricular injection of Aβ in mice (for review, see Van Dam and De Deyn, 2006). Using this model, we assessed the role of TNF-α and iNOS in Aβ-induced early impairment of learning and memory. Our data indicate that TNF-α production is one of the earliest events induced by Aβ1–40, representing an important signal for iNOS expression. The cross talk between TNF-α and iNOS is probably mediated by activation of two major intracellular pathways: c-Jun-NH2-terminal kinase (JNK)/c-Jun and NF-κB. The present results implicate TNF-α and iNOS as important mediators of Aβ-induced cognitive impairment.
Materials and Methods
Subjects.
Experiments were conducted using male Swiss, C57BL/6, TNFR1 knock-out, and iNOS knock-out mice (20–30 g) kept in a controlled room temperature (22 ± 2°C) and humidity (60–80%) under a 12 h light/dark cycle (lights on 6:00 A.M.). TNFR1 (Rothe et al., 1993) and iNOS (MacMicking et al., 1995) knock-out mice are on the C57BL/6 background, constructed as described previously. All procedures used in the present study followed the “Principles of Laboratory Animal Care” from National Institutes of Health (NIH) publication number 85-23 and were approved by the Animal Ethics Committee of the Universidade Federal de Santa Catarina.
Drug treatment protocol.
Human Aβ1–40 (Tocris, Ellisville, MO) and Aβ40–1 (Bachem, Torrance, CA) were dissolved in sterile PBS, pH 7.4, at 1 mg/ml and were incubated at 37°C for 4 d, as described previously (El Khoury et al., 1996). Control mice received sterile PBS (vehicle). The aggregated form of amyloid fragments (400 pmol per mice) and vehicle solution were administered intracerebroventricularly as described previously (Laursen and Belknap, 1986). As a positive control for molecular studies, some animals received an intracerebroventricular injection of 2.5 μg of lipopolysaccharide (LPS) from Escherichia coli (serotype 0111:B4; Sigma-Aldrich, Sao Paulo, Brazil). Briefly, each mouse was given an injection at bregma with a 5 μl Hamilton microsyringe fitted with a 26 gauge needle that was inserted to a depth of 2.4 mm. The injection volume was 3 μl. Mice exhibited normal behavior within 1 min after injection. Accurate placement of the injection or needle track was verified at the time of dissection. The present Aβ1–40 dose was selected according to previous literature data (Yan et al., 2001).
Some animals were treated with anti-TNF-α antibody (AbTNF-α; 10 ηg, i.c.v., per mouse; R & D Systems, Minneapolis, MN) 15 min before Aβ1–40 injection. The AbTNF-α dose was determined in pilot experiments (results not shown). The iNOS inhibitor aminoguanidine (AG; 100 mg/kg, i.p.; Sigma-Aldrich) was administered 1 h before an intracerebroventricular injection of Aβ1–40 and throughout consecutive days until the day of the experiment. In addition, some animals were pretreated with the selective inhibitor of JNK, SP600126 (anthra[1-9-cd]pyrazol-6(2H)-one; 50 mg/kg; i.p.; 1 h before; Tocris), or the NF-κB blocker pyrrolidine dithiocarbamate (PDTC; 100 mg/kg, i.p., 1 h before; Sigma-Aldrich).
Water maze test–memory reference test.
The Morris water maze test was performed as described previously (Morris et al., 1982). The experimental apparatus consisted of a circular water tank (diameter, 97 cm; height, 60 cm) containing water at 23 ± 2°C. The target platform (10 × 10 cm) was submerged 1 cm below the water surface and placed at the midpoint of one quadrant. The platform was located in a fixed position, equidistant from the center and the wall of the pool. The pool was located in a test room containing various prominent visual cues. Mice were submitted to a spatial reference memory version of the water maze as described previously (Prediger et al., 2007). The acquisition training session was performed 7 d after Aβ1–40 injection and consisted of 10 consecutive trials during which the animals were left in the tank facing the wall and allowed to swim freely to the escape platform. If an animal did not find the platform within a period of 60 s, it was gently guided to it. The animal was allowed to remain on the platform for 10 s after escaping to it, and it was then removed from the tank for 20 s before being placed at the next starting point in the tank. This procedure was repeated 10 times, with the starting points varying in a pseudo-randomized manner. The test session was performed 24 h after the training session (on day 8 after injection). The test session consisted of a single probe trial in which the platform was removed from the pool and each mouse was allowed to swim for 60 s in the maze. The time spent in the correct quadrant (i.e., where the platform was located on the training session) was recorded, and the percentage of the total time was calculated.
Open-field test.
To discard the possible effects on Aβ1–40 in locomotor activity, the animals were tested in the open-field paradigm. The apparatus, made of wood covered with impermeable Formica, had a black floor of 30 × 30 cm (divided by white lines into nine squares of 10 × 10 cm) and transparent walls, 15 cm high. Each mouse was placed in the center of the open field, and the total number of squares crossed with the four paws and the rearing behavior were registered for 5 min.
Histology.
Eight days after intracerebroventricular injection of aggregated Aβ1–40, mice were perfused transcardially with PBS solution containing 4% paraformaldeyde (w/v). Brain was removed and kept overnight in the same solution. Serial sections (3 μm) were selected to include the lateral ventricle areas. Sections were stained with 0.5% cresyl violet reagent (Sigma-Aldrich) according to standard procedures. Stained cells were determined on visual inspection in the hippocampal CA1, CA2, and CA3 subfields and cortical layer with a microscope (Eclipse 50I; Nikon, Melville, NY) using a counting grid at 400× magnification. The light-microscopic characteristics of the Aβ1–40 aggregates were studied using the alkaline Congo red technique according to standard procedures.
Immunohistochemistry.
Immunohistochemistry detection of apoptotic cell death, immunoreactivity of the astrocyte marker glial fibrillary acidic protein (GFAP), and synaptic changes were assessed on paraffin tissue sections 1 and 8 d after Aβ1–40 intracerebroventricular injection, using the polyclonal rabbit anti-caspase-3 (1:200; Cell Signaling Technology, Beverly, MA), monoclonal mouse anti-GFAP (1:300; Dako Cytomation, Carpinteria, CA), and monoclonal mouse anti-synaptophysin (1:400; Novocastra, Newcastle, UK), respectively. High-temperature antigen retrieval was performed by immersion of the slides in a water bath at 95–98°C in 10 mm trisodium citrate buffer, pH 6.0, for 45 min. The nonspecific binding was blocked by incubating sections for 1 h with goat normal serum diluted in PBS. After overnight incubation at 4°C with primary antibodies, the slides were washed with PBS and incubated with the secondary antibody Envision plus (Dako Cytomation), ready to use, for 1 h at room temperature. The sections were washed in PBS, and the visualization was completed by using 3,3′-diaminobenzidine (Dako Cytomation) in chromogen solution and counterstained lightly with Harris's hematoxylin solution. Images were taken with a microscope (Eclipse 50i; Nikon) and digital sight camera (DS-5M-L1; Nikon). Control and experimental tissues were placed on the same slide and processed under the same conditions. Settings for image acquisition were identical for control and experimental tissues. For each mouse, we obtained four images (one per section) of the parietal cortex and three images (one per section) of the hippocampal CA1, CA2, and CA3 subregions. Digitized, 8-bit images were transferred to a computer, and the average pixel intensity of synaptophysisn staining was calculated for each image using NIH ImageJ 1.36b imaging software (NIH, Bethesda, MD). For each mouse, the values obtained for the parietal cortex or the hippocampal subregions were averaged. This approach for the assessment of synaptic degeneration has been validated in various experimental models of neurodegeneration (Buttini et al., 1999) and in diseased human brains (Masliah et al., 1992). GFAP- and caspase-3-positive cells were determined on visual inspection in the same brain areas using a counting grid at 400× magnification.
Enzymatic studies.
Mice were anesthetized with pentobarbital (50 mg/kg, i.p.) and perfused transcardially with ice-cold 0.9% NaCl (10 ml/10 g body weight) to remove the free radical-scavenging and radical-generating sources in the brain. The prefrontal cortex and hippocampus were carefully excised and stored at −70°C. Tissues were homogenized in 20 mm HEPES, pH 7.0, and centrifuged at 20,000 × g for 30 min at 4°C. The enzyme activities were determined in the supernatant in a Varian (Palo Alto, CA) Cary 50 spectrophotometer. Glutathione reductase (GR) and glutathione peroxidase (GPx) activity was determined as described previously (Prediger et al., 2007). Briefly, GR reduces oxidized glutathione (GSSG) to glutathione (GSH) in expending NADPH, the disappearance of which can be accompanied at 340 nm. GPx activity was measured indirectly by the NADPH consumption at 340 nm. The GPx uses GSH to reduce the tert-butylhydroperoxide-producing GSSG, which is readily reduced to GSH by excess GR, consuming NADPH. The disappearance of NADPH in this reaction reflects the GPx activity. The enzyme activities were expressed in milliunits per milligram of total protein content, which was quantified using the Bio-Rad (Hercules, CA) protein assay kit.
Determination of total GSH level.
The prefrontal cortex and hippocampus were isolated and homogenized in cooled 0.5 m perchloric acid. The homogenates were centrifuged at 15,000 × g for 2 min, and the supernatant was separated, neutralized in potassium phosphate buffer (0.1 m, pH 7.4), and subsequently submitted to the assay. Total GSH, comprising the total of reduced (GSH) and oxidized (GSSG) forms, was determined by the GR–5′,5′-dithio-bis(2-nitrobenzoic acid) recycling assay as described previously (Prediger et al., 2007).
Preparation of cytosolic and nuclear extracts.
Tissues were homogenized in ice-cold 10 mm HEPES, pH 7.4, containing 1.5 mm MgCl2, 10 mm KCl, 1 mm phenylmethylsulphonyl fluoride, 5 μg/ml leupeptin, 5 μg/ml pepstatin A, 10 μg/ml aprotinin, 1 mm sodium orthovanadate, 10 mm β-glycerophosphate, 50 mm sodium fluoride, and 0.5 mm dithiothreitol (all from Sigma-Aldrich). The homogenates were chilled on ice for 15 min and vigorously shaken for 15 min in the presence of 0.1% Triton X-100. The nuclear fraction was precipitated by centrifugation at 10,000 × g for 30 min. The supernatant containing the cytosolic fraction was stored at −70°C until use. The nuclear pellet was resuspended in 500 μl of high-salt extraction buffer (20 mm HEPES, pH 7.4, 420 mm NaCl, 1.5 mm MgCl2, 0.2 mm EDTA, 25% v/v glycerol, 1 mm phenylmethylsulphonyl fluoride, 5 μg/ml pepstatin A, 5 μg/ml leupeptin, 10 μg/ml aprotinin, and 0.5 mm dithiothreitol) and incubated under continuous shaking at 4°C for 30 min. The nuclear extract was then centrifuged for 30 min at 10,000 × g, and the supernatant was aliquoted and stored at −70°C. Protein concentration was determined using the Bio-Rad protein assay kit.
Western blot analysis.
Equal protein amounts were separated on an SDS-PAGE and transferred to a polyvinylidene fluoride membrane (Immobilon P; Millipore, Bedford, MA). The membranes were saturated by incubation with 10% nonfat dry milk solution and incubated overnight with one of the following antibodies: p65 NF-κB (sc-372, 1:1000), c-jun (sc-45, 1:1000), iNOS (sc-7271, 1:200), α-actin (sc-1615, 1:2000), JNK1 (sc-571, 1:1000), phosphorylated JNK (sc-6254, 1:1000) (all from Santa Cruz Biotechnology, Santa Cruz, CA), or lamin A/C (catalog #2032; Cell Signaling Technology). After washing, the membranes were incubated with adjusted secondary antibodies coupled to horseradish peroxidase or alkaline phosphatase. The immunocomplexes were visualized using the ECL chemiluminescence detection system (GE Healthcare, Sao Paulo, Brazil) or 5-bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium color development substrate (Promega, Madison, WI). Band density measurements were made using the Scion (Frederick, MD) Image software package.
RNA preparation and reverse transcription-PCR.
Total RNA was extracted from ∼100 mg of prefrontal cortex and hippocampus using Trizol reagent (Invitrogen, Sao Paulo, Brazil) according to the manufacturer's instructions. The concentration of total RNA was determined by measuring the absorbance at 260 nm in a spectrophotometer. The quality of the preparations was checked using gel electrophoresis. One microgram of total RNA was reverse transcribed using oligo(dT) and 200 U of reverse transcriptase (Invitrogen) in 20 μl of PCR buffer containing (in mm) 0.5 dNTP, 10 DTT, 2.5 MgCl2, 50 KCl, and 20 Tris-HCl, pH 8.4. The samples were incubated for 5 min at 70°C, 5 min at 4°C, 1 h at 37°C, 5 min at 70°C, and 5 min at 4°C. Specific primers (Invitrogen) were used for TNF-α (sense, TCTCATCAGTTCTATGGCCC; antisense, GGGAGTAGACAAGGTACAAC), iNOS (sense, CAGAAGCAGAATGTGACCATC; antisense, CTTCTGGTCGATGTCATGA), and β-actin (sense, TCCTTCGTTGCCGGTCCACA; antisense, CGTCTCCGGAGTCCATCACA). β-Actin cDNA was used for standardization of the amount of RNA. Five microliters of reverse transcription (RT) aliquots were mixed in a 20 mm Tris-HCl buffer, pH 8.4, containing 1.5 mm MgCl2, 300 μm dNTP, 2 μg/ml of each primer, and 50 U/ml Taq polymerase (Invitrogen) in a final volume of 100 μl. The following cycling protocol was used: initial denaturation, 95°C for 4 min; cycling: 95°C for 30 s, 53°C for 30 s (TNF-α and iNOS) or 62°C for 30 s (β-actin), and 72°C for 1 min; and a final extension period at 72°C for 5 min. Optimal amplification was achieved at 29 cycles for TNF-α, iNOS, and β-actin. Aliquots of 25 μl were analyzed by PAGE and stained with silver salts. Band density measurements for TNF-α, iNOS, and β-actin mRNAs were made using the Scion Image software package.
Software.
All Western blot and RT-PCR experiments were scanned to acquire digital images using a Genius ColorPage Scanner (KYE Systems, Taipei, Taiwan). Digital images were processed with the Photoshop software package (Adobe Systems, Mountain View, CA), complying with strict standards (Rossner and Yamada, 2004).
Statistical analysis.
All values are expressed as means ± SEM (n is the number of mice included in each analysis). Differences between groups in total GSH levels and GR and GPx activities were analyzed using an unpaired Student's t test. Statistical analysis for the rest of the data were performed using two- or three-way ANOVA with strain (or pretreatment), treatment, or the number of trials (repeated measure) as independent variables. After significant ANOVAs, multiple post hoc comparisons were performed using the Newman–Keuls test. The accepted level of significance for the tests was p < 0.05. All tests were performed using the Statistica software package (StatSoft, Tulsa, OK).
Results
The role of TNF-α and iNOS in Aβ-induced learning and memory impairment
Learning and memory functions are vulnerable to several pathological processes, including AD (Budson and Price, 2005). To investigate some of the mechanisms involved in Aβ-induced cognitive decline, the Morris water maze task was used. We tested the ability of mice to acquire (training session) and retrieve (test session) spatial information as indicative of learning and memory functions. As expected, intracerebroventricular injection of Aβ1–40 in Swiss mice resulted in a significant decline in both learning and memory, as indicated by longer latencies (Fig. 1A) and reduced target quadrant preference (Fig. 1B) during the probe trial. No cognitive deficits were observed in Swiss mice treated with the inverse Aβ40–1 (Fig. 1). The effects of Aβ1–40 administration on water maze performance are not directly related to motor impairment, because no alterations of the swimming speed in the water maze (data not shown), or the total squares crossed and rearing behavior in the open-field arena, were observed (Table 1).
Figure 1.

Intracerebroventricular injection of Aβ1–40 disrupts learning and memory in mice. The spatial reference memory version of the Morris water maze test in Swiss mice (n = 9–10 mice per group) was used as a measure of cognition. A, Training trials were performed on day 7 after a single intracerebroventricular administration of aggregated Aβ1–40 (400 pmol per mouse), inverse peptide Aβ40–1 (400 pmol per mouse) (used as a negative control), or vehicle (PBS). Treatment with Aβ1–40 significantly increased the escape latencies to find the submerged platform during the training trials (F(2,26) = 104.32; p < 0.0001). B, The probe test session was performed 24 h after the training trials. Treatment with Aβ1–40 reduced the frequency of time spent in the correct quadrant (F(2,26) = 19.91; p < 0.0001). The values represent the mean ± SEM. *p < 0.05 compared with the vehicle-treated group (Newman–Keuls test).
Table 1.
Intracerebroventricular (i.c.v.) injection of Aβ1–40did not affect motor behavior in mice
| Treatment (i.c.v.) | Squares crossing | Rearing |
|---|---|---|
| Vehicle | 78.0 ± 4.6 | 32.5 ± 2.9 |
| Aβ1–40 | 78.6 ± 4.3 | 34.3 ± 3.2 |
| Aβ40–1 | 75.7 ± 2.4 | 27.3 ± 1.9 |
Data are expressed as the mean ± SEM of the total squares crossed and rearing (n = 9–10 animals in each group). Aβ1–40treatment did not significantly alter the number of squares crossed (F(2,26) = 0.14; p = 0.86) and rearing (F(2,26) = 1.49; p = 0.24) in the open-field arena. Total squares crossed and rearing by Swiss mice in the open-field arena (for 5 min) was used as a measure of locomotor activity. Experiments were performed on day 8 after a single intracerebroventricular administration of aggregated Aβ1–40(400 pmol per mouse), the inverse peptide Aβ40–1(400 pmol per mouse; used as a negative control), or vehicle (PBS).
To verify whether TNF-α and iNOS are required for Aβ1–40-induced cognitive decline, pharmacological and genetic approaches were applied. Swiss mice were treated with the specific antibody against mouse TNF-α (AbTNF-α), 15 min before Aβ1–40, or the preferential iNOS inhibitor AG, 1 h before Aβ1–40, and throughout 7 consecutive days, until the test day (day 8 after injection). As shown in Figure 2, A and E, both pharmacological treatments resulted in a significant improvement of spatial learning deficits induced by Aβ1–40, with a significant reduction in the final escape latency to find the platform in the training session. Additionally, the statistical analysis of the probe test scores revealed that either AbTNF-α or AG significantly prevented the memory impairment induced by Aβ1–40, increasing the time spent in the correct quadrant (Fig. 2B,F). Genetic deletion of TNFR1 or iNOS also significantly attenuated the spatial learning deficits induced by Aβ1–40, as indicated by a reduction in the final escape latency to find the platform when compared with wild-type (C57BL/6) Aβ-treated mice (Fig. 2C,G). In the test session, TNFR1−/− and iNOS−/− mice given injections of Aβ1–40 presented higher swimming scores in the correct quadrant compared with wild-type Aβ1–40-treated mice, indicating a diminished sensitivity to memory deficits (Fig. 2D,H).
Figure 2.
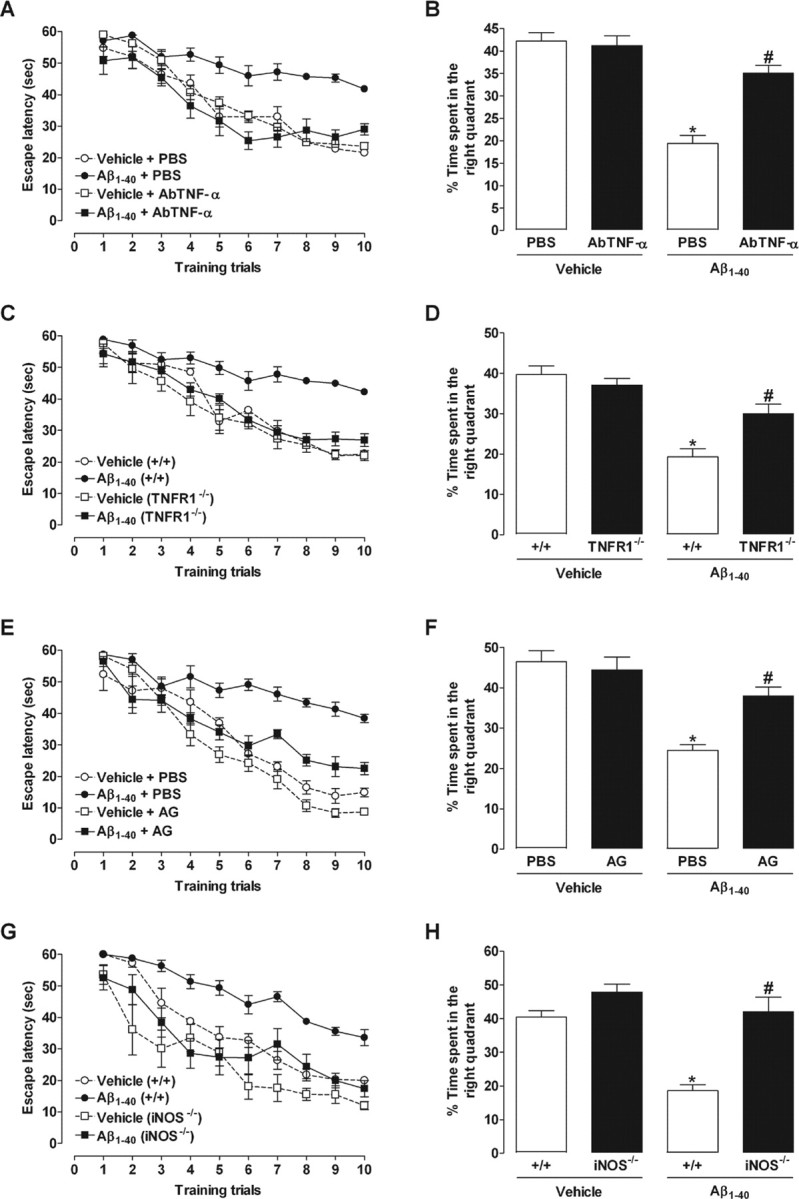
Involvement of TNF-α and iNOS in the cognitive deficits induced by Aβ1–40 in mice. The spatial reference memory version of the Morris water maze test in mice was used as a measure of cognition. Training trials were performed on day 7 after a single intracerebroventricular administration of aggregated Aβ1–40 (400 pmol per mouse) or vehicle (PBS). Data are presented as means ± SEM latency (seconds) for escape to a submerged platform (n = 8–10 mice per group). The probe test session was performed 24 h after training trials. Data are presented as means ± SEM of the frequency of time spent in the correct quadrant. A, B, E, F, Pretreatment with the specific antibody against mouse TNF-α (AbTNF-α; 10 ηg, i.c.v., per mouse) (A, B) or the preferential iNOS inhibitor AG (100 mg/kg, i.p., once per day) (E, F) improved the cognitive deficits induced by Aβ1–40 in Swiss mice during training trials (AbTNF-α: F(1,27) = 21.89, p < 0.0001; AG: F(1,33) = 82.78, p < 0.0001) and test sessions (AbTNF-α: F(1,27) = 5.26, p < 0.05; AG: F(1,33) = 29.14, p < 0.0001) of the Morris water maze. C, D, G, H, TNFR1 knock-out (TNFR1−/−; C, D) and iNOS knock-out (iNOS−/−; G, H) mice were significantly more resistant than wild-type C57BL/6 mice to the deleterious effect of Aβ1–40 in the spatial learning (TNFR1−/−: F(1,30) = 29.58, p < 0.0001; iNOS−/−: F(1,26) = 81.46, p < 0.0001) and spatial retrieval (TNFR1−/−: F(1,30) = 15.45, p < 0.001; iNOS−/−: F(1,26) = 28.46, p < 0.0001). *p < 0.05 compared with the vehicle/PBS-treated group; #p < 0.05 compared with the Aβ1–40/PBS-treated group (Newman–Keuls test).
Prevention of synaptic dysfunction by TNF-α and iNOS inhibition
To evaluate the possible amyloid deposits and/or neuronal damage after Aβ1–40 intracerebroventricular injection, we performed histological and immunohistochemistry analysis. Histological examination was performed on cresyl violet- and on Congo red-stained sections of mouse brain, 8 d after the intracerebroventricular administrations. Cresyl violet-stained sections indicated that injection by itself failed to produce any significant neuronal damage in control animals, at or distant from the injection site. The administration of Aβ1–40 peptide also failed to produce any neuronal loss in the hippocampus or cortex of treated mice (data not shown). Using Congo red staining, control brain sections or brain sections of animals treated with Aβ1–40 showed no evidence of amyloid deposits (data not shown).
To further evaluate the neuronal integrity, we performed immunohistochemistry analysis for the presynaptic protein synaptophysin and the pro-apoptotic protein caspase-3 in the parietal cortex and the hippocampal CA1, CA2, and CA3 subregions 1 and 8 d after Aβ1–40 intracerebroventricular injection. No modification in the synaptophysin levels was found 1 d after Aβ1–40 treatment (results not shown). However, a significant reduction in the synaptophysin levels was found in Aβ1–40-treated mice 8 d after intracerebroventricular injection, suggesting a decrease in synaptic density (Fig. 3 and supplemental Fig. 1, available at www.jneurosci.org as supplemental material). On the other hand, no alteration was observed for caspase-3 protein immunostaining at day 1 or 8, indicating that neuronal apoptosis was not activated, at least not up to 8 d after Aβ1–40 treatment (data not shown). Also, either AbTNF-α or AG (Fig. 3A–D) significantly prevented Aβ1–40-induced decrease in synaptophysin levels 8 d after Aβ1–40 treatment. Consistent with these findings, comparable results were seen in TNFR1−/− and iNOS−/− (Fig. 3A–D) mice. Extending previous data (Walsh and Selkoe, 2004), our results indicated that synaptic dysfunction precedes neuronal death in the AD brain through a mechanism that is dependent on TNF-α and iNOS.
Figure 3.
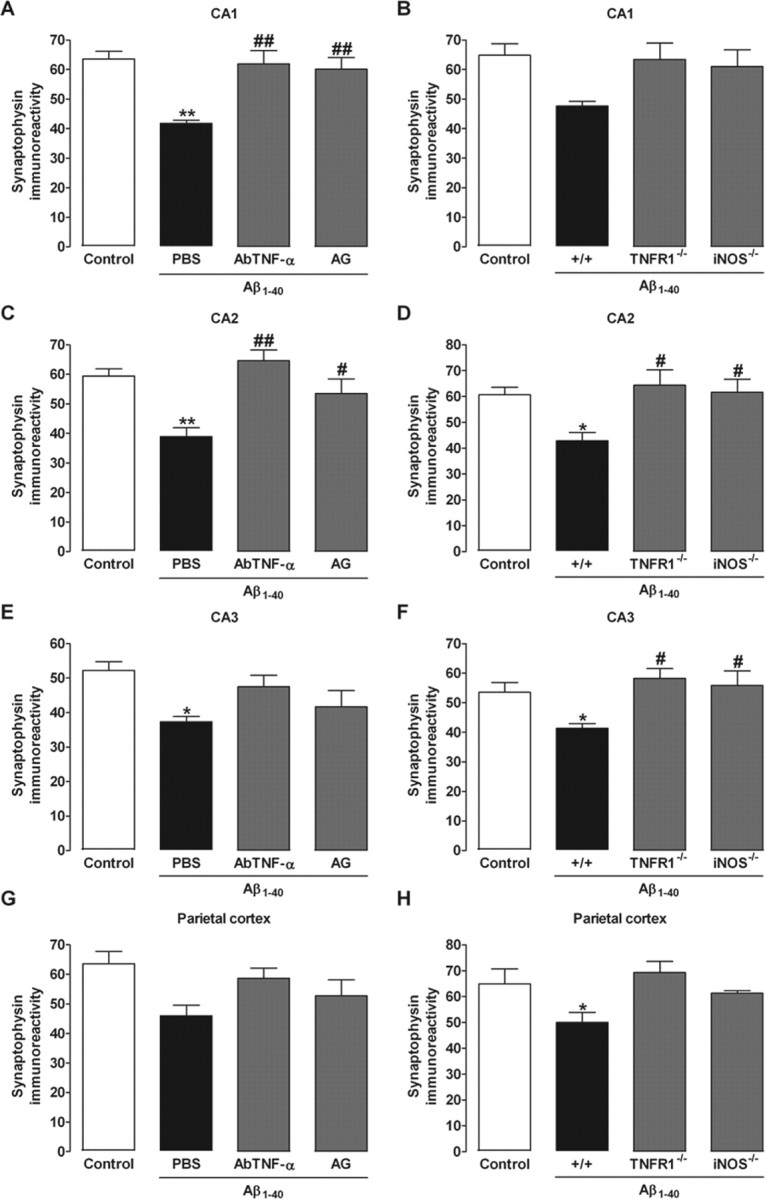
TNF-α and iNOS participate in Aβ1–40-induced synaptic disruption. Immunohistochemistry analysis for the presynaptic protein synaptophysin was performed on day 8 after aggregated Aβ1–40 (400 pmol per mouse) intracerebroventricular injection. A–H, Relative optical densitometry of synaptophysin immunostaining in the CA1 (A, B), CA2 (C, D), and CA3 (E, F) subregions of the hippocampus and parietal cortex (G, H). Synaptophysin immunoreactivity was used as a measure of synaptic density. A, C, E, G, Pretreatment with the specific antibody against mouse TNF-α (AbTNF-α; 10 ηg, i.c.v., per mouse) or with the preferential iNOS inhibitor AG (100 mg/kg, i.p., once per day) prevented the Aβ1–40-induced synaptic disruption in Swiss mice in the CA1 (F(3,8) = 9.38; p < 0.01; A) and CA2 (F(3,8) = 9.44; p < 0.01; C), but not in the CA3 (F(3,8) = 4.02; p < 0.05; E) and parietal cortex (F(3,8) = 3.26; p = 0.08; G). B, D, F, H, TNFR1 knock-out (TNFR1−/−) and iNOS knock-out (iNOS−/−) mice were significantly more resistant than wild-type C57BL/6 mice to the Aβ1–40-induced synaptic disruption in the CA2 (F(3,8) = 4.79; p < 0.05; D) and CA3 (F(3,8) = 4.63; p < 0.05; F), but not in the CA1 (F(3,8) = 3.17; p = 0.08; B) and parietal cortex (F(3,8) = 4.00; p < 0.05; H). The values represent the mean ± SEM. *p < 0.05, **p < 0.01 compared with the control group (naive); #p < 0.05, ##p < 0.01 compared with the Aβ1–40/PBS-treated group (Newman–Keuls test).
Aβ-induced astrocyte activation
Astrogliosis is one of the earliest pathological hallmarks of AD and may occur in response to the increasing number of degenerating neurons and synapses, or to the accumulation of Aβ. The injection of Aβ1–40 induced an increase in the astrocyte activation at day 1 after Aβ1–40 intracerebroventricular injection, mainly in the hippocampus, as demonstrated by a rise in the number of GFAP-positive cells (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). No significant alteration in the GFAP immunoreactivity was found in the parietal cortex, whereas the GFAP detection was reduced 8 d after Aβ1–40 treatment in the hippocampus (data not shown). The involvement of TNF-α and iNOS for the activation of astrocytes was also verified. Results show that treatment with AbTNF-α or AG failed to reduce the immunoreactivity to GFAP in Aβ-injected mice 1 d after treatment (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). These data were also confirmed by experiments using knock-out mice. TNFR1−/− or iNOS−/− mice failed to prevent GFAP immunodetection compared with wild-type (C57BL/6) Aβ-treated mice (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). These data indicate that astrogliosis present a role in the initial inflammatory response induced by Aβ1–40 in the AD model. In addition, it suggests that astrocyte activation may be involved in initial TNF-α release. However, additional studies are necessary to confirm this hypothesis.
Aβ stimulates iNOS protein expression through TNF-α production
Next, we attempted to outline the temporal sequence of intracellular events after Aβ1–40 intracerebroventricular injection. The expression of TNF-α and iNOS was assessed in the hippocampus and prefrontal cortex at different time points after Aβ1–40 injection using RT-PCR and/or Western blot techniques. Under basal conditions, very low levels of TNF-α mRNA were observed in the hippocampus or prefrontal cortex. Conversely, a rapid onset and time-dependent induction of TNF-α mRNA was observed in Aβ1–40-treated mice (Fig. 4A,B). The increase in TNF-α mRNA levels was detected after 15 min of Aβ1–40 treatment, remaining elevated through 24 h, and it was found to be diminished after 7 d in both brain structures. Similarly, the time-course analysis revealed that iNOS mRNA expression was minimal under basal conditions but increased in a time-dependent manner, peaking at 1–3 h and being reduced 24 h after Aβ1–40 treatment (Fig. 4A,B). Moreover, Western blot analysis indicated that Aβ1–40 intracerebroventricular injection led to a sustained iNOS protein expression in both brain regions. As seen in Figure 4, C and D, little alteration of iNOS expression was verified up to 60 min, but a marked increase was demonstrated at 3 h, remaining elevated for at least 7 d. These time-related changes might indicate a correlation between TNF-α mRNA induction and iNOS mRNA/protein expression (Fig. 4E,F). Thus, we examined the effects of TNF-α signaling blockage on Aβ1–40-induced iNOS expression. The treatment with AbTNF-α almost completely inhibited iNOS protein expression, without affecting the control protein α-actin (Fig. 4C,D). Last, as a control for the Aβ1–40, we treated mice with the reverse peptide Aβ40–1 and found that it had no significant effect on the TNF-α mRNA (results not shown) or iNOS expression (Fig. 4C,D). In our model, TNF-α production appears to be one of the earliest events induced by Aβ, representing a signal for iNOS protein expression.
Figure 4.

Requirement of TNF-α for Aβ1–40-induced iNOS upregulation. Swiss mice were treated with aggregated Aβ1–40 (400 pmol, i.c.v., per mouse) [except naive, untreated mice (N)], and the hippocampus and prefrontal cortex were isolated at the time points indicated. A, B, Total RNA was isolated from the hippocampus (A) and prefrontal cortex (B) for evaluation of TNF-α and iNOS expression by RT-PCR. β-Actin mRNA was assessed in all RNA samples as an internal control for the amount of RNA in each sample. TNF-α and iNOS mRNA increased in a time-dependent manner after Aβ1–40 intracerebroventricular treatment, whereas β-actin levels remained constant. C, D, Aβ1–40 induced a time-dependent and prolonged iNOS protein expression in the hippocampus (C) and prefrontal cortex (D). Western blot analysis revealed that pretreatment with the specific antibody against mouse TNF-α (AbTNF-α; 10 ηg, i.c.v., per mouse) prevented Aβ1–40-induced iNOS expression in the hippocampus (F(1,36) = 118.26; p < 0.0001) and prefrontal cortex (F(1,36) = 87.69; p < 0.0001). Immunoblot for α-actin was used as a cytosolic loading control. Treatment with the reverse peptide Aβ40–1 (6 h) had no effect on the iNOS protein expression. As a positive control, LPS (2.5 μg, i.c.v., 12 h) treatment induced a marked iNOS protein expression. E, F, Graph illustrating the temporal profile of the TNF-α and iNOS mRNA synthesis and iNOS protein expression in the hippocampus (E) and prefrontal cortex (F). Results were normalized by arbitrarily setting the densitometry from the maximal responsive group and are expressed as the mean of three to four independent experiments.
Oxidative stress induced by Aβ depends on TNF-α
Previous evidence suggests a causal linkage among Aβ, iNOS expression, and cellular redox state. NO generated by iNOS reacts with superoxide anion (O2−) producing peroxynitrite (ONOO−) that exerts cytotoxic effects (Beckman et al., 1990). Thus, it is reasonable to expect that Aβ1–40-induced iNOS expression is accompanied by oxidative alterations, in a TNF-α-dependent process. Although quantification of NO is more related with our data, it is well accepted that GSH parameters are associated with cellular oxidative state, and this might be indirectly modified by augmentation in the NO production. Hence, we evaluated the GSH status and GSH-dependent enzyme activities after Aβ1–40 treatment. As shown in supplemental Figure 3 (available at www.jneurosci.org as supplemental material), total GSH levels were reduced on days 1 and 7 after intracerebroventricular injection in the hippocampus and prefrontal cortex of Aβ1–40-treated Swiss mice, respectively. In addition, GR and GPx activities were significantly increased by Aβ1–40 in both brain structures on day 1 after injection, returning to basal values after 7 d (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). As expected, AbTNF-α treatment reverted all the oxidative alterations induced by Aβ1–40 in the hippocampus and prefrontal cortex (Fig. 5). These results establish TNF-α as a critical modulator of the oxidative responses induced by Aβ.
Figure 5.

Blockade of TNF-α prevents alterations in GSH-dependent antioxidant parameters induced by Aβ1–40. Swiss mice were treated with a specific antibody against mouse TNF-α (AbTNF-α; 10 ηg, i.c.v., per mouse) 15 min before the administration of the aggregated Aβ1–40 (400 pmol per mouse) or vehicle (PBS). A–F, Total GSH levels (GSH total; A, B) and antioxidant enzymes, GR (C, D), and GPx (E, F) were measured in the hippocampus and prefrontal cortex of Swiss mice at 24 h after Aβ1–40 administration. The values represent the mean ± SEM of GSH total levels (micromoles per gram of wet tissue) and GR and GPx activities (milliunits per milligram of protein) (n = 5–7 mice per group). A, B, Treatment with AbTNF-α prevented the reduction in GSH total levels in the hippocampus (F(3,16) = 18.04; p < 0.0001; A) and prefrontal cortex (B) of Aβ1–40-treated mice. C–F, AbTNF-α treatment prevented the increase in GR and GPx activities in the hippocampus (C, E) (F(3,16) = 2.05, p < 0.05 and F(3,16) = 5.13, p < 0.001, respectively) and (D, F) prefrontal cortex (F(3,16) = 5.68, p < 0.01 and F(3,16) = 4.17, p < 0.05, respectively) induced by Aβ1–40 injection. The values represent the mean ± SEM. *p < 0.05 compared with the vehicle/PBS-treated group; #p < 0.05 compared with the Aβ1–40/PBS-treated group (Newman–Keuls test).
JNK/c-Jun pathway involvement in Aβ-induced iNOS expression
Recent evidence suggests that JNK and its target c-Jun play an important role in AD neural dysfunction (Bozyczko-Coyne et al., 2001; Morishima et al., 2001; Troy et al., 2001). We next tested the possible requirement of JNK and c-Jun for Aβ-mediated effects in our AD paradigm. As shown in Figure 6, A and B, a low basal level of activated JNK was detected in the hippocampus and prefrontal cortex. However, an evident increase in JNK phosphorylation was found between 15 and 30 min. This reached a peak at 6 h and lasted for up to 7 d after Aβ1–40 treatment in both brain regions. The increase in JNK activation was accompanied by a rise in c-Jun translocation to the nucleus (Fig. 6C,D), which suggests a possible association between these events. To confirm this, mice were treated with the JNK inhibitor SP600125 (1 h before Aβ1–40), and the hippocampus and prefrontal cortex were isolated for Western blot analysis. As expected, nuclear c-Jun (Fig. 6E,F) was found to be reduced in both brain structures of SP600125-treated mice.
Figure 6.

Aβ1–40-induced JNK/c-Jun pathway activation. A, B, Activation of cytoplasmic JNK in response to aggregate Aβ1–40 intracerebroventricular injection was detected in the hippocampus (A) and prefrontal cortex (B) of Swiss mice, at the time points indicated, by phosphospecific antibody for diphosphorylated JNK (p-JNK; at Thr183 and Tyr185). Blots were reprobed with anti-JNK to verify equal loading. C, D, Kinetics of c-Jun protein translocation from cytoplasm into the nucleus in the hippocampus (C) and prefrontal cortex (D) after Aβ1–40 administration. Activation of the JNK pathway is dependent on TNF-α. Pretreatment with the specific antibody against mouse TNF-α (AbTNF-α; 10 ηg, i.c.v., per mouse) prevented Aβ1–40-induced JNK activation and subsequent c-Jun translocation in the hippocampus and prefrontal cortex. Immunoblot for lamin A/C was used as a nuclear loading control. N, Naïve, untreated mice. Treatment with the reverse peptide Aβ40–1 (6 h) had no significant effect on the JNK/c-Jun activation. As a positive control, LPS (2.5 μg, i.c.v., 1 h) treatment induced activation in the JNK/c-Jun pathway. E, F, Pretreatment with JNK inhibitor SP600125 (25 mg/kg, i.p., 1 h before Aβ), but not with NF-κB inhibitor PDTC (100 mg/kg, i.p., 1 h before Aβ), prevented Aβ1–40-induced c-Jun translocation in the hippocampal (E) and cortical (F) homogenates. Brain samples were collected 6 h after Aβ1–40 intracerebroventricular injection. These data indicate an association between JNK and c-Jun proteins.(C), Cytoplasmatic; (N), nuclear.
TNF-α signaling effectors include JNK and c-Jun that might also contribute to iNOS promoter activity (Eberhardt et al., 1996). Pretreatment with AbTNF-α completely blocked Aβ1–40-caused JNK phosphorylation (Fig. 6A,B) and c-Jun nuclear translocation (Fig. 6C,D) in the hippocampus and prefrontal cortex. Moreover, the Western blot analysis applied to brain samples of SP600125-treated mice revealed a partial reduction in the iNOS expression (see Fig. 8). These data indicate a sequence of events subsequent to Aβ1–40 intracerebroventricular injection that involves TNF-α production, JNK phosphorylation, c-Jun translocation, and iNOS expression.
Figure 8.

Aβ1–40-induced expression of iNOS is dependent on JNK/c-Jun and NF-κB activation. Involvement of JNK/c-Jun and NF-κB signaling pathways on iNOS expression was verified in the hippocampus (A) and prefrontal cortex (B) 24 h after Aβ1–40 intracerebroventricular administration. Top, Pretreatment with the selective inhibitor of JNK, SP600126 (50 mg/kg, i.p.), or NF-κB blocker PDTC (100 mg/kg, i.p.) 1 h before Aβ1–40 treatment reduced iNOS expression in the hippocampus (F(3,8) = 40.00; p < 0.0001) and prefrontal cortex (F(3,8) = 12.68; p < 0.01). Bottom, Graphs showing quantification of iNOS protein normalized by α-actin protein (loading control). The values represent the mean ± SEM. **p < 0.01 compared with the vehicle-treated group; #p < 0.05, ##p < 0.01 compared with the Aβ1–40-treated group (Newman–Keuls test).
TNF-α regulates Aβ-induced iNOS expression via NF-κB activation
In resting cells, NF-κB dimmers are found in the cytoplasm associated with IκB proteins. Pathological conditions (including AD) result in NF-κB translocation to the nucleus where it regulates the transcription of several genes (Ghosh and Karin, 2002). To analyze the possible involvement of NF-κB in Aβ1–40-induced iNOS expression, we first assessed the temporal profile of NF-κB activation after Aβ1–40 treatment. Immunodetection of p65 subunit performed in the cytosolic and nuclear homogenates from hippocampus and prefrontal cortex confirmed the activation of NF-κB. As shown in Figure 7, A and B, p65 was found to be increased in nuclear extracts after 15 and 30 min of Aβ1–40 treatment, respectively. After 7 d, nuclear p65 levels returned toward baseline in both brain areas. Next, we determined to what extent NF-κB activation caused by Aβ1–40 was dependent on the TNF-α. The AbTNF-α almost completely blocked the translocation of NF-κB induced by Aβ1–40 (Fig. 7A,B). To further verify how NF-κB activation could participate in the iNOS expression induced by Aβ1–40, Swiss mice were treated with the NF-κB inhibitor PDTC, at a dose that was sufficiently effective to prevent p65 translocation to the nucleus (Fig. 7C,D). PDTC was given intraperitoneally 60 min before Aβ1–40 injection, and the hippocampus and the prefrontal cortex were isolated 24 h later for Western blot analysis. Data in Figure 8, A and B, show that PDTC-treated mice presented a significant reduction in Aβ-induced iNOS expression in both brain regions. Collectively, these results suggest that Aβ1–40-induced iNOS expression is dependent, at least in part, on TNF-α-driven NF-κB activation.
Figure 7.

Aβ1–40-induced NF-κB pathway activation. A, B, Cytoplasmic [(C)] and nuclear [(N)] samples of hippocampus (A) and prefrontal cortex (B) from Swiss mice were probed with a p65 NF-κB antibody at the time points indicated. Data indicate that Aβ1–40 induced a translocation of p65 NF-κB from the cytoplasm to the nucleus. Pretreatment with the specific antibody against mouse TNF-α (AbTNF-α; 10 ηg, i.c.v., per mouse) prevented Aβ1–40 induction of p65 NF-κB translocation in hippocampus and prefrontal cortex. Immunoblot for lamin A/C was used as a nuclear loading control. Treatment with the reverse peptide Aβ40–1 (6 h) had no significant effect on the p65 NF-κB translocation. As a positive control, LPS (2.5 μg, i.c.v., 1 h) treatment resulted in p65 NF-κB migration into the nucleus. C, D, Aβ1–40-induced p65 NF-κB translocation was prevented by pretreatment with NF-κB inhibitor PDTC (100 mg/kg, i.p.), but not by pretreatment with JNK inhibitor SP600125 (25 mg/kg, i.p.), in hippocampus (C) and prefrontal cortex (D). These data indicate that JNK/c-Jun and NF-κB pathways are independently activated by Aβ1–40 intracerebroventricular injection. N, Naïve, untreated mice; C, control.
Discussion
Evidence suggests that inflammatory reaction induced by Aβ in the CNS involves the release of damaging factors, such as cytokines, NO, and ROS, that promote the activation of intracellular pathways, contributing to the progression of AD (Walsh and Selkoe, 2004; Wyss-Coray, 2006). In the present study, we demonstrate that a single intracerebroventricular injection of Aβ1–40, at a picomolar dose, induces a neuroinflammatory response that is linked to a decline in learning and memory functions. Present results are suggestive of glial activation and synaptic loss after Aβ1–40 injection; however, assessment of apoptotic protein caspase-3 and cresyl violet staining indicated that programmed cell death seems not likely involved. Of high interest, the cognitive decline found after Aβ1–40 treatment appears to be related to the synaptic loss. This supports the idea that synaptic dysfunction induced by Aβ is the primary marker of AD and precedes neuronal death (Walsh and Selkoe, 2004). In addition, by use of pharmacological and molecular tools, we have shown a connecting link between Aβ, TNF-α, iNOS, oxidative stress, and the cognitive decline. Our results suggest that TNF-α, probably through TNFR1 activation, exerts an essential role in Aβ-mediated iNOS expression and oxidative alterations, culminating in synaptic failure and, consequently, in cognitive impairment observed in the current AD model.
TNF-α is a multifunctional cytokine that triggers a wide range of cellular responses. In the CNS, TNF-α (through activation of TNFR1) disrupts learning and memory and regulates neuronal death. TNF-α has also been shown to participate in Aβ-induced inhibition of LTP, a form of synaptic plasticity closely associated with learning and memory, and it is probably dependent on mGluR5 (metabotropic glutamate receptor 5) and p38 MAPK (Wang et al., 2005). Moreover, the activation of programmed cell death seems to rely on the TNF-α signaling pathway. TNFR1 contains a cytoplasmatic death domain (DD) that binds to adaptor TRADD (TNFR-associated DD), which indirectly activates caspase cascades, causing apoptosis (Varfolomeev and Ashkenazi, 2004). In this context, it has been suggested that Aβ1–40 binds directly to TNFR1, resulting in neuronal apoptosis (Li et al., 2004). Our data clearly demonstrate that Aβ1–40 induced the expression of TNF-α in the hippocampus and prefrontal cortex, two brain areas essential for the cognitive functions. Despite mRNA, TNF-α levels started to reduce before 6 h; it is reasonable to expect that TNF-α protein remains elevated for a longer period. In fact, Song et al. (2001) have shown that Aβ1–42 intracerebroventricular injection in mice results in a rapid onset and time-dependent TNF-α protein expression, which is maximal at 4 h, returning to basal values only after 72 h. A similar effect might be expected in our experimental paradigm. Notably, pharmacological and genetic inhibition of TNF-α signaling reduced the synaptic loss and improved the learning and memory performance in Aβ-treated mice even 8 d after treatment. These findings clearly place TNF-α as one of the earliest mediators in the neurodegenerative process induced by Aβ1–40. However, additional studies are necessary to determine whether or not TNF-α effects depend on the participation of other inflammatory proteins (e.g., IL-1β, IL-6, MCP-1).
The signal transduction cascades elicited by TNF-α culminate in the activation of key transcriptional regulators, including NF-κB (Beg et al., 1993) and AP-1 (Angel and Karin, 1991). In most cell types, NF-κB complexes are found in the cytoplasm associated with IκBs, which prevent their nuclear localization. Cell stimulation and/or injury leads to the rapid degradation of IκB, allowing NF-κB translocation to the nucleus, where it regulates the transcription of several genes (Ghosh and Karin, 2002). Functional NF-κB dimers are present in essentially all cell types in the CNS, including neurons, astrocytes, microglia, and oligodendrocytes (O'Neill and Kaltschmidt, 1997). In AD brains, NF-κB immunoreactivity is greater in neurons and astrocytes surrounding amyloid plaques (Terai et al., 1996; Kaltschmidt et al., 1997). Additionally, it has been reported that Aβ stimulation leads to NF-κB activation in cultured neurons and glia (Akama et al., 1998). Corroborating these findings, we demonstrate that Aβ promotes NF-κB activation in the hippocampus and prefrontal cortex through a TNF-α dependent pathway.
AP-1 is a transcriptional factor composed of the protein products of c-jun and c-fos, and their expression is stimulated by cytokines (Angel and Karin, 1991). Phosphorylation and activation of c-Jun is catalyzed by the MAPK family members JNKs. The activation of both JNK and c-Jun has been described in cultured neurons after Aβ exposure, and their inhibition attenuates Aβ toxicity (Bozyczko-Coyne et al., 2001; Morishima et al., 2001; Troy et al., 2001). JNK activation has also been implicated in Aβ-mediated LTP decline, indicating a possible role for learning and memory (Wang et al., 2004a). In the present study, we demonstrate that Aβ1–40 produced a rapid and sustained activation of JNK and its target c-Jun, via mechanisms possibly dependent on TNF-α. These results are consistent with previous reports indicating that JNK pathways are activated in the AD brain (Anderson et al., 1996; Zhu et al., 2001).
In the CNS, iNOS is not commonly found in healthy tissues, but it can be expressed after brain insult in astrocytes, neurons, and endothelial cells, where it triggers the production of high amounts of NO (Vallance and Leiper, 2002). Overproduction of NO may lead to neuronal damage and death. The predominant mechanism by which NO promotes neuronal toxicity implicates the reaction of NO with superoxide anion to generate the cytotoxic substance peroxynitrite (Beckman et al., 1990; Vallance and Leiper, 2002). Under physiological conditions, highly reactive molecules are rapidly eliminated by antioxidant enzymes, including GPxs (Thannickal and Fanburg, 2000). However, in pathological conditions, excessively accumulated reactive species induce several cellular dysfunctions (Thannickal and Fanburg, 2000). The present data revealed that intracerebroventricular Aβ1–40 injection induced a marked iNOS expression, a phenomenon accompanied by a reduction in GSH levels, suggesting a possible increase in the production of reactive species, including NO. The iNOS inhibition or the iNOS genetic deletion significantly protected mice from Aβ1–40-induced impairment of learning and memory. These protective effects might be explained by a reduction in synaptic disruption. The expression of iNOS and the NO synthesis have been deeply associated with AD pathology. In this context, it has been demonstrated that both iNOS expression and peroxynitrite damage take place in the AD brain (Smith et al., 1997; Luth et al., 2002). A recent report showed that genetic deletion of iNOS substantially protected mice from Aβ toxicity (Nathan et al., 2005). Furthermore, it has been demonstrated that iNOS exerts an important role in Aβ-caused cognitive deficits in rats and mice (Tran et al., 2001; Wang et al., 2004b).
Production of iNOS protein is tightly regulated at the transcriptional level, but the upstream signaling events mediating Aβ-induced iNOS expression remain poorly understood. For instance, the molecular cloning and analysis of the promoter region of iNOS gene revealed the presence of binding sites to several transcriptional factors, such as AP-1, NF-κB, and TNF response element (Eberhardt et al., 1996). Of high interest, in the present study, we show for the first time that iNOS induction by Aβ1–40 depends on the TNF-α signaling, with involvement of JNK/c-Jun and NF-κB (for more details, see the proposed scheme in supplemental Fig. 4, available at www.jneurosci.org as supplemental material). These results also extend the notion that iNOS protein represents an unusual enzyme that can be modulated by highly specialized mechanisms.
Overall, our study suggests that TNF-α and iNOS signaling pathways are linked and exert an important role in the cognitive deficits observed in the earlier stages of AD. Additional characterization of a candidate substrate(s) for both proteins would help to clarify their role and potential as molecular targets for therapeutic intervention in human AD.
Footnotes
This work was supported by grants from the Conselho Nacional de Desenvolvimento Científico e Tecnológico, the Coordenacão de Aperfeiçoamento de Pessoal de Nível Superior, the Programa de Apoio aos Núcleos de Excelência, and the Fundação de Apoio a Pesquisa do Estado de Santa Catarina, all from Brazil.
References
- Anderson AJ, Su JH, Cotman CW. DNA damage and apoptosis in Alzheimer's disease: colocalization with c-Jun immunoreactivity, relationship to brain area, and effect of postmortem delay. J Neurosci. 1996;16:1710–1719. doi: 10.1523/JNEUROSCI.16-05-01710.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angel P, Karin M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta. 1991;1072:129–157. doi: 10.1016/0304-419x(91)90011-9. [DOI] [PubMed] [Google Scholar]
- Akama KT, Albanese C, Pestell RG, Van Eldik LJ. Amyloid beta-peptide stimulates nitric oxide production in astrocytes through an NFkappaB-dependent mechanism. Proc Natl Acad Sci USA. 1998;95:5795–5800. doi: 10.1073/pnas.95.10.5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci USA. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beg AA, Finco TS, Nantermet PV, Baldwin AS., Jr Tumor necrosis factor and interleukin-1 lead to phosphorylation and loss of Ikappa B alpha: a mechanism for NF-kappa B activation. Mol Cell Biol. 1993;13:3301–3310. doi: 10.1128/mcb.13.6.3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozyczko-Coyne D, O'Kane TM, Wu ZL, Dobrzanski P, Murthy S, Vaught JL, Scott RW. CEP-1347/KT-7515, an inhibitor of SAPK/JNK pathway activation, promotes survival and blocks multiple events associated with Abeta-induced cortical neuron apoptosis. J Neurochem. 2001;77:849–863. doi: 10.1046/j.1471-4159.2001.00294.x. [DOI] [PubMed] [Google Scholar]
- Budson AE, Price BH. Memory dysfunction. N Engl JMed. 2005;352:692–699. doi: 10.1056/NEJMra041071. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Drake J, Pocernich C, Castegna A. Evidence of oxidative damage in Alzheimer's disease brain: central role for amyloid beta-peptide. Trends Mol Med. 2001;7:548–554. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- Buttini M, Orth M, Bellosta S, Akeefe H, Pitas RE, Wyss-Coray T, Mucke L, Mahley RW. Expression of human apolipoprotein E3 or E4 in the brains of Apoe−/− mice: isoform-specific effects on neurodegeneration. J Neurosci. 1999;19:4867–4880. doi: 10.1523/JNEUROSCI.19-12-04867.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhardt W, Kunz D, Hummel R, Pfeilschifter J. Molecular cloning of the rat inducible nitric oxide synthase gene promoter. Biochem Biophys Res Commun. 1996;223:752–756. doi: 10.1006/bbrc.1996.0968. [DOI] [PubMed] [Google Scholar]
- El Khoury J, Hickman SE, Thomas CA, Cao L, Silverstein SC, Loike JD. Scavenger receptor-mediated adhesion of microglia to beta-amyloid fibrils. Nature. 1996;382:716–719. doi: 10.1038/382716a0. [DOI] [PubMed] [Google Scholar]
- Fillit H, Ding WH, Buee L, Kalman J, Altstiel L, Lawlor B, Wolf-Klein G. Elevated circulating tumor necrosis factor levels in Alzheimer's disease. Neurosci Lett. 1991;129:318–320. doi: 10.1016/0304-3940(91)90490-k. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109:S81–S96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- Goodwin JL, Uemura E, Cunnick JE. Microglial release of nitric oxide by the synergistic action of β-amyloid and IFN-γ. Brain Res. 1995;692:207–214. doi: 10.1016/0006-8993(95)00646-8. [DOI] [PubMed] [Google Scholar]
- Kaltschmidt B, Uherek M, Volk B, Baeuerle PA, Kaltschmidt C. Transcription factor NF-kappaB is activated in primary neurons by amyloid beta peptides and in neurons surrounding early plaques from patients with Alzheimer disease. Proc Natl Acad Sci USA. 1997;94:2642–2647. doi: 10.1073/pnas.94.6.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laursen SE, Belknap JK. Intracerebroventricular injections in mice. Some methodological refinements. J Pharmacol Methods. 1986;16:355–357. doi: 10.1016/0160-5402(86)90038-0. [DOI] [PubMed] [Google Scholar]
- Li R, Yang L, Lindholm K, Konishi Y, Yue X, Hampel H, Zhang D, Shen Y. Tumor necrosis factor death receptor signaling cascade is required for amyloid-β protein-induced neuron death. J Neurosci. 2004;24:1760–1771. doi: 10.1523/JNEUROSCI.4580-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luth HJ, Munch G, Arendt T. Aberrant expression of NOS isoforms in Alzheimer's disease is structurally related to nitrotyrosine formation. Brain Res. 2002;953:135–143. doi: 10.1016/s0006-8993(02)03280-8. [DOI] [PubMed] [Google Scholar]
- MacMicking JD, Nathan C, Hom G, Chartrain N, Fletcher DS, Trumbauer M, Stevens K, Xie QW, Sokol K, Hutchinson N, Chen H, Mudget JS. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell. 1995;81:641–650. doi: 10.1016/0092-8674(95)90085-3. [DOI] [PubMed] [Google Scholar]
- Masliah E, Achim CL, Ge N, DeTeresa R, Terry RD, Wiley CA. Spectrum of human immunodeficiency virus-associated neocortical damage. Ann Neurol. 1992;32:321–329. doi: 10.1002/ana.410320304. [DOI] [PubMed] [Google Scholar]
- Meda L, Cassatella MA, Szendrei GI, Otvos L, Jr, Baron P, Villalba M, Ferrari D, Rossi F. Activation of microglial cells by β-amyloid protein and interferon-γ. Nature. 1995;374:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- Morishima Y, Gotoh Y, Zieg J, Barrett T, Takano H, Flavell R, Davis RJ, Shirasaki Y, Greenberg ME. β-Amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J Neurosci. 2001;21:7551–7560. doi: 10.1523/JNEUROSCI.21-19-07551.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RGM, Garrud P, Rawlins JNP, O'Keefe J. Place navigation impaired in rats with hippocampal lesions. Nature. 1982;297:681–683. doi: 10.1038/297681a0. [DOI] [PubMed] [Google Scholar]
- Nathan C, Calingasan N, Nezezon J, Ding A, Lucia MS, La Perle K, Fuortes M, Lin M, Ehrt S, Kwon NS, Chen J, Vodovotz Y, Kipiani K, Beal MF. Protection from Alzheimer's-like disease in the mouse by genetic ablation of inducible nitric oxide synthase. J Exp Med. 2005;202:1163–1169. doi: 10.1084/jem.20051529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill LA, Kaltschmidt C. NF-kappa B: a crucial transcription factor for glial and neuronal cell function. Trends Neurosci. 1997;20:252–258. doi: 10.1016/s0166-2236(96)01035-1. [DOI] [PubMed] [Google Scholar]
- Perry RT, Collins JS, Wiener H, Acton R, Go RC. The role of TNF and its receptors in Alzheimer's disease. Neurobiol Aging. 2001;22:873–883. doi: 10.1016/s0197-4580(01)00291-3. [DOI] [PubMed] [Google Scholar]
- Prediger RD, Franco JL, Pandolfo P, Medeiros R, Duarte FS, Di Giunta G, Figueiredo CP, Farina M, Calixto JB, Takahashi RN, Dafre AL. Differential susceptibility following beta-amyloid peptide-(1–40) administration in C57BL/6 and Swiss albino mice: evidence for a dissociation between cognitive deficits and the glutathione system response. Behav Brain Res. 2007;177:205–213. doi: 10.1016/j.bbr.2006.11.032. [DOI] [PubMed] [Google Scholar]
- Rossner M, Yamada KM. What's in a picture? The temptation of image manipulation. J Cell Biol. 2004;166:11–15. doi: 10.1083/jcb.200406019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothe J, Lesslauer W, Lotscher H, Lang Y, Koebel P, Kontgen F, Althage A, Zinkernagel R, Steinmetz M, Bluethmann H. Mice lacking the tumor necrosis factor receptor 1 are resistant but highly susceptible to infection by Listeria monocytogenes. Nature. 1993;364:798. doi: 10.1038/364798a0. [DOI] [PubMed] [Google Scholar]
- Smith MA, Richey Harris PL, Sayre LM, Beckman JS, Perry G. Widespread peroxynitrite-mediated damage in Alzheimer's disease. J Neurosci. 1997;17:2653–2657. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song DK, Im YB, Jung JS, Cho J, Suh HW, Kim YH. Central beta-amyloid peptide-induced peripheral interleukin-6 responses in mice. J Neurochem. 2001;76:1326–1335. doi: 10.1046/j.1471-4159.2001.00121.x. [DOI] [PubMed] [Google Scholar]
- Terai K, Matsuo A, McGeer PL. Enhancement of immunoreactivity for NF-kappa B in the hippocampal formation and cerebral cortex of Alzheimer's disease. Brain Res. 1996;735:159–168. doi: 10.1016/0006-8993(96)00310-1. [DOI] [PubMed] [Google Scholar]
- Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1005–L1028. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- Tran MH, Yamada K, Olariu A, Mizuno M, Ren XH, Nabeshima T. Amyloid beta-peptide induces nitric oxide production in rat hippocampus: association with cholinergic dysfunction and amelioration by inducible nitric oxide synthase inhibitors. FASEB J. 2001;15:1407–1409. doi: 10.1096/fj.00-0719fje. [DOI] [PubMed] [Google Scholar]
- Troy CM, Rabacchi SA, Xu Z, Maroney AC, Connors TJ, Shelanski ML, Greene LA. Beta-amyloid-induced neuronal apoptosis requires c-Jun N-terminal kinase activation. J Neurochem. 2001;77:157–164. doi: 10.1046/j.1471-4159.2001.t01-1-00218.x. [DOI] [PubMed] [Google Scholar]
- Vallance P, Leiper J. Blocking NO synthesis: how, where and why? Nat Rev Drug Discov. 2002;1:939–950. doi: 10.1038/nrd960. [DOI] [PubMed] [Google Scholar]
- Van Dam D, De Deyn PP. Drug discovery in dementia: the role of rodent models. Nat Rev Drug Discov. 2006;5:956–970. doi: 10.1038/nrd2075. [DOI] [PubMed] [Google Scholar]
- Varfolomeev EE, Ashkenazi A. Tumor necrosis factor: an apoptosis JuNKie? Cell. 2004;116:491–497. doi: 10.1016/s0092-8674(04)00166-7. [DOI] [PubMed] [Google Scholar]
- Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. Deciphering the molecular basis of memory failure in Alzheimer's disease. Neuron. 2004;44:181–193. doi: 10.1016/j.neuron.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Wang Q, Walsh DM, Rowan MJ, Selkoe DJ, Anwyl R. Block of long-term potentiation by naturally secreted and synthetic amyloid β-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J Neurosci. 2004a;24:3370–3378. doi: 10.1523/JNEUROSCI.1633-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Rowan MJ, Anwyl R. β-Amyloid-mediated inhibition of NMDA receptor-dependent long-term potentiation induction involves activation of microglia and stimulation of inducible nitric oxide synthase and superoxide. J Neurosci. 2004b;24:6049–6056. doi: 10.1523/JNEUROSCI.0233-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Wu J, Rowan MJ, Anwyl R. Beta-amyloid inhibition of long-term potentiation is mediated via tumor necrosis factor. Eur J Neurosci. 2005;22:2827–2832. doi: 10.1111/j.1460-9568.2005.04457.x. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med. 2006;12:1005–1015. doi: 10.1038/nm1484. [DOI] [PubMed] [Google Scholar]
- Yan JJ, Cho JY, Kim HS, Kim KL, Jung JS, Huh SO, Suh HW, Kim YH, Song DK. Protection against beta-amyloid peptide toxicity in vivo with long-term administration of ferulic acid. Br J Pharmacol. 2001;133:89–96. doi: 10.1038/sj.bjp.0704047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Raina AK, Rottkamp CA, Aliev G, Perry G, Boux H, Smith MA. Activation and redistribution of c-jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer's disease. J Neurochem. 2001;76:435–441. doi: 10.1046/j.1471-4159.2001.00046.x. [DOI] [PubMed] [Google Scholar]