

Abstract
P/Q-type (CaV2.1) and N-type (CaV2.2) Ca2+ channels are critical to stimulus-secretion coupling in the nervous system; feedback regulation of these channels by Ca2+ is therefore predicted to profoundly influence neurotransmission. Here we report divergent regulation of Ca2+-dependent inactivation (CDI) of native N- and P/Q-type Ca2+ channels by calmodulin (CaM) in adult chromaffin cells. Robust CDI of N-type channels was observed in response to prolonged step depolarizations, as well as repetitive stimulation with either brief step depolarizations or action potential-like voltage stimuli. Adenoviral expression of Ca2+-insensitive calmodulin mutants eliminated CDI of N-type channels. This is the first demonstration of CaM-dependent CDI of a native N-type channel. CDI of P/Q-type channels was by comparison modest and insensitive to expression of CaM mutants. Cloning of the C terminus of the CaV2.1 α1 subunit from chromaffin cells revealed multiple splice variants lacking structural motifs required for CaM-dependent CDI. The physiological relevance of CDI on stimulus-coupled exocytosis was revealed by combining perforated-patch voltage-clamp recordings of pharmacologically isolated Ca2+ currents with membrane capacitance measurements of exocytosis. Increasing stimulus intensity to invoke CDI resulted in a significant decrease in the exocytotic efficiency of N-type channels compared with P/Q-type channels. Our results reveal unexpected diversity in CaM regulation of native CaV2 channels and suggest that the ability of individual Ca2+ channel subtypes to undergo CDI may be tailored by alternative splicing to meet the specific requirements of a particular cellular function.
Keywords: voltage-gated calcium channels, CaV2.1, CaV2.2, calmodulin, chromaffin cells, exocytosis
Introduction
Voltage-gated Ca2+ channels (VGCCs) mediate Ca2+ influx into excitable cells in response to membrane depolarization, thereby providing a vital link between membrane excitability, action potential firing, and fundamental processes such as neurotransmitter release (Catterall, 2000). Inactivation is an important determinant of VGCC function because it helps define the temporal properties of Ca2+ signals. Two factors controlling VGCC inactivation are voltage and Ca2+ (Cens et al., 2006). Both forms of inactivation are regulated by multiple domains within the main pore-forming α subunit and auxiliary β subunit. Investigations on heterologously expressed CaV1.2 (L-type) and CaV2.1 (P/Q-type) channels have identified calmodulin (CaM) as the Ca2+ sensor mediating Ca2+-dependent inactivation (CDI) (Petersen et al., 1999; Zuhlke et al., 1999; Halling et al., 2005). CaM binds to an IQ-like motif in the C′ tail of the α1 subunit, and Ca2+ binding to CaM produces lobe-specific effects on channel function (Pate et al., 2000; DeMaria et al., 2001; Erickson et al., 2001; Lee et al., 2003; Liang et al., 2003; Kim et al., 2004; Petegem et al., 2005). With CaV2.1, Ca2+ binding to the C-terminal lobe of CaM induces Ca2+-dependent facilitation (CDF), whereas binding to the N-terminal lobe produces CDI (DeMaria et al., 2001; Lee et al., 2003). Lobe-specific modulation has important consequences with regards to the spatiotemporal properties of the Ca2+ signals required to induce CDI/CDF. C-lobe-specific CDF is activated by Ca2+ microdomains generated near open Ca2+ channels and may regulate the duration of a single action potential. N-lobe-specific CDI is sensitive to global Ca2+ elevations and therefore develops more slowly, typically requiring a train of action potentials (Lee et al., 2000). The spatiotemporal properties of Ca2+ signals supporting exocytosis are also vitally important, with prolonged signals required to support vesicle priming (Seward et al., 1995; Seward and Nowycky, 1996) and full fusion (Elhamdani et al., 2006; Fulop and Smith, 2006). Feedback regulation of CaV2 channels by CaM may therefore be an important regulator of exocytosis.
Although CaM-dependent CDI of other members of the CaV2 family, including a cloned variant of CaV2.2 (N-type), has been reported (Patil et al., 1998; Cens et al., 1999; Jones et al., 1999; Petersen et al., 1999; Zuhlke et al., 1999; Liang et al., 2003), the prevalence and function of this form of regulation in native CaV2 channels remains unknown. Here we examined CDI of endogenous CaV2.1 and CaV2.2 channels in chromaffin cells. Because CaM-dependent CDI of CaV2 channels is sensitive to intracellular Ca2+ chelation (Liang et al., 2003; Meuth et al., 2005; Kreiner and Lee, 2006), we used the perforated-patch configuration to maintain physiological buffering conditions. We show that endogenous CaV2.2 channels undergo prominent CDI through a CaM-dependent mechanism; CaV2.1 channels lack such modulation. Cloning the C terminus of CaV2.1 α1 subunit revealed variants lacking IQ-like domains. The consequences of divergent CDI on depolarization-evoked exocytosis were examined with membrane capacitance (Cm) measurements. Because stimulus strength was increased to promote CDI, N-type channels lost their ability to support exocytosis, whereas P/Q-type channels continued to provide the Ca2+ signals required to support secretion. The results suggest that CaM-dependent CDI of CaV2 channels may be regulated in a cell- and function-specific context.
Materials and Methods
Chromaffin cell culture and adenoviral infection.
Chromaffin cells were prepared as described previously (Powell et al., 2000). Infections with recombinant adenovirus bearing enhanced green fluorescent protein (EGFP) (gift from Terry Herbert, University of Leicester, Leicester, UK), wild-type CaM (CaMWT–pIRES–GFP), or a mutant CaM incapable of binding Ca2+ (CaM1234–pIRES–GFP) (gift from David Yue, John Hopkins University School of Medicine, Baltimore, MD) were performed immediately after media replacement on the day after plating. Phosphorylated internal ribosomal entry site (pIRES) constructs were used so that wild-type or mutant CaM and EGFP were expressed as separate proteins (Alseikhan et al., 2002), permitting identification of positively infected cells, without possible perturbation of CaM function. The addition of 3 μl viral stock/100,000 cells to the culture medium resulted in a transfection efficiency of >50% 48 h after infection. Viral titer (in plaque forming units per milliliter) were determined in HEK 293 cells to be 6e+10 pfu/ml for CaMWT–pIRES–GFP, 8e+9 pfu/ml for CaM1234–pIRES–GFP, and 6e+7 pfu/ml for EGFP.
Electrophysiology.
All electrophysiological recordings were acquired at room temperature as described previously (Powell et al., 2000). Cells were continuously superfused at a high flow rate (>2 ml/min) with an external solution containing the following (in mm): 150 NaCl, 2 KCl, 5 NaHCO3, 1 MgCl2, 2.5 CaCl2, 10 glucose, and 10 HEPES, pH adjusted to 7.2 with NaOH (osmolarity ∼290–300 mOsm). After cells were voltage clamped (holding potential, −80 mV), the external solution was replaced with one in which the concentration of NaCl was dropped to 135 mm, and 10 mm tetraethylammonium was added to prevent a slowly developing Ca2+-activated current. Tetrodotoxin was omitted because sodium channels serve as an identification marker for chromaffin cells; they are notably absent from adrenal cortical cells that can make up to 10% of our cultures. ICa and Cm were recorded in either the whole-cell or perforated-patch configuration using borosilicate glass electrodes coated with Sylguard 184 (Dow Corning, Midland, MI) and fire polished to a resistance of 1.5–2.5 MΩ. Electrodes were filled with internal solution containing the following (in mm): 145 Cs-glutamate (Calbiochem, Nottingham, UK), 10 HEPES, 8.5 NaCl, 2 Mg-ATP, and 0.3 mm BAPTA (Invitrogen, Carlsbad, CA), adjusted to pH 7.3 with CsOH (ICN Biomedicals, Aurora, OH) (osmolarity ∼290 mOsm). For perforated-patch experiments, Mg-ATP was omitted from the internal solution and gramicidin D (Sigma, St. Louis, MO) at a final concentration of ∼50 μg/ml in DMSO was added. Series resistance was <17 MΩ. Voltage-clamp experiments and Cm measurements described in Figures 1–6 and 8 and supplemental Figures 1 and 2 (available at www.jneurosci.org as supplemental material) were performed using an Axopatch 200B amplifier (Molecular Devices, Palo Alto, CA) using custom acquisition software (provided by Dr. A. P. Fox, University of Chicago, Chicago, IL) running on a Pentium computer equipped with a Digidata acquisition board (Molecular Devices). Current traces were low-pass filtered at 5kHz using the four-pole Bessel filter supplied with the amplifier and digitized at 10kHz. Changes in Cm were measured using the software-based phase-tracking method relative to a 100 fF calibration signal (Powell et al., 2000). Cm were interrupted to stimulate cells with depolarizing voltage steps from a holding potential of −80 mV to a test potential of +20. Data were stored on the computer hard drive and analyzed off-line using self-written analysis software (Axobasic; Molecular Devices) and commercial software (Origin; Microcal, Northampton, MA). No corrections were made for liquid junction potentials. ICa was not leak subtracted, and only cells with a leak current <15 pA were included in the analysis. Peak ICa (in picoamperes) was detected, and calcium entry was quantified by integration. The left integration limit was set at 3 ms into the voltage pulse to exclude Na+ current, and the right limit was set to exclude tail current. Stimulations with action potential-like waveforms (see Fig. 7) were performed with an EPC-10 amplifier (HEKA Elektronik, Lambrecht, Germany) running Patchmaster version 2.2 software. Voltage-clamp records were acquired at 20 kHz and filtered at 3 kHz. Neither leak current subtraction nor series resistance compensation were performed. Data analysis was performed off-line using Fitmaster version 2.2 (HEKA Elektronik). The action potential waveform consisted of a ramp up from the holding potential of −80 to +50 mV over 2.5 ms, followed by a ramp down to −90 mV over 2.5 ms, followed by a ramp up to −80 mV over 2.5 ms. This waveform is the same as that described previously (Polo-Parada et al., 2006) and mimics native action potentials in chromaffin cells. Inward currents evoked by the action potential-like voltage protocol were biphasic, consisting of an initial, rapidly inactivating Na+ current followed by a temporally isolated ICa. Measurements of peak ICa were limited to the second component of inward current.
Figure 1.
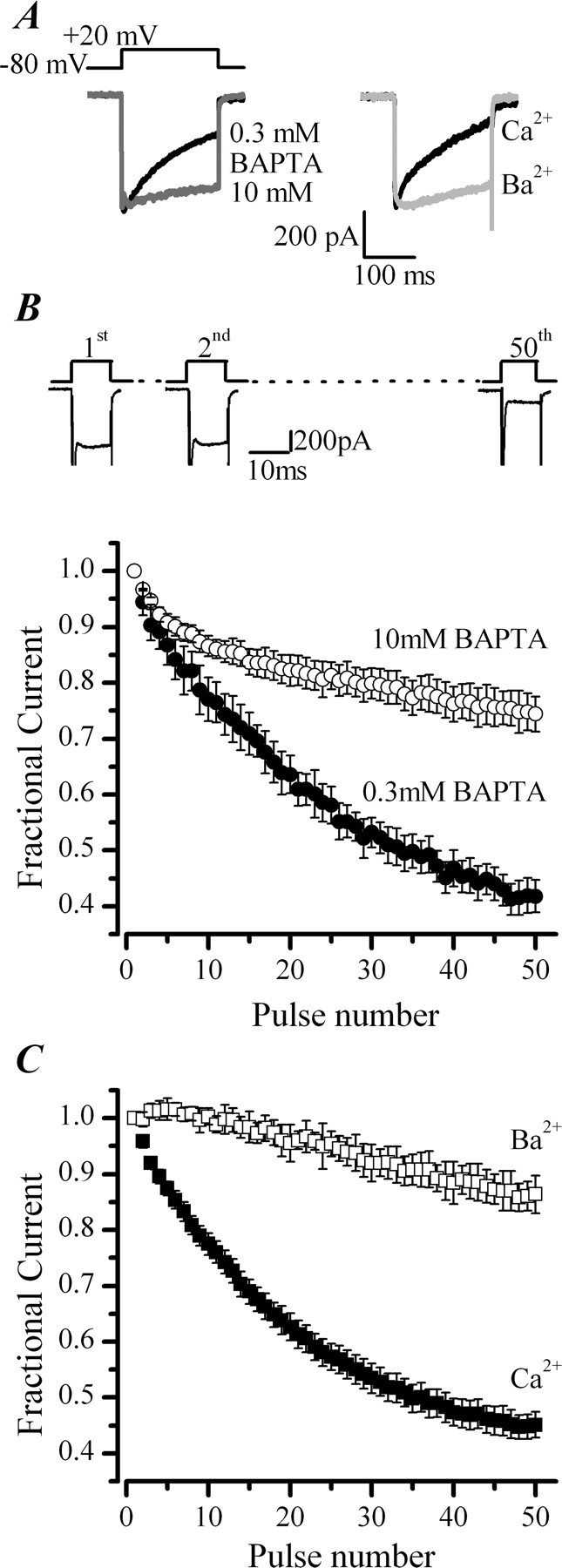
CDI of ICa in chromaffin cells. A, Left, Representative ICa recorded in the whole-cell patch configuration with low and high BAPTA added to the pipette solution. A, Right, Representative ICa recorded under perforated-patch conditions before and after equimolar replacement of extracellular Ca2+ with Ba2+. B, Top, Schematic diagram of the voltage protocol and representative ICa recorded during the first two and final depolarization of a train of 50 pulses from −80 to +20 mV delivered at 20 Hz. Fractional current was calculated from the peak ICa measured for each pulse in the train, normalized to the peak ICa elicited by the first pulse in the train. Summary plot showing the effects of raising the intracellular Ca2+ chelator BAPTA from 0.3 mm (•; n = 7) to 10 mm (○; n = 5) on ICa evoked by the train protocol. C, Effect of replacing extracellular Ca2+ (■; n = 20) with equimolar Ba2+ (□; n = 3) on ICa evoked by the train protocol.
Figure 2.

Pharmacological identification of VGCCs expressed by adult bovine chromaffin cells. A, Superimposed ICa from a representative chromaffin cell before and after 5 min application of the P/Q-type-selective inhibitor AgaIVA (300 nm) and N-type-selective inhibitor ω-CgTX (1 μm). B, Corresponding Cm recorded in the same cell. Gaps in the traces indicate timing of the depolarizing pulses during which capacitance detection is interrupted. Combined inhibition of N- and P/Q-type channels abolishes Ca2+ entry and exocytosis in adult bovine chromaffin cells. C, Mean ± SEM percentage inhibition in Ca2+ entry (determined by integration of ICa; hatched bars) and corresponding ΔCm (filled bars) resulting from combined application of AgaIVA (300 nm) and ω-CgTX (1 μm) (n = 3), the L-type channel inhibitor nimodipine (Nmdp; 10 μm, n = 3), or the N- and P/Q-type inhibitor ω-CgTX MVIIC (10 μm; n = 3).
Figure 3.
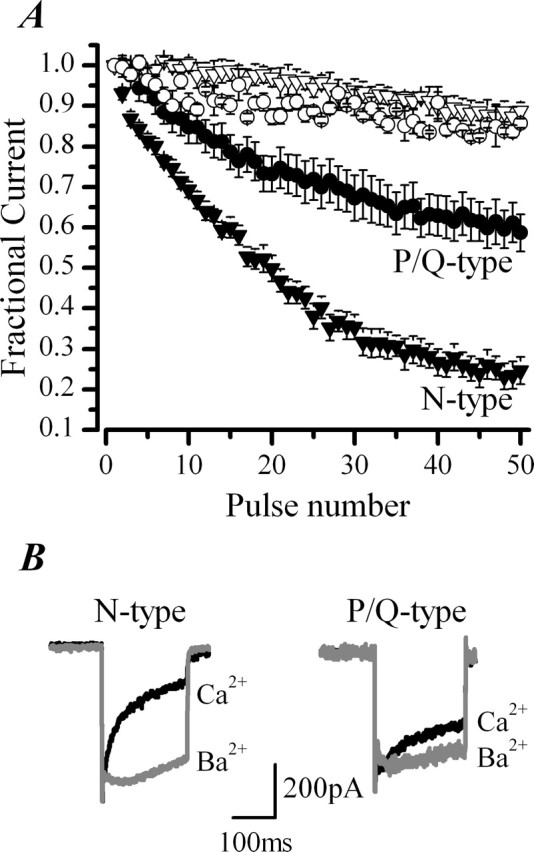
CDI is more pronounced in N-type channels than P/Q-type channels in chromaffin cells. Replacement of extracellular Ca2+ with Ba2+ was used to probe CDI of pharmacologically isolated N- and P/Q-type channels. A, ICa was evoked with a train of 50 pulses of 10 ms duration from −80 to +20 mV, delivered at 20 Hz. Mean ± SEM data are from n = 10 cells recorded in perforated-patch conditions in either the presence of 300 nm AgaIVA to isolate N-type channels (▿, ▾) or 1 μm CgTX to isolate P/Q-type channels (○, •). Cells were stimulated first in control external solution (2.5 mm Ca2+) (▾, •) and then after equimolar replacement with Ba2+ (▿, ○). The difference in CDI between N-type and P/Q-type channels at the 50th pulse in Ca2+ is significantly different (p < 0.0001). B, Superimposed current traces of pharmacologically isolated N-type and P/Q-type channels recorded in response to a 200 ms depolarization from −80 to +20 mV before and after replacement of extracellular Ca2+ (black trace) with Ba2+ (gray trace). I200/Ipeak for N-type channels in Ca2+ was 40 ± 7% (n = 15) compared with 98 ± 1% (n = 3) in Ba2+ and for P/Q-type currents I200/Ipeak increased from 53 ± 6% (n = 15) in Ca2+ to 84 ± 3% (n = 5) in Ba2 +.
Figure 4.

Adenovirus provides an efficient and nontoxic method for exogenous gene expression in chromaffin cells. A, The top shows a bright-field image of chromaffin cells 54 h after infection with CaMWT–pIRES–GFP adenovirus. Scale bar, 10 μm. Infected cells were identified by their EGFP fluorescence (middle). Dim cells (indicated by the arrow) were selected for electrophysiological experiments. Apoptosis was assessed with the DNA stain Hoechst 33342 (bottom). B, Correlation between EGFP expression levels and ICa density after adenoviral infection. Cells were infected with either EGFP adenovirus or CaMWT–pIRES–GFP adenovirus for 48 h. Tissue culture-matched uninfected cells (UIF) served as a control. EGFP fluorescence was measured and correlated to the size of the cell (brightness density in volts per picofarads). Accordingly, cells were categorized as dim (dark gray bars; <0.05 V/pF) or bright (light gray bars; >0.05 V/pF). Despite cell-to-cell variability, there is no significant difference overall in fluorescence measured between the dim EGFP and CaMWT group of cells. Thus, the mean ± SD fluorescence density for dim cells expressing EGFP was 0.016287 ± 0.00876 V/pF (range, 0.00462–0.037931 V/pF; n = 34) compared with 0.02207 ± 0.01017 V/pF (range, 0.00678–0.04286 V/pF; n = 26) for cells expressing CaMWT and EGFP. ICa density was calculated from the peak current evoked by a 10 ms pulse from a holding potential of −80 mV to a test potential of +20 mV divided by the cell size assessed from the whole-cell capacitance. Error bars represent mean ± SEM for number of cells indicated, **p < 0.01. C, Sample ICa evoked by a 200 ms depolarization from −80 to +20 mV in an uninfected and EGFP adenovirus-infected cell. I200/Ipeak was 55 ± 6%, n = 7 for control cells and 48 ± 7%, n = 4 for EGFP-expressing cells. D, Inactivation after repetitive depolarizations by a train of pulses (50 pulses for 10 ms at 20 Hz) was not significantly affected by adenovirus infection. Points represent the mean ± SEM fractional current measured in n = 7 uninfected cells and n = 4 culture-matched EGFP-infected cells.
Figure 5.
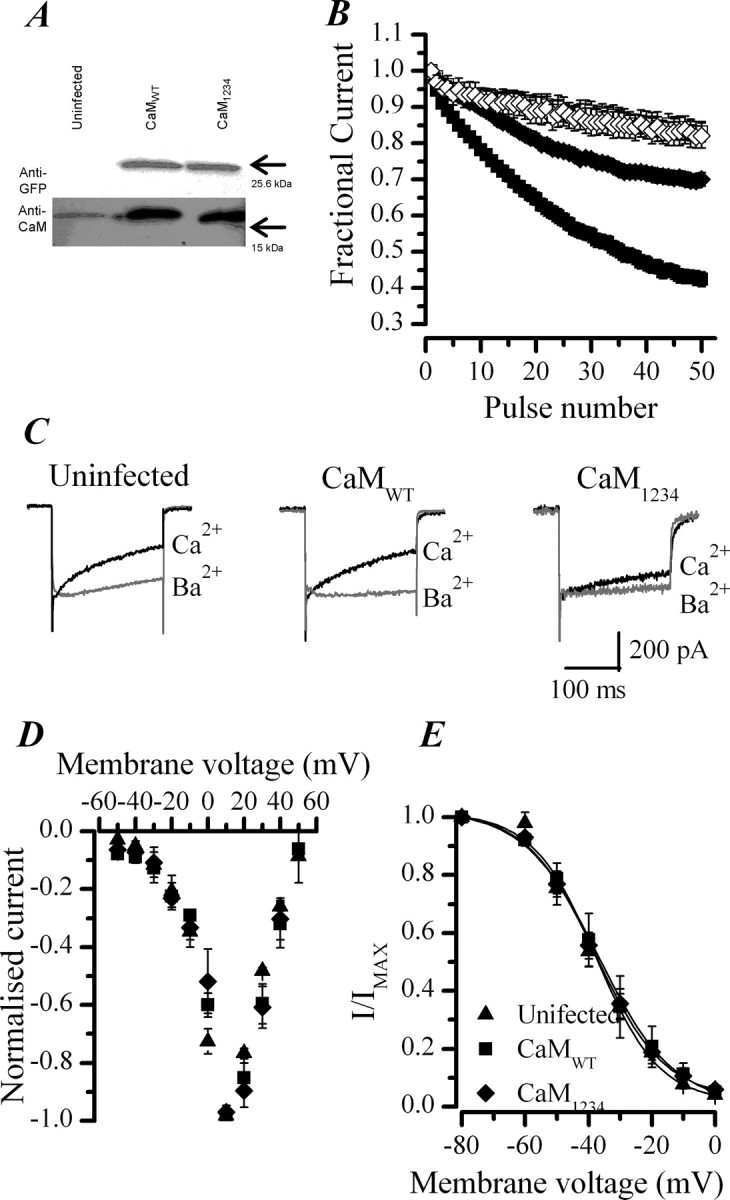
CaM regulation of CDI in chromaffin cells. A, Western blot showing the overexpression of CaM and EGFP after viral infection of chromaffin cells with CaMWT or CaM1234 adenoviruses. Results from culture-matched uninfected cells are shown for comparison. B, Effect of replacing extracellular Ca2+ (■, ♦) with Ba2+ (□, ◇) on fractional ICa recorded during a train of 50 pulses of 10 ms duration from −80 to +20 mV, delivered at 20 Hz in cells infected with CaMWT (■, □) or CaM1234 (♦, ◇) adenoviruses. Data plotted are the mean ± SEM. CaMWT, n = 39 cells in Ca2+, n = 12 in Ba2+; CaM1234, n = 25 in Ca2+, n = 9 in Ba2+. C, Superimposed ICa activated with a 200 ms voltage steps from −80 to +20 mV before and after replacement of extracellular Ca2+ with Ba2+. Data shown are representative from an uninfected, CaMWT-infected, and CaM1234-infected cell. D, Current–voltage relationship for ICa in uninfected (▴), CaMWT-infected (■), or CaM1234-infected (♦) cells. ICa measured at each potential was normalized to the peak current, and data points represent the mean ± SEM from five cells in each group. Note that, in all three groups of cells, the threshold for activation of the channels is approximately −20 mV, consistent with them being high-voltage-gated channels of the CaV2 family. E, Steady-state inactivation properties were not affected by viral infection and expression of either CaMWT or CaM1234. ICa were elicited by a 50 ms test depolarization to +20 mV after a 30 s shift in the membrane holding potential to potentials varying from −80 to 60 mV. The peak ICa of the test pulse, normalized to the value obtained from a holding potential of −80 mV are plotted against membrane potential and fitted with a Boltzmann relationship. Mean ± SEM data are plotted from five cells from each group. V50 of −38 mV for CaMWT-infected, CaM1234-infected, and uninfected cells.
Figure 6.
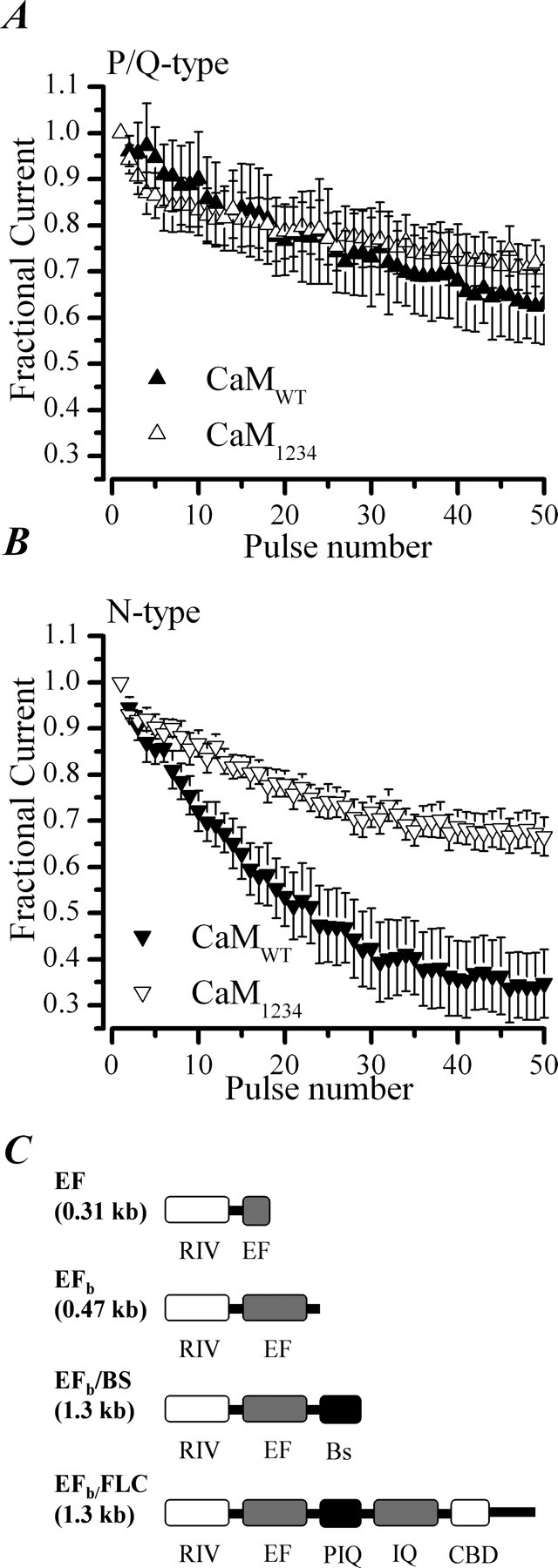
CaM regulates CDI in N-type but not P/Q-type channels in chromaffin cells. Inactivation of P/Q-type and N-type channels during a train of repetitive depolarizations from −80 to +20 mV, delivered at 20 Hz. A, Data plotted are the mean ± SEM for CaMWT-expressing (n = 5) and CaM1234-expressing (n = 6) cells and recorded with perforated patch and 1 μm ω-CgTx added to the external solution. B, Data are from CaMWT-expressing (n = 8) and CaM1234-expressing (n = 6) cells recorded in the presence of 300 nm AgaIVA. C, Schematic diagram showing the domains contained in the CaV2.1 RACE products isolated from bovine chromaffin cells. RIV, C′ terminal of domain IV including exon 36; EF, EF hand domain including exon 37; Bs, bovine-specific sequence; FLC, full-length C′ terminus; PIQ, pre-IQ domain; IQ, IQ-like domain. The sequence data and the corresponding nucleotide sequences are available from European Molecular Biology Laboratory/GenBank/DNA Data Bank of Japan under accession numbers AM421132, AM421133, AM421134, and AM421135 and in supplemental Figure 3B (available at www.jneurosci.org as supplemental material).
Figure 8.
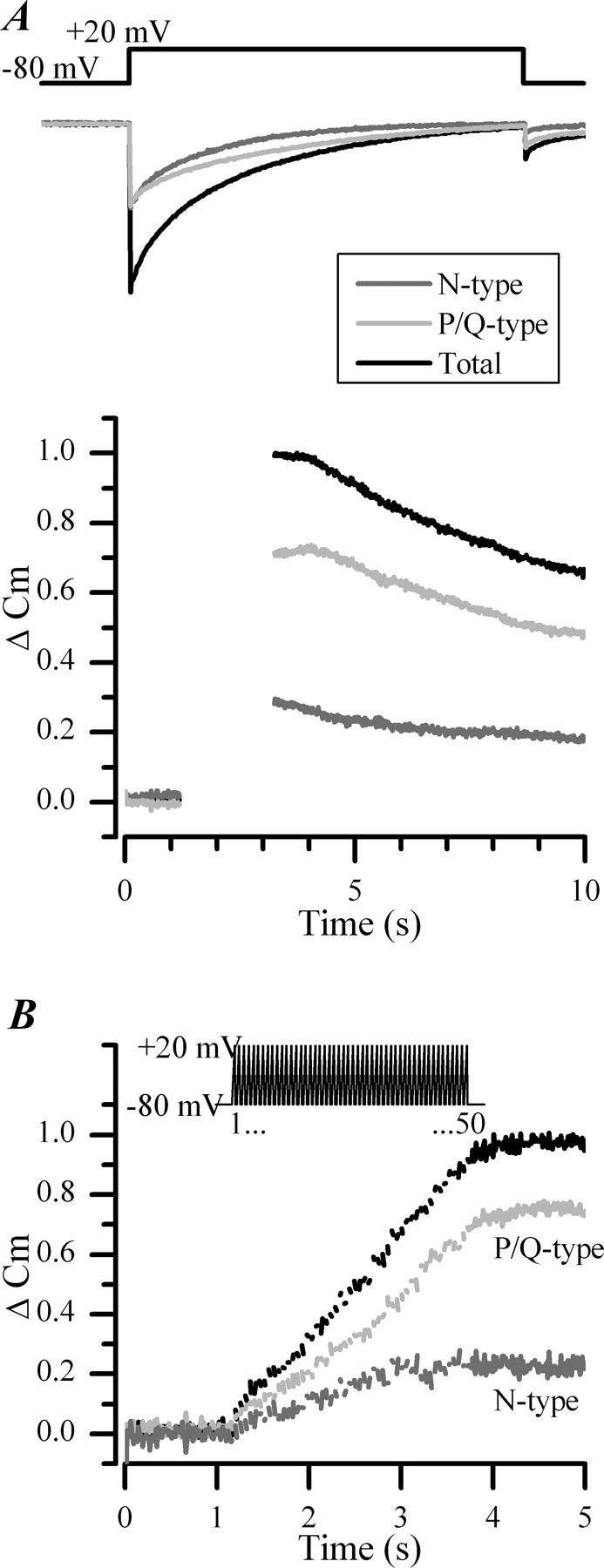
CDI of N-type channels limits their ability to support exocytosis. A, Superimposed mean ICa (top) and corresponding Cm changes (bottom) recorded in chromaffin cells (n = 15) in response to 800 ms depolarizations. Gaps in the Cm traces indicate timing of depolarizing pulses during which capacitance detection was interrupted. The contribution from each channel subtype was assessed pharmacologically by addition of ω-CgTX (1 μm; n = 9) or AgaIVA (300 nm; n = 6). ΔCm were normalized to the maximum measured before addition of toxin. B, Mean Cm changes measured in cells stimulated with a train of 50 pulses of 10 ms duration from −80 to +20 mV delivered at 20 Hz (illustrated above the panel). Gaps in the Cm traces indicate timing of depolarizing pulses during which capacitance detection was interrupted. Superimposed traces show the contribution from N-type channels (dark gray trace) and P/Q-type channels (light gray trace) to the total secretory response (black trace; n = 6).
Figure 7.

CDI of VGCCs in bovine chromaffin cells stimulated with a train of action potential-like waveforms. A, Schematic representation (top) of the action potential-like voltage protocol and superimposed representative currents recorded from a single chromaffin cell (bottom) recorded in the perforated-patch configuration before (control; black trace) and during application of the nonselective blocker of VGCCs, CdCl2 (100 μm; dashed gray trace). The stimulus train consisted of a 100 “action potentials” delivered at 100 Hz; currents shown correspond to the first two and last two evoked by a train. Note that CdCl2 blocks the second component of inward current consistent with it representing ICa, whereas the first component of current is insensitive to block as expected for current carried by voltage-gated Na channels. ap, Action potential. B, Superimposed currents from another representative cell before and after equimolar replacement of extracellular Ca2+ (black trace) with Ba2+ (dashed gray trace). Currents shown correspond to the first two and last two evoked by a train like that used in A. In Ba2+, currents do not decrease in amplitude over the course of the stimulus consistent with the abolition of CDI. C, Inactivation after repetitive depolarizations by a train of 100 action potentials (100 Hz) was significantly different between N-type (▿) and P/Q-type (▴) channels. Points represent the mean ± SEM fractional ICa measured in n = 4 cells under perforated-patch conditions. AP, Action potential.
Statistical significance was determined using either a paired or independent Student's t test, as appropriate (InStat3; GraphPad Software, San Diego, CA). All data are expressed as the mean ± SEM.
Western blotting.
Cultured chromaffin cells infected with adenovirus for 48 h were washed with ice-cold PBS and suspended in radioimmunoprecipitation assay lysis buffer containing 5 mm Tris, pH 7.4, 5 mm EDTA, 150 mm NaCl, 1% (v/v) Nonidet P-40, 0.5% (w/v) sodium deoxycholate, and 1% SDS. Aliquots of lysates from 500,000 cells were boiled in SDS sample buffer and resolved on 15% SDS-PAGE gels. Proteins were transferred to a nitrocellulose membrane and incubated at 4°C overnight with 0.5 μg/ml anti-CaM antibody (Research Diagnostics, Flanders, NJ). Membranes were washed with Tris-buffered saline with Tween 20 (TBST) before incubation with the secondary antibody goat anti-mouse horseradish peroxidase (Sigma) for 90 min at room temperature. The membrane was washed again in TBST before visualization by chemiluminescence (GE Healthcare Bio-Sciences, Little Chalfont, UK).
Quantitative GFP fluorescence intensity measurements.
Adenoviral infected cells were identified and classified by their EGFP/GFP fluorescence. EGFP fluorescence was detected using 488 nm excitation provided by a monochromator (T.I.L.L. Photonics, Martinsried, Germany) fitted to the microscope. The intensity of EGFP emission from individual chromaffin cells was measured from the output voltage of a photomultiplier tube coupled to a variable aperture acquisition window (TILL Photonics). The EGFP fluorescence intensity of a cell was correlated to its size determined from whole-cell patch-clamp recordings of cell capacitance. These measurements are referred to as brightness density (volts per picofarads). Neutral density filters were used to cut out contamination from autofluorescence. Cells with a brightness density below 0.05 V/pF were classified as dim, and cells above 0.05 V/pF classified as bright.
[Ca2+]i measurements.
Cells were loaded with Ca2+ indicator by addition of 5 μm fura-2 AM (Invitrogen) as described previously (Teschemacher and Seward, 2000). Because the intracellular environment is unknown in unpermeabilized chromaffin cells, coupled with difficulty in determining Rmin, fura-2 measurements were not calibrated in these experiments, and results are reported as ratios.
Cytotoxicity assay.
Possible cytotoxic effects of adenoviral infection were monitored by staining the DNA with the membrane-permeable dye Hoechst 33342 (200 ng/ml; Intergen, Oxford, UK). Cells were imaged for EGFP expression and nuclear fragmentation using an Axiovert 25/AxioCam microscope/camera system (Zeiss, Herts, UK).
For quantitative analysis of cell viability, a flow cytometric assay based on detection of DNA degradation using the membrane-impermeable dye propidium iodide (PI) (15 μm) was used. The number of adenoviral-infected, EGFP-expressing cells was determined in parallel. Cell viability and transfection efficiency was analyzed with a DacsCalibur flow cytometer (BD Biosciences, San Jose, CA). At 54 h after adenoviral infection, cells were resuspended and used for flow cytometry. The cell suspension without PI was measured to obtain the background fluorescence.
Molecular biology.
Total RNA was extracted from 180 × 106 bovine chromaffin cells prepared as described previously (Burgoyne, 1992), which yields very low levels of contaminating fibroblasts. TRIzol reagent (catalog number 15596-026; Invitrogen) was used according to the instructions of the manufacturer. For 3′ rapid amplification of cDNA ends (RACE), partial cDNA sequence of bovine CaV2.1 spanning the S3, S4, S5, and S6 domains of repeat IV (Garcia-Palmero et al., 2001) (GenBank accession number AF252381) was used as a template to design two gene-specific primers (GSP-1 and GSP-2) using the GeneTool Lite Program (BioTools, Edmonton, Alberta, Canada). For DNase treatment of RNA, 1 μl of RQ1 RNase-Free DNase I (catalog number M610A; Promega, Madison, WI) was added to 1 μg of total RNA along with 1 μl of 10× reaction buffer in a total volume of 10 μl with water and incubated at 30°C for 30 min, followed by heat inactivation of the enzyme at 65°C for 10 min. The RACE procedure was performed on both DNase-treated and untreated RNA samples. First-strand cDNA was generated by priming bovine chromaffin cell total RNA with the oligo-dt-based adapter primer contained in the 3′ RACE kit (catalog number 18373-019; Invitrogen). For some RNA samples, reverse transcriptase was omitted as a negative control. Two microliters of the resulting cDNA were used in an initial PCR using the gene-specific primer GSP-1 (GCCTCAGCGGGAAACCCTGT) and the abridged universal anchor primer (AUAP) from the kit. Nested PCR was performed using 5 μl of a 1:100 dilution of the first PCR as a template with the GSP-2 (CGCGGATCCTCCTTCATCTTCCTCTGCTCGTTTC) that incorporated a BamHI restriction site and modified AUAP, which incorporated an XhoI site. Conditions for both reactions were as follows: 0.2 mm dNTPs, 1 μl of 10× reaction buffer, 0.25 μl of Extensor Hi-Fidelity PCR Enzyme Mix (catalog number AB-0720a; ABgene, Epsom, UK), 0.2 μm of each primer, in a total volume of 50 μl at 94°C, 2 min (94°C, 30 s, 75°C, 30 s, 72°C, 2 min 13 s) for 10 cycles (temperature dropping by 1°C every cycle), followed by (94°C, 30 s, 65°C, 30 s, 72°C, 2 min 13 s) for 30 cycles, 72°C, 7 min final extension. Electrophoresis of PCR products was performed on 1–1.2% agarose gels, and DNA bands were viewed by UV transillumination. DNA sequencing of all of the clones was done by the Medical Research Council (Cambridge, UK) gene service. The sequences were conceptually translated using the GeneTool Lite program (BioTools). Blastx and Blastn (Altschul et al., 1997) analyses of the sequences were performed against the nucleotide and protein databases of the National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/BLAST/). Tblastn and Blastn searches of the sequences against the human and the bovine genome databases were also done on NCBI web site (http://www.ncbi.nih.gov/Genomes/). Alignments of the DNA and protein sequences were performed using the ClustalW program (Chenna et al., 2003) available on European Bioinformatics Institute web site (http://www.ebi.ac.uk/clustalw/). The sequences of the bovine clones were aligned with the conceptual translations of the bovine CACNA1A gene (gene encoding CaV2.1) exons [predicted by automated computational analysis available on NCBI Entrez Gene database (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db = gene&cmd = Retrieve&dopt = full_report&list_uids = 282648)].
Materials.
All drugs were made up as concentrated (typically 1000×) stock solutions in distilled filtered water and stored as aliquots at −20°C until use unless stated differently. Stocks were thawed and added to external solution to obtain the required concentration. ω-Conotoxin GVIA (ω-CgTX), ω-Agatoxin IVA (AgaIVA) and ω-Conotoxin MVII (ω-CgTX MVIIC) were obtained from Alomone Labs (Jerusalem, Israel). Cyclosporin A (Sigma) was dissolved in 99% ethanol and stored at −20°C. CaM inhibitory peptide (Calbiochem) was dissolved as a concentrated stock in standard internal solution, kept at −20°C until needed, and then added to fresh internal solution to obtain the required concentration.
Results
Ca2+-dependent inactivation of Ca2+ channels in chromaffin cells
The Ca2+ dependency of CDI in isolated chromaffin cells was examined using two classical tests, namely sensitivity to intracellular Ca2+ buffering and cation substitution. We examined the sensitivity of inactivation to intracellular Ca2+ chelation by increasing the concentration of BAPTA in the whole-cell recording pipette from 0.3 to 10 mm. CDI was examined with two different protocols, a 200 ms depolarization that promotes local Ca2+ buildup under the membrane (Fig. 1A) and a train of 50 depolarizations of 10 ms duration delivered at 20 Hz that promotes more global rises in Ca2+ (Fig. 1B,C). Increasing intracellular Ca2+ chelation profoundly decreased the inactivation of ICa measured with either stimulus protocol (Fig. 1A, left, B). Inactivation, measured as the current remaining at the end of the 200 ms depolarization expressed as a percentage of the peak ICa (I200/Ipeak), was increased from 55 ± 6% (n = 7) to 70 ± 2% (n = 3) with increasing BAPTA. Similarly, the fractional current remaining at the end of a train was significantly increased from 0.42 ± 0.03 (n = 7) to 0.74 ± 0.03 (n = 5) with increasing BAPTA, consistent with a reduction in cumulative CDI over the train of pulses. Replacing extracellular Ca2+ with Ba2+ also reduced current inactivation (Fig. 1A, right, C). I200/Ipeak increased from 46 ± 2% (n = 43) to 91 ± 4% (n = 5) in Ba2+, and the fractional currents remaining after a train of depolarizations increased from to 0.45 ± 0.02 (n = 20) to 0.86 ± 0.03 (n = 3). Together, the results from these two tests indicate that there is a Ca2+-dependent component to the inactivation of Ca2+ currents present in adult bovine adrenal chromaffin cells.
CDI is more prominent in N- than P/Q-type channels
Studies in heterologous expression systems show that all types of high-voltage-activated Ca2+ channels have the potential to undergo CDI (Liang et al., 2003). What form CDI takes in a native system expressing a mixture of channels has not been investigated. We determined which of the VGCCs present in chromaffin cells exhibit CDI using a pharmacological approach to isolate specific channel subtypes and Ba2+ substitution experiments to isolate the Ca2+-sensitive component of inactivation. Combined application of ω-CgTX (1 μm) to block N- and ω-AgaIVA (300 nm) to block P/Q-type channels inhibited Ca2+ entry by 94 ± 1% (n = 3) (Fig. 2A,C). The L-type-selective dihydropyridines nimodipine (10 μm) and BayK8644 (1,4-dihydro-2,6-dimethyl-5-nitro-4-[2-(trifluoromethyl)phenyl]-3-pyridinecarboxylic acid, methyl ester) (1 μm) were without effect, whereas the N- and P/Q-type channel blocker ω-CgTX MVIIC (1 μm) abolished ICa (Fig. 2C), confirming our and others' reports that adult bovine chromaffin cells express predominantly N- and P/Q-type channels in approximately a 1:1 ratio (Engisch and Nowycky, 1996; Currie and Fox, 1997; Powell et al., 2000). Cm measurements of exocytosis performed in the presence of the toxins further confirmed that Ca2+ influx through N- and P/Q-type channels also account entirely for secretory responses evoked by depolarizing stimuli in adult bovine chromaffin cells (Fig. 2B,C). For all subsequent work reported in this study, we used ω-CgTX (1 μm) to isolate P/Q-type channels and ω-AgaIVA (300 nm) to isolate N-type channels.
Ba2+ substitution experiments on pharmacologically isolated N-type channels showed these to exhibit profound CDI (Fig. 3A,B). CDI of P/Q-type channels was by comparison modest (Fig. 3A,B). Differences between inactivation of N- and P/Q-type channels was particularly striking when cells were stimulated with a train of depolarizations (Fig. 3A); the mean fractional ICa remaining at the end of the 50th pulse was 0.59 ± 0.04 (n = 10) for P/Q-type compared with 0.25 ± 0.03 (n = 10) for N-type channels. Because the amplitude of the currents generated by each channel type are approximately equal (Fig. 3B), the quantitative difference in the amount of CDI observed for each subtype of channel is unlikely to result simply from differences in the number of channels expressed (Soong et al., 2002) but rather to differences in the way that Ca2+ regulates the channels themselves.
Cyclosporine A and CaM inhibitory peptide do not inhibit CDI
The N′-terminal lobe of CaM has been shown to mediate CDI of P/Q- and N-type channels through a direct interaction with the α2.1 and α2.2 pore-forming subunits, respectively (Budde et al., 2002; Halling et al., 2005). CaM may also act indirectly to regulate inactivation through regulation of the protein phosphatase calcineurin (Burley and Sihra, 2000; Meuth et al., 2002). To distinguish between these possibilities, we treated cells for >20 min with 1 μm cyclosporine A, a calcineurin inhibitor. No significant reduction in CDI was observed (n = 11) (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material).
To further probe the role of CaM binding proteins in mediating CDI in chromaffin cells, we examined the consequences of dialyzing cells with a 17 amino acid competing CaM binding peptide based on the CaM binding domain (CBD) of myosin light chain kinase (Calbiochem). CaM is an abundant protein reported to reach concentrations as high as 1–10 μm in a cell (Charvin et al., 1997); therefore, saturating concentrations (30 μm) of the peptide were dialyzed into the cells via the recording pipette. Access to the cell interior was monitored from the series resistance (<10 MΩ); 6 min is predicted to be sufficient time for the peptide to reach equilibrium in the cell (Pusch and Neher, 1988). The mean fractional current remaining after a train of 50 pulses was 0.42 ± 0.03, n = 7 for control cells and 0.48 ± 0.04, n = 5 for culture-matched cells dialyzed with CaM inhibitory peptide (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). I200/Ipeak was similarly unaffected, being 55 ± 6%, n = 7 for control cells and 47 ± 4%, n = 5 for cells dialyzed with peptide. These data support the cyclosporine A results in showing that proteins that form reversible low-affinity complexes with CaM, such as calcineurin (Chin and Means, 2000), do not mediate CDI in chromaffin cell.
Adenoviral infection does not affect cell viability or Ca2+ channels in chromaffin cells
To assess whether preassociated CaM was mediating CDI in chromaffin cells, we adopted a dominant-negative strategy similar to that used previously (Alseikhan et al., 2002) in which mutant CaM incapable of binding Ca2+ on either the N′ or C′ lobe (CaM1234) is expressed in primary cells. However, as is often the case with nondividing cells, transfection of chromaffin cells with lipid-based methods, electroporation, or Ca2+ phosphate precipitation is inefficient. Semliki forest or adenovirus gene transfer, conversely, are very effective (Duncan et al., 2002; Thiagarajan et al., 2004). For the purposes of this study, Semliki forest virus-mediated gene expression was considered unsuitable because it is reported to have deleterious effects on Ca2+ channels and it is cytotoxic 48 h after infection (Ashery et al., 1999; Pan et al., 2002). We therefore examined the effects of adenoviral-mediated gene transfer on chromaffin cell viability and Ca2+ channel properties. Comparisons were made between cells infected with recombinant adenoviruses expressing EGFP alone or EGFP in combination with wild-type CaM (CaMWT–pIRES–EGFP). Cells infected with CaMWT–pIRES–EGFP adenovirus for 54 h had the characteristic rounded cell morphology of uninfected chromaffin cells and an intact, smooth, and phase-bright plasma membrane (Fig. 4A, top). EGFP fluorescence in infected cells was variable from cell to cell, ranging from very dim (just above autofluorescence) to very bright (Fig. 4A, middle). This variability in EGFP fluorescence was observed with all adenoviral constructs tested over the whole experimental period of 5 d. Staining with the DNA marker Hoechst 33342 showed that the nuclei were intact and that the DNA was not fragmented regardless of the EGFP expression level (Fig. 4A, bottom), indicating that adenoviral infection per se does not induce apoptosis in chromaffin cells. To quantify the overall viability of infected cells, we used a flow cytometric assay of DNA staining by propidium iodide. Uninfected cultures of chromaffin cells were found to contain 4 ± 1% necrotic and 9 ± 2% apoptotic cells (n = 3 preparations). Populations of cells selected by their EGFP fluorescence after infection with either CaM–pIRES–EGFP or EGFP adenoviruses showed no significant increase in the percentage of necrotic (0.5 ± 0.2 vs 3.9 ± 2.1%, n = 3) or apoptotic (8.6 ± 3.9 vs 3.4 ± 0.7%, n = 3) cells compared with uninfected cells. We therefore concluded that adenoviral infection per se is not cytotoxic to chromaffin cells, nor does it induce apoptosis over the experimental period we use.
To distinguish specific effects of expressed proteins of interest (CaMWT or CaM1234) from effects of the infection marker EGFP on channel function, we examined ICa densities from cells showing a wide range of EGFP fluorescence levels. The total EGFP fluorescence of individual cells was quantified with a photomultiplier and expressed as a ratio of cell size (brightness density in volts per picofarad). Cells with a brightness density below 0.05 V/pF were classified as dim and above 0.05 V/pF as bright. A significant reduction in ICa density was observed in bright cells compared with dim or uninfected control cells (Fig. 4B). However, there was no significant difference in Ca2+ channel density between uninfected cells and dim cells infected with adenovirus expressing either EGFP alone or CaMWT with EGFP (Fig. 4B), nor was there a change in inactivation (Fig. 4C,D). The data confirm that high expression levels of EGFP are associated with a loss of functional Ca2+ channels (Thiagarajan et al., 2005). All additional investigations were restricted to dim cells.
Adenoviral expression of mutant CaM reduces CDI
To determine whether preassociated CaM mediates CDI of the endogenous VGCCs found in chromaffin cells, we infected cells with recombinant adenovirus expressing EGFP in combination with mutant CaM1234 (CaM1234–pIRES–EGFP). As a control, experiments were conducted in parallel on cells infected with CaMWT–pIRES–EGFP. Western blotting confirmed that the viruses were functional and that the CaMs were being expressed (Fig. 5A). There was no significant difference between the mean peak ICa measured in the cells infected with CaMWT–pIRES–EGFP (332 ± 28 pA, n = 20) and CaM1234–pIRES–EGFP (308 ± 36 pA, n = 18). In cells expressing CaM1234, however, the inactivation of ICa evoked by either a train of 50 depolarizations or a single 200 ms depolarization was significantly reduced compared with cells made to express excess CaMWT (Fig. 5B,C). I200/Ipeak for CaMWT was 42 ± 2% (n = 33) in Ca2+ compared with 85 ± 5% in Ba2+ (n = 8, p < 0.0001). These values are not significantly different from those measured in uninfected cells (46 ± 2%, n = 43 in Ca2+ and 9 ± 4%, n = 6, in Ba2+). CaM1234-expressing cells, however, inactivated less in Ca2+ (I200/Ipeak, 66 ± 2%, n = 30). A small component of CDI remained in CaM1234-expressing cells because, after switching to Ba2+, I200/Ipeak was increased to 81 ± 2% (n = 13).
CaM1234 expression similarly reduced inactivation of ICa during a train of depolarizations. The fractional current remaining at the end of the train was 0.70 ± 0.02, n = 25 in cells expressing CaM1234 compared with 0.42 ± 0.02, n = 39 (p < 0.0001) in cells expressing excess CaMWT. The fractional current remaining in cells infected with CaMWT–pIRES–EGFP was not significantly different from that measured in uninfected cells (0.45 ± 0.02, n = 20), despite the large increase in CaM expression (Fig. 5A). This suggests that CaM is not in rate-limiting amounts in uninfected cells and also that it does not behave simply as a Ca2+ chelator (Kreiner and Lee, 2006). The lack of effect of CaM1234 expression on Ca2+ handling by the cells was verified further by monitoring cytosolic [Ca2+] with fura 2 (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Because a shift in the voltage dependence of either activation or inactivation would affect cumulative inactivation during a train of depolarizations, we also examined the current–voltage relationship and steady-state inactivation of ICa in CaMWT- and CaM1234- expressing cells. These were indistinguishable from uninfected cells (Fig. 5D,E), consistent with the idea that CaM is acting directly on the channels to regulate CDI.
N-type but not P/Q-type current inactivation is regulated by CaM
To determine whether CaM-mediated CDI of N- and/or P/Q-type channels differs in chromaffin cells, we examined the effect of CaM1234 and CaMWT on pharmacologically isolated currents (Fig. 6A,B). Fractional P/Q-type current remaining at the end of a train of depolarizations was 0.63 ± 0.09 (n = 5) for CaMWT-expressing cells and 0.70 ± 0.05 (n = 6) for CaM1234-expressing cells; the difference was not significant (p = 0.49). In contrast, fractional N-type current remaining at pulse 50 was 0.35 ± 0.07 (n = 8) for CaMWT-expressing cells compared with 0.67 ± 0.04 (n = 6) for CaM1234-expressing cells; this difference was highly significant (p = 0.004). Thus, N-type channel CDI showed a marked sensitivity to CaM1234 expression, consistent with the notion that preassociated CaM mediates inactivation of this type of channel. The inactivation of isolated P/Q-type currents, conversely, is insensitive to CaM1234 expression, suggesting that the small amount of CDI observed for P/Q-type channels in chromaffin cells (Fig. 3) is not mediated by CaM.
Bovine CaV2.1 C termini lack regions implicated in CaM-dependent regulation of the channel
The results indicating that P/Q-type channels display relatively minor CDI in our study in bovine chromaffin cells are surprising because studies with heterologously expressed human or rat P/Q-type channel cDNAs report robust CaM-dependent inactivation. To determine whether the bovine CaV2.1 subunit cDNA contains sequence corresponding to the protein structural regions implicated in CaM-dependent regulation, we cloned its 3′ end using a 3′ RACE procedure on DNaseI-treated RNA extracted from bovine chromaffin cells. Agarose gel electrophoresis of the nested PCR products revealed multiple bands of differing size (0.31, 0.47, 0.58, 0.7, and 1.3 kb), (supplemental Fig. 3A1, available at www.jneurosci.org as supplemental material). These bands were excised, and the DNA was extracted and subcloned (supplemental Fig. 3A2, available at www.jneurosci.org as supplemental material) for sequencing. Blastx and Blastn searches against nucleotide and protein databases as well as Tblastn and Blastn searches against the human and bovine genome revealed that, with the exception of a single clone (0.7 kb, which was a nonspecific amplification), all were bovine CaV2.1 clones. An alignment of the deduced amino acid sequences of the bovine CaV2.1 clones with the sequence of a human CaV2.1 clone (GenBank accession number AAB49677) and the bovine CaV2.2 subunit (GenBank accession number AAF24229) is shown in supplemental Figure 3B (available at www.jneurosci.org as supplemental material). A schematic representation of the clones is shown in Figure 6C. The largest of the four clones, EFb/full-length C′ terminus (FLC) (1.3kb) contains all of the CaV2.1 subunit domains implicated in CaM-dependent regulation (the pre-IQ domain, an IQ-like CaM interaction domain, and a CBD), the rest of the clones lack these important regions (Fig. 6C). The EF (0.31kb) clone has an incomplete EF hand, terminating with an alanine residue, a position corresponding to the end of exon 36 in the human CaV2.1 gene. The clones having complete EF hands [EFb (0.47 kb), EFbBs (0.58kb), and EFb/FLC (1.3kb)] contained the 37b version of exon 37 (Bourinet et al., 1999). The exon boundaries on the 3′ RACE clones were compared with that of the bovine CACNA1A gene; the Bs sequences present in the EFbBs clone were not represented in the predicted bovine CACNA1A gene exons. The EFb/FLC clone showed agreement with the predicted CACNA1A gene exons up to exon 43. The EFb/FLC has no sequence in it corresponding to the predicted exon 44, and its sequence diverged from the predicted bovine CACNA1A gene in the region of exon 45. Also there was no exon 46 in the predicted bovine CACNA1A gene, whereas the EFb/FLC we cloned had sequence in it that corresponded to exon 46 of the human CACNA1A gene. However EFb/FLC lacks any sequence corresponding to the exon 47 of the long isoform of the human CaV2.1 subunit. We checked whether exon 47 is represented in the bovine genome by performing 3′ RACE on bovine chromaffin cell total RNA that was not treated with DNase. This yielded two DNA bands of 0.67 and 1.68 kb. The 0.67 kb contains intronic sequence corresponding to intron 36 of human CACNA1A, suggesting that it is a genomic clone. Its translated region corresponds to exon 36 and exon 37a of CACNA1A. The 1.68 kb clone did include exon 47, but, because it also has a stop codon intervening the coding sequence, we were unable to determine whether this clone was derived from genomic DNA or represents a RACE error. Together, the data suggest that exons critical for CaM regulation of CaV2.1 may be skipped in chromaffin cells to yield channels with short C′ termini and altered regulation.
Physiological consequences of CDI on exocytosis
Finally, we investigated the potential physiological consequences of CDI in chromaffin cells. First, we examined whether stimulation of chromaffin cells with action potential-like waveforms inactivated the channels in a similar manner to that seen with the step depolarizations. As shown in Figure 7A, currents evoked by voltage stimuli designed to mimic an action potential had two distinct peaks. The first peak arises from activation of voltage-gated sodium channels and is insensitive to block by CdCl2, whereas the delayed second peak arises from activation of VGCCs and is abolished by CdCl2 (100 μm; n = 6). ICa evoked by a train of 100 action potentials (100 Hz) underwent inactivation, reducing the mean peak current from 1018 ± 108 to 507 ± 36 pA (n = 6). Inactivation was abolished after replacement of extracellular Ca2+ with Ba2+ (Fig. 7B), indicating that, as with the step depolarizations (Fig. 1C), CDI was also activated with this more physiological stimuli. Moreover, comparison of inactivation in toxin-isolated P/Q-type versus N-type channels showed that it was the latter that underwent profound CDI (Fig. 7C).
To examine the potential consequences of CDI on exocytosis from chromaffin cells, the relationship between Ca2+ entry through pharmacologically isolated N- and P/Q-type channels and exocytosis was quantified by combining voltage-clamp recordings of the currents with Cm measurements. Initially, cells were stimulated with long depolarizations (800 ms) in which there is complete inactivation of the channels (Fig. 8A). The rate of inactivation for the toxin-isolated currents activated with 800 ms depolarizations could be described with a double-exponential time course. There was no difference in the first time constant (τ ≈ 26 and 27 ms for P/Q and N-type ICa, respectively), but N-type channels displayed a faster second time constant (τ ≈ 173 ms) compared with P/Q-type channels (τ ≈ 302 ms), which resulted in significantly less Ca2+ entering the cells (158 ± 21 × 1016 Ca2+ ions, n = 14 and 231 ± 29 × 1016 Ca2+ ions, n = 11 for N- and P/Q- type channels respectively, p = 0.05). Consistent with the overall decreased Ca2+ entry through N-type channels after 800 ms depolarizations, the exocytotic responses measured in the same cells were significantly smaller (160 ± 24, n = 14, p = 0.002) for N-type compared with P/Q-type channels (390 ± 61 fF, n = 11). The exocytotic efficiency (calculated as ΔCm/∫Ca2+ influx) was also significantly different for the two channels with these long depolarizations [1.82 ± 0.23 fF/Ca2+ ion (n = 6) for P/Q-type channels compared with 1.03 ± 0.17 fF/Ca2+ ion (n = 9) for N-type channels, **p < 0.01]. Interestingly, no difference in exocytotic efficiency between the two channel types was observed when stimulus intensity was decreased to 40 ms, at which CDI is insignificant [0.59 ± 0.08 fF/Ca2+ ion (n = 4) for P/Q-type channels and 0.43 ± 0.04 fF/Ca2+ ion (n = 4) for N-type channels]. We then compared the ability of N- and P/Q-type channels to support exocytosis evoked by a train of depolarizations (Fig. 8B). Ca2+ influx through N-type channels was found to contribute progressively less to evoked secretory responses toward the end of a stimulus train as CDI developed.
Discussion
This study is the first to report CaM-dependent inactivation of a native N-type channel. The presence of CDI for this class of VGCCs has been difficult to define, with conflicting results arising from biophysical studies (Cox and Dunlap, 1994; Patil et al., 1998; Jones, 1999) and from molecular studies of recombinant channels (Liang et al., 2003). As shown here (Figs. 3, 7, 8), CDI of N-type channels in chromaffin cells is evident when cells are stimulated with long depolarizations or at high frequency with short-duration pulses and when intracellular Ca2+ buffering is kept low (0.3 mm BAPTA) or undisturbed in perforated patch. This apparent sensitivity of N-type channel CDI to the spatiotemporal features of cytosolic Ca2+ rises is similar to that described for recombinant α2.1 channels (Lee et al., 2000; Kreiner and Lee, 2006), which is driven by Ca2+ binding to the N′-terminal lobe of CaM (Lee et al., 2000; DeMaria et al., 2001; Liang et al., 2003; Kreiner and Lee, 2006). The blocking action of CaM1234 on N-type channel CDI in chromaffin cells together with the presence of an EFb-like domain (Bourinet et al., 1999), an IQ-like domain (Petersen et al., 1999), and CBD domain (Lee et al., 1999) in the C′ terminal of bovine chromaffin α2.2 sequence (Fig. 5B) strongly suggest that the CDI observed here occurs through the same pathway described for recombinant channels of the CaV2 family (Liang et al., 2003; Halling et al., 2005). Interestingly, like in the studies by Liang et al. (2003), a small component of CDI was still present in cells expressing CaM1234, which could be attributed to the low affinity of apocalmodulin (apoCaM) and CaM1234 for the IQ-motif of α2.2 (Petersen et al., 1999; Zuhlke et al., 1999; Pate et al., 2000; Kim et al., 2004; Black et al., 2005) and competing actions of endogenous wild-type CaM. Increasing expression of CaMWT with adenovirus did not alter the observed CDI, as would be expected if endogenous levels of CaM are saturating. Moreover, the lack of effect on CDI of dialyzing cells with a CaM binding peptide (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material) supports the notion that apoCaM is preassociated with N-type channels in chromaffin cells and that dissociation is slow (Liang et al., 2003). The lack of effect of CaM1234 expression on the kinetics of voltage-dependent inactivation (VDI) in chromaffin cells further supports preassociation (Liang et al., 2003). This is different from observations made on P/Q-type channels in central neurons, in which dialysis of the same peptide was found to be an effective blocker of CDI (Xu and Wu, 2005), suggesting that CaM association of some CaV2 channels may be dynamically regulated by interactions with other Ca2+ binding proteins (Lee et al., 2002; Chaudhuri et al., 2005; Lautermilch et al., 2005). Sequence variations in the CBD of α2.2 versus α2.1 may impact on this form of regulation.
In addition to CDI, N-type channels may undergo two types of VDI that are also regulated by activity (Hering et al., 2000; Cens et al., 2006); closed-state inactivation in particular may accumulate during high-frequency stimuli and, like CDI, gives rise to U-shaped inactivation (Patil et al., 1998). Unlike CDI, however, closed-state inactivation is unaffected by substituting Ba2+ for Ca2+. As can be seen in Figures 1 and 2, once CDI in chromaffin cells is blocked with Ba2+, cumulative VDI is modest (∼15%). Two factors that regulate cumulative VDI are the subunit composition of the channel, with β2a subunit association essentially occluding such regulation (Patil et al., 1998) and alternative splicing (Thaler et al., 2004). The modest cumulative VDI observed in our study, together with the separation between the voltage dependence of activation and steady-state inactivation (Fig. 4) are consistent with previous suggestions that N-type channels in chromaffin cells have β2a subunits (Cahill et al., 2000; Hurley et al., 2000). Notably, in recombinant studies, CDI is most easily observed in CaV2* channels when the α1 subunit is coexpressed with β2a (Lee et al., 2000; Liang et al., 2003). Thus, in chromaffin cells, CDI of N-type channels takes precedence over VDI in limiting Ca2+ entry during the kind of intense stimulation, associated with stress responses.
An unexpected finding from this study is the absence of significant CDI of P/Q-type channels. Comparison of our data with that of Polo-Parada et al. (2006) suggests that a similar pattern of differential CDI between N- and P/Q-type channels also occurs in mouse chromaffin cells. Two groups have published extensively on the role CaM plays in mediating CDI and CDF of recombinant P/Q-type channels (Lee et al., 1999, 2000, 2002, 2003; DeMaria et al., 2001; Chaudhuri et al., 2004), notably all of the studies have been performed in HEK293 cells transfected with either a human or rat α1A clone coexpressed with β2a. Cell-specific regulation of CaV2.1 inactivation is likely to arise from alternative splicing (Bourinet et al., 1999; Chaudhuri et al., 2004; Lipscombe and Castiglioni, 2004) and differential association with β subunits (Lee et al., 2000; Chaudhuri et al., 2004). The main cDNA clones isolated from chromaffin cells corresponding to the C terminus of P/Q channel α1 subunits are forms with short C termini lacking the crucial IQ-like and CBD domains required for CaM regulation, and two of these also lacked a full EF hand (Fig. 5). Consistent with our findings, channels lacking these motifs would not be expected to bind or be regulated by CaM (Halling et al., 2005). Variants lacking exons 36, 37, 43, 44, and/or 47 have been described previously (Ligon et al., 1998; Krovetz et al., 2000; Soong et al., 2002; Kanumilli et al., 2006); this, however, is the first report of transcripts lacking all CaM regulatory domains, which includes exons 40 and 42. Studies on recombinant channels established previously that splicing at exons 37, 44, and/or 47 alone is not sufficient to remove CDI (Soong et al., 2002; Chaudhuri et al., 2004). There are other reports of endogenous P/Q-type channels lacking CDI (Budde et al., 2002; Chaudhuri et al., 2005); whether these too lack exons 40 and 42 remains to be seen. Regulation of CDI through alternative splicing of the C′ terminus has now also been reported for CaV1.3 in the auditory system (Shen et al., 2006), suggesting that this form of regulation and fine-tuning of VGCC inactivation may be more widespread in the nervous system than previously thought.
The longest clone we obtained (EFb/FLC) has complete EF hand, pre-IQ, IQ-like, and CBD domains. This splice variant might have been expected to display some regulation by CaM. Inclusion of exon 37b would be expected to ablate CDF but should leave CDI intact (Chaudhuri et al., 2004). Four possible reasons why CDI may not have been observed are as follows: (1) these channels are associated with β subunits that attenuate CaM regulation (Lee et al., 2000; Chaudhuri et al., 2005), (2) CaM-dependent CDI is blocked by association with another Ca2+ binding protein (Weiss and Burgoyne, 2001; Lautermilch et al., 2005), (3) sequence variation at the C-terminal end of the CBD impair CDI (Lee et al., 2003), and (4) exon 47 is missing, which is reported to be among the sites of interaction for regulation by β and Gβγ subunits (Zhong et al., 1999; Dolphin, 2003). It is possible that the EFb/FLC splice variant may not have the full extent of channel regulation seen for the longer channel types. For example, the bovine N-type channel that does have a longer C terminus than EFb/FLC shows enhanced sensitivity to regulation by Gβγ (Currie and Fox, 1997, 2002).
Because the discovery that the neuronal VGCCs commonly associated with neurotransmitter release can display CDF and CDI, there has been much speculation on the impact these forms of regulation might have on exocytosis and synaptic plasticity. In sensory cell ribbon synapses, CDI of CaV1.4 channels has been found to be switched off to allow for tonic neurotransmitter release (Singh et al., 2006). Here we show that, as CDI develops, the exocytotic efficiency of N-type channels is decreased relative to P/Q-type channels, indicating that the efficacy with which Ca2+ entry through the two types of channels support exocytosis is different. This is unlikely to be attributable simply to differences in the ability of the channels to interact with the fusion machinery (Bezprozvanny et al., 1995; Rettig et al., 1996), because no differences are observed when cells are stimulated with 40 ms step depolarizations (Engisch and Nowycky, 1996). A more likely explanation is that CDI reduces the extent of Ca2+ domain overlap around N-type channels and consequently decreases the likelihood that nearby primed vesicles will undergo fusion. Because subplasmalemmal Ca2+ also regulates the distance between docked vesicles and Ca2+ channels (Becherer et al., 2003), CDI may also reduce the probability of new vesicles docking in the vicinity of N-type channels compared with P/Q-type channels during periods of intense stimulation. Interestingly, two recent studies in chromaffin cells have also established that the mode of exocytosis shifts from kiss-and-run to full fusion as stimulus intensity is increased to levels associated with activation of the stress response (Fulop et al., 2005; Elhamdani et al., 2006); whether CDI therefore limits the ability of N-type channels to support full fusion is an intriguing possibility that will require additional investigation.
Footnotes
This work was supported by a project grant from The Wellcome Trust (E.P.S.) and a studentship (R.C.E.W.) and a project grant (J.L.W.) from Biotechnology and Biological Sciences Research Council. We thank C. Heinemann from Heka Elektronik (Lambrecht, Germany) for help with the Patchmaster and Fitmaster programming and Robert Woolley for technical help with the adenovirus and chromaffin cells.
References
- Alseikhan BA, DeMaria CD, Colecraft HM, Yue DT. Engineered calmodulins reveal the unexpected eminence of Ca2+ channel inactivation in controlling heart excitation. Proc Natl Acad Sci USA. 2002;99:17185–17190. doi: 10.1073/pnas.262372999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashery U, Betz A, Xu T, Brose N, Rettig J. An efficient method for infection of adrenal chromaffin cells using the Semliki Forest virus gene expression system. Eur J Cell Biol. 1999;78:525–532. doi: 10.1016/s0171-9335(99)80017-x. [DOI] [PubMed] [Google Scholar]
- Becherer U, Moser T, Stuhmer W, Oheim M. Calcium regulates exocytosis at the level of single vesicles. Nat Neurosci. 2003;6:846–853. doi: 10.1038/nn1087. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Scheller RH, Tsien RW. Functional impact of syntaxin on gating of N-type and Q-type calcium channels. Nature. 1995;378:623–626. doi: 10.1038/378623a0. [DOI] [PubMed] [Google Scholar]
- Black DJ, Halling DB, Mandich DV, Pedersen SE, Altschuld RA, Hamilton SL. Calmodulin interactions with IQ peptides from voltage-dependent calcium channels. Am J Physiol Cell Physiol. 2005;288:C669–C676. doi: 10.1152/ajpcell.00191.2004. [DOI] [PubMed] [Google Scholar]
- Bourinet E, Soong TW, Sutton KG, Slaymaker SJ, Mathews E, Monteil A, Zamponi GW, Nargeot J, Snutch TP. Splicing of α1A subunit gene generates phenotypic variants of P- and Q-type calcium channels. Nat Neurosci. 1999;2:407–415. doi: 10.1038/8070. [DOI] [PubMed] [Google Scholar]
- Budde T, Meuth S, Pape HC. Calcium-dependent inactivation of neuronal calcium channels. Nat Rev Neurosci. 2002;3:873–883. doi: 10.1038/nrn959. [DOI] [PubMed] [Google Scholar]
- Burgoyne RD. Intracellular messengers. In: Boulton AA, Baker GB, Taylor CW, editors. Neuromethods. Vol 20. Totowa, NJ: Humana; 1992. pp. 433–470. [Google Scholar]
- Burley JR, Sihra TS. A modulatory role for protein phosphatase 2B (calcineurin) in the regulation of Ca2+ entry. Eur J Neurosci. 2000;12:2881–2891. doi: 10.1046/j.1460-9568.2000.00178.x. [DOI] [PubMed] [Google Scholar]
- Cahill AL, Hurley JH, Fox AP. Coexpression of cloned α1B, β2a, and α2/δ subunits produces non-inactivating calcium currents similar to those found in bovine chromaffin cells. J Neurosci. 2000;20:1685–1693. doi: 10.1523/JNEUROSCI.20-05-01685.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- Cens T, Restituito S, Galas S, Charnet P. Voltage and calcium use the same molecular determinants to inactivate calcium channels. J Biol Chem. 1999;274:5483–5490. doi: 10.1074/jbc.274.9.5483. [DOI] [PubMed] [Google Scholar]
- Cens T, Rousset M, Leyris JP, Fesquet P, Charnet P. Voltage- and calcium-dependent inactivation in high voltage-gated Ca2+ channels. Prog Biophys Mol Biol. 2006;90:104–117. doi: 10.1016/j.pbiomolbio.2005.05.013. [DOI] [PubMed] [Google Scholar]
- Charvin N, L'eveque C, Walker D, Berton F, Raymond C, Kataoka M, Shoji-Kasai Y, Takahashi M, De Waard M, Seagar MJ. Direct interaction of the calcium sensor protein synaptotagmin I with a cytoplasmic domain of the alpha1A subunit of the P/Q-type calcium channel. EMBO J. 1997;16:4591–4596. doi: 10.1093/emboj/16.15.4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri D, Chang SY, DeMaria CD, Alvania RS, Soong TW, Yue DT. Alternative splicing as a molecular switch for Ca2+/calmodulin-dependent facilitation of P/Q-type Ca2+ channels. J Neurosci. 2004;24:6334–6342. doi: 10.1523/JNEUROSCI.1712-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri D, Alseikhan BA, Chang SY, Soong TW, Yue DT. Developmental activation of calmodulin-dependent facilitation of cerebellar P-type Ca2+ current. J Neurosci. 2005;25:8282–8294. doi: 10.1523/JNEUROSCI.2253-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin D, Means AR. Calmodulin: a prototypical calcium sensor. Trends Cell Biol. 2000;10:322–328. doi: 10.1016/s0962-8924(00)01800-6. [DOI] [PubMed] [Google Scholar]
- Cox DH, Dunlap K. Inactivation of n-type calcium current in chick sensory neurons: calcium and voltage dependence. J Gen Physiol. 1994;104:311–336. doi: 10.1085/jgp.104.2.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currie KP, Fox AP. Comparison of N- and P/Q-type voltage-gated calcium channel current inhibition. J Neurosci. 1997;17:4570–4579. doi: 10.1523/JNEUROSCI.17-12-04570.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currie KP, Fox AP. Differential facilitation of N- and P/Q-type calcium channels during trains of action potential-like waveforms. J Physiol (Lond) 2002;539:419–431. doi: 10.1113/jphysiol.2001.013206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMaria CD, Soong TW, Alseikhan BA, Alvania RS, Yue DT. Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature. 2001;411:484–489. doi: 10.1038/35078091. [DOI] [PubMed] [Google Scholar]
- Dolphin AC. G protein modulation of voltage-gated calcium channels. Pharmacol Rev. 2003;55:607–627. doi: 10.1124/pr.55.4.3. [DOI] [PubMed] [Google Scholar]
- Duncan RR, Greaves J, Tapechum S, Apps DK, Shipston MJ, Chow RH. Efficacy of Semliki Forest virus transduction of bovine adrenal chromaffin cells: an analysis of heterologous protein targeting and distribution. Ann NY Acad Sci. 2002;971:641–646. doi: 10.1111/j.1749-6632.2002.tb04543.x. [DOI] [PubMed] [Google Scholar]
- Elhamdani A, Azizi F, Artalejo CR. Double patch clamp reveals that transient fusion (kiss-and-run) is a major mechanism of secretion in calf adrenal chromaffin cells: high calcium shifts the mechanism from kiss-and-run to complete fusion. J Neurosci. 2006;26:3030–3036. doi: 10.1523/JNEUROSCI.5275-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engisch KL, Nowycky MC. Calcium dependence of large dense-cored vesicle exocytosis evoked by calcium influx in bovine adrenal chromaffin cells. J Neurosci. 1996;16:1359–1369. doi: 10.1523/JNEUROSCI.16-04-01359.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson MG, Alseikhan BA, Peterson BZ, Yue DT. Preassociation of calmodulin with voltage-gated Ca2+ channels revealed by FRET in single living cells. Neuron. 2001;31:973–985. doi: 10.1016/s0896-6273(01)00438-x. [DOI] [PubMed] [Google Scholar]
- Fulop T, Smith C. Physiological stimulation regulates exocytic mode through calcium activation of protein kinase C in mouse chromaffin cells. Biochem J. 2006;399:111–119. doi: 10.1042/BJ20060654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulop T, Radabaugh S, Smith C. Activity-dependent differential transmitter release in mouse adrenal chromaffin cells. J Neurosci. 2005;25:7324–7332. doi: 10.1523/JNEUROSCI.2042-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Palomero E, Renart J, Andres-Mateos E, Solis-Garrido LM, Matute C, Herrero CJ, Garcia AG, Montiel C. Differential expression of calcium channel subtypes in the bovine adrenal medulla. Neuroendocrinology. 2001;74:251–261. doi: 10.1159/000054692. [DOI] [PubMed] [Google Scholar]
- Halling DB, Racena-Parks P, Hamilton SL. Regulation of voltage-gated Ca2+ channels by calmodulin. Sci STKE. 2005;315:re15. doi: 10.1126/stke.3152005re15. [DOI] [PubMed] [Google Scholar]
- Hering S, Berjukow S, Sokolov S, Marksteiner R, Regina G, Kraus R, Timin EN. Molecular determinants of inactivation in voltage-gated Ca2+ channels. J Physiol (Lond) 2000;528:237–249. doi: 10.1111/j.1469-7793.2000.t01-1-00237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley JH, Cahill AL, Currie KP, Fox AP. The role of dynamic palmitoylation in Ca2+ channel inactivation. Proc Natl Acad Sci USA. 2000;97:9293–9298. doi: 10.1073/pnas.160589697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones LP, DeMaria CD, Yue DT. N-type calcium channel inactivation probed by gating-current analysis. Biophys J. 1999;76:2530–2552. doi: 10.1016/S0006-3495(99)77407-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SW. Inactivation of N-type Ca2+ channels: Ca2+ vs. voltage. J Physiol (Lond) 1999;518:630. doi: 10.1111/j.1469-7793.1999.0630p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanumilli S, Tringham EW, Payne CE, Dupere JR, Venkateswarlu K, Usowicz MM. Alternative splicing generates a smaller assortment of CaV2.1 transcripts in cerebellar Purkinje cells than in the cerebellum. Physiol Genomics. 2006;24:86–96. doi: 10.1152/physiolgenomics.00149.2005. [DOI] [PubMed] [Google Scholar]
- Kim J, Ghosh S, Nunziato DA, Pitt GS. Identification of the components controlling inactivation of voltage-gated Ca2+ channels. Neuron. 2004;41:745–754. doi: 10.1016/s0896-6273(04)00081-9. [DOI] [PubMed] [Google Scholar]
- Kreiner L, Lee A. Endogenous and exogenous Ca2+ buffers differentially modulate Ca2+-dependent inactivation of CaV2.1 Ca2+ channels. J Biol Chem. 2006;281:4691–4698. doi: 10.1074/jbc.M511971200. [DOI] [PubMed] [Google Scholar]
- Krovetz HS, Helton TD, Crews AL, Horne WA. C-Terminal alternative splicing changes the gating properties of a human spinal cord calcium channel alpha 1A subunit. J Neurosci. 2000;20:7564–7570. doi: 10.1523/JNEUROSCI.20-20-07564.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lautermilch NJ, Few AP, Scheuer T, Catterall WA. Modulation of CaV2.1 channels by the neuronal calcium-binding protein visinin-like protein-2. J Neurosci. 2005;25:7062–7070. doi: 10.1523/JNEUROSCI.0447-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A, Wong ST, Gallagher D, Li B, Storm DR, Scheuer T, Catterall WA. Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature. 1999;399:155–159. doi: 10.1038/20194. [DOI] [PubMed] [Google Scholar]
- Lee A, Scheuer T, Catterall WA. Ca2+/calmodulin-dependent facilitation and inactivation of P/Q-type Ca2+ channels. J Neurosci. 2000;20:6830–6838. doi: 10.1523/JNEUROSCI.20-18-06830.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A, Westenbroek RE, Haeseleer F, Palczewski K, Scheuer T, Catterall WA. Differential modulation of Cav2.1 channels by calmodulin and Ca2+-binding protein 1. Nat Neurosci. 2002;5:210–217. doi: 10.1038/nn805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A, Zhou H, Scheuer T, Catterall WA. Molecular determinants of Ca2+/calmodulin-dependent regulation of Cav2.1 channels. Proc Natl Acad Sci USA. 2003;100:16059–16064. doi: 10.1073/pnas.2237000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, DeMaria CD, Erickson MG, Mori MX, Alseikhan BA, Yue DT. Unified mechanisms of Ca2+ regulation across the Ca2+ channel family. Neuron. 2003;39:951–960. doi: 10.1016/s0896-6273(03)00560-9. [DOI] [PubMed] [Google Scholar]
- Ligon B, Boyd AE, Dunlap K. Class A calcium channel variants in pancreatic islets and their role in insulin secretion. J Biol Chem. 1998;273:13905–13911. doi: 10.1074/jbc.273.22.13905. [DOI] [PubMed] [Google Scholar]
- Lipscombe D, Castiglioni AJ. Alternative splicing in voltage-gated calcium channels. In: McDonough SI, editor. Calcium channel pharmacology. London: Kluwer Academic/Plenum; 2004. pp. 369–409. [Google Scholar]
- Meuth S, Pape HC, Budde T. Modulation of Ca2+ currents in rat thalamocortical relay neurons by activity and phosphorylation. Eur J Neurosci. 2002;15:1603–1614. doi: 10.1046/j.1460-9568.2002.01999.x. [DOI] [PubMed] [Google Scholar]
- Meuth SG, Kanyshkova T, Landgraf P, Pape HC, Budde T. Influence of Ca2+-binding proteins and the cytoskeleton on Ca2+-dependent inactivation of high-voltage activated Ca2+ currents in thalamocortical relay neurons. Pflügers Arch. 2005;450:111–122. doi: 10.1007/s00424-004-1377-z. [DOI] [PubMed] [Google Scholar]
- Pan CY, Jeromin A, Lundstrom K, Yoo SH, Roder J, Fox AP. Alterations in exocytosis induced by neuronal Ca2+ sensor-1 in bovine chromaffin cells. J Neurosci. 2002;22:2427–2433. doi: 10.1523/JNEUROSCI.22-07-02427.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pate P, Mochca-Morales J, Wu Y, Zhang JZ, Rodney G, Serysheva II, Williams BY, Anderson ME, Hamilton SL. Determinants for calmodulin binding on voltage dependent calcium channels. J Biol Chem. 2000;275:39786–39792. doi: 10.1074/jbc.M007158200. [DOI] [PubMed] [Google Scholar]
- Patil PG, Brody DL, Yue DT. Preferential closed-state inactivation of neuronal calcium channels. Neuron. 1998;20:1027–1038. doi: 10.1016/s0896-6273(00)80483-3. [DOI] [PubMed] [Google Scholar]
- Petegem FV, Chatelain FC, Minor DL. Insights into voltage-gated calcium channel regulation from the structure of the CaV1.2 IQ domain-Ca2+/calmodulin complex. Nat Struct Mol Biol. 2005;12:1108–1115. doi: 10.1038/nsmb1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen BZ, DeMaria CD, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron. 1999;22:549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- Polo-Parada L, Chan SA, Smith C. An activity-dependent increased role for L-type calcium channels in exocytosis is regulated by adrenergic signaling in chromaffin cells. Neuroscience. 2006;143:445–459. doi: 10.1016/j.neuroscience.2006.08.001. [DOI] [PubMed] [Google Scholar]
- Powell AD, Teschemacher AG, Seward EP. P2Y purinoceptors inhibit exocytosis in adrenal chromaffin cells via modulation of voltage-operated calcium channels. J Neurosci. 2000;20:606–616. doi: 10.1523/JNEUROSCI.20-02-00606.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch M, Neher E. Rates of diffusional exchange between small cells and a measuring patch pipette. Pflügers Arch. 1988;411:204–211. doi: 10.1007/BF00582316. [DOI] [PubMed] [Google Scholar]
- Rettig J, Sheng ZH, Kim DK, Hodson CD, Snutch TP, Catterall WA. Isoform-specific interaction of the alpha(1A) subunits of brain Ca2+ channels with the presynaptic proteins syntaxin and SNAP-25. Proc Natl Acad Sci USA. 1996;93:7363–7368. doi: 10.1073/pnas.93.14.7363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seward EP, Nowycky MC. Kinetics of stimulus-coupled secretion in dialyzed bovine chromaffin cells in response to trains of depolarizing pulses. J Neurosci. 1996;16:553–562. doi: 10.1523/JNEUROSCI.16-02-00553.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seward EP, Chernevskaya NI, Nowycky MC. Exocytosis in peptidergic nerve terminals exhibits two calcium-sensitive phases during pulsatile calcium entry. J Neurosci. 1995;15:3390–3399. doi: 10.1523/JNEUROSCI.15-05-03390.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Yu D, Hiel H, Liao P, Yue DT, Fuchs PA, Soong TW. Alternative splicing of the Cav1.3 channel IQ domain, a molecular switch for Ca2+-dependent inactivation within auditory hair cells. J Neurosci. 2006;26:10690–10699. doi: 10.1523/JNEUROSCI.2093-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Hamedinger D, Hoda JC, Gebhart M, Koschak A, Romanin C, Striessnig J. C-terminal modulator controls Ca2+-dependent gating of Cav1.4 L-type Ca2+ channels. Nat Neurosci. 2006;9:1108–1116. doi: 10.1038/nn1751. [DOI] [PubMed] [Google Scholar]
- Soong TW, DeMaria CD, Alvania RS, Zweifel LS, Liang MC, Mittman S, Agnew WS, Yue DT. Systematic identification of splice variants in human P/Q-type channel α12.1 subunits: implications for current density and Ca2+-dependent inactivation. J Neurosci. 2002;22:10142–10152. doi: 10.1523/JNEUROSCI.22-23-10142.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teschemacher AG, Seward EP. Bidirectional modulation of exocytosis by angiontensin II involves multiple G-protein-regulated transduction pathways in adrenal chromaffin cells. J Neurosci. 2000;20:4776–4785. doi: 10.1523/JNEUROSCI.20-13-04776.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaler C, Gray AC, Lipscombe D. Cumulative inactivation of N-type CaV2.2 calcium channels modified by alternative splicing. Proc Natl Acad Sci USA. 2004;101:5675–5679. doi: 10.1073/pnas.0303402101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiagarajan R, Tewolde T, Li Y, Becker PL, Rich MM, Engisch KL. Rab3A negatively regulates activity-dependent modulation of exocytosis in bovine adrenal chromaffin cells. J Physiol (Lond) 2004;555:439–457. doi: 10.1113/jphysiol.2003.056333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiagarajan R, Wilhelm J, Tewolde T, Li Y, Rich MM, Engisch KL. Enhancement of asynchronous and train-evoked exocytosis in bovine adrenal chromaffin cells infected with a replication deficient adenovirus. J Neurophysiol. 2005;94:3278–3291. doi: 10.1152/jn.00336.2005. [DOI] [PubMed] [Google Scholar]
- Weiss JL, Burgoyne RD. Voltage-independent inhibition of P/Q-type Ca2+ channels in adrenal chromaffin cells via a neuronal Ca2+ sensor-1-dependent pathway involves Src family tyrosine kinase. J Biol Chem. 2001;276:44804–44811. doi: 10.1074/jbc.M103262200. [DOI] [PubMed] [Google Scholar]
- Xu J, Wu LG. The decrease in the presynaptic calcium current is a major cause of short-term depression at a calyx-type synapse. Neuron. 2005;46:633–645. doi: 10.1016/j.neuron.2005.03.024. [DOI] [PubMed] [Google Scholar]
- Zhong H, Yokoyama CT, Scheuer T, Catterall WA. Reciprocal regulation of P/Q-type Ca2+ channels by SNAP-25, syntaxin and synaptotagmin. Nat Neurosci. 1999;2:939–941. doi: 10.1038/14721. [DOI] [PubMed] [Google Scholar]
- Zuhlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999;399:159–162. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]