

Abstract
β-Site amyloid precursor protein cleaving enzyme 1 (BACE1) (β-secretase) initiates generation of β-amyloid (Aβ), which plays an early role in Alzheimer's disease (AD). BACE1 levels are increased in postmortem AD brain, suggesting BACE1 elevation promotes Aβ production and AD. Alternatively, the BACE1 increase may be an epiphenomenon of late-stage AD. To distinguish between these possibilities, we analyzed BACE1 elevation using a highly specific BACE1 antibody, BACE-Cat1, made in BACE1−/− mice, which mount a robust anti-BACE1 immune response. Previous BACE1 immunohistochemical studies lack consistent results because typical BACE1 antibodies produce nonspecific background, but BACE-Cat1 immunolabels BACE1 only. BACE1 elevation was recapitulated in two amyloid precursor protein (APP) transgenic mouse lines. 5XFAD mice form amyloid plaques at young ages and exhibit neuron loss. In contrast, Tg2576 form plaques at a more advanced age and do not show cell death. These two mouse lines allow differentiation between early Aβ-induced events and late phenomena related to neuron death. BACE1 levels became elevated in parallel with amyloid burden in each APP transgenic, starting early in 5XFAD and late in Tg2576. The increase in BACE1 protein occurred without any change in BACE1 mRNA level, indicating a posttranscriptional mechanism. In APP transgenic and AD brains, high BACE1 levels were observed in an annulus around Aβ42-positive plaque cores and colocalized with neuronal proteins. These results demonstrate that amyloid plaques induce BACE1 in surrounding neurons at early stages of pathology before neuron death occurs. We conclude that BACE1 elevation is most likely triggered by the amyloid pathway and may drive a positive-feedback loop in AD.
Keywords: APP transgenic, BACE-Cat1, β-secretase, neurodegeneration, Tg2576, 5XFAD
Introduction
Amyloid plaques, deposits of β-amyloid (Aβ), are defining lesions in Alzheimer's disease (AD) brain (Selkoe, 2000). The etiology of AD is poorly understood, however accumulation of Aβ is likely to play an early role in AD pathogenesis (Naslund et al., 2000; Hardy and Selkoe, 2002). Aβ is a proteolytic product of the amyloid precursor protein (APP) and is generated through sequential cleavages by enzymes called β- and γ-secretase. β-Secretase was identified as the transmembrane aspartic protease β-site APP cleaving enzyme 1 (BACE1) (Hussain et al., 1999; Sinha et al., 1999; Vassar et al., 1999; Yan et al., 1999; Haniu et al., 2000). Because BACE1 initiates formation of Aβ, factors that elevate BACE1 levels may promote AD.
Recent studies demonstrate that BACE1 levels and activity are increased in postmortem AD brains (Fukumoto et al., 2002; Holsinger et al., 2002; Tyler et al., 2002; Yang et al., 2003; Li et al., 2004; Harada et al., 2006), suggesting a role in AD. However, numerous proteins become elevated in AD (for review, see Fountoulakis and Kossida, 2006) and the AD brain undergoes massive cell death (for review, see Morrison and Hof, 1997), so it is difficult to determine from postmortem brain whether any given change is an epiphenomenon in late-stage AD, or whether it is an early event directly involved in pathogenesis. To address this problem, we investigated whether the BACE1 elevation could be recapitulated in two APP transgenic mouse lines, 5XFAD (Oakley et al., 2006) and Tg2576 (Hsiao et al., 1996), which develop amyloid plaques at young and old ages, and do and do not exhibit neuron loss, respectively. In contrast to postmortem AD, these APP transgenic lines permitted us to analyze the effects of amyloid pathology on BACE1 elevation in the presence or absence of cell death.
Our analysis required immunohistochemistry (IHC) and we needed a BACE1 antibody that did not cross-react with other polypeptides. Previous BACE1 immunolocalization studies in AD lacked consistency, variously reporting BACE1 in neuron cell bodies (Fukumoto et al., 2002; Sun et al., 2002), neurites (Sheng et al., 2003), tangle-bearing neurons (Leuba et al., 2005), and astrocytes around plaques (Rossner et al., 2001; Heneka et al., 2005). We found that typical BACE1 antibodies produce nonspecific backgrounds in immunohistochemistry and bind to numerous non-BACE1 polypeptides on immunoblots. Therefore, we generated a highly specific anti-BACE1 monoclonal antibody, named BACE-Cat1, by immunizing BACE1−/− mice, which are immunologically naive to BACE1 and develop a robust immune response against BACE1 immunogen. Immunoblots demonstrated that BACE-Cat1 was monospecific for BACE1. Furthermore, BACE1 elevation occurred without increased BACE1 mRNA levels, paralleled amyloid load, and rose early in 5XFAD and late in Tg2576. Importantly, BACE1 elevation appeared in an annulus around Aβ42-positive plaques and colocalized with neuronal, but not astrocytic, markers in APP transgenic and AD brains. These results strongly suggest that BACE1 elevation in AD (1) is triggered by amyloid pathology, (2) is an early event before neuron loss, (3) is posttranscriptional, and (4) involves a positive-feedback loop that drives Aβ production and promotes AD.
Materials and Methods
Animals and human brain tissues.
The BACE1 knock-out mice (129/Black Swiss or C57BL/6J background) were generated as described previously (Luo et al., 2001). The APP/PS1 double transgenic Tg6799 mice (C57BL/6J and SJL hybrid background) containing five familial AD mutations (henceforth called “5XFAD” mice) were created by coinjecting APP K670N/M671L plus I716V plus V717I and PS1 M146L plus L286V transgenes (both driven by the mouse Thy1 promoter) into single-cell embryos, thus allowing cointegration, so that mice breed as single transgenics (Oakley et al., 2006). The C57BL/6J wild-type and Tg2576 (APPsw, K670N/M671L) (Hsiao et al., 1996) mice were purchased from Taconic (Hudson, NY). All mice were maintained in microisolator cages in the Barrier Facilities of Northwestern University Center for Comparative Medicine. All animal procedures were in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Northwestern University Animal Care and Use Committee.
Temporal lobe of postmortem brain cortex tissue (frozen samples and 4% paraformaldehyde-fixed/cryopreserved) from two human AD (Braak stage VI; 71-year-old male and 89-year-old female) and one non-demented (ND) (Braak stage IV; 85-year-old female) subjects were provided by Northwestern University Cognitive Neurology and Alzheimer's Disease Center Brain Bank. In addition, tissue from inferior temporal gyrus (cryopreserved 40 μm sections) from one AD subject and one ND subject were obtained from Rush Alzheimer's Disease Center at the Rush University Medical Center (Chicago, IL).
Production of human BACE1 N terminus protein.
DNA encoding the BACE1 N terminus catalytic domain (amino acids 46–460) was amplified from the human BACE1 cDNA (Vassar et al., 1999) by PfuUltra High-Fidelity DNA Polymerase-Based PCR (Stratagene, La Jolla, CA) with the oligonucleotide forward primer 5′-CACCGAGACCGACGAAGAGCC-3′ and the reverse primer 5′-CTAATAGGCTATGGTCATGAGGGT-3′ (Integrated DNA Technologies, Coralville, IA). After purification using the QIAquick PCR Purification kit from Qiagen (Valencia, CA), BACE1 N terminus DNA was subcloned in-frame with the N-terminal 6xHis tag of pET100 using the pET100/D-TOPO Expression kit (Invitrogen, Carlsbad, CA) following the manufacturer's instructions. The resulting plasmid was verified with DNA sequencing (SeqWright DNA Technology Services; Fisher Scientific, Houston, TX) before purification using the QIAfilter Maxi kit (Qiagen) and transformation into One Shot TOP10 chemically competent cells. Expression of the 6xHis-BACE1 N terminus protein was induced in BL21 Star (DE3) Escherichia coli by 1 m isopropyl β-d-thiogalactoside (Invitrogen), and then protein purified by Ni-NTA agarose (Qiagen) chromatography following the manufacturer's instructions. Purified proteins were dialyzed against a buffer containing 10 mm Tris-HCl with 0.1% SDS, pH 7.0, at 4°C using D-Tube Dialyzer Maxi of MWCO 12–14 kDa (Novagen/EMD Biosciences, Madison, WI) to remove small protein contaminants, and then concentrated in Millipore Amicon Ultra-4 centrifugal filter units (Fisher Scientific) at 3000 rpm for 5 min at 4°C in the Beckman Allegra 25R centrifuge (Beckman Coulter, Fullerton, CA). Concentrated recombinant proteins were stored at −80°C until used.
Immunization of BACE1−/− mice and antibody production.
The monoclonal anti-BACE1 antibodies were generated by immunizing BACE1−/− mice with 6xHis-BACE1 N-terminal protein consisting of amino acid residues 46–460. Procedures for mouse immunization and antibody production were as described previously (Binder et al., 1985; Gamblin et al., 2003) with some modifications. Briefly, five BACE1−/− mice (129/Black Swiss background) received a regimen of six immunizations at 3 week intervals, with each immunization including two subcutaneous injections of 150 μg of recombinant protein in 1:1 mixture with Freund's adjuvant (the first injection contained immunogen in Freund's complete adjuvant, whereas all subsequent immunizations used Freund's incomplete adjuvant). The last immunization included an additional intraperitoneal injection of 150 μg of immunogen. Anti-BACE1 antibodies in the serum of each immunized mouse were titered by ELISA, and 4 d after the last immunization, the spleen from the mouse with highest BACE1 antibody titer was isolated and splenocytes were fused with SP2/o myeloma cells to generate hybridomas.
Anti-BACE1-positive hybridoma clones were initially screened by ELISA based on their ability to bind to recombinant BACE1 N terminus (46–460) protein. Hybridoma culture supernatants from positive clones were further assessed by immunoblot analysis of wild-type and BACE1−/− mouse brain homogenates. Clone 3D5, among all tested, exhibited the highest avidity for BACE1 (see Fig. 2B) and hence was subcloned four times and designated BACE-Cat1. The isotype of BACE-Cat1 was determined to be IgG1 by a Mouse Monoclonal Antibody Isotyping kit (Roche, Nutley, NJ). Antibody production was performed by adapting the hybridoma secreting the BACE-Cat1 antibody to serum-free medium, before placing it in a miniPERM Bioreactor (Greiner Bio-One, Monroe, NC) at 37°C with 5% CO2 and 70% relative humidity. The BACE-Cat1 antibody was then harvested and purified to homogeneity by affinity chromatography using protein A-Sepharose 4B (Sigma, St. Louis, MO) before concentration to 1 mg/ml and storage in HEPES buffered saline containing 50% glycerol. Either affinity-purified BACE-Cat1 antibody, or the culture supernatant directly harvested from the stationary-phase hybridoma, was used in the ELISA, immunoblot, or IHC experiments described below.
Figure 2.

Immunoblotting with BACE-Cat1 reveals elevated BACE1 levels in human AD and APP transgenic mouse brains. A, Human autopsy brain tissues (temporal lobe cortex) of clinically diagnosed AD (n = 3) and non-demented controls (ND) (n = 3) were homogenized and analyzed by BACE-Cat1 immunoblot. Lanes with BACE1−/− (KO) brain homogenate and lysate from BACE1-overexpressing HEK293 cells (+) were included as negative and positive controls, respectively. Molecular mass markers are on the left. Immunoblot analysis for β-actin served as a loading control. B, Brain homogenates from representative 2- to 12-month-old 5XFAD (n = 3–5 per age) and age-matched normal control (Non-Tg) (n = 3–5 per age) mice were subjected to immunoblot analysis using BACE-Cat1 antibody. The BACE1 band migrates at ∼70 kDa, and the occasional band at ∼60 kDa represents endogenous mouse IgG (ms IgG) (detected by goat anti-mouse IgG secondary antibody) present in blood of brains from mice that were not transcardially perfused with buffer. Note that the lane with the homogenate from perfused KO brain does not have the background band. The intensities of the BACE1 bands in 5XFAD lanes are significantly greater than those of age-matched Non-Tg BACE1 bands. C, Quantification of 5XFAD and Non-Tg BACE1 signals detected by BACE-Cat1 immunoblot analysis is represented as percentage of 2 month Non-Tg control (100%). Note that BACE1 levels in 5XFAD brains reach ∼190% of control at 9 months and level off and that Non-Tg BACE1 levels appear to decrease slightly with age. Error bars indicate SEM. ***p < 0.001.
ELISA.
ELISA screening of anti-BACE1-positive hybridoma clones was performed as reported previously (Binder et al., 1985). Briefly, purified BACE1 N terminus (46–460) protein was coated on 96-well microtiter plates at 100 ng/well. After blocking for 1 h, hybridoma supernatants were added to the plates and allowed to incubate for 2 h at room temperature (RT). The plates were then washed and incubated in the presence of horseradish peroxidase (HRP)-conjugated goat anti-mouse secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) for 1 h at RT. After addition of the substrate tetramethyl benzidine (Sigma), the reaction was stopped with an equal volume of 0.65 m H2SO4, and the plates were read immediately on an ELISA reader at 450 nm. Clones with significantly positive absorbance readings (>2.0) were then chosen for additional screening by immunoblot analysis.
To verify specificity of the affinity-purified BACE-Cat1 antibody by ELISA, BACE1 N terminus (46–460) protein was coated on a 96-well plate as above, followed by addition of serial dilutions of BACE-Cat1 or the commercial BACE1 antibody PA1-757 (Affinity Bioreagents, Golden, CO) (supplemental Table 1, available at www.jneurosci.org as supplemental material), which served as the negative control [PA1-757 recognizes BACE1 amino acids 485–501, which are not present in BACE1 N terminus (46–460) immunogen]. With the exception that goat anti-rabbit secondary antibody was used to detect PA1-757, incubation of secondary antibodies, substrate reaction, termination, and absorbance readings were performed as above.
Immunoblotting.
Protein concentration of brain tissue homogenates was measured using a BCA protein assay kit from Pierce (Rockford, IL). Equal amounts (15 μg of protein) of homogenates from wild-type, BACE1−/−, and/or normal and AD human brain tissues were heated at 70°C for 10 min in SDS sample loading buffer before being separated on 4–12% NuPAGE Bis-Tris gels in MOPS [3-(N-morpholino)propanesulfonic acid] buffer (Invitrogen) and transferred to Millipore (Bedford, MA) Immobilon-P polyvinylidene difluoride membranes. The blots were blocked at RT in 5% nonfat dry milk made in Tris-buffered saline with 0.1% Tween 20 (TBST) (Sigma; T9039; modified form), pH 8.0, and then incubated with various commercial primary antibodies against human BACE1 (supplemental Table 1, available at www.jneurosci.org as supplemental material) diluted in 5% nonfat milk in TBST at the concentrations recommended by the manufacturers. For initial screening of mouse monoclonal anti-BACE1 antibodies, blots were incubated in hybridoma culture supernatant from each clone at 1:50 or 1:100 dilution. Immunoblotting of brain homogenates of Tg6799, Tg2576, AD, and controls was performed using affinity-purified BACE-Cat1 antibody (1:1000 or 1:2000) at 4°C overnight. Blots were then washed with TBST and incubated in HRP-conjugated goat anti-rabbit or goat anti-mouse secondary antibodies (1:10,000; 1 h at RT). Finally, blots were developed using enhanced chemiluminescence (ECL) Plus detection reagents (Amersham Biosciences, Piscataway, NJ) and immunosignals captured digitally using the Kodak (Rochester, NY) Image Station 440C. Some blots were processed in stripping buffer containing 62.5 mm Tris-HCl, pH 6.7, 2% SDS, and 115 mm β-mercaptoethanol at 55°C for 30 min, and then reprobed with anti-β-actin antibody (Sigma), which was used as loading control, followed by incubation in HRP-conjugated goat anti-mouse (1:10,000) secondary antibody.
RNA isolation and real-time PCR.
Hemibrains from 9-month-old 5XFAD mice were homogenized in 2.5 ml of QIAzol Lysis Reagent and RNA was isolated from 0.5 ml of lysate using the RNeasy Lipid Mini kit (Qiagen). After RNA concentration determination, 1 μg of total RNA from each sample was used for first-strand cDNA synthesis using SuperScript III (Invitrogen). cDNA was amplified by quantitative real-time PCR with Assays-on-Demand premixed Taqman primer/probe set for mouse BACE1 mRNA (catalog #Mm00478664_m1; Applied Biosystems, Foster City, CA) and analyzed using an Applied Biosystems 7900HT sequence analyzer with the relative quantification method normalized against 18S rRNA (catalog #4333760F; Applied Biosystems). Samples were run in triplicate, sample averages were determined, and then means and SEMs were calculated based on the sample averages. All procedures were performed according to the recommendations of the manufacturer.
Immunohistochemistry.
Wild-type and BACE1−/− mice were deeply anesthetized with intraperitoneal injection of pentobarbital (100 mg/kg), and then transcardially perfused with HEPES buffer containing protease inhibitors [20 μg/ml phenylmethylsulphonyl fluoride, 0.5 μg/ml leupeptin, 20 μm sodium orthovanadate, and 100 μm dithiothreitol (DTT)] before removal of brains and postfixation in either 4% paraformaldehyde (PFA) in PBS or 10% Neutral Buffered Formalin (VWR Scientific, West Chester, PA). Brains from 5XFAD and Tg2576 mice together with their wild-type controls were drop-fixed in 4% PFA for 24 h without perfusion. All mouse brain tissues were then cryopreserved in 20% (w/v) sucrose for >24 h and 30 μm frozen coronal sections were cut on a sliding microtome and collected in 0.1 m PBS with 0.01% sodium azide and processed for IHC analyses as described below.
Evaluation of commercial BACE1 antibody immunostaining of BACE1+/+ and −/− brain sections by IHC was conducted using a series of dilutions for each primary antibody (supplemental Table 1, available at www.jneurosci.org as supplemental material), following standard protocols. Omission of primary antibody during the staining procedure was also performed as a control. Antigen retrieval as described previously (Jiao et al., 1999) was performed for immunostaining with BACE-Cat1 antibody. Briefly, free-floating sections were incubated in TBS containing 0.1% SDS and 5 mm DTT for 30 min before heating in 0.1 m sodium citrate, pH 9.0, at 80°C in a water bath for 3 h. The sections were further permeabilized with 0.25% Triton X-100 in TBS. Sections were blocked in 5% horse serum, and then incubated in 1% nonfat dry milk in Triton X-100/TBS before being placed overnight in the BACE-Cat1 primary antibody diluted at 1:100 (hybridoma culture supernatant) or 1:2500 (affinity-purified antibody) in Triton X-100/TBS containing 1% nonfat milk at 4°C. Sections were incubated in biotinylated goat anti-mouse secondary antibody (Jackson ImmunoResearch Laboratories) at 1:2000 for 2 h followed by incubation in the avidin–biotin complex (Vectastain ABC kit; Vector Laboratories, Burlingame, CA) for 1 h, and finally developed in the chromogen diaminobenzidine containing H2O2. Adjacent brain sections from 5XFAD, Tg2576, and AD were probed with the mouse monoclonal anti-human Aβ antibody 4G8 (1:5000; Chemicon International, Temecula, CA) for the detection of amyloid plaques. Sections were counterstained with hematoxylin and micrographed with a Nikon (Tokyo, Japan) Eclipse E800 microscope and Spot advanced digital camera (Diagnostic Instruments, Sterling Heights, MI).
Double-label immunofluorescence and confocal microscopy.
5XFAD and AD brain sections were prepared as described above. After antigen retrieval, floating sections were blocked and incubated overnight at 4°C with shaking with the BACE-Cat1 antibody (1:250 for 5XFAD; 1:100 for AD) together with each of the other primary antibodies listed in supplemental Table 2 (available at www.jneurosci.org as supplemental material). After washes, sections were then incubated in the appropriate secondary antibodies conjugated with Alexa Fluor 488 or 594 corresponding to the respective primary antibody (also listed in supplemental Table 2, available at www.jneurosci.org as supplemental material). Sections were imaged with a Zeiss (Oberkochen, Germany) LSM 510 META confocal laser-scanning microscope with a 40×, 1.4 numerical aperture oil immersion objective.
Statistical analysis.
Densitometric analyses of BACE1 immunoblots were performed using Kodak 1D 3.6 image analysis software. Statistical differences for 5XFAD, Tg2576, and AD versus their respective controls were determined using two-tailed t test. Data are presented as the mean ± SEM, and p < 0.05 was considered significant.
Results
BACE1 levels are elevated in APP transgenic mouse and human AD brains
To investigate the BACE1 elevation in AD, we required a monospecific anti-BACE1 antibody appropriate for immunohistochemistry. However, all commercial antibodies that we tested labeled non-BACE1 bands in immunoblot analysis and produced nonspecific backgrounds on immunostained BACE1−/− brain sections (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The failure of these antibodies to bind only BACE1 suggested that the enzyme was not particularly immunogenic. To generate monospecific monoclonal antibodies against BACE1, the catalytic domain of human BACE1 (residues 46–460) (Fig. 1A) was used to immunize BACE1−/− mice, which are immunologically naive with regard to BACE1 and therefore mount a robust immune response against BACE1 antigen. Immunoblot screening of hybridoma culture media identified two clones (3D5 and 2D5) that produce antibodies labeling a strong ∼70 kDa BACE1 band in homogenates of wild-type mouse and human AD brain and in extracts of BACE1-overexpressing cells (Fig. 1B). Clone 3D5 was monospecific for BACE1, so it was chosen for antibody production and renamed BACE-Cat1. To characterize the binding properties of BACE-Cat1, we measured its affinity for BACE1(46–460) immunogen by ELISA (Fig. 1C). The dilution curve of BACE-Cat1 revealed that ∼50% of immunogen was bound by it at an antibody dilution of ∼4 × 104 (∼25 ng/ml), demonstrating high affinity for BACE1.
Figure 1.

Characterization of anti-BACE1 monoclonal antibodies generated in BACE1−/− mice. A, Schematic diagram of BACE1. BACE1 is a type I transmembrane glycoprotein with a signal sequence (SP) (residues 1–21) and a short prodomain (PD) (residues 22–45) that are cleaved during maturation, a long catalytic domain (residues 46–460), a single transmembrane domain (TM), and a short cytoplasmic tail (CD) (residues 478–501). The entire N-terminal catalytic domain (46–460) was expressed in E. coli and used as the antigen to immunize BACE1−/− mice and generate anti-BACE1 monoclonal antibodies. B, Screening and analysis of initial anti-BACE1 monoclonal antibodies in hybridoma culture supernatants by immunoblotting. Results for four representative hybridoma clones are shown. Protein extracts (15 μg/lane) of brain tissues from C57BL/6J wild-type (WT) mouse, BACE1−/− (KO) mouse, and non-demented aged human (Human) were immunoblotted and incubated with hybridoma culture media from clones 3D5, 2D5, 5C4, and 2D2. Compared with the others, supernatant staining by clone 3D5, subsequently named BACE-Cat1, displays the strongest BACE1 signal (arrow at left), has no nonspecific background bands in the BACE1 WT lane, and has no signal in the KO lane. Hybridoma supernatant dilutions are indicated in parentheses at the bottom of each blot. C, ELISA characterization of the BACE-Cat1 antibody. A dilution series of either affinity-purified BACE-Cat1 (1 mg/ml) or PA1-757 (1 mg/ml; ABR) antibodies were added to wells of a 96-well plate that were precoated with recombinant BACE1 catalytic domain (residues 46–460). Wells were then incubated with either HRP-conjugated goat anti-mouse or anti-rabbit secondary antibodies, developed, and optical densities (O. D.) read at 450 nm. Note the dose-dependent reduction in binding of BACE-Cat1 to BACE1 with increasing antibody dilutions, whereas PA1-757 (which recognizes the BACE1 C terminus) shows no detectable reading at any dilution and served as a negative control in the ELISA. D, The BACE-Cat1 antibody identifies authentic BACE1 immunoreactivity in mouse brain by immunohistochemistry. Coronal sections of C57BL/6J wild-type (WT) and BACE1−/− (KO; C57BL/6J background) brains were immunostained under identical conditions with BACE-Cat1 hybridoma culture supernatant and counterstained with hematoxylin after antigen retrieval (see Materials and Methods). In the WT brain section, diffuse BACE1 immunoreactivity is present throughout the entire section, but is higher in certain brain regions such as the mossy fiber pathway of the hippocampus (hatched arrow), the endopiriform nucleus/claustrum (open arrow), and globus pallidus/amygdala (closed arrow). In contrast, BACE-Cat1 signal does not appear in the KO brain section. Scale bar, 400 μm.
To determine whether the BACE-Cat1 antibody accurately labeled BACE1 by immunohistochemistry, we stained wild-type and BACE1−/− mouse brain sections. The strongest BACE-Cat1 signal was observed in the terminal fields of the hippocampus mossy fiber output pathway (Fig. 1D, hatched arrow in WT), the endopiriform nucleus/claustrum (Fig. 1D, open arrow in WT), and globus pallidus/amygdala (Fig. 1D, closed arrow in WT). In support of these results, BACE1 was previously shown to localize to mossy fiber terminals (Laird et al., 2005). No brain regions were stained in BACE1−/− sections incubated under identical conditions with BACE-Cat1 (Fig. 1D, KO). We also observed a less intense BACE-Cat1 signal throughout gray and white matter in wild-type brain that was absent in BACE1−/− brain. These results demonstrate that antibody BACE-Cat1 recognizes no other protein than BACE1 in the brain and therefore faithfully identifies BACE1.
Next, we wanted to determine whether we could repeat the work of others demonstrating that BACE1 levels were elevated in AD brain. To accomplish this, we obtained postmortem brain tissue (temporal lobe cortex) from three AD and three ND individuals and performed immunoblot analysis of brain homogenates using antibody BACE-Cat1. As with initial BACE-Cat1 immunoblots of brain homogenates, a single ∼70 kDa BACE1 band was observed (Fig. 2A). Quantification of the band intensities indicated that BACE1 levels in AD brain were elevated to 163.8% (±14.7%) of those found in ND brain (n = 3 per group; t test, p < 0.05) (data not shown), similar in magnitude to the BACE1 increase reported by other groups (Fukumoto et al., 2002; Holsinger et al., 2002; Tyler et al., 2002; Li et al., 2004). These results confirmed that BACE1 is elevated in AD brain and demonstrated that BACE-Cat1 is ideal for specific, unambiguous detection and measurement of BACE1.
Changes in numerous proteins have been reported in postmortem AD brain (for review, see Fountoulakis and Kossida, 2006), and it has been challenging to determine whether a particular protein alteration plays an active role in the disease process, or whether it is a passive bystander caused by end-stage neuronal degeneration and death. One way to address this question is to use an AD animal model that recapitulates critical aspects of disease over time. Therefore, we chose to analyze two APP-overexpressing transgenic (Tg) mice that develop AD-like amyloid plaque pathology with age: (1) the well characterized Tg2576 mouse (Hsiao et al., 1996) that expresses APP with the Swedish mutation (Mullan et al., 1992), and (2) a Tg mouse that we recently developed called “5XFAD” (Oakley et al., 2006) that expresses five familial AD mutations [APP K670N/M671L (Swedish) plus I716V (Florida) plus V717I (London) and PS1 M146L plus L286V)]. Tg2576 develops plaques starting at ∼9 months of age. In contrast, the additive effect of five familial AD mutations in 5XFAD mice elevates Aβ42 production and initiates plaque formation at ∼2 months. Importantly, large pyramidal neurons in cortical layer 5 and the subiculum are lost in 5XFAD, whereas cell death is minimal in Tg2576. Therefore, analysis of these two APP Tg lines allowed us to compare the effects of amyloid pathology on BACE1 levels in both the presence and absence of neuron loss.
Immunoblot analysis with BACE-Cat1 on whole brain homogenates from 5XFAD and nontransgenic (non-Tg) mice of ages ranging from 2 to 12 months showed a gradual increase in cerebral BACE1 levels (Fig. 2B) that paralleled the age-dependent rise of Aβ42 level and amyloid plaque load in this Tg line (Oakley et al., 2006). Quantification of BACE1 band intensities demonstrated that BACE1 levels in 5XFAD brain were increased up to ∼190% of those found in non-Tg brain by 9 months of age, and then leveled off (Fig. 2C) (n = 3–5 per genotype and age; t test, p < 0.001). BACE1 elevation was also observed in brains of 4- to 6-month-old 5XFAD mice by immunoblot using a different anti-BACE1 antibody directed against the C terminus of BACE1 (Ohno et al., 2006). Interestingly, TaqMan real-time PCR analysis revealed that BACE1 mRNA levels in 5XFAD brains (108.3 ± 6.6%) (data not shown) were not increased relative to age-mated non-Tg (100 ± 6.4%), indicating that the BACE1 elevation was not the result of increased BACE1 gene transcription or mRNA stability. Tg2576 also exhibited a small (∼15%) but significant increase in BACE1 level at 12 and 18 months of age (data not shown), reflecting the lower amyloid load and slower plaque onset in Tg2576. The BACE1 elevation occurred either in the absence of (i.e., Tg2576) or before (i.e., 5XFAD) major cell loss, indicating that the BACE1 increase was not the end product of late-stage degeneration and neuron death. Together, these data demonstrate that elevated BACE1 levels (1) are found in both AD and APP transgenic brain, (2) follow amyloid burden but not neuron loss, and (3) are likely the result of a posttranscriptional mechanism.
BACE1 levels are elevated around amyloid plaques in APP transgenic and AD brain
The increase of BACE1 level with amyloid load in two APP Tg mouse models demonstrated that the BACE1 elevation was triggered by Aβ or amyloid plaques. To initially investigate the relationship of the BACE1 increase to plaques, we stained alternate brain sections from APP Tg (5XFAD, 9 months; Tg2576, 18 months) and non-Tg mice with BACE-Cat1. Interestingly, in addition to the normal BACE1 staining pattern found in the mossy fiber pathway of the hippocampus (Fig. 3A, Non-Tg), we observed aberrant BACE1 localization having a plaque-like distribution in the brains of 5XFAD (Fig. 3, compare B, D) and Tg2576 (Fig. 3, compare C, E) mice. BACE1-positive accumulations were present in cortex, hippocampus (Fig. 3), and other brain regions with amyloid plaques in 5XFAD and Tg2576 mice, but were completely absent in Non-Tg brain (Fig. 3A,F). Of the commercial anti-BACE1 antibodies tested, only one (B0681; Sigma) labeled ring-like BACE1 accumulations similar to BACE-Cat1 in 5XFAD mouse brain, although it had a much higher background than BACE-Cat1 (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Adjacent APP Tg brain sections stained with the anti-total Aβ antibody 4G8 displayed patterns of amyloid plaques that appeared similar to those identified by BACE-Cat1 staining. Because 5XFAD mice generate higher levels of Aβ42 and have greater amyloid burdens than Tg2576 mice (Oakley et al., 2006), 4G8 detected numerous amyloid plaques in 5XFAD (Fig. 3D) but few by comparison in Tg2576 (Fig. 3E). The numbers of BACE-Cat1-positive accumulations and 4G8-positive plaques were proportional within each APP Tg line. When comparing the morphologies of BACE-Cat1- and 4G8-positive deposits at higher magnification, we observed that BACE1 immunoreactivity frequently formed ring-like structures with clear cores (Fig. 3G,H), whereas the Aβ-positive plaques typically had solid cores (Fig. 3I,J).
Figure 3.

BACE-Cat1 reveals plaque-like staining in APP transgenic brains by immunohistochemistry. A–J, Coronal brain sections from 5XFAD and Tg2576 mice were immunostained with BACE-Cat1 antibody (A–C, F–H) or anti-total Aβ-amyloid antibody 4G8 (D, E, I, J) and counterstained with hematoxylin. Only images of the hippocampus are shown. A, F, Nontransgenic (Non-Tg) brain section from 9-month-old mouse stained with BACE-Cat1 shows the normal BACE1 immunoreactivity pattern with strongest labeling in the hippocampal mossy fiber pathway. B, G, 5XFAD brain section from 9-month-old mouse stained with BACE-Cat1 displays plaque-like BACE1 immunoreactivity. Note that BACE1-positive deposits typically have unstained cores. D, I, Adjacent 5XFAD brain section stained with 4G8 antibody reveals amyloid plaques that have a similar spatial distribution as BACE-Cat1-positive deposits. C, H, Tg2576 brain section from 18-month-old mouse stained with BACE-Cat1 also shows a plaque-like pattern of BACE1-positive deposits, although the number of deposits is less than in 5XFAD mice. E, J, Adjacent Tg2576 brain section immunostained with 4G8 antibody shows a similar distribution of amyloid plaques as that of BACE1-positive deposits. F–J, Higher magnification images of boxed areas in A–E. BACE1 immunoreactivity typically forms a ring-like structure with a clear center in both 5XFAD and Tg2576 transgenic mice (G, H), whereas amyloid plaques have a more uniformly stained core (I, J). Scale bars: A–E, 200 μm; F–J, 20 μm.
To determine the pattern of BACE1 localization in human AD brain, we performed BACE-Cat1 and 4G8 immunohistochemistry on sections of temporal lobe cortex from the three AD patients and ND control analyzed previously by immunoblot. Similar to the labeling in APP Tg brain sections, the BACE-Cat1 antibody stained BACE1-positive concentrations in AD brain that typically had ring-like profiles with clear cores (Fig. 4B–D,I–K). BACE1 accumulations were not apparent in ND brain (Fig. 4A,H). We observed that the number of BACE-Cat1-positive deposits was typically less than that of 4G8-positive plaques in AD brain (Fig. 4E–G). The reason for the smaller number of BACE1 accumulations relative to plaques is not completely clear, although we speculate that the BACE1 elevation occurs in only a subpopulation of plaques.
Figure 4.
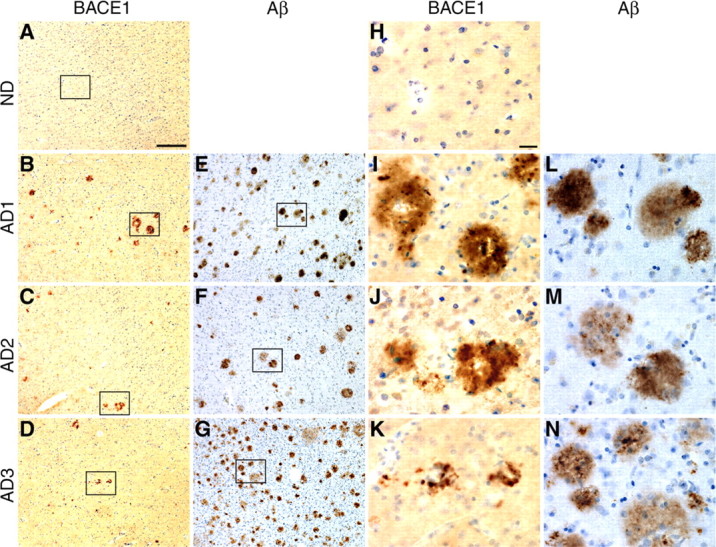
BACE-Cat1 reveals plaque-like staining in human AD brains by immunohistochemistry. A–N, Brain sections (inferior temporal gyrus or temporal lobe cortex) from normal (ND) and AD age-matched humans were immunostained with BACE-Cat1 (A–D, H–K) or anti-total Aβ antibody 4G8 (E–G, L–N) and counterstained with hematoxylin. A, H, Diffuse low-level BACE1 immunoreactivity with BACE-Cat1 is observed in ND cortex, but no deposits are apparent. B–D, Brain sections from three AD subjects all exhibit plaque-like BACE1-immunopositive deposits with BACE-Cat1, although numbers of BACE1 deposits in different patients vary considerably. I–K, As in APP transgenic brains, BACE1 immunoreactivity in AD brains is typically ring-like in appearance surrounding a relatively unstained core. E–G, AD brain sections stained with anti-total Aβ antibody 4G8 show numerous amyloid plaques in all three specimens. L–N, In contrast to BACE1-positive ring-like deposits, most amyloid plaques in AD brain exhibit stained cores with 4G8. Note that the number of BACE1-positive deposits is considerably less than that of amyloid plaques in the section of the same brain region and that many 4G8-positve deposits appear to be diffuse plaques. Higher-magnification images of boxed areas in A–G are shown in H–N. Scale bars: A–G, 60 μm; H–N, 20 μm.
The plaque-like characteristics of the BACE1 accumulations suggested that they might be physically associated with amyloid deposits. To investigate this, we performed double immunofluorescence staining and confocal microscopy on APP Tg and AD brain sections incubated with BACE-Cat1 in combination with anti-Aβ antibodies. First, we did double staining of 5XFAD (9 month) and AD (AD1) brain sections with BACE-Cat1 plus anti-Aβ42 C-terminal-specific antibody. Interestingly, we typically observed a ring of BACE1 immunoreactivity surrounding an Aβ42-positive core in both human AD and 5XFAD brains (Fig. 5A). BACE1 immunostaining around deposits had similar morphologies in both human and mouse, although the BACE1 labeling in 5XFAD was usually stronger and had a more globular appearance than in AD. The majority of Aβ42-positive plaques in 5XFAD also stained for BACE1, but a lower proportion of Aβ42 plaques displayed BACE1 immunoreactivity in AD.
Figure 5.
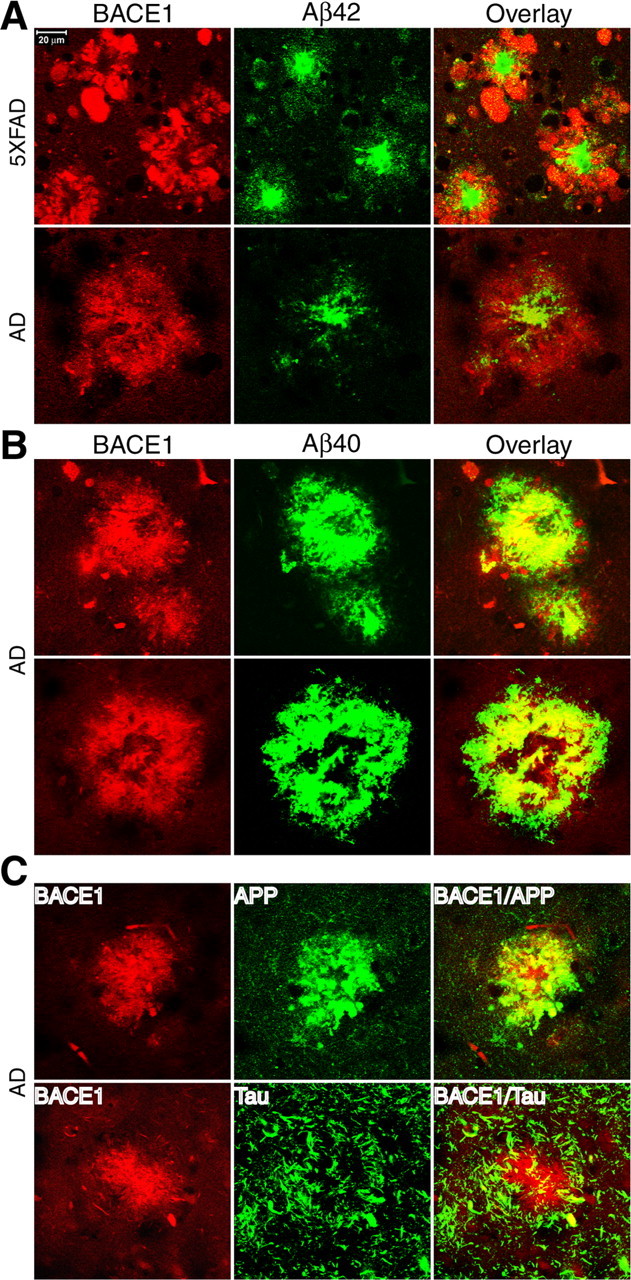
Immunofluorescence double labeling of BACE1 together with Aβ, APP, or tau in 5XFAD and AD brains. Brain sections from 5XFAD or AD brains were costained with BACE-Cat1 and either antibodies against Aβ42 (A), Aβ40 (B), APP (C, top), or tau (C, bottom). A, 5XFAD (top) and AD (bottom) brain sections were double stained with BACE-Cat1 (red) and anti-Aβ42 C-terminal specific antibody (green). Note that BACE1 immunoreactivity surrounds the Aβ42 core of the plaques in both APP Tg and AD brains. B, AD brain section was double stained with BACE-Cat1 (red) and anti-Aβ40 C-terminal-specific antibody (green). Two representative amyloid plaques are shown. Note the high degree of colocalization between BACE1 and Aβ40 immunoreactivities. C, Top, AD brain section was double stained with BACE-Cat1 (red) and anti-APP C-terminal antibody (green). There is extensive colocalization of BACE1 and APP immunoreactivities within the deposit. C, Bottom, AD brain section was double stained with BACE-Cat1 (red) and anti-tau R1 antibody (green). Filamentous tau-positive structures tend to be concentrated around and partially overlap with the BACE1 deposit. Scale bar: A, Top left, 20 μm.
Next, we double labeled AD brain sections with BACE-Cat1 and anti-Aβ40 C-terminal-specific antibody (Fig. 5B). In this case, BACE1 and Aβ40 immunoreactivities mostly overlapped, with both signals often assuming ring-like profiles, implying that an Aβ42 core was surrounded by a halo of BACE1 and Aβ40. Together, our results with double immunofluorescence labeling for BACE1 and either Aβ42 or Aβ40 demonstrate that the BACE1 elevation in APP Tg and AD brain is physically associated with amyloid plaques.
The BACE1 elevation occurs predominantly within neurons surrounding plaques
We were interested in determining the cell type in which BACE1 becomes elevated in the brain, so we incubated 5XFAD and AD brain sections with BACE-Cat1 plus antibodies against neuronal and glial markers and performed double immunofluorescence microscopy. Labeling of AD brain sections with BACE-Cat1 and an antibody that recognizes the C terminus of APP (amino acids 676–695) (Cole et al., 2005) revealed robust colocalization of BACE1 and APP (Fig. 5C, top row). Thus, accumulation of both APP substrate and BACE1 enzyme occurred in plaques, a condition that could contribute to amyloid deposition. Double immunostaining of AD brain sections for BACE1 and tau (which comprises neurofibrillary tangles) showed tau-positive filamentous structures (presumably neurites or neuropil threads) encircling BACE1-positive deposits in AD brain (Fig. 5C, bottom row). Although some tau immunoreactivity penetrated into BACE-Cat1-labeled deposits and colocalized with BACE1, the amount of overlap was relatively low.
Although APP and tau are highly expressed in neurons, they are found in other cell types as well. Therefore, we performed double immunofluorescence labeling of 5XFAD and AD brain sections with BACE-Cat1 plus the neuron markers neuron-specific enolase (NSE), synaptophysin, microtubule-associated protein 2 (MAP2), or β-III tubulin. BACE1-positive deposits mostly costained for NSE (Fig. 6A) and the presynaptic marker synaptophysin (Fig. 6B) in 5XFAD brain, although colocalization appeared less in AD because of partial loss of NSE and synaptophysin immunoreactivity in AD plaques. MAP2, a somatodendritic marker, did not exhibit significant colocalization with BACE1 around 5XFAD plaques (Fig. 6C). BACE1 colocalized with β-III tubulin to a small extent in 5XFAD and even less in AD (Fig. 6D), but this was mainly the result of diminished β-III tubulin staining in the plaque. Finally, very little BACE1 colocalization was observed with glial fibrillary acidic protein (GFAP), an astrocyte marker, in either 5XFAD or AD brain (Fig. 6E), indicating that astrocytes are not the major source of the BACE1 elevation in AD or APP Tg brain. Together, our double immunofluorescence staining results indicate that neurons are the primary cell type that harbors the BACE1 increase in both AD and APP Tg brain, and that the BACE1 elevation appears predominantly in presynaptic structures.
Figure 6.

Immunofluorescence double labeling of BACE1 with neuronal and astrocytic markers in 5XFAD and AD brains. Brain sections from 5XFAD or AD brains were costained with BACE-Cat1 and either antibodies against NSE (A), synaptophysin (presynaptic marker) (B), MAP2 (postsynaptic marker) (C), β-III tubulin (neuronal marker) (D), or GFAP (astrocyte marker) (E). A, 5XFAD (top) and AD (bottom) brain sections were double stained with BACE-Cat1 (red) and anti-NSE antibody (green). Note that, within BACE1-positive deposits, BACE1 immunoreactivity colocalizes extensively (5XFAD) and moderately (AD) with NSE staining. B, 5XFAD (top) and AD (bottom) brain sections were double stained with BACE-Cat1 (red) and anti-synaptophysin antibody (green). Within deposits, BACE1 immunoreactivity colocalizes moderately well with synaptophysin staining in both 5XFAD and AD brains. C, 5XFAD brain sections were double stained with BACE-Cat1 (red) and anti-MAP2 antibody (green). BACE1 and MAP2 immunoreactivities do not colocalize to any significant extent. D, 5XFAD (top) and AD (bottom) brain sections were double stained with BACE-Cat1 (green) and anti-β-III tubulin antibody (red). BACE1 colocalization with β-III tubulin is present but relatively low within BACE1-positive deposits in 5XFAD and AD brains. E, 5XFAD (top) and AD (bottom) brain sections were double stained with BACE-Cat1 (red) and anti-GFAP antibody (green). Note that the overlap between BACE1 and GFAP immunoreactivities in 5XFAD and AD deposits is low. Scale bar: A, Top left, 20 μm.
Discussion
BACE1 localization in brain
Several groups have reported that BACE1 levels and activity are increased in AD brain (Fukumoto et al., 2002; Holsinger et al., 2002; Tyler et al., 2002; Yang et al., 2003; Li et al., 2004; Harada et al., 2006). The cause of this increase and whether it plays a role in disease has been difficult to determine because postmortem brain tissue does not allow investigation of the evolution of pathology. Fortunately, transgenic mouse models are available that recapitulate aspects of AD pathology, thus allowing analysis of disease initiation and progression. In this study, we show with a new highly specific anti-BACE1 monoclonal antibody, BACE-Cat1, that BACE1 levels become elevated in the brains of two APP transgenic mouse lines, 5XFAD and Tg2576, and human AD. Overall, the biochemical and histological characteristics of the BACE1 elevation in the two APP transgenics matched well with those in AD. To our knowledge, this is the first demonstration that BACE1 levels begin to increase early and in parallel with amyloid pathology in AD mouse models. Importantly, the BACE1 elevation was directly proportional to the amyloid burden of a given APP transgenic line. Because the BACE1 increase occurred either in the absence of (Tg2576) or before (5XFAD) major neuron loss in APP transgenics, the rise in BACE1 levels appeared to be the result of amyloid pathology rather than of cell death. Immunostaining revealed that BACE1 accumulated in ring-like deposits that mirrored the distribution of amyloid plaques. BACE1 was concentrated in neurons, rather than astrocytes, surrounding Aβ42-containing plaque cores in both APP transgenic and AD brains. The BACE1-positive deposits colocalized mainly with synaptophysin, but not MAP2 (a somatodendritic marker), suggesting presynaptic BACE1 localization, although a more definitive EM immunolocalization study is required to demonstrate this conclusively. In addition, BACE1 elevation occurred in AD deposits containing APP and tau. Our results are supported by those of Sheng et al. (2003), showing BACE1 immunoreactivity around Aβ deposits in APP/PS1 transgenic mice. Together, our data suggest that the BACE1 elevation occurs in presynaptic neuronal structures around neuritic plaques and that Aβ42 may cause the increase.
Mouse and human BACE1 are highly homologous (Luo et al., 2001), and therefore it was not surprising that BACE-Cat1 recognized a single ∼70 kDa band representing the mature BACE1 protein (Vassar et al., 1999; Haniu et al., 2000) in both mouse and human brain homogenates. Immunohistochemistry using BACE-Cat1 on mouse brain sections revealed robust anti-BACE1 staining in terminal fields and lighter labeling in neuronal cell bodies throughout the brain. Importantly, BACE1−/− brain sections were negative after BACE-Cat1 staining. The mossy fiber staining pattern for BACE1 was similar to that reported by Laird et al. (2005), who used a rabbit antibody against amino acids 46–163 of BACE1 (Cai et al., 2001). Other groups have also reported cerebral BACE1 immunolocalization in normal and AD humans, as well as in wild-type and APP transgenic mice (Hussain et al., 1999; Rossner et al., 2001; Fukumoto et al., 2002; Sun et al., 2002; Hartlage-Rubsamen et al., 2003; Heneka et al., 2005; Leuba et al., 2005; Singer et al., 2005; Harada et al., 2006). However, a consensus BACE1 immunostaining pattern has not emerged. The reports have variously noted BACE1 immunostaining in neuron cell bodies, neurites, tangle-bearing neurons, and astrocytes around amyloid plaques. Some results are consistent, but most are not. We attribute these disparate findings to nonspecific labeling of the different BACE1 antibodies used in these studies. Because immunostaining of BACE1−/− brain sections has not been routinely used as a negative control, validation of BACE1 labeling patterns has been lacking. Our current study was rigorously controlled, using BACE1−/− brain sections and homogenates and BACE1–293 cell lysates for negative and positive controls, respectively. Moreover, BACE-Cat1 labeled only a single ∼70 kDa band of the correct size for mature BACE1 in human or mouse brain and no bands were detected in BACE1−/− brain. Therefore, we conclude that the BACE-Cat1 immunostaining pattern reflects genuine BACE1 localization in the brain.
In wild-type brain, BACE1 immunolocalization was highest within terminal fields, including those of the hippocampal mossy fiber pathway (giant boutons), the endopiriform nucleus/claustrum, and the globus pallidus/amygdala. We recently confirmed presynaptic BACE1 localization within the giant boutons of the mossy fiber pathway (J. Zhao and R. Vassar, unpublished observations). Our work, and that of others (Lazarov et al., 2002; Sheng et al., 2003), supports the notion that the terminal is a major site of amyloidogenic APP processing. Aβ may act as a negative regulator of excitatory transmission at the synapse (Kamenetz et al., 2003), adding functional significance to the localization of BACE1 in terminals. Other BACE1 substrates have been reported, such as APLPs (APP-like proteins) (Li and Sudhof, 2004), Na+ channel β-subunit (Wong et al., 2005), LDL (low-density lipoprotein)-receptor related protein (von Arnim et al., 2005), sialyltransferase (Kitazume et al., 2001), and P-selectin glycoprotein ligand-1 (Lichtenthaler et al., 2003), and some may have terminal localization, suggesting that BACE1 cleavage of certain substrates may be required for normal synaptic function.
Role of BACE1 elevation in AD
By the time of death, the AD brain has been devastated by neurodegeneration and cell loss. Postmortem AD brain is severely reduced in mass, and the number of neurons in the hippocampus and neocortex is decreased by ∼30–60% (for review, see Morrison and Hof, 1997). Many biochemical parameters deviate from normal in AD, and a large number of proteins undergo a plethora of changes, including increased or decreased expression, fragmentation, accumulation, phosphorylation, oxidation, etc. Recent proteomic analyses estimate that ∼100 different proteins have deranged levels or abnormal modifications in AD (for review, see Fountoulakis and Kossida, 2006). Some of these protein alterations may play a role in AD pathogenesis; however, it is exceedingly difficult to determine whether a particular change actively participates in pathophysiology, or whether it is a passive end product of advanced cellular degeneration and death. Our observation that BACE1 levels become elevated around amyloid plaques in two APP transgenic mouse lines, one with neuron death and one without, demonstrates that the BACE1 increase is a pre-cell loss event that is triggered by plaque pathology. Although the two mouse models express transgenes with different FAD mutations, promoters, and integration sites, they share the common phenotypes of Aβ overproduction and plaque formation, indicating that these properties must be responsible for the BACE1 elevation. Therefore, it is likely that the BACE1 elevation in human postmortem AD brain is not just one of numerous degenerative changes in terminal brains but may be actively involved in disease progression.
Our data suggest that a positive feedback loop starts in AD in which Aβ42 deposition causes BACE1 (and possibly APP) levels to rise in nearby neurons. Increased Aβ production may then ensue, initiating a vicious cycle of additional amyloid deposition followed by higher BACE1 levels. Although the total BACE1 increase in AD is relatively small (∼1.6-fold), BACE1 concentrations in neurons near plaques appear high and may stimulate local Aβ production. In support of this hypothesis, modestly elevated BACE1 levels cause increased Aβ production in vivo in BACE1 transgenic mice (Bodendorf et al., 2002; Chiocco et al., 2004; Mohajeri et al., 2004; Willem et al., 2004; Chiocco and Lamb, 2005; Lee et al., 2005; Ozmen et al., 2005) and in APP transgenic mice treated with energy inhibitors (Velliquette et al., 2005). Because perforant pathway axons are a major source of Aβ for plaque formation in the outer molecular layer of the hippocampus (Lazarov et al., 2002; Sheng et al., 2002), we speculate that BACE1 and APP accumulation in dystrophic neurites around plaques could drive local Aβ overproduction. However, we emphasize it is currently unclear whether the BACE1 elevation in AD leads to increased Aβ generation and disease progression. An AD feedback loop has long been surmised, and the BACE1 elevation in neurons around plaques suggests a plausible mechanism for it.
The molecular mechanism of BACE1 elevation around plaques is unknown. BACE1 mRNA levels are unchanged in 5XFAD brain, even in mice with extensive amyloid and an approximately twofold rise in BACE1 protein. These results suggest translational or protein stability mechanisms and are supported by recent studies showing control of BACE1 levels through posttranscriptional processes such as translational efficiency (Lammich et al., 2004), lysosomal targeting (Koh et al., 2005), and proteosomal degradation (Qing et al., 2004). Because plaques show BACE1 elevation around an Aβ42 core, it is likely that Aβ42 triggers the BACE1 increase. Aβ42 is highly toxic to neurons, and we favor the notion that Aβ42 neurotoxicity directly induces the BACE1 elevation. However, indirect effects of Aβ are also possible, such as the involvement of inflammatory cells recruited to the plaque. Alternatively, reduced BACE1 degradation as a result of disrupted trafficking in dystrophic neurites could underlie BACE1 accumulation around plaques.
Interestingly, nearly all deposits were BACE1 positive in 5XFAD brains, whereas only a subset of plaques exhibited BACE1 immunoreactivity in AD brains. As mentioned above, we speculate that BACE1-positive deposits represent neuritic plaques, although unequivocal demonstration of this requires additional investigation. Conversely, diffuse plaques lacking a compact Aβ42 core may not trigger BACE1 elevation. The number of BACE1-positive plaques appears to be proportional to Aβ42 ratio: 5XFAD mice generate Aβ42 almost exclusively (Oakley et al., 2006), whereas in AD Aβ42 is made at lower levels. Alternatively, the BACE1 elevation may occur at an early/middle stage of plaque evolution preferentially modeled in APP transgenics. Because AD specimens usually represent end-stage disease, it is possible that many plaques are “burned out” as a result of degeneration, as indicated by relatively reduced immunostaining of neuronal markers around plaques in AD compared with 5XFAD (Fig. 6A–D), and therefore loose BACE1 immunoreactivity. Although we favor the notion that Aβ42 plaque cores trigger BACE1 elevation, the possibility exists that Aβ oligomers (for review, see Walsh et al., 2005; Glabe and Kayed, 2006), rather than amyloid plaques, may be responsible for increased BACE1 levels. Additional investigation will be required to fully understand which form of Aβ causes the BACE1 elevation in AD.
Cerebral BACE1 levels increase after injury or stress (Apelt et al., 2004; Blasko et al., 2004; Chen et al., 2004). Recently, we reported that energy inhibitor treatment causes BACE1 elevation in Tg2576 brain to promote Aβ production (Velliquette et al., 2005). Thus, aging-related stresses in pre-AD brain may cause BACE1 levels and Aβ production to increase. Short-term elevation of BACE1 may be beneficial under conditions of acute injury or stress. However, under chronic conditions of disease, as occurs in AD, elevated BACE1 may exacerbate amyloid pathology and accelerate AD progression.
In conclusion, we showed that amyloid plaques appear to induce a dramatic increase in BACE1 levels in surrounding neurons and that this increase parallels amyloid burden but not neuron loss. Therefore, BACE1 elevation in AD is likely triggered by the amyloid pathway and is probably an early pre-cell death event, suggesting that once amyloid deposition begins, BACE1 levels increase and accelerate Aβ production. BACE1 is a prime therapeutic target for AD, although it has been challenging to develop BACE1 inhibitor drugs and understanding the mechanism of amyloid-induced BACE1 elevation may uncover novel targets more amenable to drug development than BACE1 itself.
Footnotes
This work was supported by National Institutes of Aging Grants P01 AG021184 and R01 AG022560. We thank the following individuals for their generous assistance: Dr. C. J. Han for her technical assistance on subcloning and Nichole LaPointe for her assistance on protein induction in Escherichia coli and protein purification for BACE1 monoclonal antibody production; Drs. Marsel Mesulam and Eileen Bigio of the Northwestern University Cognitive Neurology and Alzheimer's Disease Center (Chicago, IL) and Dr. David Bennett of the Rush University Medical School Alzheimer's Disease Center (Chicago, IL) for human brain tissue; and Drs. Teng-leong Chew and Paul Cheresh for their excellent assistance on confocal microscopy.
References
- Apelt J, Bigl M, Wunderlich P, Schliebs R. Aging-related increase in oxidative stress correlates with developmental pattern of beta-secretase activity and beta-amyloid plaque formation in transgenic Tg2576 mice with Alzheimer-like pathology. Int J Dev Neurosci. 2004;22:475–484. doi: 10.1016/j.ijdevneu.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Binder LI, Frankfurter A, Rebhun LI. The distribution of tau in the mammalian central nervous system. J Cell Biol. 1985;101:1371–1378. doi: 10.1083/jcb.101.4.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasko I, Beer R, Bigl M, Apelt J, Franz G, Rudzki D, Ransmayr G, Kampfl A, Schliebs R. Experimental traumatic brain injury in rats stimulates the expression, production and activity of Alzheimer's disease beta-secretase (BACE-1) J Neural Transm. 2004;111:523–536. doi: 10.1007/s00702-003-0095-6. [DOI] [PubMed] [Google Scholar]
- Bodendorf U, Danner S, Fischer F, Stefani M, Sturchler-Pierrat C, Wiederhold KH, Staufenbiel M, Paganetti P. Expression of human beta-secretase in the mouse brain increases the steady-state level of beta-amyloid. J Neurochem. 2002;80:799–806. doi: 10.1046/j.0022-3042.2002.00770.x. [DOI] [PubMed] [Google Scholar]
- Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, Wong PC. BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat Neurosci. 2001;4:233–234. doi: 10.1038/85064. [DOI] [PubMed] [Google Scholar]
- Chen XH, Siman R, Iwata A, Meaney DF, Trojanowski JQ, Smith DH. Long-term accumulation of amyloid-beta, beta-secretase, presenilin-1, and caspase-3 in damaged axons following brain trauma. Am J Pathol. 2004;165:357–371. doi: 10.1016/s0002-9440(10)63303-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiocco MJ, Lamb BT. Spatial and temporal control of age-related APP processing in genomic-based beta-secretase transgenic mice. Neurobiol Aging. 2005;28:75–84. doi: 10.1016/j.neurobiolaging.2005.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiocco MJ, Kulnane LS, Younkin L, Younkin S, Evin G, Lamb BT. Altered amyloid-beta metabolism and deposition in genomic-based beta-secretase transgenic mice. J Biol Chem. 2004;279:52535–52542. doi: 10.1074/jbc.M409680200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole SL, Grudzien A, Manhart IO, Kelly BL, Oakley H, Vassar R. Statins cause intracellular accumulation of amyloid precursor protein, beta-secretase-cleaved fragments, and amyloid beta-peptide via an isoprenoid-dependent mechanism. J Biol Chem. 2005;280:18755–18770. doi: 10.1074/jbc.M413895200. [DOI] [PubMed] [Google Scholar]
- Fountoulakis M, Kossida S. Proteomics-driven progress in neurodegeneration research. Electrophoresis. 2006;27:1556–1573. doi: 10.1002/elps.200500738. [DOI] [PubMed] [Google Scholar]
- Fukumoto H, Cheung BS, Hyman BT, Irizarry MC. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch Neurol. 2002;59:1381–1389. doi: 10.1001/archneur.59.9.1381. [DOI] [PubMed] [Google Scholar]
- Gamblin TC, Chen F, Zambrano A, Abraha A, Lagalwar S, Guillozet AL, Lu M, Fu Y, Garcia-Sierra F, LaPointe N, Miller R, Berry RW, Binder LI, Cryns VL. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer's disease. Proc Natl Acad Sci USA. 2003;100:10032–10037. doi: 10.1073/pnas.1630428100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glabe CG, Kayed R. Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology. 2006;66:S74–S78. doi: 10.1212/01.wnl.0000192103.24796.42. [DOI] [PubMed] [Google Scholar]
- Haniu M, Denis P, Young Y, Mendiaz EA, Fuller J, Hui JO, Bennett BD, Kahn S, Ross S, Burgess T, Katta V, Rogers G, Vassar R, Citron M. Characterization of Alzheimer's beta-secretase protein BACE. A pepsin family member with unusual properties. J Biol Chem. 2000;275:21099–21106. doi: 10.1074/jbc.M002095200. [DOI] [PubMed] [Google Scholar]
- Harada H, Tamaoka A, Ishii K, Shoji S, Kametaka S, Kametani F, Saito Y, Murayama S. Beta-site APP cleaving enzyme 1 (BACE1) is increased in remaining neurons in Alzheimer's disease brains. Neurosci Res. 2006;54:24–29. doi: 10.1016/j.neures.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hartlage-Rubsamen M, Zeitschel U, Apelt J, Gartner U, Franke H, Stahl T, Gunther A, Schliebs R, Penkowa M, Bigl V, Rossner S. Astrocytic expression of the Alzheimer's disease beta-secretase (BACE1) is stimulus-dependent. Glia. 2003;41:169–179. doi: 10.1002/glia.10178. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Sastre M, Dumitrescu-Ozimek L, Dewachter I, Walter J, Klockgether T, Van Leuven F. Focal glial activation coincides with increased BACE1 activation and precedes amyloid plaque deposition in APP[V717I] transgenic mice. J Neuroinflammation. 2005;2:22. doi: 10.1186/1742-2094-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holsinger RM, McLean CA, Beyreuther K, Masters CL, Evin G. Increased expression of the amyloid precursor beta-secretase in Alzheimer's disease. Ann Neurol. 2002;51:783–786. doi: 10.1002/ana.10208. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Hussain I, Powell D, Howlett DR, Tew DG, Meek TD, Chapman C, Gloger IS, Murphy KE, Southan CD, Ryan DM, Smith TS, Simmons DL, Walsh FS, Dingwall C, Christie G. Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol Cell Neurosci. 1999;14:419–427. doi: 10.1006/mcne.1999.0811. [DOI] [PubMed] [Google Scholar]
- Jiao Y, Sun Z, Lee T, Fusco FR, Kimble TD, Meade CA, Cuthbertson S, Reiner A. A simple and sensitive antigen retrieval method for free-floating and slide-mounted tissue sections. J Neurosci Methods. 1999;93:149–162. doi: 10.1016/s0165-0270(99)00142-9. [DOI] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Kitazume S, Tachida Y, Oka R, Shirotani K, Saido TC, Hashimoto Y. Alzheimer's beta-secretase, beta-site amyloid precursor protein-cleaving enzyme, is responsible for cleavage secretion of a Golgi-resident sialyltransferase. Proc Natl Acad Sci USA. 2001;98:13554–13559. doi: 10.1073/pnas.241509198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh YH, von Arnim CA, Hyman BT, Tanzi RE, Tesco G. BACE is degraded via the lysosomal pathway. J Biol Chem. 2005;280:32499–32504. doi: 10.1074/jbc.M506199200. [DOI] [PubMed] [Google Scholar]
- Laird FM, Cai H, Savonenko AV, Farah MH, He K, Melnikova T, Wen H, Chiang HC, Xu G, Koliatsos VE, Borchelt DR, Price DL, Lee HK, Wong PC. BACE1, a major determinant of selective vulnerability of the brain to amyloid-β amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J Neurosci. 2005;25:11693–11709. doi: 10.1523/JNEUROSCI.2766-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammich S, Schobel S, Zimmer AK, Lichtenthaler SF, Haass C. Expression of the Alzheimer protease BACE1 is suppressed via its 5′-untranslated region. EMBO Rep. 2004;5:620–625. doi: 10.1038/sj.embor.7400166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarov O, Lee M, Peterson DA, Sisodia SS. Evidence that synaptically released β-amyloid accumulates as extracellular deposits in the hippocampus of transgenic mice. J Neurosci. 2002;22:9785–9793. doi: 10.1523/JNEUROSCI.22-22-09785.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EB, Zhang B, Liu K, Greenbaum EA, Doms RW, Trojanowski JQ, Lee VM. BACE overexpression alters the subcellular processing of APP and inhibits Abeta deposition in vivo. J Cell Biol. 2005;168:291–302. doi: 10.1083/jcb.200407070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuba G, Wernli G, Vernay A, Kraftsik R, Mohajeri MH, Saini KD. Neuronal and nonneuronal quantitative BACE immunocytochemical expression in the entorhinohippocampal and frontal regions in Alzheimer's disease. Dement Geriatr Cogn Disord. 2005;19:171–183. doi: 10.1159/000083496. [DOI] [PubMed] [Google Scholar]
- Li Q, Sudhof TC. Cleavage of amyloid-beta precursor protein and amyloid-beta precursor-like protein by BACE 1. J Biol Chem. 2004;279:10542–10550. doi: 10.1074/jbc.M310001200. [DOI] [PubMed] [Google Scholar]
- Li R, Lindholm K, Yang LB, Yue X, Citron M, Yan R, Beach T, Sue L, Sabbagh M, Cai H, Wong P, Price D, Shen Y. Amyloid beta peptide load is correlated with increased beta-secretase activity in sporadic Alzheimer's disease patients. Proc Natl Acad Sci USA. 2004;101:3632–3637. doi: 10.1073/pnas.0205689101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenthaler SF, Dominguez DI, Westmeyer GG, Reiss K, Haass C, Saftig P, De Strooper B, Seed B. The cell adhesion protein P-selectin glycoprotein ligand-1 is a substrate for the aspartyl protease BACE1. J Biol Chem. 2003;278:48713–48719. doi: 10.1074/jbc.M303861200. [DOI] [PubMed] [Google Scholar]
- Luo Y, Bolon B, Kahn S, Bennett BD, Babu-Khan S, Denis P, Fan W, Kha H, Zhang J, Gong Y, Martin L, Louis JC, Yan Q, Richards WG, Citron M, Vassar R. Mice deficient in BACE1, the Alzheimer's beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat Neurosci. 2001;4:231–232. doi: 10.1038/85059. [DOI] [PubMed] [Google Scholar]
- Mohajeri MH, Saini KD, Nitsch RM. Transgenic BACE expression in mouse neurons accelerates amyloid plaque pathology. J Neural Transm. 2004;111:413–425. doi: 10.1007/s00702-003-0057-z. [DOI] [PubMed] [Google Scholar]
- Morrison JH, Hof PR. Life and death of neurons in the aging brain. Science. 1997;278:412–419. doi: 10.1126/science.278.5337.412. [DOI] [PubMed] [Google Scholar]
- Mullan M, Crawford F, Axelman K, Houlden H, Lilius L, Winblad B, Lannfelt L. A pathogenic mutation for probable Alzheimer's disease in the APP gene at the N-terminus of beta-amyloid. Nat Genet. 1992;1:345–347. doi: 10.1038/ng0892-345. [DOI] [PubMed] [Google Scholar]
- Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA. 2000;283:1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M, Chang L, Tseng W, Oakley H, Citron M, Klein WL, Vassar R, Disterhoft JF. Temporal memory deficits in Alzheimer's mouse models: rescue by genetic deletion of BACE1. Eur J Neurosci. 2006;23:251–260. doi: 10.1111/j.1460-9568.2005.04551.x. [DOI] [PubMed] [Google Scholar]
- Ozmen L, Woolley M, Albientz A, Miss MT, Nelboeck P, Malherbe P, Czech C, Gruninger-Leitch F, Brockhaus M, Ballard T, Jacobsen H. BACE/APPV717F double-transgenic mice develop cerebral amyloidosis and inflammation. Neurodegener Dis. 2005;2:284–298. doi: 10.1159/000092314. [DOI] [PubMed] [Google Scholar]
- Qing H, Zhou W, Christensen MA, Sun X, Tong Y, Song W. Degradation of BACE by the ubiquitin-proteasome pathway. FASEB J. 2004;18:1571–1573. doi: 10.1096/fj.04-1994fje. [DOI] [PubMed] [Google Scholar]
- Rossner S, Apelt J, Schliebs R, Perez-Polo JR, Bigl V. Neuronal and glial beta-secretase (BACE) protein expression in transgenic Tg2576 mice with amyloid plaque pathology. J Neurosci Res. 2001;64:437–446. doi: 10.1002/jnr.1095. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Toward a comprehensive theory for Alzheimer's disease. Hypothesis: Alzheimer's disease is caused by the cerebral accumulation and cytotoxicity of amyloid beta-protein. Ann NY Acad Sci. 2000;924:17–25. doi: 10.1111/j.1749-6632.2000.tb05554.x. [DOI] [PubMed] [Google Scholar]
- Sheng JG, Price DL, Koliatsos VE. Disruption of corticocortical connections ameliorates amyloid burden in terminal fields in a transgenic model of Aβ amyloidosis. J Neurosci. 2002;22:9794–9799. doi: 10.1523/JNEUROSCI.22-22-09794.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng JG, Price DL, Koliatsos VE. The beta-amyloid-related proteins presenilin 1 and BACE1 are axonally transported to nerve terminals in the brain. Exp Neurol. 2003;184:1053–1057. doi: 10.1016/j.expneurol.2003.08.018. [DOI] [PubMed] [Google Scholar]
- Singer O, Marr RA, Rockenstein E, Crews L, Coufal NG, Gage FH, Verma IM, Masliah E. Targeting BACE1 with siRNAs ameliorates Alzheimer disease neuropathology in a transgenic model. Nat Neurosci. 2005;8:1343–1349. doi: 10.1038/nn1531. [DOI] [PubMed] [Google Scholar]
- Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, Doan M, Dovey HF, Frigon N, Hong J, Jacobson-Croak K, Jewett N, Keim P, Knops J, Lieberburg I, Power M, Tan H, Tatsuno G, Tung J, Schenk D, et al. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature. 1999;402:537–540. doi: 10.1038/990114. [DOI] [PubMed] [Google Scholar]
- Sun A, Koelsch G, Tang J, Bing G. Localization of beta-secretase memapsin 2 in the brain of Alzheimer's patients and normal aged controls. Exp Neurol. 2002;175:10–22. doi: 10.1006/exnr.2002.7875. [DOI] [PubMed] [Google Scholar]
- Tyler SJ, Dawbarn D, Wilcock GK, Allen SJ. Alpha- and beta-secretase: profound changes in Alzheimer's disease. Biochem Biophys Res Commun. 2002;299:373–376. doi: 10.1016/s0006-291x(02)02635-9. [DOI] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, et al. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- Velliquette RA, O'Connor T, Vassar R. Energy inhibition elevates β-secretase levels and activity and is potentially amyloidogenic in APP transgenic mice: possible early events in Alzheimer's disease pathogenesis. J Neurosci. 2005;25:10874–10883. doi: 10.1523/JNEUROSCI.2350-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Arnim CA, Kinoshita A, Peltan ID, Tangredi MM, Herl L, Lee BM, Spoelgen R, Hshieh TT, Ranganathan S, Battey FD, Liu CX, Bacskai BJ, Sever S, Irizarry MC, Strickland DK, Hyman BT. The low density lipoprotein receptor-related protein (LRP) is a novel beta-secretase (BACE1) substrate. J Biol Chem. 2005;280:17777–17785. doi: 10.1074/jbc.M414248200. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Shankar GM, Townsend M, Fadeeva JV, Betts V, Podlisny MB, Cleary JP, Ashe KH, Rowan MJ, Selkoe DJ. The role of cell-derived oligomers of Abeta in Alzheimer's disease and avenues for therapeutic intervention. Biochem Soc Trans. 2005;33:1087–1090. doi: 10.1042/BST20051087. [DOI] [PubMed] [Google Scholar]
- Willem M, Dewachter I, Smyth N, Van Dooren T, Borghgraef P, Haass C, Van Leuven F. Beta-site amyloid precursor protein cleaving enzyme 1 increases amyloid deposition in brain parenchyma but reduces cerebrovascular amyloid angiopathy in aging BACE × APP[V717I] double-transgenic mice. Am J Pathol. 2004;165:1621–1631. doi: 10.1016/s0002-9440(10)63419-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong HK, Sakurai T, Oyama F, Kaneko K, Wada K, Miyazaki H, Kurosawa M, De Strooper B, Saftig P, Nukina N. beta Subunits of voltage-gated sodium channels are novel substrates of beta-site amyloid precursor protein-cleaving enzyme (BACE1) and gamma-secretase. J Biol Chem. 2005;280:23009–23017. doi: 10.1074/jbc.M414648200. [DOI] [PubMed] [Google Scholar]
- Yan R, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, Brashier JR, Stratman NC, Mathews WR, Buhl AE, Carter DB, Tomasselli AG, Parodi LA, Heinrikson RL, Gurney ME. Membrane-anchored aspartyl protease with Alzheimer's disease beta-secretase activity. Nature. 1999;402:533–537. doi: 10.1038/990107. [DOI] [PubMed] [Google Scholar]
- Yang LB, Lindholm K, Yan R, Citron M, Xia W, Yang XL, Beach T, Sue L, Wong P, Price D, Li R, Shen Y. Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med. 2003;9:3–4. doi: 10.1038/nm0103-3. [DOI] [PubMed] [Google Scholar]