

Abstract
Spinal and bulbar muscular atrophy (SBMA) is an inherited motor neuron disease caused by the expansion of a polyglutamine tract within the androgen receptor (AR). The pathologic features of SBMA are motor neuron loss in the spinal cord and brainstem and diffuse nuclear accumulation and nuclear inclusions of the mutant AR in the residual motor neurons and certain visceral organs. Many components of the ubiquitin-proteasome and molecular chaperones are also sequestered in the inclusions, suggesting that they may be actively engaged in an attempt to degrade or refold the mutant AR. C terminus of Hsc70 (heat shock cognate protein 70)-interacting protein (CHIP), a U-box type E3 ubiquitin ligase, has been shown to interact with heat shock protein 90 (Hsp90) or Hsp70 and ubiquitylates unfolded proteins trapped by molecular chaperones and degrades them. Here, we demonstrate that transient overexpression of CHIP in a neuronal cell model reduces the monomeric mutant AR more effectively than it does the wild type, suggesting that the mutant AR is more sensitive to CHIP than is the wild type. High expression of CHIP in an SBMA transgenic mouse model also ameliorated motor symptoms and inhibited neuronal nuclear accumulation of the mutant AR. When CHIP was overexpressed in transgenic SBMA mice, mutant AR was also preferentially degraded over wild-type AR. These findings suggest that CHIP overexpression ameliorates SBMA phenotypes in mice by reducing nuclear-localized mutant AR via enhanced mutant AR degradation. Thus, CHIP overexpression would provide a potential therapeutic avenue for SBMA.
Keywords: CHIP, polyglutamine, SBMA, transgenic mice, protein degradation, androgen receptor
Introduction
Polyglutamine (polyQ) diseases are inherited neurodegenerative disorders caused by the expansion of trinucleotide CAG repeats in the causative genes (Gatchel and Zoghbi, 2005). To date, nine polyQ diseases have been identified (Di Prospero and Fischbeck, 2005). One of these is spinal and bulbar muscular atrophy (SBMA), characterized by premature muscular exhaustion, progressive muscular weakness, atrophy, and fasciculation in bulbar and limb muscles (Kennedy et al., 1968; Sobue et al., 1993; Sperfeld et al., 2002; Atsuta et al., 2006). In SBMA, a polymorphic CAG repeat with 14–32 CAGs expands to 40–62 CAGs in the first exon of the androgen receptor (AR) gene (La Spada et al., 1991; Tanaka et al., 1996). CAG repeat size is inversely correlated with the age at onset and positively correlated with disease severity in SBMA (Doyu et al., 1992; Igarashi et al., 1992; La Spada et al., 1992). The histopathologic hallmarks of SBMA are lower motor neuronal loss (Sobue et al., 1989), diffuse nuclear accumulation, and nuclear inclusions (NIs) of expanded polyQ mutant AR in the residual motor neurons in brainstem and spinal cord as well as in some other visceral organs (Li et al., 1998a,b; Adachi et al., 2005). Such NIs are common pathological features in polyQ diseases and also colocalize with many components of the ubiquitin-proteasome and molecular chaperones (Adachi et al., 2001; Schmidt et al., 2002; Ross and Poirier, 2004), raising the possibility that the ubiquitin-proteasome system and molecular chaperones may actively attempt to degrade or refold components of the inclusions (Stenoien et al., 1999; Ross and Pickart, 2004). Furthermore, these proteasomes and chaperones should also facilitate refolding or proteolysis of toxic misfolded proteins (McClellan et al., 2005) and may play a role in protecting neuronal cells against the toxic properties of expanded polyQ (Cummings et al., 1998; Kobayashi et al., 2000).
C terminus of heat shock cognate protein 70 (Hsc70)-interacting protein (CHIP) has three tetratricopeptide repeat (TPR) domains that interact with the molecular chaperones heat shock protein 70 (Hsp70) and Hsp90 (Ballinger et al., 1999; Connell et al., 2001) and a U-box domain that interacts with the proteasome, conferring CHIP with E3 ubiquitin ligase activity (Hatakeyama et al., 2001; Jiang et al., 2001). Wild-type AR is one of the CHIP substrates (Cardozo et al., 2003; He et al., 2004). CHIP also interacts with misfolded proteins trapped by molecular chaperones and degrades them, thus acting as a “quality control” E3 (Cyr et al., 2002; Murata et al., 2003). In fact, CHIP suppressed inclusion formation and cellular toxicity in cell, zebrafish, and Drosophila polyQ disease models (Jana et al., 2005; Miller et al., 2005; Al-Ramahi et al., 2006).
In this study, we examine whether CHIP exerts therapeutic effects on a cultured cell model and a transgenic mouse model expressing the mutant AR to explore a potential strategy for SBMA therapy. We report that CHIP markedly ameliorated motor and pathological phenotypes and that this amelioration was correlated with the reduction of monomeric mutant AR and mutant AR protein complexes in the SBMA models.
Materials and Methods
Cell culture.
SH-SY5Y cells were transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) with plasmids encoding ARs containing normal (24 CAGs) or expanded (65 CAGs) polyQ repeats (Waza et al., 2005). Stable clones expressing these normal and mutant ARs were established by selection with the antibiotic G418 (0.4 mg/ml final concentration). The androgen receptor is not expressed in untransfected SH-SY5Y cells. All cell cultures were propagated in the absence of androgen. In Western blots from these cultures, we detected a band of monomeric mutant AR in the separating gel but could hardly detect the high-molecular-weight mutant AR protein complex, which was retained in the stacking gel. Therefore, this cultured cell model is better suited for estimating the change in monomeric mutant AR expression. There was no difference in viability between cells expressing the wild-type and mutant ARs in the absence of androgen using the CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI).
DNA transfection.
Plasmid pcDNA3-CHIP, encoding FLAG-tagged human CHIP, was kindly provided by Dr. Keiji Tanaka (Laboratory of Frontier Science, Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan) (Murata et al., 2001). AR stable cells were plated in six-well dishes in 2 ml of DMEM/F-12 containing 10% charcoal-stripped fetal bovine serum with penicillin and streptomycin, and each dish was transfected with 4 μg of the vector containing CHIP or mock (negative control) using Lipofectamine 2000 according to the manufacturer's instructions. Transfection efficiency was 60–70%. The cells were cultured for 48 h at 37°C under 5% CO2.
Transgene construction.
Full-length human CHIP cDNA was generated from total RNA extracted from SH-SY5Y cells by reverse transcription-PCR. Full-length human CHIP was constructed by subcloning CHIP inserts derived from the full-length human CHIP cDNA into the pcDNA3.1-myc-his mammalian expression vector (Invitrogen) using PCR. Then, the myc-tagged CHIP fragments were subcloned into the pCAGGS vector (Niwa et al., 1991). All constructs were confirmed by DNA sequence analysis. The final plasmids were digested to remove the transgene.
Generation and maintenance of Tg mice and genotyping.
We generated CHIP overexpression mice by microinjection of the transgene into BDF1 fertilized eggs and obtained four founders. BDF1 homozygous CHIP transgenic females were mated with BDF1/B6 male mice expressing full-length human AR with 24 (AR-24Q mice, 5-5 line) or 97-polyQ tracts (AR-97Q mice, 7-8 line), producing a mixed BDF1 and B6 genetic background. First-generation AR-24Q/CHIP(tg/−) or AR-97Q/CHIP(tg/−) mice were mated with either CHIP(tg/−) or CHIP(tg/tg) mice to produce all AR or AR/CHIP double-transgenic mice for each analysis. We screened mouse tail DNA by PCR for the presence of the transgene using the primers 5′-CATCTCAGAAGAGGATCTGTG-3′ and 5′-GGTCGAGGGATCTTCATAAG-3′.
Neurological and behavioral assessment of SBMA model mice.
The AR-24Q and AR-97Q mice were generated and maintained as described previously (Katsuno et al., 2002). All animal experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and under the approval of the Nagoya University Animal Experiment Committee. The AR-97Q male mice showed progressive muscular atrophy and weakness as well as diffuse nuclear staining and NIs of the mutant AR. These phenotypes were very pronounced in male transgenic mice, similar to the situation in SBMA patients. The mouse rotarod task (Economex Rotarod; Ugo Basile, Comerio, Italy) was performed on a weekly basis, and cage activity was measured weekly with the AB system (Neuroscience, Tokyo, Japan) as described previously (Katsuno et al., 2002; Minamiyama et al., 2004). Spontaneous motor activity was monitored for 24 h periods; all spontaneous movements, both vertical and horizontal, including locomotion, rearing, and head movements, were counted and automatically totaled.
Immunohistochemistry and histopathology.
Mice were deeply anesthetized with ketamine–xylazine and transcardially perfused with 20 ml of 4% paraformaldehyde fixative in phosphate buffer, pH 7.4. Spinal cord and skeletal muscle tissues were removed, postfixed overnight in 10% phosphate-buffered formalin, and processed for paraffin embedding. Sections (6 μm thick) of the above tissues were deparaffinized, dehydrated with alcohol, and treated in formic acid for 5 min at room temperature. For the immunohistochemical studies, the paraffin sections were preheated in a microwave oven for 10 min. The sections were blocked with normal animal serum (1:20) and incubated with mouse anti-expanded polyQ antibody (1:10,000; 1C2; Millipore, Billerica, MA), anti-CHIP antibody (1:1000; Medical and Biological Laboratories, Nagoya, Japan), and mouse anti-glial fibrillary acidic protein (GFAP) antibody (1:1000; Roche Diagnostics, Mannheim, Germany). Primary antibodies were probed with a biotinylated anti-species-specific IgG (Vector Laboratories, Burlingame, CA), and the immune complexes were visualized using streptavidin-horseradish peroxidase (Dako, Glostrup, Denmark) and 3,3′-diaminobenzidine (Dojindo, Kumamoto, Japan) as a substrate. Sections were counterstained with Mayer's hematoxylin. Paraffin-embedded sections (6 μm thick) of the gastrocnemius muscles were air dried and stained with hematoxylin and eosin. For double-immunofluorescence staining of the spinal cord, sections were blocked with 5% normal goat serum and then sequentially incubated with anti-CHIP antibody (1:1000; Medical and Biological Laboratories) and 1C2 antibody (1:10,000; Millipore) at 4°C overnight. The sections were then incubated with Alexa 488-conjugated goat anti-chicken IgG (1:1000; Invitrogen) and Alexa 568-conjugated goat anti-mouse IgG (1:1300; Invitrogen) for 8 h at 4°C. The stained sections were examined and photographed with a confocal laser-scanning microscope (LSM 5 PASCAL; Carl Zeiss MicroImaging, Tokyo, Japan).
Patients.
Tissue from nine patients with clinicopathologically and genetically confirmed SBMA (51–84 years of age; mean, 64.3 years), and three non-neurological controls (51–76 years of age; mean, 64.0 years) were also used in the present study. These patients had been hospitalized and followed up at Nagoya University Hospital and its affiliated hospitals during the past 25 years. Informed consent was obtained to use the tissues for research purposes. Paraffin-embedded sections of the spinal cord and brain were obtained and examined in the same way as those from transgenic mice.
Quantification of 1C2-positive cells.
For assessment of 1C2-positive cells in the ventral horn of the spinal cord, 50 consecutive transverse sections of the thoracic spinal cord were prepared from each individual mouse, and 1C2-positive cells within the ventral horn of every fifth section were counted as described previously (Adachi et al., 2001). Populations of 1C2-positive cells were expressed as number/mm2. For assessment of 1C2-positive cells in muscle, the number of 1C2-positive cells was calculated from counts of >500 fibers in randomly selected areas of individual mice and expressed as the number per 100 muscle fibers. The quantitative data of six individual mice were expressed as mean ± SEM.
Protein expression analysis and ubiquitination assay.
Forty-eight hours after transfection, cells were lysed in CelLytic-M Mammalian Cell Lysis/Extraction Reagent (Sigma, St. Louis, MO) with 1 mm PMSF and 6 μg/ml aprotinin and centrifuged at 15,000 × g for 15 min at 4°C. Sixteen-week-old mice were exsanguinated under ketamine–xylazine anesthesia, and tissues were snap frozen with powdered CO2 in acetone. The tissues were homogenized in CelLytic-M Mammalian Cell Lysis/Extraction Reagent (Sigma) with 1 mm PMSF and 6 μg/ml aprotinin and centrifuged at 2500 × g for 15 min at 4°C. Supernatant fraction protein concentrations were determined using the DC protein assay (Bio-Rad, Hercules, CA). Aliquots of supernatant fractions were loaded on 5–20% SDS-PAGE gels, each lane containing 10 μg of protein for cells, 160 μg for neural tissue, and 80 μg for muscle tissue, and then transferred to Hybond-P membranes (GE Healthcare, Buckinghamshire, UK), using 25 mm Tris, 192 mm glycine, 0.1% SDS, and 10% methanol as transfer buffer. Primary antibodies were used at the following concentrations: rabbit anti-AR, 1:1000 (N20; Santa Cruz Biotechnology, Santa Cruz, CA); rabbit anti-AR, 1:1000 (H280; Santa Cruz Biotechnology); mouse anti-Hsp70, 1:1000 (SPA-810; Stressgen Biotechnologies, San Diego, CA); rabbit anti-Hsp40, 1:5000 (SPA-400; Stressgen Biotechnologies); mouse anti-Hsp90, 1:1000 (F8; Santa Cruz Biotechnology); and mouse anti-α-tubulin, 1:5000 (T9026; Sigma). Primary antibodies were probed using HRP-conjugated anti-rabbit Ig F(ab′)2 and anti-mouse Ig F(ab′)2 (1:5000; GE Healthcare) secondary antibodies and detected with the ECL Plus kit (GE Healthcare). An LAS-3000 imaging system was used to produce digital images and to quantify band intensities, which were then analyzed with Image Gauge software version 4.22 (Fujifilm, Tokyo, Japan). Densitometric values of AR were normalized to those of endogenous α-tubulin. Relative signal intensity (RSI) was computed as the signal intensity of each sample divided by that of mock-transfected cells (see Fig. 1) or AR-24Q/CHIP(−/−) or AR-97Q/CHIP(−/−) mice (see Fig. 6).
Figure 1.

Effect of CHIP on the AR or chaperones in cultured cells. A, Although anti-AR (N20) immunoblotting and densitometry demonstrated a dose-dependent decline of both wild-type (24Q) and mutant (65Q) AR expression in response to CHIP overexpression, the mutant AR decreased more than did the wild type. Mean levels of AR-24Q and AR-65Q expression were relatively compared between CHIP-transfected cells and mock-transfected cells. CHIP overexpression did not increase the expression of Hsp70, Hsp40, and Hsp90. *p < 0.005. B, The decrease in mutant AR in response to CHIP overexpression was much higher than that of the wild type (18.8% vs 38.0%; 6 μg of CHIP). *p < 0.05. C, Pulse-chase analysis of two forms of AR. Data are from one representative experiment for wild-type and mutant AR. D, Pulse-chase assessment of the half-life of wild-type (left) and mutant (right) AR. The percentages of AR-24Q and AR-65Q remaining in the absence (●) and presence (○) of overexpressed CHIP are indicated. Mutant AR was degraded more rapidly than the wild-type AR in the presence of overexpressed CHIP. E, Real-time RT-PCR of wild-type and mutant AR mRNA normalized to GAPDH levels. The wild-type and mutant AR mRNA levels were similar under CHIP overexpression. F, Ubiquitination of AR in control, AR-24Q-, and AR-97Q-transfected cells in the absence (−) or presence (+) of CHIP cotransfection. No ubiquitination was detected in control cells without expression of AR. Although both AR-24Q and AR-97Q were ubiquitinated without coexpression of CHIP, AR-97Q was strongly ubiquitinated. CHIP significantly enhanced the level of mutant AR ubiquitination but only slightly enhanced that of wild-type AR. A, B, D, E, Values represent means ± SEM (n = 5). IP, Immunoprecipitation; Ub, ubiquitin.
Figure 6.

CHIP decreases mutant AR protein complexes as well as monomeric mutant AR. A, B, Western blot analysis of total tissue homogenates from the spinal cord (A) and muscle (B) of AR-24Q/CHIP(−/−), AR-24Q/CHIP(tg/tg), AR-97Q/CHIP(−/−), and AR-97Q/CHIP(tg/tg) mice (16-week-old) probed with an AR-specific antibody (H280). The mutant AR complex appears in the stacking gel, and the monomeric mutant AR appears in the separating gel. Values are expressed as mean ± SEM for six mice. *p < 0.001; **p < 0.0001. C, Real-time RT-PCR of wild-type (AR-24Q) and mutant AR (AR-97Q) mRNA in transgenic mouse spinal cord and skeletal muscle in the absence (wt) and presence (CHIP) of CHIP overexpression. Values are expressed as means ± SE (n = 6). D, Filter trap assay of total tissue homogenates from the spinal cord and muscle of AR-97Q/CHIP(−/−) and AR-97Q/CHIP(tg/tg) mice (16 weeks of age), in the absence and presence of CHIP overexpression. Homogenates were filtrated and immunolabeled with an antibody against AR (H280). Large aggregated mutant AR complexes were trapped by the cellulose acetate membrane; soluble monomeric mutant AR passed through the cellulose acetate membrane and was trapped by the nitrocellulose membrane. Endogenous α-tubulin was used as a loading control.
Immunoprecipitation from mouse tissues was performed using 1 mg of total protein lysed in CelLytic-M Mammalian Cell Lysis/Extraction Reagent (Sigma), 15 μl of protein G Sepharose (GE Healthcare) and 2.5 μl of anti-myc antibody (Medical and Biological Laboratories). Protein was eluted from beads by boiling for 5 min in 15 μl of elution buffer (50 mm Tris-HCl, pH 6.8, 2% SDS, 60 μl/ml 2-mercaptoethanol, 10% glycerol) and loaded on SDS-polyacrylamide gels.
For the AR ubiquitination assay, full-length ARs were constructed by subcloning AR inserts derived from pCR-AR24 (24 CAG repeats) or pCR-AR97 (97 CAG repeats) into the pDsRed monomer mammalian expression vector (Takara Bio, Otsu, Japan). SH-SY5Y cells were seeded into 60 mm plates and cotransfected with plasmids encoding DsRed-AR and either CHIP or mock. Cells were exposed to MG132 (5 μm) for a 24 h period. Extracts were prepared, and AR was immunoprecipitated with anti-DsRed antibody. Blots were probed as described for Western blots with ubiquitin antibody (1B3; Medical and Biological Laboratories).
Filter trap assay.
To quantify the large-molecular aggregated and soluble forms of the mutant AR protein, filter trap assays of total tissue homogenates from the spinal cord and muscle of male AR-24Q or 97Q mice (16 weeks of age) were performed as described previously (Adachi et al., 2003). Proteins were filtered through a 0.2 μm cellulose acetate membrane (Sartorius, Goettingen, Germany) using a slot-blot apparatus (Bio-Rad). We also put 0.45 μm nitrocellulose membranes (Bio-Rad) under the cellulose acetate membrane to capture the monomeric AR protein passing through this membrane. Only the larger-sized mutant AR protein was retained on the cellulose acetate membrane (pores 0.2 μm in diameter), whereas the nitrocellulose membrane captured protein of all sizes. Samples of protein, 200 μg for spinal cord, and 80 μg for muscle, were prepared in a final volume of 200 μl of lysis buffer, loaded, and gently vacuumed. Slot blots were probed as described for Western blots by an antibody against AR (H-280; Santa Cruz Biotechnology) or α-tubulin (T9026; Sigma).
Pulse-chase labeling assay.
Cells were transfected as described above, starved for 1 h in methionine- and cysteine-free DMEM containing 10% dialyzed fetal calf serum, and then labeled for 1 h with 150 μCi of Redivue Pro-Mix L-[35S] in vitro cell-labeling mix (GE Healthcare) per milliliter. After washing in PBS, the cells were chased for the indicated time intervals in complete medium. Immunoprecipitation was performed using equivalent amounts of protein lysates, 10 μl of protein G Sepharose (GE Healthcare), and 5 μl of anti-AR antibody (N20; Santa Cruz Biotechnology) as described above. Each sample was separated by 5–20% SDS-PAGE, and analyzed by phosphorimaging (Typhoon 8600 PhosphorImager; GE Healthcare) and Image Gauge software version 4.22 (Fujifilm).
Quantitative real-time reverse transcription-PCR.
The levels of AR mRNA were determined by real-time Taqman PCR as described previously (Ishigaki et al., 2002). Total RNA was isolated from SH-SY5Y cells using the RNeasy Mini kit (Qiagen, Valencia, CA) and from transgenic mouse spinal cord and muscle by homogenizing in Trizol (Invitrogen) according to the manufacturer's instructions. Total RNA (5 μg) from cells and mouse spinal cord and muscle were reverse transcribed using SuperScript III reverse transcriptase (Invitrogen). Real-time Taqman reverse transcription (RT)-PCR was performed in a total volume of 50 μl, containing 25 μl of 2× QuantiTect SYBR Green PCR Master Mix (Qiagen) and 10 μm each primer. PCR products were detected by the iCycler system (Bio-Rad). The reaction conditions were 95°C for 15 min and then 45 cycles of 15 s at 94°C, 30 s at 55°C, and 30 s at 72°C. As an internal standard control, the expression level of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was simultaneously quantified. PCR primers were designed as described previously (Waza et al., 2005). RSI was computed as the signal intensity of each sample divided by that of mock-transfected cells (see Fig. 1 E) or AR-24Q/CHIP(−/−) or AR-97Q/CHIP(−/−) mice (see Fig. 6 C).
Statistical analysis.
Data were analyzed by unpaired t tests in the experiments shown in Figures 1, B and E, and 6 and log-rank tests for survival rate in that shown in Figure 4 D using Statview software version 5 (Hulinks, Tokyo, Japan). Statistical significance of the data shown in Figures 1 A, 4, A–C and F, and 5G–H was examined by the Williams test for multiple comparisons using Microsoft Excel 2004 (Microsoft, Redmond, WA).
Figure 4.
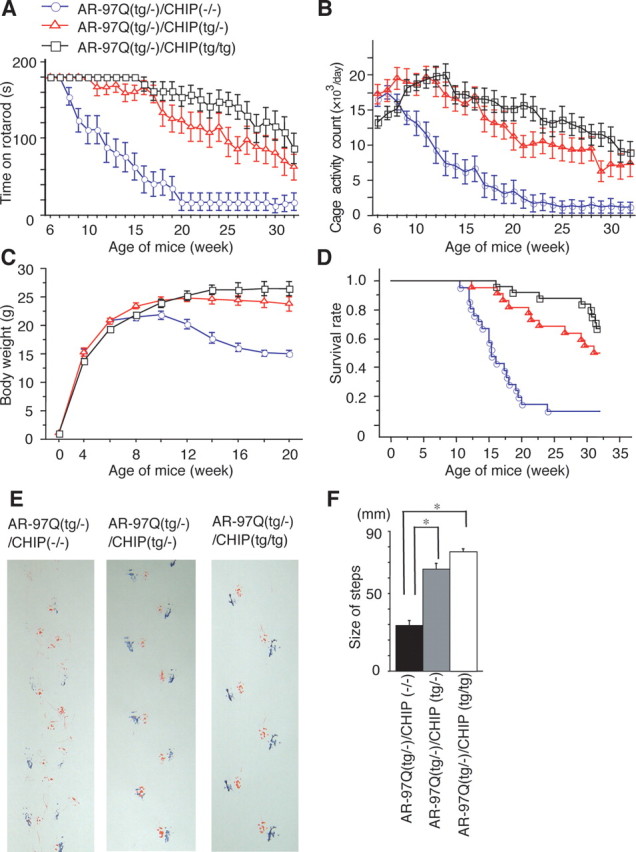
Effects of human CHIP overexpression on the behavioral phenotypes in male AR-97Q mice. A–D, Rotarod task (A; n = 22), cage activity (B; n = 22), body weight (C; n = 26), survival rate (D; n = 26) of the AR-97Q/CHIP(−/−) (○), AR-97Q/CHIP(tg/−) (□), and AR-97Q/CHIP(tg/tg) mice (▵). AR-97Q mice overexpressing human CHIP remained longer on the rotarod and showed higher cage activity than the AR-97Q/CHIP(−/−). The AR-97Q/CHIP(−/−) lost weight earlier than the other two double transgenics. D, A Kaplan-Meier plot shows the prolonged survival of AR-97Q/CHIP(tg/−) and AR-97Q/CHIP(tg/tg) mice compared with the AR-97Q/CHIP(−/−). The AR-97Q/CHIP(−/−) mice were significantly different from either of the other two in all parameters tested. Moreover, the AR-97Q/CHIP(tg/−) mice were worse off than the AR-97Q/CHIP(tg/tg) in all parameters tested. E, Footprints of representative 16-week-old AR-97Q/CHIP(−/−), AR-97Q/CHIP(tg/−), and AR-97Q/CHIP(tg/tg) mice. Front paws are indicated in red, and hindpaws are indicated in blue. AR-97Q/CHIP(−/−) mice exhibit motor weakness with dragging of the legs, AR-97Q/CHIP(tg/tg) mice walk almost normally, and AR-97Q/CHIP(tg/−) mice walk with somewhat shorter steps. F, The average length of hindpaw steps in 16-week-old AR-97Q/CHIP(−/−), AR-97Q/CHIP(tg/−), and AR-97Q/CHIP(tg/tg) mice. Values are expressed as means ± SEM (n = 6). *p < 0.005.
Results
Effect of CHIP overexpression on expression and ubiquitination of AR in vitro
CHIP directly interacts with and degrades the wild-type AR through its N-terminal conserved motif (He et al., 2004) and induces wild-type AR ubiquitination (Cardozo et al., 2003). To address the question of whether CHIP overexpression promotes the degradation of polyQ-expanded AR, we transfected SH-SY5Y cells stably expressing the wild-type (AR-24Q) or mutant (AR-65Q) AR with varying amounts of CHIP or mock as control. Although immunoblot analysis demonstrated a dose-dependent decline in both wild-type and mutant AR expression after CHIP overexpression (Fig. 1 A), the monomeric mutant AR decreased significantly more than did the wild type (p < 0.05) (Fig. 1 B), suggesting that the mutant AR is more sensitive to CHIP than is the wild type. CHIP was reported to interact with HSF1 and increase Hsp chaperone levels (Dai et al., 2003; Qian et al., 2006); however, the expression levels of Hsp90, Hsp70, and Hsp40 were not changed after CHIP overexpression (Fig. 1 A). This finding is consistent with a previous report (Miller et al., 2005) and suggests that the stress-induced response is different among different cell types.
To determine whether the enhanced degradation of mutant AR was attributable to protein degradation or to changes in RNA expression, the turnover of wild-type and mutant AR was then assessed with a pulse-chase labeling assay. SH-SY5Y stable cells were transiently transfected with mock or CHIP constructs. Without CHIP, the wild-type and mutant ARs were degraded almost equally, as reported previously (Bailey et al., 2002; Lieberman et al., 2002). In the presence of overexpressed CHIP, however, the wild-type and mutant ARs had half-lives of 3.6 and 2.7 h, respectively (Fig. 1 D), whereas mRNA levels for both the wild-type and mutant AR were quite similar (Fig. 1 E). These data indicate that CHIP preferentially degrades the mutant AR protein without altering mRNA levels.
The preferential degradation of mutant AR by CHIP suggests that CHIP may promote mutant AR ubiquitination, thereby targeting it for degradation. To assess this possibility, SH-SY5Y cells were transiently cotransfected with CHIP and DsRed-tagged AR-24Q or AR-97Q, and the cell lysates were immunoprecipitated with anti-DsRed. In this experiment, blots were probed with anti-ubiquitin antibody. No ubiquitination was detected in control cells without expression of AR. Although both AR-24Q and AR-97Q were ubiquitinated without coexpression of CHIP, AR-97Q was more strongly ubiquitinated. The levels of ubiquitinated AR were further increased in cells simultaneously expressing mutant AR and CHIP (Fig. 1 F). CHIP expression significantly enhanced the level of ubiquitinated mutant AR but only slightly enhanced that of the wild-type AR (Fig. 1 F).
Colocalization of CHIP with mutant AR in the nuclei
Next, we evaluated the colocalization of endogenous CHIP and mutant AR in AR-97Q mice and in SBMA patients. Double-immunofluorescence staining with chicken anti-CHIP and mouse anti-expanded polyQ (1C2) antibodies revealed that the endogenous CHIP (Fig. 2 A,D,G,J) and mutant AR (Fig. 2 B,E,H,K) were colocalized diffusely in the nuclei (Fig. 2 C,I) and NIs (Fig. 2 F,L) in the spinal anterior horn neurons of the AR-97Q mice and in the hypoglossal nucleus cells and spinal anterior horn neurons of SBMA patients, suggesting that the endogenous CHIP coexists with mutant AR and exerts its function in both AR-97Q mice and SBMA patients.
Figure 2.
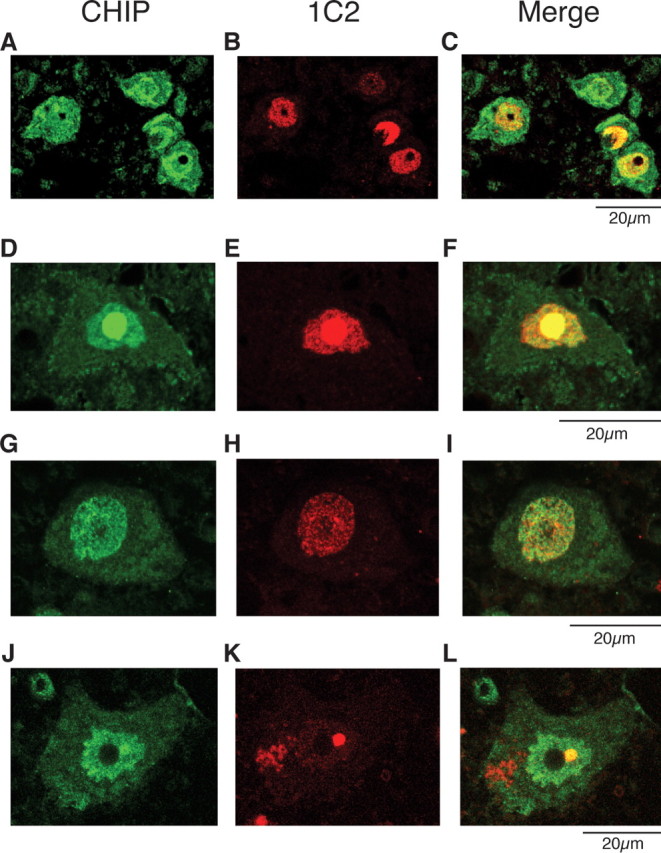
Colocalization of nuclear-localized CHIP with mutant AR. A–L, Anti-CHIP and anti-polyQ immunohistochemistry in spinal cords of 16-week-old AR-97Q mice (A–F) and an SBMA patient (G–L). A–C, Double-immunofluorescence staining for CHIP (A; green), expanded-polyQ (B; red), and overlay of the two signals (C; yellow) in the spinal anterior horn cells. D–F, CHIP (green; D) and mutant AR (red; E) are colocalized in nuclear inclusions (shown in yellow; F) in the spinal anterior horn cell. G–I, Double-immunofluorescence staining in cells of the hypoglossal nucleus of an SBMA patient revealed diffuse nuclear colocalization of CHIP (G) and mutant AR (H, I). J–L, CHIP (green; J) and mutant AR (red; K) were also colocalized in NIs (shown in yellow; L) in the spinal anterior horn cell of SBMA patients.
Nondeleterious effects of CHIP overexpression and generation of double-transgenic mice
Because CHIP colocalizes with polyQ-expanded AR, we further tested the effects of CHIP overexpression in the SBMA transgenic mouse model to explore a potential strategy for SBMA therapy. We generated transgenic mice expressing full-length human CHIP under the control of a cytomegalovirus enhancer and a chicken β-actin promoter (Fig. 3 A). From 14 available lines, we established four that express CHIP in the brain and skeletal muscle and examined the effects of overexpressed CHIP on mouse phenotypes. Through 50 weeks of age, none of the hemizygous or homozygous transgenic mice overexpressing CHIP showed any neurological phenotypes assessed using the rotarod task; they did, however, display slightly delayed weaning. Histological examination at 50 weeks did not show any detectable effects on neuronal cell morphology, neuronal cell number, or muscle structure (data not shown). These studies indicated that overexpression of human CHIP alone does not impair neuronal development or motor function.
Figure 3.

Increased CHIP expression in double-transgenic mice. A, Schematic view of the transgene construct. The microinjected fragment was composed of a cytomegalovirus enhancer (E), a chicken β-actin promoter (Pro), full-length human CHIP with a myc tag, and a rabbit β-globin polyadenylation signal sequence (polyA). B, Western blot analysis of total spinal cord and muscle protein lysates from AR-97Q/CHIP(−/−), AR-97Q/CHIP(tg/−), and AR-97Q/CHIP(tg/tg) mice immunolabeled with antibodies against CHIP, Hsp90, Hsp70, and Hsp40. C, Coimmunoprecipitation Western blots for CHIP. Soluble fractions were collected from the spinal cord of AR-97Q/CHIP(−/−) and AR-97Q/CHIP(tg/tg) mice. Equal amounts of protein were immunoprecipitated with an antibody to myc and immunoblotted for AR and Hsp70. Coimmunoprecipitation of CHIP and the polyQ-expanded mutant AR or the Hsp70 chaperone was detected. D, Western blot analysis of CHIP expression in total spinal cord and muscle protein lysates from CHIP(tg/tg) and AR-97Q/CHIP(tg/tg) mice of the indicated ages, immunolabeled with antibodies against myc. E, F, CHIP immunohistochemistry in spinal anterior horn and skeletal muscle of 16-week-old AR-97Q/CHIP(tg/tg) mice counterstained with Mayer's hematoxylin. E, CHIP immunoreactivity is localized to the nuclei and cytoplasm, with intense and diffuse staining in the anterior horn cells. F, Skeletal muscle showed diffuse nuclear and cytoplasmic staining. IP, Immunoprecipitation.
To determine whether overexpression of human CHIP could ameliorate the disease phenotype of the SBMA transgenic mouse model, we crossed the AR-24Q mice and the AR-97Q mice (Katsuno et al., 2002) with mice overexpressing human CHIP (CHIP2 line). The AR-97Q mice (SBMA model) are small and have short lifespans, progressive muscle atrophy, and weakness, as well as reduced cage activity (Katsuno et al., 2002). Because these phenotypes are markedly pronounced in the males, similar to SBMA patients (Katsuno et al., 2002), we used male transgenic mice in this study. We generated AR-24Q/CHIP(tg/tg) and AR-97Q/CHIP(tg/tg) mice as homozygotes, the AR-24Q/CHIP(tg/−) and AR-97Q/CHIP(tg/−) mice as hemizygotes, and the AR-24Q/CHIP(−/−) and AR-97Q/CHIP(−/−) mice as controls. The AR transgene expression was at the hemizygous level in all AR-24Q/CHIP and AR-97Q/CHIP double transgenics.
Expression of CHIP in double-transgenic mice
We examined whether the AR/CHIP double-transgenic mice express increased levels of the CHIP protein in the spinal cord and skeletal muscle. Western blot analysis revealed that CHIP expression in the spinal cords of AR-97Q/CHIP(tg/−) and AR-97Q/CHIP(tg/tg) mice was three and six times as high, respectively, as endogenous CHIP in the AR-97Q/CHIP(−/−) mice. In muscle, it was six times as high in the AR-97Q/CHIP(tg/−) mice and 12 times as high in the AR-97Q/CHIP(tg/tg) mice (Fig. 3 B). As in transfected cells, the expression levels of Hsp90, Hsp70, and Hsp40 were not changed after CHIP overexpression in the double-transgenic mice (Fig. 3 B). The increased CHIP was coimmunoprecipitated with polyQ-expanded AR and Hsp70, suggesting that CHIP may recognize AR either directly or indirectly through association with Hsp70 (Fig. 3 C). We also performed Western blotting analysis using 8-, 16-, 24-, and 32-week-old CHIP(tg/tg) and AR-97Q/CHIP(tg/tg) mice to examine the effect of age on the expression level of CHIP in spinal cord and muscle. We found that the expression levels of CHIP did not change even in the 32-week-old mice (Fig. 3 D). Immunohistochemical studies of double-transgenic mice tissue stained with the CHIP-specific antibody confirmed that spinal anterior horn neurons and muscle cells expressed the CHIP (Fig. 3 E,F). CHIP protein was diffusely distributed in the nuclei and cytoplasm (Fig. 3 E,F). Glial cells also showed diffuse nuclear staining of CHIP protein (Fig. 3 E).
Human CHIP overexpression ameliorates phenotypic expression of SBMA mice
To determine whether CHIP overexpression has an ameliorative effect on the motor phenotypes in the double-transgenic mice, we performed the rotarod task and measured locomotor cage activity with an infrared sensor system (Fig. 4 A,B). Motor impairment on the rotarod task was evident in the AR-97Q/CHIP(−/−) mice as early as 8 weeks after birth but was seen, to a lesser degree, in the AR-97Q/CHIP(tg/tg) mice beginning at only 16 weeks (Fig. 4 A). Although both the AR-97Q/CHIP(tg/tg) and AR-97Q/CHIP(tg/−) mice performed significantly better than the AR-97Q/CHIP(−/−) mice (p < 0.005 and p < 0.025, respectively) (Fig. 4 A), the AR-97Q/CHIP(tg/tg) mice were on the rod longer than the AR-97Q/CHIP(tg/−) mice during the trial. The locomotor cage activity of the AR-97Q/CHIP(−/−) mice was also significantly decreased at 32 weeks compared with the other two double transgenics (p < 0.005, respectively) (Fig. 4 B). Although there were no differences in body weight at birth among the various lines, the AR-97Q/CHIP(−/−) mice lost weight significantly earlier than did the AR-97Q/CHIP(tg/−) and AR-97Q/CHIP(tg/tg) mice (p < 0.005) (Fig. 4 C). The survival rate was significantly higher in the AR-97Q/CHIP(tg/−) and AR-97Q/CHIP(tg/tg) mice than in the AR-97Q/CHIP(−/−) mice (p < 0.0001) (Fig. 4 D). Because the decrease in ameliorative effects of CHIP overexpression in the aged mice is not attributable to decreased CHIP expression (Fig. 3 D), it is probably caused by the progressive nuclear accumulation of toxic mutant AR in the aged mice (Katsuno et al., 2003). The affected AR-97Q/CHIP(−/−) mice exhibited motor weakness, took short steps, or dragged their legs, whereas the AR-97Q/CHIP(tg/tg) mice moved almost normally, and the AR-97Q/CHIP(tg/−) mice only took somewhat shorter steps (Fig. 4 E). Both the AR-97Q/CHIP(tg/−) and AR-97Q/CHIP(tg/tg) mice took significantly longer steps than the AR-97Q/CHIP(−/−) mice (p < 0.005) (Fig. 4 F). Although the SBMA phenotypes were ameliorated in both the AR-97Q/CHIP(tg/tg) and AR-97Q/CHIP(tg/−) mice, the AR-97Q/CHIP(tg/tg) mice were better than the AR-97Q/CHIP(tg/−) mice in most of the parameters, suggesting that the improved motor phenotype depended on the CHIP expression level rather than the genetic background.
Immunohistochemical staining for mutant AR using the 1C2 antibody showed a marked reduction in diffuse nuclear staining and NIs in spinal cord (Fig. 5 A–C) and muscle (Fig. 5 D–F) of the AR-97Q/CHIP(tg/−) and AR-97Q/CHIP(tg/tg) mice compared with the AR-97Q/CHIP(−/−) mice. In the AR-97Q/CHIP(−/−) mice, intense staining was frequently seen in the nuclei (Fig. 5 A,D), whereas staining was infrequent in the AR-97Q/CHIP(tg/−) mice (Fig. 5 B,E) and much less frequent in the AR-97Q/CHIP(tg/tg) mice (Fig. 5 C,F). There were significantly more 1C2-positive cells in spinal cord (Fig. 5 G) and muscle (Fig. 5 H) of the AR-97Q/CHIP(−/−) mice than in the AR-97Q/CHIP(tg/−) and AR-97Q/CHIP(tg/tg) mice. The 1C2-positive cell populations were not, however, statistically different in the AR-97Q/CHIP(tg/−) and AR-97Q/CHIP(tg/tg) mice. GFAP-specific antibody staining showed an apparent reduction in reactive astrogliosis in the AR-97Q/CHIP(tg/tg) mice compared with the AR-97Q/CHIP(−/−) mice in the spinal anterior horn (Fig. 5 I). Muscle histology also demonstrated marked amelioration of muscle atrophy in the AR-97Q/CHIP(tg/tg) mice (Fig. 5 J). The AR-24Q/CHIP mice displayed no altered phenotypes (data not shown). The numbers of neuronal cells in the spinal ventral horns of AR-97Q/CHIP(−/−), AR-97Q/CHIP(tg/−), and AR-97Q/CHIP(tg/tg) mice were not significantly decreased compared with those in the wild-type mice (data not shown).
Figure 5.

CHIP decreases nuclear-localized mutant AR in double-transgenic mice. A–F, PolyQ immunohistochemistry (1C2) in the spinal anterior horn (A–C) and muscle (D–F) of 16-week-old AR-97Q/CHIP(−/−) and AR-97Q/CHIP double-transgenic mice. AR-97Q/CHIP(−/−) mice have intense and frequent staining for 1C2 in the nucleus (A, D). B, C, E, F, AR-97Q/CHIP(tg/−) (B, E) and AR-97Q/CHIP(tg/tg) (C, F) mice exhibit low levels of 1C2 staining in the nucleus. G, H, Quantitative assessment of diffuse nuclear staining for 1C2 in the spinal ventral horn (G) and muscle (H). Bars represent the density of 1C2-positive cells in the AR-97Q/CHIP(−/−), AR-97Q/CHIP(tg/−), and AR-97Q/CHIP(tg/tg) mice. There are significantly more 1C2-positive cells in AR-97Q/CHIP(−/−) mice than in AR-97Q/CHIP(tg/−) mice or AR-97Q/CHIP(tg/tg) mice in both tissues. Results are expressed as mean ± SEM for six mice. *p < 0.025; **p < 0.005. I, Immunohistochemical staining with GFAP-specific antibody also showed an obvious reduction in reactive astrogliosis in the spinal anterior horn of AR-97Q/CHIP(tg/tg) mice. J, Hematoxylin and eosin (HE) staining of muscle tissue in AR-97Q/CHIP(−/−) mice revealed obvious atrophy and small-angulated fibers, which were not seen in AR-97Q/CHIP(tg/tg) mice. No., Number.
Overexpression of CHIP decreases the high-molecular-weight mutant AR protein and monomeric mutant AR protein
Because the mutant AR was preferentially degraded compared with the wild-type AR when CHIP was overexpressed in vitro, we also examined levels of AR in the SBMA mouse model. Western blot analysis from lysates of the spinal cord and muscle of AR-97Q mice revealed high-molecular-weight mutant AR protein complex retained in the stacking gel as well as a band of monomeric mutant AR, whereas only the band of wild-type monomeric AR was visible in tissues from the AR-24Q mice (Fig. 6 A,B). CHIP overexpression notably diminished both the high-molecular-weight complex and the monomer of mutant AR in the spinal cord and muscle of the AR-97Q mice but only slightly diminished the wild-type monomeric AR in AR-24Q mice (Fig. 6 A,B). CHIP overexpression decreased the amount of the monomeric AR in AR-97Q mice by 50% in the spinal cord and 75% in the skeletal muscle but only by 8% and 5%, respectively, in AR-24Q mice (Fig. 6 A,B). The levels of wild-type and mutant AR mRNA were similar in both AR-24Q and AR-97Q mice under CHIP overexpression (Fig. 6 C). These observations suggest that overexpression of CHIP markedly decreases not only the monomeric mutant AR protein but also the high-molecular-weight mutant AR protein.
We also performed filter trap assays to quantitatively analyze the effects of CHIP overexpression on levels of both the large-molecular aggregated and soluble forms of mutant AR (Wanker et al., 1999). Only the larger-sized mutant AR protein was retained on the upper, cellulose acetate membrane (pores 0.2 μm in diameter), whereas the lower nitrocellulose membrane captured all proteins that passed through the upper membrane (Fig. 6 D). Values were normalized to endogenous α-tubulin trapped on the nitrocellulose membrane. Both forms of trapped AR-97Q protein were markedly reduced in the spinal cord and muscle of mice overexpressing CHIP, whereas levels of soluble monomeric AR protein from the AR-24Q mice were only slightly reduced (Fig. 6 D). The endogenous AR protein was not retained on the cellulose acetate membrane in wild-type mice (data not shown). These results strongly indicate that CHIP markedly reduces not only the monomeric mutant AR protein but also the high-molecular-weight mutant AR complex, by preferentially degrading the mutant AR. These observations also suggest that overexpression of CHIP enhanced the function of the ubiquitin-proteasome pathway and subsequently accelerated degradation of monomeric mutant AR protein.
Discussion
CHIP is a U-box-dependent E3 ubiquitin ligase that associates with the Hsp70 and Hsp90 molecular chaperones and targets folded or toxic misfolded proteins for degradation (McDonough and Patterson, 2003). A wide range of different proteins have been identified as CHIP substrates, including members of the steroid hormone receptor family (Connell et al., 2001; Tateishi et al., 2004; Wang and DeFranco, 2005), the cystic-fibrosis transmembrane-conductance regulator (Meacham et al., 2001; Younger et al., 2006), E2A transcription factors (Huang et al., 2004), raf-1 protein kinase (Demand et al., 2001), ErbB2 (Zhou et al., 2003), nucleophosmin-anaplastic lymphoma kinase (Bonvini et al., 2004), dual leucine zipper-bearing kinase (Daviau et al., 2006), caytaxin (Grelle et al., 2006), αB-crystallin (Chavez Zobel et al., 2003), tau (Hatakeyama et al., 2004; Petrucelli et al., 2004; Sahara et al., 2005; Dickey et al., 2006), α-synuclein (Shin et al., 2005), the p53 tumor suppressor (Esser et al., 2005), apoptosis signal-regulating kinase 1 (Hwang et al., 2005), and polyQ-disease causative proteins (Jana et al., 2005; Miller et al., 2005; Al-Ramahi et al., 2006). CHIP can directly interact with and degrade the wild-type AR in a phosphorylation-dependent or -independent manner (Cardozo et al., 2003; Rees et al., 2006) and can repress AR transcriptional activity, suggesting that CHIP may play a role in regulating AR function in the cell (He et al., 2004). CHIP also has been shown to associate with the polyQ-expanded AR (Thomas et al., 2004). In this study, we addressed the question of whether CHIP overexpression promotes the degradation of mutant AR and exerts therapeutic effects on the SBMA phenotype. In a cultured neuronal cell model of SBMA, we demonstrated that increasing levels of CHIP more effectively ubiquitinated and degraded the monomeric mutant AR than the wild-type AR, suggesting that the mutant AR is more sensitive to CHIP than is the wild type. Overexpression of CHIP strongly inhibited nuclear accumulation of the mutant AR and markedly ameliorated motor impairments in SBMA transgenic mice in a dose-dependent manner. Mutant AR and CHIP were colocalized diffusely in the nuclei and in the NIs in neurons of the AR-97Q/CHIP double-transgenic mice. More importantly, CHIP also colocalized with mutant AR aggregates present in the anterior horn cells from postmortem tissues of SBMA patients. Western blot analysis showed that both a band of monomeric mutant AR and the high-molecular-weight form of mutant AR protein complexes retained in the stacking gel were diminished in the spinal cord and muscle of the double-transgenic mice, suggesting that the degradation of mutant AR may have been accelerated by overexpression of CHIP.
Our AR-97Q transgenic mice display progressive muscular atrophy and weakness, as well as diffuse nuclear staining and NIs of the mutant AR (Katsuno et al., 2002). These phenotypes are very pronounced in male transgenic mice, similar to human SBMA patients. The fact that AR has a specific ligand (i.e., testosterone), renders the pathogenesis of SBMA unique among polyQ diseases (Poletti et al., 2005). There is increasing evidence that the AR ligand (Katsuno et al., 2003; Chevalier-Larsen et al., 2004; Sopher et al., 2004; Katsuno et al., 2006; Yu et al., 2006) and molecular chaperones (Kobayashi et al., 2000; Bailey et al., 2002; Adachi et al., 2003) play a crucial role in the pathogenesis of SBMA. The success of androgen deprivation therapy in SBMA mouse models has been translated into clinical trials (Banno et al., 2006). In addition, elucidation of its pathophysiology using SBMA animal models has led to the development of other chaperone-related disease-modifying drugs, an Hsp90 inhibitor (Waza et al., 2005) and a heat shock protein inducer (Katsuno et al., 2005), which inhibit the pathogenic process of neuronal degeneration. Recent studies suggested that soluble causative protein species, not insoluble protein aggregates, might be toxic and thus targets in treatments of neurodegenerative disorders (Slow et al., 2006). Here, we demonstrated that overexpression of human CHIP exerts therapeutic effects on motor dysfunction in the AR-97Q mouse. Overexpression of CHIP served to decrease monomeric mutant AR in the double-transgenic mice. The large aggregated mutant AR protein complexes were also significantly reduced by CHIP overexpression, suggesting that CHIP accelerated the turnover of mutant AR. Together, these data suggest that targeting mutant AR for proteasomal degradation by overexpression of CHIP could be a disease-modifying therapeutic strategy in SBMA neuropathology. These findings are consistent with previous studies showing that CHIP serves as a protective factor in other polyQ diseases by promoting reduced aggregation of disease proteins (Jana et al., 2005; Miller et al., 2005) and proteasomal degradation (Al-Ramahi et al., 2006). In contrast to symptom-relief therapies, such as l-DOPA for Parkinson's disease, these disease-modifying therapies inhibit or slow down the pathogenic processes of neuronal degeneration.
CHIP interacts with Hsp90 or Hsp70, ubiquitylates unfolded proteins trapped by molecular chaperones, and degrades them, thus acting as a quality control E3 ubiquitin ligase (Murata et al., 2001). The remarkable reduction of monomeric mutant AR in the AR-97Q/CHIP mice may reflect accelerated degradation of mutant AR through the CHIP-mediated E3-proteasome system. CHIP also ubiquitinated the AR protein in a polyQ length-dependent manner, suggesting that overexpression of CHIP enhances the degradation of monomeric mutant AR by activating the Hsp70-interacting, quality control E3 system. This subsequently reduces the amount of nuclear-localized mutant AR, resulting in amelioration of phenotypic expression induced by mutant AR. Interaction between Hsp70 and CHIP, detected by coimmunoprecipitation and Western blot analysis, in the double-transgenic mice supports this view. Increased activity of CHIP was reported to modify a neurodegenerative phenotype caused by expanded ataxin-1 and huntingtin in a chaperone-dependent manner in cellular and Drosophila models (Jana et al., 2005; Al-Ramahi et al., 2006). CHIP lacking the normal TPR domain did not ubiquitinate the ataxin-1 protein (Al-Ramahi et al., 2006). These results suggest that chaperone interaction is essential for CHIP-dependent ubiquitination. Hsp70 overexpression in cell culture and mouse models of SBMA enhanced degradation of mutant AR-polyQ protein via its interaction with the ubiquitin-proteasome system (Bailey et al., 2002; Adachi et al., 2003). CHIP might be one such coupling factor between the Hsp70 chaperone system and the machinery responsible for degrading mutant AR. Another possibility is that overexpression of CHIP may accelerate chaperone-independent interaction with mutant AR and its degradation through the proteasome pathway. CHIP directly interacts with and degrades the AR protein (He et al., 2004; Rees et al., 2006). Thus, the interactions we demonstrated between CHIP and AR may be either direct or mediated by chaperones that are known to interact with CHIP. This direct interaction of CHIP with the AR protein might promote mutant AR degradation through the proteasome system.
Accumulation of misfolded proteins is causally related to many age-related neurodegenerative diseases (Muchowski and Wacker, 2005; Bates, 2006). Prompt removal and/or refolding may be required more in aged or damaged cells than in young healthy cells in which appropriate protein quality control systems function (Wickner et al., 1999). In SBMA patients, diffuse nuclear accumulation of mutant AR is frequent and extensive, being distributed in a wide array of CNS nuclei and in visceral organs (Adachi et al., 2005). In this study, we demonstrated that overexpression of CHIP significantly ameliorates the phenotypes of SBMA transgenic mice, by reducing the amount of both the monomeric and large aggregated forms of nuclear-accumulated mutant AR protein, suggesting that CHIP may change the triage of mutant AR and promote its degradation by the proteasome system (Marques et al., 2006). Thus, CHIP overexpression might provide a potential therapeutic avenue for SBMA and other polyQ diseases.
Footnotes
This work was supported by a Center of Excellence grant and KAKENHI (17025020) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan; by Special Coordination Funds for Promoting Science and Technology from the Ministry of Education, Culture, Sports, Science, and Technology, Japan; and by grants from the Ministry of Health, Labor, and Welfare, Japan. We have no financial conflict of interest that might be construed to influence the results or interpretation of this manuscript. We thank Jun-ichi Miyazaki for kindly providing the pCAGGS vector and Keiji Tanaka for the pcDNA3-CHIP vector.
References
- Adachi H, Kume A, Li M, Nakagomi Y, Niwa H, Do J, Sang C, Kobayashi Y, Doyu M, Sobue G. Transgenic mice with an expanded CAG repeat controlled by the human AR promoter show polyglutamine nuclear inclusions and neuronal dysfunction without neuronal cell death. Hum Mol Genet. 2001;10:1039–1048. doi: 10.1093/hmg/10.10.1039. [DOI] [PubMed] [Google Scholar]
- Adachi H, Katsuno M, Minamiyama M, Sang C, Pagoulatos G, Angelidis C, Kusakabe M, Yoshiki A, Kobayashi Y, Doyu M, Sobue G. Heat shock protein 70 chaperone overexpression ameliorates phenotypes of the spinal and bulbar muscular atrophy transgenic mouse model by reducing nuclear-localized mutant androgen receptor protein. J Neurosci. 2003;23:2203–2211. doi: 10.1523/JNEUROSCI.23-06-02203.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adachi H, Katsuno M, Minamiyama M, Waza M, Sang C, Nakagomi Y, Kobayashi Y, Tanaka F, Doyu M, Inukai A, Yoshida M, Hashizume Y, Sobue G. Widespread nuclear and cytoplasmic accumulation of mutant androgen receptor in SBMA patients. Brain. 2005;128:659–670. doi: 10.1093/brain/awh381. [DOI] [PubMed] [Google Scholar]
- Al-Ramahi I, Lam YC, Chen HK, de Gouyon B, Zhang M, Perez AM, Branco J, de Haro M, Patterson C, Zoghbi HY, Botas J. CHIP protects from the neurotoxicity of expanded and wild-type ataxin-1 and promotes their ubiquitination and degradation. J Biol Chem. 2006;281:26714–26724. doi: 10.1074/jbc.M601603200. [DOI] [PubMed] [Google Scholar]
- Atsuta N, Watanabe H, Ito M, Banno H, Suzuki K, Katsuno M, Tanaka F, Tamakoshi A, Sobue G. Natural history of spinal and bulbar muscular atrophy (SBMA): a study of 223 Japanese patients. Brain. 2006;129:1446–1455. doi: 10.1093/brain/awl096. [DOI] [PubMed] [Google Scholar]
- Bailey CK, Andriola IF, Kampinga HH, Merry DE. Molecular chaperones enhance the degradation of expanded polyglutamine repeat androgen receptor in a cellular model of spinal and bulbar muscular atrophy. Hum Mol Genet. 2002;11:515–523. doi: 10.1093/hmg/11.5.515. [DOI] [PubMed] [Google Scholar]
- Ballinger CA, Connell P, Wu Y, Hu Z, Thompson LJ, Yin LY, Patterson C. Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol Cell Biol. 1999;19:4535–4545. doi: 10.1128/mcb.19.6.4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banno H, Adachi H, Katsuno M, Suzuki K, Atsuta N, Watanabe H, Tanaka F, Doyu M, Sobue G. Mutant androgen receptor accumulation in spinal and bulbar muscular atrophy scrotal skin: a pathogenic marker. Ann Neurol. 2006;59:520–526. doi: 10.1002/ana.20735. [DOI] [PubMed] [Google Scholar]
- Bates GP. BIOMEDICINE: One misfolded protein allows others to sneak by. Science. 2006;311:1385–1386. doi: 10.1126/science.1125246. [DOI] [PubMed] [Google Scholar]
- Bonvini P, Dalla Rosa H, Vignes N, Rosolen A. Ubiquitination and proteasomal degradation of nucleophosmin-anaplastic lymphoma kinase induced by 17-allylamino-demethoxygeldanamycin: role of the co-chaperone carboxyl heat shock protein 70-interacting protein. Cancer Res. 2004;64:3256–3264. doi: 10.1158/0008-5472.can-03-3531. [DOI] [PubMed] [Google Scholar]
- Cardozo CP, Michaud C, Ost MC, Fliss AE, Yang E, Patterson C, Hall SJ, Caplan AJ. C-terminal Hsp-interacting protein slows androgen receptor synthesis and reduces its rate of degradation. Arch Biochem Biophys. 2003;410:134–140. doi: 10.1016/s0003-9861(02)00680-x. [DOI] [PubMed] [Google Scholar]
- Chavez Zobel AT, Loranger A, Marceau N, Theriault JR, Lambert H, Landry J. Distinct chaperone mechanisms can delay the formation of aggresomes by the myopathy-causing R120G alphaB-crystallin mutant. Hum Mol Genet. 2003;12:1609–1620. doi: 10.1093/hmg/ddg173. [DOI] [PubMed] [Google Scholar]
- Chevalier-Larsen ES, O'Brien CJ, Wang H, Jenkins SC, Holder L, Lieberman AP, Merry DE. Castration restores function and neurofilament alterations of aged symptomatic males in a transgenic mouse model of spinal and bulbar muscular atrophy. J Neurosci. 2004;24:4778–4786. doi: 10.1523/JNEUROSCI.0808-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell P, Ballinger CA, Jiang J, Wu Y, Thompson LJ, Hohfeld J, Patterson C. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat Cell Biol. 2001;3:93–96. doi: 10.1038/35050618. [DOI] [PubMed] [Google Scholar]
- Cummings CJ, Mancini MA, Antalffy B, DeFranco DB, Orr HT, Zoghbi HY. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet. 1998;19:148–154. doi: 10.1038/502. [DOI] [PubMed] [Google Scholar]
- Cyr DM, Hohfeld J, Patterson C. Protein quality control: U-box-containing E3 ubiquitin ligases join the fold. Trends Biochem Sci. 2002;27:368–375. doi: 10.1016/s0968-0004(02)02125-4. [DOI] [PubMed] [Google Scholar]
- Dai Q, Zhang C, Wu Y, McDonough H, Whaley RA, Godfrey V, Li HH, Madamanchi N, Xu W, Neckers L, Cyr D, Patterson C. CHIP activates HSF1 and confers protection against apoptosis and cellular stress. EMBO J. 2003;22:5446–5458. doi: 10.1093/emboj/cdg529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daviau A, Proulx R, Robitaille K, Di Fruscio M, Tanguay RM, Landry J, Patterson C, Durocher Y, Blouin R. Down-regulation of the mixed-lineage dual leucine zipper-bearing kinase by heat shock protein 70 and its co-chaperone CHIP. J Biol Chem. 2006;281:31467–31477. doi: 10.1074/jbc.M607612200. [DOI] [PubMed] [Google Scholar]
- Demand J, Alberti S, Patterson C, Hohfeld J. Cooperation of a ubiquitin domain protein and an E3 ubiquitin ligase during chaperone/proteasome coupling. Curr Biol. 2001;11:1569–1577. doi: 10.1016/s0960-9822(01)00487-0. [DOI] [PubMed] [Google Scholar]
- Dickey CA, Yue M, Lin WL, Dickson DW, Dunmore JH, Lee WC, Zehr C, West G, Cao S, Clark AM, Caldwell GA, Caldwell KA, Eckman C, Patterson C, Hutton M, Petrucelli L. Deletion of the ubiquitin ligase CHIP leads to the accumulation, but not the aggregation, of both endogenous phospho- and caspase-3-cleaved tau species. J Neurosci. 2006;26:6985–6996. doi: 10.1523/JNEUROSCI.0746-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Prospero NA, Fischbeck KH. Therapeutics development for triplet repeat expansion diseases. Nat Rev Genet. 2005;6:756–765. doi: 10.1038/nrg1690. [DOI] [PubMed] [Google Scholar]
- Doyu M, Sobue G, Mukai E, Kachi T, Yasuda T, Mitsuma T, Takahashi A. Severity of X-linked recessive bulbospinal neuronopathy correlates with size of the tandem CAG repeat in androgen receptor gene. Ann Neurol. 1992;32:707–710. doi: 10.1002/ana.410320517. [DOI] [PubMed] [Google Scholar]
- Esser C, Scheffner M, Hohfeld J. The chaperone-associated ubiquitin ligase CHIP is able to target p53 for proteasomal degradation. J Biol Chem. 2005;280:27443–27448. doi: 10.1074/jbc.M501574200. [DOI] [PubMed] [Google Scholar]
- Gatchel JR, Zoghbi HY. Diseases of unstable repeat expansion: mechanisms and common principles. Nat Rev Genet. 2005;6:743–755. doi: 10.1038/nrg1691. [DOI] [PubMed] [Google Scholar]
- Grelle G, Kostka S, Otto A, Kersten B, Genser KF, Muller EC, Walter S, Boddrich A, Stelzl U, Hanig C, Volkmer-Engert R, Landgraf C, Alberti S, Hohfeld J, Strodicke M, Wanker EE. Identification of VCP/p97, carboxyl terminus of Hsp70-interacting protein (CHIP), and amphiphysin II interaction partners using membrane-based human proteome arrays. Mol Cell Proteomics. 2006;5:234–244. doi: 10.1074/mcp.M500198-MCP200. [DOI] [PubMed] [Google Scholar]
- Hatakeyama S, Yada M, Matsumoto M, Ishida N, Nakayama KI. U box proteins as a new family of ubiquitin-protein ligases. J Biol Chem. 2001;276:33111–33120. doi: 10.1074/jbc.M102755200. [DOI] [PubMed] [Google Scholar]
- Hatakeyama S, Matsumoto M, Kamura T, Murayama M, Chui DH, Planel E, Takahashi R, Nakayama KI, Takashima A. U-box protein carboxyl terminus of Hsc70-interacting protein (CHIP) mediates poly-ubiquitylation preferentially on four-repeat Tau and is involved in neurodegeneration of tauopathy. J Neurochem. 2004;91:299–307. doi: 10.1111/j.1471-4159.2004.02713.x. [DOI] [PubMed] [Google Scholar]
- He B, Bai S, Hnat AT, Kalman RI, Minges JT, Patterson C, Wilson EM. An androgen receptor NH2-terminal conserved motif interacts with the COOH terminus of the Hsp70-interacting protein (CHIP) J Biol Chem. 2004;279:30643–30653. doi: 10.1074/jbc.M403117200. [DOI] [PubMed] [Google Scholar]
- Huang Z, Nie L, Xu M, Sun XH. Notch-induced E2A degradation requires CHIP and Hsc70 as novel facilitators of ubiquitination. Mol Cell Biol. 2004;24:8951–8962. doi: 10.1128/MCB.24.20.8951-8962.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang JR, Zhang C, Patterson C. C-terminus of heat shock protein 70-interacting protein facilitates degradation of apoptosis signal-regulating kinase 1 and inhibits apoptosis signal-regulating kinase 1-dependent apoptosis. Cell Stress Chaperones. 2005;10:147–156. doi: 10.1379/CSC-90R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi S, Tanno Y, Onodera O, Yamazaki M, Sato S, Ishikawa A, Miyatani N, Nagashima M, Ishikawa Y, Sahashi K, Ibi T, Miyatake T, Tsuji S. Strong correlation between the number of CAG repeats in androgen receptor genes and the clinical onset of features of spinal and bulbar muscular atrophy. Neurology. 1992;42:2300–2302. doi: 10.1212/wnl.42.12.2300. [DOI] [PubMed] [Google Scholar]
- Ishigaki S, Liang Y, Yamamoto M, Niwa J, Ando Y, Yoshihara T, Takeuchi H, Doyu M, Sobue G. X-linked inhibitor of apoptosis protein is involved in mutant SOD1-mediated neuronal degeneration. J Neurochem. 2002;82:576–584. doi: 10.1046/j.1471-4159.2002.00998.x. [DOI] [PubMed] [Google Scholar]
- Jana NR, Dikshit P, Goswami A, Kotliarova S, Murata S, Tanaka K, Nukina N. Co-chaperone CHIP associates with expanded polyglutamine protein and promotes their degradation by proteasomes. J Biol Chem. 2005;280:11635–11640. doi: 10.1074/jbc.M412042200. [DOI] [PubMed] [Google Scholar]
- Jiang J, Ballinger CA, Wu Y, Dai Q, Cyr DM, Hohfeld J, Patterson C. CHIP is a U-box-dependent E3 ubiquitin ligase: identification of Hsc70 as a target for ubiquitylation. J Biol Chem. 2001;276:42938–42944. doi: 10.1074/jbc.M101968200. [DOI] [PubMed] [Google Scholar]
- Katsuno M, Adachi H, Kume A, Li M, Nakagomi Y, Niwa H, Sang C, Kobayashi Y, Doyu M, Sobue G. Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron. 2002;35:843–854. doi: 10.1016/s0896-6273(02)00834-6. [DOI] [PubMed] [Google Scholar]
- Katsuno M, Adachi H, Doyu M, Minamiyama M, Sang C, Kobayashi Y, Inukai A, Sobue G. Leuprorelin rescues polyglutamine-dependent phenotypes in a transgenic mouse model of spinal and bulbar muscular atrophy. Nat Med. 2003;9:768–773. doi: 10.1038/nm878. [DOI] [PubMed] [Google Scholar]
- Katsuno M, Sang C, Adachi H, Minamiyama M, Waza M, Tanaka F, Doyu M, Sobue G. Pharmacological induction of heat-shock proteins alleviates polyglutamine-mediated motor neuron disease. Proc Natl Acad Sci USA. 2005;102:16801–16806. doi: 10.1073/pnas.0506249102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuno M, Adachi H, Minamiyama M, Waza M, Tokui K, Banno H, Suzuki K, Onoda Y, Tanaka F, Doyu M, Sobue G. Reversible disruption of dynactin 1-mediated retrograde axonal transport in polyglutamine-induced motor neuron degeneration. J Neurosci. 2006;26:12106–12117. doi: 10.1523/JNEUROSCI.3032-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy WR, Alter M, Sung JH. Progressive proximal spinal and bulbar muscular atrophy of late onset. A sex-linked recessive trait. Neurology. 1968;18:671–680. doi: 10.1212/wnl.18.7.671. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Kume A, Li M, Doyu M, Hata M, Ohtsuka K, Sobue G. Chaperones Hsp70 and Hsp40 suppress aggregate formation and apoptosis in cultured neuronal cells expressing truncated androgen receptor protein with expanded polyglutamine tract. J Biol Chem. 2000;275:8772–8778. doi: 10.1074/jbc.275.12.8772. [DOI] [PubMed] [Google Scholar]
- La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352:77–79. doi: 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- La Spada AR, Roling DB, Harding AE, Warner CL, Spiegel R, Hausmanowa-Petrusewicz I, Yee WC, Fischbeck KH. Meiotic stability and genotype-phenotype correlation of the trinucleotide repeat in X-linked spinal and bulbar muscular atrophy. Nat Genet. 1992;2:301–304. doi: 10.1038/ng1292-301. [DOI] [PubMed] [Google Scholar]
- Li M, Miwa S, Kobayashi Y, Merry DE, Yamamoto M, Tanaka F, Doyu M, Hashizume Y, Fischbeck KH, Sobue G. Nuclear inclusions of the androgen receptor protein in spinal and bulbar muscular atrophy. Ann Neurol. 1998a;44:249–254. doi: 10.1002/ana.410440216. [DOI] [PubMed] [Google Scholar]
- Li M, Nakagomi Y, Kobayashi Y, Merry DE, Tanaka F, Doyu M, Mitsuma T, Hashizume Y, Fischbeck KH, Sobue G. Nonneural nuclear inclusions of androgen receptor protein in spinal and bulbar muscular atrophy. Am J Pathol. 1998b;153:695–701. doi: 10.1016/S0002-9440(10)65612-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman AP, Harmison G, Strand AD, Olson JM, Fischbeck KH. Altered transcriptional regulation in cells expressing the expanded polyglutamine androgen receptor. Hum Mol Genet. 2002;11:1967–1976. doi: 10.1093/hmg/11.17.1967. [DOI] [PubMed] [Google Scholar]
- Marques C, Guo W, Pereira P, Taylor A, Patterson C, Evans PC, Shang F. The triage of damaged proteins: degradation by the ubiquitin-proteasome pathway or repair by molecular chaperones. FASEB J. 2006;20:741–743. doi: 10.1096/fj.05-5080fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan AJ, Tam S, Kaganovich D, Frydman J. Protein quality control: chaperones culling corrupt conformations. Nat Cell Biol. 2005;7:736–741. doi: 10.1038/ncb0805-736. [DOI] [PubMed] [Google Scholar]
- McDonough H, Patterson C. CHIP: a link between the chaperone and proteasome systems. Cell Stress Chaperones. 2003;8:303–308. doi: 10.1379/1466-1268(2003)008<0303:calbtc>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meacham GC, Patterson C, Zhang W, Younger JM, Cyr DM. The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat Cell Biol. 2001;3:100–105. doi: 10.1038/35050509. [DOI] [PubMed] [Google Scholar]
- Miller VM, Nelson RF, Gouvion CM, Williams A, Rodriguez-Lebron E, Harper SQ, Davidson BL, Rebagliati MR, Paulson HL. CHIP suppresses polyglutamine aggregation and toxicity in vitro and in vivo . J Neurosci. 2005;25:9152–9161. doi: 10.1523/JNEUROSCI.3001-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamiyama M, Katsuno M, Adachi H, Waza M, Sang C, Kobayashi Y, Tanaka F, Doyu M, Inukai A, Sobue G. Sodium butyrate ameliorates phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Hum Mol Genet. 2004;13:1183–1192. doi: 10.1093/hmg/ddh131. [DOI] [PubMed] [Google Scholar]
- Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- Murata S, Minami Y, Minami M, Chiba T, Tanaka K. CHIP is a chaperone-dependent E3 ligase that ubiquitylates unfolded protein. EMBO Rep. 2001;2:1133–1138. doi: 10.1093/embo-reports/kve246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata S, Chiba T, Tanaka K. CHIP: a quality-control E3 ligase collaborating with molecular chaperones. Int J Biochem Cell Biol. 2003;35:572–578. doi: 10.1016/s1357-2725(02)00394-1. [DOI] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Petrucelli L, Dickson D, Kehoe K, Taylor J, Snyder H, Grover A, De Lucia M, McGowan E, Lewis J, Prihar G, Kim J, Dillmann WH, Browne SE, Hall A, Voellmy R, Tsuboi Y, Dawson TM, Wolozin B, Hardy J, Hutton M. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet. 2004;13:703–714. doi: 10.1093/hmg/ddh083. [DOI] [PubMed] [Google Scholar]
- Poletti A, Negri-Cesi P, Martini L. Reflections on the diseases linked to mutations of the androgen receptor. Endocrine. 2005;28:243–262. doi: 10.1385/ENDO:28:3:243. [DOI] [PubMed] [Google Scholar]
- Qian SB, McDonough H, Boellmann F, Cyr DM, Patterson C. CHIP-mediated stress recovery by sequential ubiquitination of substrates and Hsp70. Nature. 2006;440:551–555. doi: 10.1038/nature04600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees I, Lee S, Kim H, Tsai FT. The E3 ubiquitin ligase CHIP binds the androgen receptor in a phosphorylation-dependent manner. Biochim Biophys Acta. 2006;1764:1073–1079. doi: 10.1016/j.bbapap.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Ross CA, Pickart CM. The ubiquitin-proteasome pathway in Parkinson's disease and other neurodegenerative diseases. Trends Cell Biol. 2004;14:703–711. doi: 10.1016/j.tcb.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10(Suppl):S10–S17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- Sahara N, Murayama M, Mizoroki T, Urushitani M, Imai Y, Takahashi R, Murata S, Tanaka K, Takashima A. In vivo evidence of CHIP up-regulation attenuating tau aggregation. J Neurochem. 2005;94:1254–1263. doi: 10.1111/j.1471-4159.2005.03272.x. [DOI] [PubMed] [Google Scholar]
- Schmidt T, Lindenberg KS, Krebs A, Schols L, Laccone F, Herms J, Rechsteiner M, Riess O, Landwehrmeyer GB. Protein surveillance machinery in brains with spinocerebellar ataxia type 3: redistribution and differential recruitment of 26S proteasome subunits and chaperones to neuronal intranuclear inclusions. Ann Neurol. 2002;51:302–310. doi: 10.1002/ana.10101. [DOI] [PubMed] [Google Scholar]
- Shin Y, Klucken J, Patterson C, Hyman BT, McLean PJ. The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates alpha-synuclein degradation decisions between proteasomal and lysosomal pathways. J Biol Chem. 2005;280:23727–23734. doi: 10.1074/jbc.M503326200. [DOI] [PubMed] [Google Scholar]
- Slow EJ, Graham RK, Hayden MR. To be or not to be toxic: aggregations in Huntington and Alzheimer disease. Trends Genet. 2006;22:408–411. doi: 10.1016/j.tig.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Sobue G, Hashizume Y, Mukai E, Hirayama M, Mitsuma T, Takahashi A. X-linked recessive bulbospinal neuronopathy. A clinicopathological study. Brain. 1989;112:209–232. doi: 10.1093/brain/112.1.209. [DOI] [PubMed] [Google Scholar]
- Sobue G, Doyu M, Kachi T, Yasuda T, Mukai E, Kumagai T, Mitsuma T. Subclinical phenotypic expressions in heterozygous females of X-linked recessive bulbospinal neuronopathy. J Neurol Sci. 1993;117:74–78. doi: 10.1016/0022-510x(93)90157-t. [DOI] [PubMed] [Google Scholar]
- Sopher BL, Thomas PS, Jr, LaFevre-Bernt MA, Holm IE, Wilke SA, Ware CB, Jin LW, Libby RT, Ellerby LM, La Spada AR. Androgen receptor YAC transgenic mice recapitulate SBMA motor neuronopathy and implicate VEGF164 in the motor neuron degeneration. Neuron. 2004;41:687–699. doi: 10.1016/s0896-6273(04)00082-0. [DOI] [PubMed] [Google Scholar]
- Sperfeld AD, Karitzky J, Brummer D, Schreiber H, Haussler J, Ludolph AC, Hanemann CO. X-linked bulbospinal neuronopathy: Kennedy disease. Arch Neurol. 2002;59:1921–1926. doi: 10.1001/archneur.59.12.1921. [DOI] [PubMed] [Google Scholar]
- Stenoien DL, Cummings CJ, Adams HP, Mancini MG, Patel K, DeMartino GN, Marcelli M, Weigel NL, Mancini MA. Polyglutamine-expanded androgen receptors form aggregates that sequester heat shock proteins, proteasome components and SRC-1, and are suppressed by the HDJ-2 chaperone. Hum Mol Genet. 1999;8:731–741. doi: 10.1093/hmg/8.5.731. [DOI] [PubMed] [Google Scholar]
- Tanaka F, Doyu M, Ito Y, Matsumoto M, Mitsuma T, Abe K, Aoki M, Itoyama Y, Fischbeck KH, Sobue G. Founder effect in spinal and bulbar muscular atrophy (SBMA) Hum Mol Genet. 1996;5:1253–1257. doi: 10.1093/hmg/5.9.1253. [DOI] [PubMed] [Google Scholar]
- Tateishi Y, Kawabe Y, Chiba T, Murata S, Ichikawa K, Murayama A, Tanaka K, Baba T, Kato S, Yanagisawa J. Ligand-dependent switching of ubiquitin-proteasome pathways for estrogen receptor. EMBO J. 2004;23:4813–4823. doi: 10.1038/sj.emboj.7600472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas M, Dadgar N, Aphale A, Harrell JM, Kunkel R, Pratt WB, Lieberman AP. Androgen receptor acetylation site mutations cause trafficking defects, misfolding, and aggregation similar to expanded glutamine tracts. J Biol Chem. 2004;279:8389–8395. doi: 10.1074/jbc.M311761200. [DOI] [PubMed] [Google Scholar]
- Wang X, DeFranco DB. Alternative effects of the ubiquitin-proteasome pathway on glucocorticoid receptor down-regulation and transactivation are mediated by CHIP, an E3 ligase. Mol Endocrinol. 2005;19:1474–1482. doi: 10.1210/me.2004-0383. [DOI] [PubMed] [Google Scholar]
- Wanker EE, Scherzinger E, Heiser V, Sittler A, Eickhoff H, Lehrach H. Membrane filter assay for detection of amyloid-like polyglutamine-containing protein aggregates. Methods Enzymol. 1999;309:375–386. doi: 10.1016/s0076-6879(99)09026-6. [DOI] [PubMed] [Google Scholar]
- Waza M, Adachi H, Katsuno M, Minamiyama M, Sang C, Tanaka F, Inukai A, Doyu M, Sobue G. 17-AAG, an Hsp90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration. Nat Med. 2005;11:1088–1095. doi: 10.1038/nm1298. [DOI] [PubMed] [Google Scholar]
- Wickner S, Maurizi MR, Gottesman S. Posttranslational quality control: folding, refolding, and degrading proteins. Science. 1999;286:1888–1893. doi: 10.1126/science.286.5446.1888. [DOI] [PubMed] [Google Scholar]
- Younger JM, Chen L, Ren HY, Rosser MF, Turnbull EL, Fan CY, Patterson C, Cyr DM. Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell. 2006;126:571–582. doi: 10.1016/j.cell.2006.06.041. [DOI] [PubMed] [Google Scholar]
- Yu Z, Dadgar N, Albertelli M, Gruis K, Jordan C, Robins DM, Lieberman AP. Androgen-dependent pathology demonstrates myopathic contribution to the Kennedy disease phenotype in a mouse knock-in model. J Clin Invest. 2006;116:2663–2672. doi: 10.1172/JCI28773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Fernandes N, Dodge IL, Reddi AL, Rao N, Safran H, DiPetrillo TA, Wazer DE, Band V, Band H. ErbB2 degradation mediated by the co-chaperone protein CHIP. J Biol Chem. 2003;278:13829–13837. doi: 10.1074/jbc.M209640200. [DOI] [PubMed] [Google Scholar]