

Abstract
Farnesoid X receptor (FXR) agonism is emerging as an important potential therapeutic mechanism of action for multiple chronic liver diseases. The bile acid‐derived FXR agonist obeticholic acid (OCA) has shown promise in a phase 2 study in patients with nonalcoholic steatohepatitis (NASH). Here, we report efficacy of the novel nonbile acid FXR agonist tropifexor (LJN452) in two distinct preclinical models of NASH. The efficacy of tropifexor at <1 mg/kg doses was superior to that of OCA at 25 mg/kg in the liver in both NASH models. In a chemical and dietary model of NASH (Stelic animal model [STAM]), tropifexor reversed established fibrosis and reduced the nonalcoholic fatty liver disease activity score and hepatic triglycerides. In an insulin‐resistant obese NASH model (amylin liver NASH model [AMLN]), tropifexor markedly reduced steatohepatitis, fibrosis, and profibrogenic gene expression. Transcriptome analysis of livers from AMLN mice revealed 461 differentially expressed genes following tropifexor treatment that included a combination of signatures associated with reduction of oxidative stress, fibrogenesis, and inflammation. Conclusion: Based on preclinical validation in animal models, tropifexor is a promising investigational therapy that is currently under phase 2 development for NASH.
Abbreviations
- Acta2
α‐smooth muscle actin
- Aif1
allograft inflammatory factor 1
- ALT
alanine aminotransferase
- AMLN
amylin liver nonalcoholic steatohepatitis model
- ANOVA
analysis of variance
- AST
aspartate aminotransferase
- BSEP
bile salt export pump
- Ccl2
chemokine (C-C motif) ligand 2
- CD
clusters of differentiation
- COL1A1
collagen, type I, alpha 1
- CYP
cytochrome P450
- DEG
differentially expressed gene
- EC50
half‐maximal effective concentration
- FGF
fibroblast growth factor
- FXR
farnesoid X receptor
- H&E
hematoxylin and eosin
- HFD
high‐fat diet
- IBA1
ionized calcium‐binding adaptor molecule 1
- mRNA
messenger RNA
- NAFLD
nonalcoholic fatty liver disease
- NAS
nonalcoholic fatty liver disease activity score
- NASH
nonalcoholic steatohepatitis
- OCA
obeticholic acid
- qRT‐PCR
quantitative real‐time polymerase chain reaction
- RNAseq
RNA sequencing
- SHP
small heterodimer partner
- STAM
Stelic animal model
- STZ
streptozotocin
- TIMP1
tissue inhibitor of metalloproteinase 1
- vol
volume
Nonalcoholic fatty liver disease (NAFLD) is a hepatic manifestation of the metabolic syndrome and is becoming an epidemic; its prevalence in Western countries ranges from 20% to 33%.1, 2 Approximately 2% to 3% of patients with NAFLD progress to nonalcoholic steatohepatitis (NASH), which is characterized by lipid accumulation in the liver (steatosis), hepatocellular inflammation (steatohepatitis), injury (ballooning), and fibrosis, which may eventually lead to cirrhosis, hepatocellular carcinoma (HCC), and liver‐related mortality.3, 4 NASH is the most rapidly increasing etiology among patients wait listed for liver transplant and is expected to become the leading indication for liver transplantation in the near future.5, 6 There is a clear lack of effective therapies targeting the underlying pathophysiology, symptoms, and complications of NASH, and as its global prevalence continues to rise, therapies that can reduce the progression to cirrhosis are urgently needed.7
The farnesoid X receptor (FXR) is an attractive therapeutic target for NASH1 due to its role as a master metabolic regulator and sensor of elevated bile acid levels.8, 9 FXR is expressed in the liver, intestine, and kidneys10 and is activated by bile acids.11, 12 FXR initiates homeostatic responses to control bile acid levels by inducing genes involved in basolateral bile acid efflux, conjugation, detoxification, and renal excretion.1, 8 In addition, FXR reduces hepatic bile acids by down‐regulating expression of genes involved in hepatic bile acid uptake and synthesis through small heterodimer partner (SHP)13 in the liver.14 Some of the effects of FXR are indirectly mediated by inducing the ileal enterokine fibroblast growth factor 15 (FGF15) in rodents and FGF19 in humans. In the liver, FGF19 inhibits gluconeogenesis, stimulates glycogen synthesis, and regulates bile acid synthesis through down‐regulation of cytochrome P450 family 7 subfamily A member 1 (CYP7A1) expression in an SHP‐dependent manner.15
Several studies point toward a benefit of FXR as a therapeutic target for NASH.16 In patients with NAFLD, hepatic FXR expression is down‐regulated and its levels are inversely correlated with disease severity.17 FXR‐null mice display increased levels of hepatic inflammation and fibrosis.18, 19 FXR agonism protects against the development of inflammation and fibrosis in various rodent models of liver disease.20, 21, 22 FXR activation also reduces lipogenesis and promotes fatty acid β‐oxidation,23 thereby reducing hepatic steatosis.24, 25, 26
A phase 2 study with the bile acid‐derived FXR agonist obeticholic acid (OCA) provided clinical validation for FXR agonists as therapies for NASH.27 In the FXR Ligand Obeticholic Acid in NASH Treatment (FLINT) trial, 35% of patients treated with OCA showed improvement in fibrosis compared with 19% of patients with placebo; however, OCA caused significant elevation in low‐density lipoprotein cholesterol and reduction in high‐density lipoprotein cholesterol. Furthermore, pruritus was more common in patients treated with OCA (23%) compared to patients with placebo (6%),28 possibly due to off‐target activation of cell surface G protein‐coupled bile acid receptor 1 (GPBAR1) by OCA or its metabolites.29, 30
Novel nonbile acid FXR agonists, which retain efficacy while lacking activity on GPBAR1, may reduce side effects and provide promising treatment options for NASH. Hence, these results have stimulated the search for effective nonbile acid FXR agonists. Here, we report the efficacy of tropifexor (the most potent FXR agonist undergoing clinical investigation31) in preclinical models of NASH and provide new insights into its potentially beneficial molecular mechanisms.
Materials and Methods
Test Substances
For in vitro assays, tropifexor was dissolved in dimethyl sulfoxide (DMSO) followed by intermediate dilutions in Williams’ E medium (Life Technologies) to obtain final concentrations (1×) with 0.1% DMSO. For animal experiments, tropifexor was formulated in 0.5% methylcellulose/0.5% Tween 80 in water, and OCA was formulated in a 20% polyethylene glycol 300, 30% tocopheryl polyethylene glycol (10%), 50% water solution.
In Vitro Experiments
Cryopreserved human hepatocytes (CellzDirect, Life Technologies) were cultured for 24 hours and then incubated with compounds or vehicle for 48 hours. RNA was isolated using the RNeasy 96 kit (Qiagen). RNA was isolated from frozen tissues homogenized in TRIzol (Invitrogen) and used for gene expression analysis. Expression of FXR target genes was analyzed by quantitative real‐time polymerase chain reaction (qRT‐PCR) using the SuperScript III One‐Step RT‐PCR System on the 7900HT from Applied Biosystems with the following settings: 1 cycle of 52°C for 25 minutes, 1 cycle of 95°C for 10 minutes, and 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. The primer pairs and probes used are described in Supporting Table S1.
Animal Experiments
Experimental protocols were approved by the local Animal Care and Use Committee and were in compliance with Animal Welfare Act regulations and U.S. regulations (Guide for the Care and Use of Laboratory Animals).
Experiments in the Stelic animal model (STAM) were performed at SMC Laboratories (Tokyo, Japan). Briefly, 2‐day‐old male C57BL/6J mice were injected with streptozotocin (STZ) and placed on a high‐fat diet (HFD) from weeks 4 to 12 (HFD‐32; CLEA Japan, Tokyo, Japan). From weeks 9 to 12, STAM mice received 0.03, 0.1, or 0.3 mg/kg of tropifexor; 25 mg/kg of OCA; or corresponding vehicles once a day (qd). The animals were maintained in a specific‐pathogen‐free facility under controlled conditions of temperature (23°C ± 2°C), humidity (45% ± 10%), lighting (12‐hour artificial light and dark cycles; light from 8:00 am to 8:00 pm), and air exchange. A high pressure was maintained in the experimental room to prevent contamination of the facility. The animals were housed in transparent polymer X (TPX) cages (CLEA Japan) with a maximum of four mice per cage. Hematoxylin and eosin (H&E)‐stained sections were evaluated for the NAFLD activity score (NAS) according to defined criteria.32 Total lipid extracts were isolated from livers of treated mice33 by homogenizing in chloroform–methanol (2:1 volume [vol]/vol) and incubating overnight at room temperature. After washing with chloroform–methanol–water (8:4:3 vol/vol/vol), the extracts were air dried and dissolved in isopropanol. Liver triglycerides were measured using the Triglyceride E‐test (Wako Pure Chemical Industries, Ltd., Japan).
A separate model for diet‐induced NASH (amylin liver NASH model [AMLN]) was employed as described by Trevaskis et al.34 Male C57BL/6 mice approximately 6 weeks of age were maintained on a high‐fat (40% kcal; Primex), high‐fructose (22% by weight), and high‐cholesterol (2% by weight) diet (D09100301; Research Diets, Inc., New Brunswick, NJ) for 26 weeks to induce NASH. Control animals received a low‐fat diet (10% kcal) with no fructose or cholesterol (D09100304; Research Diets). From week 26, animals received tropifexor (0.1, 0.3, or 0.9 mg/kg) or OCA (25 mg/kg) qd orally for 4 weeks. Collagen type I alpha 1 (Col1a1) and tissue inhibitor of metalloproteinase 1 (Timp1) messenger RNA (mRNA) were analyzed by qRT‐PCR.
Histopathology
Liver sections were fixed in 4% paraformaldehyde for 48 hours for histopathologic analysis. Liver damage and collagen deposition were assessed by H&E staining and picrosirius red staining, respectively. In the diet‐driven NASH model, liver sections were stained with Masson trichrome stain (Sigma‐Aldrich, St Louis, MO) and for ionized calcium‐binding adaptor molecule 1 (IBA1; 019‐19741; Wako). Image analysis was performed with a positive pixel count algorithm using Aperio software (Aperio, Inc., Vista, CA).
RNA Sequencing
Livers from AMLN mice were used for RNA sequencing (RNAseq). RNAseq reads in FASTQ format were aligned to the mouse genome (mm10) using STAR 2.5.1. Counts for each gene were quantified and normalized across all samples using Cuffnorm, version 2.2.1.35 Differentially expressed genes (DEGs) were identified by Cuffdiff with the following cutoffs unless specified otherwise: log2 fold change >1 or <−1, false discovery rate <5%, and reads per kilobase per million >1 in at least one treatment group. Gene ontology analysis was performed, following which RNAseq data were deposited into the Gene Expression Omnibus database (accession number GSE129389).
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 5.02 (GraphPad Software, San Diego, CA). For efficacy studies in diet‐driven NASH models, analysis of variance (ANOVA) followed by Dunnett's multiple comparison posttest was used to determine specific differences between treated and vehicle groups.
Results
Tropifexor Is a Highly Potent Agonist of the Bile Acid Receptor FXR
We previously reported the discovery and optimization of the highly potent and selective FXR agonist tropifexor (Fig. 1A).31 Tropifexor showed a half‐maximal effective concentration (EC50) in the subnanomolar range in several functional assays, including an assay measuring the interaction of FXR with steroid receptor coactivator 1 (SRC1; mean EC50, 0.20 nM) and a transcriptional activity assay measuring the promoter activity of a canonical FXR target gene, bile salt export pump (BSEP; EC50, 0.26 nM; Fig. 1A). In agreement with its high potency in the biochemical and cell‐based assays, tropifexor induced the expression of BSEP and SHP at concentrations as low as 0.1 nM in human primary hepatocytes (Fig. 1B). Furthermore, in vivo target engagement by tropifexor was confirmed by induction of FXR target genes in the liver (Shp and Bsep) and ileum (Shp and Fgf15) and an increase in circulating FGF15 protein.31 Taken together, these data demonstrate that tropifexor is an orally bioavailable potent FXR agonist that is able to regulate FXR in the liver and intestine, both in vitro and in vivo.
Figure 1.
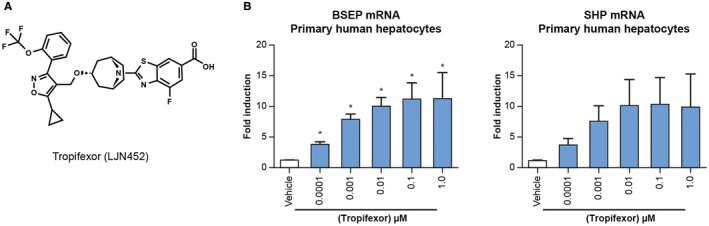
Tropifexor regulates FXR target genes in human hepatocytes. (A) Chemical structure of tropifexor. (B) Tropifexor dose dependently induced the expression of FXR target genes SHP and BSEP in primary human hepatocytes. Data are representative of three independent experiments using hepatocytes from three independent donors. Values are displayed as mean ± SEM; n = 3. *P < 0 .05 versus vehicle using one‐way ANOVA with Dunnett's test.
Tropifexor Reduces Hepatic Steatosis, Inflammation, and Fibrosis in Mice With NASH
We evaluated the ability of tropifexor to improve the characteristic pathophysiologic features of NASH in two distinct mouse models. The first is an insulin‐deficient model in which diabetes was induced by an early injection of STZ followed by feeding an HFD from weeks 4 to 12 (STAM model). After NASH was established at week 9, some animals were killed as the disease comparator group (baseline) whereas the remaining mice were treated with vehicle, tropifexor, or OCA (Fig. 2A). Compared with normal mice, STAM mice showed a marked increase in NAS, liver triglyceride levels, and percentage of fibrotic areas (Fig. 2B‐D). At week 12, NASH control (untreated animals) or vehicle‐treated animals did not display significant differences from the baseline group. Treatment with tropifexor showed a significant decrease in NAS at doses 0.1 and 0.3 mg/kg due to reductions in all three components of NAS (steatosis, lobular inflammation, and hepatocyte ballooning; Fig. 2B). Steatosis improvement was demonstrated by histopathology and reduction in liver triglycerides (Fig. 2B,C). Importantly, these changes were observed relative to the baseline group, indicating a regression in NASH from the baseline (Fig. 2B‐D).
Figure 2.
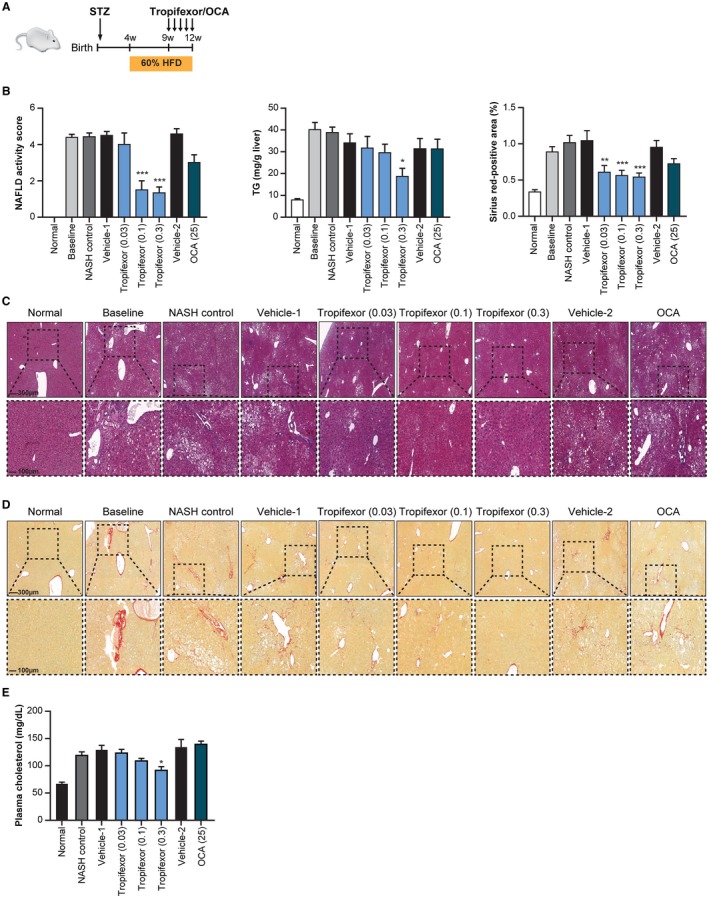
Tropifexor ameliorates NASH‐like symptoms in the STAM model. (A) Schematic representation of induction of STAM disease in mice. Normal group represents animals that received neither STZ nor high fat and were killed at week 9; baseline group represents animals that developed the disease up to week 9 and were killed before treatment initiation. NASH control group represents animals that developed the disease up to week 9 but were killed at week 12 without receiving either vehicle or drug. NASH treatment groups include animals that developed the disease up to week 9 and were treated from weeks 9 to 12 with tropifexor (0.03 to 0.3 mg/kg), OCA (25 mg/kg), or respective vehicles. (B) NAFLD activity score, hepatic triglycerides, and sirius red‐positive areas were significantly reduced with tropifexor treatment (n = 7). Values are represented as mean ± SEM. *P < 0.01, **P < 0.001, and ***P < 0.0001 compared to vehicle control by one‐way ANOVA with Dunnett's test. (C) H&E‐stained and (D) sirius red‐stained (without counterstain) liver sections from control or STAM disease animals show improvement in liver damage and fibrosis with tropifexor treatment, respectively. (E) Plasma cholesterol levels were significantly reduced in tropifexor‐treated mice only at the 0.3‐mg/kg dose but were not significantly reduced in OCA‐treated STAM mice. Values are represented as mean ± SEM. *P < 0.01 compared to vehicle control by one‐way ANOVA with Dunnett's test. Abbreviations: TG, triglyceride; w, weeks.
The percentage of sirius red‐positive areas within liver sections was higher in STAM mice relative to normal mice, demonstrating the presence of pericellular fibrosis in both baseline and NASH control groups. Mice treated with tropifexor showed a statistically significant dose‐dependent reduction of the fibrotic areas in comparison with those of the baseline group (Fig. 2B,D), indicating regression of the fibrotic phenotype of NASH by tropifexor. It is noteworthy that a high concentration of OCA (25 mg/kg) did not result in a significant decrease in liver triglycerides but tended toward reduction in NAS (P > 0.05). Additionally, serum cholesterol levels were reduced with tropifexor but not with OCA (Fig. 2E).
Because many individuals with NASH are obese and diabetic, we evaluated the effects of tropifexor in an obese insulin‐resistant model of NASH. NASH was established in mice by feeding a high‐trans fat, high‐fructose, and high‐cholesterol diet (AMLN) for 26 weeks36 followed by compound treatment for 4 weeks (Fig. 3A). Consistent with results from the STAM model, tropifexor could resolve liver inflammation, steatosis, and fibrosis in the therapeutic mode in this diet‐driven model of NASH (Fig. 3). Markers of liver damage alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were elevated in NASH mice compared to control animals that were fed with a low‐fat diet. Tropifexor‐treated NASH mice showed a dose‐dependent reduction of ALT and AST relative to vehicle‐treated controls (Fig. 3B). Importantly, the mid‐level dose of tropifexor normalized ALT and AST levels to that of control animals, whereas the high dose reduced ALT/AST to an even greater extent. By contrast, vehicle‐ and OCA‐treated mice showed similar plasma levels of ALT and AST in NASH mice. This finding agrees with reports that showed that OCA failed to reduce the levels of circulating liver enzymes in HFD‐fed rodent models of NASH.26, 37
Figure 3.
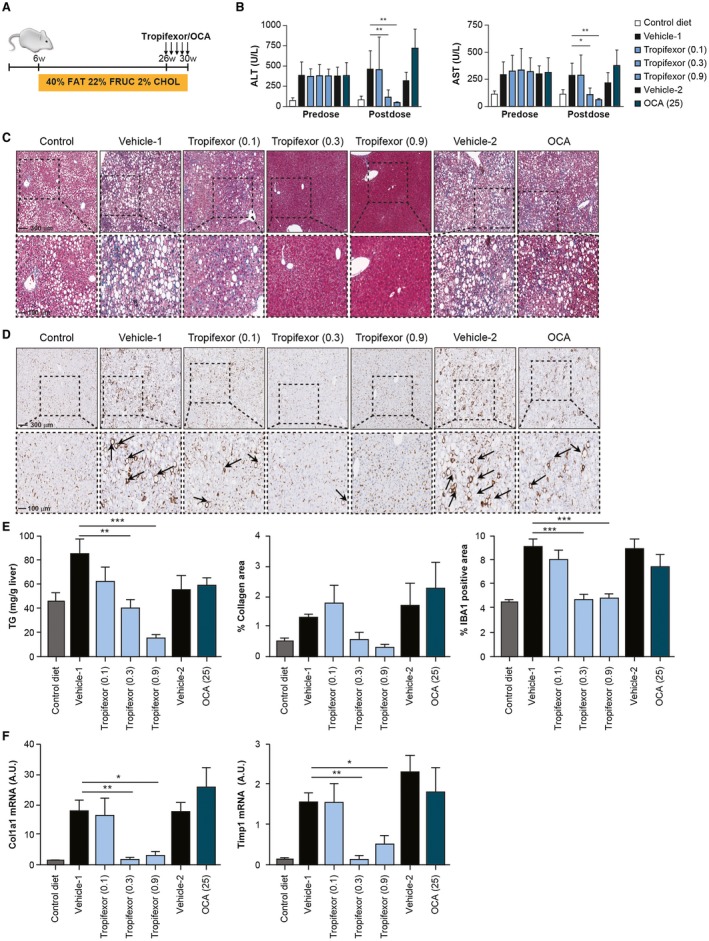
Tropifexor reverses fibrosis in a diet‐driven insulin‐resistant model of NASH. (A) Schematic representation of the diet‐driven mouse model for induction of NASH. Control animals received standard chow (10% kcal from fat, with no fructose or cholesterol), and NASH animals received high fat (40% kcal), high fructose (20% by weight), and high cholesterol (2% by weight) from 6 to 26 weeks. NASH animals were subsequently treated for 4 weeks with tropifexor (0.1, 0.3, or 0.9 mg/kg), OCA (25 mg/kg), or respective vehicles. (B) Serum markers AST and ALT were markedly elevated in NASH animals before dosing and were reduced in a dose‐dependent manner following 4 weeks of dosing with tropifexor but not OCA. *P < 0.05, **P < 0.01 compared to vehicle control by two‐way ANOVA with Dunnett's test. (C) Trichrome‐stained liver sections show dose‐dependent improvement in liver damage, and (D) IBA1‐stained liver sections show dose‐dependent decrease in infiltration of crown‐like macrophages (arrowheads) following treatment with tropifexor. (E) Hepatic triglyceride levels, collagen ratio as determined by trichrome‐positive areas, and IBA1‐positive areas were significantly reduced with tropifexor. **P < 0.01, ***P < 0.001 compared to vehicle control by one‐way ANOVA with Dunnett's test. (F) Expression levels of profibrogenic markers Col1a1 and Timp1 were also reduced in livers of tropifexor‐treated animals in a dose‐dependent manner. *P < 0.05, **P < 0.01 compared to vehicle control by one‐way ANOVA with Dunnett's test. Abbreviations: CHOL, cholesterol; FRUC, fructose; TG, triglyceride; w, weeks.
Liver histology analysis showed that the key phenotypic NASH features found in vehicle‐treated NASH mice (i.e., steatosis and ballooning) were completely reverted by tropifexor at 0.3 and 0.9 mg/kg doses (Fig. 3C). Dose‐dependent reduction in steatosis by tropifexor was further confirmed by quantification of liver triglycerides (Fig. 3E), with no statistically significant differences in hepatic cholesterol levels in any of the treated groups (data not shown).
In addition to steatosis, vehicle‐treated NASH groups displayed significant hepatic inflammation, as shown by the staining of macrophages and Kupffer cells (ionized calcium‐binding adapter molecule 1 [IBA1]‐positive cells; Fig. 3D). In these groups, macrophages form the characteristic crown‐like structures described in human and rodent NASH livers.38 Interestingly, hepatic crown‐like structures were completely eliminated in the livers of mid‐ and high‐dose tropifexor‐treated NASH mice but not in OCA‐treated mice (Fig. 3D). Quantification of IBA1‐positive staining further confirmed reduction of inflammation by tropifexor that was normalized to the same level of the control diet group with the 0.3‐mg/kg and 0.9‐mg/kg tropifexor dose groups (Fig. 3E).
Consistent with other studies, the AMLN diet induced hepatic fibrosis in this model of NASH34, 36 (Fig. 3C,E). Trichrome staining showed that tropifexor strongly abrogated collagen deposition in the liver (Fig. 3C,E) and dramatically reduced mRNA levels of the canonical profibrogenic markers Col1a1 and Timp1 (Fig. 3F). Notably, mice treated with a high dose of OCA (25 mg/kg) showed only a slight reduction in steatosis and hepatic triglyceride content as well as a minor reduction in inflammation and IBA1‐positive crown‐like structures. Consequently, extracellular matrix deposition and fibrosis were also slightly reduced in OCA‐treated mice. These results are consistent with reports in which OCA alone was not able to completely reduce steatosis, inflammation, and fibrosis in AMLN‐fed mice.37, 39
Taken together, both STAM and AMLN in vivo studies demonstrate that tropifexor has the potential to markedly improve NASH through reduction of liver steatosis, inflammation, and fibrosis.
Tropifexor Abrogates Inflammation and Fibrotic Signatures and Induces the Antioxidant Gene Expression Profile in NASH Livers
In order to obtain insights into the molecular mechanism of tropifexor, we performed a genome‐wide transcriptome analysis of livers from vehicle‐, tropifexor‐, and OCA‐treated AMLN mice. Interestingly, the gene signature regulated by tropifexor was found to be broader than that of OCA, with tropifexor not only regulating >90% of the DEGs by OCA but also showing a specific tropifexor gene signature. We defined a NASH signature comprising 588 DEGs by comparing livers of vehicle‐treated NASH mice relative to normal mice (Fig. 4A; Supporting Fig. S1, left panel). Gene ontology analysis confirmed that proinflammatory and fibrotic pathways were up‐regulated whereas pathways involved in steroid and lipid biosynthesis were down‐regulated in NASH livers. In line with amelioration of the NASH phenotype with tropifexor and consistent with the histology results in AMLN mice, RNAseq confirmed that dysregulation of NASH genes was largely reversed with mid‐dose (0.3 mg/kg) or high‐dose (0.9 mg/kg) tropifexor and only partially reverted by OCA. Furthermore, tropifexor was able to revert proinflammatory and fibrotic NASH signatures in a dose‐dependent manner (Fig. 4A), whereas OCA at 25 mg/kg only had a weak effect on NASH genes, consistent with its modest effect on the NASH phenotype in the AMLN model (Fig. 3).
Figure 4.
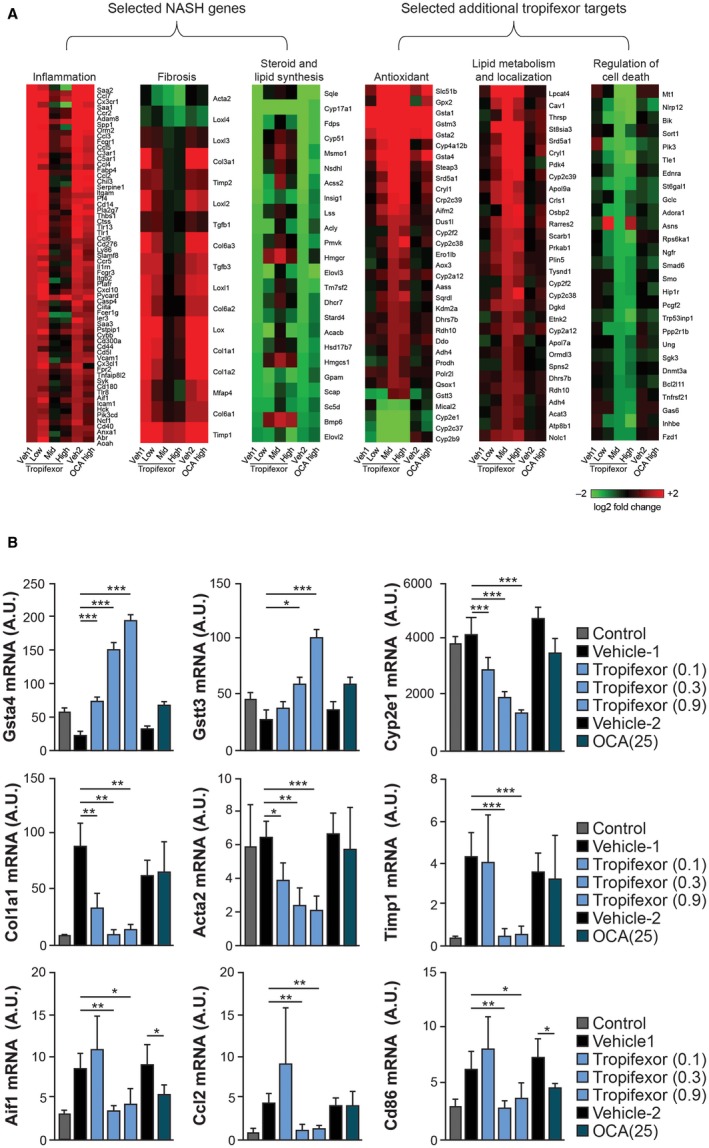
RNA sequencing identifies unique NASH‐ and tropifexor‐mediated signatures in NASH livers. The AMLN model was used for RNAseq. (A) Heat maps of selected DEGs in NASH livers treated with vehicle; low (0.1 mg/kg), mid (0.3 mg/kg), or high (0.9 mg/kg) dose of tropifexor; or 25 mg/kg of OCA (left panel). Heat maps of a set of unique DEGs in response to tropifexor treatment in NASH livers (right panel). (B) qRT‐PCR analysis to validate the differential expression of select genes identified by RNAseq. Expression levels of Gsta4, Gstt3, and Cyp2e1 as markers of oxidative stress; Col1a1, Acta2, and Timp1 as markers of fibrosis; and Aif1, Ccl2, and Cd86 as markers of inflammation were quantified. Values are represented as mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 compared to vehicle control. Abbreviations: Acta2, α‐smooth muscle actin; Aif1, allograft inflammatory factor 1; A.U., arbitrary units; Ccl2, chemokine (C‐C motif) ligand 2; Cd86, clusters of differentiation 86; Gsta4, glutathione S‐transferase alpha 4; Gstt3, glutathione S‐transferase theta 3; Veh, vehicle.
Interestingly, we also identified 461 additional DEGs in response to tropifexor treatment (Fig. 4; Supporting Fig. S1, right panel). Because these genes had similar expression levels in normal and NASH mice (data not shown), they may not be directly responsible for the NASH phenotype. Instead, they could belong to pathways indirectly contributing to the amelioration of the NASH phenotype. Gene ontology analysis suggested that the tropifexor signature genes belonged to pathways involved in the reduction of oxidative stress, lipid metabolism, and regulation of cell death (Fig. 4A). Notably, OCA only showed weak effects on these genes.
RNAseq results were validated using qRT‐PCR by analyzing the expression of select genes belonging to different biological processes (Fig. 4B). Tropifexor dose dependently up‐regulated antioxidant genes glutathione S‐transferase α4 and glutathione S‐transferase θ3 and down‐regulated the oxidative stress marker Cyp2e1. Tropifexor also dose dependently suppressed the expression of the canonical fibrotic markers Col1a1, α‐smooth muscle actin (Acta2), and Timp1 and reduced the expression of several proinflammatory genes, such as allograft inflammatory factor 1 (Aif1), chemokine (C‐C motif) ligand 2 (Ccl2), and clusters of differentiation 86 (Cd86), to the levels found in normal livers.
Discussion
The prevalence of NAFLD and NASH has increased over recent decades, and these conditions now manifest as a worldwide epidemic. The development and progression of hepatic fibrosis from NAFLD through NASH may ultimately lead to cirrhosis. In this advanced fibrotic stage, normal liver function is impaired to an extent requiring liver transplantation or can eventually cause HCC, for which liver transplantation remains the only available intervention. Currently, there are no approved pharmacologic therapies for NASH, and new therapies that reduce the progression of hepatic fibrosis may provide significant benefit in a wide variety of chronic liver diseases, including NASH. FXR agonists have emerged as potential treatment options for chronic liver disease, with the bile acid‐derived OCA showing efficacy in NASH.27, 28
Here, we described the preclinical efficacy of tropifexor, a novel nonbile acid FXR agonist, and its effect on gene expression in rodent models of NASH. Tropifexor is a highly potent FXR agonist that demonstrates remarkable efficacy in preclinical NASH models. In rodents, tropifexor potently and dose dependently induced (Fgf15, Bsep, and Shp) or repressed (Cyp8b1) FXR target genes in the ileum and liver.31 Notably, we observed increased circulating levels of the biomarker FGF15 in rodents31 and FGF19 in humans with tropifexor treatment.40
Given the complex pathophysiology of NASH, we evaluated the efficacy of tropifexor in disease reversal in two distinct rodent models. The mode of inducing fatty liver changes varies among different animal models, thereby causing high phenotypic variability. Thus, no single model can exhibit the full spectrum of NASH pathophysiology in humans, and any single model can limit the translation of preclinical efficacy to human disease.41, 42
The STAM model is a chemical and dietary model in which NASH is induced by an HFD in mice with STZ‐driven diabetes; this model is thought to be useful for investigating late‐stage NASH endpoints, namely, steatosis, inflammation, ballooning, and fibrosis, in a relatively short time frame.41 In STAM mice, tropifexor showed a significant reduction in NAS (which quantifies the extent of steatosis, inflammation, and ballooning) and liver triglycerides and was able to reverse liver fibrosis with a dose as low as 0.1 mg/kg.
We also evaluated plasma cholesterol levels following compound treatment in STAM mice. Although cholesterol levels were down‐regulated with tropifexor, no effect was observed with OCA. These findings are inconsistent with the effect of OCA observed in humans28 wherein cholesterol levels were elevated with OCA treatment. Thus, the use of this rodent model may not be an appropriate tool to predict the effects of FXR agonists on cholesterol metabolism in humans.
The dietary AMLN NASH model uses high trans fat in combination with high fructose and high cholesterol34, 36 and is characterized by obesity, insulin resistance, steatosis, fibrosis, and increased plasma AST and ALT. The AMLN model recapitulates several aspects of NASH in humans given that dietary fructose specifically increases de novo lipogenesis, promotes dyslipidemia, and increases insulin resistance and visceral adiposity in overweight/obese individuals.43 We used this model to test the efficacy of tropifexor in alleviating disease symptoms in the setting of obesity and insulin resistance by providing the NASH‐inducing diet for 26 weeks prior to compound treatment. Tropifexor showed exceptional efficacy on all parameters, including steatosis, inflammation, and fibrosis, with only 4 weeks of treatment. Tropifexor potently lowered ALT and AST levels within 2 weeks of treatment and continued to 4 weeks (Fig. 3; data not shown), and tropifexor reduced steatosis as determined by both histopathology and liver triglycerides. Marked changes in inflammation were demonstrated by a reduction in macrophages and the elimination of crown‐like structures in tropifexor‐treated mice. Importantly, tropifexor decreased fibrosis in a dose‐dependent manner as revealed by trichrome staining and normalization of mRNA levels of the fibrotic markers Col1a1 and Timp1 in tropifexor‐treated NASH animals compared to control animals. In this context, OCA is the only other FXR agonist to have recently shown improvement in NAS, steatosis, ballooning, and histologic markers of NASH in Lepobese [ ob ]/ ob mice maintained on the AMLN diet but not on the purely dietary AMLN model.26, 37, 44, 45
Liver fibrosis is a key hallmark of advanced NASH.3 In particular, fibrosis defines the prognosis in NASH and is associated with overall and liver‐related morbidity and mortality.46 Currently, there are no direct antifibrotic therapies approved for liver fibrosis; hence, resolution of fibrosis remains a key unmet need in the NASH disease landscape. We found that tropifexor significantly reduced liver fibrosis in a dose‐dependent manner in two distinct chronic liver disease models. Genome‐wide transcriptome analysis revealed two distinct gene signatures regulated by tropifexor: a panel of 588 genes that are differentially expressed between normal and NASH livers (also common to the OCA signature) and a panel of 461 genes that are strongly regulated by tropifexor regardless of whether the liver is in a normal or NASH state.
Mid and high doses of tropifexor were able to reverse the 588‐gene signature that comprised genes involved in steroid and lipid biosynthesis, inflammation, and fibrosis, suggesting that tropifexor could be effective against hepatic fibrosis in NASH. Additionally, the tropifexor‐specific 461‐gene signature included genes associated with lipid metabolism, regulation of cell death, and oxidative stress, which are highly prevalent in patients with NASH.47 We hypothesize that the combination of reduction in oxidative stress and down‐regulation of fibrotic and inflammatory genes could be responsible for the remarkable efficacy of tropifexor in the reversal of NASH in preclinical mouse models.
The differential regulation of gene expression patterns of tropifexor and OCA may be attributed to the differences in the chemical structures of these two FXR agonists. Fiorucci et al.48 first described that different structural FXR agonists exhibit differential gene expression profiles. In this regard, it is well known that distinct ligands of the nuclear vitamin D receptor are able to generate differential gene signatures,49 probably due to minor conformational changes induced in the receptor by the various ligands. We hypothesize that the tropifexor backbone allows a structural conformation of FXR that generates a more favorable interaction with coactivators and/or chromatin remodelers, including histone acetyltransferases (SRC1)16 and methyltransferases (protein arginine methyl‐transferase type I [PRMT1]),50 than the OCA backbone. Further studies will be needed to confirm this hypothesis. Based on the RNAseq findings, we conclude that the remarkable efficacy of tropifexor in reversal of NASH in preclinical mouse models may be attributed to its ability to regulate NASH‐dependent and NASH‐independent unique signature genes involved in oxidative stress, fibrosis, and inflammation.
In summary, tropifexor showed remarkable efficacy in two distinct preclinical models that recapitulated the characteristic features of human NASH, indicating a potentially superior translatability to human disease. Detailed analysis of the tropifexor‐induced transcriptome revealed a potential role of antioxidant pathways that could contribute to the superior efficacy of tropifexor across preclinical models. These findings warrant further clinical investigation of tropifexor, and a phase 2 trial (NCT02855164) in NASH is currently under way.
Supporting information
Acknowledgment
We thank Wendy Richmond, Perry Gordon, Kevin Johnson, Robbin Newlin, Liane Yanas, and Yenyen Yu for technical assistance and Ignacio Sancho‐Martinez for assistance with histology experiments and for providing inputs on the manuscript draft. We also thank Prachiti Narvekar (Novartis Healthcare Pvt., Ltd.) for medical writing support and editorial assistance.
Potential conflict of interest: Dr. Zheng, Dr. Fang, Dr. Dietrich, Dr. Bao, Dr. Klickstein, Dr. Molteni, Dr. McNamara, Dr. Tully, Dr. Badman, and Dr. Xu own stock in and are employed by Novartis; Dr. Hernandez, Dr. Kim, Dr. Rucker, Dr. Chianelli, Dr. Schmeits, Dr. Zoll, Dr. Federe, Dr. Chen, Dr. Walker and Dr. Meeusen are employed by Novartis; Dr. Liu and Dr. Laffitte owns stock in Novartis; the other authors have nothing to report.
References
- 1. Musso G, Cassader M, Gambino R. Non‐alcoholic steatohepatitis: emerging molecular targets and therapeutic strategies. Nat Rev Drug Discov 2016;15:249‐274. [DOI] [PubMed] [Google Scholar]
- 2. Loomba R, Sanyal AJ. The global NAFLD epidemic. Nat Rev Gastroenterol Hepatol 2013;10:686‐690. [DOI] [PubMed] [Google Scholar]
- 3. Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018;67:328‐357. [DOI] [PubMed] [Google Scholar]
- 4. Younossi ZM, Stepanova M, Rafiq N, Makhlouf H, Younoszai Z, Agrawal R, et al. Pathologic criteria for nonalcoholic steatohepatitis: interprotocol agreement and ability to predict liver‐related mortality. Hepatology 2011;53:1874‐1882. [DOI] [PubMed] [Google Scholar]
- 5. Wong RJ, Aguilar M, Cheung R, Perumpail RB, Harrison SA, Younossi ZM, et al. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 2015;148:547‐555. [DOI] [PubMed] [Google Scholar]
- 6. Cholankeril G, Wong RJ, Hu M, Perumpail RB, Yoo ER, Puri P, et al. Liver transplantation for nonalcoholic steatohepatitis in the US: temporal trends and outcomes. Dig Dis Sci 2017;62:2915‐2922. [DOI] [PubMed] [Google Scholar]
- 7. Williams R, Horton R. Liver disease in the UK: a Lancet Commission. Lancet 2013;382:1537‐1538. [DOI] [PubMed] [Google Scholar]
- 8. Calkin AC, Tontonoz P. Transcriptional integration of metabolism by the nuclear sterol‐activated receptors LXR and FXR. Nat Rev Mol Cell Biol 2012;13:213‐224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Matsubara T, Li F, Gonzalez FJ. FXR signaling in the enterohepatic system. Mol Cell Endocrinol 2013;368:17‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Forman BM, Goode E, Chen J, Oro AE, Bradley DJ, Perlmann T, et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell 1995;81:687‐693. [DOI] [PubMed] [Google Scholar]
- 11. Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, et al. Bile acids: natural ligands for an orphan nuclear receptor. Science 1999;284:1365‐1368. [DOI] [PubMed] [Google Scholar]
- 12. Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, et al. Identification of a nuclear receptor for bile acids. Science 1999;284:1362‐1365. [DOI] [PubMed] [Google Scholar]
- 13. Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, et al. A regulatory cascade of the nuclear receptors FXR, SHP‐1, and LRH‐1 represses bile acid biosynthesis. Mol Cell 2000;6:517‐526. [DOI] [PubMed] [Google Scholar]
- 14. Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab 2005;2:217‐225. [DOI] [PubMed] [Google Scholar]
- 15. Schaap FG, Trauner M, Jansen PL. Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol 2014;11:55‐67. [DOI] [PubMed] [Google Scholar]
- 16. Pellicciari R, Fiorucci S, Camaioni E, Clerici C, Costantino G, Maloney PR, et al. 6alpha‐ethyl‐chenodeoxycholic acid (6‐ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J Med Chem 2002;45:3569‐3572. [DOI] [PubMed] [Google Scholar]
- 17. Yang ZX, Shen W, Sun H. Effects of nuclear receptor FXR on the regulation of liver lipid metabolism in patients with non‐alcoholic fatty liver disease. Hepatol Int 2010;4:741‐748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang YD, Chen WD, Wang M, Yu D, Forman BM, Huang W. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology 2008;48:1632‐1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang YD, Yang F, Chen WD, Huang X, Lai L, Forman BM, et al. Farnesoid X receptor protects liver cells from apoptosis induced by serum deprivation in vitro and fasting in vivo. Mol Endocrinol 2008;22:1622‐1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang S, Wang J, Liu Q, Harnish DC. Farnesoid X receptor agonist WAY‐362450 attenuates liver inflammation and fibrosis in murine model of non‐alcoholic steatohepatitis. J Hepatol 2009;51:380‐388. [DOI] [PubMed] [Google Scholar]
- 21. Verbeke L, Mannaerts I, Schierwagen R, Govaere O, Klein S, Vander Elst I, et al. FXR agonist obeticholic acid reduces hepatic inflammation and fibrosis in a rat model of toxic cirrhosis. Sci Rep 2016;6:33453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fiorucci S, Antonelli E, Rizzo G, Renga B, Mencarelli A, Riccardi L, et al. The nuclear receptor SHP mediates inhibition of hepatic stellate cells by FXR and protects against liver fibrosis. Gastroenterology 2004;127:1497‐1512. [DOI] [PubMed] [Google Scholar]
- 23. Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, et al. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP‐1c. J Clin Invest 2004;113:1408‐1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jin L, Wang R, Zhu Y, Zheng W, Han Y, Guo F, et al. Selective targeting of nuclear receptor FXR by avermectin analogues with therapeutic effects on nonalcoholic fatty liver disease. Sci Rep 2015;5:17288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang Y, Lee FY, Barrera G, Lee H, Vales C, Gonzalez FJ, et al. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc Natl Acad Sci U S A 2006;103:1006‐1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cipriani S, Mencarelli A, Palladino G, Fiorucci S. FXR activation reverses insulin resistance and lipid abnormalities and protects against liver steatosis in Zucker (fa/fa) obese rats. J Lipid Res 2010;51:771‐784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mudaliar S, Henry RR, Sanyal AJ, Morrow L, Marschall HU, Kipnes M, et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 2013;145:574‐582.e571. [DOI] [PubMed] [Google Scholar]
- 28. Neuschwander‐Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al.; NASH Clinical Research Network . Farnesoid X nuclear receptor ligand obeticholic acid for non‐cirrhotic, non‐alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo‐controlled trial. Lancet 2015;385:956‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pellicciari R, Sato H, Gioiello A, Costantino G, Macchiarulo A, Sadeghpour BM, et al. Nongenomic actions of bile acids. Synthesis and preliminary characterization of 23‐ and 6,23‐alkyl‐substituted bile acid derivatives as selective modulators for the G‐protein coupled receptor TGR5. J Med Chem 2007;50:4265‐4268. [DOI] [PubMed] [Google Scholar]
- 30. Rizzo G, Passeri D, De Franco F, Ciaccioli G, Donadio L, Rizzo G, et al. Functional characterization of the semisynthetic bile acid derivative INT‐767, a dual farnesoid X receptor and TGR5 agonist. Mol Pharmacol 2010;78:617‐630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tully DC, Rucker PV, Chianelli D, Williams J, Vidal A, Alper PB, et al. Discovery of tropifexor (LJN452), a highly potent non‐bile acid FXR agonist for the treatment of cholestatic liver diseases and nonalcoholic steatohepatitis (NASH). J Med Chem 2017;60:9960‐9973. [DOI] [PubMed] [Google Scholar]
- 32. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al.; Nonalcoholic Steatohepatitis Clinical Research Network . Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313‐1321. [DOI] [PubMed] [Google Scholar]
- 33. Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 1957;226:497‐509. [PubMed] [Google Scholar]
- 34. Trevaskis JL, Griffin PS, Wittmer C, Neuschwander‐Tetri BA, Brunt EM, Dolman CS, et al. Glucagon‐like peptide‐1 receptor agonism improves metabolic, biochemical, and histopathological indices of nonalcoholic steatohepatitis in mice. Am J Physiol Gastrointest Liver Physiol 2012;302:G762‐G772. [DOI] [PubMed] [Google Scholar]
- 35. Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA‐seq experiments with TopHat and Cufflinks. Nat Protoc 2012;7:562‐578. Erratum in: Nat Protoc 2014;9:2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Clapper JR, Hendricks MD, Gu G, Wittmer C, Dolman CS, Herich J, et al. Diet‐induced mouse model of fatty liver disease and nonalcoholic steatohepatitis reflecting clinical disease progression and methods of assessment. Am J Physiol Gastrointest Liver Physiol 2013;305:G483‐G495. [DOI] [PubMed] [Google Scholar]
- 37. Jouihan H, Will S, Guionaud S, Boland ML, Oldham S, Ravn P, et al. Superior reductions in hepatic steatosis and fibrosis with co‐administration of a glucagon‐like peptide‐1 receptor agonist and obeticholic acid in mice. Mol Metab 2017;6:1360‐1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ioannou GN, Haigh WG, Thorning D, Savard C. Hepatic cholesterol crystals and crown‐like structures distinguish NASH from simple steatosis. J Lipid Res 2013;54:1326‐1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Makri E, Cholongitas E, Tziomalos K. Emerging role of obeticholic acid in the management of nonalcoholic fatty liver disease. World J Gastroenterol 2016;22:9039‐9043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Badman MK, Desai S, Laffitte B, Decris‐tofaro M, Lin TH, Chen J, et al. First‐in‐human experience with LJN452, an orally available non‐bile acid FXR agonist, demonstrates potent activation of FXR in healthy subjects. Hepatology 2016;64(Suppl. 1):A32. [Google Scholar]
- 41. Cole BK, Feaver RE, Wamhoff BR, Dash A. Non‐alcoholic fatty liver disease (NAFLD) models in drug discovery. Expert Opin Drug Discov 2018;13:193‐205. [DOI] [PubMed] [Google Scholar]
- 42. Santhekadur PK, Kumar DP, Sanyal AJ. Preclinical models of non‐alcoholic fatty liver disease. J Hepatol 2018;68:230‐237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stanhope KL, Schwarz JM, Keim NL, Griffen SC, Bremer AA, Graham JL, et al. Consuming fructose‐sweetened, not glucose‐sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J Clin Invest 2009;119:1322‐1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Roth J, Feigh M, Veidal S, Rigbolt K, Young M. Obeticholic acid improves histological, biochemical and gene expression profiles in Gubra AMLN mice with biopsy‐confirmed NASH. J Hepatol 2017;66(Suppl.):S431‐S432. [Google Scholar]
- 45. Tolbol KS, Kristiansen MN, Hansen HH, Veidal SS, Rigbolt KT, Gillum MP, et al. Metabolic and hepatic effects of liraglutide, obeticholic acid and elafibranor in diet‐induced obese mouse models of biopsy‐confirmed nonalcoholic steatohepatitis. World J Gastroenterol 2018;24:179‐194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Singh S, Allen AM, Wang Z, Prokop LJ, Murad MH, Loomba R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta‐analysis of paired‐biopsy studies. Clin Gastroenterol Hepatol 2015;13:643‐654.e641‐e649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Grohmann M, Wiede F, Dodd GT, Gurzov EN, Ooi GJ, Butt T, et al. Obesity drives STAT‐1‐dependent NASH and STAT‐3‐dependent HCC. Cell 2018;175:1289‐1306.e1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fiorucci S, Mencarelli A, Palladino G, Cipriani S. Bile‐acid‐activated receptors: targeting TGR5 and farnesoid‐X‐receptor in lipid and glucose disorders. Trends Pharmacol Sci 2009;30:570‐580. [DOI] [PubMed] [Google Scholar]
- 49. Ito I, Waku T, Aoki M, Abe R, Nagai Y, Watanabe T, et al. A nonclassical vitamin D receptor pathway suppresses renal fibrosis. J Clin Invest 2013;123:4579‐4594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rizzo G, Renga B, Antonelli E, Passeri D, Pellicciari R, Fiorucci S. The methyl transferase PRMT1 functions as co‐activator of farnesoid X receptor (FXR)/9‐cis retinoid X receptor and regulates transcription of FXR responsive genes. Mol Pharmacol 2005;68:551‐558. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials